
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling conjugated polymer morphology by precise oxygen position in single-ether side chains†
Pablo
Durand
 a,
Huiyan
Zeng
b,
Badr
Jismy
a,
Olivier
Boyron
c,
Benoît
Heinrich
d,
Laurent
Herrmann
b,
Olivier
Bardagot
*a,
Ioannis
Moutsios
e,
Alina V.
Mariasevskaia
f,
Alexey P.
Melnikov
f,
Dimitri A.
Ivanov
ef,
Martin
Brinkmann
*b and
Nicolas
Leclerc
*a
a,
Huiyan
Zeng
b,
Badr
Jismy
a,
Olivier
Boyron
c,
Benoît
Heinrich
d,
Laurent
Herrmann
b,
Olivier
Bardagot
*a,
Ioannis
Moutsios
e,
Alina V.
Mariasevskaia
f,
Alexey P.
Melnikov
f,
Dimitri A.
Ivanov
ef,
Martin
Brinkmann
*b and
Nicolas
Leclerc
*a
aUniversité de Strasbourg, CNRS, ICPEES UMR 7515, 67087 Strasbourg, France. E-mail: olivier.bardagot@cnrs.fr; leclercn@unistra.fr
bUniversité de Strasbourg, CNRS, ICS UPR 22, 67000 Strasbourg, France. E-mail: martin.brinkmann@ics-cnrs.unistra.fr
cUniversité de Lyon, CNRS, Laboratoire CP2M, UMR 5128, 69100 Villeurbanne, France
dUniversité de Strasbourg, CNRS, IPCMS UMR 7504, F-67034 Strasbourg, France
eUniversité de Mulhouse, CNRS, IS2M, UMR 7361, 15 Jean Starcky, Mulhouse 68057, France
fFaculty of Chemistry, Lomonosov Moscow State University, GSP-1, 1-3 Leninskiye Gory, 119991 Moscow, Russia
First published on 10th July 2024
Abstract
Recently, polar side chains have emerged as a functional tool to enhance conjugated polymer doping properties by improving the polymer miscibility with polar chemical dopants and facilitate solvated ion uptake. In this work, we design and investigate a novel family of side chains containing a single ether function, enabling the modulation of the oxygen atom position along the side chain. A meticulous investigation of this new polymer series by differential scanning calorimetry, fast scanning chip calorimetry and X-ray scattering shows that polymers bearing single-ether side chains can show high degree of crystallinity under proper conditions. Importantly, due to a gauche effect allowing the side chain to bend at the oxygen atom, the degree of crystallinity of polymers can be controlled by the position of the oxygen atom along the side chain. The further the oxygen atom is from the conjugated backbone, the more crystalline the polymer becomes. In addition, for all new polymers, high thermomechanical properties are demonstrated, leading to remarkable electrical conductivities and thermoelectric power factors in rub-aligned and sequentially doped thin films. This work confirms the potential of single-ether side chains to be used as polar solubilizing side chains for the design of a next generation of p- and n-type semiconducting polymers with increased affinity to polar dopants while maintaining high molecular order.
New conceptsThe development of semiconducting polymers with polar side chains is gaining considerable momentum, driven by thermoelectric, storage and bioelectronic applications. To date, most molecular designs are focusing on oligo(ethylene glycol) (OEG) side chains, containing multiple oxygen atoms. While OEG side chains effectively promote dopant/ion uptake, they also suffer from a low crystallinity, which limits the ordering of the polymer and hence its charge transport properties. Recently, single-ether side chains, containing a single oxygen atom, have emerged as an alternative. They offer a compromise between polarity, to promote doping, and self-assembly order, to promote transport properties. In this work, we provide a full understanding of the impact of the position of the oxygen atom along the side chain on the morphological and transport properties of novel high-performance PBTTT polymers. By using nanocalorimetric measurements coupled with X-ray scattering, we show that single-ether side chains are crystalline and, more importantly, that the degree of morphological order of the polymers can be controlled by varying the position of the oxygen atom (crystallinity index). This work showcases a new design tool to guide chemists in the development of high-performance doped semiconductors and organic mixed ionic-electronic conductors with optimal balance between polymer polarity and molecular order. |
Introduction
Organic conjugated polymers (CPs) are intriguing but complex synthetic materials. Their highly desirable mechanical and optoelectronic properties, resulting from the combination of both polymeric and specific organic functionalities, make them highly attractive candidates for application in energy and healthcare devices.1–5 CPs are composed of conjugated units which govern most of their intrinsic optoelectronic properties. However, their final performance in devices are also highly dependent on their processability as well as solid-state self-assembly, which vary significantly with the nature of the solubilizing side chains.6–10Recently, polar side chains have emerged as a functional tool to enhance CP doping properties by improving the polymer miscibility with polar chemical dopants (such as TCNQ derivatives) and facilitate ionic uptake,10 which considerably improved performance in organic electrochemical transistors (OECT),11 water splitting photocatalysis,12,13 and organic thermoelectric (OTE) devices.14 Polar side chains are most often based on the use of short chains of the oligo(ethylene glycol) (OEG) type, known for their hydrophilicity. Numerous variations of these OEG side chains have been proposed, including tuning their length (number of ethylene glycol repeating units),15 their position,16 and the introduction of spacers.17,18 Although, significant progress in functional devices have been achieved using CPs carrying OEG side chains, such side chains are known to be much less crystalline and ordered than the corresponding alkyl counterparts.19–22 As a result, they lead to less ordered CP structures in the solid state and reduced charge transport properties.17 Recently, the Luscombe's team proposed to reduce the number of oxygen atoms in order to find a compromise between polymer self-assembly order and polarity for application in OECT.23,24
In this context, simultaneously to Luscombe's work, we demonstrated the beneficial use of a single ether-based side chain as solubilizing polar substituents for the standard high-mobility PBTTT polymer (named PBTTT-8O).25 In contrast to the usual OEG side chains,26 we have shown that the introduction of single-ether side chains improves the affinity between polymer and dopant, without disrupting the beneficial lamellar order of PBTTT.27 Furthermore, enhanced thermomechanical properties are observed for PBTTT-8O compared to PBTTT-C12, the reference polymer bearing apolar dodecyl side chains. As a result, polymers with single-ether side chains can be effectively processed from solution and subjected to mechanical high-temperature rubbing (HTR) to produce highly oriented thin films. Notably, much higher CP orientation and lamellar order have been obtained compared to PBTTT-C12, assigned to stronger cohesive forces between oxygens from the ether functions and neighboring CH2 units within the side chain sublayers (dichroic ratio of 20 for PBTTT-8O vs. 14 for PBTTT-C12).25 In addition, this single ether-based design proved particularly relevant for inducing a polar dopant pinpoint close to the ether function. Consequently, such highly ordered and uniaxially aligned doped-CPs lead to a metallic-like electrical conductivity in the 1–5 × 104 S cm−1 range. This is particularly relevant for OTE applications as the dominant strategy to enhance the CP performances is based on the improvement of the electrical conductivity that is intimately related to the doping process.25,28
Importantly, a specific advantage of the single ether-based design is the ability to adjust the position of the oxygen atom along the linear side chain. In particular, one can vary the distance between the polar ether function and the conjugated backbone at will. By doing so, one can finely tune the order/disorder ratio of the side chains and ultimately control the CP degree of crystallinity, self-assembly and electronic properties in the solid state.
In this contribution, we report the synthesis and characterization of a new series of single-ether side chain PBTTTs with a variable position ‘x’ of the oxygen atom along the side chain from the backbone (PBTTT-xO, x = 3, 5, 8, 11) (Scheme 1). We locate the ether function at a range of positions that do not impact the energy levels of the polymer backbone. We hence specifically avoid the first position, which would result in an alkoxy side chain with a donor mesomeric effect on the backbone. Note that we use a constant side chain length of 12 atoms to allow relevant comparisons to the reference alkyl PBTTT-C12 polymer.
 | ||
| Scheme 1 General synthetic route towards the ether-based PBTTT-xO. In inset, PBTTT-C12 structure. | ||
We study the influence of the oxygen position along the side chain, by evaluating the thermophysical properties, the structural properties before and after HTR and upon chemical doping. By way of possible application, the evolution of the thermoelectric performance of the doped films are finally investigated. Using a combination of characterization tools, including differential scanning calorimetry (DSC), fast scanning chip calorimetry (FSC also called nanocalorimetry), X-ray diffraction, transmission electron microscopy (TEM) and UV-Vis-NIR spectroscopy, we demonstrate that regardless of the oxygen position, all polymers display lamellar ordering. However, the polymorphism and semi-crystallinity of the polymers (including side chain ordering) are strongly determined by the position of the ether function. Finally, when chemically doped, an optimal oxygen position between the fifth and eighth atomic position in the side chain appears to maximize the TE performance for the PBTTT-xO series. Despite being remarkably high, we would like to stress that the main finding of this study is not the device performance, but the report of a full understanding of how single-ether side chains can be synthetized, how they impact the structure of the polymers, and ultimately how the charge properties are impacted upon doping.
Results and discussion
Polymers synthesis and characterizations
In order to synthesize the new polymers of this series, we used the synthesis sequence previously described for PBTTT-8O (Scheme 1).25 As a reminder, the chemical route consists of a first Sonogashira coupling followed by mild reduction to give the corresponding thienyl alcohol. Then, a bromination with NBS gives the 2-bromothiophene intermediate which is homocoupled by palladium catalyzed CH activation to form the dihydroxy bithiophene. This dihydroxy bithiophene is cautiously recrystallized to get rid of unwanted stereoisomers (Head-to-Tail and Tail-to-Tail). A final Williamson reaction give the ether functionalized bithiophene with high purity. All 5,5′-dibromo-2,2′-bithiophenes are then copolymerized with the 2,5-bis(trimethylstannyl)thieno[3,2-b]thiophene comonomer by Stille cross-coupling to afford the desired PBTTT-xO polymers.The polymer series was characterized by size exclusion chromatography in trichlorobenzene at 150 °C and NMR spectroscopy (Fig. S1–S18, ESI†). The reference PBTTT-C12, PBTTT-11O and PBTTT-8O show similar Mn molar masses of around 30 kg mol−1, while PBTTT-5O and PBTTT-3O show slightly higher molar masses of ∼39 and ∼50 kg mol−1, respectively (Fig. S19, ESI,† and Table 1). Note that all polymers display molar masses in the range previously considered as optimal for high charge carrier mobilities (30–50 kg mol−1) in PBTTT,29 allowing for fair comparisons.
| M n (kg mol−1) | Đ |
T
d
5%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (°C)
(°C) |
IPPESA (eV) | ΔEoptgc (eV) |
|
|---|---|---|---|---|---|
| a Measured by high-temperature SEC. b Measured by TGA. c Measured from thin-film absorbance. | |||||
| PBTTT-C12 | 29.6 | 1.8 | 386 | 4.8 | 1.87 |
| PBTTT-11O | 30.0 | 1.8 | 387 | 4.8 | 1.91 |
| PBTTT-8O | 30.2 | 1.8 | 376 | 4.8 | 1.91 |
| PBTTT-5O | 38.8 | 1.6 | 371 | 4.8 | 1.91 |
| PBTTT-3O | 49.5 | 1.4 | 353 | 4.8 | 1.97 |
Chemical stabilities were characterized by thermogravimetric analysis (TGA, Fig. S20, ESI† and Table 1). The resulting thermograms show a first interesting result: the oxygen position along the side chain impacts the polymer degradation temperature Td. Indeed, if we consider the PBTTT-C12 as reference material, one can see a clear trend that the closer the oxygen is to the conjugated backbone, the lower the degradation temperature, decreasing stepwise from 386 °C for PBTTT-11O down to 353 °C for PBTTT-3O. However, even the PBTTT-3O, with a Td of 353 °C remains more stable than OEG side chains based PBTTT.16
Cyclic voltammetry (CV) and photoelectron spectroscopy in air (PESA) were employed to gain insights into the highest occupied molecular orbital (HOMO) energy levels (Fig. S21 and S22, ESI†). As expected, by PESA on thin films, all these single ether PBTTTs display the same HOMO energy levels at −4.8 eV, identical to the reference PBTTT-C12. This result confirms that the ether functions are far enough from the backbones to not impact the energetic levels of the polymers. All polymers are therefore expected to be easily doped with F6TCNNQ.
Optical absorbance properties are studied in chlorobenzene solutions and in thin films (Fig. S23, ESI†). Solution UV-visible absorbance spectra exhibit almost identical Gaussian-like line shapes with a maximum at ∼470 nm for all polymers, suggesting appropriate solubility in chlorobenzene. They also show similar optical bandgap of about 1.9 eV, in accordance with identical HOMO levels found in PESA. Similarly, high similarities are found in solid-state absorbance spectra.
Structure and self-assembly
The electronic and thermal performance of CP thin films are vastly driven by morphology and structure. To better rationalize the performance presented in the next section, the melting and crystallization properties ruling the solid-state morphology of each polymer are assessed by DSC (Fig. 1(A)).30 To highlight the impact of the oxygen position, the thermo-structural properties of PBTTT-xO polymers are compared to reference PBTTT-C12. In the solid state, PBTTT-C12 is known to exhibit polymorphism with crystalline phases (Crys) and smectic liquid-crystal phases (Lam).31 Upon heating, first the side chains melt at around 140 °C (1st endothermic peak, crystal melting, Crys–Lam transition) followed by the thiophene backbone at around 230 °C (2nd peak, isotropization, Lam–Iso transition). Upon cooling the DSC curve shows two exotherms corresponding to the liquid-crystal phase formation and crystallization, respectively. PBTTTs with alkyl side chains, including PBTTT-C12, are considered as crystalline below the melting temperature of side chains and liquid-crystalline above that temperature and up to the isotropic transition at around 230 °C.32 At high temperatures, associated to the backbone behavior (220–250 °C), all PBTTT-xO polymers exhibit an endothermic and an exothermic peak, like PBTTT-C12. We can therefore first deduce that the introduction of the ether function preserves the ordering of the π-conjugated backbone. Note that this result highlights one benefit of single-ether side chains compared to OEG side chains, this latter being reported as tending to reduce macromolecular order.16,17 At low temperature ranges, corresponding to the side-chain ordering (100–150 °C), the PBTTT-xO series can be divided in two categories. PBTTT-11O behaves as PBTTT-C12, with endo/exothermic peaks of similar intensities and at similar temperatures, indicating alike melting and crystallization behaviors of the -11O and -C12 side chains. When the oxygen is positioned closer to the backbone, such as for PBTTT-8O, -5O, and -3O, no endothermic or exothermic peak could be detected by conventional DSC. This observation suggests that the distance of oxygen atom to the backbone is a key parameter controlling the side-chain ordering. However, interpretation of conventional DSC curves performed at slow scan speed (5 °C min−1 ≃0.08 °C s−1) can be difficult if thermal transitions occur at fast kinetics. Therefore, using fast scanning rates is beneficial for DSC measurements as it allows to kinetically bypass these processes. Thus, to probe the behavior of -8O, -5O, and -3O side chains, FSC with cooling and heating ramps on the order of 1000 °C s−1 has been applied (Fig. 1(B) and Table 2).33–37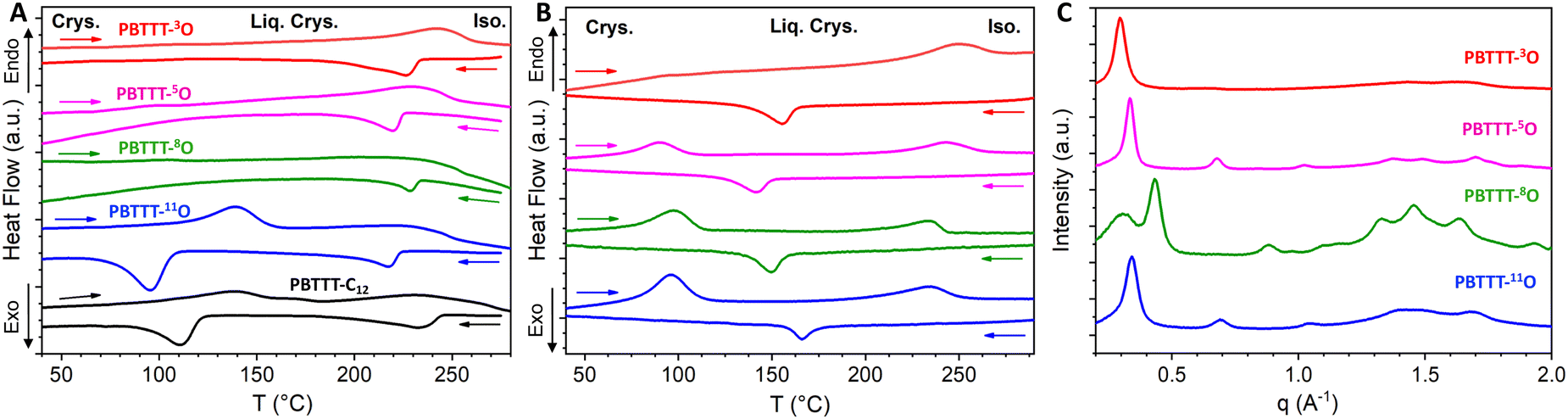 | ||
| Fig. 1 Color code: PBTTT-C12, PBTTT-11O, PBTTT-8O, PBTTT-5O, PBTTT-3O, respectively; (A) conventional DSC curves recorded at 5 °C min−1 (≃0.08 °C s−1); (B) fast scanning chip calorimetry curves recorded during heating at 1000 °C s−1 for samples crystallized at 40 °C and during cooling at 1000 °C s−1 from the isotropic melt for pristine samples; (C) X-ray micro-focus scattering patterns measured on a few nanogram-size samples after 2 days annealing at 40 °C for PBTTT-11O, PBTTT-8O and 5 days annealing at 40 °C PBTTT-5O, PBTTT-3O. | ||
| DSC | FSC | C (%) | |||
|---|---|---|---|---|---|
| Heating Tonset (Tmax) (°C) | Cooling Tonset (Tmax) (°C) | Heating Tonset (Tmax) (°C) | Cooling Tonset (Tmax) (°C) | ||
| a C is the crystallinity index as measured by decomposition of the X-ray scattering profiles in a sum of crystalline peak intensities and amorphous halo. | |||||
| PBTTT-C12 | Crys–Lam 103 (139) | Iso–Lam 244 (232) | — | — | — |
| Lam–Iso 190 (234) | Lam–Crys 121 (112) | ||||
| PBTTT-11O | Crys–Lam 113 (139) | Iso–Lam 244 (232) | Crys–Lam 75 (92) | Iso–Lam 178 (166.0) | 53 |
| Lam–Iso 202 (231) | Lam–Crys 121 (112) | Lam–Iso 215 (237) | |||
| PBTTT-8O | Crys–Lam 89 | Iso–Lam 233 (228) | Crys–Lam 71 (96) | Iso–Lam 158 (150) | 56 |
| Lam–Iso 225 (241) | Lam–Iso 214 (241) | ||||
| PBTTT-5O | Crys–Lam 83 | Iso–Lam 225 (220) | Crys–Lam 63 (89) | Iso–Lam 153 (146) | 37 |
| Lam–Iso 206 (232) | Lam–Iso 216 (245) | ||||
| PBTTT-3O | Crys–Lam 86 | Iso–Lam 233 (226) | Crys–Lam 73 (95) | Iso–Lam 164 (155) | 26 |
| Lam–Iso 212 (244) | Lam–Iso 216 (250) | ||||
Fig. 1(B) shows PBTTT-xO nanocalorimetric curves recorded at about 1000 °C s−1. As for conventional DSC, the formation of the liquid-crystal phase, originating from the backbone crystallization (Iso–Lam transition) is observed during fast cooling for all PBTTT-xO polymers. No specific trend with the oxygen position is found. However, the onset temperatures of the exothermic peak are shifted from 220–250 °C for conventional DSC, down to 165–175 °C for FSC, suggesting a higher degree of supercooling of the Iso–Lam transition with faster cooling rate. More importantly, regardless of the oxygen position, none of the samples exhibits formation of the crystalline phase from the mesophase upon cooling (Fig. S24, ESI†) indicating that side chains have difficulties to order at fast scanning rates. Therefore, prior to the FSC heating scan, the samples were subjected to isothermal crystallization to promote side chain ordering. A crystallization temperature of 40 °C was selected to be between the glass transition temperature (from −30 °C to −20 °C for PBTTT-xO as seen in Fig. S24 (ESI†) and reported at −80 °C for PBTTT-C12)38 and the onset of side-chain melting (from +80 °C to +140 °C) was selected to mostly enhance the side-chain crystallinity (and not the backbone).39 The annealing time was adapted for each polymer. Upon fast heating at 1000 °C s−1, all isothermally crystallized polymers show two endothermic peaks (Fig. 1(B)). The low-temperature transition being assigned to side chain melting.30,40 Interestingly, both the transition temperature and the enthalpy of the peaks vary with the distance between the oxygen and the backbone. To take things a step further, we used nanocalorimetric analysis coupled with small- and wide-angle X-ray scattering (SAXS-WAXS) to evaluate a crystallinity index (C) for each polymer. Synchrotron micro-focus SAXS and WAXS experiments were conducted in situ on nanocalorimetric sensors using a custom-built device described previously.33,41
The X-ray scattering patterns of all PBTTT-xO isothermally crystallized samples are given in Fig. 1(C). All polymers exhibit the most intense peak at 18.5–21.5 Å, attributed to periodic inter-lamellar distance. Among the series, PBTTT-8O stands out clearly as it shows polymorphism with coexistence of the crystalline phase with d100 = 14.6 Å and the liquid crystalline phase with d100 = 19.5 Å. To evaluate the crystallinity index C, the scattering profiles are decomposed in a sum of crystalline peak intensities and amorphous halo as shown in Fig. S25 in ESI.† The crystallinity index is then computed as the ratio of all crystalline peak intensities over the total scattering intensity. The shape of the amorphous halo is measured for each sample above the isotropization temperature (230–250 °C). The obtained values are the following: 26% < 37% < 56% ≃53% for PBTTT-3O/5O/8O/11O, respectively. Remarkably, a clear trend of increasing crystallinity index with the oxygen distance from the backbone is found. The further the ether function is from the backbone, the higher the crystallinity of polymers. In other words, PBTTT-11O and PBTTT-8O are more crystalline than PBTTT-5O, while PBTTT-3O is the least crystalline. We believe that this phenomenon originates from hyperconjugation effects (also known as the gauche effect) which tend to bend the side chains due to the synclinal conformation of the C–C alpha-beta bond next to the ether function.26,42–44 The closer the oxygen atom to the conjugated backbone, the longer the alkyl segment that adopts a non-straight conformation (not antiperiplanar), resulting in a decrease in side-chain order and overall macromolecular crystallinity (see Fig. 3(A)).
In a nutshell, the polymer morphology, and in particular the semi-crystalline/amorphous domain ratio, can be finely controlled by the position of the oxygen along single-ether side chains. Single-ether side chains therefore represent a tunable trade-off between the polarity of OEG side chains and the crystallinity of alkyl side chains. The impacts of these balanced properties on the polymer propension to align upon mechanical rubbing and to chemically dope is presented in the following section in views of applications of PBTTT-xO as TE materials.
Thin film alignment
We have previously shown that HTR is a very effective method to orient CP thin films.45 In particular, with an adequate rubbing temperature TR, PBTTT films of remarkable crystallinity and uniaxial alignment can be generated.46 Polarized UV-Vis spectroscopy is used to evaluate the alignment quality, in particular through the calculation of the dichroic ratio (DR, defined as the absorbance in parallel divided by the absorbance in perpendicular at the wavelength corresponding to the maximum λmax) and the 3D order parameter (OP = (DR − 1)/(DR + 2)). Alignment conditions were first optimized as function of TR (Fig. 2(A) and Fig. S26, ESI†). A previous study has shown that the highest alignment of PBTTT-C12 is reached for TR = 125 °C, just below the side-chain melting temperature (140 °C), for PBTTT-C12, giving DR and OP of around 14 and 0.80, respectively.25 Above this temperature, films of PBTTT-C12 were found to delaminate from the substrate upon rubbing. In stark contrast, it is possible to align all the PBTTT-xO polymers with rubbing temperatures up to TR = 240 °C (around the isotropization temperatures) while retaining film integrity. We speculate that the enhanced thermo-mechanical stability of PBTTT-xO polymers results from a higher lamellar cohesion thanks to dipolar inter-chain interactions between ether-based side chains. Interestingly, the higher optimized rubbing temperatures systematically lead to higher DR and OP compared to PBTTT-C12, synonymous of higher polymer chain alignment. Indeed, the best alignment is achieved for PBTTT-11O (DR = 19, OP = 0.86), followed by PBTTT-8O/5O (DR = 18/17 and OP = 0.85/0.84) and finally PBTTT-3O (DR = 15, OP = 0.83), still higher than PBTTT-C12. The variation of DR with TR reveals two remarkable trends. Firstly, the closer the ether function to the backbone, the lower the maximum DR. Secondly, the closer the ether function to the backbone, the lower the crystalline index, the broader the range of rubbing temperatures leading to high DR offering a larger tolerance during the film processing (Fig. S26B, ESI†). In the following, to evaluate the thermoelectric properties of rub-aligned and doped films, the different polymers were rubbed at temperatures corresponding to their maximum alignment (DR), i.e. 160 °C and 170 °C for PBTTT-3O and PBTTT-8O, respectively, while PBTTT-5O and PBTTT-11O, are both rubbed at 200 °C.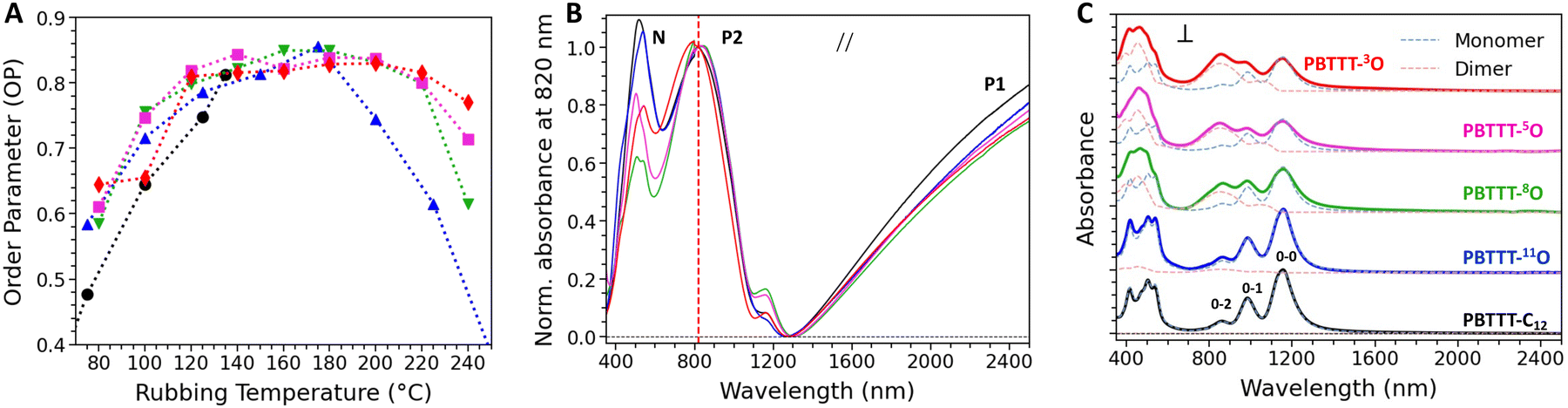 | ||
| Fig. 2 (A) Plot of the order parameter for each polymer as function of the rubbing temperature. (B) and (C) Polarized UV-vis-NIR spectra of oriented thin films of PBTTT-C12, PBTTT-11O, PBTTT-8O, PBTTT-5O, PBTTT-3O, respectively, doped with F6TCNNQ in acetonitrile ([F6TCNNQ] = 2 mg mL−1); (B) the light is polarized parallel to the rubbing direction. The main polaronic bands (P1 and P2) and the neutral band (N) are assigned. All spectra are normalized by the maximum absorbance of the P2 band at 830 nm, highlighting the variation of the P2/N ratio and hence of the doping extent; (C) the light is polarized perpendicular to the rubbing direction. All spectra are stacked to highlight the change in monomer/dimer dopant ratio with the oxygen position. Solid lines = experimental data. Dashed lines = Relative absorbance of F6TCNNQ˙− monomer (blue) and (F6TCNNQ˙−)2 dimer (pink) found by MCR deconvolution. Details on MCR fitting in Fig. S29 in (ESI†). | ||
Polymer order and chemical doping
The possibility to achieve very high alignment and to tune the crystallinity index in the PBTTT-xO series by changing the position of the oxygen along the side chain is used as an opportunity to probe the influence on the charge transport and TE properties of doped polymer films oriented by HTR. The open question was to know what is the optimal distance of the oxygen from the conjugated backbone to achieve the best TE power factors. The films are doped using the incremental concentration doping (ICD) method, which helps preserving the pristine structure of the polymer thin film by progressively inserting dopant molecules in the polymer matrix (see ESI†). In case of oriented thin films, ICD allows to retain a very high crystallinity and alignment quality after doping (Fig. S27, S28 and S30, ESI†).47The doping extent of the oriented PBTTT thin films and the localization of F6TCNNQ dopants are studied by polarized UV-vis-NIR absorbance spectroscopy. The Fig. 2(B) and (C) display the UV-vis-NIR absorbance spectra of oriented thin films of all polymers doped with F6TCNNQ at a concentration of 2 mg mL−1 for a light polarized parallel (B) and perpendicular (C) to the rubbing direction. All UV-vis-NIR absorbance spectra recorded for lower dopant concentrations are reported in EI† (Fig. S27).
The doping of all PBTTT-C12 and PBTTT-xO polymers with F6TCNNQ is efficient, as evidenced by a strong bleaching of the neutral absorption band in the visible domain inducing the generation of two polaronic bands (P1 and P2) mainly in the NIR domain (Fig. 2(B)). The polaronic bands are polarized in the chain direction (rubbing direction) and their absorbance increases with dopant concentration, reflecting the gradual increase of charge carrier density in the polymers (Fig. S28, ESI†). On the other hand, when the light polarization is perpendicular to the chain direction, the spectrum is dominated by the signature of the radical anion F6TCNNQ˙− (Fig. 2(C)). In agreement with previous studies, this indicates that a large amount of dopant molecules are located inside the side chain layers and preferentially oriented with the long molecular axis perpendicular to the PBTTT backbone, regardless of the nature of the side chains.46,48 Nonetheless, two categories of polymers can be distinguished, just as evidenced by DSC studies. Indeed, both the polaronic absorbance signatures of P1 and P2 bands as well as the signature of F6TCNNQ˙− radical anions are similar for PBTTT-C12 and PBTTT-11O (Fig. 2(B) and (C)). This means that despite the presence of the ether function in the side chain, the intercalation of F6TCNNQ inside the side chain layers is close to that observed for PBTTT-C12 (the long axis of the dopant is oriented in a plane perpendicular to the PBTTT backbone).
For PBTTT-8O/5O/3O, the scenario differs. In particular, the line shape of the absorbance assigned to the reduced dopants is more complex. As one can notice in the spectrum recorded with the light polarization perpendicular to the rubbing direction, the 0–0 band at ∼1150 nm decreases while the 0–2 band at ∼850 nm apparently increases (Fig. 2(C)). A spectral deconvolution based on the multivariate curve resolution (MCR) analysis indicates that the vibronic signature of the F6TCNNQ˙− radical anion overlaps with at least one additional signature with a characteristic band centered at ∼850 nm (Fig. S29, ESI†). MCR analysis of the spectra at 2 mg mL−1 allows to isolate two distinct species, exhibiting high similitudes with the experimental spectra reported in literature for monomeric TCNQ˙− anion and dimeric (TCNQ˙−)2, respectively.49 We therefore conclude that both monomers and (F6TCNNQ˙−)2 dimers (or clusters) of ionized dopants are formed when PBTTT-8O/5O/3O are doped. In addition, the ratio of monomer/dimer oriented perpendicular to the polymer backbone scales with the position of the oxygen along the side chain. As shown in Fig. S29 in ESI,† an equilibrated proportion of monomers and dimers (52%/48%) is found for PBTTT-8O, while the fraction of dimers increases to ∼60% for PBTTT-5O, and ∼65% for PBTTT-3O. In other words, the closer the ether function to the backbone, the lower the crystallinity index, the higher the dimer content. A possible explanation could be related to more spatial freedom for the dopant to dimerize due to the disorder induced by the gauche effect, previously described, within the side chain sublayer (Fig. 3(A)).
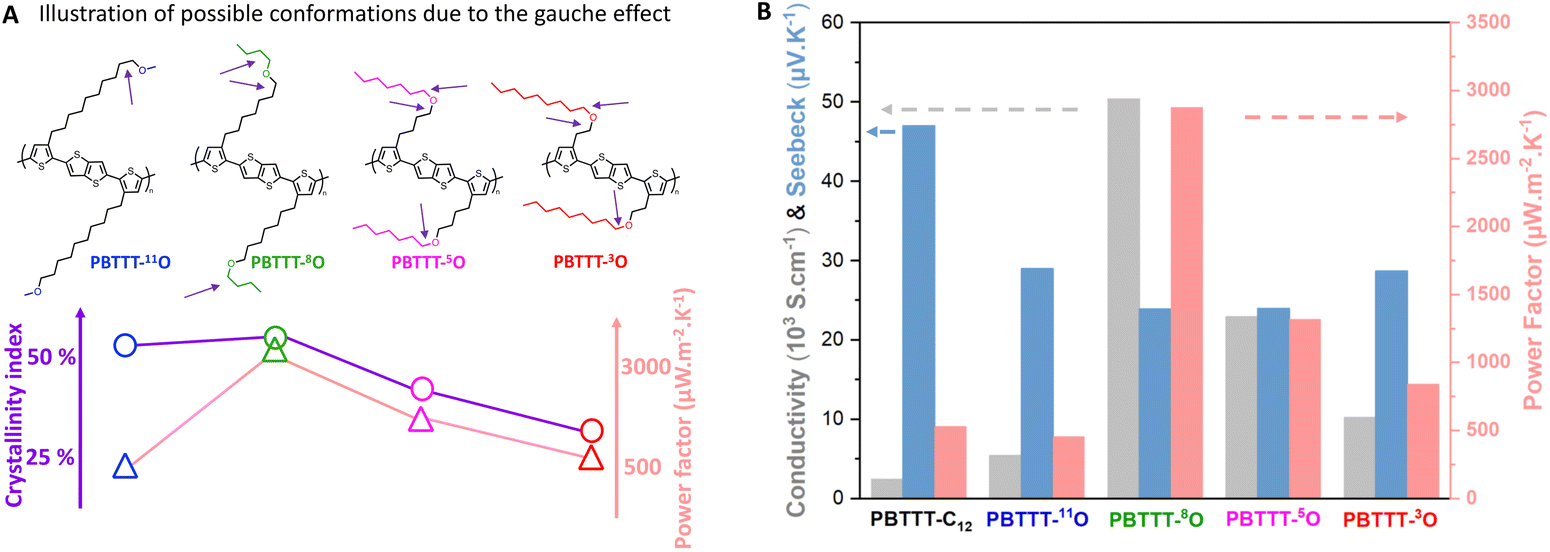 | ||
| Fig. 3 (A) Illustration of possible conformations related to the gauche effect, showing the evolution of the disorder associated with the position of the oxygen along the side chain. The arrows indicate the gauche effects represented on each side chain. Evolution of crystallinity index and power factors are illustrated as function of the position of the oxygen along the side chain (B) TE properties of doped polymer thin-films ([F6TCNNQ] = 5 mg mL−1) measured in parallel to the rubbing direction (color code: electrical conductivity, Seebeck coefficient and power factor). | ||
The impact of the oxygen position, and therefore the crystallinity index, on the doping level can be qualitatively estimate by calculating the ratio between the neutral band intensity (at 510 nm) and the P1 and P2 polaronic band intensities (820 and 2450 nm, respectively) in the UV-Vis-NIR absorbance spectra with light polarized parallel to the rubbing direction (Fig. S28, ESI†). Single ether-based side chains systematically lead to a higher bleaching of the neutral band, indicating a higher overall oxidation of the PBTTT-backbone compared to PBTTT-C12, thereby confirming the expected higher dopant/polymer miscibility offered by the ether function, as known for polar OEG side chains.10 The facilitated dopant insertion for PBTTT-xO polymers is also highlighted by a more pronounced doping level at lower dopant concentration (Fig. S28, ESI†). Again, a trend in doping level with the oxygen position is found. But in this case, an optimum is observed with the doping level being maximal for PBTTT-8O. The doping level of PBTTT-5O/-3O are similar and remain higher than for PBTTT-11O/-C12.
The influence of the ether position in the side chain on doping is also manifested in the variation of the unit cell parameters versus doping concentration (Fig. S31 and S32, ESI†). In our previous work, we demonstrated that doping PBTTT-8O with F6TCNNQ led to very peculiar structural change different from PBTTT-C12. Instead of a continuous expansion of the lattice in the side chain direction (d100) and a contraction along the π-stacking direction (d020) with increasing doping concentration, the lattice of PBTTT-8O first expands for F6TCNNQ ≤ 0.1 g L−1 and contracts back to a value d100 close to the original undoped lattice when doping up to 2 g L−1. This behavior was associated to the dimer formation and/or the rejection of dopants to amorphous zones.25 Interestingly, the same type of behavior is seen for PBTTT-5O whereas all other polymers show the classical continuous expansion of d100 and contraction of d020 with increasing dopant concentration.50–52 Overall, these results underlines again the major impact of the structure of side-chain layers on the doping of the polymers: side chain layers control orientation and clustering of ionized dopant molecules as well as maximum amount of intercalated dopants. In this perspective, the position of the ether in the side chain plays a key role.
Thermoelectric properties
The reduced lattice modification observed upon doping of PBTTT-5O and PBTTT-8O is interesting as these two polymers show incidentally the best TE performances. In strong contrast, the poorest transport properties are found for the polymers PBTTT-11O and PBTTT-3O featuring the largest lattice expansion upon doping (Δd100 variation of almost 1 Å and 2 Å, respectively). In other words, doping PBTTT with F6TCNNQ while maintaining the original lattice seems the optimal doping condition to obtain the best TE properties in the PBTTT-xO series. Note that a similar trend was already evidenced for the case of P3HT when doped with magic blue as the dopant molecules locate essentially in the amorphous phase of the polymer.53Fig. 3(B) reports the TE parameters (charge conductivity, Seebeck coefficient and thermoelectric power factor) along the alignment direction measured for the thin films doped with F6TCNNQ at a concentration of 5 mg mL−1. Complete data for all investigated concentrations along and perpendicular to the alignment direction can be found in ESI† (Fig. S33–S35).
In our previous article, we have shown a remarkable improvement of TE properties for PBTTT-8O over PBTTT-C12, with a near 20-fold increase in electrical conductivity (σ reaching about 5 × 104 S cm−1) and a 6-fold increase in power factor (PF of almost 3000 μW m−2 K−1). This trend applies to the whole single-ether series since, compared to PBTTT-C12, all the PBTTT-xO polymers show significant improvements in electrical conductivities (Fig. 3(B)). For PBTTT-5O, the conductivity achieved at high doping concentration (5 mg mL−1) along the alignment direction is up to 2 × 104 S cm−1 (a 10-fold improvement compared to PBTTT-C12). For PBTTT-11O and PBTTT-3O, the conductivity improvement is less significant but still reaching conductivities as high as 104 S cm−1 and 5000 S cm−1, respectively. It is worth noting here that the best thermoelectric performances do not correlate with the maximum crystallinity index, the maximum dichroic ratio or the largest amount of monomeric dopant. For instance, the most crystalline and aligned polymer PBTTT-11O (high crystallinity and high DR/OP) performs less than PBTTT-3O (lower crystallinity, high dimeric dopant content) which in turns performs less than PBTTT-5O and PBTTT-8O. These observations highlight the complex interplay between morphology, doping process, thin film processing and resulting charge transport properties. Notably, the two polymers showing the highest conductivities are PBTTT-5O and PBTTT-8O, which display high to moderate crystallinity (37% and 56%) and balance amount of dimeric/monomeric dopants (60%/40% and 52%/48%, respectively). The common point of these polymers is that both have ether functions located around the center of the side chains.
Generally, the TE properties probed in the rub-aligned PBTTT-xO series confirm previous findings that alignment is able to increase both electrical conductivity and Seebeck coefficient (S) in the alignment direction, demonstrating a simple means to enhance the power factor PF of doped CPs.48 Since Seebeck coefficients show limited dependence on the oxygen position, the evolution of PF in the series reflects mainly the trend of the charge conductivities, with PBTTT-5O and PBTTT-8O, performing well above the other polymers (Fig. 3(B)). These PF values, 1300 and 2900 μW m−2 K−1, respectively, are still the highest in the literature for doped p-type CPs (see Table S2, ESI†). Interestingly, the best TE properties are observed for PBTTT-8O, which exhibits a mixed morphology with coexistence of crystalline and liquid-crystalline phases due to its polymorphism (Fig. 1(C)).
Conclusion
We recently demonstrated that grafting a polar single-ether side chain (-xO) to an organic CP allows to reach higher electrical conductivity and thermoelectric performance in doped rub-aligned thin films compared to standard alkyl side chains.25,28Interestingly, this novel single-ether side-chain design allows to modulate the position of the oxygen atom along the side chain. In this work, by combining controlled synthesis, advanced structural characterizations and doping studies of a new series of PBTTT-xO, we demonstrate that not only can these single-ether chains be used to finely control the structural properties of polymers, but that they also represent a viable alternative to polar (ethylene glycol)-based side chains.
A meticulous investigation by DSC, FSC and WAXS shows that polymers bearing single-ether side chains can crystallize under proper conditions (isothermal crystallization at 40 °C). Moreover, the crystallinity index of the polymers depends on the position of the oxygen atom in the side chain. The further the oxygen atom from the conjugated backbone, the more crystalline the side chain. This phenomenon is attributed to a hyperconjugation effect (also known as the gauche effect), which tends to bend the side chain due to the synclinal conformation of the C–C α-β bond next to the ether function (Fig. 3(A)).
Interestingly, despite a reduction in the crystallinity of the -xO side chains compared to a standard dodecyl (–C12) side chain, the CP backbone cohesion is preserved leading to cohesive lamellar stacks. Furthermore, the single-ether chains improve the thermomechanical properties of PBTTT, enabling the polymers to be processed by HTR up to very high temperatures, reaching very high alignment levels.
This enhanced ordering strongly impacts the TE performance with record electrical conductivities and PFs. The best TE properties in the series are observed for PBTTT-8O, which exhibits a unique polymorphic structure with coexistence of crystalline and liquid-crystalline phases.
This study showcases the potential of single-ether side chains to be used as polar solubilizing side chains for the design of a next generation of solution-processable p- and n-type CPs and mixed ionic-electronic conductors with high doping properties. In particular, single-ether side chains are also expected to promote the development of organic electronics which require a balanced polarity, for high dopant uptake, and a mixed polymorphic morphology in the solid state, for high electrical properties and rapid (de)doping kinetics (e.g. OTEs, Supercapacitors, batteries, OECTs…).
Author contributions
P. D. and B. J. synthesized and characterized the materials. P. D. and H. Z. prepared rubbed and doped thin films and characterized their polarized and thermoelectric optical properties. O. Boyron characterized the polymer properties by SEC. B. H. carried out the DSC and TGA characterizations. L. H. contributed to thin film alignment. I. M. performed the FSC measurements. A. V. M. and A.P. M. performed the FSC measurements coupled with XRD measurements. D. I. conceived and supervised the FSC and XRD measurements. M. B. conceived and supervised the rubbing and doping process of thin films. He additionally carried out the TEM and ED measurements. N. L. supervised the materials design and synthesis. O. Bardagot, P. D., M. B., D. I., B. H. and N. L. contributed to the manuscript preparation.Data availability
The authors declare that all data supplementary to the findings of this study are available within the article and its ESI.† Any further relevant data are available from the authors upon reasonable request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge Dr F. Richard and Prof. P. Samori (ISIS, Strasbourg) for carrying out PESA measurements. The authors thank the Agence Nationale de la Recherche for financial support through projects ANR-17-CE05-0012, ANR-22-CE50-0020, region Grand’Est for the co-funding of Pablo Durand and University of Strasbourg for the funding of Badr Jismy. A. V. M. and D. A. I. acknowledge financial support from the Russian Science Foundation (grant number 23-73-30005). The authors acknowledge perfect technical support from the personnel of the ID13 beamline of the ESRF (Grenoble, France).References
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, Nat. Rev. Chem., 2020, 4, 66–77 CrossRef CAS.
- L. Ding, Z.-D. Yu, X.-Y. Wang, Z.-F. Yao, Y. Lu, C.-Y. Yang, J.-Y. Wang and J. Pei, Chem. Rev., 2023, 123, 7421–7497 CrossRef CAS PubMed.
- L. Cao, C. Ren and T. Wu, J. Mater. Chem. C, 2023, 11, 3428–3447 RSC.
- I. B. Dimov, M. Moser, G. G. Malliaras and I. McCulloch, Chem. Rev., 2022, 122, 4356–4396 CrossRef CAS PubMed.
- D. Luo, W. Jang, D. D. Babu, M. S. Kim, D. H. Wang and A. K. K. Kyaw, J. Mater. Chem. A, 2022, 10, 3255–3295 RSC.
- H. Bin, Y. Yang, Z. Peng, L. Ye, J. Yao, L. Zhong, C. Sun, L. Gao, H. Huang, X. Li, B. Qiu, L. Xue, Z. Zhang, H. Ade and Y. Li, Adv. Energy Mater., 2018, 8, 1702324 CrossRef.
- O. A. Ibraikulov, C. Ngov, P. Chávez, I. Bulut, B. Heinrich, O. Boyron, K. L. Gerasimov, D. A. Ivanov, S. Swaraj, S. Méry, N. Leclerc, P. Lévêque and T. Heiser, J. Mater. Chem. A, 2018, 6, 12038–12045 RSC.
- N. Kamatham, O. A. Ibraikulov, P. Durand, J. Wang, O. Boyron, B. Heinrich, T. Heiser, P. Lévêque, N. Leclerc and S. Méry, Adv. Funct. Mater., 2021, 31, 2007734 CrossRef CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- Y. He, N. A. Kukhta, A. Marks and C. K. Luscombe, J. Mater. Chem. C, 2022, 10, 2314–2332 RSC.
- A. Giovannitti, D.-T. Sbircea, S. Inal, C. B. Nielsen, E. Bandiello, D. A. Hanifi, M. Sessolo, G. G. Malliaras, I. McCulloch and J. Rivnay, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12017–12022 CrossRef CAS.
- D. J. Woods, S. A. J. Hillman, D. Pearce, L. Wilbraham, L. Q. Flagg, W. Duffy, I. McCulloch, J. R. Durrant, A. A. Y. Guilbert, M. A. Zwijnenburg, R. S. Sprick, J. Nelson and A. I. Cooper, Energy Environ. Sci., 2020, 13, 1843–1855 RSC.
- J. Neu, S. Samson, K. Ding, J. J. Rech, H. Ade and W. You, Macromolecules, 2023, 56, 2092–2103 CrossRef CAS.
- A. Giovannitti, I. P. Maria, D. Hanifi, M. J. Donahue, D. Bryant, K. J. Barth, B. E. Makdah, A. Savva, D. Moia, M. Zetek, P. R. F. Barnes, O. G. Reid, S. Inal, G. Rumbles, G. G. Malliaras, J. Nelson, J. Rivnay and I. McCulloch, Chem. Mater., 2018, 30, 2945–2953 CrossRef CAS PubMed.
- M. Moser, L. R. Savagian, A. Savva, M. Matta, J. F. Ponder, T. C. Hidalgo, D. Ohayon, R. Hallani, M. Reisjalali, A. Troisi, A. Wadsworth, J. R. Reynolds, S. Inal and I. McCulloch, Chem. Mater., 2020, 32, 6618–6628 CrossRef CAS.
- R. K. Hallani, B. D. Paulsen, A. J. Petty, R. Sheelamanthula, M. Moser, K. J. Thorley, W. Sohn, R. B. Rashid, A. Savva, S. Moro, J. P. Parker, O. Drury, M. Alsufyani, M. Neophytou, J. Kosco, S. Inal, G. Costantini, J. Rivnay and I. McCulloch, J. Am. Chem. Soc., 2021, 143, 11007–11018 CrossRef CAS.
- P. Schmode, A. Savva, R. Kahl, D. Ohayon, F. Meichsner, O. Dolynchuk, T. Thurn-Albrecht, S. Inal and M. Thelakkat, ACS Appl. Mater. Interfaces, 2020, 12, 13029–13039 CrossRef CAS.
- Y. Wang, E. Zeglio, H. Liao, J. Xu, F. Liu, Z. Li, I. P. Maria, D. Mawad, A. Herland, I. McCulloch and W. Yue, Chem. Mater., 2019, 31, 9797–9806 CrossRef CAS.
- C.-A. Chen, S.-C. Wang, S.-H. Tung and W.-F. Su, Soft Matter, 2019, 15, 9468–9473 RSC.
- X. Zhao, G. Xue, G. Qu, V. Singhania, Y. Zhao, K. Butrouna, A. Gumyusenge, Y. Diao, K. R. Graham, H. Li and J. Mei, Macromolecules, 2017, 50, 6202–6209 CrossRef CAS.
- S. Zhang, J. Gao, W. Wang, C. Zhan, S. Xiao, Z. Shi and W. You, ACS Appl. Energy Mater., 2018, 1, 1276–1285 CrossRef CAS.
- Y. Zhang, Y. He, L. Zeng, L. Lüer, W. Deng, Y. Chen, J. Zhou, Z. Wang, C. J. Brabec, H. Wu, Z. Xie and C. Duan, Small, 2023, 19, 2302314 CrossRef CAS.
- J. W. Onorato, Z. Wang, Y. Sun, C. Nowak, L. Q. Flagg, R. Li, B. X. Dong, L. J. Richter, F. A. Escobedo, P. F. Nealey, S. N. Patel and C. K. Luscombe, J. Mater. Chem. A, 2021, 9, 21410–21423 RSC.
- S. E. Chen, L. Q. Flagg, J. W. Onorato, L. J. Richter, J. Guo, C. K. Luscombe and D. S. Ginger, J. Mater. Chem. A, 2022, 10, 10738–10749 RSC.
- P. Durand, H. Zeng, T. Biskup, V. Vijayakumar, V. Untilova, C. Kiefer, B. Heinrich, L. Herrmann, M. Brinkmann and N. Leclerc, Adv. Energy Mater., 2022, 12, 2103049 CrossRef CAS.
- S. Moro, N. Siemons, O. Drury, D. A. Warr, T. A. Moriarty, L. M. A. Perdigão, D. Pearce, M. Moser, R. K. Hallani, J. Parker, I. McCulloch, J. M. Frost, J. Nelson and G. Costantini, ACS Nano, 2022, 16, 21303–21314 CrossRef CAS.
- H. Zeng, P. Durand, S. Guchait, L. Herrmann, C. Kiefer, N. Leclerc and M. Brinkmann, J. Mater. Chem. C, 2022, 10, 15883–15896 RSC.
-
O. Bardagot, P. Durand, S. Guchait, G. Rebetez, P. Cavassin, J. Réhault, M. Brinkmann, N. Leclerc and N. Banerji, Ultra-high μC* beyond 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 000 F cm−1V−1s−1in organic electrochemical transistors, In Review, 2023.
000 F cm−1V−1s−1in organic electrochemical transistors, In Review, 2023. - I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328–333 CrossRef CAS.
- I. Botiz, M. M. Durbin and N. Stingelin, Macromolecules, 2021, 54, 5304–5320 CrossRef CAS.
- D. M. DeLongchamp, R. J. Kline, Y. Jung, D. S. Germack, E. K. Lin, A. J. Moad, L. J. Richter, M. F. Toney, M. Heeney and I. McCulloch, ACS Nano, 2009, 3, 780–787 CrossRef CAS.
- S. E. Root, M. A. Alkhadra, D. Rodriquez, A. D. Printz and D. J. Lipomi, Chem. Mater., 2017, 29, 2646–2654 CrossRef CAS.
- R. K. Verma, I. M. Szilagyi, K. Pielichowska, K. N. Raftopoulos, P. Šimon, A. P. Melnikov and D. A. Ivanov, J. Therm. Anal. Calorim., 2023, 148, 2211–2231 CrossRef CAS.
- E. V. Komov, A. P. Melnikov, A. A. Piryazev, A. V. Maryasevskaya, A. O. Petrov, G. V. Malkov, A. V. Shastin, D. V. Anokhin and D. A. Ivanov, Thermochim. Acta, 2023, 728, 179577 CrossRef CAS.
- D. Doblas, M. Rosenthal, M. Burghammer, D. Chernyshov, D. Spitzer and D. A. Ivanov, Cryst. Growth Des., 2016, 16, 432–439 CrossRef CAS.
- S.-F. Yang, Z.-T. Liu, Z.-X. Cai, M. J. Dyson, N. Stingelin, W. Chen, H.-J. Ju, G.-X. Zhang and D.-Q. Zhang, Adv. Sci., 2017, 4, 1700048 CrossRef PubMed.
- P. Boufflet, Y. Han, Z. Fei, N. D. Treat, R. Li, D. Smilgies, N. Stingelin, T. D. Anthopoulos and M. Heeney, Adv. Funct. Mater., 2015, 25, 7038–7048 CrossRef CAS.
- V. Pirela, A. J. Müller and J. Martín, J. Mater. Chem. C, 2024, 12, 4005–4012 RSC.
- T. Qu, Y. Li, L. Li, C. Zhang, X. Wang, W. Chen, G. Xue, E. Zhuravlev, S. Luo and D. Zhou, Macromolecules, 2023, 56, 6407–6418 CrossRef CAS.
- E. M. Thomas, K. A. Peterson, A. H. Balzer, D. Rawlings, N. Stingelin, R. A. Segalman and M. L. Chabinyc, Adv. Electron. Mater., 2020, 6, 2000595 CrossRef CAS.
- A. P. Melnikov, M. Rosenthal and D. A. Ivanov, ACS Macro Lett., 2018, 7, 1426–1431 CrossRef CAS.
- I. V. Alabugin, G. dos Passos Gomes and M. A. Abdo, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1389 Search PubMed.
- W. Zierkiewicz, D. Michalska and T. Zeegers-Huyskens, Phys. Chem. Chem. Phys., 2010, 12, 13681 RSC.
- D. M. DeLongchamp, R. J. Kline, Y. Jung, E. K. Lin, D. A. Fischer, D. J. Gundlach, S. K. Cotts, A. J. Moad, L. J. Richter, M. F. Toney, M. Heeney and I. McCulloch, Macromolecules, 2008, 41, 5709–5715 CrossRef CAS.
- M. Brinkmann, Mater. Chem. Front., 2020, 4, 1916–1929 RSC.
- V. Vijayakumar, E. Zaborova, L. Biniek, H. Zeng, L. Herrmann, A. Carvalho, O. Boyron, N. Leclerc and M. Brinkmann, ACS Appl. Mater. Interfaces, 2019, 11, 4942–4953 CrossRef CAS.
- V. Vijayakumar, Y. Zhong, V. Untilova, M. Bahri, L. Herrmann, L. Biniek, N. Leclerc and M. Brinkmann, Adv. Energy Mater., 2019, 9, 1900266 CrossRef.
- A. Hamidi-Sakr, L. Biniek, J.-L. Bantignies, D. Maurin, L. Herrmann, N. Leclerc, P. Lévêque, V. Vijayakumar, N. Zimmermann and M. Brinkmann, Adv. Funct. Mater., 2017, 27, 1700173 CrossRef.
- R. H. Boyd and W. D. Phillips, J. Chem. Phys., 1965, 43, 2927–2929 CrossRef CAS.
- B. D. Paulsen, R. Wu, C. J. Takacs, H.-G. Steinrück, J. Strzalka, Q. Zhang, M. F. Toney and J. Rivnay, Adv. Mater., 2020, 32, 2003404 CrossRef CAS.
- E. M. Thomas, M. A. Brady, H. Nakayama, B. C. Popere, R. A. Segalman and M. L. Chabinyc, Adv. Funct. Mater., 2018, 28, 1803687 CrossRef.
- S. Nagamatsu and S. S. Pandey, Sci. Rep., 2020, 10, 20020 CrossRef CAS PubMed.
- Y. Zhong, V. Untilova, D. Muller, S. Guchait, C. Kiefer, L. Herrmann, N. Zimmermann, M. Brosset, T. Heiser and M. Brinkmann, Adv. Funct. Mater., 2022, 32, 2202075 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, chemical characterization, energy level characterization, sample preparation, UV/Vis-NIR absorbance, thermal characterization, X-ray characterization, doping methods, electron transmission microscopy and electrical property measurements. See DOI: https://doi.org/10.1039/d4mh00492b |
| This journal is © The Royal Society of Chemistry 2024 |