
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-healing, stretchable and recyclable polyurethane-PEDOT:PSS conductive blends†
Jinsil
Kim
 ,
Jiaxin
Fan
,
Gayaneh
Petrossian
,
Xin
Zhou
,
Pierre
Kateb
,
Noemy
Gagnon-Lafrenais
and
Fabio
Cicoira
*
,
Jiaxin
Fan
,
Gayaneh
Petrossian
,
Xin
Zhou
,
Pierre
Kateb
,
Noemy
Gagnon-Lafrenais
and
Fabio
Cicoira
*
Department of Chemical Engineering, Polytechnique Montréal, Montréal, QC H3C 3A7, Canada. E-mail: fabio.cicoira@polymtl.ca
First published on 13th June 2024
Abstract
Future electronics call for materials with mechanical toughness, flexibility, and stretchability. Moreover, self-healing and recyclability are highly desirable to mitigate the escalating environmental threat of electronic waste (e-waste). Herein, we report a stretchable, self-healing, and recyclable material based on a mixture of the conductive polymer poly(3,4-ethylenedioxythiophene) doped with polystyrene sulfonate (PEDOT:PSS) with a custom-designed polyurethane (PU) and polyethylene glycol (PEG). This material showed excellent elongation at brake (∼350%), high toughness (∼24.6 MJ m−3), moderate electrical conductivity (∼10 S cm−1), and outstanding mechanical and electrical healing efficiencies. In addition, it demonstrated exceptional recyclability with no significant loss in the mechanical and electrical properties after being recycled 20 times. Based on these properties, as a proof of principle for sustainable electronic devices, we demonstrated that electrocardiogram (ECG) electrodes and pressure sensors based on this material could be recycled without significant performance loss. The development of multifunctional electronic materials that are self-healing and fully recyclable is a promising step toward sustainable electronics, offering a potential solution to the e-waste challenge.
New conceptsIn this manuscript, we present a novel conductive blend of poly(3,4-ethylenedioxythiophene) doped with polystyrene sulfonate (PEDOT:PSS), polyurethane (PU), and polyethylene glycol (PEG), which exhibits outstanding electrical and mechanical self-healing properties, recyclability, flexibility, stretchability, and toughness. While previous research has predominantly focused on enhancing the stretchable, flexible, and self-healing properties of PEDOT:PSS, our aim is to surpass these features and enhance the material's toughness and recyclability. Materials that encompass all the aforementioned properties, while recovering their mechanical and electrical attributes after damage through self-healing, have rarely been explored. These multifunctional materials not only exhibit durability, but also contribute to sustainability, offering the potential to prolong the lifespan of electronic devices and mitigate electronic waste. In this study, we achieved all of these requirements by utilizing hydrogen bonding interactions among custom-designed PU, PEDOT:PSS, and PEG. The versatility of this novel combination extends to electrocardiogram (ECG) electrodes and pressure sensors, which consistently maintain performance throughout self-healing and recycling. Thus, this multifunctional material opens new possibilities for electronic devices that adhere to environmentally friendly practices. |
1. Introduction
Blends of conductive and insulating polymers can yield materials with tunable electrical and mechanical properties,1–4 which can find applications in bioelectronics, wearable electronics, and energy-storage devices.5–14 For future electronics, materials with mechanical toughness, flexibility, stretchability, high electrical conductivity, self-healing, and recyclability are in high demand. Although soft electronic materials are presently not a relevant component of e-waste, we believe they can contribute to sustainable electronics by replacing critical materials in targeted applications, such as sensing, bioelectronics, wearable electronics, and soft robotics.15 Self-healing and recycling are equally important for future sustainable electronic materials, as they share the ultimate goals of extending material lifespan and reducing their environmental impact. Although self-healing electronic materials can withstand several damage-healing cycles, recycling is still needed to recover and reuse materials.Poly(3,4-ethylenedioxythiophene) doped with polystyrene sulfonate (PEDOT:PSS) is the most investigated conducting polymer, because it combines high electrical conductivity, ease of processing in aqueous media, environmental stability, biocompatibility, and self-healing properties, especially when combined with other polymers.11,16,17 For instance, blending PEDOT:PSS with polyethylene glycol (PEG),18 polyurethane diol (PUD),4 tannic acid19 and TritonX-10020 resulted in improved flexibility, stretchability, and electrical self-healing properties. Improved mechanical self-healing was achieved by combining PEDOT:PSS with polyvinyl alcohol (PVA)-borax21 and polyglutamic acid (PGA)-trimethoxysilane (GPTMS).22 Nonetheless, obtaining materials encompassing high stretchability, flexibility, toughness, and self-healing capabilities remains challenging and requires further investigation of PEDOT:PSS-based blends.23
Polyurethanes (PUs), with their tunable structures and well-established synthesis, are promising candidates for improving the mechanical and self-healing properties of PEDOT: PSS.24,25 PUs are typically prepared via addition polymerization of different polyols, diisocyanates, and chain extenders.26 Generally, polyols (e.g., polyether, polyester, and polycarbonate diols) constitute the soft segments of the chain, which ensure elastic behavior, whereas diisocyanates (aromatic or aliphatic) and chain extenders constitute the hard segments,27,28 which provide mechanical strength. Therefore, PUs can be tailored to achieve toughness, self-healing capabilities, and recyclability by selecting suitable polyols, diisocyanates, and chain extenders.29 A low degree of cross-linking and linear structure leads to thermoplastic elastomers with flexible chains, whereas a high degree leads to thermosetting and rigid polymers. This versatility makes PUs appealing for various applications.30–32 For instance, thermoplastic PUs (TPUs), which combine plastic and elastic properties, are employed in packaging, tubing, footwear, and medical devices; rigid or flexible PU foams are widely used in automotive components and construction insulators, and PU sprays can be used as waterproof coatings. To promote self-healing and recyclability of PUs, dynamic covalent (Diels–Alder bonds and disulfide bonds) and noncovalent bonds (hydrogen bonds, host–guest interaction, and metal–ligand coordination) can be introduced in their polymer chain.33,34 The reversibility of these bonds allows for recombination after bond breakage, making PUs suitable as recyclable materials.35–37
In this study, we report a novel self-healing and recyclable conductive material with excellent mechanical properties. This material was obtained by combining a custom-designed PU, PEDOT:PSS, and PEG, which is expected to enable dynamic hydrogen-bonding interactions. The resulting material exhibited remarkable toughness, stretchability, and simultaneous electrical and mechanical healing. Furthermore, it could be recycled multiple times without significant deterioration of its mechanical and electrical properties. As a proof of principle for sustainable electronic devices, we demonstrated electrocardiogram (ECG) electrodes and pressure sensors based on this material, which could be self-healed and recycled without significant performance loss.
2. Results and discussion
2.1. Synthesis of PU and preparation of PEDOT:PSS/PU-based films
The PU used in this study (shown in Fig. 1a) is based on poly(ε-caprolactone) diol (PCL diol), isophorone diisocyanate (IPDI), and 1,3-propanediol (PDO) as the chain extender. The linear PCL, diol acting as the soft segment, promotes chain mobility, thus facilitating dynamic intermolecular interactions between the polymer chains, resulting in improved self-healing and recyclability.38–41 IPDI and PDO, which serve as hard segments, contribute to the strength and durability of the material.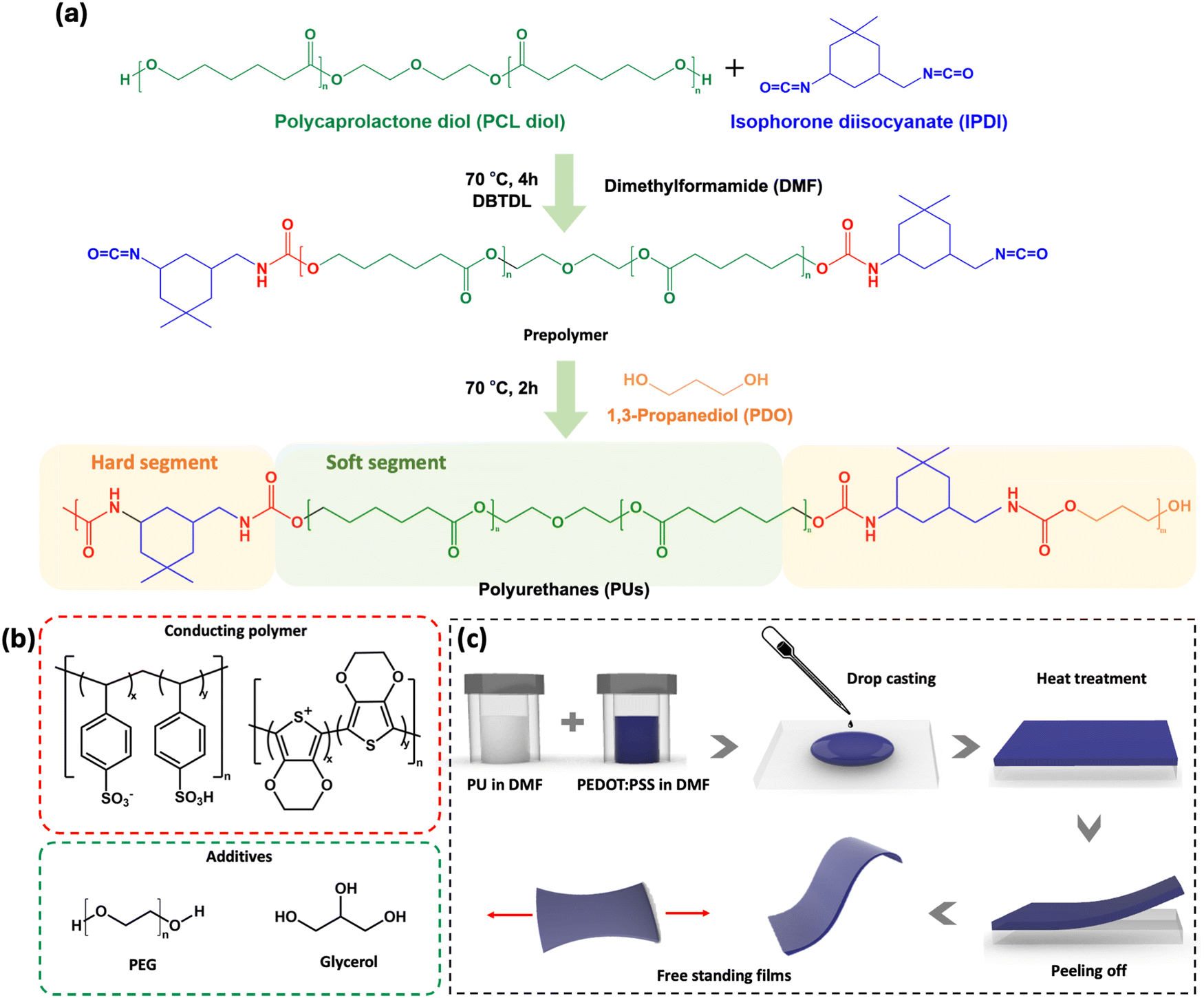 | ||
| Fig. 1 (a) Scheme of the synthesis of the polyurethane (PU) used in this study. (b) Chemical structures of PEDOT:PSS, PEG, and glycerol. (c) Process flow for film preparation. The synthesis is described in the experimental section. | ||
To produce conductive, stretchable, and self-healing films (Fig. 1c), PU was mixed with PEDOT:PSS, PEG and glycerol (the chemical structures are shown in Fig. 1b). Based on preliminary electrical and mechanical tests, the optimal PU concentration was found to be 16 wt%. PEG 400 was added to the mixture as it is known to promote autonomous self-healing of PEDOT:PSS.18 Glycerol, which has been shown to increase PEDOT:PSS conductivity by promoting PEDOT chain alignment, was added as the conductivity enhancer.42,43 The hydroxyl and ether groups of PEG are expected to interact via hydrogen bonds with both PU and PEDOT:PSS, leading to a looser packing within the PU and PEDOT:PSS chains, resulting in increased chain mobility.44 This also leads to a higher toughness, as it increases the ability of the material to plastically deform and absorb energy before breaking.41,45–47
2.2. Mechanical and electrical properties
The electrical conductivity of different PEDOT:PSS/PU-based films ranged between ∼5 and ∼15 S cm−1 (Table 1), i.e., slightly lower than that of films obtained from the pure PEDOT:PSS screen printing formulation (∼60 S cm−1).48| Samples | Thickness (μm) | Young's modulus (MPa) | Elongation at break (%) | Toughness (MJ m−3) | Conductivity (S cm−1) |
|---|---|---|---|---|---|
| PEDOT:PSS/PU | 24 ± 2 | 35.8 ± 3.5 | 30 ± 5 | 0.37 ± 0.03 | 5.2 ± 0.3 |
| PEDOT:PSS/PU/PEG | 25 ± 3 | 50.5 ± 2.5 | 350 ± 30 | 24.60 ± 0.23 | 9.4 ± 0.5 |
| PEDOT:PSS/PU/glycerol | 22 ± 2 | 21.5 ± 1.5 | 20 ± 4 | 0.26 ± 0.05 | 15.4 ± 1.2 |
| PEDOT:PSS/PU/PEG/glycerol | 25 ± 1 | 28.3 ± 2.6 | 112 ± 13 | 2.35 ± 0.14 | 11.5 ± 0.7 |
Materials for soft bioelectronics, specifically wearable devices interfacing with tissues, need to achieve a balance between elasticity, stretchability, and toughness. For instance, they require stretchability and toughness to accommodate various movements, and simultaneously high Young's modulus to maintain their structural shape. Stress–strain measurements of samples with different compositions (Fig. 2a) showed a linear relationship up to ∼30% strain. For samples containing PEG, plastic deformation was observed at higher stains. The PEDOT:PSS/PU/PEG samples exhibited the most interesting mechanical properties (Table 1, Fig. 2b and Fig. S1a, ESI†), i.e., an exceptional elongation at break (∼350%), high toughness (∼24.6 MJ m−3), and relatively high Young's modulus (∼50 MPa). The presence of 2 wt% PEG led to a 10-fold increase in elongation at break and a 65-fold increase in toughness. The addition of glycerol, although leading to a small increase in conductivity, caused deterioration of the mechanical properties, likely due to the disruption of hydrogen bonding between the polymer chains, resulting in lower mechanical strength. The toughness of PEDOT:PSS/PU/PEG calculated using the method of Rivlin and Thomas (Fig. S1b, ESI†)49 exhibited about 30% reduction (16.54 ± 1.24 MJ m−3) (Fig. S1c and d, ESI†).
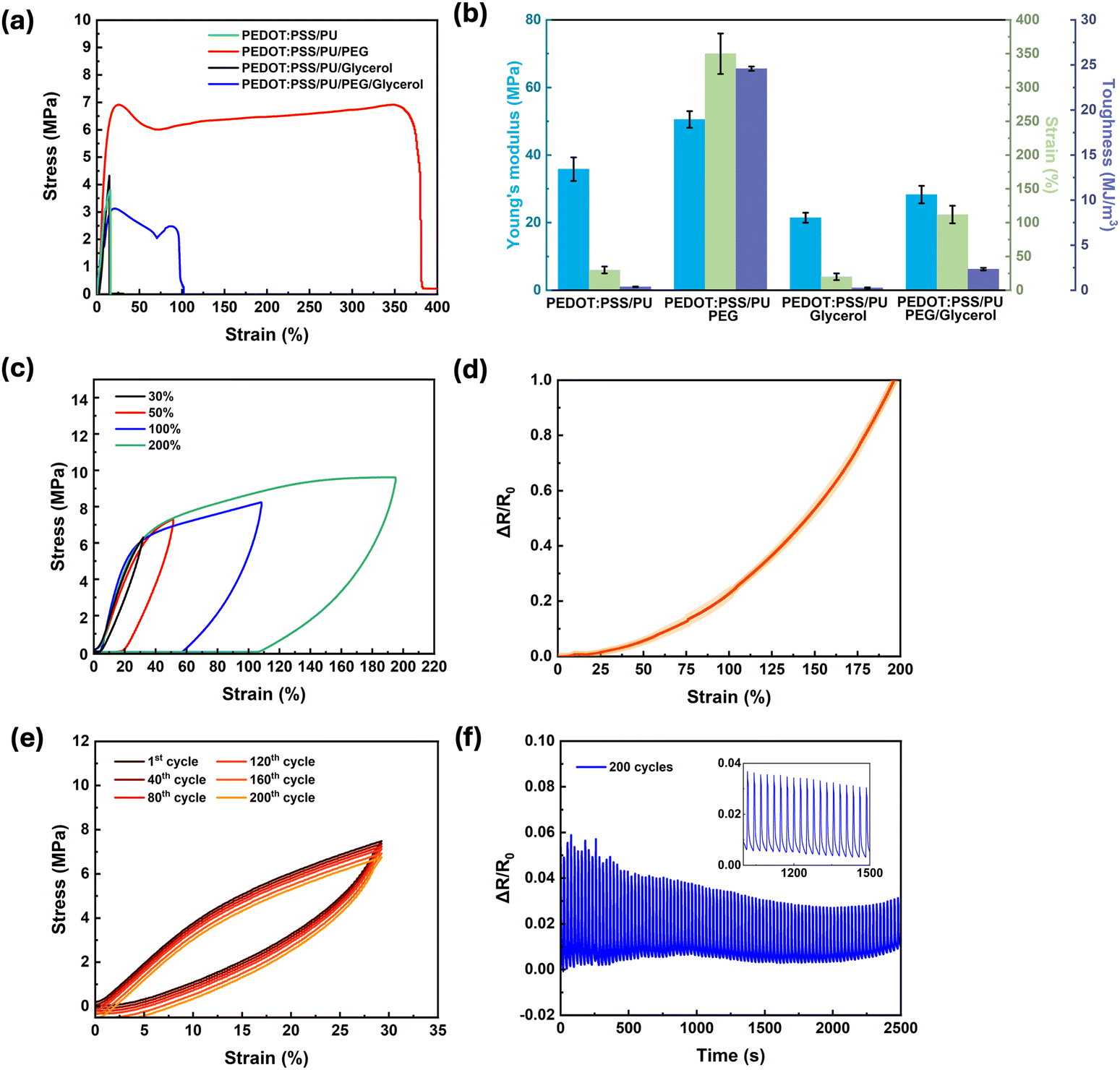 | ||
| Fig. 2 Mechanical properties of PEDOT:PSS/PU-based films. (a) Tensile stress–strain curves. (b) Comparison of the Young's moduli in the range of 0–10% strain, elongation at break, and toughness. Data (n = 3) are reported as mean ± standard deviation. (c) Stress–strain hysteresis curves obtained from cyclic tests of PEDOT:PSS/PU/PEG at different strain ranges (30, 50, 100, and 200%). (d) Resistance change (ΔR/R0) vs. strain. Data (n = 3) are reported as mean ± standard deviation. The yellow-shaded area around the curve represents the standard deviation. (e) Tensile loading–unloading plots for the PEDOT:PSS/PU/PEG samples at the 1st, 40th, 80th, 120th, 160th, and 200th cycles at a strain range of 0–30%. (f) Resistance change (ΔR/R0) vs. time during 200 cyclic strains from 0 to 30%. The Inset is a magnification from 1000 to 1500s showing the resistance change with cyclic strain. | ||
Given its outstanding mechanical properties, PEDOT:PSS/PU/PEG was selected for further investigation.
To assess the durability of PEDOT:PSS/PU/PEG, the deformation and recovery behavior of the films were studied via tensile loading–unloading tests at different strains (Fig. 2c). The films could withstand elastic deformation of up to 30% strain without notable energy dissipation (area inside the hysteresis loop). Higher strains led to higher energy dissipation (from 0.24 MJ m−3 at 30% strain to 6.32 MJ m−3 at 200% strain) and plastic deformation. Similarly, the electromechanical behavior of the films, that is, the resistance changes up to 200% strain (Fig. 2d), revealed minimal changes up to 30% (∼4%), while a steep rise occurred between 30 and 200%. At a strain of 200%, the resistance almost doubled. This increase is due to the change of the polymer network at higher strains.
Cyclic measurements performed in the elastic region (0–30% strain, Fig. 2e), revealed a very small change in dissipation energy upon 200 cycles (0.21 ± 0.02 MJ m−3 for the 1st cycle and 0.27 ± 0.04 MJ m−3 for the 200th cycle). To evaluate the electrical stability of the PEDOT:PSS/PU/PEG films during cyclic stretching, their resistance was recorded during stress–strain cyclic tests (0–30% strain) (Fig. 2f). A minor change (∼6%) was observed during the first 40 cycles, which stabilized at ∼3% for the successive cycles (inset of Fig. 2f). This behavior indicates that the material retained its arrangement during the loading–unloading cycles, resisting conformational changes, structural breakdown, and microstructural alterations.50
The PEDOT:PSS/PU/PEG films showed notable performance, as evidenced by the results of mechanical and electrical characterization. Specifically, they exhibited remarkable electrical and mechanical stability at 30% strain over repeated loading–unloading cycles. Therefore, the addition of PEG to PEDOT:PSS/PU-based films is a promising approach to enhance their mechanical properties, making them suitable for developing wearable and flexible electronics with high durability.51–53
Hydrogen bonds play a crucial role in the interactions between PEG, PEDOT:PSS, and PU, thereby influencing the mechanical properties of the films. To elucidate which of these bonds play a relevant role, we performed liquid-state FTIR measurements of a simplified model system, showing hydrogen bonding (H-bonding) similar to our materials. The samples consisted of liquid mixtures of poly(4-styrenesulfonic acid) (PSS), ethyl-N-carbamate (urethane, U), and PEG (detailed compositions are listed in Table 3). A schematic representation of the interactions between these components is shown in Fig. 3a. Based on their chemical structures, five types of hydrogen bonds are possible ((i)–(v) in Fig. 3b), which are expected to lead to a red-shift in the FTIR peaks of the pure materials. For the characteristic peaks of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (Fig. 3c) and SO3–H (Fig. S2a, ESI†) in PSS, as well as CO (Fig. 3d) and C–O–C (Fig. S2b, ESI†) in urethane, we noticed a consistent shift with increasing PEG content (Table S1, ESI†), which can be attributed to the formation of H-bonding with the hydroxyl and ether groups in PEG. The peaks corresponding to N–H bonds in urethane and O–H bonds in PEG overlap, making it difficult to analyze the H-bonding. In contrast, the positions of the characteristic peaks did not change for the different ratios of PSS and U (Fig. S2c and d, ESI†), revealing no significant H-bonding interactions between PSS and U. When PEG was added to the PSS-U mixture, the shift of the peaks of PSS (Fig. S2e, ESI†) and U (Fig. S2f, ESI†) follows the same trends as in the binary mixtures. This confirms that H-bonding does not occur between PSS and U. Overall, the additional H-bonds induced by the presence of PEG may explain the observed improvements in stretchability and toughness.
O (Fig. 3c) and SO3–H (Fig. S2a, ESI†) in PSS, as well as CO (Fig. 3d) and C–O–C (Fig. S2b, ESI†) in urethane, we noticed a consistent shift with increasing PEG content (Table S1, ESI†), which can be attributed to the formation of H-bonding with the hydroxyl and ether groups in PEG. The peaks corresponding to N–H bonds in urethane and O–H bonds in PEG overlap, making it difficult to analyze the H-bonding. In contrast, the positions of the characteristic peaks did not change for the different ratios of PSS and U (Fig. S2c and d, ESI†), revealing no significant H-bonding interactions between PSS and U. When PEG was added to the PSS-U mixture, the shift of the peaks of PSS (Fig. S2e, ESI†) and U (Fig. S2f, ESI†) follows the same trends as in the binary mixtures. This confirms that H-bonding does not occur between PSS and U. Overall, the additional H-bonds induced by the presence of PEG may explain the observed improvements in stretchability and toughness.
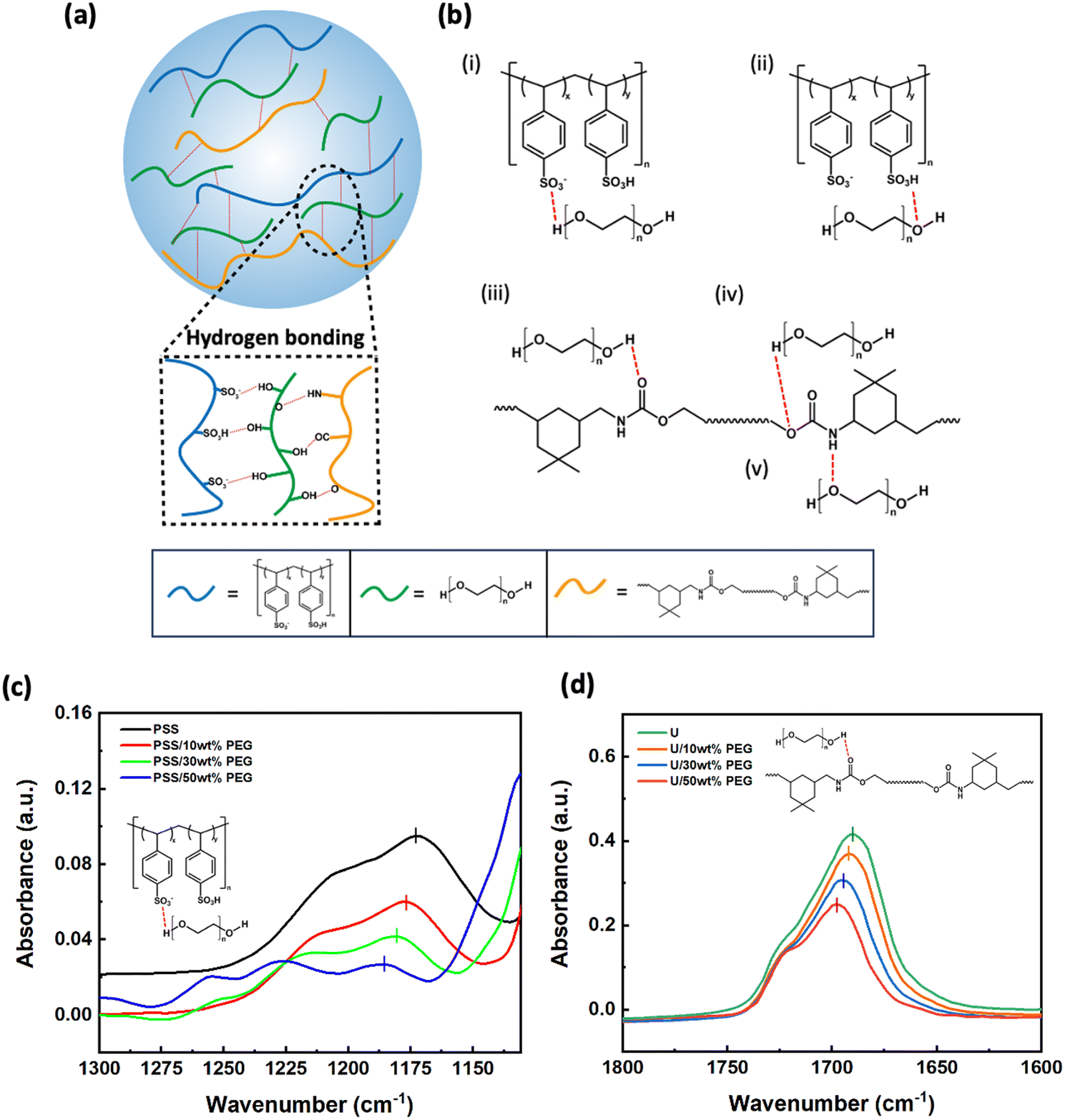 | ||
| Fig. 3 (a) Scheme illustrating the hydrogen bonding (H-bonding) interactions within PEG, PSS, and U. (b) Types of H-bonding within PEG, PSS, and U: (i) O–H in hydroxyl groups of PEG and OS in the sulfonate groups of PSS, (ii) SO3–H in the sulfonic acid groups and O–H in the hydroxyl groups; (iii) O–H in hydroxyl groups and OC in urethane groups, (iv) –O– in urethane groups and O–H in hydroxyl groups, and (v) N–H in urethane groups and –O– in ether groups. (c) FTIR spectra of pristine PSS and mixtures of PSS/PEG with different percentages (10%, 30%, 50%). (d) FTIR spectra of pristine urethane (U) and mixture of U/PEG with different percentages (10%, 30%, 50%). | ||
2.3. Electrical and mechanical self-healing
All PEDOT:PSS/PU-based films demonstrated autonomous electrical self-healing when cut several times with a razor blade (average gap size of 20 μm), unlike the pure PEDOT:PSS (Fig. S3a, ESI†). In particular, the PEDOT:PSS/PU/PEG and PEDOT:PSS/PU films exhibited a rapid autonomous healing response, recovering their electrical properties within ∼0.1s (Fig. 4a and Fig. S3b, ESI†). Optical imaging of the cut-healing process of the PEDOT:PSS/PU/PEG film shows that the gap (∼20 μm) left by the blade (Fig. 4b) closes after healing, although the scar left by the cut remains visible (Fig. 4c). The PEDOT:PSS/PU/glycerol and PEDOT:PSS/PU/PEG/glycerol films displayed longer recovery times of ∼5 s and ∼0.5 s, respectively (Fig. S3c and d, ESI†).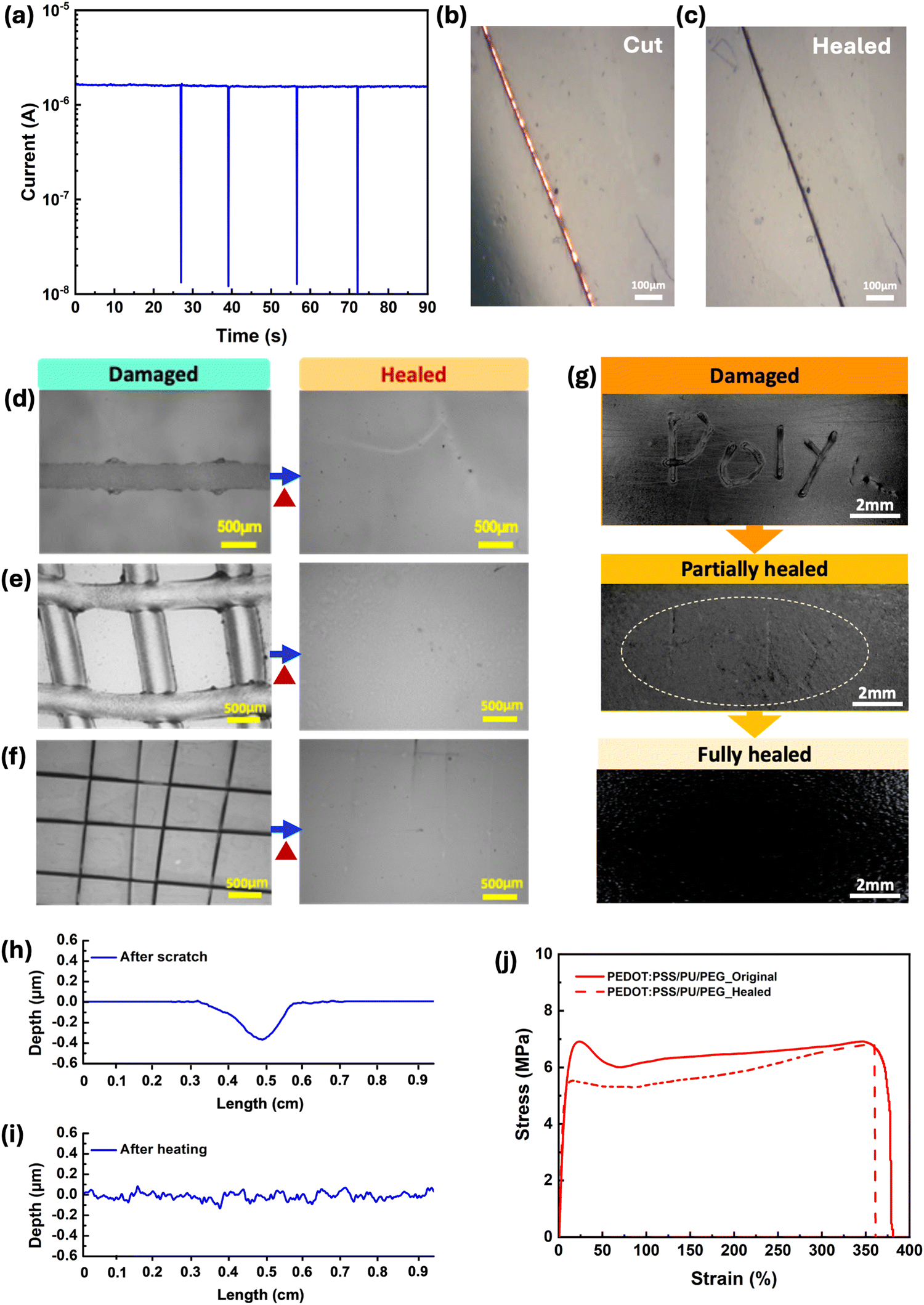 | ||
| Fig. 4 (a) Current vs. time plot for PEDOT:PSS/PU/PEG films during several cut/healing cycles upon application of 0.2 V. The current drops correspond to sequential cuts. Optical microscope images of the PEDOT:PSS/PU/PEG film: (b) after cutting and (c) after autonomous healing (∼40 s). Optical microscope images of PEDOT:PSS/PU/PEG film after scratching with the pencil and healing: (d) single scratching and (e) grid scratching. (f) Optical microscope images of PEDOT:PSS/PU/PEG film after scratching with a blade on the surface and healing. (g) Digital pictures of the healing process of PEDOT:PSS/PU/PEG film after irregular damaging by pencil. (h) Depth profile of PEDOT:PSS/PU/PEG films (h) after surface scratching by pencil and (i) healing. (j) Stress–strain curves of PEDOT:PSS/PU/PEG films as-prepared (solid line) and after cut-stick self-healing at 50 °C (dashed line). | ||
Mechanical self-healing was studied in response to the scratching (Fig. 4d–i) and cutting (Fig. 4j) of the films. As no healing was observed at room temperature within three days, the experiments were carried out at 50 °C, which is close to the glass transition temperature (Tg) of PU (Fig. S4c, ESI†). At around Tg, the polymer transitions from a glassy to a rubbery phase, improving chain mobility. This transition is favorable to the reformation of dynamic bonds, such as hydrogen bonds, which plays a significant role in the self-healing process.54
Optical microscopy images revealed complete recovery of single scratches on the surfaces of PEDOT:PSS/PU/PEG and PEDOT:PSS/PU after heating at 50 °C for 10 min (Fig. 4d and e and Fig. S5b, ESI†), unlike PEDOT:PSS (Fig. S5a, ESI†). Moreover, we performed two different types of scratches on the surface to test the material's ability to heal different types of wounds. Both PEDOT:PSS/PU/PEG and PEDOT:PSS/PU showed complete healing of pencil scratched single lines and girds after heating (Fig. 4e and Fig. S5f, ESI†). The PEDOT:PSS/PU/PEG film was further subjected to grid-shape surface damage with a blade, demonstrating complete healing (Fig. 4f). Even for more irregular surface scratches created with the pencil, the film exhibited partial healing after 2 min heating, and complete healing after 10 min heating (Fig. 4g). Both the PEDOT:PSS/PU/PEG and PEDOT:PSS/PU films exhibited 100% healing efficiencies (Fig. 4h, i and Fig. S6a, ESI†), unlike pure PEDOT:PSS film (Fig. S6c, ESI†). We also evaluated the self-healing properties of pure PU and observed effective healing of surface scratches (Fig. S7, ESI†), although at a significantly slower rate (∼50 min) and slightly higher temperature (60 °C). Samples containing glycerol exhibited less efficient healing (Fig. S5c–e, g, h, S6b and S8, ESI†).
The free-standing PEDOT:PSS/PU-based films demonstrated cut-stick mechanical self-healing (Movie S1, ESI†). Tensile stress–strain tests performed on intact and healed PEDOT:PSS/PU/PEG showed a high average healing efficiency of ∼90% of its toughness after undergoing the cut-stick healing test (Fig. 4j). Additionally, the electrical properties after cut-stick healing of the PEDOT:PSS/PU/PEG films were tested using a simple electric circuit connected to a light-emitting diode (LED) (Fig. S9 and Movie S2, ESI†). As shown in the video, cutting the film causes the LED to switch off. When the film was healed by sticking the pieces together and heating at 50 °C, the LED was immediately switched on. The self-healing behavior of the PEDOT:PSS/PU/PEG blends is attributed to a mechanism based on dynamic non-covalent interactions, as indicated by FTIR results.
2.4. Recyclability of PEDOT:PSS/PU/PEG
The ability to recycle or reuse electronic materials without compromising their electrical and mechanical properties is crucial to reduce e-waste. PEDOT:PSS/PU/PEG was selected for recyclability tests due to its exceptional mechanical and self-healing properties. The samples were cut into small pieces and heated (Fig. 5a) at 100 °C to improve the fluidity of the material and to facilitate rapid molding. As shown in Movie S3 (ESI†), small pieces of PEDOT:PSS/PU/PEG could be easily remolded by hand pressing after removal from the hot plate at 100 °C. Stress–strain measurements carried out on pristine and remolded dogbone-shaped samples revealed an 85% recovery of toughness after the 1st recycling, which remained almost unchanged for the following cycles (Fig. 5b). Moreover, there was no significant deterioration in the electrical properties even after 20 remolding cycles (Fig. 5c). The stable electrical and mechanical properties of the recycled material indicate that heating above the Tg of PU improved the chain mobility,54 facilitating the reformation of hydrogen bonds among PEG, PEDOT:PSS, and PU. The reversible dissociation and reformation of hydrogen bonds within the material ensured its high recyclability, allowing consistent performance even after 20 reprocessing cycles. Hence, our results suggest that PEDOT:PSS/PU/PEG holds great promise for application in sustainable and recyclable electronics. The PEDOT:PSS/PU/PEG films displayed a range of advantageous properties including notable stretchability, toughness, electrical and mechanical healing, and recyclability (Table S2, ESI†). While previous studies have addressed higher electrical conductivity, stretchability, and self-healing,4,11,17,18,21 in this study, we have comprehensively reported on multiple features, including recyclability, high toughness, and the simultaneous recovery of mechanical and electrical properties.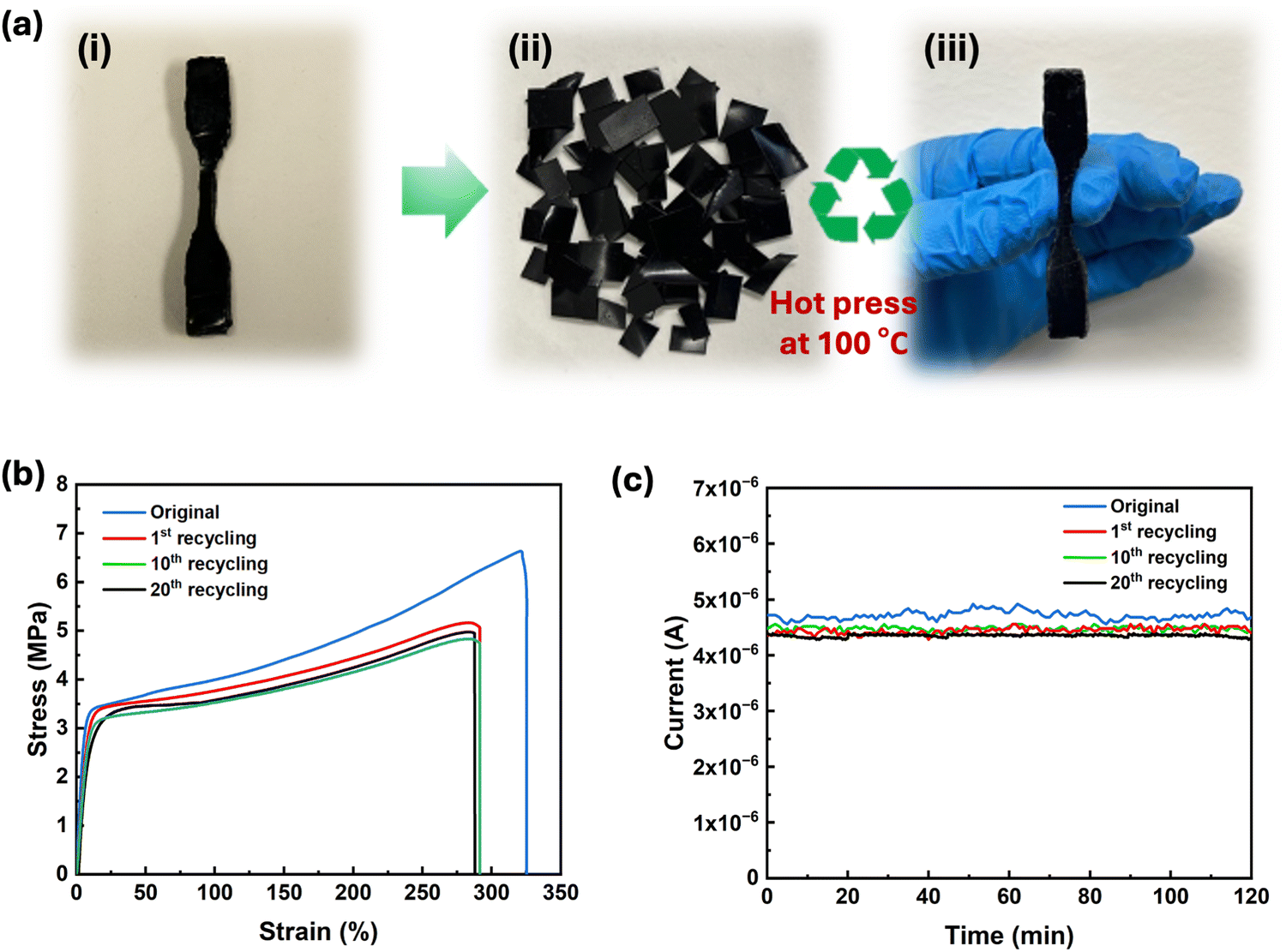 | ||
| Fig. 5 Recycling of PEDOT:PSS/PU/PEG using a hot press at 100 °C. (a) Photographs of the original PEDOT: PSS/PU/PEG sample (i) after being cut into small pieces (ii) and after remolding by hot-pressing (iii). (b) Comparison of tensile stress–strain curves for the PEDOT:PSS/PU/PEG sample before and after recycling (1, 10 and 20 times). (c) Current vs. time plot before and after recycling (1, 10 and 20 times) (applied voltage of 0.2 V). | ||
2.5. Self-healable and recyclable skin-electrode impedance, ECG signal monitoring and pressure sensor
To demonstrate potential applications of the PEDOT:PSS/PU/PEG blend, we fabricated an epidermal electrode and a resistive pressure sensor using our materials. When tested on skin, the electrodes exhibited an impedance (103–106 Ω), similar to commercial electrodes (Fig. 6a), which remained stable after self-healing and recycling (Fig. 6a). The ECG recording performance of our electrodes was found to be similar to the commercial electrode (Fig. 6b and Fig. S11, ESI†), with clearly identifiable ECG peaks, including P waves, QRS complexes, and T waves (Fig. 6c).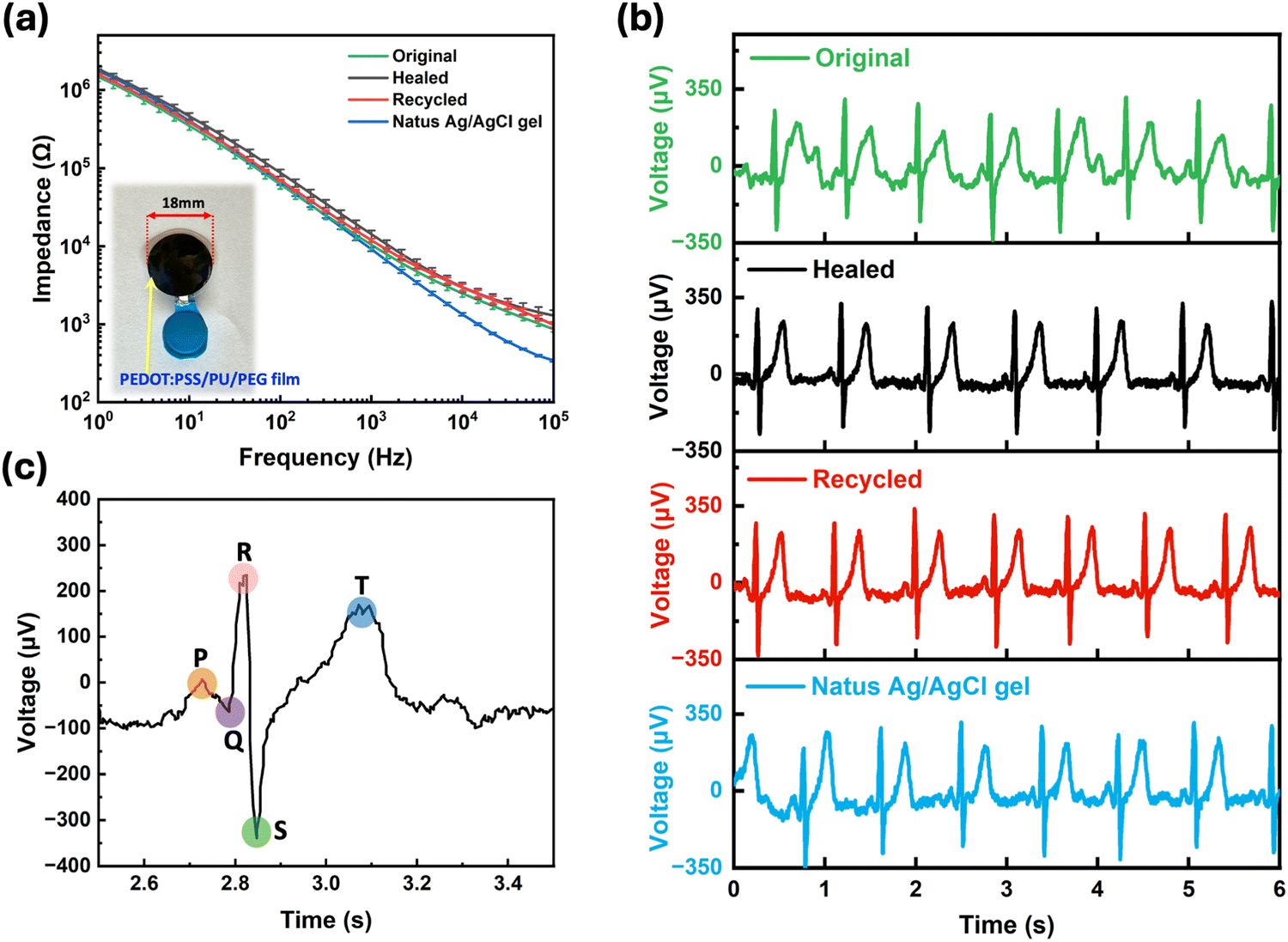 | ||
| Fig. 6 Skin-electrode impedance measurement and ECG signal recording with PEDOT:PSS/PU/PEG film electrodes. (a) Skin-electrode impedance versus frequency for commercial Natus® Ag/AgCl gel electrodes, original, healed, and recycled PEDOT:PSS/PU/PEG film electrodes. The Inset is an electrode. (b) ECG signals recorded using PEDOT:PSS/PU/PEG film electrodes before (green) and after self-healing (black) and recycling (red) and commercial Natus® Ag/AgCl gel (blue) electrodes. (c) A zoom-in plot of the ECG signal from the original PEDOT:PSS/PU/PEG film plot, indicating depolarization of atrium represented by the P wave, depolarization wave of ventricular indicated by the QRS complex, and repolarization wave of ventricular depicted by the T wave. | ||
We also fabricated a resistive pressure sensor by combining the PEDOT:PSS/PU/PEG film with printed interdigitated Ag electrodes on a flexible substrate (Fig. S12a, ESI†). To assess the performance of the sensor, we applied different weights to the active region (PEDOT:PSS/PU/PEG) and monitored the changes in resistance. A consistent reduction in resistance was observed when the same weight was applied to the film, and the signal consistency was maintained even after self-healing and recycling (Fig. S12b–g, ESI†). The PEDOT:PSS/PU/PEG sample responded to different weights with varying resistance changes, both in its original state and after healing and recycling (Fig. S12b–g, ESI†). We calculated the relative resistance change with respect to pressure from these measurements to extract the sensitivity (S) (Fig. S12h, ESI†). The sensor originally showed two distinctive sensitivity regions with a higher sensitivity of 1.25 kPa−1 for the pressure range 0.002–0.2 kPa, and a lower sensitivity of 0.06 kPa−1 was observed for 0.2–1.2 kPa. The sensitivity remained nearly unchanged after mechanical self-healing and recycling. Hence, these proof of principle devices results demonstrate the excellent properties of the PEDOT:PSS/PU/PEG film, highlighting its potential for applications in sustainable electronics.
3. Conclusions
In summary, a material obtained from a blend of PEDOT:PSS, PU and PEG exhibited excellent mechanical and electrical self-healing properties and could be recycled via moderate pressure and heat treatment. PEG was found to improve the mechanical properties through H-bonding with PEDOT:PSS and PU, resulting in a substantial increase in the material toughness (∼25 MJ m−3) and elongation at break (∼350%). This material also exhibited excellent durability and maintained its mechanical and electrical properties under 30% stretching for at least 200 cycles. As a proof of concept, the PEDOT:PSS/PU/PEG film was used to develop an ECG electrode and a resistive pressure sensor, which maintained their responses and pressure sensitivity after self-healing and recycling. We believe this PEDOT:PSS/PU/PEG blend paves the way for the development of sustainable wearable electronics, offering a promising solution for e-waste. In an era characterized by rapid technological advancements, our research represents a crucial step toward a more environmentally conscious future for electronic devices, in the ongoing pursuit of innovative and sustainable technologies.4. Experimental section
4.1. Materials
Poly(3,4-ethylenedioxythiophene) doped with polystyrene sulfonate (PEDOT:PSS) in the formulation of a screen-printing paste (CleviosTM SV3 STAB) was purchased from Heraeus Precious Metals (Germany). The paste contained diethylene glycol (DEG) and propylene glycol (PG). Poly(caprolactone) diol (average Mw of 2000 g mol−1), polyethylene glycol (PEG, average molecular weight of 400 g mol−1), glycerol, dimethylformamide (DMF), poly(4-styrenesulfonic acid) (PSS, Mw = ∼75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, 18 wt% in H2O), and eutectic gallium–indium (EGaIn) were purchased from Millipore Sigma (St. Louis, MO, USA). Isophorone diisocyanate (IPDI), 1,3-propanediol (PDO), dibutyltin dilaurate (DBTDL), and ethyl N-carbamate were purchased from Tokyo Chemical Industry (USA). Glass slides were obtained from Corning (USA). A TeflonTM plate was purchased from McMaster Carr. The dog bone mold was 3D printed using a stereolithography printer (Form 3 printer, Formlabs) and high-temperature resin (Formlabs). Silver ink (520 EI) was donated by Chimet S.p.A. TegadermTM adhesive films were purchased from 3M. Thermoplastic polyurethane (TPU) on removable silicon paper (Elecrom Stretch White, thickness of 80 μm) was purchased from Policrom Screens (Italy).
000 g mol−1, 18 wt% in H2O), and eutectic gallium–indium (EGaIn) were purchased from Millipore Sigma (St. Louis, MO, USA). Isophorone diisocyanate (IPDI), 1,3-propanediol (PDO), dibutyltin dilaurate (DBTDL), and ethyl N-carbamate were purchased from Tokyo Chemical Industry (USA). Glass slides were obtained from Corning (USA). A TeflonTM plate was purchased from McMaster Carr. The dog bone mold was 3D printed using a stereolithography printer (Form 3 printer, Formlabs) and high-temperature resin (Formlabs). Silver ink (520 EI) was donated by Chimet S.p.A. TegadermTM adhesive films were purchased from 3M. Thermoplastic polyurethane (TPU) on removable silicon paper (Elecrom Stretch White, thickness of 80 μm) was purchased from Policrom Screens (Italy).
4.2. Synthesis of self-healing polyurethane
The two-step synthesis (Fig. 1a) for preparing polyurethane (PU) was conducted at 70 °C in a three-necked round-bottomed flask sealed with a rubber septum. IPDI (diisocyanate) and the chain extender PDO constituted the hard segments, whereas the soft segments were made of PCL diol. The molar ratios were 2:1 for IPDI/PCL diol and 1:2:1 for PCL diol/IPDI/PDO according to the reaction stoichiometry. The first step of the synthesis reaction was carried out in a DMF solution (66.20 g, 70 wt%) containing PCL diol (22.51 g, 10 mmol, 23.80 wt%), IPDI (5 g, 20 mmol, 5.30 wt%) and the DBTDL catalyst (0.09 g). The PCL diol was dissolved in a DMF aliquot, and the solution was stirred at room temperature for 10 min with a magnet, then heated at 70 °C in an oil bath and stirred for an additional 30 min to achieve complete dissolution. The IPDI and DBTDL catalysts were then added dropwise to the flask. The reaction proceeded for 4 h to allow the PCL diol to react with the IPDI to form an –NCO-terminated prepolymer. For the second step of the synthesis, the chain-extender PDO (0.86 g, 10 mmol, 0.90 wt%) was added to the prepolymer, and the reaction proceeded for additional 2 hours to achieve the final PU. Finally, the solution was allowed to cool to the ambient temperature. This synthesis yielded a solution of 30 wt% PU in DMF. The average molecular weight of the synthesized PU was approximately 10000 g mol−1, as determined by gel permeation chromatography (GPC) (Fig. S13a, ESI†). The chemical structure of PU was confirmed by FTIR and H1NMR. The FTIR spectrum of PU (Fig. S13b, ESI†) did not show the characteristic –NCO peak at 2270 cm−1, indicating the completion of the reaction. Characteristic peaks for CO and N–H were observed at 1700 cm−1 and 3200 cm−1, respectively. The NMR spectrum of PU shows peaks associated with the proton signals of the –NH bonds at 7.0 ppm (Fig. S13c, ESI†).
4.3. Fabrication of PEDOT:PSS/PU-based films
The PEDOT:PSS paste (Clevios™ S V3) was homogenized using a centrifugal mixer (ARM-310, Thinky, USA) at 2000 rpm for 5 minutes and mixed with DMF (50:50 weight ratio) to ensure its miscibility with the PU solution, using a vortex mixer (VWR). The mixtures for film deposition (compositions in Table 2) were prepared by mixing the PEDOT:PSS and PU solutions in different proportions using a vortex mixer. Films containing additives were prepared by adding PEG and/or glycerol to the PU solution, followed by mixing with PEDOT:PSS. The films were obtained by drop-casting on glass slides (5.0 × 7.0 cm2) or Teflon plates and baking on a hot plate with the following sequence: 50 °C for 16 hours, 80 °C for 1 hour, 120 °C for 1 hour, 150 °C for 1 hour, and 100 °C for 24 hours (Fig. 1b and c). This procedure yielded homogeneous films without the formation of bubbles which, once fully dried, were removed from the substrates using a razor blade. For preliminary tests with different PEDOT:PSS/PU ratios, we assessed the mechanical performance by manually stretching the films and identifying the compositions that yielded the best stretchability. Films with an 8 wt% PU concentration exhibited poor stretchability; increasing the PU concentration to 25 wt% and 30 wt% led to films with lower conductivities (∼1 S cm−1) without notable improvement in mechanical properties compared to the 16 wt% PU films. Preliminary tests also revealed that PEG 400 concentrations exceeding 2 wt% were unsuitable, as they led to the formation of oily residues on the film surfaces. Glycerol was added as the conductivity enhancer. PEG 400 was chosen based on preliminary tests, revealing no significant differences in mechanical properties compared to PEG 200. Notably, previous research in PEDOT:PSS demonstrated similar electrical conductivity upon addition of either PEG 400 or PEG 200.18
| Samples | PEDOT:PSS solution (wt%) | PU (wt%) | PEG (wt%) | Glycerol (wt%) |
|---|---|---|---|---|
| PEDOT:PSS | 100 | — | — | — |
| PEDOT:PSS/PU | 84 | 16 | — | — |
| PEDOT:PSS/PU/PEG | 82 | 16 | 2 | — |
| PEDOT:PSS/PU/glycerol | 82 | 16 | — | 2 |
| PEDOT:PSS/PU/PEG/glycerol | 80 | 16 | 2 | 2 |
Thermogravimetric analysis (TGA) was performed using a TG Q500 analyzer (TA Instruments, USA) to evaluate the stability of the material at the temperatures required for the self-healing and recycling tests, as well as to verify the residual solvent content after processing. Samples (∼15 mg) were placed in an open platinum crucible and heated under a nitrogen atmosphere (40 mL min−1) from 40 to 600 °C at a ramping rate of 10 °C min−1. The percentage weight loss as a function of temperature (Fig S4a, ESI†) indicated that all samples exhibited an onset decomposition temperature of approximately 300 °C, exceeding the temperatures at which the self-healing (50 °C) and recycling (100 °C) processes were carried out, excluding any thermal decomposition occurring during these processes. Additionally, all the TGA curves showed no noticeable change in weight within the temperature range where the DMF solvent typically evaporated, revealing that minimal residual solvent remained in the film after processing.
Differential scanning calorimetry (DSC Q2000, TA Instruments, USA) was used to study the thermal properties, specifically focusing on confirming the glass transition temperatures (Tg) of PEDOT:PSS/PU-based films and pure PU samples. PEDOT:PSS/PU-based film displayed no clear Tg (Fig. S4b, ESI†). The samples (∼5 mg) were placed in a closed-lid standard aluminum pan and subjected to constant nitrogen flow. To eliminate any thermal history and ensure that subsequent heating and cooling cycles accurately represented the true thermal behavior, the samples were first cooled to −30 °C and then heated to 250 °C at a heating rate of 10 °C min−1. Subsequently, a second cycle was performed by heating the samples from −30 °C to 250 °C at 10 °C min−1. The data collected during the second cycle were used for analysis. No melting point was observed in the DSC curve, which confirms that the PU synthesized in this work was thermosetting. Additionally, DSC was performed in modulated mode (MDSC) to increase sensitivity for clear verification of Tg. Approximately 15 mg of sample in a closed-lid standard Al pan was used, operating at a heating rate of 2 °C min−1 with a modulated period of 60 s. The Tg was determined using the built-in functions of TA Instruments software.
4.4. Fourier-transform infrared (FTIR) spectroscopy
Fourier-transform infrared (FTIR) spectroscopy was performed in the absorption mode on the liquid samples specified in Table 3, using a PerkinElmer Spectrum 65 spectrometer. The background spectrum was recorded prior to sample data acquisition. The spectra of the solutions were collected at room temperature with a scan range of 4000–600 cm−1 and a resolution of 4 cm−1 for 16 scans. Each sample was subjected to three scans using identical parameters. The liquid samples for FTIR measurements were prepared using PSS solutions consisting of 18 wt% PSS in H2O, PEG 400 (>99%), and ethyl-N-carbamate (>99%, urethane, U) monomers. We prepared 14 different samples to isolate and study the hydrogen-bonding interactions for four different material combinations: PSS and PEG, U and PEG, PSS and U, and PSS, U, and PEG. The detailed compositions are listed in Table 3.| FTIR samples | PSS | PEG | U | |||
|---|---|---|---|---|---|---|
| (wt%) | (g) | (wt%) | (g) | (wt%) | (g) | |
| Aqueous PSS | 100 | 1 | — | 0 | — | 0 |
| PSS/10 wt% PEG | 90 | 1 | 10 | 0.1 | — | 0 |
| PSS/30 wt% PEG | 70 | 1 | 30 | 0.43 | — | 0 |
| PSS/50 wt% PEG | 50 | 1 | 50 | 1 | — | 0 |
| U | — | 0 | — | 0 | 100 | 1 |
| U/10 wt% PEG | — | 0 | 10 | 0.1 | 90 | 1 |
| U/30 wt% PEG | — | 0 | 30 | 0.43 | 70 | 1 |
| U/50 wt% PEG | — | 0 | 50 | 1 | 50 | 1 |
| PSS/10 wt% U | 90 | 1 | — | 0 | 10 | 0.1 |
| PSS/30 wt% U | 70 | 1 | — | 0 | 30 | 0.43 |
| PSS/50 wt% U | 50 | 1 | — | 0 | 50 | 1 |
| PSS/10 wt% U/10 wt% PEG | 80 | 1 | 10 | 0.1 | 10 | 0.1 |
| PSS/30 wt% U/10 wt% PEG | 60 | 1 | 10 | 0.1 | 30 | 0.43 |
| PSS/50 wt% U/10 wt% PEG | 40 | 1 | 10 | 0.1 | 50 | 1 |
4.5. 1H-nuclear magnetic resonance (NMR) spectroscopy
1H NMR spectra, obtained using a Bruker AVANCE II 400 spectrometer operating at 400 MHz, were used to verify the chemical structure of the synthesized PU. 1H NMR analysis was performed at room temperature using a 5 wt% PU solution in dimethyl sulfoxide-d6 (DMSO-d6) as the NMR solvent.4.6. Gel permeation chromatography (GPC)
Gel permeation chromatography (GPC) was used to determine the average molecular weight of the synthesized PU. The PU solution (30 wt% in DMF) was diluted with tetrahydrofuran (THF) to a concentration of 0.2 wt%. The solution was then filtered through a 0.2 μm PTFE syringe filter prior to injection. Measurements were conducted using a 1260 infinity GPC system equipped with a WAT044228-styragel HR 5E column manufactured by Waters. THF was employed as the eluent at 30 °C and a flow rate of 1 mL min−1. A refractive index detector was used for data collection. Polystyrene (PS) standards in THF were employed to calibrate the GPC system.4.7. Electrical measurements
The sheet resistance (Rs) of the PEDOT:PSS/PU-based films was recorded using a four-point probe system (Jandel Engineering) connected to an Agilent B2902A source measure unit (SMU). The thickness of the PEDOT:PSS/PU-based films was measured using a profilometer (DEKTAK 150, Veeco). The average resistivity (ρ) of the five films was obtained based on their thickness (t) using the formula ρ = Rst. Conductivity (σ) was calculated as σ = 1/ρ.4.8. Mechanical and electromechanical characterization
Tensile tests were performed using a Mach-1 V500csst MA009 mechanical tester (Biomomentum Inc., Canada) at a strain rate of 10 mm min−1. The stress–strain curves allowed the extraction of the elongation at break and modulus of elasticity (Young's modulus). The latter was calculated as the slope of the linear section (up to 10% strain) of the strain–stress curve. The tensile tests following the Rivlin and Thomas method were conducted on the samples with notches of 0.5 mm (half width of the film), created by using a razor blade. Mechanical cyclic loading tests (stretching–unloading-releasing) were performed up to a strain of 200% at a rate of 10 mm min−1 using a 70 N load cell. The film toughness was calculated as the integral of the stress–strain curve using the OriginLab built-in functions. Three samples of each film type were measured, and the results were reported as mean ± standard deviation.Electromechanical tests were performed on PEDOT:PSS/PU/PEG films up to 200% strain while monitoring the resistance using an Agilent B2902A SMU and a custom-made 4-point probe setup. The resistances were normalized as R/R0, where R is the measured resistance and R0 is the initial resistance measured at 0% strain.
4.9. Self-healing
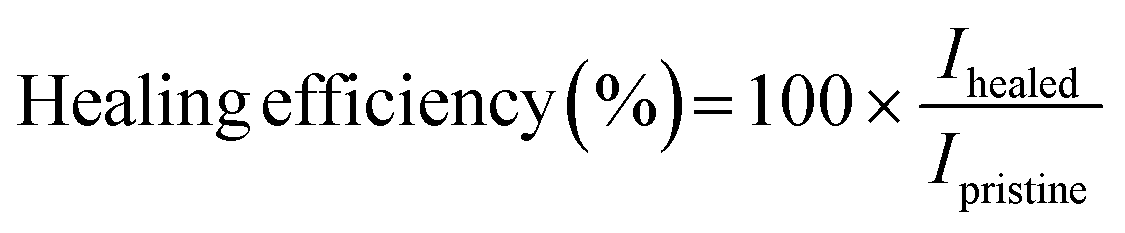 | (1) |
For the scratch test, a 5N or 3N force was applied to the films using a scratch pencil (Erichsen SmartPen, test tip ISO1518; diameter of 1.0 mm). The scratch pencil was employed to create the single, grid, and variously shaped scratches. Additional grid patterns were scratched on the PEDOT:PSS/PU/PEG film manually with a razor blade (Feather Hi Stainless Double Edge, Japan). Irregular shape damages were created on the PEDOT:PSS/PU/PEG film with the scratch pencil. The films were then heated to 50 °C for 10 min on a hot plate to induce self-healing. A profilometer (DEKTAK 150, Veeco) was used to measure the depth of the scratched region (single scratch with pencil) before and after healing, and images were obtained using an inverted optical microscope (Carl Zeiss, Primovert, Germany) and a digital microscope (Dino-Lite Edge 3.0 AM73915MT8, USA). The mechanical healing efficiency for scratching (η) was calculated using eqn (2):
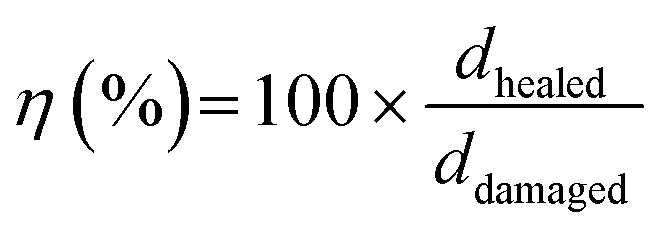 | (2) |
For the cut-stick test, a pristine (uncut) film (1.0 × 5.0 cm2, thickness ∼25 μm) was cut into two pieces with a stainless-steel razor blade, and the two pieces were placed on a hot plate at 50 °C for 10 min with the fractured ends in contact. A small pressure was manually applied (as indicated by the red arrows in Fig. S14b, ESI†) to ensure contact between the pieces. Tensile tests were performed before and after healing at a constant rate of 10 mm min−1 with a 70N load cell using a mechanical tester. The mechanical healing efficiency was calculated using eqn (3)
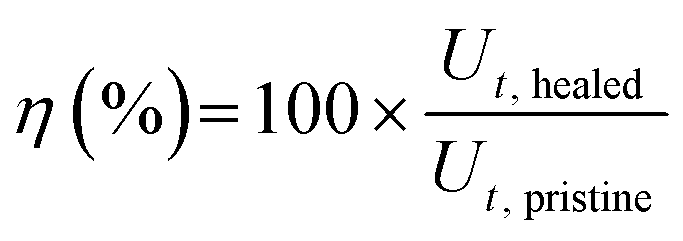 | (3) |
4.10. Recycling
The PEDOT:PSS/PU/PEG mixture was poured into a dog-bone mold (ASTM D638, Fig. S15, ESI†) and heated in the following sequence: 50 °C for 24 h, 80 °C for 3 h, 100 °C for 2 h, 120 °C for 2 h, and 150 °C for 2 h. This gradual increase in temperature allowed the samples to dry completely without forming any bubbles. The samples were then removed from the mold and subjected to tensile testing after cooling to room temperature. They were then cut into small pieces, which were manually dispersed in a dog-bone mold. The mold was then transferred to a hot press (Combo heat press machine) set at 100 °C, and a pressure of ∼3 GPa was applied to the sample for 20 min to remold the pieces into one. This heating step was necessary to facilitate the remolding required for recycling. The recycling was completed by cooling for 2 h at room temperature and removing the samples from the mold. The process was repeated 20 times, and the mechanical and electrical properties were evaluated after the first, the tenth and the twentieth time.4.11. Electrodes preparation and ECG signal monitoring
The PEDOT:PSS/PU/PEG electrodes were fabricated through the following procedure (Fig. S10a and b, ESI†): PEDOT:PSS/PU/PEG films (5.0 × 7.0 cm2) were cut into 18 mm diameter disks using a Brother ScanNCut (model SDX225), and placed on to a thermoplastic polyurethane (TPU) substrate (disk with 20 mm diameters). Then, electrical contacts were established through Ag traces connected to a snap button of a commercial electrode (Ambu®). A Tegaderm™ tape (3M) was used to secure the electrode on the skin during the measurements.Skin-electrode impedance and ECG measurements were performed following previously reported protocols.4,21 The ECG signal monitoring was conducted using commercial Ag/AgCl gel electrodes, PEDOT:PSS/PU/PEG film electrodes, as well as electrodes made from healed and recycled films. The PEDOT:PSS/PU/PEG films were subjected to cut-stick healing and recycling following the same procedures as described earlier. All data were collected from the same volunteer on the same day (Fig. S10c and d, ESI†).
4.12. PEDOT:PSS/PU/PEG film-based pressure sensor
PEDOT:PSS/PU/PEG films (1 × 2 cm2, thickness ∼25 μm) were placed on interdigitated silver patterns printed on Tegaderm films using a direct-ink write-printed circuit board printer (Voltera V-One). Adhesive Tegaderm tape was used to secure the connections between the films and silver patterns. Two alligator clips were used to establish an electrical connection with the SMU (Agilent B2902A). A constant potential of 0.2 V was applied to monitor the resistance change upon application of different pressures. Various weights, including 1, 5, 10, 50, 100, and 500 g, and a finger touch, were tested to determine the pressure sensitivity, and the changes in resistance were calculated. The sensitivity (S) was determined by analyzing the slope of the relative resistance change versus applied pressure plots and was calculated based on the following formula: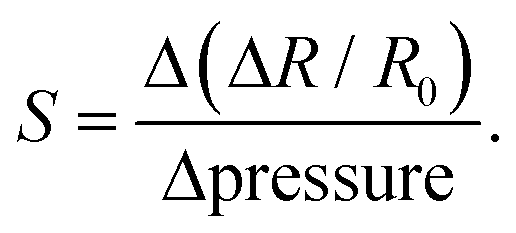 | (4) |
Author contributions
Jinsil Kim: conceptualization, methodology, validation, formal analysis, investigation, visualization, and writing – original draft. Jiaxin Fan: analysis, investigation, and writing – review and editing. Gayaneh Petrossian: analysis, writing – review and editing. Xin Zhou: analysis, writing – review and editing. Pierre Kateb: resources, writing – review and editing. Noemy Gagnon-Lafrenais: resource. Fabio Cicoira: writing – review and editing, supervision, conceptualization, methodology.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research was supported by the Natural Science and Engineering Council Canada (NSERC) through a Discovery Grant awarded to FC and National Defense Canada (IDEas project CFPMN1-008, awarded to FC). The equipment and infrastructure used in this study were acquired and maintained by the Canada Foundation for Innovation. JK acknowledges the Trottier Energy Institute for a PhD scholarship. JF acknowledges support from the NSERC postdoctoral fellowship. GP acknowledges support from the NSERC Vanier Canada Graduate Scholarship. XZ is grateful to the China Scholarship Council for a PhD scholarship. PK acknowledges the Pierre Arbour Foundation and NSERC for a Master's scholarship.References
- N. A. M. Radzuan, A. B. Sulong and J. Sahari, Int. J. Hydrogen Energy, 2017, 42, 9262–9273 CrossRef.
- T. K. Das and S. Prusty, Polym.-Plast. Technol. Eng., 2012, 51, 1487–1500 CrossRef CAS.
- M. H. Naveen, N. G. Gurudatt and Y.-B. Shim, Appl. Mater. Today, 2017, 9, 419–433 CrossRef.
- P. Kateb, J. Fan, J. Kim, X. Zhou, G. A. Lodygensky and F. Cicoira, Flexible Printed Electron., 2023, 8, 045006 CrossRef.
- J. Kang, J. B.-H. Tok and Z. Bao, Nat. Electron., 2019, 2, 144–150 CrossRef.
- S. J. Benight, C. Wang, J. B. Tok and Z. Bao, Prog. Polym. Sci., 2013, 38, 1961–1977 CrossRef CAS.
- Y. Cao, Y. J. Tan, S. Li, W. W. Lee, H. Guo, Y. Cai, C. Wang and B. C.-K. Tee, Nat. Electron., 2019, 2, 75–82 CrossRef.
- X. M. An, J. Liu, J. H. Zhang, X. X. Huang, T. S. Zhu, H. P. Yan, X. D. Jia and Q. H. Zhang, Mater. Chem. Front., 2022, 6, 1779–1787 RSC.
- B. Q. Wan, X. Yang, X. D. Dong, M. S. Zheng, Q. L. Zhao, H. K. Zhang, G. R. Chen and J. W. Zha, Adv. Mater., 2023, 35, 2304175 CrossRef PubMed.
- T. Wang, Y. Zhang, Q. C. Liu, W. Cheng, X. R. Wang, L. J. Pan, B. X. Xu and H. X. Xu, Adv. Funct. Mater., 2018, 28 Search PubMed.
- Y. Li, S. M. Zhang, N. Hamad, K. Kim, L. Liu, M. Lerond and F. Cicoira, Macromol. Biosci., 2020, 20, 2000146 CrossRef CAS PubMed.
- Y. Lu, Z. Q. Liu, H. M. Yan, Q. Peng, R. G. Wang, M. E. Barkey, J. W. Jeon and E. K. Wujcik, ACS Appl. Mater. Interfaces, 2019, 11, 20453–20464 CrossRef CAS PubMed.
- F. Y. Sun, Z. X. Li, S. L. Gao, Y. Y. He, J. C. Luo, X. Zhao, D. D. Yang, T. Gao, H. B. Yang and P. F. Cao, ACS Appl. Mater. Interfaces, 2022, 14, 26014–26023 CrossRef CAS PubMed.
- H. Wang, B. H. Zhao, W. J. Dong, Y. Zhong, X. R. Zhang, Y. L. Gong, R. X. Zhan, M. Xing, J. X. Zhang, G. X. Luo and W. Qian, Chem. Eng. J., 2020, 393 Search PubMed.
- J. M. R. Tan, Y. Farraj, A. Kamyshny and S. Magdassi, ACS Appl. Electron Mater., 2023, 5, 1376–1393 CrossRef CAS.
- Y. Li, X. Zhou, B. Sarkar, N. Gagnon-Lafrenais and F. Cicoira, Adv. Mater., 2022, 34, 2108932 CrossRef CAS PubMed.
- S. Zhang and F. Cicoira, Adv. Mater., 2017, 29, 1703098 CrossRef PubMed.
- Y. Li, X. Li, S. Zhang, L. Liu, N. Hamad, S. R. Bobbara, D. Pasini and F. Cicoira, Adv. Funct. Mater., 2020, 30, 2002853 CrossRef CAS.
- X. Zhou, P. Kateb, J. X. Fan, J. Kim, G. A. Lodygensky, B. Amilhon, D. Pasini and F. Cicoira, J. Mater. Chem. C, 2024, 12, 5708–5717 RSC.
- J. Y. Oh, S. Kim, H. K. Baik and U. Jeong, Adv. Mater., 2016, 28, 4455–4461 CrossRef CAS PubMed.
- X. Zhou, A. Rajeev, A. Subramanian, Y. Li, N. Rossetti, G. Natale, G. A. A. Lodygensky and F. Cicoira, Acta Biomater., 2022, 139, 296–306 CrossRef CAS PubMed.
- C. Y. Zhang, M. X. Wang, C. H. Jiang, P. Z. Zhu, B. Q. Sun, Q. Gao, C. X. Gao and R. Y. Liu, Nano Energy, 2022, 95, 106991 CrossRef CAS.
- H. G. Liao, S. L. Liao, X. L. Tao, C. Liu and Y. P. Wang, J. Mater. Chem. C, 2018, 6, 12992–12999 RSC.
- P. J. Taroni, G. Santagiuliana, K. N. Wan, P. Calado, M. T. Qiu, H. Zhang, N. M. Pugno, M. Palma, N. Stingelin-Stutzman, M. Heeney, O. Fenwick, M. Baxendale and E. Bilotti, Adv. Funct. Mater., 2018, 28, 1704285 CrossRef.
- M. Z. Seyedin, J. M. Razal, P. C. Innis and G. G. Wallace, Adv. Funct. Mater., 2014, 24, 2957–2966 CrossRef CAS.
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- Y. Li, Y. Jin, W. Fan and R. Zhou, J. Leather Sci. Eng., 2022, 4, 24 CrossRef CAS.
- F. W. Xie, T. L. Zhang, P. Bryant, V. Kurusingal, J. M. Colwell and B. Laycock, Prog. Polym. Sci., 2019, 90, 211–268 CrossRef CAS.
- Y. Yao, M. Xiao and W. G. Liu, Macromol. Chem. Phys., 2021, 222, 2100002 CrossRef CAS.
- X. H. Chen, X. Y. Zeng, K. Luo, T. Chen, T. Zhang, G. L. Yan and L. Wang, Small, 2022, 18, 2205286 CrossRef CAS PubMed.
- S. W. Yang, S. Wang, X. S. Du, Z. L. Du, X. Cheng and H. B. Wang, Chem. Eng. J., 2020, 391, 123544 CrossRef CAS.
- Z. Q. Zhou, S. Y. Chen, X. M. Xu, Y. Chen, L. P. Xu, Y. N. Zeng and F. A. Zhang, Prog. Org. Coat., 2021, 154, 106213 CrossRef CAS.
- S. H. Lee, S. R. Shin and D. S. Lee, Mater. Design, 2019, 172, 107774 CrossRef CAS.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed.
- B. Willocq, J. Odent, P. Dubois and J. M. Raquez, RSC Adv., 2020, 10, 13766–13782 RSC.
- R. H. Aguirresarobe, S. Nevejans, B. Reck, L. Irusta, H. Sardon, J. M. Asua and N. Ballard, Prog. Polym. Sci., 2021, 114, 101362 CrossRef CAS.
- Z. W. An, R. Xue, K. Ye, H. Zhao, Y. Liu, P. Li, Z. M. Chen, C. X. Huang and G. H. Hu, Nanoscale, 2023, 15, 6505–6520 RSC.
- Z. X. Fei, C. X. Yin, J. R. Sun, L. Yuan and L. Y. Shi, Polymer, 2023, 289, 126467 CrossRef CAS.
- S. Yu, R. C. Zhang, Q. Wu, T. H. Chen and P. C. Sun, Adv. Mater., 2013, 25, 4912–4917 CrossRef CAS PubMed.
- F. T. Sai, H. T. Zhang, J. B. Qu, J. Y. Wang, X. Z. Zhu, Y. Bai and P. Ye, Appl. Surf. Sci., 2022, 573 Search PubMed.
- Z. W. Guo, X. Y. Lu, X. H. Wang, X. Li, J. Li and J. Q. Sun, Adv. Mater., 2023, 35 Search PubMed.
- H. Shi, C. C. Liu, Q. L. Jiang and J. K. Xu, Adv. Electron. Mater., 2015, 1, 1500017 CrossRef.
- S. L. Lai, M. Y. Chan, M. K. Fung, C. S. Lee and S. T. Lee, Mater. Sci. Eng., B, 2003, 104, 26–30 CrossRef.
- E. Mehrbakhsh, M. Rezaei, R. Lotfi Mayan Sofla and A. Babaie, Int. J. Polym. Mater. Polym. Biomater., 2024, 73, 239–249 CrossRef CAS.
- A. X. Chen, A. T. Kleinschmidt, K. Choudhary and D. J. Lipomi, Chem. Mater., 2020, 32, 7582–7601 CrossRef CAS.
- X. L. Niu, L. X. Huo, C. T. Cai, J. S. Guo and H. Zhou, Ind. Eng. Chem. Res., 2014, 53, 16359–16365 CrossRef CAS.
- J. W. Chen, J. Dai, J. H. Yang, N. Zhang, T. Huang and Y. Wang, Chin. J. Polym. Sci., 2013, 31, 232–241 CrossRef CAS.
- C. Kim, P. Kateb, J. Yeu, N. Gagnon-Lafrenais, E. Gee, S. Audry and F. Cicoira, Flexible Printed Electron., 2022, 7, 014008 CrossRef.
- J. Y. Sun, X. H. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. G. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- M. Uchida, K. Kamimura, T. Yoshida and Y. Kaneko, Int. J. Plasticity, 2022, 153 Search PubMed.
- Y. R. Jeong, G. Lee, H. Park and J. S. Ha, Acc. Chem. Res., 2019, 52, 91–99 CrossRef CAS PubMed.
- X. H. Ma, Z. F. Jiang and Y. J. Lin, J. Semicond., 2021, 42, 101602 CrossRef CAS.
- H. W. Sheng, X. T. Zhang, J. Liang, M. J. Shao, E. Q. Xie, C. J. Yu and W. Lan, Adv. Healthcare Mater., 2021, 10, 2100199 CrossRef CAS PubMed.
- J. H. Xu, J. Y. Chen, Y. N. Zhang, T. Liu and J. J. Fu, Angew. Chem., Int. Ed., 2021, 60, 7947–7955 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh00203b |
| This journal is © The Royal Society of Chemistry 2024 |