
Development of immunoliposomes containing cytotoxic gold payloads against HER2-positive breast cancers†‡
Afruja
Ahad
abcf,
Fatima
Aftab
ab,
Alexa
Michel
f,
Jason S.
Lewis
 *fgh and
Maria
Contel
*abcde
*fgh and
Maria
Contel
*abcde
aDepartment of Chemistry, Brooklyn College, The City University of New York, Brooklyn, NY, USA. E-mail: mariacontel@brooklyn.cuny.edu
bBrooklyn College Cancer Center, Brooklyn College, The City University of New York, Brooklyn, NY, USA
cBiology PhD Program, The Graduate Center, The City University of New York, New York, NY, USA
dChemistry PhD Program, The Graduate Center, The City University of New York, New York, NY, USA
eBiochemistry PhD Program, The Graduate Center, The City University of New York, New York, NY, USA
fRadiology, Memorial Sloan Kettering Cancer Center, New York, NY, USA
gMolecular Pharmacology Program, Memorial Sloan Kettering Cancer Center, New York, NY, USA
hRadiochemistry and Molecular Imaging Probes Core, Memorial Sloan Kettering Cancer Center, New York, NY, USA
First published on 21st November 2023
Abstract
Overexpression of the human epidermal growth factor receptor 2 (HER2) is found in 20–30% of breast cancer tumors (HER2-positive breast cancers) and is associated with more aggressive onset of disease, higher recurrence rate and increased mortality. Monoclonal antibodies (mAb) like trastuzumab and pertuzumab in combination with chemotherapeutics, and trastuzumab-based antibody drug conjugates (ADCs) are used in the clinic to treat these cancers. An alternative targeted strategy (not yet in clinical use) is the encapsulation of chemotherapeutic drugs in immunoliposomes. Such systems may not only facilitate targeted delivery to the tumor and improve intracellular penetration, but also override some of the resistance developed by tumors in response to cytotoxic loads. As a supplement to classical chemotherapeutics (based on organic compounds and conventional platinum-based derivatives), gold compounds are emerging as potential anticancer agents due to their high cytotoxicity and capacity for immunogenic cell death. Here, we describe the development of immunoliposomes functionalized with trastuzumab and pertuzumab; containing simple gold(I) neutral compounds ([AuCl(PR3)] (PR3 = PPh3 (1), PEt3 (2))) generated by the thin-film method to afford Lipo-1–Lipo-2. Trastuzumab and pertuzumab were engrafted onto these liposomes to generate gold-based immunoliposomes (Immunolipo-Tras-1, Immunolipo-Tras-2, Immunolipo-Per-1, Immunolipo-Per-2). We have characterized all liposomal formulations and demonstrated that the immunoliposomes (190 nm) are stable, have high binding affinity for HER2, and display selective cytotoxicity towards HER2-positive breast cancer cell lines. Trastuzumab-based immunoliposomes of a smaller size (100 nm) – encapsulating [AuCl(PEt3)] (2) – have been generated by an extrusion homogenization method. These optimized immunoliposomes (Opt-Immunolipo-Tras-2) have a trastuzumab engraftment efficiency, encapsulation efficiency for 2, and affinity for HER-2 similar to the immunoliposomes obtained by sonication (Immunolipo-Tras-2). While the amount of Au encapsulated is slightly lower, they display almost identical cytotoxicity and selectivity profiles. Moreover, the fluorescently-labeled phosphane drug [AuCl(PPh2-BODIPY)] (3) was encapsulated in both larger (Immunolipo-Tras-3) and smaller (Opt-Immunolipo-Tras-3) immunoliposomes and used to visualize the intracellular localization of the payload. Fluorescent imaging studies found that Opt-Immunolipo-Tras-3 accumulates in the cells more than 3 and that the unencapsulated payload accumulates primarily in lysosomes, while targeted liposomal 3 localizes in mitochondria and ER, hinting at different possibilities for modes of action.
Introduction
Enhancing drug delivery is a promising approach to maximize the effectiveness of anticancer drugs while minimizing negative side effects caused by the treatment.1 There is much interest in nanocarriers as delivery vehicles, especially those containing targeting vectors, or that respond to stimuli at cancer sites.2–4 The enhanced permeation and retention (EPR) effect allows nanoparticles to gather in and around the tumor, enabling cytotoxic drugs to reach tumor tissues more effectively.5,6 This effect is observed for liposomal treatments. Liposomes offer improved pharmacokinetics and higher blood circulation times, providing for better accumulation to the site of the tumor and preventing toxicity in normal tissue.5–7 In addition, liposomes – biocompatible hollow vesicles made up of phospholipids – are particularly attractive as delivery vehicles for a variety of drugs due to their amphipathic nature.7–9 Monoclonal antibodies (mAb) can be engrafted onto the surface of these liposomes to generate delivery vehicles with targeting properties.10Studies have found that immunoliposomes encapsulated with conventional anticancer agents – docetaxel,10,11 doxorubicin,12 paclitaxel and rapamycin13 – and engrafted with trastuzumab show efficient drug encapsulation and delivery, longer circulation times and enhanced anti-tumor efficacy in vitro and in vivo against HER2-positive breast cancers relative to free drug, free mAb, the antibody–drug conjugate ado-trastuzumab emtansine,11 and control liposomes.12 To date, there is only one example of immunoliposomes encapsulating a metal-based compound (oxaliplatin) and based on the mAb cetuximab for which selective cytotoxicity against overexpressed EGFR in colorectal (CRC) cell lines has been demonstrated.14
In addition to classical chemotherapeutics (based on organic compounds and conventional platinum-based derivatives), gold compounds are emerging as potential anticancer agents due to their high cytotoxicity, unique mode of action, and immunogenic cell death generation. Nanoparticle-based delivery systems for gold chemotherapeutics have been recently reviewed,15,16 but there are very few examples of gold-compound-based liposome systems.17,18
We are interested in the development of targeted therapies for HER2-positive breast cancer. In addition to monoclonal antibodies such as trastuzumab and pertuzumab, antibody–drug conjugates containing trastuzumab, like KADCYLA® (ado-trastuzumab emtansine)19–21 and ENHERTU® (fam-trastuzumab deruxtecan-nxki),22 have been successfully used in the clinic. We have used gold(I) compounds as cytotoxic payloads for trastuzumab-based ADCs and found these systems to be cytotoxic and selective for HER-2 overexpressing cells.23,24 As mentioned, a potential alternative targeting strategy for HER2-positive cancers is to encapsulate cytotoxic payloads into liposomes engrafted with HER2-targeting monoclonal antibodies. This strategy eliminates the need for complex linker chemistry and allows for the encapsulation of a higher number of drug molecules.25,26
We report here on the development of optimized targeted therapies for HER2-positive breast cancers based on immunoliposomes functionalized with trastuzumab and pertuzumab and containing coordination gold(I)-phosphane neutral compounds.
Results and discussion
Preparation and characterization of liposomes and immunoliposomes containing gold(I) compounds
The study of gold(I) compounds containing phosphanes as anticancer agents27 has been revisited, highly cytotoxic and selective compounds recently demonstrating efficacy in vitro and in vivo.28 These studies have been motivated by the success of auranofin (a gold(I)-phosphane containing a thiosugar as a negatively charged ligand) against a variety of cancers,29 which led to recent clinical trials for its use in combination therapy for chronic lymphocytic leukemia, lung and ovarian cancer.30 Some reports have suggested that the pharmacological efficacy of this drug is due to the [Au(PEt3)]+ fragment.31,32 We have used [AuPR3]+ fragments as components of titanium(IV)-based heterometallic complexes, with extremely efficacious results in renal cancer in vivo.33–35 [AuPPh3] was also the cytotoxic payload of choice in antibody–drug conjugates targeting HER2-positive breast cancer cell lines.23,24Here, we chose gold(I) chloro derivatives [AuCl(PR3)] containing PR3 as ligands: PPh3 (1) and PEt3 (2),36 as the compounds to be encapsulated into liposomes (structures in Fig. S1‡). The liposomal formulation consists of a phosphatidylcholine (PC) and phosphatidylethanolamine (PE) mix, cholesterol and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-poly(ethylene glycol)-maleimide (DSPE-PEG-maleimide) at a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.1 with 20 mM of gold complexes. The lipids were generated by the thin-film method (see ESI‡ for details).10,37
:1:0.1 with 20 mM of gold complexes. The lipids were generated by the thin-film method (see ESI‡ for details).10,37
Dynamic light scattering (DLS) was used to assess the size, polydispersity index (PDI) and zeta potential of the liposomes and immunoliposomes (Table 1 and Fig. S2‡). Liposomes encapsulating 1 or 2 (Lipo-1 or Lipo-2) were of 172 ± 7 nm and 188 ± 7 nm diameter in size. The PDI was observed to be 0.1 for both, an acceptable value of sample uniformity for sonicated liposomes. The zeta potential was neutral, given the neutral charges of the PC and the PE composition of the liposomes. While cholesterol maintains a net negative charge and often reduces the surface charge of lipid systems,38 the reduced concentration used in our experiments allowed for the overall zeta potential to remain neutral. High-resolution particle visualization revealed liposomal concentrations to be between 1010 and 1011 liposomes per mL of saline solution. Immunoliposomes generated with trastuzumab encapsulating 1 or 2 (Immunolipo-Tras-1 and Immunolipo-Tras-2) or pertuzumab (Immunolipo-Per-1 and Immunolipo-Per-2) were slightly larger in size than Lipo-1 or Lipo-2 (Table 1), possibly due to the antibody attachments on the surface. PDI, zeta potential and encapsulation efficiency were all comparable to that of the gold-containing liposomes. Lastly, the engraftment efficiency (percentage of starting antibody concentration which bound to the liposome surface) was quantified by protein concentration measurements and observed by SDS-PAGE (Fig. S3‡). The engraftment of trastuzumab was observed to be marginally higher than that of pertuzumab (33.2% and 37.0% vs. 30.1 and 34.2%) for both immunoliposomes (Table 1). These engraftment efficiencies are like that described for trastuzumab-based immunoliposomes containing docetaxel.10,11 Immunoliposomes containing pertuzumab have not been described to date.
| Lipo- 1 | Lipo- 2 | Immunolipo-Tras- 1 | Immunolipo-Per- 1 | Immunolipo-Tras- 2 | Immunolipo-Per- 2 | |
|---|---|---|---|---|---|---|
| a Compound 2 shows higher encapsulation, Au content and engraftment efficiency. Stability measurements of size, PDI, zeta potential, encapsulation and engraftment remain unchanged in storage in PBS at 4 °C for up to 60 days. | ||||||
| Size (nm) | 172 ± 7 | 168 ± 10 | 188 ± 7 | 189 ± 9 | 187 ± 9 | 192 ± 6 |
| Polydispersity index | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 |
| Zeta potential (mV) | Neutral | Neutral | Neutral | Neutral | Neutral | Neutral |
| Concentration (particles per mL) | 0.92 × 1011 | 0.78 × 1010 | 0.78 × 109 | 0.97 × 109 | 0.86 × 109 | 0.75 × 109 |
| Encapsulation (%) | 55 ± 6 | 57 ± 6 | 52 ± 6 | 49 ± 5 | 59 ± 7 | 51 ± 4 |
| Au content (μM) | 277.7 | 282.4 | 262.6 | 247.5 | 400.7 | 376.7 |
| Engraftment efficiency (%) | — | — | 33.2 | 30.1 | 37.0 | 34.2 |
The morphology of liposomes and immunoliposomes was further visualized by transmission electron microscopy (TEM), a strategy often used for the visualization of nanoparticles such as liposomes39 (Fig. 1). TEM images showed spherical structures with distinct lipid membranes and confirmed the sizes of the nanocarriers as measured by DLS as shown in Table 1.
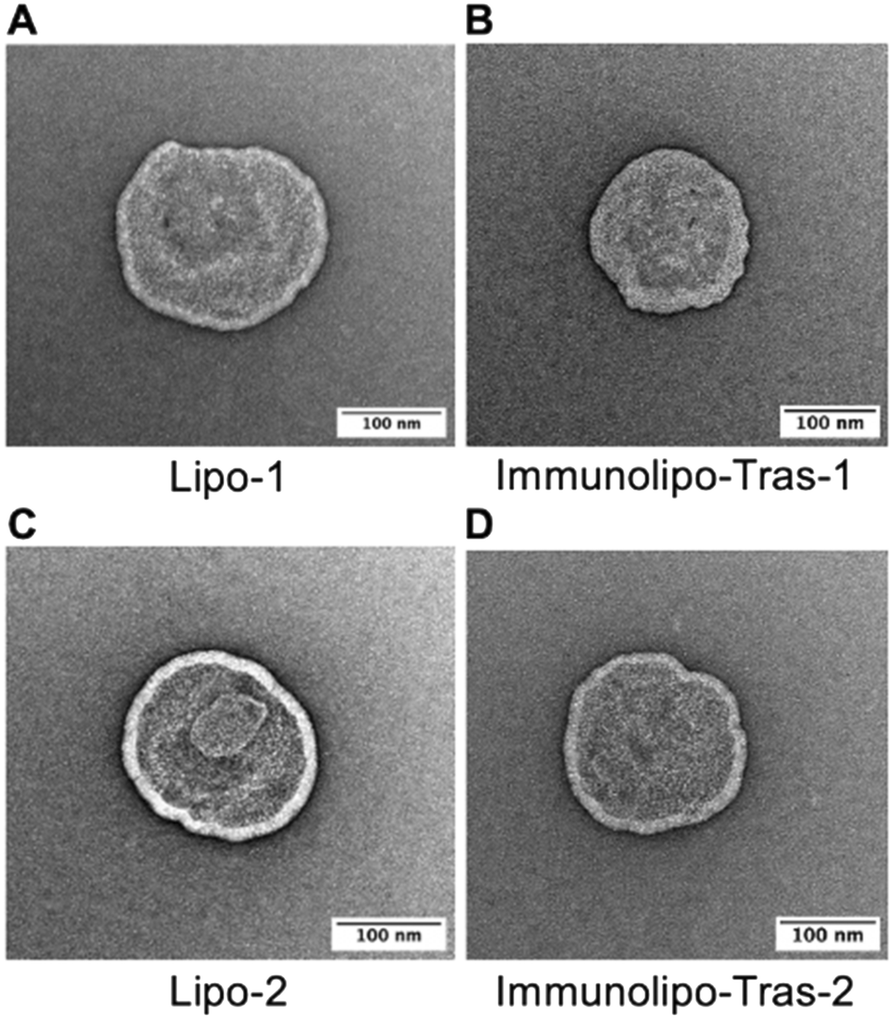 | ||
| Fig. 1 TEM images of liposomes or immunoliposomes encapsulating [AuCl(PPh3)] (1) (A and B) and [AuCl(PEt3)] (2) (C and D) following uranyl acetate staining, 150000–200000× magnification. | ||
To assess the encapsulation efficiency, liposomes were prepared for measurement of Au content by atomic absorption spectrometry (AAS). Lipo-1 presented with an encapsulation efficiency of 55% while Lipo-2 showed 57%. The slight increase in encapsulation of 2 may be attributed to the less bulky phosphane PEt3 in 2 relative to the bulkier PPh3 in 1. Because both compounds are hydrophobic, in solution they embed themselves into the lipid membrane; it is hypothesized that a higher amount of 2 may be encapsulated for this reason. Likely for the same reason, immunoliposomes encapsulating 2 (Immunolipo-Tras-2 and Immunolipo-Per-2) show higher encapsulation efficiency than immunoliposomes encapsulating 1 (400.7 μM and 376.7 μM vs. 262.6 μM and 247.5 μM, respectively). The stability of these nanocarriers was tested by assessing the presence of leakage of the compounds over a period of 60 days with samples in storage in PBS at 4 °C. The encapsulation efficiency of 1 and 2 were maintained at ∼50% during this period for both liposomes and immunoliposomes containing 1 and 2 (Fig. S4A‡). Similarly, size measurements by DLS over 60 days revealed little change for liposomes and immunoliposomes containing 1 as well as vehicle carriers, with liposomes steady at 170 nm, immunoliposomes at 190 nm and vehicle carriers at 160 nm, indicating size and drug loading stability (Fig. S4B‡).
Drug release, HER-2 affinity, and anticancer activity in cells of gold-containing liposomes and immunoliposomes
Having prepared and characterized the gold-containing immunoliposomes, we next wanted to elucidate properties of these drug vehicles for their use against HER2-positive breast cancer cells. First, we studied the binding affinity of the immunoliposomes for HER2, as it is crucial to know the specificity and affinity of therapeutic antibody-containing candidates.40 The affinity of Immunolipo-Tras-1, Immunolipo-Tras-2, Immunolipo-Per-1 and Immunolipo-Per-2 as well as the native antibodies trastuzumab and pertuzumab were assessed with the use of the enzyme-linked immunosorbent assay (ELISA). From our experiments, we observed that the EC50 of each of the immunoliposomes were 4–5 times higher than that of the native mAbs: 0.24 ± 0.05 nM for trastuzumab and 0.33 ± 0.05 nM for pertuzumab vs. 0.88 ± 0.09 nM for Immunolipo-Tras-1, 1.05 ± 0.07 nM for Immunolipo-Tras-2, 1.07 ± 0.12 nM for Immunolipo-Per-1, and 1.11 ± 0.08 nM for Immunolipo-Per-2 (Fig. 2A). These values indicate that native mAbs have a higher affinity for HER2, as expected, with EC50 values in the sub-nanomolar range; and that immunoliposomes decrease the affinity while still maintaining high affinity at low nanomolar and sub-nanomolar values. This is comparable to the affinity of native antibodies and their respective immunoliposomes described.41 Because drug encapsulation and affinity were higher for trastuzumab-based immunoliposomes, we selected Immunolipo-Tras-1 and Immunolipo-Tras-2 for further testing. | ||
| Fig. 2 (A) ELISA binding affinity assay showing EC50 values between free antibodies and immunoliposomes to HER2 after 2.5 h incubation on pre-coated HER2 plate followed by colorimetric quantification of TMB substrate. Abs measured at 450 nm. p > 0.95 for affinity of free antibody vs. respective IL. (B) EC50 quantification from ELISA (nM). IL: immunoliposome. (C) Drug release of gold-based payload from liposomes vs. trastuzumab-engrafted immunoliposomes in human serum at 37 °C for up to 72 hours. Comparable drug release for both drugs with <90% released for Immunolipo-Tras-2 at 72 h. SD represents analysis of three independent experiments. | ||
We then proceeded to evaluate drug release from the liposomes and immunoliposomes when the samples were incubated in serum at 37 °C (Fig. 2B). The drug release experiment was designed to mimic the conditions of a cell-based experiment to assess the release effect of these carriers at various time points in the presence of serum at a temperature at which cells would proliferate. This proliferation derives from the proteins present in the serum of cell environments, which can coat the liposomes and thereby alter the membrane bilayer, causing encapsulated content to leak out.42 Such leakage may present clinical challenges, as drug contents could be released before reaching the site of disease. However, it has been found that liposomes of smaller size, neutral charge and steric stability can circumvent the rapid protein coating and thus circulate for longer in the blood.42 The immunoliposomes described here were observed to be neutral; due to the presence of polyethylene glycol (PEG) on the DSPE lipids, they are also sterically stable. In the drug release experiments, we observed that Lipo-1, Lipo-2, Immunolipo-Tras-1 and Immunolipo-Tras-2 all had similar release patterns, with <40% of 1 and 2 being released by 6 hours followed by a steady increase at each subsequent time point. By 72 hours of incubation, all samples had released almost 90% of their drug contents, with Immunolipo-Tras-2 releasing 92% (Fig. 2B). As such, these systems present significantly better release dynamics than EGFR-targeted immunoliposomes (35% of oxaliplatin released at 24 h with no further release).14 In addition to informing the dosing incubation times for cell-based experiments, this data indicates that the immunoliposomes described here would have sufficient time in future experiments to reach the site of a tumor with most of the drug contents intact.
Once ideal immunoliposomes were identified based on their binding affinity and drug release dynamics, the next step was to evaluate their anti-cancer activity in cells in an in vitro cell viability assay.
For encapsulated compounds, we chose cell viability instead of IC50 assays (Fig. 3B and C and 5) due to the large number of compounds/formulations/controls for each (free drug, liposomal drug, immunoliposomal drug, empty liposomes, empty immunoliposomes, and free antibodies) as well as the increased number of concentrations for each drug and each cell line all at the same time needed for an IC50 study. Cell viability studies are usually employed to asses toxicity of drugs encapsulated in nanocarriers.43–46 we selected four cell lines for this study: two HER2-positive breast cancer cell lines, BT-474 and SKBR-3 (both with equal overexpression of HER242); a triple-negative breast cancer line, MDA-MB-231; and a non-cancerous lung fibroblast line, IMR-90. We first measured the IC50 value of both 1 and 2 at 72 hours and found low micromolar values for all cell lines, with slightly higher toxicity for MDA-MB-231 (Fig. 3A). This may be explained by the high toxicity displayed by several gold(I) phosphane-containing compounds against triple-negative breast cancers.47–49 To assess targeting selectivity, we tested Immunolipo-Tras-1 and Immunolipo-Tras-2, as well as their respective controls: gold compounds 1, 2, gold-containing liposomes Lipo-1, Lipo-2, liposomes containing no drug (empty lipo), immunoliposomes containing no drug (empty immunolipo) and free mAb trastuzumab.
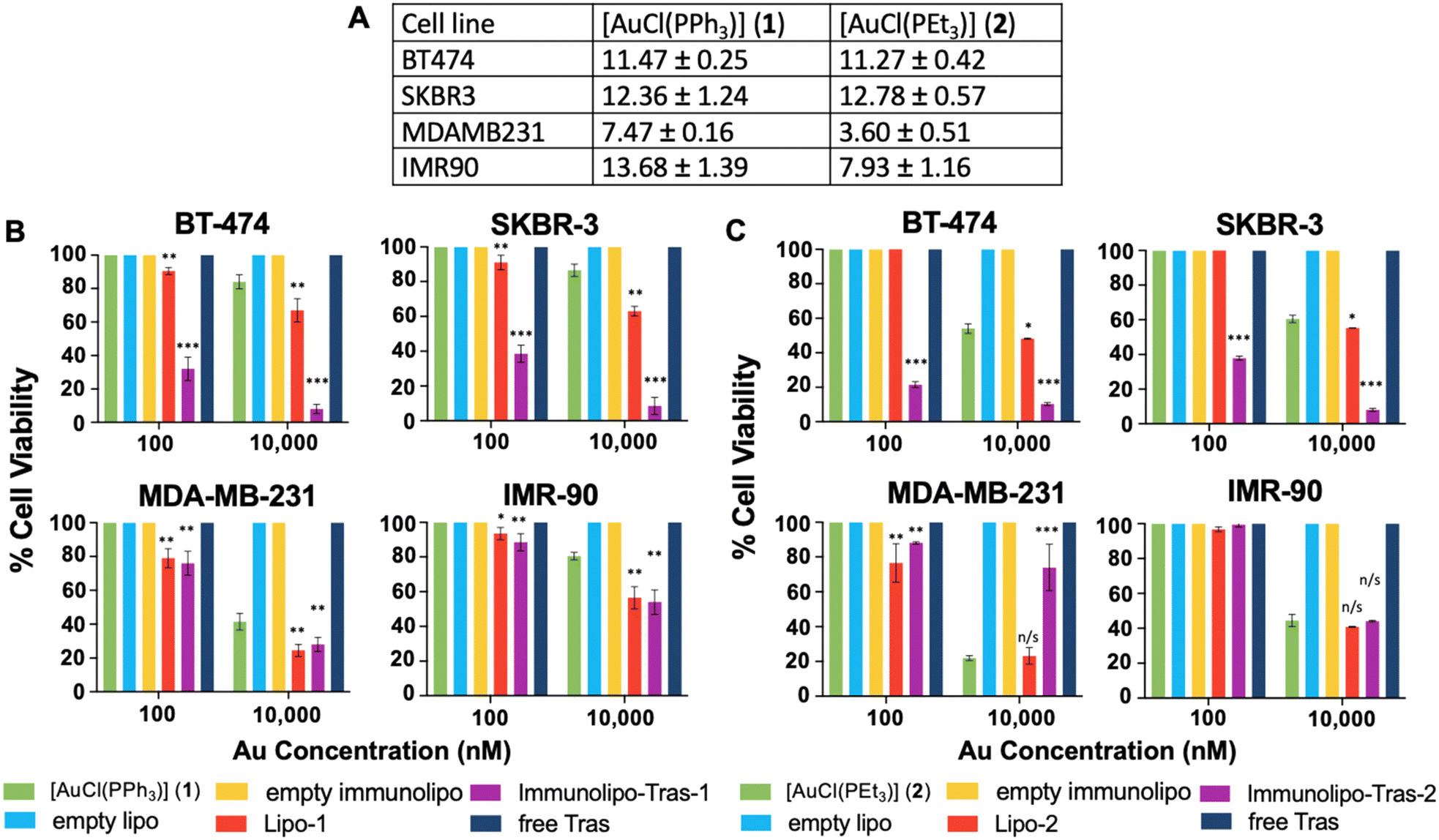 | ||
| Fig. 3 (A) IC50 values (μM) of cell lines dosed with 1 or 2 dissolved in DMSO (>1% final concentration). Presto Blue assay, 72-hour dose. Cell viability of cell lines with varying HER2 status dosed with 100 and 10000 nM of free or encapsulated [AuCl(PPh3)] (1) (B) or [AuCl(PEt3)] (2) (C) in liposomes or trastuzumab-based immunoliposomes. Controls without Au were dosed with equivalent volumes of solution. Dosing with 10 nM of all compounds showed 100% viable cells in all cell lines. SD represents analysis of three independent experiments. n/s = not significant, *p < 0.05, **p < 0.01, ***p < 0.001 of viability vs. dosing with 1 or 2 at respective dose. | ||
After cells were plated and adhered, they were dosed with 10, 100 and 10000 nM of each Au drug or equivalent volume and allowed to incubate for 72 hours. At this time point, the plates were quantified for viable cells at each dose. HER2-positive breast cancer BT-474 and SKBR-3 cells showed targeted toxicity, with only 32% and 38.5% of cells remaining viable after a 100 nM dose of Immunolipo-Tras-1, respectively. In contrast, MDA-MB-231 produced 76% cell viability and IMR-90 100% (Fig. 3B). At the higher dose of 10000 nM, there was cell death for all Immunolipo-Tras-1 lines as well as for unencapsulated 1 (as expected, due to its IC50 values, Fig. 3A). At the lower dose of 10 nM, cells remained 100% viable. There was no cell death observed for cells treated with other controls. A similar pattern was observed for cells dosed with Immunolipo-Tras-2 and its respective controls (Fig. 3C). In fact, the viability of BT-474 and SKBR-3 was 21.5% and 35%, respectively, indicating an increased toxicity for Immunolipo-Tras-2 in comparison to Immunolipo-Tras-1. This combined data demonstrates the targeting selectivity of trastuzumab-engrafted immunoliposomes – their ability to recognize, bind to and treat HER2-positive cancers, while maintaining basal levels of toxicity toward cells not overexpressing HER2. Unencapsulated drugs appear to have no effect on cell viability at the 100 nM dose, while encapsulating them in liposomes increases toxicity marginally at 100 nM and more drastically at 10000 nM. However, the greatest effect is seen when the drugs are encapsulated and the liposomes engrafted with antibodies, in which case viability decreases from 100% to 18% at the same dose (BT-474, Immunolipo-Tras-2). After a 10000 nM dose of either Immunolipo-Tras-1 or Immunolipo-Tras-2, <10% of HER2-positive cells are left viable. No dose produced cell death for wells dosed with free trastuzumab. This is expected, as the IC50 of trastuzumab against all cell lines easily exceeds 50 μM.23,50
Optimization of trastuzumab-based immunoliposome size for specific gold compounds
With the previous experiments described, we have shown the facile preparation of gold-containing liposomes and demonstrated that engrafting specific mAbs onto these vehicles can induce targeting selectivity and toxicity toward cells overexpressing an antigen recognized by these mAbs. We showed these gold-containing lipo- and immunoliposomes to be <200 nm in diameter, a precondition for the EPR effect that allows nanoparticles to accumulate in tumor environments.51 However, we decreased this size further, since smaller liposomes have longer blood circulation times and less degradation in circulation.41 Furthermore, nanocarriers >250 nm are unable to pass through the vasculature in and around the tumor environment as a result of ongoing angiogenesis, while those <50 nm are likely to penetrate the blood vessels.52,53 For this reason, we aimed to reduce the size of the gold-containing immunoliposomes to 100 nm in diameter, within the optimal range for nanocarrier drug delivery in an in vivo model of cancer.54We were able to develop 100 nm diameter gold-containing liposomes using a liposome extruder. Extrusion passes the heterogeneous liposomal sample through a series of polycarbonate membranes with designated pore sizes, filtering out all but liposomes of the desired size. After passing gold-containing liposomes through these membranes, we were able to reduce the size to ∼100 nm (Fig. S5‡). We proceeded on to engraftment, as described earlier. The optimized immunoliposomes underwent further characteristic evaluation and experimentation to compare their anti-cancer properties to that of the larger immunoliposomes. Since we found that Immunolipo-Tras-2 was the most efficacious in the cell viability studies, we decided to optimize this system. After extrusion, we obtained Opt-Immunolipo-Tras-2, whose size was measured to be 104 nm in diameter (TEM in Fig. S6‡). The PDI was enhanced as well, with a value of 0.06 (compared to 0.1 for Immunolipo-Tras-2), indicating a more homogenous sample (Table 2). The zeta potential was neutral, as expected. The concentration was lowered by a magnitude of 102, possibly because of the extruder, which leaves behind debris and lipid aggregates; it is possible that, in the sonicated sample, these aggregates made more liposomes, increasing the concentration. It was most interesting to observe that the encapsulation efficiency was only decreased from 59% to 51%, despite the surface of the liposomes being reduced by half, suggesting that, despite a reduction in size, the amount of drug loaded is not affected significantly. Similarly, the engraftment efficiency was 29.7% (in comparison to 33.2% for Immunolipo-Tras-2); so, despite the surface area being reduced, the number of antibodies engrafted was not drastically affected.
| Immunolipo-Tras- 2 | Opt-Immunolipo-Tras- 2 | |
|---|---|---|
| a Stability measurements of size, PDI, zeta potential, encapsulation and engraftment remain unchanged in storage in PBS at 4 °C for up to 60 days. | ||
| Size (nm) | 190 ± 10 | 104 ± 5 |
| Polydispersity index | 0.1 | 0.06 |
| Zeta potential (mV) | Neutral | Neutral |
| Concentration (particles per mL) | 0.78 × 109 | 0.47 × 107 |
| Encapsulation (%) | 59 ± 7 | 51 ± 4 |
| Au content (μM) | 400.7 | 374.6 |
| Engraftment efficiency (%) | 33.2 | 29.7 |
We next studied the binding affinity and drug release properties of Opt-Immunolipo-Tras-2. We found that the affinity of Opt-Immunolipo-Tras-2 improved significantly relative to Immunolipo-Tras-2 (EC50 of 0.49 ± 0.02 nM vs. 1.08 ± 0.08 nM, respectively) (Fig. 4A and B). In addition, the drug release experiment showed that both Immunolipo-Tras-2 and Opt-Immunolipo-Tras-2 released >90% of their drug content, indicating no change in drug release dynamics despite the decrease in overall size (Fig. 4B).
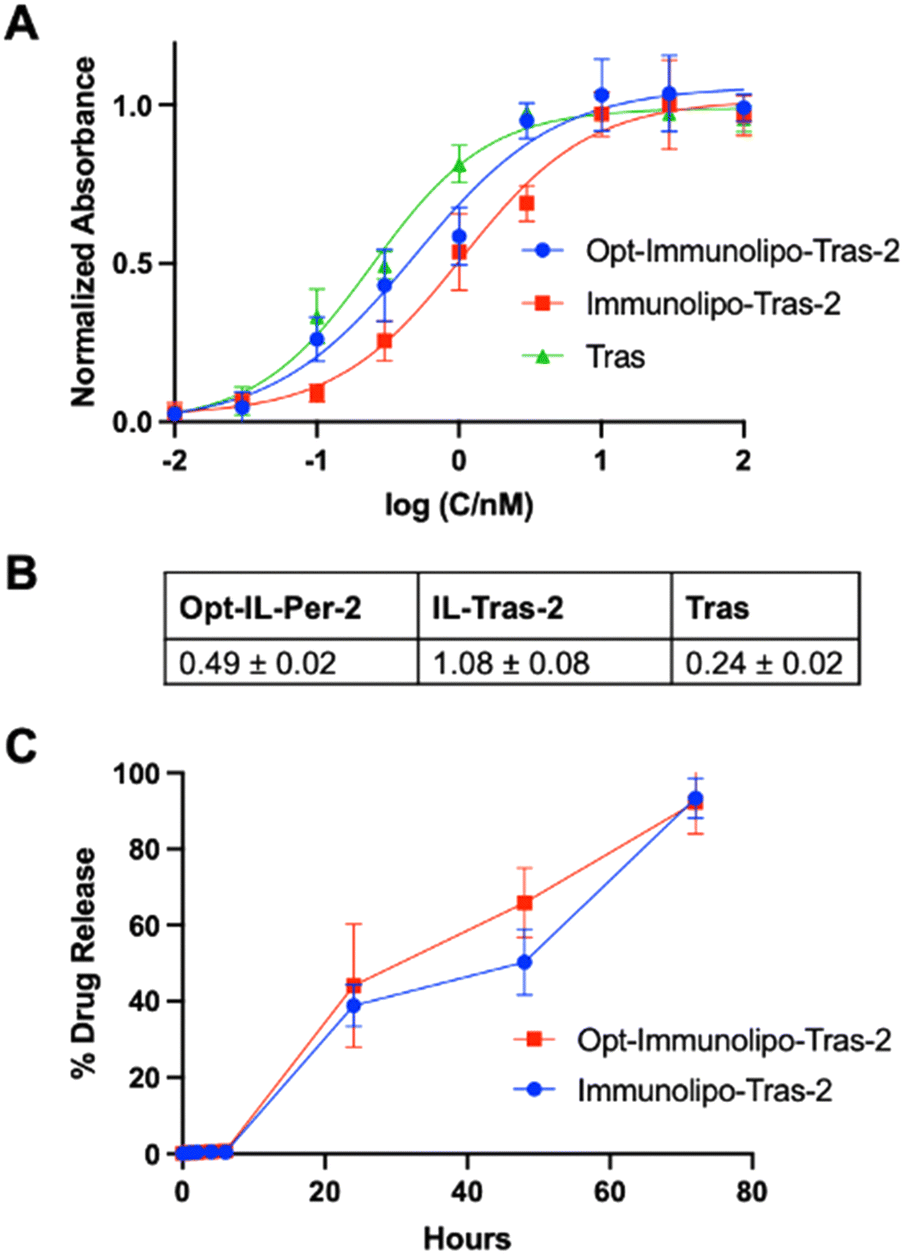 | ||
| Fig. 4 (A) ELISA binding affinity assay between free trastuzumab and immunoliposomes extruded (Opt-Immunolipo-Tras-2) or sonicated (Immunolipo-Tras-2) to HER2 (2 = [AuCl(PEt3)]). Extruded smaller size ILs show greater binding affinity than larger sonicated immunoliposomes. p > 0.80 for affinity of free antibody vs. ILs. (B) EC50 quantification from ELISA. IL: immunoliposome. (C) Comparable drug release for both drugs with <90% released for both extruded and sonicated samples. SD represents analysis of three independent experiments. | ||
The cell viability experiment comparing Immunolipo-Tras-2 and Opt-Immunolipo-Tras-2 showed no significant difference in toxicity between the two constructs in all cell lines studied (Fig. 5): 21.5% of HER2-positive breast cancer BT-474 cells were viable at a 100 nM dose of Immunolipo-Tras-2vs. 23% for Opt-Immunolipo-Tras-2. For HER2-breast cancer SKBR-3 cells, the viabilities found were 35% and 32.4% for the same two constructs, respectively. Cells treated with Lipo-2 and Opt-Lipo-2 showed no cell death except when dosed at 10000 nM. At this dose, both BT-474 and SKBR-3 showed <10% viability for both sizes of immunoliposomes. As observed earlier, the effect of the encapsulated gold content can be seen in MDA-MB-231, as cell death occurs at both 100 nM and 10000 nM for liposomes and immunoliposomes. In IMR-90, an effect is only seen at 10000 nM, at which dose all carriers show ∼40% viability. These results suggest that reducing the size of our immunoliposomes was successful: increasing binding affinity while maintaining drug loading capacity, antibody engraftment, and anti-cancer activity.
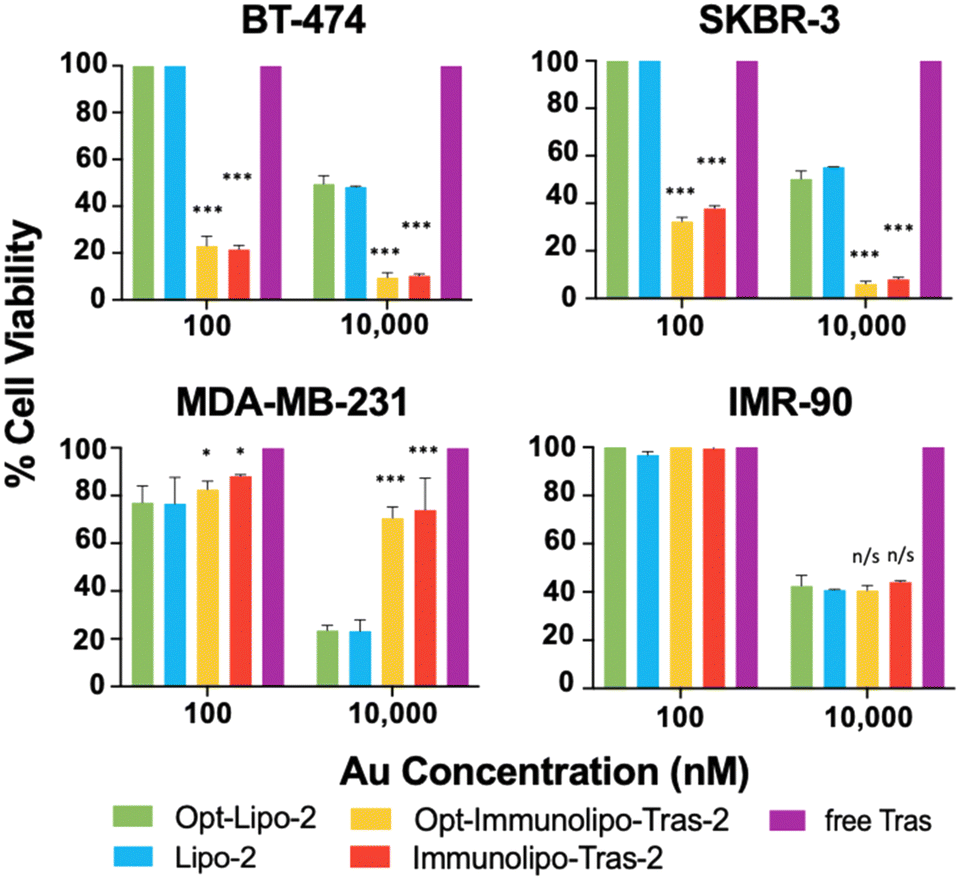 | ||
| Fig. 5 Cell viability comparing extruded (Opt-Immunolipo-Tras-2) and sonicated (Immunolipo-2) immunoliposomes (2 = [AuCl(PEt3)]). Controls without Au were dosed with equivalent volumes of solution. Dosing with 10 nM of all compounds showed 100% viable cells in all cell lines. Presto Blue assay, 72-hour dose. SD represents analysis of three independent experiments. n/s = not significant, *p < 0.05, **p < 0.01, ***p < 0.001 of viability vs. dosing with 2 at respective dose. | ||
Generation of liposomes and immunoliposomes containing luminescent cytotoxic gold payloads [AuCl(PPh2-BODIPY)]
[AuCl(PPh2-BODIPY)] (3) is a gold compound containing a fluorescent phosphane (Fig. S1‡); in a previous study, A2780S human ovarian cancer cells showed cellular uptake of 3 with high quantum yields as visualized by confocal microscopy.55 We have previously reported on the intracellular localization of a ruthenium-based anti-cancer drug with the fluorescent BODIPY moiety.56 We used 3 to elucidate the intracellular localization the Au-immunoliposomes via fluorescent imaging. These immunoliposomes were produced in both 200 nm and 100 nm diameters (Immunolipo-Tras-3 and Opt-Immunolipo-Tras-3) (Table S1, Fig. S7 and S8‡) and showed comparable encapsulation and engraftment efficiency to the phosphane payloads (35% and 32% for encapsulation efficiency and 30% and 31% for engraftment efficiency, for Immunolipo-Tras-3 and Opt-Immunolipo-Tras-3, respectively). As the latter showed comparable drug release, binding affinity and in vitro activity (for BT474) at a smaller size, it was chosen for further study (Fig. S9‡). In terms of drug release, 87% of Opt-Immunolipo-Tras-3's contents were released by 72 h. Binding affinity was found to be an EC50 of 0.57 ± 0.08 nM, indicating decreasing affinity (from 0.24 ± 0.05 nM of native trastuzumab), possibly due to the bulkier nature of the phosphane in the compound. The IC50 values of 3 against the different HER2+ and HER2− cancer cell lines and IMR-90 were observed to be high (>60 μM), as previously reported for other cancer cell lines.50 Encapsulating 3 into liposomes and immunoliposomes did not significantly affect viability. High IC50 values of 54.7 ± 7.3 μM for Lipo-3 and 52.6 ± 5.4 μM for Opt-Immunolipo-Tras-3 were obtained. IC50 values were calculated to obtain dosing values for imaging.Fluorescent imaging experiments were performed on BT-474 cells, which grow in clusters, as visualized in the background fluorescence (BF) panel in Fig. 6. Imaging studies showed that cells dosed with free [AuCl(PPh2-BODIPY)] (3) (as seen previously55) or Opt-Immunolipo-Tras-3 were taken up by the cells, as detected by the fluorescent signal of the compound (λabs = 526 nm and λem = 539 nm), as seen previously. Colocalization of 3 in cellular organelles was detected using commercial organelle markers and analysed using Manders' colocalization coefficients.57 Cells dosed with 3 were found to accumulate in lysosomes (M1 = 0.24 and M2 = 0.99 for 3 and Lyso Tracker Red, respectively) indicating that 24 ± 5% of the fluorescent signal emitted by 3 coincides with areas stained for lysosomes (Fig. S10‡). In addition, 3 also accumulated in the ER (M1 = 0.79 and M2 = 0.97 for 3 and ER Tracker Red, respectively). Mitochondrial colocalization was found to be 34 ± 5% for 3. When the cells were dosed with Opt-Immunolipo-Tras-3, it was observed that the distribution of organelle uptake of 3 shifted from lysosomes to accumulation in all three detected organelles. Colocalization was 81 ± 7%, 13 ± 6%, and 98 ± 9% for mitochondria, lysosomes, and ER, respectively (Fig. 6). This data indicates that the encapsulation of 3 into targeted liposomes prevents the drug from being subjected to lysosomal sequestration and degradation. This, in turn, protects the drug until it reaches its intracellular target site. In addition, Opt-Immunolipo-Tras-3 accumulated better in cells than did 3. Fluorescent signal measurements indicated a 164.58% greater mean fluorescence intensity for whole cells treated with Opt-Immunolipo-Tras-3 than with 3. These combined results suggest that encapsulating and targeted these drugs into immunoliposomes allows for not only higher cellular accumulation but also protection from degradation en route to target sites.
 | ||
| Fig. 6 Confocal microscopy images of live BT-474 cells treated with Opt-Immunolipo-Tras-3 (IC10) and MitoTracker Red FM (top), LysoTracker Red DND-99 (middle), and ER-Tracker Red (bottom). 3 = [AuCl(PPh2-BODIPY)]. Manders' colocalization coefficient M1 reflects the fraction of total signal emitted by Opt-Immunolipo-Tras-3 that overlaps with the organelle tracker, and M2 reflects the fraction of the total signal emitted by the organelle tracker that overlaps with that of Opt-Immunolipo-Tras-3. Images are a representation of three independent experiments. | ||
Conclusions
Antibody–drug conjugates have become a targeted drug of choice to treat HER2-positive cancers, including breast cancers. Nevertheless, the high costs associated with linker technology and the complex synthesis of highly toxic payloads must be offset to expand access.In this study, we demonstrated that high affinity and selectivity for HER2-postive breast cancer cell lines can be achieved with immunoliposomes containing commercially available gold(I)-chloro-phosphane derivatives (including the fragment [Au(PEt3)] contained in auranofin). Our system combines the selective targeting of the mAbs trastuzumab and pertuzumab, the cytotoxicity of gold-based payloads, and a protective and versatile liposome system. Combining the three constructs into one highly targeted and toxic immunoliposome produces a system capable of encapsulating several other metal-based drugs and of being engrafted with mAbs that recognize other antigens. Whereas mAbs are sometimes unable to diffuse into tissues and tumors, engrafting them onto a liposome allows the system to penetrate a tumor mass to release cytotoxic contents. The smaller size of our optimized immunoliposomes, their other desirable characteristics, and our promising in vitro data makes this Au-immunoliposome system a good candidate for further preclinical evaluation.
Materials and methods
Materials
L-α-Phosphatidylcholine (PC), phosphatidylethanolamine (PE), cholesterol, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG(2000) maleimide), and polycarbonate membranes were all purchased from Avanti Lipids. Compounds ([AuCl(PPh3)] (1), [AuCl(PEt3)] (2) and [AuCl(BODIPY-PPh2)] (3) respectively), were all synthesized as reported.36,55,58 2-Iminothiolane (Traut's reagent) and the ELISA kit were purchased from Sigma-Aldrich. Trastuzumab and pertuzumab were purchased in-house at Memorial Sloan Kettering Cancer Center. Four human cell lines were used for experimentation: BT-474 and SKBR-3 (both HER2-overexpressing breast cancer), MDA-MB-231 (triple-negative breast cancer) and IMR-90 (non-cancerous lung fibroblasts). BT-474, MDA-MB-231 and IMR-90 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Fisher Scientific) with 10% fetal bovine serum (FBS) (Fisher Scientific), 1% minimum essential media (MEM) nonessential amino acids (NEAA) (Fisher Scientific), and 1% penicillin–streptomycin (PenStrep) (Fisher Scientific). SKBR-3 cells were cultured in McCoy's medium and supplemented with the additives previously mentioned plus 1% bovine pancreas insulin (Sigma-Aldrich). All cells were purchased from the American Type Culture Collection and grown in a humidified CO2 incubator at 37 °C.Immunoliposome preparation
Lipids were dissolved in chloroform in a fixed mixture of PC/PE, cholesterol and DSPE-PEG(2000) maleimide (2:1:0.1) with 20 mM of Au compound (500 μL total volume). The lipids were generated by the thin-film method9,14 (Scheme 1). Briefly, the lipids were dissolved in chloroform and evaporated under N2 (gas) for 3–4 hours. Once a lipid film was obtained, it was hydrated with 5% glucose w/v. Liposomes were extracted and homogenized by two methods: first, the hydrated sample was sonicated at 30 kHz on ice for 10 minutes. Following this, the sample was centrifuged using 100 kDa MWCO filters to obtain 200 nm liposomes. Secondly, the hydrated samples underwent lipid extrusion using 200 nm, 100 nm and 50 nm polycarbonate membranes to yield 100 nm liposomes. For antibody engraftment, the monoclonal antibodies trastuzumab or pertuzumab were thiolated by addition of Traut's reagent (1:10 molar ratio of Traut's:antibody) in 0.15 M sodium borate buffer and gently shaken at room temperature for 2 hours. The thiolated antibody was then purified from free Traut's reagent by centrifugation and gently mixed with the liposomal sample (1:20 molar ratio of antibody:MAL-PEG) overnight at 4 °C. The following day, the sample was centrifuged at 14000 rpm for 60 minutes using 300 kDa MWCO filters to remove unbound antibody and obtain immunoliposomes.
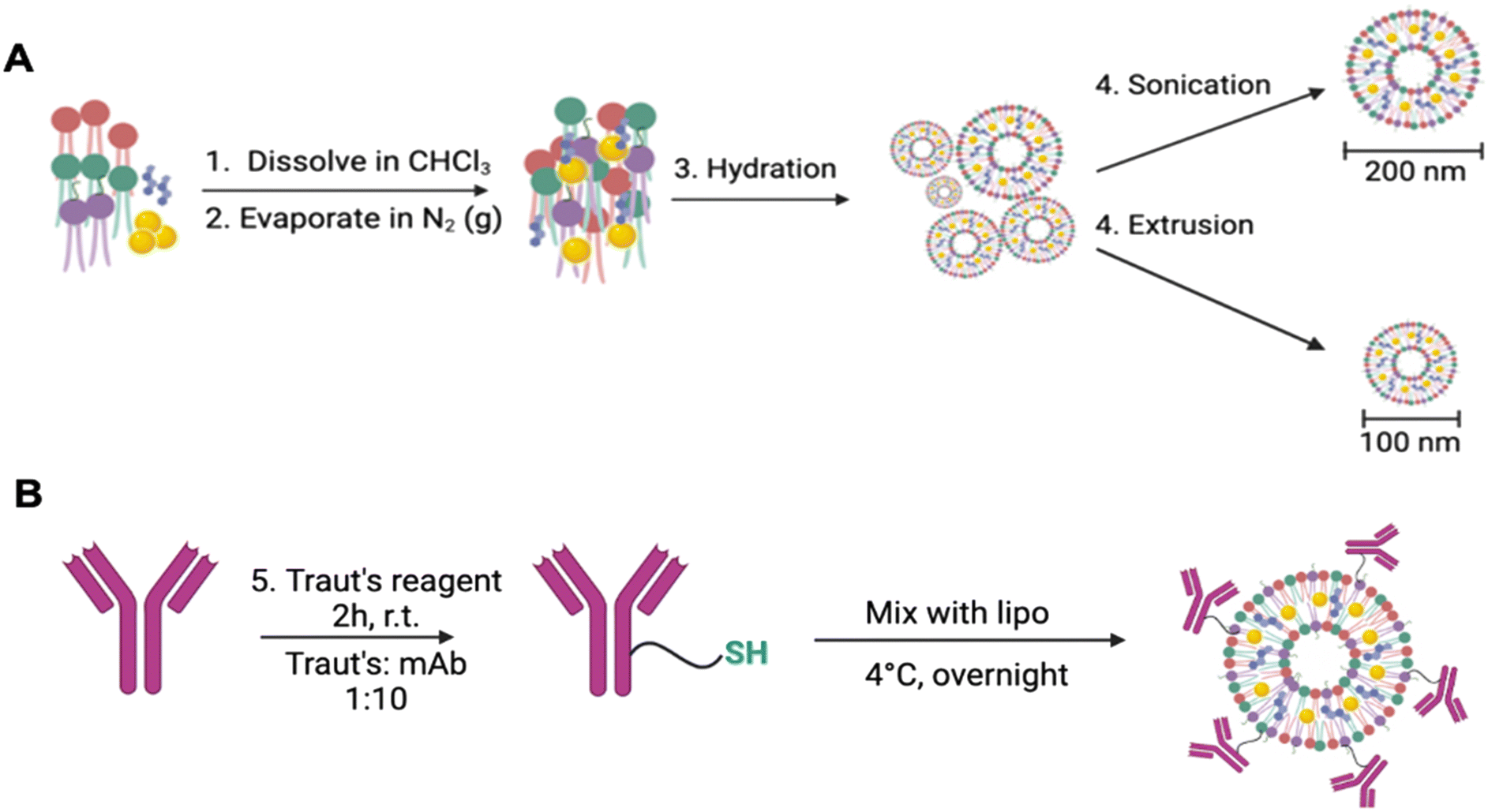 | ||
| Scheme 1 (A) Liposomes generated using PC (red), PE (green), DSPE-PEG-maleimide (purple), and cholesterol (blue) as a stabilizing lipid and encapsulating gold payloads (yellow) by sonication or extrusion, followed by antibody engraftment with a thiolate (B). | ||
Immunoliposome characterization and stability
DLS measurements were taken to measure size, PDI and zeta potential. Measurements were conducted in 1:100 diluted immunoliposomes in ddH2O using a Zetasizer Nano ZS size analyzer (Malvern Instruments). Particle concentration was assessed on the NanoSight NS300 (Malvern Instruments) by measuring the average number of tracks on multiple runs of 1:100, 1:10000 and 1:100000 diluted particles in ddH2O. Quantification of gold content was measured by AAS. Briefly, liposomal samples were digested in 10× volume of concentrated HNO3 and reconstituted in 5% HNO3/10% HCl. Elemental gold content was assessed at a wavelength of 242.8 nm on the AAnalyst 400 (Perkin Elmer). Engraftment efficiency of antibodies to the immunoliposomes was measured by protein concentration measurements using a NanoDrop at absorbance of A280 and by the Bradford assay. Stability was evaluated by repeated measurements of size, PDI and zeta potential on the DLS and drug leakage was measured by AAS. Immunoliposomes were stored in PBS at 4 °C for up to 60 days through the duration of stability measurements. Antibody engraftment was visualized via sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis using polyacrylamide 10% gels and a Tris/glycine/SDS buffer at a voltage of 100 V. The gels were then stained with Coomassie blue (Bio-Rad) and rinsed overnight prior to analysis.
Transmission electron microscopy
Immunoliposome samples were diluted 1:100 in PBS, transferred in drops onto a copper grid, and stained in 5% uranyl acetate for 5 minutes prior to imaging with a JEOL JEM 1400 transmission electron microscope at the Weill Cornell Medicine, Microscopy and Imaging Core Facility.
ELISA: binding affinity quantification
The ELISA kit for HER2 was purchased from Sigma-Aldrich. The antibodies and immunoliposomes were diluted in PBS to 300, 100, 30, 10, 3, 1, 0.3, 0.1, 0.03 and 0.01 nM concentrations in triplicates. 100 μL of each sample was pipetted into the pre-coated 96-well plate to be incubated at room temperature for 2.5 hours. The wells were then washed in wash buffer (100 μL, 3×) and 1× detection antibody (100 μL) was added to each well and incubated at room temperature for 1 hour with gentle shaking. The wells were again washed (100 μL, 3×) and streptavidin (100 μL) was added, followed for 45 minutes of incubation with gentle shaking. After another washing step, 100 μL of prepared TMB substrate was added to each well and the plate was incubated in the dark for 30 minutes with gentle shaking. Finally, 50 μL of the Stop solution was added, and the plate was read at 450 nm absorbance. GraphPad Prism 9 was used to analyze data.Drug release
Samples of immunoliposomes were mixed in a 1:1 HEPES:serum buffer (pH 7.4) and gently shaken in a 37 °C water bath. At various time points, ranging from 0–72 hours, samples were taken from the water bath and centrifuged through 30 kDa MWCO filters to isolate the encapsulated Au remains in the upper fraction and the released drug in the lower fraction. The fractions were separated and frozen at −20 °C until all samples were obtained. Samples were then digested by HNO3 (as mentioned previously) and quantified by AAS.
In vitro anti-cancer activity
Cytotoxic profiles (IC50) of all compounds (1, 2, and 3) were obtained by measuring the viability of human breast cancer cell lines BT-474, SKBR-3 and MDA-MB-231 and IMR-90 with the appropriate culture medium containing the specific compounds. Cells were plated at a density of 5600 cells per well on a cell-culture-treated 96-well plate and allowed to seed for 24 hours at 37 °C and then treated with 50 μM to 1 μM of 1, 2, or 3. All compounds were dissolved in DMSO and diluted in cell medium maintaining less than 1% solvent in the final concentrations. After 72 hours, cell viability was determined by use of the colorimetric cell viability assay PrestoBlue™ (Thermo Fisher Scientific). IC50 were fit using GraphPad Prism 7 using the appropriate curve. Statistical analysis was determined by one-way ANOVA GraphPad Prism 7 following three independent experiments.Cell viability experiments were also performed on BT-474, SKBR-3, MDA-MB-231 and IMR-90 cells. Following seeding, cells were treated with 10, 100 and 10000 nM concentrations of liposomes or immunoliposomes with encapsulated drug (100 μL) in PBS in triplicate. Different conditions were tested: drug-only, drug encapsulated in liposomes or immunoliposomes, free antibody, and empty liposomes or immunoliposomes. Controls containing no drug were dosed with equivalent volumes of solution. Cells were then incubated for 72 hours, prior to assessment of viability by PrestoBlue™. Statistical analysis was determined by one-way ANOVA GraphPad Prism 7 following three independent experiments.
Immunolipo-Tras- 3 confocal imaging
BT-474 cells were plated in 8-well chambered coverslips (Ibidi) in DMEM supplemented with 10% FBS at 37 °C in a 5% CO2 incubator. After 24 hours, the medium was replaced with IC10 concentrations of 3 or Opt-Immunolipo-Tras-3 and incubated at 37 °C for 2 hours. The organelle trackers MitoTracker Red FM (100 nM, Thermo Fisher) and LysoTracker Red DND-99 (100 nM, Thermo Fisher) were added in the final 30 minutes of incubation. Cells to be visualized for ER were washed with Hank's balanced salt solution (HBSS) prior to staining of 500 nM ER-Tracker (Thermo Fisher) in HBSS for 30 minutes. All wells were washed in phenol-red-free DMEM prior to visualization. Fluorescent imaging was performed with the Leica Stellaris 8 confocal microscope (Leica Microsystems). For the green channel, excitation was conducted at λabs = 526 nm and emission at λem = 539 nm. For the red channel, MitoTracker Red FM was detected at λabs = 581 nm and emission at λem = 644 nm; LysoTracker Red DND-99 at λabs = 577 nm and emission at λem = 590 nm; and ER-Tracker Red at λabs = 587 nm and emission at λem = 615 nm. Image processing and colocalization analysis was performed with FIJI.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institute of General Medical Sciences through Grant 2SC1GM127278 (to M. C.) and R35CA232130 (to J. S. L.). We thank the Graduate Research Technology Initiative Fund Round 21 Supplement from CUNY for funds to purchase a plate reader (to M. C.). A. A. thanks the Tow Foundation Fellowship at MSK for a doctoral grant. F. A. thanks the Tow Mentoring Initiative at Brooklyn College (Fall 2022) and the Brooklyn College Cancer Center Stacy and Michael Garil Summer Research Internship (Summer of 2022). We would like to thank the Microscopy and Imaging Core Facility at Weill Cornell Medicine for use of the Transmission Electron Microscope as well as the Molecular Cytology Core Facility at Memorial Sloan Kettering Cancer Center for use of the confocal microscope.Notes and references
- G. Liu, L. Yang, G. Chen, F. Xu, F. Yang, H. Yu, L. Li, X. Dong, J. Han, C. Cao, J. Qi, J. Su, X. Xu, X. Li and B. Li, Front. Pharmacol., 2021, 12, 735446 CrossRef CAS PubMed.
- S. N. Bhatia, X. Chen, M. A. Dobrovolskaia and T. Lammers, Nat. Rev. Cancer, 2022, 22, 550–556 CrossRef CAS PubMed.
- W. Mu, Q. Chu, Y. Liu and N. Zhang, Nano-Micro Lett., 2020, 12, 142 CrossRef CAS PubMed.
- N. Kaushik, S. B. Borkar, S. K. Nandanwar, P. K. Panda, E. H. Choi and N. K. Kaushik, J. Nanobiotechnol., 2022, 20, 152 CrossRef CAS PubMed.
- P. Liu, G. Chen and J. Zhang, Molecules, 2022, 27, 1372 CrossRef CAS PubMed.
- J. Wang, J. Gong and Z. Wei, AAPS PharmSciTech, 2022, 23, 27 CrossRef PubMed.
- V. V. S. N. L. Andra, S. V. N. Pammi, L. V. K. P. Bhatraju and L. K. Ruddaraju, BioNanoScience, 2022, 12, 660–660 CrossRef.
- K. Park, J. Controlled Release, 2017, 250, 116 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- A. Rodallec, J.-M. Brunel, S. Giacometti, H. Maccario, F. Correard, E. Mas, C. Orneto, A. Savina, F. Bouquet, B. Lacarelle, J. Ciccolini and R. Fanciullino, Int. J. Nanomed., 2018, 13, 3451–3465 CrossRef CAS PubMed.
- A. Rodallec, G. Sicard, S. Giacometti, M. Carré, B. Pourroy, F. Bouquet, A. Savina, B. Lacarelle, J. Ciccolini and R. Fanciullino, Int. J. Nanomed., 2018, 13, 6677–6688 CrossRef CAS PubMed.
- J. W. Park, K. Hong, D. B. Kirpotin, G. Colbern, R. Shalaby, J. Baselga, Y. Shao, U. B. Nielsen, J. D. Marks, D. Moore, D. Papahadjopoulos and C. C. Benz, Clin. Cancer Res., 2002, 8, 1172–1181 CAS.
- J. O. Eloy, R. Petrilli, D. L. Chesca, F. P. Saggioro, R. J. Lee and J. M. Marchetti, Eur. J. Pharm. Biopharm., 2017, 115, 159–167 CrossRef CAS PubMed.
- S. Zalba, A. M. Contreras, A. Haeri, T. L. M. Ten Hagen, I. Navarro, G. Koning and M. J. Garrido, J. Controlled Release, 2015, 210, 26–38 CrossRef CAS PubMed.
- R. T. Mertens, S. Gukathasan, A. S. Arojojoye, C. Olelewe and S. G. Awuah, Chem. Rev., 2023, 123, 6612–6667 CrossRef CAS PubMed.
- G. Moreno-Alcántar, P. Picchetti and A. Casini, Angew. Chem., 2023, 135, e202218000 CrossRef.
- P. J. Konigsberg, J. E. Debrick, T. J. Pawlowski and U. D. Staerz, Biochim. Biophys. Acta, Biomembr., 1999, 1421, 149–162 CrossRef CAS PubMed.
- S. Venkatraman, Y. Huang, A. Nagle, J. V. Natarajan, N. D. M. Foin, T. T. Ching and J. L. Fua, Liposomal Formulations, WO2018/052373A1, (03/22/ 2018).
- J. M. Lambert and R. V. J. Chari, J. Med. Chem., 2014, 57, 6949–6964 CrossRef CAS PubMed.
- L. Amiri-Kordestani, G. M. Blumenthal, Q. C. Xu, L. Zhang, S. W. Tang, L. Ha, W. C. Weinberg, B. Chi, R. Candau-Chacon, P. Hughes, A. M. Russell, S. P. Miksinski, X. H. Chen, W. D. McGuinn, T. Palmby, S. J. Schrieber, Q. Liu, J. Wang, P. Song, N. Mehrotra, L. Skarupa, K. Clouse, A. Al-Hakim, R. Sridhara, A. Ibrahim, R. Justice, R. Pazdur and P. Cortazar, Clin. Cancer Res., 2014, 20, 4436–4441 CrossRef CAS PubMed.
- F. Montemurro, P. Ellis, A. Anton, R. Wuerstlein, S. Delaloge, J. Bonneterre, N. Quenel-Tueux, S. C. Linn, N. Irahara, M. Donica, N. Lindegger and C. H. Barrios, Eur. J. Cancer, 2019, 109, 92–102 CrossRef CAS PubMed.
- X. Nguyen, M. Hooper, J. P. Borlagdan and A. Palumbo, Ann. Pharmacother., 2021, 55, 1410–1418 CrossRef CAS.
- N. Curado, G. Dewaele-Le Roi, S. Poty, J. S. Lewis and M. Contel, Chem. Commun., 2019, 55, 1394–1397 RSC.
- M. Contel, N. Curado, J. S. Lewis and S. Poty, Antibody Drug Conjugates Based on Gold Compounds, US Pat., 11141490 (10/12/ 2021) Search PubMed.
- M. Merino, S. Zalba and M. J. Garrido, J. Controlled Release, 2018, 275, 162–176 CrossRef CAS PubMed.
- J. Di, F. Xie and Y. Xu, Adv. Drug Delivery Rev., 2020, 154–155, 151–162 CrossRef CAS PubMed.
- J. C. Lima and L. Rodriguez, Anti-Cancer Agents Med. Chem., 2011, 11, 921–928 CrossRef CAS PubMed.
- J. H. Kim, E. Reeder, S. Parkin and S. G. Awuah, Sci. Rep., 2019, 9, 12335 CrossRef PubMed.
- F. H. Abdalbari and C. M. Telleria, Discover Oncol., 2021, 12, 42 CrossRef CAS PubMed.
- https://clinicaltrials.gov, (accessed on 05/25/ 2023).
- T. Marzo, D. Cirri, C. Gabbiani, T. Gamberi, F. Magherini, A. Pratesi, A. Guerri, T. Biver, F. Binacchi, M. Stefanini, A. Arcangeli and L. Messori, ACS Med. Chem. Lett., 2017, 8, 997–1001 CrossRef CAS PubMed.
- T. Marzo, L. Massai, A. Pratesi, M. Stefanini, D. Cirri, F. Magherini, M. Becatti, I. Landini, S. Nobili, E. Mini, O. Crociani, A. Arcangeli, S. Pillozzi, T. Gamberi and L. Messori, ACS Med. Chem. Lett., 2019, 10, 656–660 CrossRef CAS PubMed.
- J. Fernández-Gallardo, B. T. Elie, T. Sadhukha, S. Prabha, M. Sanaú, S. A. Rotenberg, J. W. Ramos and M. Contel, Chem. Sci., 2015, 6, 5269–5283 RSC.
- B. T. Elie, K. Hubbard, B. Layek, W. S. Yang, S. Prabha, J. W. Ramos and M. Contel, ACS Pharmacol. Transl. Sci., 2020, 3, 644–654 CrossRef CAS PubMed.
- J. E. López-Hernández and M. Contel, Curr. Opin. Chem. Biol., 2023, 72, 102250 CrossRef PubMed.
- A. Heinrich, Synlett, 2015, 26, 1135–1136 CrossRef CAS.
- H. Zhang, in Liposomes, ed. G. G. M. D'Souza, Springer New York, New York, NY, 2017, vol. 1522, pp. 17–22 Search PubMed.
- A. Magarkar, V. Dhawan, P. Kallinteri, T. Viitala, M. Elmowafy, T. Róg and A. Bunker, Sci. Rep., 2014, 4, 5005 CrossRef CAS.
- U. Baxa, in Characterization of Nanoparticles Intended for Drug Delivery, ed. S. E. McNeil, Springer New York, New York, NY, 2018, vol. 1682, pp. 73–88 Search PubMed.
- B. Wang, S. Gallolu Kankanamalage, J. Dong and Y. Liu, Antibody Ther., 2021, 4, 45–54 CrossRef CAS PubMed.
- S. Biswas, N. S. Dodwadkar, R. R. Sawant and V. P. Torchilin, Bioconjugate Chem., 2011, 22, 2005–2013 CrossRef CAS PubMed.
- M. P. Hashmi and T. M. Koester, in Reference Module in Materials Science and Materials Engineering, Elsevier, 2018, p. B9780128035818102589 Search PubMed.
- Y. Marciano, N. Nayeem, D. Dave, R. V. Ulijn and M. Contel, ACS Biomater. Sci. Eng., 2023, 9, 3379–3389 CrossRef CAS PubMed.
- Y. Marciano, V. Del Solar, N. Nayeem, D. Dave, J. Son, M. Contel and R. V. Ulijn, J. Am. Chem. Soc., 2023, 145, 234–246 CrossRef CAS PubMed.
- A. Elamir, S. Ajith, N. A. Sawaftah, W. Abuwatfa, D. Mukhopadhyay, V. Paul, M. H. Al-Sayah, N. Awad and G. A. Husseini, Sci. Rep., 2021, 11, 7545 CrossRef CAS PubMed.
- L. Matusewicz, J. Podkalicka and A. Sikorski, Cancers, 2018, 10, 418 CrossRef CAS PubMed.
- V. A. Gall, A. V. Philips, N. Qiao, K. Clise-Dwyer, A. A. Perakis, M. Zhang, G. T. Clifton, P. Sukhumalchandra, Q. Ma, S. M. Reddy, D. Yu, J. J. Molldrem, G. E. Peoples, G. Alatrash and E. A. Mittendorf, Cancer Res., 2017, 77, 5374–5383 CrossRef CAS PubMed.
- N. Nayeem and M. Contel, Chem. – Eur. J., 2021, 27, 8891–8917 CrossRef CAS PubMed.
- M. V. Babak, K. R. Chong, P. Rapta, M. Zannikou, H. M. Tang, L. Reichert, M. R. Chang, V. Kushnarev, P. Heffeter, S. M. Meier-Menches, Z. C. Lim, J. Y. Yap, A. Casini, I. V. Balyasnikova and W. H. Ang, Angew. Chem., Int. Ed., 2021, 133, 13517–13525 CrossRef.
- R. Galassi, L. Luciani, J. Wang, S. Vincenzetti, L. Cui, A. Amici, S. Pucciarelli and C. Marchini, Biomolecules, 2022, 12, 80 CrossRef CAS.
- M. Marcinkowska, E. Sobierajska, M. Stanczyk, A. Janaszewska, A. Chworos and B. Klajnert-Maculewicz, Polymer, 2018, 10, 187 Search PubMed.
- H. Maeda, Adv. Enzyme Regul., 2001, 41, 189–207 CrossRef CAS PubMed.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- S. Tasan, O. Zava, B. Bertrand, C. Bernhard, C. Goze, M. Picquet, P. Le Gendre, P. Harvey, F. Denat, A. Casini and E. Bodio, Dalton Trans., 2013, 42, 6102–6109 RSC.
- K. Miachin, V. Del Solar, E. El Khoury, N. Nayeem, A. Khrystenko, P. Appelt, M. C. Neary, D. Buccella and M. Contel, Inorg. Chem., 2021, 60, 19152–19164 CrossRef CAS PubMed.
- E. M. M. Manders, F. J. Verbeek and J. A. Aten, J. Microsc., 1993, 169, 375–382 CrossRef CAS PubMed.
- A. K. Al-Sa'Ady, C. A. Mcauliffe, R. V. Parish, J. A. Sandbank, R. A. Potts and W. F. Schneider, Inorg. Synth., 1985, 23, 191–194 CrossRef.
Footnotes |
| † We dedicate this article to Dr. Leonard Tow (Brooklyn College distinguished alumni 2022), a long-time supporter of Brooklyn College and Memorial Sloan Kettering Cancer Center students, and faculty, and champion philanthropist for cancer research. |
| ‡ Electronic supplementary information (ESI) available: Details of the experimental procedures and ESI figures. See DOI: https://doi.org/10.1039/d3md00334e |
| This journal is © The Royal Society of Chemistry 2024 |