
Discovery of novel β-elemene hybrids with hydrogen sulfide-releasing moiety possessing cardiovascular protective activity for the treatment of atherosclerosis†
Wenjian
Zhu‡
a,
Hongyu
Wu‡
a,
Chen
He
a,
Huajian
Zhu
a,
Hong
Yao
a,
Yun
Cao
b,
Yueman
Shi
c,
Xiaotong
Chen
c,
Xue
Feng
c,
Shengtao
Xu
*a,
Zheying
Zhu
d and
Jinyi
Xu
 *a
*a
aState Key Laboratory of Natural Medicines and Department of Medicinal Chemistry, China Pharmaceutical University, 24 Tong Jia Xiang, Nanjing 210009, PR China. E-mail: cpuxst@cpu.edu.cn; jinyixu@china.com
bJinling High School, Nanjing, Jiangsu 210005, China
c50 Dongbei Street Development Zone, CSPC Yuanda (Dalian) Pharmaceutical Co., Ltd, Dalian, Liaoning 116600, China
dDivision of Molecular Therapeutics & Formulation, School of Pharmacy, The University of Nottingham, University Park Campus, Nottingham NG7 2RD, UK
First published on 16th October 2023
Abstract
Herein, a series of novel β-elemene hybrids with different types of hydrogen sulfide (H2S) donors was designed and synthesized for the first time. In addition, all compounds were tested for H2S release in phosphate buffer solution assay, among which the derivatives with 5-p-hydroxyphenyl-3H-1,2-dithiole-3-thione (ADT-OH) as the H2S donor released the best level. The results of the isolated vasodilation assay revealed that all the compounds exhibited a degree of vasodilatory effect, and the representative compound “β-elemene-H2S gas donor” hybrid L13-2h produced more than 50% vasodilatory activity at a concentration of 20 μM. Furthermore, L13-2h possessed good concentration dependence and significantly better vasodilatory activity than the lead compound L13. In the RAW 264.7 cellular lipid inhibition against oxidized low-density lipoprotein (ox-LDL) stimulation assay, eight compounds, including L13-2g and L13-2h, produced significant cellular lipid-lowering activity. The results of the further antioxidant activity study showed that the representative compounds L13-2g and L13-2h improved H2O2-induced oxidative damage in HUVEC cells and compound L13-2h exhibited excellent antioxidant damage protection activity compared to the positive control. Moreover, none of the target compounds appeared to be significantly cytotoxic at the tested concentrations. These results suggest that the hybridization of hydrogen sulfide donors with β-elemene provides a promising approach for the discovery of novel anti-atherosclerotic drugs from natural products.
1. Introduction
Cardiovascular diseases seriously threaten human health, and atherosclerosis (AS) is one of the most common potential causes of cardiovascular diseases.1,2 Although statin lipid-lowering drugs have shown good results in clinical treatment, these drugs are also associated with side effects, such as muscle pain and abnormal liver function.3,4 Additionally, as a chronic and complex pathological process, AS is commonly treated clinically with a multi-drug combination strategy. However, the drug combination strategy inevitably involves varying degrees of adverse effects and unacceptable risks from drug–drug interactions. Alternatively, with the profound understanding of active natural products and endogenous gas molecules, novel dual-functional “natural product-active gas donor” hybrid molecules offer a strategy for synergistic pharmacological drug discovery. Therefore, it is beneficial to develop safe and effective anti-AS drugs with novel molecule skeletons using this strategy.Hydrogen sulfide (H2S) is an important endogenous signaling molecule that plays numerous vital regulatory roles, particularly as an anti-atherosclerotic agent, in cardiovascular diseases.5–8 H2S acting as a vasodilator has been reported to improve vascular disorders and alleviate the progression of AS.9 Disturbances in the vascular cystathionine γ-lyase (CSE)/H2S pathway were found in the studies on apolipoprotein E (ApoE) knockout mice.10 Another study showed that decreased endogenous H2S production accelerated AS in CSE knockout mice.11 However, despite its important role in physiology, H2S has been shown to be undesirable for direct clinical treatment because it is a gaseous molecule with uncontrollable dosage and has toxic properties at high concentrations. Accordingly, in recent years, exogenous H2S donors have been widely reported as substitutes for H2S gas donors, such as p-hydroxybenzothionamide (TBZ), 5-p-hydroxyphenyl-3H-1,2-dithiole-3-thione (ADT-OH), sulfenylTCMs, and α-thioctic acid (Fig. 1).12–15 For instance, two hydrogen sulfide donors, TBZ and ADT-OH, were each linked to antihistamine drugs to design bifunctional hybrid molecules, which showed improved efficacy and reduced side effects in itchy mouse models.16
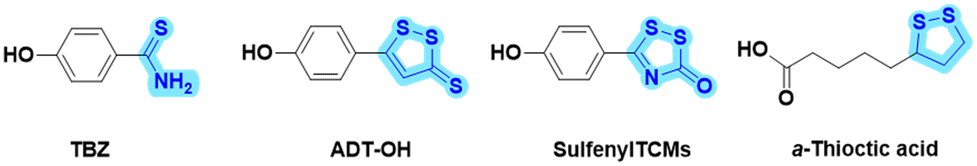 | ||
| Fig. 1 Structures of representative H2S-donating molecules. | ||
Elemene, a mixture of active sesquiterpenes, is extracted from Curcuma aromatica Salisb., where β-elemene is one of the primary active components.17–19 A study revealed that β-elemene displays activities such as inhibition of oxidative damage in endothelial cells, inhibition of pro-inflammatory cytokine (TNF-α, IL-6) production, anti-AS, and possesses low toxicity.20 A further study showed that β-elemene inhibited oxidative damage in vascular endothelial cells, suppressed the growth of vascular smooth muscle cells (VSMCs) and prevented neointimal hyperplasia by regulating oxidation-related active factors such as superoxide dismutase (SOD), malondialdehyde (MDA), catalase (CAT) and glutathione peroxidase (GSH-Px).21 In a study on a rabbit AS model, β-elemene dramatically reduced the thickness of the atherosclerotic lesion fraction.20 Thus, the above-mentioned evidence adequately demonstrates the potential significance of β-elemene in anti-AS.
The natural product β-elemene exhibits anti-atherosclerotic and cardioprotective activity and is characterized by low toxicity.20–23 Furthermore, the anti-atherosclerotic effect of natural β-elemene has been reported in pharmacological experiments; however, no novel β-elemene derivatives with enhanced activity have been reported to date. Alternatively, the discovery of drug molecules as gas donors has received increasing attention, while gas donor drugs have entered clinical research. H2S is the third gasotransmitter to be discovered after nitric oxide (NO) and carbon monoxide (CO), which has important regulatory effects in cardiovascular diseases, especially anti-atherosclerotic effects.24 Therefore, to develop new molecular skeletons and further improve the anti-AS pharmacological activity of β-elemene, we applied the combination principle of drug design to introduce the H2S donor fragment with anti-AS activity in the β-elemene molecule to achieve a synergistic pharmacological effect, and thus enhance the cardiovascular protection in vivo.
In the previous work by our group, a series of novel H2S-releasing isochroman derivatives was designed and synthesized by combining H2S donors with a natural product, (±) 7,8-dihydroxy-3-methyl-isochroman-4-one analogs (XJP), with antihypertensive activity, which exhibited synergistic antihypertensive activity.25 In this work, we continued our efforts to confirm the rationality of the synergistic pharmacological strategy of “natural product-active gas donor” heterogeneous molecules. Using this strategy, we isolated and identified the excellent antioxidant activities of the lead compounds L13 and L14, which were further used to design and synthesize a series of novel β-elemene hybrids. Consequently, we developed a series of new dual-functional “β-elemene-H2S gas donor” hybrid molecules with different H2S-releasing donors to exert enhanced pharmacological effects for the first time, and further evaluated the biological properties of these hybrids as anti-AS drug candidates (Fig. 2). The comprehensive pharmacological evaluation results indicated that compound L13-2h is the valuable lead for further study as an anti-AS candidate.
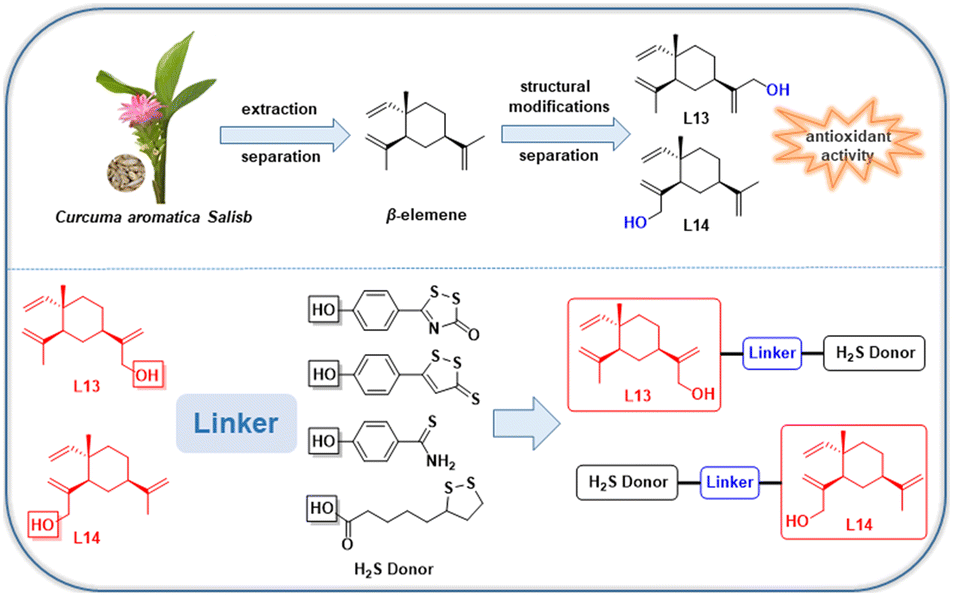 | ||
| Fig. 2 Design strategy for “natural product-active gas donor” hybrid molecules. | ||
2. Results and discussion
2.1 Chemistry
The chemical synthesis of the target compounds L13-1a and L13-1b was performed according to the procedure shown in Scheme 1. Commercial p-hydroxybenzonitrile (W1a) was reacted with thioacetamide to produce p-hydroxythiobenzamide (TBZ). The cyclization reaction of TBZ with chlorocarbonyl sulfenyl chloride gave the sulfenylTCM donor (W1c). The reaction of W1c with bromoethanol, and subsequently succinic anhydride gave intermediate W1e, which underwent esterification with L13 to afford the target product L13-1a. Target compound L13-1b was prepared in a similar manner as described above.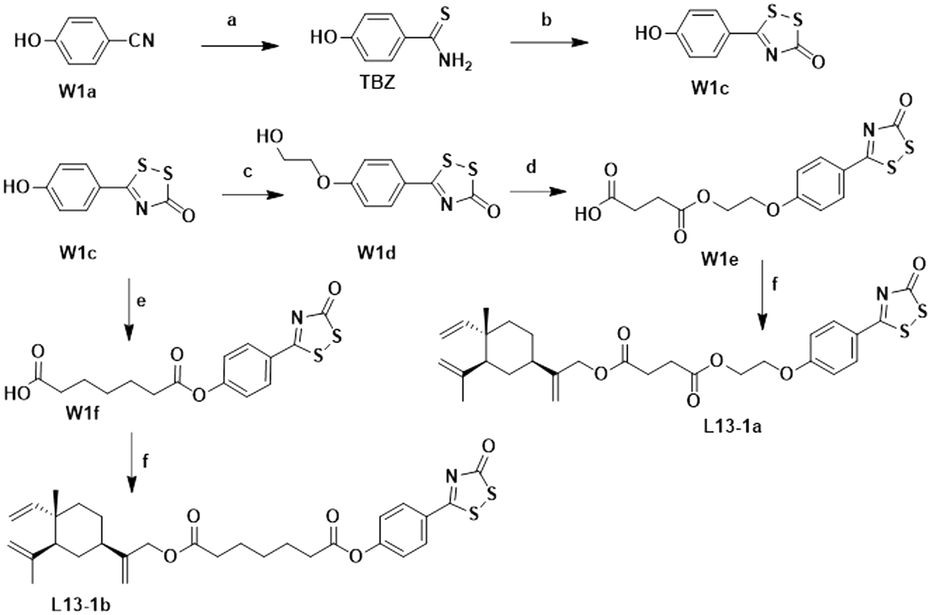 | ||
| Scheme 1 Synthesis of compounds L13-1a and L13-1b. Reagents and conditions: (a) thioacetamide, HCl, 50 °C, 4 h, 95%; (b) chlorocarbonyl sulfenyl chloride, THF, rt, 16 h, 36%; (c) ethylene bromohydrin, K2CO3, DMF, 90 °C, 6 h, 47%; (d) succinic anhydride, DMAP, TEA, DCM, 100 °C, 4 h, 84%; (e) heptanedioic acid, EDCI, DMAP, DCM, rt, 3 h, 58%; and (f) L13, EDCI, DMAP, DCM, rt, 2 h, 55–60%. | ||
W2a was demethylated by the reaction of boron tribromide, and subsequently reacted with bromoethanol to yield intermediate W2c. This intermediate was esterified with succinic anhydride, maleic anhydride, phthalic anhydride and 3-fluorophthalic anhydride to obtain intermediates W-L1–W-L4 with extended carbon chains, respectively. Intermediates W-L6 and W-L7 were obtained by reacting W2b with heptanedioic acid or octanedioic acid, respectively. Intermediate W-5L was prepared in a similar manner to W-L1 (Scheme 2). The esterification reactions of the above-mentioned intermediates with the lead compound (L13) produced the target compounds L13-2a–L13-2d and L13-2g–L13-2h, respectively (Scheme 3).
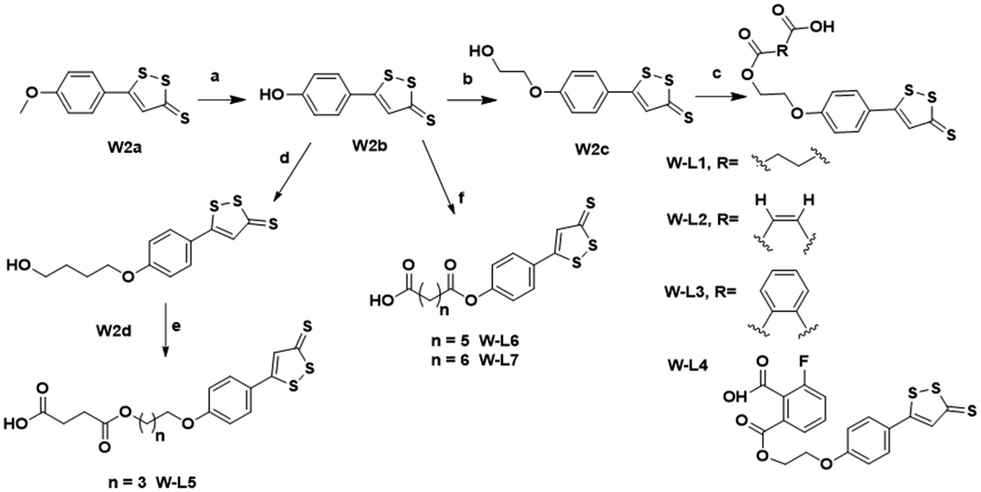 | ||
| Scheme 2 Synthesis of compounds W-L1–W-L7. Reagents and conditions: (a) BBr3, DCM, −78 °C, 1 h, 90%; (b) ethylene bromohydrin, K2CO3, DMF, 90 °C, 6 h, 31%; (c) succinic anhydride/maleic anhydride/phthalic anhydride/3-fluorophthalic anhydride, DMAP, TEA, DCM, 100 °C, 4 h, 39–80%; (d) 4-bromo-1-butanol, K2CO3, DMF, 90 °C, 6 h, 31%; (e) succinic anhydride, DMAP, TEA, DCM, 100 °C, 4 h, 39%; (f) heptanedioic acid/suberic acid, EDCI, DMAP, DCM, rt, 3 h, 51–59%. | ||
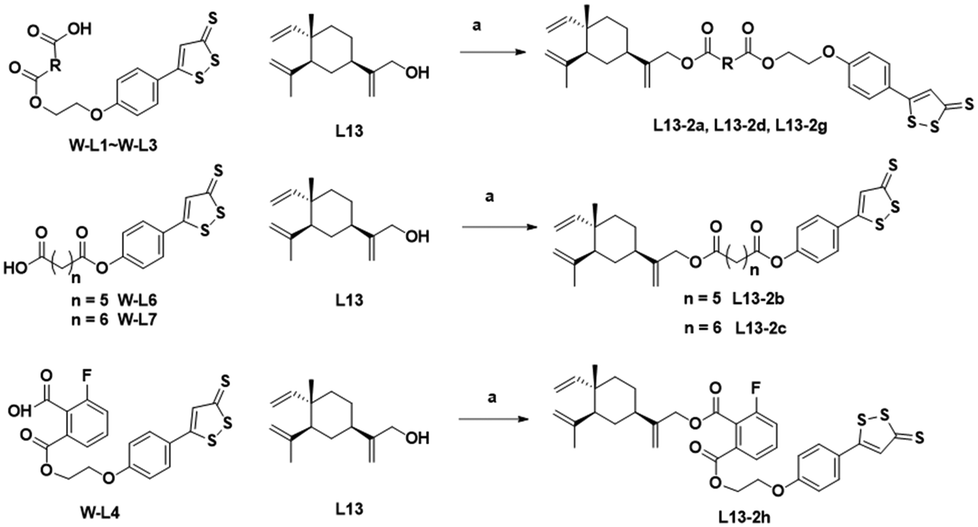 | ||
| Scheme 3 Synthesis of compounds L13-2a–L13-2d and L13-2g–L13-2h. Reagents and conditions: (a) EDCI, DMAP, DCM, rt, 2 h, 22–61%. | ||
The lead compound (L13) underwent esterification with Boc-L-isoleucine to obtain intermediate W4a, in which the Boc protecting group was deprotected with trifluoroacetic acid to give compound W-L8. Intermediates W-L1 and W-L5 yielded the target compounds L13-2e and L13-2f by the amide condensation reaction with intermediate W-L8, respectively. Intermediate W-L1 was ester condensed with the lead compound (L14) to afford the target compound L14-2a. Target compound L14-2b was prepared in a similar manner to L13-2f (Scheme 4).
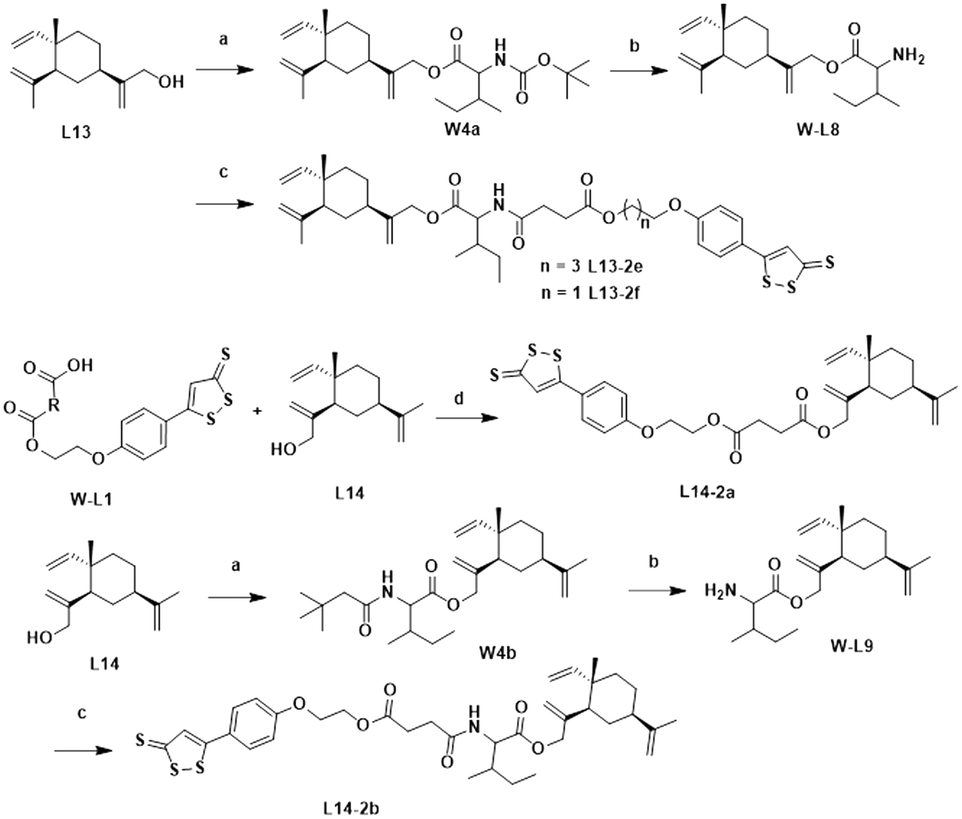 | ||
| Scheme 4 Synthesis of compounds L13-2e–L13-2f and L14-2a–L14-2b. Reagents and conditions: (a) Boc-L-isoleucine, EDCI, DMAP, DCM, rt, 4–10 h, 82–91%; (b) TFA, DCM, rt, 30 min; 77–84%; (c) W-L1/W-L5, EDCI, DMAP, DCM, rt, 2 h, 39–45% and (d) EDCI, DMAP, DCM, rt, 2 h, 61%. | ||
The synthesis of the target products L13-3a–L13-3d with different chain lengths was completed according to Scheme 5. Target compound L13-3b was obtained by linking L13 with TBZvia heptanedioic acid. Then, TBZ was linked with dibromoalkanes of different chain lengths, and these compounds underwent ether-forming reactions with L13 to obtain target compounds L13-3a, L13-3c and L13-3d. As depicted in Scheme 6, target compounds L13-4a and L14-4a were obtained by ester condensation of α-thioctic acid (W6a) with L13 and L14. Similarly, target compounds L13-4b and L14-4b were obtained as described above.
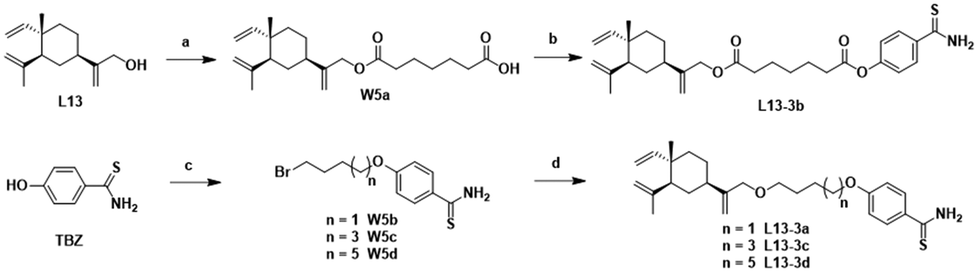 | ||
| Scheme 5 Synthesis of compounds L13-3a–L13-3d. Reagents and conditions: (a) heptanedioic acid, EDCI, DMAP, DCM, rt, 3 h, 61%; (b) TBZ, EDCI, DMAP, DCM, rt, 2 h, 36%; (c) dibromoalkanes, K2CO3, acetone, 80 °C, 6 h, 27–41%; and (d) L13, K2CO3, acetonitrile, 80 °C, 6 h, 33–57%. | ||
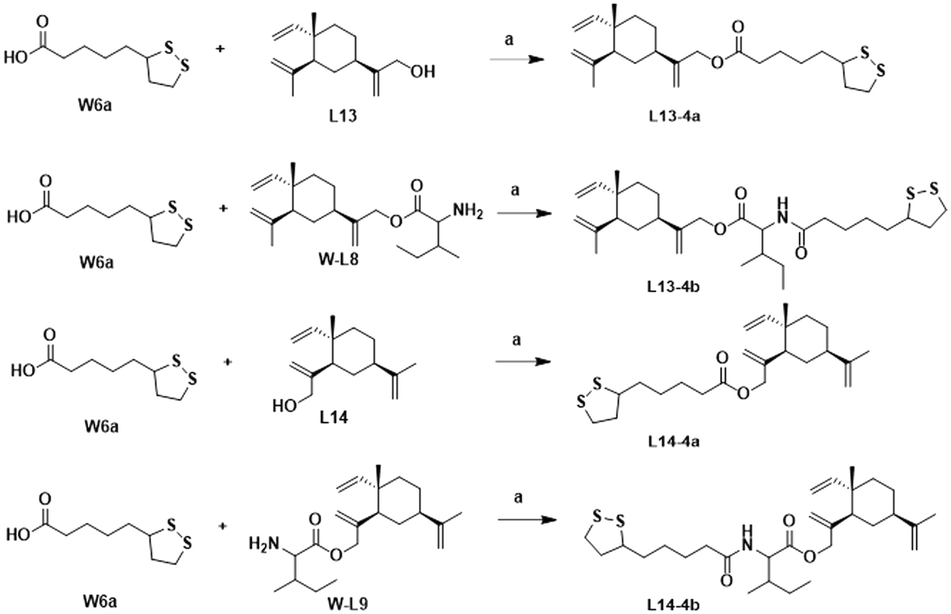 | ||
| Scheme 6 Synthesis of compounds L13-4a–L13-4b and L14-4a–L14-4b. Reagents and conditions: (a) EDCI, DMAP, DCM, rt, 2–6 h, 33–75%. | ||
2.2 H2S-releasing test for target compounds in phosphate buffer solution
To date, the widely used H2S detection techniques include electrochemical, bioluminescence, colorimetric and chromatographic methods. Accordingly, we detected the H2S-release levels of the target compounds using the methylene blue colorimetric assay. A standard curve was established using NaHS to test the H2S release from 100 μM of compound in phosphate buffer at 37 °C. The results showed that all the tested compounds could release H2S under L-cysteine trigger, and the maximum release concentration was reached in about 2 min, basically ensuring a sustained release time of about 40 min. Among them, the H2S-release concentration of α-thioctic acid as the H2S donor was generally relatively low, with 100 μM α-thioctic acid only achieving about 20 μM H2S release. The H2S release concentration of sulfenylTCMs as the H2S donor was significantly better for the donor directly linked by an ester bond than that linked by an ether bond. The maximum release concentration of H2S obtained by TBZ as the H2S donor was stable between 50 and 100 μM, with compound 13-3b possessing the best release concentration. Although the best H2S-release level was achieved with ADT-OH as the H2S donor, compounds L13-2a, L13-2f, L13-2g and L13-2h with an ether bond linkage could release an H2S concentration of almost 100 μM. Comparing the target compounds L13-2f and L14-2b with the lead compounds, their H2S release concentration was 2-fold higher than that of lead compounds L13 and L14, and the results were consistent with the comparison of target compounds L13-2a and L14-2a with the lead compounds (Fig. 3).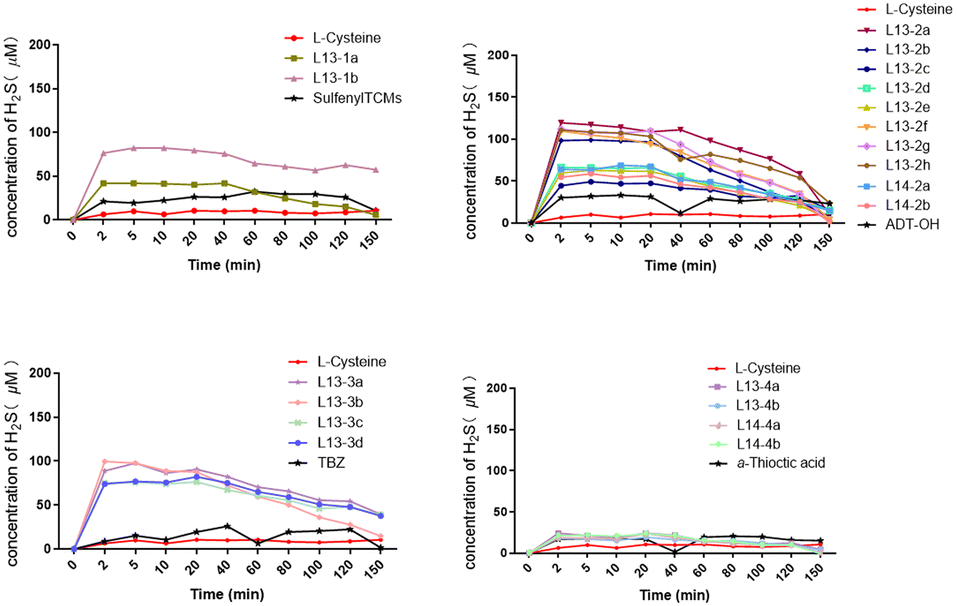 | ||
| Fig. 3 H2S release from the target compounds in the phosphate buffer solution assay. | ||
2.3 In vitro vasodilator activity assay of target compounds
The regulation of vasodilation prevents the formation of atherosclerotic plaques, which can be controlled and reversed. Thus, to confirm the synergistic effect of the “β-elemene-H2S donor” hybrid molecular on in vitro vasodilation, we tested the effect of the compounds on isolated vascular vessels and performed tension analysis. We explored the vasoconstrictor inhibitory effect of the target compounds using the classical KCl-induced vasoconstriction assay and ADT-OH was used as the control. Freshly obtained rat thoracic aorta was suspended from the vascular tone measurement system and the vascular tone data were read in real time under incubation with different compound concentrations. The results of the vasodilation rate calculation showed that all the β-elemene H2S donor-type derivatives exhibited different degrees of vasodilatory activity and showed good dose dependence compared to the control (Table 1). Among them, compound L13-2h was the most effective, producing 5–6 times higher vasodilatory activity than L13 and ADT-OH at low concentrations, and over 50% vasodilatory activity at a concentration of 20 μM. However, the vasodilatory activity of the target compounds with both α-thioctic acid and TZB as H2S donors showed poor vasodilatory activity and lacked any apparent vasodilatory effect at low concentrations. Comparing compounds L13-1a and L13-1b, by introducing an ether bond to connect the H2S donor in the target compounds apparently enhanced their vasodilatory activity. The derivatives of the two lead compounds (L13 and L14) showed no apparent difference in vasodilatory activity at the tested concentrations.| Compd. | Relative vasodilation percentagea (RVP, %) | ||||
|---|---|---|---|---|---|
| 1 μM | 5 μM | 10 μM | 20 μM | 40 μM | |
| a Vasodilatation rate (%) is the ratio of the maximum vascular tension with the addition of KCl minus the vascular tension upon administration to the maximum vascular tension with the addition of KCl minus the vascular ring preload. The data are expressed as the mean of four independent experiments. Values are expressed as mean ± SD, n = 4. | |||||
| ADT-OH | 3.08 ± 5.24 | 5.39 ± 12.56 | 42.24 ± 9.73 | 61.69 ± 8.97 | 75.01 ± 9.14 |
| L13 | 0.09 ± 0.43 | 4.63 ± 1.20 | 5.63 ± 0.88 | 27.81 ± 20.10 | 53.06 ± 13.51 |
| L13-1a | −0.71 ± 3.73 | 10.11 ± 8.05 | 17.84 ± 11.51 | 26.82 ± 7.47 | 34.29 ± 4.65 |
| L13-1b | −0.40 ± 3.74 | 4.76 ± 8.49 | 2.38 ± 3.72 | 7.08 ± 8.72 | 8.22 ± 13.94 |
| L13-2a | 1.27 ± 3.99 | 6.87 ± 4.46 | 11.63 ± 2.89 | 12.67 ± 4.48 | 12.14 ± 4.52 |
| L13-2b | −1.15 ± 7.07 | 6.64 ± 4.82 | 6.99 ± 11.96 | 16.08 ± 9.00 | 21.17 ± 6.00 |
| L13-2c | 12.81 ± 15.64 | 5.04 ± 18.73 | 0.55 ± 13.72 | 7.96 ± 8.26 | 14.68 ± 8.10 |
| L13-2d | −4.07 ± 8.67 | 1.54 ± 7.71 | 7.28 ± 6.35 | 12.57 ± 7.28 | 10.78 ± 11.82 |
| L13-2e | 9.16 ± 6.44 | 11.89 ± 7.09 | 16.62 ± 8.06 | 15.38 ± 5.25 | 18.44 ± 5.27 |
| L13-2f | 1.70 ± 5.38 | 3.38 ± 11.41 | −6.76 ± 12.03 | 4.83 ± 8.09 | 14.29 ± 6.52 |
| L13-2g | −2.99 ± 13.31 | 10.01 ± 18.25 | 17.00 ± 14.09 | 17.46 ± 8.75 | 28.25 ± 10.21 |
| L13-2h | 18.12 ± 9.23 | 25.08 ± 9.79 | 38.87 ± 7.96 | 51.85 ± 7.02 | 61.41 ± 7.75 |
| L13-3a | 4.61 ± 2.89 | 4.13 ± 13.54 | 17.37 ± 11.52 | 15.38 ± 9.92 | 30.23 ± 8.96 |
| L13-3b | 1.20 ± 1.41 | 4.53 ± 2.02 | 2.67 ± 4.23 | 4.19 ± 4.42 | 6.38 ± 4.32 |
| L13-3c | 6.68 ± 7.68 | 4.72 ± 7.22 | 0.07 ± 10.57 | 10.31 ± 3.22 | 12.18 ± 11.15 |
| L13-3d | −4.54 ± 9.52 | 1.71 ± 2.99 | 8.86 ± 3.21 | 12.44 ± 7.80 | 19.64 ± 6.42 |
| L13-4a | −3.41 ± 6.59 | −5.69 ± 18.59 | 6.38 ± 2.39 | 8.99 ± 1.26 | 11.51 ± 1.94 |
| L13-4b | −5.90 ± 13.31 | −8.16 ± 13.91 | 2.19 ± 13.31 | 1.21 ± 11.07 | 14.47 ± 12.33 |
| L14-2a | 7.47 ± 7.81 | −8.64 ± 41.15 | 14.98 ± 17.17 | 13.20 ± 18.34 | 7.43 ± 10.73 |
| L14-2b | 0.64 ± 3.11 | 2.75 ± 4.03 | 6.75 ± 6.54 | 7.93 ± 5.68 | 13.00 ± 5.68 |
| L14-4a | −19.80 ± 41.52 | −2.40 ± 29.49 | 20.75 ± 11.79 | 24.52 ± 17.62 | 17.27 ± 32.01 |
| L14-4b | 1.10 ± 2.69 | 2.78 ± 6.91 | −3.60 ± 12.56 | 5.16 ± 13.36 | 14.67 ± 10.58 |
2.4 Validation of the mechanism of potassium channel activation by L13-2h
Glyburide is an ATP-sensitive potassium channel (KATP) blocker that antagonizes the vasodilatory effect induced by potassium channel opening. Therefore, we used glyburide-pretreated aortic vascular rings to verify whether the “β-elemene-H2S donor” hybrid molecular compounds produce vasodilatory activity by activating the KATP channel. We validated the potassium channel mechanism with the H2S donor ADT-OH as well as the optimal compound L13-2h (Fig. 4). The results showed a reduction in ADT-OH vasodilatory activity after its co-incubation with glyburide, while there was no significant difference in the activity of L13-2h. It is speculated that this class of compounds may involve a mechanism of action that is less likely to produce vasodilatory activity by opening potassium channels on vascular smooth muscle, and thus other mechanisms of action need to be investigated.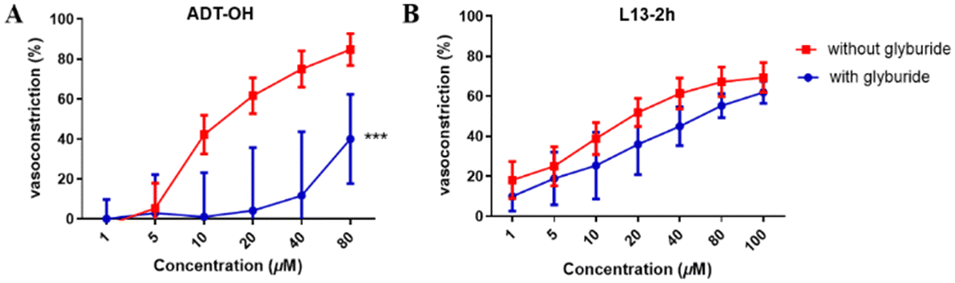 | ||
| Fig. 4 (A) Effect of compound ADT-OH on vasodilation with and without glyburide treatment. (B) Effect of compound L13-2h on vasodilation before and after glyburide treatment. Values are expressed as mean ± SD, n = 4; ***p < 0.001 compared with the respective control. | ||
2.5 Effect of compounds on ox-LDL-stimulated lipolysis in macrophages
Oxidized low-density lipoprotein (ox-LDL) is an important factor in the pathogenesis of AS, which induces macrophages to form foam cells and destroy endothelial cells, ultimately leading to plaque accumulation in the lumen of blood vessels.26 The compound to be tested (50 μM) was incubated with ox-LDL-stimulated macrophages RAW 264.7 for 24 h. Lipids were stained with oil red O (ORO), and then observed under a microscope (Fig. 5). The ImagePro Plus 6.0 software was employed to quantify the percentage of ORO-positive regions, which yielded information on the cellular lipid-lowering activity of the compounds (Fig. 6). As an illustration of the results of the studies induced by 50 μg mL−1 ox-LDL, significant lipid-lowering activity was present for the lead compound L13 and the other thirteen compounds. Compared with the lead compound L13, compounds L13-1a, L13-1b, L13-2g, L13-3d and L13-2h showed superior or similar lipid-lowering activity. The lipid-lowering activity of the compounds was correlated with their chain length, where in the case of a carbon chain length of 7–8 atoms, it was found that the activity of the compounds was significantly potent.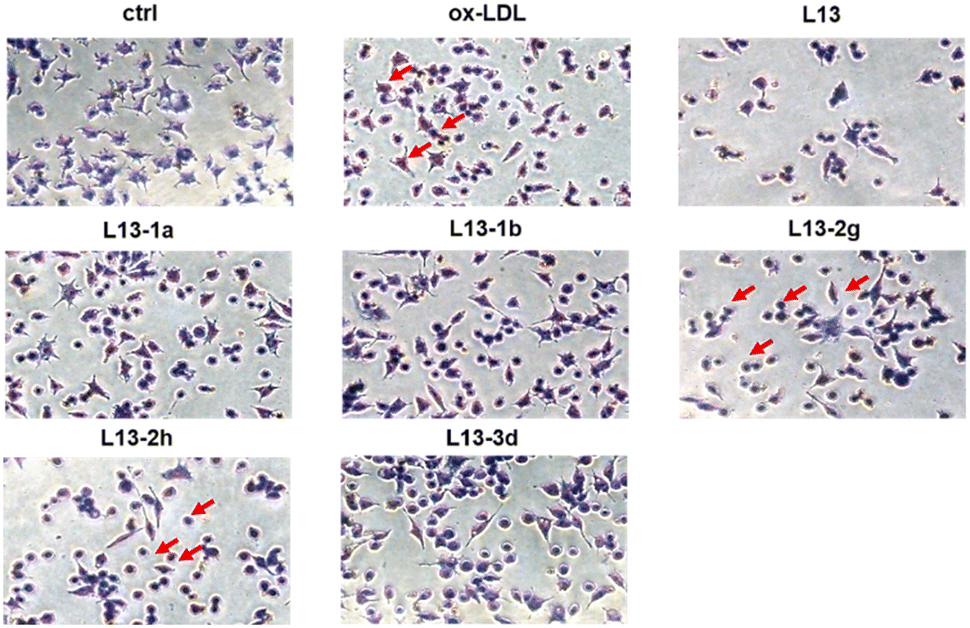 | ||
| Fig. 5 Inhibitory effect of compounds L13, L13-1a, L13-1b, L13-2g, L13-2h, and L13-3d on lipidswallow in macrophages stimulated by ox-LDL at 50 μg mL−1. Oil red stains lipids in cells. Oxidized low-density lipoprotein-induced model groups have a large accumulation of intracellular lipids, and lipid content decreases after treatment with the target compounds. | ||
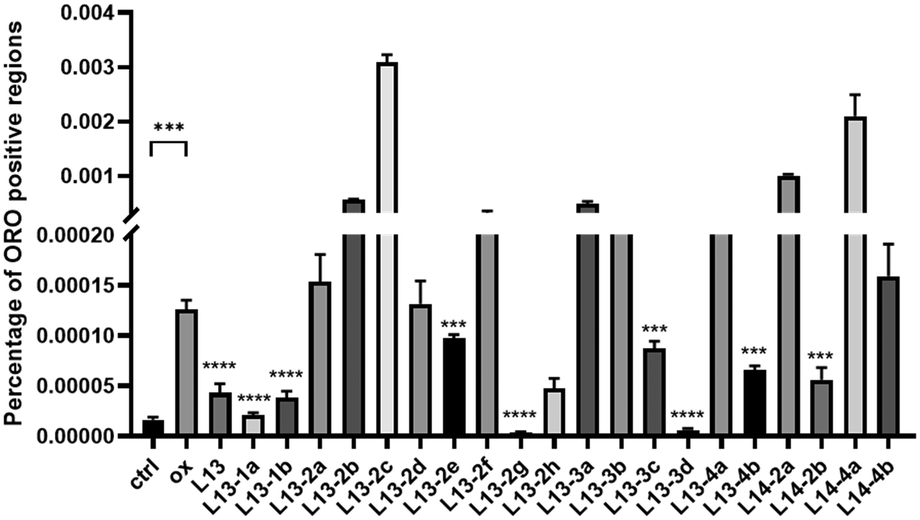 | ||
| Fig. 6 Inhibitory effect of target compound on lipidswallow in macrophages stimulated by ox-LDL at 50 μg mL−1, ORO positive region ratio. Values are expressed as mean ± SD, n = 3; compared with ox, ***p < 0.001, ****p < 0.0001. | ||
2.6 Antioxidant activity of representative compounds L13-2g and L13-2h
Based on the results of the above-mentioned experiments, compounds L13-2g and L13-2h were selected as representative compounds to further test the antioxidant damage effect at the cellular level with an H2O2-induced oxidative damage model in human umbilical vein endothelial cells (HUVECs) (Fig. 7). The compounds were preincubated with HUVECs at different concentrations (0.5 μM, 5 μM and 20 μM) for 24 h. The cells were stimulated with 1 mM H2O2 for 2 h, and then their absorbance was measured using the MTT method to calculate the cell viability. Trolox was used as the positive control, and the results showed that representative compounds L13-2g, L13-2h, and L13 and the H2S donor ADT-OH all ameliorated H2O2-induced oxidative damage in the HUVEC cells. Surprisingly, the representative compound L13-2h maintained antioxidant damage activity comparable to that of lead compound L13 and superior to the positive control after the hydroxyl group of lead compound L13 was substituted. | ||
| Fig. 7 Effect of target compounds on the survival rate (%) of HUVEC cells induced by H2O2 injury; values are expressed as mean ± SD, n = 5; *p < 0.05 compared with the model group. | ||
2.7 Cytotoxicity of the compounds
We expected the target compounds to have a favourable cellular lipid-lowering effect without affecting the growth of normal cells, and thus we tested the cytotoxicity of the target compounds. Mouse bone marrow-derived macrophages (BMDM) and HUVECs were used as the test cells (Table 2 and Fig. 8). BMDM was selected as the test cells and L13 was employed as the control, and the cytotoxicity of the compounds was measured by the MTT method after 24 h of administration. The values showed that compounds L13-3a, L13-3c and L13-3d with TBZ as the H2S donor exhibited low cytotoxicity at a concentration of 100 μM, while all the compounds presented no significant cytotoxicity at low concentrations (Table 2). Simultaneous cytotoxicity assays showed that the compounds under test displayed no significant cell-killing effect against HUVECs at the starting concentration and higher concentrations, indicating a certain degree of safety (Fig. 8).| Compd. | BMDM cell viabilitya (%) | |
|---|---|---|
| 50 μM | 100 μM | |
| a Values are expressed as mean ± SD, n = 4. | ||
| L13 | 274.83 ± 7.96 | 281.38 ± 6.82 |
| L13-1a | 265.86 ± 7.32 | 253.79 ± 5.77 |
| L13-1b | 322.76 ± 8.04 | 390.69 ± 4.83 |
| L13-2a | 264.48 ± 4.87 | 348.96 ± 4.74 |
| L13-2b | 232.07 ± 4.61 | 458.62 ± 3.35 |
| L13-2c | 188.62 ± 3.14 | 202.41 ± 7.93 |
| L13-2d | 98.97 ± 2.59 | 89.66 ± 2.99 |
| L13-2e | 265.52 ± 2.54 | 207.59 ± 1.81 |
| L13-2f | 295.02 ± 4.32 | 583.80 ± 7.38 |
| L13-2g | 349.22 ± 5.24 | 454.21 ± 4.62 |
| L13-2h | 192.84 ± 3.43 | 347.35 ± 4.39 |
| L13-3a | 163.24 ± 3.21 | 14.95 ± 0.69 |
| L13-3b | 176.95 ± 3.89 | 218.69 ± 5.59 |
| L13-3c | 279.44 ± 3.75 | 17.13 ± 0.87 |
| L13-3d | 74.46 ± 1.51 | 27.10 ± 0.59 |
| L13-4a | 296.26 ± 1.39 | 377.57 ± 2.26 |
| L13-4b | 207.72 ± 1.65 | 268.42 ± 8.49 |
| L14-2a | 243.86 ± 5.34 | 262.46 ± 4.79 |
| L14-2b | 268.77 ± 7.73 | 271.58 ± 5.50 |
| L14-4a | 156.84 ± 3.41 | 335.79 ± 7.26 |
| L14-4b | 671.58 ± 16.79 | 882.11 ± 14.29 |
 | ||
| Fig. 8 Effect of representative compounds on the survival rate (%) of normal HUVEC cells; values are expressed as mean ± SD, n = 3. | ||
2.8 Water solubility test for compound L13-2h
Natural products usually have poor water solubility, and in this case, structural modifications may improve their water solubility. Accordingly, we determined the water solubility of the optimal compound L13-2h using HPLC, and the result showed that its water solubility was 40 μg mL−1, which has certain water solubility. Meanwhile, compound L13-2h significantly increased the water solubility compared to the parent compound β-elemene.273. Conclusion
In this work, we combined two β-elemene derivatives, 13-β-elemene and 14-β-elemene, with four types of H2S donors as novel hybrid molecules, respectively. Also, for the first time, we designed and synthesized twenty novel β-elemene derivatives with different H2S donors. The pharmacological studies verified that all the target compounds were effective in releasing H2S in phosphate buffer solution. Among them, the best H2S-releasing level was achieved with ADT-OH as the H2S donor, where compounds L13-2a, L13-2f, L13-2g and L13-2h with an ether bond linkage could release an H2S concentration of almost 100 μM. Meanwhile, the results of the isolated vasodilation assay showed that the representative compound L13-2h exhibited the best vasodilatory activity with good dose dependence, and at a concentration of 20 μM, more than 50% vasodilator activity was achieved, which was significantly better than that of lead compound L13. Meantime, the derivatives of the two lead compounds (L13 and L14) showed no apparent difference in vasodilatory activity at the tested concentrations. Moreover, thirteen β-elemene derivatives showed significant cellular lipid-lowering activity. Compared with lead compound L13, compounds L13-2h, L13-2g, L13-1a, L13-1b and L13-3d showed superior or similar lipid-lowering activity, among which, L13-2g possessed the best activity. The results of the further antioxidant activity study indicated that representative compounds L13-2g and L13-2h could improve H2O2-induced oxidative damage in HUVEC cells, and compound L13-2h showed a superior protective effect against oxidative damage compared to the positive control. Meanwhile, none of the compounds showed significant cytotoxicity at the tested concentrations. In summary, the novel dual-functional “β-elemene-H2S gas donor” hybrid molecules exerted synergistic pharmacological activities and significantly enhanced the activity of the lead compound L13, which rationally demonstrated the potential significance of β-elemene and its “β-elemene-H2S gas donor” hybrid molecules in the treatment of AS.4. Experimental
4.1 Chemistry
In the experiments, the NMR instrument was a BRUKER AV-300 or BRUKER AV-400, the solvent was CDCl3 or DMSO-d6, TMS was the internal standard, the chemical shift unit is ppm, the coupling constant unit is Hz, and the signal peaks are expressed as single (s), double (d), triple (t), quadruple (q), broad single (brs), and multiple (m) peaks. The mass spectrometer was an Agilent 1100-LC-MSD-Trap/SL model or FTMS-2000 model. Thin layer chromatography plates were made of commercially available Yantai Chemical Research Institute silica gel GF254 prefabricated plates with 254 nm UV light color development. The reagents were commercially available chemically pure or analytically pure products, and anhydrous treatment was performed when necessary.2-((1R,3S,4S)-4-Methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl (2-(4-(3-oxo-3H-1,2,4-dithiazol-5-yl)phenoxy)ethyl) succinate (L13-1a). Yellow oil, yield 55%. 1H NMR (300 MHz, chloroform-d) δ 7.60 (d, J = 8.8 Hz, 2H), 6.96 (d, J = 8.8 Hz, 2H), 5.81 (dd, J = 17.7, 10.5 Hz, 1H), 5.03 (s, 1H), 5.00 (s, 1H), 4.93 (d, J = 4.2 Hz, 1H), 4.88 (s, 1H), 4.83 (s, 1H), 4.59 (s, 3H), 4.46 (d, J = 4.8 Hz, 2H), 4.22 (d, J = 4.8 Hz, 2H), 2.69 (s, 4H), 2.01 (t, J = 8.0 Hz, 2H), 1.71 (s, 3H), 1.50–1.44 (m, 2H), 1.28 (s, 2H), 1.25 (s, 2H), 1.00 (s, 3H). 13C NMR (75 MHz, chloroform-d) δ 172.1, 171.8, 161.6, 149.9, 148.1, 147.3, 134.0, 119.0, 115.2, 112.2, 110.9, 110.0, 104.4, 66.43, 66.0, 62.5, 52.6, 41.7, 39.7, 39.7, 33.0, 29.0, 28.9, 27.0, 24.8, 16.5. ESI-MS m/z: 580.2 [M + Na]+.
1-(2-((1R,3S,4S)-4-Methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl) 7-(4-(3-oxo-3H-1,2,4-dithiazol-5-yl)phenyl) heptanedioate (L13-1b). Yellow solid, yield 60%, m.p. 58–60 °C. 1H NMR (300 MHz, chloroform-d) δ 8.19–8.08 (m, 2H), 7.33–7.26 (m, 2H), 5.78 (d, J = 10.7 Hz, 1H), 5.03 (d, J = 11.7 Hz, 2H), 4.96–4.90 (m, 1H), 4.88 (s, 1H), 4.83 (t, J = 1.8 Hz, 1H), 4.59 (d, J = 3.4 Hz, 3H), 2.62 (t, J = 7.4 Hz, 2H), 2.40 (t, J = 7.4 Hz, 2H), 2.02 (dt, J = 10.9, 5.5 Hz, 2H), 1.71 (s, 7H), 1.48 (d, J = 2.9 Hz, 6H), 1.27 (d, J = 6.3 Hz, 2H), 1.01 (s, 3H); 13C NMR (75 MHz, chloroform-d) δ 186.8, 185.8, 173.2, 171.1, 155.8, 150.02, 148.5, 147.4, 129.8, 128.8, 122.8, 112.3, 111.0, 110.1, 52.7, 41.9, 39.8, 39.7, 34.1, 34.1, 33.1, 28.5, 27.1, 24.8, 24.5, 24.4, 16.6. ESI-MS m/z: 578.3 [M + Na]+.
2-((1R,3S,4S)-4-Methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl (2-(4-(3-thioxo-3H-1,2-dithiol-5-yl)phenoxy)ethyl) succinate (L13-2a). Orange solid, yield 55%, m.p. 60–62 °C. 1H NMR (400 MHz, chloroform-d) δ 7.62 (d, J = 8.8 Hz, 2H), 7.40 (s, 1H), 6.99 (d, J = 8.8 Hz, 2H), 5.81 (dd, J = 17.8, 10.5 Hz, 1H), 5.04 (d, J = 1.3 Hz, 1H), 5.00 (s, 1H), 4.95–4.91 (m, 1H), 4.89 (t, J = 1.3 Hz, 1H), 4.83 (t, J = 1.7 Hz, 1H), 4.60 (s, 2H), 4.59–4.57 (m, 1H), 4.50–4.46 (m, 2H), 4.24 (dd, J = 5.4, 4.1 Hz, 2H), 2.70 (s, 4H), 2.01 (dd, J = 10.1, 5.9 Hz, 2H), 1.71 (d, J = 0.7 Hz, 3H), 1.60–1.57 (m, 2H), 1.46 (dd, J = 5.9, 3.1 Hz, 2H), 1.29–1.24 (m, 2H), 1.00 (s, 3H); 13C NMR (75 MHz, chloroform-d) δ 215.1, 172.8, 172.1, 171.9, 161.7, 149.9, 148.2, 147.3, 134.7, 128.6, 124.6, 115.5, 112.3, 110.9, 110.1, 66.4, 66.1, 62.6, 52.6, 41.7, 39.8, 39.7, 33.0, 29.0, 28.9, 27.0, 24.8, 16.5. ESI-MS m/z: 595.2 [M + Na]+.
1-(2-((1R,3S,4S)-4-Methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl) 7-(4-(3-thioxo-3H-1,2-dithiol-5-yl)phenyl) heptanedioate (L13-2b). Orange oil, yield 55%. 1H NMR (300 MHz, chloroform-d) δ 7.68 (d, J = 8.7 Hz, 2H), 7.40 (s, 1H), 7.22 (d, J = 8.7 Hz, 2H), 5.81 (dd, J = 17.8, 10.5 Hz, 1H), 5.12–4.97 (m, 2H), 4.92 (d, J = 3.6 Hz, 1H), 4.88 (d, J = 1.0 Hz, 1H), 4.82 (s, 1H), 4.59 (s, 2H), 4.58 (s, 1H), 2.60 (t, J = 7.4 Hz, 2H), 2.40 (t, J = 7.4 Hz, 2H), 2.01 (dd, J = 10.4, 5.6 Hz, 2H), 1.78 (dd, J = 13.7, 6.1 Hz, 4H), 1.70 (s, 3H), 1.65–1.57 (m, 4H), 1.53–1.42 (m, 4H), 1.00 (s, 3H). 13C NMR (75 MHz, chloroform-d) δ 215.4, 173.2, 171.7, 171.4, 153.6, 149.9, 148.4, 147.3, 136.0, 129.1, 128.2, 122.9, 112.3, 110.9, 110.1, 66.0, 52.6, 41.8, 39.8, 39.7, 34.1, 33.0, 28.5, 27.0, 24.8, 24.5, 24.4, 16.6. ESI-MS m/z: 593.2 [M + Na]+.
1-(2-((1R,3S,4S)-4-Methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl) 8-(4-(3-thioxo-3H-1,2-dithiol-5-yl)phenyl) octanedioate (L13-2c). Orange solid, yield 22%, m.p. 40–42 °C. 1H NMR (300 MHz, chloroform-d) δ 7.67 (d, J = 8.7 Hz, 2H), 7.40 (s, 1H), 7.22 (d, J = 8.7 Hz, 2H), 5.80 (dd, J = 17.8, 10.5 Hz, 1H), 5.04 (d, J = 1.4 Hz, 1H), 5.00 (s, 1H), 4.95–4.90 (m, 1H), 4.89–4.85 (m, 1H), 4.82 (t, J = 1.7 Hz, 1H), 4.58 (d, J = 3.1 Hz, 3H), 2.59 (t, J = 7.5 Hz, 2H), 2.37 (t, J = 7.5 Hz, 2H), 2.05–1.95 (m, 2H), 1.83–1.72 (m, 2H), 1.70 (t, J = 1.0 Hz, 3H), 1.68 (s, 2H), 1.61 (s, 2H), 1.58 (d, J = 1.5 Hz, 2H), 1.43 (s, 2H), 1.41 (d, J = 3.2 Hz, 2H), 1.34 (t, J = 7.3 Hz, 2H), 1.00 (s, 3H). 13C NMR (75 MHz, chloroform-d) δ 215.5, 173.4, 171.8, 171.6, 153.6, 150.0, 148.4, 147.4, 136.0, 129.1, 128.2, 122.9, 112.3, 110.9, 110.1, 66.0, 64.9, 52.6, 41.8, 39.8, 34.2, 33.0, 28.7, 27.0, 24.8, 24.7, 24.6, 19.0, 16.5, 13.7. ESI-MS m/z: 607.2 [M + Na]+.
2-((1R,3S,4S)-4-Methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl (2-(4-(3-thioxo-3H-1,2-dithiol-5-yl)phenoxy)ethyl) maleate (L13-2d). Orange solid, yield 61%, m.p. 40–42 °C. 1H NMR (300 MHz, chloroform-d) δ 7.63 (d, J = 8.8 Hz, 2H), 7.39 (s, 1H), 6.99 (d, J = 8.8 Hz, 2H), 6.94–6.90 (m, 2H), 5.81 (dd, J = 17.8, 10.5 Hz, 1H), 5.10–5.00 (m, 2H), 4.96–4.91 (m, 1H), 4.88 (q, J = 1.4 Hz, 1H), 4.82 (t, J = 1.7 Hz, 1H), 4.71 (s, 2H), 4.63–4.56 (m, 3H), 4.30 (dd, J = 5.5, 3.7 Hz, 2H), 2.11–1.97 (m, 2H), 1.70 (t, J = 1.0 Hz, 3H), 1.50–1.43 (m, 2H), 1.42–1.31 (m, 2H), 1.25 (s, 2H), 1.00 (s, 3H). 13C NMR (100 MHz, chloroform-d) δ 215.1, 172.7, 164.7, 164.4, 161.5, 149.9, 147.7, 147.3, 134.8, 134.2, 133.0, 128.6, 124.7, 115.5, 112.3, 111.7, 110.1, 67.0, 65.9, 63.2, 52.3, 41.7, 39.7 (d, J = 1.6 Hz), 33.0, 29.7, 27.0, 24.8, 16.5. ESI-MS m/z: 593.2 [M + Na]+.
2-((1R,3S,4S)-4-Methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl 3-methyl-2-(4-oxo-4-(4-(4-(3-thioxo-3H-1,2-dithiol-5yl)phenoxy)butoxy)butanamido) pentanoate (L13-2e). Orange solid, yield 40%, m.p. 40–42 °C. 1H NMR (300 MHz, chloroform-d) δ 7.61 (d, J = 8.8 Hz, 2H), 7.40 (s, 1H), 6.96 (d, J = 8.8 Hz, 2H), 6.17 (d, J = 8.7 Hz, 1H), 5.81 (dd, J = 17.8, 10.5 Hz, 1H), 5.05 (d, J = 10.9 Hz, 2H), 4.96–4.92 (m, 1H), 4.90–4.87 (m, 1H), 4.83 (t, J = 1.7 Hz, 1H), 4.65 (dd, J = 8.5, 5.2 Hz, 3H), 4.58 (d, J = 1.7 Hz, 1H), 4.18 (t, J = 5.9 Hz, 2H), 4.05 (t, J = 5.7 Hz, 2H), 2.67 (t, J = 6.6 Hz, 2H), 2.56 (d, J = 6.9 Hz, 2H), 2.00–1.93 (m, 2H), 1.90–1.83 (m, 3H), 1.71 (d, J = 1.2 Hz, 3H), 1.63 (s, 3H), 1.60 (s, 2H), 1.47 (d, J = 2.8 Hz, 2H), 1.41 (q, J = 4.4 Hz, 2H), 1.17 (ddd, J = 13.6, 6.8, 2.9 Hz, 1H), 1.00 (s, 3H), 0.93 (dd, J = 7.1, 5.0 Hz, 6H). 13C NMR (100 MHz, chloroform-d) δ 215.0, 173.0, 172.8, 171.7, 171.1, 162.3, 149.9, 147.7, 147.2, 134.5, 128.5, 124.1, 115.4 (d, J = 1.9 Hz), 112.3, 111.8, 110.1, 67.7, 66.9, 64.2, 56.5, 52.6, 41.5, 39.7, 39.7, 38.0, 32.9, 30.8, 29.4, 26.9, 25.6, 25.2, 25.1, 24.8, 16.5, 15.5, 11.6. ESI-MS m/z: 736.3 [M + Na]+.
2-((1R,3S,4S)-4-Methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl 3-methyl-2-(4-oxo-4-(2-(4-(3-thioxo-3H-1,2-dithiol-5-yl)phenoxy)ethoxy)butanamido) pentanoate (L13-2f). Orange solid, yield 45%, m.p. 42–44 °C. 1H NMR (300 MHz, chloroform-d) δ 7.63 (d, J = 8.9 Hz, 2H), 7.40 (s, 1H), 7.00 (d, J = 8.9 Hz, 2H), 5.80 (dd, J = 17.8, 10.5 Hz, 1H), 4.94–4.91 (m, 1H), 4.87 (q, J = 1.4 Hz, 1H), 4.82 (d, J = 1.9 Hz, 1H), 4.60–4.57 (m, 1H), 4.49 (dd, J = 5.5, 3.8 Hz, 3H), 4.24 (dd, J = 5.4, 4.0 Hz, 3H), 4.08 (dd, J = 14.1, 6.1 Hz, 3H), 2.72 (s, 4H), 2.20–2.06 (m, 1H), 2.01–1.90 (m, 2H), 1.70 (s, 3H), 1.67–1.59 (m, 2H), 1.53 (d, J = 12.5 Hz, 2H), 1.46–1.43 (m, 2H), 1.31–1.25 (m, 2H), 1.18 (d, J = 1.3 Hz, 3H), 0.98 (s, 6H). 13C NMR (75 MHz, chloroform-d) δ 215.3, 173.1, 173.0, 172.0, 171.3, 162.0, 150.2, 148.0, 147.6, 135.0, 128.9, 124.8, 115.8, 112.6, 112.1, 110.4, 67.3, 66.4, 62.9, 56.8, 52.9, 41.8, 40.0, 40.0, 38.3, 33.2, 31.0, 29.6, 27.3, 25.4, 25.1, 16.9, 15.8, 12.0. ESI-MS m/z: 708.2 [M + Na]+.
2-((1R,3S,4S)-4-Methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl (2-(4-(3-thioxo-3H-1,2-dithiol-5-yl)phenoxy)ethyl) phthalate (L13-2g). Orange solid, yield 22%, m.p. 70–72 °C. 1H NMR (300 MHz, chloroform-d) δ 7.79 (dd, J = 6.0, 3.1 Hz, 1H), 7.72 (dd, J = 5.7, 3.3 Hz, 1H), 7.61 (d, J = 8.6 Hz, 2H), 7.57 (dd, J = 5.7, 3.3 Hz, 2H), 7.39 (s, 1H), 7.03–6.98 (m, 2H), 5.80 (dd, J = 17.8, 10.5 Hz, 1H), 5.10 (d, J = 1.3 Hz, 1H), 5.03 (s, 1H), 4.92 (q, J = 1.4 Hz, 1H), 4.90–4.86 (m, 1H), 4.82 (t, J = 1.8 Hz, 1H), 4.77 (s, 2H), 4.72–4.65 (m, 2H), 4.57 (d, J = 2.0 Hz, 1H), 4.38–4.29 (m, 2H), 2.12–1.96 (m, 2H), 1.70 (d, J = 1.3 Hz, 3H), 1.66–1.59 (m, 2H), 1.52–1.41 (m, 2H), 1.29 (t, J = 11.1 Hz, 2H), 0.99 (s, 3H). 13C NMR (100 MHz, chloroform-d) δ 215.0, 172.7, 167.6, 166.8, 161.7, 149.9, 147.9, 147.3, 134.7, 132.0, 131.6, 131.4, 131.2, 129.1, 128.9, 128.6, 124.5, 115.5, 112.3, 111.3, 110.1, 67.2, 66.0, 63.4, 52.6, 41.7, 39.7, 39.7, 33.0, 27.0, 24.8, 16.6. ESI-MS m/z: 621.2 [M + H]+.
2-(2-((1R,3S,4S)-4-Methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl) 1-(2-(4-(3-thioxo-3H-1,2-dithiol-5-yl)phenoxy)ethyl) 3-fluorophthalate (L13-2h). Orange solid, yield 61%, m.p. 88–90 °C. 1H NMR (300 MHz, chloroform-d) δ 7.81 (d, J = 7.8 Hz, 1H), 7.60 (d, J = 8.8 Hz, 2H), 7.50 (td, J = 8.1, 5.3 Hz, 1H), 7.38 (s, 1H), 7.33 (t, J = 8.7 Hz, 1H), 7.00 (d, J = 8.8 Hz, 2H), 5.80 (dd, J = 17.8, 10.5 Hz, 1H), 5.07 (d, J = 13.3 Hz, 2H), 4.92 (d, J = 2.6 Hz, 1H), 4.87 (d, J = 2.6 Hz, 1H), 4.82 (t, J = 1.8 Hz, 1H), 4.76 (s, 1H), 4.74 (s, 3H), 4.57 (s, 1H), 4.36 (t, J = 4.7 Hz, 2H), 2.00 (dd, J = 11.0, 5.2 Hz, 2H), 1.70 (s, 3H), 1.65–1.59 (m, 2H), 1.52–1.43 (m, 2H), 1.32–1.23 (m, 2H), 0.99 (s, 3H). 13C NMR (100 MHz, chloroform-d) δ 215.1, 172.8, 164.8, 164.2 (d, J = 3.3 Hz), 161.8, 160.4, 157.9, 149.8, 147.5 (d, J = 35.3 Hz), 134.7, 131.2 (d, J = 8.2 Hz), 130.0 (d, J = 3.3 Hz), 128.6, 125.7 (d, J = 3.3 Hz), 124.5, 123.3 (d, J = 19.3 Hz), 120.3 (d, J = 21.4 Hz), 115.6, 112.3, 111.7, 110.2, 67.4, 65.9, 63.7, 52.6, 41.7, 39.7 (d, J = 1.9 Hz), 33.0, 27.0, 24.8, 16.5, 14.1. ESI-MS m/z: 639.2 [M + H]+. HPLC purity 98.4% (tR = 8.22 min).
2-((1R,2S,5R)-2-Methyl-5-(prop-1-en-2-yl)-2-vinylcyclohexyl)allyl (2-(4-(3-thioxo-3H-1,2-dithiol-5-yl)phenoxy)ethyl) succinate (L14-2a). Orange oil, yield 61%. 1H NMR (300 MHz, chloroform-d) δ 7.62 (d, J = 8.8 Hz, 2H), 6.99 (d, J = 8.8 Hz, 2H), 5.75 (dd, J = 17.8, 10.5 Hz, 1H), 5.13 (d, J = 1.5 Hz, 1H), 5.00–4.92 (m, 1H), 4.92–4.84 (m, 2H), 4.71 (t, J = 1.5 Hz, 2H), 4.50 (s, 2H), 4.47 (d, J = 4.8 Hz, 2H), 4.24 (dd, J = 5.4, 4.0 Hz, 2H), 2.69 (s, 4H), 1.98 (dd, J = 21.8, 6.8 Hz, 2H), 1.74 (d, J = 1.3 Hz, 3H), 1.64–1.55 (m, 2H), 1.46 (d, J = 7.5 Hz, 2H), 1.34–1.25 (m, 2H), 0.99 (s, 3H). 13C NMR (75 MHz, chloroform-d) δ 215.1, 172.7, 172.1, 171.7, 161.7, 149.9, 149.2, 145.9, 134.7, 128.6, 124.6, 115.5, 113.4, 111.0, 108.4, 68.4, 66.1, 62.6, 60.3, 48.3, 45.6, 39.6, 32.9, 29.5, 26.5, 21.7, 15.9, 14.2. ESI-MS m/z: 595.2 [M + Na]+.
2-((1R,2S,5R)-2-Methyl-5-(prop-1-en-2-yl)-2-vinylcyclohexyl)allyl 3-methyl-2-(4-oxo-4-(2-(4-(3-thioxo-3H-1,2-dithiol-5-yl)phenoxy)ethoxy)butanamido) pentanoate (L14-2b). Orange oil, yield 45%. 1H NMR (400 MHz, chloroform-d) δ 7.61 (d, J = 8.8 Hz, 2H), 7.38 (s, 1H), 6.98 (d, J = 8.8 Hz, 2H), 6.18 (d, J = 8.6 Hz, 1H), 5.72 (td, J = 17.2, 11.5 Hz, 1H), 5.17 (t, J = 4.8 Hz, 1H), 4.96–4.87 (m, 4H), 4.66–4.59 (m, 1H), 4.58–4.53 (m, 1H), 4.46 (dd, J = 5.7, 4.1 Hz, 3H), 4.23 (dd, J = 5.3, 4.2 Hz, 2H), 2.76–2.67 (m, 2H), 2.60–2.52 (m, 3H), 2.04–1.92 (m, 2H), 1.75–1.61 (m, 2H), 1.55 (s, 3H), 1.44–1.32 (m, 2H), 1.18 (dddd, J = 13.7, 9.4, 7.5, 6.4 Hz, 2H), 0.97–0.96 (m, 3H), 0.92 (d, J = 2.3 Hz, 3H), 0.90 (s, 3H). 13C NMR (75 MHz, chloroform-d) δ 215.1, 172.8, 172.7, 171.7, 170.9, 161.7, 148.4, 145.4, 134.7, 128.6, 124.6, 120.5, 116.6, 116.0, 115.5, 114.1, 66.1, 62.6, 56.4, 47.3, 44.4, 39.5, 38.5, 38.0, 34.9, 30.7, 29.2, 25.1, 23.2, 22.7, 21.5, 15.5, 11.6. ESI-MS m/z: 708.3 [M + Na]+.
4-(4-((2-((1R,3S,4S)-4-Methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl) allyl)oxy)butoxy)benzothioamide (L13-3a). Yellow solid, yield 33%, m.p. 66–68 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.61 (s, 1H), 9.30 (s, 1H), 8.06–7.84 (m, 2H), 6.99–6.83 (m, 2H), 5.80 (dd, J = 17.8, 10.4 Hz, 1H), 5.00–4.82 (m, 4H), 4.80–4.76 (m, 1H), 4.57 (d, J = 2.1 Hz, 1H), 4.04 (t, J = 6.5 Hz, 2H), 3.92 (s, 2H), 3.41 (t, J = 6.2 Hz, 2H), 2.89 (s, 1H), 2.73 (s, 1H), 1.99 (dt, J = 6.5, 3.9 Hz, 2H), 1.66 (s, 3H), 1.62–1.53 (m, 2H), 1.52–1.46 (m, 2H), 1.45–1.37 (m, 2H), 1.35–1.21 (m, 2H), 0.96 (s, 3H). 13C NMR (75 MHz, chloroform-d) δ 162.5, 150.7, 150.2, 147.6, 143.3, 131.0, 129.1, 114.0, 112.1, 109.9, 73.2, 69.7, 68.0, 52.7, 41.4, 39.8 (d, J = 8.8 Hz), 33.1, 27.0, 26.2, 26.1, 24.8, 16.6. ESI-MS m/z: 450.3 [M + Na]+.
1-(4-Carbamothioylphenyl) 7-(2-((1R,3S,4S)-4-methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl) heptanedioate (L13-3b). Yellow solid, yield 40%, m.p. 76–78 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.87 (s, 1H), 9.51 (s, 1H), 8.01–7.83 (m, 2H), 7.15 (d, J = 8.5 Hz, 2H), 5.81 (dd, J = 17.8, 10.4 Hz, 1H), 5.00 (s, 2H), 4.90 (d, J = 3.2 Hz, 1H), 4.85 (d, J = 1.7 Hz, 1H), 4.79 (s, 1H), 4.58 (d, J = 3.6 Hz, 1H), 4.57 (s, 2H), 2.59 (t, J = 7.3 Hz, 2H), 2.37 (t, J = 7.3 Hz, 2H), 2.06–1.96 (m, 2H), 1.66 (s, 3H), 1.60 (t, J = 8.1 Hz, 4H), 1.54 (d, J = 11.5 Hz, 2H), 1.51–1.44 (m, 2H), 1.43–1.32 (m, 4H), 0.96 (s, 3H). 13C NMR (100 MHz, chloroform-d) δ 201.4, 173.2, 171.6, 153.4, 149.9, 148.4, 147.3, 136.8, 128.6, 121.4, 112.2, 110.9, 110.0, 66.0, 52.6, 41.8, 39.8, 39.7, 34.0, 33.0, 28.5, 27.0, 24.8, 24.5, 24.4, 16.6. ESI-MS m/z: 520.2 [M + Na]+.
4-((6-((2-((1R,3S,4S)-4-Methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl) allyl)oxy)hexyl)oxy)benzothioamide (L13-3c). Yellow solid, yield 57%, m.p. 78–88 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.64 (s, 1H), 9.32 (s, 1H), 7.95 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 5.80 (dd, J = 17.9, 10.5 Hz, 1H), 4.96 (d, J = 1.8 Hz, 1H), 4.92 (s, 1H), 4.89 (q, J = 1.5 Hz, 1H), 4.86–4.83 (m, 1H), 4.79–4.76 (m, 1H), 4.57 (d, J = 2.1 Hz, 1H), 4.01 (t, J = 6.4 Hz, 2H), 3.89 (s, 2H), 3.34 (d, J = 6.6 Hz, 2H), 2.06–1.95 (m, 2H), 1.77–1.67 (m, 2H), 1.65 (s, 3H), 1.62–1.52 (m, 2H), 1.50 (d, J = 4.1 Hz, 2H), 1.46 (t, J = 4.5 Hz, 2H), 1.44–1.32 (m, 6H), 0.96 (s, 3H). 13C NMR (75 MHz, chloroform-d) δ 201.4, 162.6, 150.8, 150.2, 147.7, 131.0, 129.1, 114.1, 112.2, 109.9, 109.9, 73.2, 70.1, 68.2, 52.8, 41.4, 40.0, 39.9, 33.2, 29.7, 29.1, 27.1, 26.2, 25.9, 24.8, 16.6. ESI-MS m/z: 478.3 [M + Na]+.
4-((8-((2-((1R,3S,4S)-4-Methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl) allyl)oxy)octyl)oxy)benzothioamide (L13-3d). Yellow solid, yield 35%, m.p. 62–64 °C. 1H NMR (400 MHz, chloroform-d) δ 7.91 (d, J = 8.8 Hz, 2H), 7.60 (s, 1H), 7.17 (s, 1H), 6.90 (d, J = 8.9 Hz, 2H), 5.92–5.78 (m, 1H), 5.04 (d, J = 1.6 Hz, 1H), 4.97 (s, 1H), 4.96–4.92 (m, 1H), 4.90 (s, 1H), 4.84 (p, J = 1.6 Hz, 1H), 4.60 (d, J = 2.1 Hz, 1H), 4.02 (t, J = 6.5 Hz, 2H), 3.97 (s, 2H), 3.41 (t, J = 6.6 Hz, 2H), 2.04 (dt, J = 9.7, 4.0 Hz, 2H), 1.85–1.77 (m, 2H), 1.74–1.72 (m, 3H), 1.69–1.64 (m, 4H), 1.63–1.57 (m, 4H), 1.50–1.44 (m, 4H), 1.38–1.35 (m, 4H), 1.03 (s, 3H). 13C NMR (100 MHz, chloroform-d) δ 201.3, 162.6, 150.8, 150.2, 147.6, 130.9, 129.1, 114.0, 112.1, 109.9, 109.8, 73.1, 70.2, 68.3, 52.8, 41.4, 39.9, 39.8, 33.1, 29.7, 29.4, 29.2, 29.0, 27.0, 26.2, 25.9, 24.8, 16.6. ESI-MS m/z: 506.3 [M + Na]+.
2-((1R,3S,4S)-4-Methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl 5-(1,2-dithiolan-3-yl)pentanoate (L13-4a). Yellow oil, yield 33%. 1H NMR (400 MHz, chloroform-d) δ 5.95 (d, J = 8.7 Hz, 1H), 5.80 (dd, J = 17.8, 10.5 Hz, 1H), 5.10–4.99 (m, 1H), 4.93–4.90 (m, 1H), 4.88 (q, J = 1.4 Hz, 1H), 4.82 (t, J = 1.7 Hz, 1H), 4.69–4.65 (m, 1H), 4.63 (dd, J = 7.5, 1.3 Hz, 1H), 4.57 (dt, J = 1.9, 0.9 Hz, 1H), 3.61–3.51 (m, 1H), 3.21–3.05 (m, 2H), 2.50–2.36 (m, 1H), 2.24 (td, J = 7.5, 1.4 Hz, 2H), 1.99 (dd, J = 11.8, 4.2 Hz, 2H), 1.95–1.86 (m, 2H), 1.70 (t, J = 1.1 Hz, 3H), 1.68–1.59 (m, 4H), 1.59–1.52 (m, 2H), 1.51–1.47 (m, 2H), 1.45–1.39 (m, 2H), 1.22–1.12 (m, 1H), 1.00 (s, 3H). 13C NMR (75 MHz, chloroform-d) δ 173.2, 150.0, 148.4, 147.4, 112.3, 111.0, 110.0, 66.0, 56.3, 52.6, 41.8, 40.2, 39.8, 38.5, 34.6, 34.1, 33.0, 29.7, 28.8, 27.0, 24.8, 24.7, 16.6. ESI-MS m/z: 431.2 [M + Na]+.
2-((1R,3S,4S)-4-Methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl 2-(5-(1,2-dithiolan-3-yl)pentanamido)-3-methylpentanoate (L13-4b). Yellow oil, yield 75%. 1H NMR (300 MHz, chloroform-d) δ 5.80 (dd, J = 17.8, 10.5 Hz, 1H), 4.92 (q, J = 1.5 Hz, 1H), 4.85 (dt, J = 15.8, 1.6 Hz, 1H), 4.61–4.56 (m, 1H), 4.16–3.98 (m, 2H), 3.89 (dd, J = 3.6, 1.3 Hz, 1H), 3.67–3.50 (m, 1H), 3.29–3.05 (m, 2H), 2.48 (dt, J = 12.5, 6.2 Hz, 1H), 2.40 (t, J = 7.3 Hz, 2H), 2.21–2.04 (m, 1H), 2.00–1.92 (m, 2H), 1.92–1.79 (m, 2H), 1.71 (s, 3H), 1.70–1.59 (m, 4H), 1.58–1.49 (m, 4H), 1.45 (dt, J = 7.2, 3.2 Hz, 4H), 1.39–1.32 (m, 2H), 1.24 (d, J = 9.7 Hz, 2H), 1.18 (d, J = 1.3 Hz, 3H), 0.99 (t, J = 3.4 Hz, 6H). 13C NMR (75 MHz, chloroform-d) δ 172.4, 171.9, 149.9, 147.7, 147.3, 112.2, 111.7, 110.1, 66.9, 56.3, 56.2, 52.6, 41.5, 40.2, 39.7, 38.4, 38.0, 36.4, 36.3, 34.6, 32.9, 28.8, 26.9, 25.3, 25.1, 24.8, 16.5, 15.5, 11.6. ESI-MS m/z: 544.3 [M + Na]+.
2-((1R,2S,5R)-2-Methyl-5-(prop-1-en-2-yl)-2-vinylcyclohexyl)allyl 5-(1,2-dithiolan-3-yl)pentanoate (L14-4a). Yellow oil, yield 60%. 1H NMR (300 MHz, chloroform-d) δ 5.76 (dd, J = 17.8, 10.4 Hz, 1H), 5.13 (q, J = 1.4 Hz, 1H), 4.96–4.95 (m, 1H), 4.93–4.87 (m, 2H), 4.71 (d, J = 1.7 Hz, 2H), 4.51–4.44 (m, 2H), 3.57 (dd, J = 7.9, 6.2 Hz, 1H), 3.27–3.07 (m, 2H), 2.46 (dd, J = 12.6, 6.2 Hz, 1H), 2.36 (t, J = 7.4 Hz, 2H), 2.09–2.00 (m, 1H), 1.92 (dt, J = 12.9, 6.9 Hz, 2H), 1.75 (s, 3H), 1.73–1.67 (m, 4H), 1.65–1.56 (m, 4H), 1.48 (s, 2H), 1.46–1.40 (m, 2H), 1.00 (s, 3H). 13C NMR (101 MHz, chloroform-d) δ 173.0, 150.0, 149.2, 146.1, 113.3, 111.0, 108.4, 68.0, 56.3, 48.3, 45.6, 40.2, 39.6, 38.5, 34.6, 34.1, 32.9, 28.8, 26.6, 24.7, 21.1, 16.0. ESI-MS m/z: 431.2 [M + Na]+.
2-((1R,2S,5R)-2-Methyl-5-(prop-1-en-2-yl)-2-vinylcyclohexyl)allyl 2-(5-(1,2-dithiolan-3-yl)pentanamido)-3-methylpentanoate (L14-4b). Yellow oil, yield 55%. 1H NMR (400 MHz, chloroform-d) δ 6.10–6.00 (m, 1H), 5.84–5.67 (m, 1H), 5.19 (t, J = 4.9 Hz, 1H), 5.03–4.88 (m, 3H), 4.65 (dd, J = 8.6, 4.8 Hz, 1H), 4.60–4.54 (m, 1H), 4.49 (d, J = 4.9 Hz, 1H), 3.57 (dd, J = 8.0, 6.2 Hz, 1H), 3.25–3.06 (m, 2H), 2.46 (dd, J = 12.6, 6.3 Hz, 1H), 2.25 (t, J = 7.4 Hz, 2H), 2.10–1.99 (m, 2H), 1.96–1.84 (m, 3H), 1.84–1.61 (m, 6H), 1.58 (s, 3H), 1.54–1.34 (m, 7H), 1.19 (d, J = 6.8 Hz, 1H), 1.03–1.00 (m, 1H), 0.98 (d, J = 4.7 Hz, 3H), 0.95–0.93 (m, 3H), 0.92 (s, 3H). 13C NMR (75 MHz, chloroform-d) δ 172.4, 171.9, 148.5, 145.2, 127.9, 114.7, 108.5, 91.4, 69.2, 60.4, 56.3, 47.7, 46.7, 40.2, 39.4, 39.0, 38.4, 38.0, 36.3, 34.6, 28.8, 25.3, 23.3, 21.7, 21.0, 15.5, 14.2, 11.6. ESI-MS m/z: 544.3 [M + Na]+.
4.2 Biology
Author contributions
The project and experiments were designed by W. Z., H. W., S. X. and J. X. The chemistry was performed by W. Z., H. W., C. H., H. Z., Y. C. and H. Y. The biological research was carried out by W. Z., H. W., Y. S., X. C., X. F. and Z. Z. All authors contributed to the manuscript writing and review process.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This study was supported from the “Double First-Class” University project CPU2018GY04, CPU2018GY35, China Pharmaceutical University.References
- A. V. Poznyak, A. V. Grechko, R. Wetzker and A. N. Orekhov, In Search for Genes Related to Atherosclerosis and Dyslipidemia Using Animal Models, Int. J. Mol. Sci., 2020, 21, 2097 CrossRef CAS PubMed.
- I. Tabas, G. García-Cardeña and G. K. Owens, Recent Insights into the Cellular Biology of Atherosclerosis, J. Cell Biol., 2015, 209, 13–22 CrossRef CAS PubMed.
- C. Reith and J. Armitage, Management of Residual Risk after Statin Therapy, Atherosclerosis, 2016, 245, 161–170 CrossRef CAS.
- H. I. Keen, J. Krishnarajah, T. R. Bates and G. F. Statin, Myopathy: the Fly in the Ointment for the Prevention of Cardiovascular Disease in the 21st Century?, Expert Opin. Drug Saf., 2014, 13, 1227–1239 CrossRef CAS PubMed.
- R. Wang, Physiological Implications of Hydrogen Sulfide: A Whiff Exploration That Blossomed, Physiol. Rev., 2012, 92, 791–896 CrossRef CAS PubMed.
- L. A. Barr and J. W. Calvert, Discoveries of Hydrogen Sulfide as a Novel Cardiovascular Therapeutic, Circ. J., 2014, 78, 2111–2118 CrossRef CAS.
- B. Lv, S. Chen, C. Tang, H. Jin, J. Du and Y. Huang, Hydrogen Sulfide and Vascular Regulation - An Update, J. Adv. Res., 2020, 27, 85–97 CrossRef PubMed.
- L. Zhang, Y. Wang, Y. Li, L. Li, S. Xu, X. Feng and S. Liu, Hydrogen Sulfide (H2S)-Releasing Compounds: Therapeutic Potential in Cardiovascular Diseases, Front. Pharmacol., 2018, 9, 1066 CrossRef.
- H. Zhang, Z. Bai, L. Zhu, Y. Liang, X. Fan, J. Li, H. Wen, T. Shi, Q. Zhao and Z. Wang, Hydrogen Sulfide Donors: Therapeutic Potential in Anti-atherosclerosis, Eur. J. Med. Chem., 2020, 205, 112665 CrossRef CAS.
- Y. Wang, X. Zhao, H. Jin, H. Wei, W. Li, D. Bu, X. Tang, Y. Ren, C. Tang and J. Du, Role of Hydrogen Sulfide in the Development of Atherosclerotic Lesions in Apolipoprotein E Knockout Mice, Arterioscler., Thromb., Vasc. Biol., 2009, 29, 173–179 CrossRef CAS PubMed.
- S. Mani, H. Li, A. Untereiner, L. Wu, G. Yang, R. C. Austin, J. G. Dickhout, Š. Lhoták, Q. H. Meng and R. Wang, Decreased Endogenous Production of Hydrogen Sulfide Accelerates Atherosclerosis, Circulation, 2013, 127, 2523–2534 CrossRef CAS.
- C. R. Powell, K. M. Dillon and J. B. Matson, A Review of Hydrogen Sulfide (H2S) Donors: Chemistry and Potential Therapeutic Applications, Biochem. Pharmacol., 2018, 149, 110–123 CrossRef CAS.
- Y. Zhao, H. Wang and M. Xian, Cysteine-activated Hydrogen Sulfide (H2S) Donors, J. Am. Chem. Soc., 2011, 133, 15–17 CrossRef CAS PubMed.
- Y. Zhao, A. K. Steiger and M. D. Pluth, Cyclic Sulfenyl Thiocarbamates Release Carbonyl Sulfide and Hydrogen Sulfide Independently in Thiol-Promoted Pathways, J. Am. Chem. Soc., 2019, 141, 13610–13618 CrossRef CAS PubMed.
- M. M. Cerda, Y. Zhao and M. D. Pluth, Thionoesters: A Native Chemical Ligation-Inspired Approach to Cysteine-Triggered H2S Donors, J. Am. Chem. Soc., 2018, 140, 12574–12579 CrossRef CAS.
- I. Antoniadou, M. Georgiou, Y. Dotsikas, A. Lamprou, N. Lougiakis, N. Pouli, N. Karousis, Y. Loukas, I. Tseti, P. Marakos and A. Papapetropoulos, Novel H2S-Releasing Bifunctional Antihistamine Molecules with Improved Antipruritic and Reduced Sedative Actions, J. Med. Chem., 2023, 66, 9607–9621 CrossRef CAS PubMed.
- T. Tan, J. Li, R. Luo, R. Wang, L. Yin, M. Liu, Y. Zeng, Z. Zeng and T. Xie, Recent Advances in Understanding the Mechanisms of Elemene in Reversing Drug Resistance in Tumor Cells: A Review, Molecules, 2021, 26, 5792 CrossRef CAS.
- X. S. Wu, T. Xie, J. Lin, H. Z. Fan, H. J. Huang-Fu, L. F. Ni and H. F. Yan, An Investigation of the Ability of Elemene to Pass Through the Blood-brain Barrier and Its Effect on Brain Carcinomas, J. Pharm. Pharmacol., 2009, 61, 1653–1656 CrossRef CAS.
- Y. J. Tian Ren, Clinical Observations of Gastric Cancer Patients Treated with Chemotherapy and Elemene as a MDR Mediator, Zhongliu Fangzhi Yanjiu, 1999, 26, 215–216 Search PubMed.
- Y. Zhong, J. Liu, W.-M. Huo, W.-L. Duan, X. Wang and J. Shang, β-Elemene Reduces the Progression of Atherosclerosis in Rabbits, Chin. J. Nat. Med., 2015, 13, 415–420 CAS.
- L. Wu, G. Wang, S. Tang, G. Long and T. Yin, Protection of Endothelial Cells, Inhibition of Neointimal Hyperplasia by β-elemene in an Injured Artery, Cardiovasc. Drugs Ther., 2011, 25, 233–242 CrossRef CAS PubMed.
- Q. Q. Li, G. Wang, M. Zhang, C. F. Cuff, L. Huang and E. Reed, β-Elemene, a Novel Plant-derived Antineoplastic Agent, Increases Cisplatin Chemosensitivity of Lung Tumor Cells by Triggering Apoptosis, Oncol. Rep., 2009, 22, 161–170 CAS.
- Q. Q. Li, R. X. Lee, H. Liang, G. Wang, J. M. Li, Y. Zhong and E. Reed, β-Elemene Enhances Susceptibility to Cisplatin in Resistant Ovarian Carcinoma Cells Via Downregulation of ERCC-1 and XIAP and Inactivation of JNK, Int. J. Oncol., 2013, 43, 721–728 CrossRef CAS PubMed.
- R. Wang, Two's Company, Three's a Crowd: Can H2S be the Third Endogenous Gaseous Transmitter?, FASEB J., 2002, 16, 1792–1798 CrossRef CAS PubMed.
- H. Wu, X. Li, C. He, Y. Liu, C. Wang, X. Yang, F. Ma, J. Liu and J. Xu, Design, Synthesis and Biological Evaluation of Hydrogen Sulfide-releasing Isochroman-4-one Derivatives as New Antihypertensive Agent Candidates, Bioorg. Med. Chem., 2022, 64, 116776 CrossRef CAS PubMed.
- C.-P. Zhang, X.-X. Ding, T. Tian, B.-J. Li, C.-Y. Wang, S.-S. Jiang, J.-Q. Shao, Y.-L. Yuan, Y. Tian, M. Zhang and S.-Y. Long, Impaired lipophagy in endothelial cells with prolonged exposure to oxidized low-density lipoprotein, Mol. Med. Rep., 2020, 22, 2665–2672 CAS.
- J. Chen, T. Wang, S. Xu, A. Lin, H. Yao, W. Xie, Z. Zhu and J. Xu, Novel hybrids of natural β-elemene bearing isopropanolamine moieties: Synthesis, enhanced anticancer profile, and improved aqueous solubility, Fitoterapia, 2017, 120, 117–125 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00447c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |