
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnravelling the environmental degradation mechanism of perovskite thin films†
Nalini
V‡
 a,
Gergely N.
Nagy‡
bc,
Ariful
Rahaman
d,
Sreeram K.
Kalpathy
e,
Tiju
Thomas
e,
Sumangala
T. P.
*a and
Mousumi Upadhyay
Kahaly
*bc
a,
Gergely N.
Nagy‡
bc,
Ariful
Rahaman
d,
Sreeram K.
Kalpathy
e,
Tiju
Thomas
e,
Sumangala
T. P.
*a and
Mousumi Upadhyay
Kahaly
*bc
aDepartment of Physics, School of Advanced Sciences, Vellore Institute of Technology, Vellore, Tamil Nadu 632014, India. E-mail: sumangala.tp@vit.ac.in
bELI ALPS, ELI-HU Non-Profit Ltd, Wolfgang Sandner utca 3., Szeged, H-6728, Hungary. E-mail: Mousumi.upadhyaykahaly@eli-alps.hu
cInstitute of Physics, University of Szeged, Dóm tér 9, H-6720, Szeged, Hungary
dDepartment of Manufacturing Engineering, School of Mechanical Engineering, Vellore Institute of Technology, Vellore, Tamil Nadu 632014, India
eDepartment of Metallurgical and Materials Engineering, Indian Institute of Technology, Madras, Chennai, 600 036, Tamil Nadu, India
First published on 3rd July 2024
Abstract
Despite having remarkable efficiency, solar cells made of metal halide perovskites – such as methylammonium lead iodide (MAPbI3) – are detrimental because of their innate instability. Since the precursor materials may have an impact on the degradation of MAPbI3, the degradation needs to be studied at the precursor level. If they are not chemically stable or contain components that are prone to degradation, these effects can be felt on the resulting perovskite. For comprehending the degradation cycle, aging studies were conducted on precursors – methylammonium iodide (MAI) and lead iodide (PbI2) powders, as well as on MAPbI3 thin films. We perform aging studies starting from freshly synthesized samples over a span of 60 days on the precursors and over a span of 21 days on MAPbI3 thin films. The degradation pathways of the precursors and MAPbI3 are characterized by utilizing X-ray diffraction (XRD), FTIR spectroscopy, UV-Vis-NIR spectroscopy and field emission scanning electron microscopy (FESEM). The aging studies reveal that the synthesized organic precursors are unstable under ambient conditions and that the hybridized valence states of MAPbI3 exhibit higher sensitivity under ambient conditions compared to that of their precursors. The material is observed to decompose into PbI2 over the span of 21 days. We find that the structure of the MAI precursor degrades over time due to photooxidation-induced disintegration of the organic compound, while the PbI2 precursor undergoes agglomeration while the structure is preserved. We explore the origin of the observed degradation and identify that the hybridized orbitals between MAI and PbI2 initiate a sequence of chemical reactions responsible for this instability. Through ab initio simulations, we identify H2O as the atmospheric molecule most readily incorporated in MAPbI3, and this effect is corroborated by experimental observation of water-related degradation in the samples. On the other hand, incorporation of O2 is shown to cause the most significant change to the electronic structure.
Introduction
Perovskites are emerging candidates for optoelectronic devices with novel functionalities, such as high-efficiency photovoltaic solar cells,1 light-emitting diodes (LEDs),2 and photodetectors.3 Organic–inorganic hybrid perovskites (HOIPs), especially organometallic halide perovskites with an AMX3 structure, are emerging candidates for next-generation optoelectronic devices.Among the plethora of organic–inorganic perovskites, methylammonium lead iodide (CH3NH3PbI3 or MAPbI3) has attracted remarkable attention due to its excellent photovoltaic properties for next-generation mass-produced high-efficiency solar cells. Its favourable properties include a direct bandgap,4,5 great band offset,6 high photoluminescence quantum efficiency,7 large oscillator strengths,8 strong visible-range absorption coefficient,5 long charge carrier diffusion lengths (up to ∼1 μm),9,10 weak exciton binding energy of ∼45 meV,11,12 ambipolar transport properties,13 a high degree of defect tolerance in the range of 1010–1016 cm−1,14 and a high carrier mobility of ∼25 cm2 V−1 S−1.15 It also demonstrates unique structural and optical tunability like popular 2D materials.16,17 Thus, MAPbI3 has potentially groundbreaking applications as a light-harvesting metal-halide perovskite.
Though the power conversion efficiency of perovskite solar cells has grown from 3.8% to 26.1% in just a few years,18 thanks to advances in the fabrication process (e.g., polymer-catalyzed nucleation) and the introduction of tandem architectures and multilayer 2D structures (e.g., BA-MAPbI3),19,20 the stability of these cells remains very far behind that of commercial solar cells. Therefore, despite these appealing perspectives of organic–inorganic perovskites, industrial implementation is still hampered by the structural instability of the perovskite photoactive layer upon exposure to ambient conditions, such as moisture, oxygen, heat, ultraviolet radiation, or electric field.21 This issue, coupled with the strong structural dependence on electronic properties, poses a significant challenge that remains unsolved.22 For instance, the phase of the pristine material – tetragonal between 160–330 K, and cubic above,16,23 determined by the orientation of the MA B-site cation – already plays a significant role. While this reorganization can be utilized to enhance electronic transport by the generation of charged domain walls,22,24 it is often undesirable as it can induce structural damage upon ambient heating. Therefore, understanding the degradation pathways of perovskite materials under such ambient conditions is crucial to appraising their practical performance and durability. Multiple studies have confirmed that the efficiency rapidly degrades upon exposure to air (except in a few special cases, such as in the case of layered systems).19 A satisfying solution to these stability-related issues remains unfound, even though it is required to render commercial applications and large-scale implementation feasible. Identifying the mechanisms of this degradation and a robust solution for the same is therefore a key challenge.
Looking closer at the perovskite structure, we find that the chemical compounds in perovskites are only weakly bonded together by van der Waals forces and hydrogen bonds, making them vulnerable to various atmospheric molecules in air.25 Exposure to oxygen and light, as well as the presence of a hydrogen bond between an organic amine in the perovskite crystal and an external water molecule, contributes to the degradation of perovskites.26 Systematic studies regarding the factors affecting the stability of perovskites help us to understand the origin of their inherent instability. To mimic certain conditions of degradation, controlled environmental facilities are often used, where the samples can be exposed to an ultra-high vacuum environment,27 a dark and nitrogen atmosphere,28 high temperatures29 humidity,30 UV light and oxygen.31,32 The innate instability of MAPbI3 is detrimental to its practical application, so efforts are taken to comprehend its degradation cycle, under ambient conditions.33 However, to understand the inherent instability of the MAPbI3, its degradation pathway and the chemical bonding should be studied from the precursor level.
Previous studies have shown that the degradation of the source material of the perovskite has a direct impact on the composition, morphology, and quality of the perovskite film or crystal itself.31,34 An understanding of its degradation is important to control the synthesis process and achieve the desired perovskite properties. Precursors that are synthesized in-house for small-scale research purposes may provide more versatility and cost-effectiveness than those purchased in bulk, which might be very expensive. Motivated by this, in the present work, perovskite precursor powders are synthesized in the lab, and their stability and reaction pathways under ambient conditions are investigated in-depth, corroborated by ab initio simulations. One important mechanism of this is the incorporation of ambient gaseous molecules into the lattice. In this work, we explore the effect of atmospheric absorption of the five most abundant atmospheric molecules31,35 into bulk MAPbI3: N2, O2, H2O, Ar, and CO2. In addition, we also consider the incorporation of atomistic O, which may be generated by UV-dissociation of O2 under strong environmental illumination. First, we determine the expected incorporation rates and their effects on the crystal structure and the electronic structure using ab initio calculations. The simulation-based predictions are then experimentally confirmed by the significant presence of both incorporated H2O, along with atomic oxygen in the samples after exposure to ambient conditions. This study on chemical kinetics will enable us to explore the effect of the incorporation of various atmospheric molecules on the synthesized precursors – methylammonium iodide (MAI) and lead iodide (PbI2).
In what follows, the first section provides a detailed description of the experimental methods employed in the preparation of the precursor materials and the perovskite thin films for solar cells. It is followed by an overview of the characterization techniques. The next section discusses the results of the ab initio simulations, complemented by the experimental characterization and conclusions made from the X-ray diffraction (XRD), Fourier-transform infrared (FTIR) spectroscopy, ultraviolet/visible/near infrared (UV-Vis-NIR) spectroscopy, field emission scanning electron microscopy (FESEM) and energy dispersive spectroscopy (EDS), respectively. This is followed by a consolidation of our findings and a discussion of their significance as we draw the study to a close in the last section.
Experimental procedure
Materials
Lead(II) nitrate (Pb (NO3)2) with a purity of 99% was purchased from Sisco Research Laboratories Pvt. Ltd (SRL), India. Potassium iodide (KI) with a purity of 99.8% was purchased from Chemico Glass and Scientific Company (SDFCL), India. Methylamine (CH3NH2) 40% w/w aqueous solution and hydroiodic acid (HI) extra pure 99% were purchased from Avra Synthesis Private Limited, India and diethyl ether was purchased from Merck. γ-Butyrolactone (C4H6O2) was purchased from Tokyo Chemical Industry TCI (India). Isopropanol (IPA), extra pure AR with a purity of 99.5% was purchased from Sisco Research Laboratories Pvt. Ltd (SRL), India. Acetone was purchased from Avra Synthesis Private Limited, India. Distilled water was utilized from the chemistry lab. Deionized water of analytical grade used in the experimental process was procured from LaboStar TWF (UV) 7 ultrapure water systems, Siemens (Germany) The water produced has a conductivity of 0.055 μS cm−1 (equivalent to 18.2 MΩ cm) and a TOC value of <10 ppb. All these chemicals were used without further purification.Synthesis of methylammonium iodide
Methylammonium iodide was synthesized as follows. 5 ml of HI acid was added dropwise into 12 ml of CH3NH2-40% w/w aqueous solution in a double neck round bottom flask. The solution was continuously stirred at 550 rpm and maintained at 0 °C using an ice-bath for 2 h. Then, the mixture was poured into a Petri dish and evaporated at 80 °C using a hot air oven, until a white precipitate was obtained. This was repeatedly rinsed with diethyl ether to produce a white crystalline powder. The prepared powder was kept at 60 °C for 24 h. This as-synthesized methylammonium iodide (MAI) powder was utilized for experiments. The as-prepared MAI is referred to as fresh MAI, whereas old (degraded) MAI denotes the sample stored in a desiccator in the dark.Synthesis of lead iodide
Lead iodide (PbI2) powder was synthesized using lead(II) nitrate and potassium iodide in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio. Separately, the reactants were dissolved in 100 ml of distilled water and stirred until the mixture was completely dissolved. Then the two solutions were mixed and stirred at 70 °C for 3 h. Then, the obtained yellow solution was centrifuged several times using DI water until most of the potassium nitrate was separated. The final product was kept in an oven overnight at 100 °C to evaporate the solvent. The as-prepared PbI2 is referred to as fresh PbI2, whereas old PbI2 denotes the sample stored in a desiccator in the dark.
:2 molar ratio. Separately, the reactants were dissolved in 100 ml of distilled water and stirred until the mixture was completely dissolved. Then the two solutions were mixed and stirred at 70 °C for 3 h. Then, the obtained yellow solution was centrifuged several times using DI water until most of the potassium nitrate was separated. The final product was kept in an oven overnight at 100 °C to evaporate the solvent. The as-prepared PbI2 is referred to as fresh PbI2, whereas old PbI2 denotes the sample stored in a desiccator in the dark.
Preparation of perovskite ink
The perovskite ink was prepared using freshly synthesized MAI powder and freshly synthesized PbI2 powder in stoichiometric ratios. They were dissolved in γ-butyrolactone and stirred for 12 h. The prepared ink was deposited onto soda lime glass substrates (2 cm × 2 cm) using a spin coater to form thin films. Before depositing the film, the substrates were cleaned sequentially with soap solution, deionized water, acetone, and isopropanol in an ultrasonic bath for 15 min each. Furthermore, the substrates were dried in a hot air oven at 150 °C for 30 min. Then, 100 μL of perovskite ink was poured on the cleaned substrate, rotated at 3000 rpm for 30 s using a spin coater, and dried in an oven at 110 °C for 15 min.Characterization
The phase and crystallinity of the synthesized samples and the grown thin films were analysed through the PANalytical X’Pert Pro X-ray diffractometer (Germany) equipped with Cu-Kα radiation (wavelength 1.5406 Å). The scan speed used was 1° per min corresponding to the step size of 0.05° in the 2θ range 10°–70°. The surface morphology and the elemental composition were studied using FEI quanta 200 FEG – field emission scanning electron microscopy (FESEM) and energy dispersive X-ray spectroscopy (EDX) (USA) operated at an accelerating voltage of 10 kV. The particle size distribution was calculated using the open-source software ImageJ. To identify chemical bonds, Fourier transform infrared spectroscopy (FTIR) absorption spectra were recorded using a Thermo Nicolet iS50 (Shimadzu, Japan) FTIR spectrophotometer with inbuilt attenuated total reflectance (ATR) measurement mode, in the range 500–4000 cm−1. The optical transmission spectra were recorded using the JASCO V570 double-beam spectrophotometer (Shimadzu, Japan), in the wavelength range 200–2000 nm with a step size of 1 nm and a scan speed of 200 nm min−1.Computational details
Ab initio modelling of atmospheric incorporation was carried out using density functional theory (DFT) calculations.36,37 We conducted simulations on both the pristine and post-incorporation structures of MAPbI3, using a 2 × 2 × 2 supercell of the conventional cell. These calculations were performed using the BANDS engine of the Amsterdam Molecular Suite (AMS-BAND).38,39 We used the Generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional40 for the potential approximation along with a Double Zeta Polarized (DZP) basis set. The sampling of the Brillouin cell was carried out using a 3 × 3 × 3 Monkhorst–Pack k-point grid. All non-valence electrons have been treated as frozen. Besides these explicit parameters, the “Normal” numerical quality configuration of AMS-BAND has been used. Given the possibility of unpaired spins arising after incorporation, all calculations were spin unrestricted.Results and discussion
Ab initio simulations
The cell parameters of the pristine MAPbI3 system have been obtained through variable-cell geometry optimization. The optimized unit cell geometry was then employed for both the pristine and the incorporated case, justified by the rationale that the effect of the incorporation is localized, and the strain caused by the incorporation is physically present in the real material due to the surrounding undisturbed lattice around the affected unit cell.Following the determination of the lattice parameters, force-minimized structures of both the pristine and post-incorporation structures were obtained using a fixed-cell DFT optimization algorithm. Finally, on the optimized structures, a final self-consistent DFT calculation was executed to obtain the band structure and the electronic ground-state energy (EGSE) of the lattice.
The energy of incorporation, Einc, is calculated as the difference between the EGSE of the cell after incorporation (EGS), and the sum of EGSE of the pristine cell (EprGS) and that of the isolated incorporated molecule (EmolGS),41,42
| Einc = EGS − (EprGS + EmolGS) | (1) |
After structural optimization, the pristine and post-incorporation structures adopt the geometry shown in Fig. 1. It can be observed that the incorporation causes bending in the Pb–I–Pb angle, an effect most prominent in the case of H2O and least prominent in the case of Ar. This bending may decrease the overlap between the Pb 6p and I 5p orbitals, increasing the band gap. The presence of the H2O molecule also causes a significant misalignment of the MA molecules, probably due to its polarized nature. While other molecules leave the neighboring unit cells relatively intact, water causes a dislocation of the I atom even in the neighboring unit cell, thanks to the mediating role of the re-oriented MA molecule. Therefore, the overall structural disturbance is significant, which could lead to structural instabilities upon water incorporation. Indeed, it is known that exposure of MAPbI3 to humid atmospheric conditions usually leads to structural degradation.43
 | ||
| Fig. 1 DFT energy optimized MAPbI3 2 × 2 × 2 supercells in the pristine case and after incorporation of different atmospheric molecules, viewed from the x (top) and from the z (bottom) axis. The incorporated molecules are marked with a yellow dashed circle. | ||
The incorporation energies, calculated according to eqn (1), are shown in Table 1. Our result shows that all molecules except O2 are incorporated exothermically. Accordingly, we can assume that the material readily incorporates atmospheric water and—to a lesser degree—N2 and CO2. The highly favorable incorporation of the water molecule may be attributed to its affinity to adhesion to the MA molecule. This is following its polar nature and readiness to form H-bonds, which promotes adhesion to the ionic lattice of the perovskite, especially to the MA cation.
| Molecule | E inc (eV) |
|---|---|
| H2O | −1.00 |
| N2 | −0.21 |
| CO2 | −0.15 |
| Ar | −0.02 |
| O2 | 0.52 |
| O (atomic) | −0.90 |
The incorporation of O2 is particularly interesting due to its strong oxidative properties. Previous experimental studies have demonstrated that incorporation of O2 into MAPbI3 is notably poor in the absence of external illumination,44 although possible thanks to the small molecular radius of O2 (appr. 1.45 Å). Furthermore, this incorporation is reversible45 and does not damage the crystal structure directly. This aligns with our prediction that the incorporation of atmospheric oxygen in molecular form (i.e., O2 molecule) is endothermic, and happens by physisorption. However, under strong illumination and atmospheric conditions, the presence of atomistic O, generated either by UV-induced photodissociation or environmental photoreactions, may become non-negligible. Therefore, in addition to the incorporation of O2, we also assess the potential incorporation of atomic O. In this case, we find that unlike the case of O2, the incorporation of atomic O mirrors the exothermic behavior of H2O incorporation, with a highly favorable incorporation energy of Einc = −0.90 eV. Additionally, our simulation results suggest that atomic oxygen can be located much closer to the Pb ion (resulting Pb–O bond length ∼ 2.28 Å) compared to the O2 molecule (nearest distance to Pb ion ∼ 2.46 Å), suggesting a chemical bonding, which hints towards a degrading effect of the incorporation of atomic O on the perovskite. Thus, upon exposure to light, the possibility of atomic oxygen becoming more readily available increases and they may easily integrate into the MAPbI3 structure, inducing chemical alterations and potential degradation. This is indicated by our FTIR spectra, as discussed later.
The calculated band structure and electronic density of states (DOS) are shown in Fig. 2. We can observe that despite the structural modifications, the band structure remains relatively intact, except upon incorporation of O2. While all simulations were performed without spin restriction, only the case of O2 incorporation shows a polarized band structure. In this case, new defect bands appear in the band gap, one occupied spin-down and two new unoccupied spin-up. These bands show weak dispersion and correspondingly can be attributed to the molecular orbitals of the incorporated O2. It shows that the incorporation of oxygen leads to a significant degradation of the electronic properties and to the appearance of localized trap states within the band gap with low dispersion and a corresponding very low carrier mobility. The low dispersion of this band, together with the endothermic incorporation, suggests that the incorporation of molecular oxygen happens by physisorption, and the chemical bonds in the perovskite are not modified significantly. The resolution of degeneration between spin-up and spin-down states suggests the formation of long-range spin pairs with the unpaired electrons of O2.
 | ||
| Fig. 2 Band structures and DOS of the material without absorption (a) and with the absorbed atmospheric gases ((b) H2O, (c) N2, (d) CO2, (e) Ar, (f) O2). | ||
As far as the rest of the molecules are concerned, the disturbance of the electronic properties is less significant than in the case of O2. Notably, the incorporation of every molecule except H2O causes a direct–indirect transition of the band gap. The incorporation of Ar causes only a minimal alteration of the band structure, as can be expected from a noble gas. Overall, the slight changes in these cases other than O2 can be mostly attributed to structural deformations.
Structural analysis using XRD
X-ray diffraction was used to study the structural properties of the synthesized MAI and PbI2 powder. Fig. 3(a) and (b) represent the diffraction pattern of fresh and old MAI powder. The observed planes in both patterns are responsible for the tetragonal structured MAI and are in agreement with JCPDS card number 10-0737. As observed in Fig. 3(b), some orientations (102), (201), (113) and (005) disappear from the XRD pattern. This may be due to the disintegration of the organic compound which changes the orientation and decreases the crystallite size. The corresponding average crystallite size (D) was estimated using the Scherrer equation given by eqn (2): | (2) |
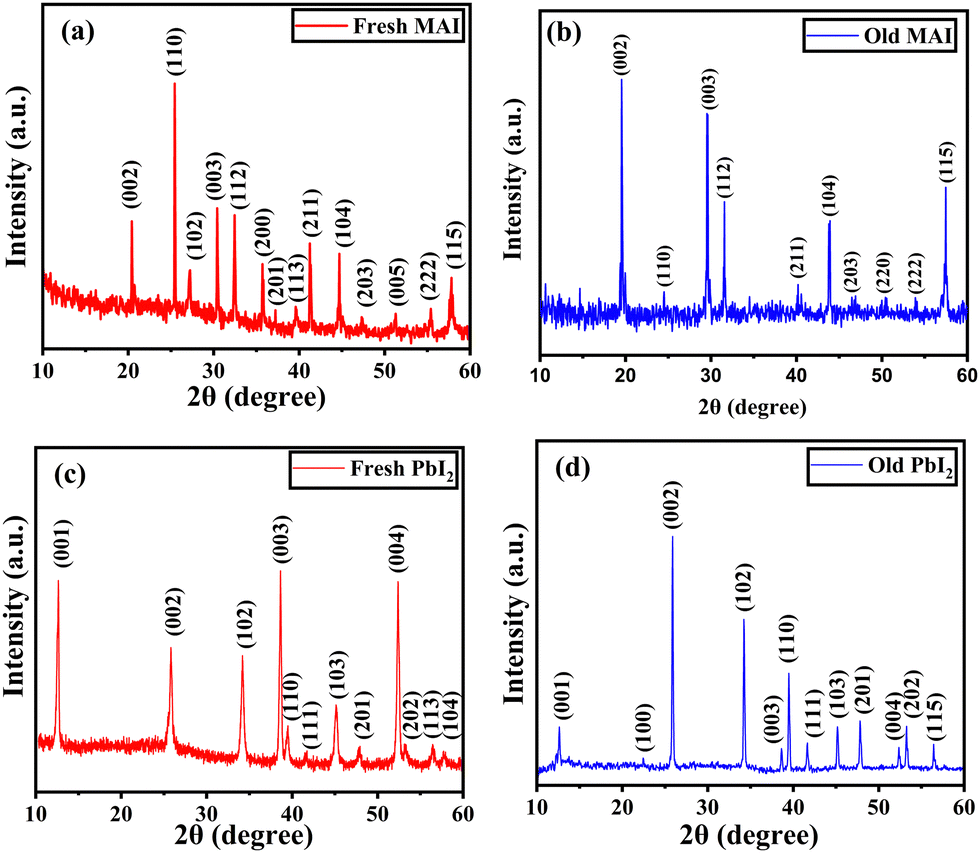 | ||
| Fig. 3 XRD pattern of the synthesized MAPbI3 perovskite precursor powders (MAI and PbI2) for both fresh and old samples which are stored in the dark for about 60 days. (a) Fresh MAI; (b) old MAI; (c) fresh PbI2 and (d) old PbI2. | ||
The lattice parameters were calculated from the following equations:
For tetragonal MAI,
 | (3) |
 | (4) |
For fresh MAI, the calculated lattice parameters were 5.12(1) Å and 9.034(1) Å, respectively, but for old MAI, they were found to be 5.11(3) Å and 9.058(8) Å respectively. Similarly, for fresh PbI2, the calculated lattice parameters were 4.571(7) Å and 6.98(1) Å, respectively, but for old PbI2, they were found to be 4.556(5) Å and 6.99(1) Å respectively as shown in Table 2. These values are in good agreement with the bulk values.46,47 In both cases, a slight change in the lattice parameter and the relative intensities of fresh and old samples causes no significant structural change.
| Synthesized precursor powders | D (nm) | a (Å) | c (Å) | Relative intensity of (002) and (202) peaks |
|---|---|---|---|---|
| MAI (fresh) | ∼106 | 5.12 (1) | 9.034 (1) | 2.03 |
| MAI (old) | ∼57 | 5.11 (3) | 9.058 (8) | 3.62 |
| PbI2 (fresh) | ∼24 | 4.571 (7) | 6.98 (1) | 1.46 |
| PbI2 (old) | ∼47 | 4.556 (5) | 6.99 (1) | 2.79 |
XRD patterns obtained for 1, 11, and 21 days after exposure of the MAPbI3 samples to ambient conditions were analysed to study the structural evolution of the MAPbI3 thin film during degradation. These patterns are shown in Fig. 4. All the peaks observed for the XRD pattern pertaining to day 1 correspond to the MAPbI3 peak except the one at around 12.7° which corresponds to the (001) plane of PbI2. The observed planes are attributed to the tetragonal phase of perovskite and are matched with the JCPDS file 83-7583. For day 11, the intensity of the PbI2 (110) peak is found to increase with the onset of new PbI2 peaks at different 2θ values. This goes along with the disappearance of prominent peaks in the MAPbI3 thin film for degrading samples.
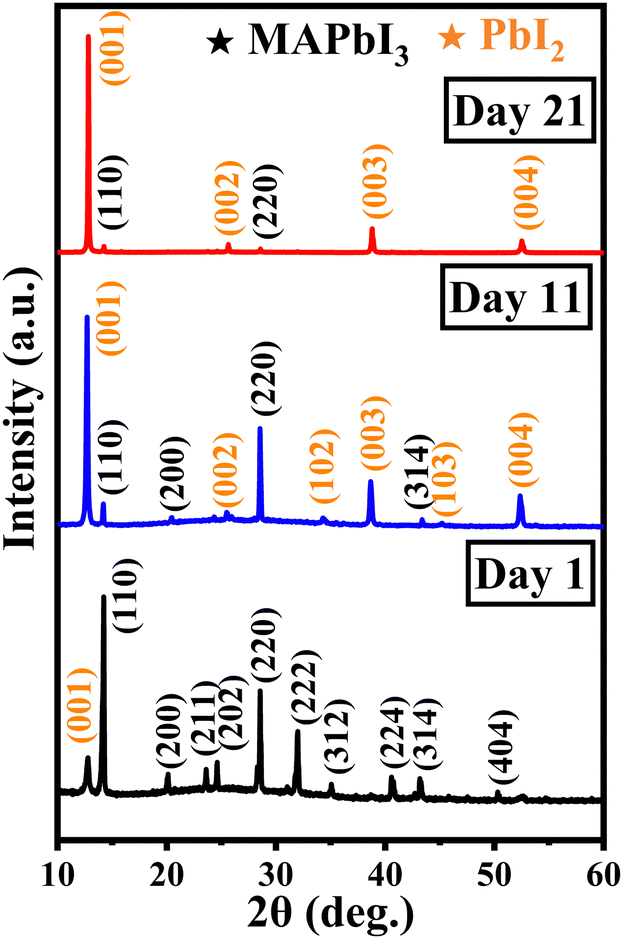 | ||
| Fig. 4 XRD pattern of MAPbI3 thin films subjected to ambient exposure for 1, 11, and 21 days in the dark. | ||
On day 21, the majority of the MAPbI3-related peaks had disappeared and only a few extremely low-intensity peaks were still visible. The (102) and (103) orientations corresponding to PbI2 were suppressed, while the emergence of other planes was observed. Thus, the evaporation of methylamine, which has a low boiling point (−6.6 °C), leaves behind the stable inorganic PbI2 phase in the day 21 degraded thin film. The grown colour of the thin film changes from black to yellow over the period of 21 days as depicted in Fig. S1 (ESI†).
In order to investigate the evolution of PbI2, the relative intensity was calculated between ∼12.7° (001) and ∼14.2° (110) peaks. It was found that the ratio had increased 30 times on day 11 and 150 times on day 21 after being exposed to the ambient conditions as mentioned in Table 3. Thus, the aging study reveals that PbI2 is the main degradation product when MAPbI3 perovskite degrades.
| Days | MAPbI3 | PbI2 | Relative intensity, (001) of PbI2/(110) of MAPbI3 | ||||
|---|---|---|---|---|---|---|---|
| a (Å) | c (Å) | D (nm) | a (Å) | c (Å) | D (nm) | ||
| 1 | 8.84 (1) | 12.54 (7) | ∼44 | — | 6.93 (1) | ∼27 | 0.23 |
| 11 | 8.82 (1) | 13.07 (1) | ∼84 | — | 6.976 (7) | ∼46 | 7.03 |
| 21 | 8.81 (1) | — | ∼35 | — | 6.93 (2) | ∼80 | 35.71 |
Using eqn (4), the lattice parameters for fresh MAPbI3, a and c were found to be 8.84(1) Å and 12.54(7) Å respectively and it matches with the previously reported values.48 These values for day 11 MAPbI3 thin film were 8.82(1) Å and ∼13.07(1) Å respectively. Similarly, for day 21, the value of a had increased to 8.81(1) Å, but the value of c could not be determined since the corresponding peaks [(110), (112), (312), (330)] required for calculating the c value had diminished during the degradation process. For the PbI2 phase on days 11 and 21, we were only able to determine the c value 6.976(7) Å and 6.93(2) Å, respectively for the planes (001), (002), (003), and (004), since the growth of PbI2 originates from the same plane of symmetry that is perpendicular to the c-axis.48 There is a minor decrease in the lattice parameter of the PbI2 phase with aging which shows a minor change in its structure.
The corresponding crystallite size was calculated for day 1, day 11, and day 21 using eqn (1). The MAPbI3 crystallite size was found to decrease from ∼44 nm to ∼35 nm for day 1 and day 21, but on day 11 the unusual increase may be due to the existence of intermediate MAPbI3/PbI2. Similarly, the PbI2 crystallite size was calculated and found to increase gradually from ∼27 nm to ∼80 nm from day 1 to day 21. This is because residual nanofragments of PbI2 cluster together and grow into larger crystallites.
Chemical bond analysis using FTIR
The FTIR spectra for the fresh and old (degraded) methylammonium iodide (MAI) precursor powders were obtained under ambient conditions (in the presence of oxygen and light) and are shown in Fig. 5(a). The vibrational bands of fresh MAI observed at 914 cm−1, 1147 cm−1, 1244 cm−1, 1410 cm−1, 1567 cm−1, 2701 cm−1, 2782 cm−1, and 3460 cm−1 can be attributed to CH3–NH3 rock, C–H twist, CH3–NH3 rock, NH3 bend, asymmetric NH3 bend, C–H stretch, N–CH3 stretch and O–H stretch respectively and match with the literature.49 In the degraded MAI, the C–H stretch widens at 2701 cm−1 and the N–CH3 stretch exhibits a slight kink at 2782 cm−1. Another notable change is the increase in transmission corresponding to the O–H stretch present at 3460 cm−1. This could be due to the inclusion of surrounding water molecules which results in the hydroxylation of the organic precursor during the degradation process. These differences in the chemical bonds can be observed by the relative area vs. chemical bond plot in Fig. 5(b). The relative area of O–H stretch for degraded MAI is significantly more prominent than that of all other chemical bonds. This is also evident from the colour change of the sample from white to brownish-yellow colour, indicating that an organic precursor was degrading in the presence of light and oxygen, releasing iodine ions as a result. In Fig. 5(c), the FTIR spectra of fresh and old PbI2 powder show no significant change in the Pb–I bond at 1670 cm−1.50 This emphasizes the stable nature of the inorganic compound PbI2. | ||
| Fig. 5 FTIR spectra of synthesized precursor powders (MAI and PbI2) kept in the dark for about 60 days under ambient conditions. The FTIR spectra of the MAPbI3 thin film were obtained for fresh and degrading films for days 1, 5, 10, 15 and 21. (a) FTIR spectra for MAI (fresh and degraded); (b) The change in the relative area of chemical bonds in MAI as a function of degradation; (c) FTIR spectra for PbI2 (fresh and old); (d) FTIR spectra for the degradation study at various intervals on MAPbI3 thin film; (e) the change in the relative area of chemical bonds in MAPbI3 as a function of degradation. | ||
Likewise, a degradation study for MAPbI3 thin film was performed at regular intervals for 21 days to understand the inherent instability of MAPbI3. From Fig. 5(d), the IR spectra of MAPbI3 thin film reveals various vibrational bands such as C–O stretch, CH3 rock, C–N stretch, C–H twist, NH3 scissor, asymmetric NH3 bend, C–H stretch, N–H stretch and NH3 stretch at 660 cm−1, 906 cm−1, 982 cm−1, 1078 cm−1, 1478 cm−1, 1588 cm−1, 2813 cm−1, 3074 cm−1 and 3461 cm−1, respectively. Fig. 5(e) shows that the NH3 scissor, C–N stretch, C–H twist and C–O stretch have the same absorption peak features both before and after the degradation process. On the other hand, significant changes in the vibrational bands were observed for the methyl group linked to ammonia groups and the ammonia groups themselves (CH3 rocking, NH3 scissoring, NH3 bending, N–H stretching and NH3 stretching) during the degradation process. As the number of days of exposure to ambient conditions increases, the binding strength of these chemical bonds is observed to decrease. Consequently, Fig. 5(e) shows that their relative areas are decreased over the degradation process.
It is evident from Fig. 5(b) that the degraded MAI precursor has a broadening O–H group. On the other hand, Fig. 5(e) makes it clear that the NH3 stretching is steadily decreasing. This indicates that MAPbI3 is being degraded by the O–H group in MAI. An additional noteworthy observation is that the smaller relative peak area indicates, the increase of the O–H group in MAI precursor occurs at a slower rate than the degradation of MAPbI3. Despite the increase in the net O–H group in MAI, the NH3 stretching in MAPbI3 exhibits a reverse trend.
It is also essential to highlight that the NH3 stretching of the pristine MAPbI3 was found to be at the same wavenumber as the O–H stretch for the degraded MAI. Thus, it becomes apparent that these two stretches merge and cause the broadening of the spectrum. This emphasizes the fact that the two stretches O–H and NH3 strongly couple and result in hybridization when MAI is exposed to PbI2. This makes the final product (MAPbI3) more sensitive to H2O, causing degradation thereby reducing the photoactivity so soon. Thus, the prominent impact of the H2O incorporation on the vibrational spectra can also be anticipated from our simulations which show maximal structural modification of the perovskite when H2O is incorporated.
The organic group (MAI) in MAPbI3 may interact with free radicals such as superoxide (O2), as shown in eqn (5), and water molecules may be formed and contribute to the MAPbI3 degradation pathway. Eqn (5) and (6) illustrate how MAI and MAPbI3 can degrade in the presence of light and surrounding atmospheric gas molecules like oxygen.32,51
 | (5) |
 | (6) |
The simulation results in Section 3 showed that the incorporation of O2 into MAPbI3 is unfavourable in the dark. On the other hand, the experiments conducted in the presence of light demonstrate oxygen and moisture-induced degradation. Therefore, it becomes obvious that MAPbI3 experiences light-driven degradation.
Optical performance using UV-Vis-NIR spectroscopy
The absorption spectra for the fresh and old methylammonium iodide (MAI) powders are shown in Fig. 6(a). The fresh MAI only shows any significant absorption in the UV region with characteristic absorption around ∼260 nm. However, the old MAI shows a significant increase in the absorbance between 260–660 nm. This may be attributed to the exposure of the MAI to ambient conditions which lead to the formation of iodine species, including iodide ion (I−), iodine (I2), and triiodide (I3−). The characteristic peaks of iodine species are observed at ∼290 nm for I2 and ∼360 nm for I3− ions.52 The release of iodine ions can also be observed by the change in colour of the sample from white to light brown as shown in the inset in Fig. 6(a). This degradation of MAI is due to the photo-oxidation of the organic group under ambient conditions. The Tauc equation, given by eqn (7), is used to calculate band gap values for the synthesized precursor powders:| (αhv)m = A(hv−Eg) | (7) |
 | ||
| Fig. 6 UV-Vis-NIR absorption spectra for the synthesized fresh and old powders of MAI and PbI2. (a) MAI and (b) PbI2 (the inset shows the corresponding Tauc plot, and digital photographs of both the fresh and old samples). | ||
From the Tauc plot shown in the inset of Fig. 6(a) the band gaps of fresh MAI powder and degraded MAI powder were found to be 3.6 eV and 1.98 eV, respectively, and the obtained value for the fresh MAI was in close agreement with the previous reports.53 The lower band gap for the degraded MAI is attributed to the structural fluctuations and cationic orientation of MAI.54 The free rotation of cations (MA+) causes polarization of the lattice and promotes charge separation. The resulting orientation of the dipoles causes significant variation in the band gap.55 Besides, the FTIR of degraded MAI showed that the incorporation of a water molecule breaks the chemical bonds in MAI. This may lead to the occurrence of defect states that alter the electrical structure of the material and ultimately affect the band gap. Thus, the water molecule incorporation can result in charge transfer, which influences the optical bandgap of the material.56
The absorption spectra of fresh and old PbI2 powders are shown in Fig. 6(b). A sharp absorption edge was observed at ∼580 nm and there are no significant changes in the absorbance for both the samples. This indicates that PbI2 maintains its optical properties throughout the aging studies. The inset in Fig. 6(b) shows the direct band gap of PbI2 for the fresh and old samples, which were found to be 2.28 eV and agrees well with previously reported values.57
To demonstrate the effect of precursor aging to the resulting thin films its optical properties are studied using UV-Vis-NIR spectroscopy. The absorption spectra for the MAPbI3 thin films prepared from the fresh and old precursors are depicted in Fig. 7(a) and (b). It shows a broad absorption in the visible region up to 780 nm for all the films. It is noteworthy to mention that the films made from fresh MAI and fresh/old PbI2 exhibit the same optical properties as those made from degraded MAI and fresh/old PbI2 (Fig. 7(b)). The energy band gap for these films ranges from 1.48 eV to 1.55 eV (shown in the inset of Fig. 7(a) and (b)). The band edge of the films with fresh MAI appeared sharp because of well-defined energy levels. This is a sign of good crystallinity and low defect density in the vicinity of the band edge. However, the band edge of the films made with degraded MAI appears to be tailed due to the imperfections or irregularities in the crystal lattice of MAI. Although all the thin films have comparable optical properties, thin films made with fresh MAI are more suited to optoelectronic devices.
 | ||
| Fig. 7 (a) and (b) UV-Vis-NIR absorption spectra for MAPbI3 thin films formed using fresh and aged precursors (the inset shows the respective band gap of the thin film samples); (c) absorption spectra for MAPbI3 thin film for various days; (d) Tauc plot for the MAPbI3 thin film for day 1; (e) band gap and the ratio of max/min absorbance vs. days. | ||
To investigate the degradation of MAPbI3 thin film in the ambient environment, the absorption spectra obtained from day 1 to day 21 are shown in Fig. 7(a) and (b). In the broad visible region up to 780 nm, the absorption spectra exhibit the absorbance at a threshold of about 340 nm (Fig. 7(a)). When compared to the day 1 thin film, the old films show a gradual decrease in the absorption intensity. Thus, the aging studies reveal significant changes in the optical properties of the degrading thin film.
From the Tauc plot the corresponding band gap for the day 1 thin film was found to be 1.52 eV as depicted in Fig. 7(d). The calculated absorption edge is found to match with the reported value in the literature.58 The band gap values for the degrading thin films were also calculated and are shown in Fig. S2(a)–(d) (ESI†). The band gap variation is fairly monotonic as shown in Fig. 7(d). It is found that the band gap increases appreciably with aging and approaches the PbI2 band gap value (∼2.48 eV), highlighting the fact that the thin film has degraded to the stable inorganic PbI2 phase. To reveal the significant variation in the absorption intensity during aging, the ratio of absorbance vs. the number of days the film is exposed to degradation was plotted and is shown in Fig. 7(d). As the thin film's absorbance decreases after being exposed to ambient conditions it appears that MAPbI3 is degrading to PbI2, which is consistent with energy band shifting from 1.52 (MAPbI3) to 2.48 eV (PbI2) in the aging studies. Thus, the PbI2 phase is observed after degradation.
The structural disorders present in the system is formed as defect bands in the band gap. This can be evaluated by the extended absorption tail called Urbach band tail and the energy associated with this defect tail is called Urbach energy (Eu). This Eu can be calculated by the reciprocal of the slope of linearly fitted lines according to eqn (8).
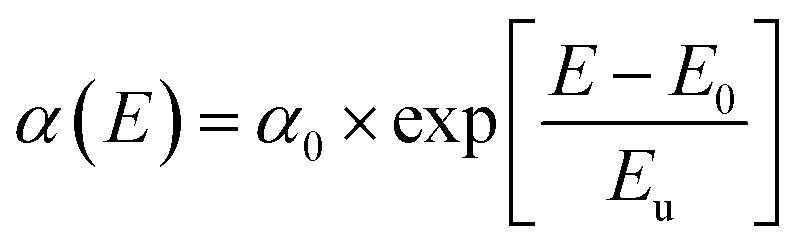 | (8) |
| Sample | Band gap (Eg) (eV) | Urbach energy (Eu) (eV) | |
|---|---|---|---|
| Precursors | Fresh MAI | 3.60 | 1.005 |
| Degraded MAI | 1.96 | 1.3054 | |
| Fresh PbI2 | 2.28 | 0.0908 | |
| Old PbI2 | 2.28 | 0.0906 | |
| Thin film | Fresh MAI + Fresh PbI2 | 1.55 | 0.4255 |
| Fresh MAI + Old PbI2 | 1.48 | 0.4508 | |
| Degraded MAI + Fresh PbI2 | 1.48 | 0.7189 | |
| Degraded MAI + Fresh PbI2 | 1.51 | 0.6633 |
Structural, morphological, and compositional analysis using FESEM and EDS
Fig. 8(a)–(d) show the surface morphology of fresh and old MAI and PbI2 powders. The fresh MAI exhibited an irregularly shaped discrete grain as shown in Fig. 8(a), with a particle size distribution within a narrow range of 46 ± 10 μm (Fig. S5(a), ESI†). When MAI is exposed to ambient conditions, the particles tend to agglomerate as observed in Fig. 8(b) which may be due to its hygroscopic nature.61 Besides, a significant increase in particle size of 80 ± 30 μm is noted. In the case of PbI2, for both fresh and old samples, the hexagonal-shaped morphology with smooth edges was observed (Fig. 8(c) and (d)). The particle size distribution of fresh (40 μm ± 10 μm) and old PbI2 (60 μm ± 30 μm) samples were calculated and it was found that it shows relatively discrete particles with a random crystallite size distribution. It is also observed that the particles of both fresh and old PbI2 range from 40 μm to 70 μm on average. In contrast to MAI, both fresh and old PbI2 show no agglomeration. | ||
| Fig. 8 (a)–(g): SEM images of the fresh and old synthesized MAI and PbI2 precursor powders and the MAPbI3 thin films obtained before and after degradation. (a) Fresh MAI; (b) old MAI (after degradation); (c) fresh PbI2; (d) old PbI2; (e) fresh MAPbI3 thin film; (f) degraded MAPbI3 thin film and (g) elemental composition obtained from the EDS spectrum for all the samples. | ||
Fig. 8(e) shows the surface morphology of the fresh MAPbI3 thin film and Fig. S6 (ESI†) shows the histogram of grain size distributions. The top-view micrograph of fresh MAPbI3 thin film (Fig. 8(e)) illustrates the smooth and compact thin film. The solvent used may greatly influence perovskite crystallization during annealing, resulting in a higher grain size.62 Particle sizes for DMF, DMSO and GBL range from 46–90 nm, 17–45 nm and 200–600 nm, respectively. GBL solvent has a high dipole moment, yet its limited solubility compared to other solvents results in grains that are quite large.63 The fresh MAPbI3 film had an average grain size of 237 ± 70 nm (Fig. S6, ESI†) since we used γ-butyrolactone solvent to dissolve perovskite precursors in the experiment. The degraded MAPbI3 thin film shown in Fig. 8(f) depicts the layered hexagonal morphology of residual PbI2. Consequently, Fig. S7 (ESI†) shows a flake-like structure in the degraded film which is consistent with previously reported literature.64
The elemental composition of fresh and old MAI and PbI2 powders and MAPbI3 films shown in Fig. 8(g) was obtained from the EDS spectrum (Fig. S8, ESI†). It reveals that the oxygen content in old MAI tends to increase in the degraded sample while the iodine level is found to drop when compared to its fresh counterpart. This loss in iodine may be due to the degradation of the organic group.
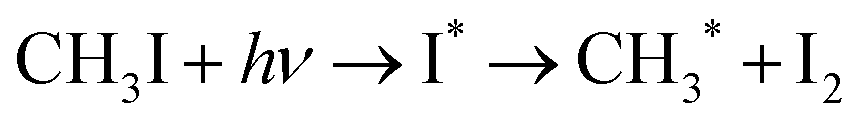 | (9) |
| CH3 + OH− → CH3OH + I− | (10) |
The EDS also shows an increase in carbon content in the old MAI and degraded MAPbI3 thin film. The residual carbon content is found to increase in the degraded samples.67 Eqn (11) illustrates the irreversible formation of CH2, NH3, HI and PbI2 due to water absorption.64
| CH3NH3PbI3(s) → (–CH2–) + NH3(g) + HI(g) + PbI2(s) | (11) |
To link the aging of perovskite precursors to the resulting thin films, surface morphology and EDS analysis were carried out on thin films made from fresh and aged precursor powders. The top-view FESEM micrograph in Fig. 9(a) and (b) shows the surface morphology of the thin films prepared using fresh MAI and fresh PbI2, and fresh MAI and old PbI2. Both the films show complete coverage of the substrate with similar morphology. The films appeared to be devoid of cracks and voids. It has irregular crystal domains and average-sized grains ranging from 237 ± 70 to 234 ± 53 nm (Fig. S9, ESI†). Thus, the high-quality thin films with fresh MAI and fresh/old PbI2 prove the stability of the PbI2 precursor under ambient conditions. Similar investigations using old (degraded) MAI with both fresh and old PbI2 revealed that the films are non-uniform and are found with more pin holes and voids that result in poor surface coverage with a deformed morphology, as seen in Fig. 9(c) and (d). Void spaces act as a place for charge carrier recombination and act as short-circuit pathways. As a result, the performance and stability of solar cells can be severely compromised if voids are present in the perovskite active layer.68 Though the morphology of all the thin films was found to be similar, the quality of the thin film made with fresh MAI and fresh/old PbI2 was found to be way better than the films prepared from degraded MAI with fresh/old PbI2.
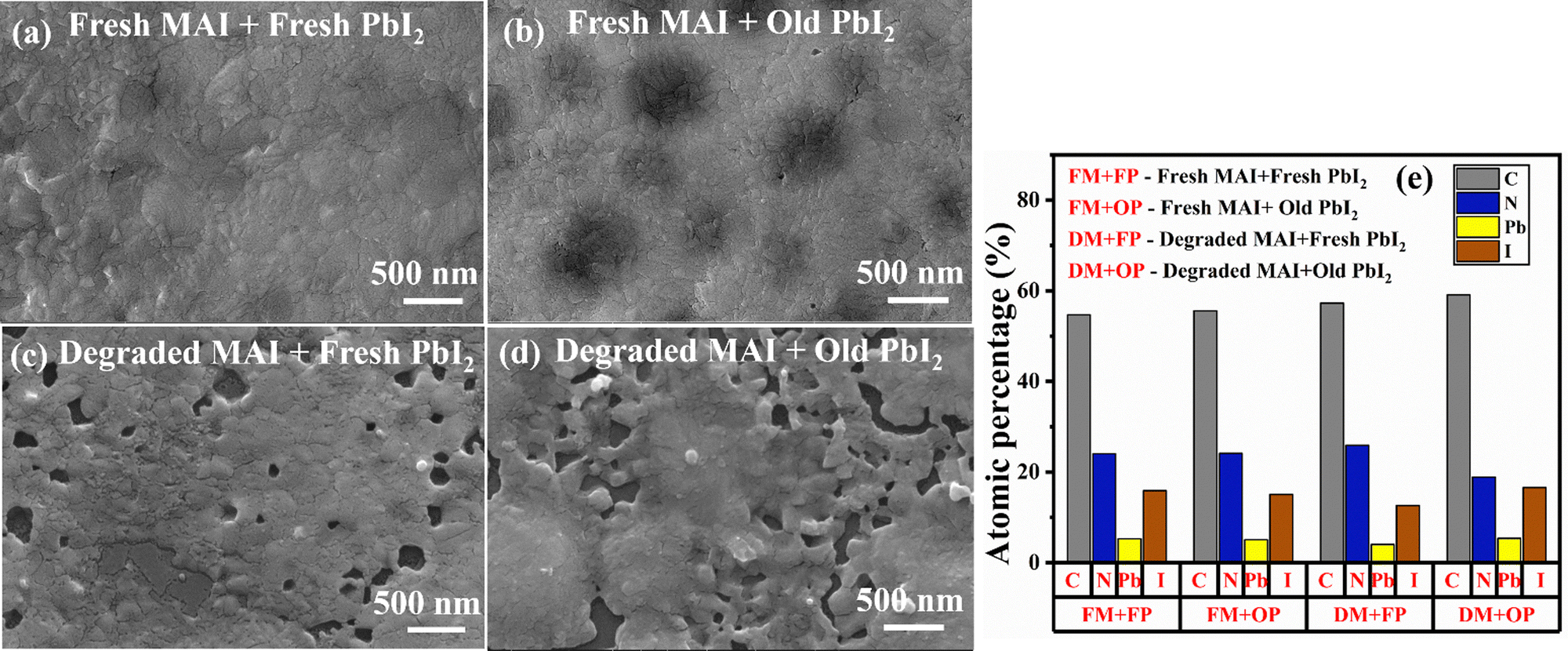 | ||
| Fig. 9 (a)–(e): SEM images of the thin films prepared using fresh and old MAI and PbI2 powders. (a) fresh MAI + fresh PbI2; (b) fresh MAI + old PbI2; (c) degraded MAI + fresh PbI2 and (d) degraded MAI + old PbI2. (e) Elemental composition of all the films obtained from the EDS spectrum. | ||
The elemental composition of the thin films prepared using fresh and old MAI and PbI2 powders shown in Fig. 9(e) was obtained from the EDS spectrum (Fig. S10, ESI†). Unlike the trend observed for the MAPbI3 degradation, the elemental analysis for the films made with degraded MAI shows no signs of iodine loss. All the elemental composition remains intact for all the films. So, the degradation of MAI impacts the quality of the MAPbI3 thin films produced from it. Thus, the degraded MAI may not directly alter the ratio of atomic arrangement, it can indirectly impact stability by producing poor-quality thin films.
Conclusions
We conducted ab initio DFT simulations using a 2 × 2 × 2 supercell to investigate the impact of the five most prevalent atmospheric gases (N2, O2, H2O, Ar and CO2) on bulk MAPbI3. We identified the structural alterations and calculated the formation energies of various species that form during degradation. Our simulations reveal that among all molecules with relevant atmospheric presence, perovskites are the most susceptible to H2O, which causes prominent distortion in their lattice structure. Addressing this issue may be feasible through the incorporation of a larger cation to strengthen the bond between the MAI and the Pb–I octahedra.Our aging studies carried out on the precursors under ambient conditions reveal that while the inorganic PbI2 remains stable, the organic group (MAI) undergoes noticeable degradation. This is evident from the Urbach energy (Eu) values obtained for fresh and old precursors. This value remains the same for PbI2, while it is found to increase for degraded MAI when compared to its fresh counterparts. The visible signs of MAI degradation, such as the change to brownish colour, suggest that the loss of iodine species contributes to the degradation process. Similar aging studies on the MAPbI3 thin films reveal that when atmospheric molecules (H2O and O2) interact with the organic group on the thin film, H2O forms a strong hydrogen bond with them, weakening the initial bond caused by the hybridized orbitals between MAI and PbI2. This interaction initiates a sequence of chemical reactions leading to the disintegration of the film: deprotonation (of CH3NH3I), volatilization of the organic group (methylamine – CH3NH2) and oxidation (of iodine species).
Experimentally, we have studied the degradation pathways of the MAPbI3 thin film and its precursors to understand their inherent instability. Our FTIR results revealed that the interaction of the organic group with the atmospheric molecules, particularly O2 and H2O, utilizes O–H radicals in oxidizing I− to I2, thus contributing to the degradation of MAI and MAPbI3. Our results also show that the degradation of MAPbI3 thin films progresses five times more quickly than that of its precursor materials, indicating that the hydrogen bond between the organic group and atmospheric molecules is a key factor in the disintegration process.
While investigating the precursor stability with the resulting thin films, the optical properties are found to be comparable while the SEM results suggest that the thin films obtained from fresh MAI and fresh/old PbI2 is of high quality while the films with degraded MAI exhibit poor quality. Therefore, the distorted structure of MAI is responsible for the non-uniformity in the film. Thus, it is evident that the synthesized inorganic precursor exhibits greater stability compared to the thin films produced from them. The significant higher rate of degradation observed in MAPbI3 thin films is attributed to the presence of hybridized orbitals within the thin film structure. Also, the calculated Eu for the system with degraded MAI shows a higher value which is due to the prevalence of more defect states. Concurrently, the degradation of the precursor powders can be prevented by storing them in a dry, dark environment. Consequently, the instability in the PSCs initiated from their precursor materials can be ameliorated by employing suitable storage techniques and the instability of the thin films may be mitigated through sufficient encapsulation.
Based on the insights acquired from our aging studies, various strategies can be developed to prevent the degradation of organic–inorganic metal halide perovskites. However, the predominant emphasis lies on the crystallization, growth of the thin films and induced defects. However, often the actual effect of the precursors is overlooked. Conventional understanding postulates the stability of precursors, for example, MAI and PbI2, in powder. In reality, however, out of the two precursors the organic (MAI) do not exhibit the presumed stability in their powder form. Deciphering the primary chemical interactions driving its degradation holds the key to addressing stability issues and enhancing the reproducibility of high-efficiency perovskite thin film-based devices.
Author contributions
Nalini V: methodology, formal analysis, and data curation, writing: original draft, writing: review and editing, collection of relevant literature, investigation, and resources. Gergely N. Nagy: software (Implementation of computer program), formal analysis, data curation, writing: original draft, writing: review and editing, collection of relevant literature, investigation, and resources. Ariful Rahaman: review and editing, resources, and visualization. Sreeram K. Kalpathy: writing: review and editing, visualization. Tiju Thomas: conceptualization, formal analysis, investigation, writing: review and editing, project administration, and supervision. Sumangala T. P: methodology, conceptualization, formal analysis, investigation, writing: review and editing, project administration, supervision, and resources. Mousumi Upadhyay Kahaly: software (implementation of computer program), formal analysis, investigation, writing: review and editing, project administration, supervision, and funding acquisition.Data availability
All ab initio simulations were performed using Amsterdam Modeling Suite computational chemistry tool https://www.scm.com/. All data that support the findings (both theoretical and experimental) of this study are either already included within the article and supplementary files, or are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge financial support from the Department of Science and Technology (DST), India, through grant number DST/INT/HUN-29/2020. The authors also thank VIT-Vellore for extending their characterization facilities for carrying out the research work. We acknowledge project No. 2019-2.1.13-TÉT-IN-2020-00059, which has been implemented with support provided by the National Research, Development and Innovation Fund of Hungary, and financed under the 2019-2.1.13-TÉT-IN funding scheme. IMPULSE has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 871161. The ELI ALPS project (GINOP-2.3.6-15-2015-00001) is supported by the European Union and co-financed by the European Regional Development Fund.References
- S. P. Feng, Y. Cheng, H. L. Yip, Y. Zhong, P. W. K. Fong, G. Li, A. Ng, C. Chen, L. A. Castriotta, F. Matteocci, L. Vesce, D. Saranin, A. Di Carlo, P. Wang, J. Wei Ho, Y. Hou, F. Lin, A. G. Aberle, Z. Song, Y. Yan, X. Chen, Y. M. Yang, A. A. Syed, I. Ahmad, T. Leung, Y. Wang, J. Y. Lin, A. M. C. Ng, Y. Li, F. Ebadi, W. Tress, G. Richardson, C. Ge, H. Hu, M. Karimipour, F. Baumann, K. Tabah, C. Pereyra, S. R. Raga, H. Xie, M. Lira-Cantu, M. V. Khenkin, I. Visoly-Fisher, E. A. Katz, Y. Vaynzof, R. Vidal, G. Yu, H. Lin, S. Weng, S. Wang and A. B. Djurišic, JPhys Mater., 2023, 6, 032501 CrossRef.
- G. J. A. H. Wetzelaer, M. Scheepers, A. M. Sempere, C. Momblona, J. Ávila and H. J. Bolink, Adv. Mater., 2015, 27, 1837–1841 CrossRef CAS PubMed.
- L. Su, Z. X. Zhao, H. Y. Li, J. Yuan, Z. L. Wang, G. Z. Cao and G. Zhu, ACS Nano, 2015, 9, 11310–11316 CrossRef CAS.
- M. E. Laamari, A. Cheknane, A. Benghia and H. S. Hilal, Sol. Energy, 2019, 182, 9–15 CrossRef CAS.
- W. J. Yin, T. Shi and Y. Yan, Appl. Phys. Lett., 2014, 104, 063903 CrossRef.
- K. G. Lim, S. Ahn, Y. H. Kim, Y. Qi and T. W. Lee, Energy Environ. Sci., 2016, 9, 932–939 RSC.
- K. Lin, J. Xing, L. N. Quan, F. P. G. de Arquer, X. Gong, J. Lu, L. Xie, W. Zhao, D. Zhang, C. Yan, W. Li, X. Liu, Y. Lu, J. Kirman, E. H. Sargent, Q. Xiong and Z. Wei, Nature, 2018, 562, 245–248 CrossRef CAS.
- S. Zhang, Q. Shang, W. Du, J. Shi, Z. Wu, Y. Mi, J. Chen, F. Liu, Y. Li, M. Liu and Q. Zhang, Adv. Opt. Mater., 2018, 6(2), 1701032 CrossRef.
- D. A. Egger, A. M. Rappe and L. Kronik, Acc. Chem. Res., 2016, 49, 573–581 CrossRef CAS PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013,(342), 341–344 CrossRef CAS.
- J. A. Sichert, Y. Tong, N. Mutz, M. Vollmer, S. Fischer, K. Z. Milowska, R. García Cortadella, B. Nickel, C. Cardenas-Daw, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2015, 15, 6521–6527 CrossRef CAS PubMed.
- E. A. Muljarov, S. G. Tikhodeev, N. A. Gippius and T. Ishihara, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 14370 CrossRef CAS.
- P. Basumatary and P. Agarwnmal, Mater. Res. Bull., 2022, 149, 111700 CrossRef CAS.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed.
- C. S. Ponseca, T. J. Savenije, M. Abdellah, K. Zheng, A. Yartsev, T. Pascher, T. Harlang, P. Chabera, T. Pullerits, A. Stepanov, J. P. Wolf and V. Sundström, J. Am. Chem. Soc., 2014, 136, 5189–5192 CrossRef CAS.
- M. Upadhyay Kahaly, S. Misra and S. K. Mishra, J. Appl. Phys., 2017, 121, 205110 CrossRef.
- T. P. Kaloni, M. Upadhyay Kahaly, Y. C. Cheng and U. Schwingenschlögl, EPL, 2012, 99, 57002 CrossRef.
- NREL, https://www.nrel.gov/pv/interactive-cell-efficiency.html, (accessed July 2024).
- Y. Duan, F. E. Oropeza, X. Jin, O. Amargós-Reyes, Y. Atoini, L. M. Cavinato, G. N. Nagy, M. U. Kahaly, V. A. de la Peña O’Shea, D. Y. Wang and R. D. Costa, Adv. Funct. Mater., 2023, 33, 2209249 CrossRef CAS.
- P. Roy, N. Kumar Sinha, S. Tiwari and A. Khare, Sol. Energy, 2020, 198, 665–688 CrossRef CAS.
- S. Zhang and G. Han, Prog. Energy, 2020, 2, 022002 CrossRef.
- F. F. Targhi, Y. S. Jalili and F. Kanjouri, Results Phys., 2018, 10, 616–627 CrossRef.
- B. Kim, J. Kim and N. Park, Sci. Rep., 2020, 10, 19635 CrossRef CAS PubMed.
- G. R. Berdiyorov, F. El-Mellouhi, M. E. Madjet, F. H. Alharbi and S. N. Rashkeev, Appl. Phys. Lett., 2016, 108, 053901 CrossRef.
- Y. T. Huang, S. R. Kavanagh, D. O. Scanlon, A. Walsh and R. L. Hoye, Nanotechnology, 2021, 32, 132004 CrossRef CAS PubMed.
- J. Yang, Z. Yuan, X. Liu, S. Braun, Y. Li, J. Tang, F. Gao, C. Duan, M. Fahlman and Q. Bao, ACS Appl. Mater. Interfaces, 2018, 10, 16225–16230 CrossRef CAS PubMed.
- J. Yang, Q. Hong, Z. Yuan, R. Xu, X. Guo, S. Xiong, X. Liu, S. Braun, Y. Li, J. Tang, C. Duan, M. Fahlman and Q. Bao, Adv. Opt. Mater., 2018, 6, 1800262 CrossRef.
- J. D. McGettrick, K. Hooper, A. Pockett, J. Baker, J. Troughton, M. Carnie and T. Watson, Mater. Lett., 2019, 251, 98–101 CrossRef CAS.
- N. K. Kim, Y. H. Min, S. Noh, E. Cho, G. Jeong, M. Joo, S. W. Ahn, J. S. Lee, S. Kim, K. Ihm, H. Ahn, Y. Kang, H. S. Lee and D. Kim, Sci. Rep., 2017, 7, 4645 CrossRef PubMed.
- G. N. Liu, R. Y. Zhao, R. D. Xu, Q. Liu, B. Xu, Y. Y. Wang, Q. Wu, J. N. Wang, Y. Nie and C. Li, J. Mater. Chem. C, 2019, 7, 7700–7707 RSC.
- A. Senocrate, T. Acartürk, G. Y. Kim, R. Merkle, U. Starke, M. Grätzel and J. Maier, J. Mater. Chem. A, 2018, 6, 10847–10855 RSC.
- N. Aristidou, I. Sanchez-Molina, T. Chotchuangchutchaval, M. Brown, L. Martinez, T. Rath and S. A. Haque, Angew. Chem., Int. Ed., 2015, 54, 8208–8212 CrossRef CAS.
- J. Zhao, Y. Deng, H. Wei, X. Zheng, Z. Yu, Y. Shao, J. E. Shield and J. Huang, Sci. Adv., 2017, 3, 5616 CrossRef.
- M. Li, L. Zhang, C. Chen, J. Chen and L. Ding, J. Semicond., 2023, 44, 010201-1 Search PubMed.
- W. M. Haynes, Handbook of Physics and Chemistry, 2016 Search PubMed.
- M. U. Kahaly and U. V. Waghmare, Appl. Phys. Lett., 2007, 91, 023112 CrossRef.
- S. Nazir, M. Upadhyay Kahaly and U. Schwingenschlögl, Appl. Phys. Lett., 2012, 100, 201607 CrossRef CAS.
- P. H. T. Philipsen, et al., BAND 2023.1, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com.
- G. Te Velde and E. J. Baerends, Precise density-functional method for periodic structures, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 7888 CrossRef CAS.
- M. U. Kahaly, K. Ozdogan and U. Schwingenschlögl, J. Mater. Chem. A, 2013, 1, 8406–8410 RSC.
- R. W. Grimes and C. R. A. Catlow, Philos. Trans. R. Soc. London, Ser. A, 1991, 335(1639), 609–634 CrossRef CAS.
- D. Gryaznov, E. Heifets and E. Kotomin, Phys. Chem. Chem. Phys., 2009, 11(33), 7241–7247 RSC.
- N. Vanni, D. Morelli Venturi, E. Radicchi, G. Quaglia, E. Cambiotti, L. Latterini, F. De Angelis and F. Costantino, Adv. Mater. Interfaces, 2021, 8, 2101888 CrossRef CAS.
- A. Senocrate, T. Acartürk, G. Y. Kim, R. Merkle, U. Starke, M. Grätzel and J. Maier, J. Mater. Chem. A, 2018, 6(23), 10847–10855 RSC.
- W. Kong, A. Rahimi-Iman, G. Bi, X. Dai and H. Wu, J. Phys. Chem. C, 2016, 120(14), 7606–7611 CrossRef CAS.
- C. M. Sterling, C. Kamal, A. García-Fernández, G. J. Man, S. Svanström, P. K. Nayak, S. M. Butorin, H. sRensmo, U. B. Cappel and M. Odelius, J. Phys. Chem. C, 2022, 126, 20143–20154 CrossRef CAS.
- D. Y. Lin, B. C. Guo, Z. Y. Dai, C. F. Lin and H. P. Hsu, Crystals, 2019, 9, 589 CrossRef CAS.
- D. A. A. Leal, B. Krishnan, S. Shaji and D. A. Avellaneda, Mater. Chem. Phys., 2018, 215, 137–147 CrossRef CAS.
- T. Glaser, C. Müller, M. Sendner, C. Krekeler, O. E. Semonin, T. D. Hull, O. Yaffe, J. S. Owen, W. Kowalsky, A. Pucci and R. Lovrinčić, J. Phys. Chem. Lett., 2015, 6, 2913–2918 CrossRef CAS.
- H. Huang, X. Chen and K. Huang, Open Chem. J., 2019, 6, 52–65 CrossRef.
- G. Abdelmageed, L. Jewell, K. Hellier, L. Seymour, B. Luo, F. Bridges, J. Z. Zhang and S. Carter, Appl. Phys. Lett., 2016, 109, 233905 CrossRef.
- C. Du, B. Liu, J. Hu and H. Li, Mater. Lett., 2021, 285, 129137 CrossRef CAS.
- K. A. Khalaph, Z. J. Shanan, A. M. Jafar and F. M. Al-Attar, Defect Diffus. Forum, 2020, 398, 140–146 Search PubMed.
- A. M. S. Riel, R. K. Rowe, E. N. Ho, A. C. C. Carlsson, A. K. Rappé, O. B. Berryman and P. S. Ho, Acc. Chem. Res., 2019, 52, 2870–2880 CrossRef CAS.
- S. Maheshwari, S. Patwardhan, G. C. Schatz, N. Renaud and F. C. Grozema, Phys. Chem. Chem. Phys., 2019, 21, 16564–16572 RSC.
- W. Wang, M. Favaro, E. Chen, L. Trotochaud, H. Bluhm, K. S. Choi, R. Van De Krol, D. E. Starr and G. Galli, J. Am. Chem. Soc., 2022, 144, 17173–17185 CrossRef CAS.
- N. Awol, C. Amente, G. Verma and J. Y. Kim, J. Alloys Compd., 2020, 829, 154486 CrossRef CAS.
- J. Yan, X. Song, Y. Chen and Y. Zhang, Opt. Mater., 2020, 99, 109513 CrossRef CAS.
- Q. Niu, L. Zhang, Y. Xu, C. Yuan, W. Qi, S. Fu, Y. Ma, W. Zeng, R. Xia and Y. Min, Polymers, 2022, 14(3), 398 CrossRef CAS PubMed.
- V. S. Katta, M. Velpandian, S. Challapalli, P. Meduri and S. S. K. Raavi, Sustainable Energy Fuels, 2022, 6(24), 5539–5556 RSC.
- M. Acik, T. M. Alam, F. Guo, Y. Ren, B. Lee, R. A. Rosenberg, J. F. Mitchell, I. K. Park, G. Lee and S. B. Darling, Adv. Energy Mater., 2018, 8, 1701726 CrossRef.
- S. A. Fateev, A. A. Petrov, V. N. Khrustalev, P. V. Dorovatovskii, Y. V. Zubavichus, E. A. Goodilin and A. B. Tarasov, Chem. Mater., 2018, 30, 5237–5244 CrossRef CAS.
- S. M. Qaid, H. M. Ghaithan, B. A. Al-Asbahi and A. S. Aldwayyan, Coatings, 2022, 12(5), 549 CrossRef CAS.
- P. Toloueinia, H. Khassaf, A. Shirazi Amin, Z. M. Tobin, S. P. Alpay and S. L. Suib, ACS Appl. Energy Mater., 2020, 3, 8240–8248 CrossRef CAS.
- D. M. Haaland and R. T. Meyer, Int. J. Chem. Kinet., 1974, 6, 297–308 CrossRef CAS.
- M. Adachi, W. Eguchi and T. Haoka, J. Chem. Eng. Jpn., 1974, 7, 364–367 CrossRef CAS.
- W. C. Lin, W. C. Lo, J. X. Li, P. C. Huang and M. Y. Wang, ACS Omega, 2021, 6, 34606–34614 CrossRef CAS PubMed.
- M. Wang, C. Fei, A. Uddin and J. Huang, Sci. Adv., 2022, 8, 5977 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00574k |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |