
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the mechanical properties of molecular perovskites by controlling framework distortions via A-site substitution†
Silva M.
Kronawitter
 a,
Shinjoo
Park
a,
Sebastian A.
Hallweger
a,
Emily
Myatt
b,
Jem
Pitcairn
b,
Matthew J.
Cliffe
b,
Dominik
Daisenberger
c,
Markus
Drees
a and
Gregor
Kieslich
*a
a,
Shinjoo
Park
a,
Sebastian A.
Hallweger
a,
Emily
Myatt
b,
Jem
Pitcairn
b,
Matthew J.
Cliffe
b,
Dominik
Daisenberger
c,
Markus
Drees
a and
Gregor
Kieslich
*a
aDepartment of Chemistry, TUM School of Natural Sciences, Technical University of Munich, Lichtenbergstraße 4, 85748 Garching, Germany. E-mail: gregor.kieslich@tum.de
bSchool of Chemistry, University of Nottingham, Nottingham NG7 2RD, UK
cDiamond Light Source Ltd., Diamond House, Harwell Campus, Didcot OX11 ODE, UK
First published on 8th July 2024
Abstract
Molecular perovskites are important materials in the area of barocalorics, improper ferroelectrics and ferroelastics, where the search for principles that link composition, structure and mechanical properties is a key challenge. Herein, we report the synthesis of a new series of dicyanamide-based molecular perovskites [A]Ni(C2N3)3, where the A-site cation (A+) is a range of alkylated piperidinium cations. We use this new family to explore how A+ cations determine their mechanical response by measuring the bulk modulus (B) – using high-pressure powder X-ray diffraction. Within the series, we find a positive correlation between the network distortions of the pseudocubic [Ni(C2N3)3]− network and B. Furthermore, we show that we can tune framework distortions, and therefore B, by synthesising A-site solid solutions. The applied methodology is a blueprint for linking framework distortions and mechanical properties in network materials and guides us toward principles for designing macroscopic properties via systematic compositional changes in molecular perovskites.
Introduction
The substitution of atoms by molecules in the structure motifs of all-inorganic materials is a powerful approach for designing new materials. It opens up a large chemical design space for studying application-oriented1 and fundamental2 structure–property relationships, while still allowing the use of established principles3 from inorganic solid-state chemistry to guide materials discovery. For instance, in coordination polymers that crystallise in an AMX3 perovskite motif, such as [CH3NH3]Mn(N3)34 and [C10H20N]Cd(C2N3)3,5 here referred to as molecular perovskites, properties such as ferroelastic,6 (improper) ferroelectric1,7,8 and multiferroic9,10 behaviour, their glass-forming abilities11,12 and their application as barocalorics13,14 are linked to the molecular nature of the A- and X-site. In molecular perovskites, a ReO3-type [MX3]− network is formed by the M-site cation (M2+) and X-site anion (X−) with the A-site cation (A+) sitting in the void of the pseudocubic [MX3]− network for charge balance (Fig. 1). | ||
| Fig. 1 Schematic of the perovskite structure of [A]Ni(C2N3)3 materials studied in this work. Emphasis is given to the various A+ cations [DIP]+ (a), [PEP]+ (b), [DEP]+ (c) and [DPP]+ (d), as incorporated in the distorted 3D ReO3-type [Ni(C2N3)3]− network. C atoms are shown in the darker colour, N atoms in the lighter colour, distorted NiN6 octahedra in grey. The X− = [C2N3]− are simplified as linear linker shown as grey bars and H atoms are not shown for clarity. | ||
In molecular perovskites, the use of molecular ions as building units introduces new chemical and structural degrees of freedom that can couple to external stimuli,15,16 producing phenomena such as temperature and pressure-driven A-site order–disorder phase transitions,17,18 unconventional octahedral tilts and shifts that can couple to break inversion symmetry,1,19,20 melting processes as a response to heating11 and a new type of polymorphism.21 Given their role as model systems for understanding structure–property relationships,1,22 investigating their mechanical properties, such as the bulk modulus (B), Young's modulus, hardness and material stability, becomes increasingly important. Since the material's crystal chemistry ultimately determines its properties, studies related to the mechanical properties address both our fundamental interest in the search for structure–property principles and operational concerns such as material processability and material longevity. Furthermore, a low B has recently been implicated to improve the barocaloric performance in molecular materials.23 The compositional variability of molecular perovskites enables us,24 therefore, to identify the underlying structure chemical principles that determine B, and therewith, to enhance a material's solid-state cooling performance.
A few principles of how composition determines the mechanical properties of molecular perovskites have already been established. For instance, nanoindentation and high-pressure diffraction studies on materials such as [(CH3)2NH2]M(HCOO)3 (with M2+ = Ni2+, Co2+, Zn2+ and Mn2+),25,26 [C(NH2)3]M(HCOO)3 (with M2+ = Co2+ and Mn2+)27 and [Pr4N]M(C2N3)3 (with [Pr4N]+ = tetrapropylammonium, M2+ = Ni2+, Co2+ and Mn2+)28 have demonstrated that a smaller size of M2+ paired with a larger ligand field stabilisation energy (LFSE) leads to a larger Young's modulus and B. These findings agree with our recent results, where we observed a decreasing B as a function of increasing Shannon ionic radius of M2+.29
Looking at the role of A+, its capability to form hydrogen bonding interactions20,30 (HBIs) to the [MX3]− network has been found to be an important factor. For instance, the mechanical properties are tunable across the solid solution [NH3NH2]1−x[NH3OH]xZn(HCOO)331via HBIs, i.e. the choice of A+.32,33 More recently, this strategy has been used to introduce ferroelasticity in [Et3P(CH2)2Cl]1−x[Et3P(CH2)2F]xMn(C2N3)3.34 Yet, our understanding of the interplay between the A+ and active framework distortions of the [MX3]− is only developing.35–38
In the pursuit to fill this gap, we have synthesised a new series of [A]Ni(C2N3)3 molecular perovskites with A+ = diisopropyl-piperidinium ([DIP]+ = [C11H24N]+), ethyl-propyl-piperidinium ([PEP]+ = [C10H22N]+), diethyl-piperidinium ([DEP]+ = [C9H20N]+) and dipropyl-piperidinium ([DPP]+ = [C11H24N]+) (Fig. 1). The absence of acidic hydrogen atoms in all A+ excludes contributions from HBIs. Furthermore, by keeping M2+ = Ni2+ in the series, we have minimised the effect of M2+ cation size and LFSE, and hence, the chemical bond strength in the [Ni(C2N3)3]− network on the structure and properties of the molecular perovskites. Therefore, this series allows us to isolate the impact of the size and shape of the A+ cation on the distortions of the 3D [MX3]− network and their ramifications on the mechanical properties, which we measured via high-pressure powder X-ray diffraction (HPPXRD) by using B as a proxy. In addition – by employing a solid solution approach – we investigated to which extent B can be fine-tuned via A+ substitution.
Results and discussion
Synthesis and characterisation
In the first step, we designed a range of A+ to produce a series of molecular perovskites that can be used to determine how structural and pressure responsive properties are altered by small chemical changes. In this pursuit, we synthesised a series of piperidinium derivatives ([DIP]+, [PEP]+, [DEP]+ and [DPP]+), where different alkyl chain modifications of the piperidinium nitrogen enable studying the impact of small variations in size and/or shape (Fig. 1). All A+ were synthesised via nucleophilic substitution of halides, see ESI† for details (Fig. S1–S5 and S11–S18). For instance, [DEP]+ was obtained from the reaction of diethylamine and 1,5-dibromopentane in acetonitrile. Single crystals of the [A]Ni(C2N3)3 series were obtained by a mild-solution crystallisation route that has previously been used for the synthesis of related materials,5,21 see ESI† for details (S-5). In a typical reaction, the precursor materials [A]Br, Na(C2N3) and Ni(NO3)2·6H2O are dissolved in water, mixed and then left to crystallise overnight in a refrigerator. An overview of all synthesised compounds in this work is given in Table 1.| [A]Ni(C2N3)3 | A+ = [DIP]+ | [PEP]+ | [DEP]+ | [DPP]+ |
|---|---|---|---|---|
| V A+/Å3 | 227.25 | 202.18 | 190.56 | 225.87 |
| G | 0.87 | 0.83 | 0.88 | 0.81 |
| Space group | P21/c | P21/c | P21/c | C2/c |
| V cell/Å3 | 2163.4(5) | 2028.08(14) | 2021.16(14) | 2079.38(10) |
| A p/Å | 1.81 | 2.01 | 2.10 | 4.23 |
| B(2nd)/GPa | 8.2 ± 0.1 | 8.5 ± 0.1 | 8.8 ± 0.2 | 10.5 ± 0.2 |
| V 0(2nd)/Å3 | 2157.1 ± 0.5 | 2030.3 ± 0.6 | 2025.9 ± 0.2 | 2073.8 ± 0.6 |
Single crystal X-ray diffraction (SCXRD) at 100 and 300 K was used to determine the structure of all perovskite materials. All compounds in the [A]Ni(C2N3)3 series crystallise with the targeted AMX3 perovskite structure (Fig. 1). [DIP]Ni(C2N3)3, [PEP]Ni(C2N3)3 and [DEP]Ni(C2N3)3 crystallise in the monoclinic space group P21/c, see ESI† for details of data collection and crystal structure solution (S-7, Tables S1–S3 and Fig. S7–S9). In contrast, [DPP]Ni(C2N3)3 crystallises in the space group C2/c (Table S4 and Fig. S10, ESI†). To quantify the extent of network distortions across the series, we have applied an established group theoretical analysis39–41 to SCXRD data of 300 K for all compounds. In this approach, the structures are first simplified to the underlying octahedral network and subsequently compared to an idealised cubic parent network. By using ISODISTORT,40 the active distortions are then identified and described via their irreducible representation and amplitude. The latter enables the calculation of the overall parent-cell normalised distortion amplitude of the network (Ap), providing a rigorous measure for network distortions across our series, see Table 1 for Ap values of the different compounds at 300 K and ESI† for further tilt analysis details (S-69 and Table S22–S24).
To determine relationships between the size and shape of A+ and the resulting network distortions (Fig. 1), atomic coordinates from SCXRD data at 300 K were used to extract the geometry of A+. Following previous reports,42 we use the globularity (G) as a measure for the asphericity of A+, where G = 1 for a sphere. For this, we used DFT calculations to obtain the A+ surface (Sm,A+), the A+ volume (VA+) as enclosed by Sm,A+, and the surface of an idealised sphere (Ssp,A+) with VA+ (Table S27, ESI†). Then, the A+ globularity (G = Ssp,A+/Sm,A+) was calculated (Table 1). By using CrystalExplorer17,43 we obtained G > 0.8 for all A+, which agrees with previous results from the literature for A+ of dicyanamide-based molecular perovskites.42 A similar trend in G with slightly smaller values is obtained when using Gaussian1644 for calculating Sm,A+, Ssp,A+ and VA+, see ESI† for details (Tables S27 and S30). No correlation between Ap, VA+ and G is observed (Table 1), while the importance of G in dictating Ap becomes apparent when considering [DPP]+ and [DIP]+. Both cations have a similar VA+ but are different in G. [DPP]+ shows the significantly smaller G, and among the here investigated series of [A]Ni(C2N3)3, the Ap of [DPP]Ni(C2N3)3 is by far the largest. Therefore, we hypothesise that the observed network distortions in [DPP]Ni(C2N3)3 are a direct ramification of the interplay between A+ and the 3D [Ni(C2N3)3]− network, highlighting the templating role of A+ and the possibility of tuning network distortions via chemical modifications on A+. For instance, for symmetric functionalisation of the piperidine backbone, we find Ap increasing with the length of the alkyl chains, e.g. from ethyl ([DEP]+) to propyl ([DPP]+). Hence, small alkyl chain modifications on the nitrogen atom of piperidinium, and thus its shape, evidently have an impact on Ap of the 3D [Ni(C2N3)3]− network (Fig. 1) and, as we will see in the following, on the resulting pressure responsiveness.
The phase purity of powder samples of [A]Ni(C2N3)3 was verified using laboratory PXRD and Pawley analysis (Fig. S28–S31, ESI†). Furthermore, thermal analysis (TGA/DSC) showed decomposition temperatures (Td) between 570 and 600 K (Fig. S20–S23, ESI†). These are at the upper end of previously reported Td of dicyanamide-based molecular perovskites.21,45,46 In parts, we rationalise this by the stability of the here employed A+, which has previously been suggested as an important factor determining Td;47 however, Td is a generally complex quantity, which depends upon many parameters, including crystal chemistry factors and analytic concerns.45,48,49 In the DSC curves, several heat events related to 1st and 2nd order phase transitions were observed (Fig. S24–S27, ESI†), while no evidence of melting before Td was found;11 however, the nature of these phase transitions is beyond the focus of this study. Here, we employ our new [A]Ni(C2N3)3 family as model compounds to study the relation between A+ and B.
Molecular perovskites under high pressure
HPPXRD was used to obtain B for all compounds (Table 1). Given the expected subtle differences in the materials' pressure response, we have used a custom-built HPPXRD setup50 to collect HPPXRD data with finely spaced pressure points, step size of Δp = 0.02 GPa, up to a maximum pressure of pmax. = 0.4 GPa (Fig. 2 and Fig. S34–S37, ESI†). Visual inspection of the HPPXRD data (Fig. 2) reveals pressure-driven phase transitions at pressures of ptrs. = 0.3 GPa ([DIP]Ni(C2N3)3), ptrs. = 0.28 GPa ([PEP]Ni(C2N3)3) and ptrs. = 0.3 GPa ([DEP]Ni(C2N3)3). Accordingly, Pawley profile fits51 were performed up to ptrs., or pmax. = 0.4 GPa for [DPP]Ni(C2N3)3, for obtaining lattice parameters and V(p) data, see ESI† for full details of the Pawley profile fits (Tables S11–S14 and Fig. S47–S50). B was calculated for each [A]Ni(C2N3)3 compound by fitting a Birch–Murnaghan (B–M) equation of state to the V(p) data. For a better comparison with literature data, both 2nd and 3rd order B–M equation of state fits were performed using EOSFit,52 see ESI† for the results and all fits of the V(p) and F(f) plots (Fig. S64–S67). Similar results were obtained by using PASCal (Table S28, ESI†).53 Only fits using 2nd order B–M equation of state show standard errors in fitted B comparable to those observed for the standard material Ni(dmgH)2 (dmgH− = dimethylglyoximate), σB = ± 0.09 GPa. Much larger uncertainties for a 3rd order B–M fit suggest overparametrisation, see ESI† (S-66).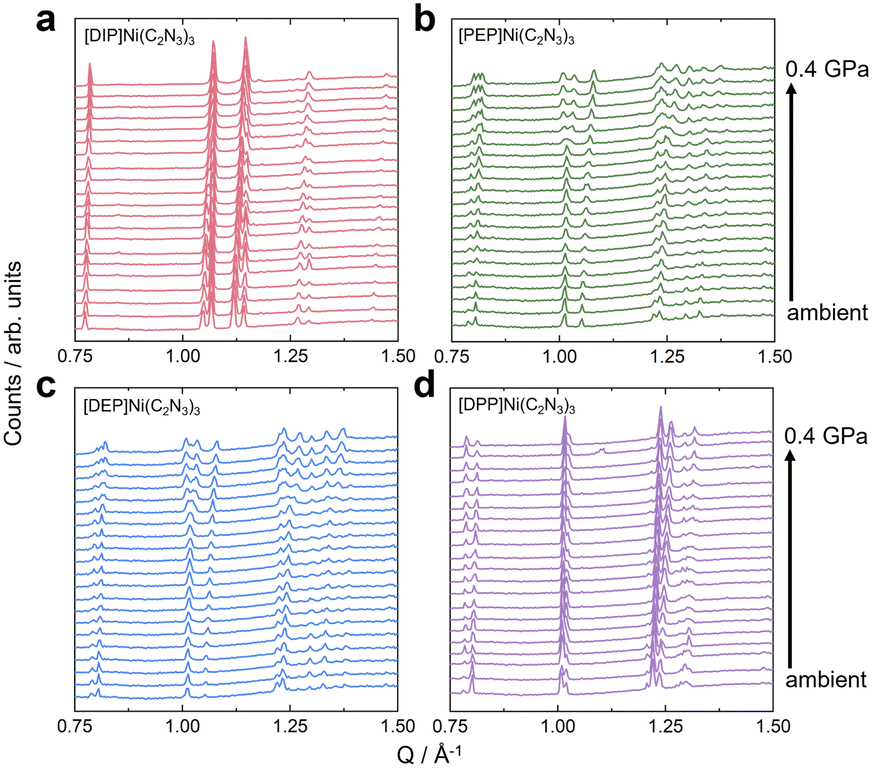 | ||
| Fig. 2 HPPXRD data (λ = 0.4246 Å) of [A]Ni(C2N3)3 with A+ = [DIP]+ (a), [PEP]+ (b), [DEP]+ (c), and [DPP]+ (d), collected in the pressure range between ambient pressure and 0.4 GPa. The data are shown as stacking plots, where intensities have been normalised. | ||
We observe a monotonically decreasing B along the series B (A+ = [DPP]+) = 10.5 ± 0.2 GPa, B (A+ = [DEP]+) = 8.8 ± 0.2 GPa, B (A+ = [PEP]+) = 8.5 ± 0.1 GPa and B (A+ = [DIP]+) = 8.2 ± 0.1 GPa. We would like to note that deriving B by using the same pressure range for each compound results in the same trend but with a slightly larger σ. Across the here investigated series of compounds, we find a correlation between Ap and B (Fig. 3). The more distorted the framework is at p = ambient, the stiffer it is (Table 1). Since framework distortions summarised in Ap are some of the lowest-energy pathways for pressure-induced structural changes, a smaller Ap provides a larger margin for pressure-induced network distortions as a response to hydrostatic pressure, and thus a larger compressibility, i.e. a smaller B. This interpretation links Ap(↑) and B(↑) along the [A]Ni(C2N3)3 series, providing a heuristic understanding of the observed pressure response within this series (Fig. 3). Given that Ap is very sensitive to chemical functionalisation of the piperidine backbone, we here show that chemical changes of A+ are highly effective for manipulating B.
 | ||
| Fig. 3 Bulk modulus (B) including error bars as a function of the parent-cell normalised amplitude over all active framework distortion modes (Ap) for the [A]Ni(C2N3)3 family with varying A+ and molecular solid solutions [PEP]1-x[DPP]xNi(C2N3)3. | ||
We note that using Ap as a measure for available distortion pathways only accounts for distortion energies when treating the 3D framework isolated from A+. Any directional interactions, such as HBIs between A+ and the 3D network, give A+ a more active role and thus produce larger variability in B, as found in [(CH)3NH2]M(HCOO)3 (M2+ = Mn2+, Fe2+ and Cu2+)54 and [C(NH2)3]M(HCOO)3 (M2+ = Mn2+, Co2+ and Cd2+),55,56 with B varying between 21.3 and 27.2 GPa. HBIs thus strengthen the overall framework by making any network distortions energetically more demanding, resulting in an increase of B overall.
Furthermore, considering the series [(C3H7)3CH3N]M(C2N3)3 with varying framework cation M2+ (M2+ = Mn2+, Co2+ and Ni2+) where Ap decreases but B increases,29e.g. B (M2+ = Mn2+) = 7.1 ± 0.2 GPa and Ap (M2+ = Mn2+) = 2.36, B (M2+ = Co2+) = 9.9 ± 0.3 GPa and Ap (M2+ = Co2+) = 2.17 and B (M2+ = Ni2+) = 10.9 ± 0.2 GPa and Ap (M2+ = Ni2+) = 2.09, it is evident that crystal chemistry factors other than Ap can be decisive in determining B. For instance, when going from Mn2+ to Co2+ and Ni2+, the 3D network is stiffened and overall, B increases. Any network distortions increase in energy, and thus, Ap is reduced, making the change in chemical bond strength the dominating factor in this comparison.
Bulk moduli of A-site solid solutions
To further explore the tunability of B via targeted changes of Ap, we have synthesised the solid solutions [PEP]1−x[DPP]xNi(C2N3)3 with x = 0, 0.45, 0.69, 0.85, 0.96 and 1. For their synthesis, the stoichiometry of precursor salts [PEP]Br and [DPP]Br has been varied and mixed with Na(C2N3) and Ni(NO3)2·6H2O in water as a solvent, see ESI† (S-6). Independent of the applied precursor stoichiometry, we only obtained members of the solid solution with x ≥ 0.45. In previous studies, a full solid solution, i.e. 0 ≤ x ≤ 1, has only been found in one example where chemically similar A+ were used.34 Therefore, we rationalise the here observed x range by the difference in crystallisation kinetics due to the chemical difference of A+. 1H NMR spectroscopy on acid-digested samples has been used to determine the chemical composition x (Table S9 and Fig. S19, ESI†). We confirmed the formation of a solid solution using laboratory PXRD measurements of a mixture of the parent materials [PEP]Ni(C2N3)3 and [DPP]Ni(C2N3)3 (Fig. S32, ESI†). We determined the lattice parameters using Pawley refinements, using P21/c for x = 0 – 0.96 (Fig. S44 and S51–S54, ESI†) and C2/c for x = 1 (Fig. S46, ESI†). We observe a monotonic expansion of the volume from 2029.7(4) Å3 (x = 0) to 2076.9(4) Å3 (x = 1) with increasing x at ambient pressure (Table S15, ESI†).We then used HPPXRD, using the same methodology, to determine B as a function of x (Fig. S68–S71, ESI†). Within standard errors, we observe an increasing B as a function of x in [PEP]1−x[DPP]xNi(C2N3)3 (Tables S20 and S29, ESI†). This points at a close to ideal solid solution behaviour57 and suggests the absence of strong coupling effects,58e.g. behaviour that could arise from the ordering of [PEP]+ and [DPP]+.
Furthermore, we have performed Rietveld-based structural refinements for x = 0.45, 0.69, 0.85 and 0.96 based on a simplified structural model for extracting the network geometry and framework distortion modes, see ESI† for the employed strategy of structure refinement (S-59) and for results (Table S26). Visual inspection of the PXRD pattern and differences curve of the Rietveld fits suggests that the results should be treated with care; however, for all compounds, Ap lies between 2.013 (x = 0) and 4.232 (x = 1), suggesting tunability of Apvia A+ substitution (Fig. 3). Thus, by employing the solid solution approach, it is possible to fine-tune B in a window as fine as B = 8.5 (x = 0) − 10.5 (x = 1) GPa.
Conclusion
In conclusion, we have synthesised a new series of [A]Ni(C2N3)3 molecular perovskites, showing that framework distortions are sensitive towards small chemical changes of A+. Subsequently, we applied this series to establish a link between the size and globularity of A+, the active distortion modes in the [Ni(C2N3)3]− framework and the pressure responsiveness of the molecular perovskites for the first time. In the absence of HBIs and changes in chemical bond strength within the network, we conclude that Ap is the decisive parameter in defining the compressibility of a molecular perovskite. Additionally, we show tunability of Ap, and thus B, through the synthesis of A-site solid solutions. Such a tunability of B via chemical modifications advances our understanding of underlying composition–structure–property relations and paves the way for the targeted design of molecular perovskites’ mechanical properties for applications such as barocalorics.13,23 Additionally, the here applied methodology to link network distortions and the pressure responsiveness is of general nature, with implications for the analysis of the high-pressure responsiveness of closely related material families, including metal–organic frameworks, covalent organic frameworks and hydrogen-bonded frameworks.Focusing on molecular perovskites, our results have important ramifications on how to think and approach the challenge of designing molecular perovskites with targeted mechanical properties and, more generally, targeted network distortions. Evidently, A+ templates framework distortions, and when aiming for large distortions, A+ should be chosen that largely diverges from a sphere. For A+ based on alkylated ammonium or phosphonium, a chemical functionalisation that enables the formation of conformers with large anisotropies seems fruitful. This suggests the use of a more rigorous conformational analysis prior to the synthesis of A+ cations.59 In parallel, robust criteria for describing the asphericity of A+ should be tested, where we consider G as an important yet only qualitative measure since any templating effect can be expected to be related to asphericity and volume. Such developments have the potential to bring related fields to fruition, such as the targeted design of active distortion modes, with implications for the synthesis of improper ferroelectrics.1
Author contributions
S. M. Kronawitter: methodology, validation, formal analysis, investigation, data curation, writing – original draft, review and editing, visualisation. S. Park: data curation, investigation. S. A. Hallweger, E. Myatt, J. Pitcairn, M. J. Cliffe, D. Daisenberger: investigation, review and editing. G. Kieslich: conceptualisation, writing – review and editing, supervision, resources, project administration.Data availability
The data supporting this article have been included as part of the ESI.† Furthermore, all crystallographic data have been deposited at the CCDC, see 2309035, 2309036, 2309038, 2309039, 2309042, 2309043, 2309049 and 2309050.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We want to acknowledge beamtime at the Diamond Light Source (UK), Beamline I15 (CY30815-2). S. M. K., S. A. H. and G. K. acknowledge support from the DFG through research grants (450070835, 493871295) and the Heisenberg program (524525093). Thanks to Melvin Resch for his contributions to the synthesis of the solid solutions.Notes and references
- H. L. B. Boström, M. S. Senn and A. L. Goodwin, Nat. Commun., 2018, 9, 2380 CrossRef PubMed.
- S. Yuan, A. Stroppa and A. E. Phillips, APL Mater., 2023, 11, 061114 CrossRef CAS.
- G. Kieslich, S. Sun and A. K. Cheetham, Chem. Sci., 2014, 5, 4712–4715 RSC.
- X.-H. Zhao, X.-C. Huang, S.-L. Zhang, D. Shao, H.-Y. Wei and X.-Y. Wang, J. Am. Chem. Soc., 2013, 135, 16006–16009 CrossRef CAS.
- S. Burger, S. Kronawitter, H. L. B. Boström, J. K. Zaręba and G. Kieslich, Dalton Trans., 2020, 49, 10740–10744 RSC.
- Z.-B. Liu, L. He, P.-P. Shi, Q. Ye and D.-W. Fu, J. Phys. Chem. Lett., 2020, 11, 7960–7965 CrossRef CAS.
- K. Li, Z.-G. Li, J. Xu, Y. Qin, W. Li, A. Stroppa, K. T. Butler, C. J. Howard, M. T. Dove, A. K. Cheetham and X.-H. Bu, J. Am. Chem. Soc., 2022, 144, 816–823 CrossRef CAS PubMed.
- A. Sieradzki, M. Mączka, M. Simenas, J. K. Zaręba, A. Gągor, S. Balciunas, M. Kinka, A. Ciupa, M. Nyk, V. Samulionis, J. Banys, M. Paluch and S. Pawlus, J. Mater. Chem. C, 2018, 6, 9420–9429 RSC.
- P. Jain, V. Ramachandran, R. J. Clark, H. D. Zhou, B. H. Toby, N. S. Dalal, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 13625–13627 CrossRef CAS.
- A. Stroppa, P. Barone, P. Jain, J. M. Perez-Mato and S. Picozzi, Adv. Mater., 2013, 25, 2284–2290 CrossRef CAS.
- B. K. Shaw, A. R. Hughes, M. Ducamp, S. Moss, A. Debnath, A. F. Sapnik, M. F. Thorne, L. N. McHugh, A. Pugliese, D. S. Keeble, P. Chater, J. M. Bermudez-Garcia, X. Moya, S. K. Saha, D. A. Keen, F.-X. Coudert, F. Blanc and T. D. Bennett, Nat. Chem., 2021, 13, 778–785 CrossRef CAS.
- S. M. Kronawitter, S. A. Hallweger, J. Meyer, C. Pedri, S. Burger, A. Alhadid, S. Henke and G. Kieslich, APL Mater., 2023, 11, 31119 CrossRef CAS.
- J. M. Bermúdez-García, M. Sánchez-Andújar, S. Castro-García, J. López-Beceiro, R. Artiaga and M. A. Señarís-Rodríguez, Nat. Commun., 2017, 8, 15715 CrossRef.
- J. Salgado-Beceiro, A. Nonato, R. X. Silva, A. García-Fernández, M. Sánchez-Andújar, S. Castro-García, E. Stern-Taulats, M. A. Señarís-Rodríguez, X. Moya and J. M. Bermúdez-García, Mater. Adv., 2020, 1, 3167–3170 RSC.
- H. L. B. Boström and A. L. Goodwin, Acc. Chem. Res., 2021, 54, 1288–1297 CrossRef.
- C. L. Hobday and G. Kieslich, Dalton Trans., 2021, 50, 3759–3768 RSC.
- W. Wei, W. Li, K. T. Butler, G. Feng, C. J. Howard, M. A. Carpenter, P. Lu, A. Walsh and A. K. Cheetham, Angew. Chem., Int. Ed., 2018, 57, 8932–8936 CrossRef CAS.
- R. X. Silva, R. R. Hora, A. Nonato, A. García-Fernández, J. Salgado-Beceiro, M. A. Señarís-Rodríguez, M. S. Andújar, A. P. Ayala and C. W. A. Paschoal, Spectrochim. Acta, Part A, 2023, 289, 122198 CrossRef CAS PubMed.
- S. G. Duyker, J. A. Hill, C. J. Howard and A. L. Goodwin, J. Am. Chem. Soc., 2016, 138, 11121–11123 CrossRef CAS PubMed.
- M. M
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) czka, M. Kryś, S. Sobczak, D. L. M. Vasconcelos, P. T. C. Freire and A. Katrusiak, J. Phys. Chem. C, 2021, 125, 26958–26966 CrossRef.
czka, M. Kryś, S. Sobczak, D. L. M. Vasconcelos, P. T. C. Freire and A. Katrusiak, J. Phys. Chem. C, 2021, 125, 26958–26966 CrossRef. - S. Burger, S. Grover, K. T. Butler, H. L. B. Boström, R. Grau-Crespo and G. Kieslich, Mater. Horiz., 2021, 8, 2444–2450 RSC.
- D. Boldrin, Appl. Phys. Lett., 2021, 118, 170502 CrossRef CAS.
- B. Li, Y. Kawakita, S. Ohira-Kawamura, T. Sugahara, H. Wang, J. Wang, Y. Chen, S. I. Kawaguchi, S. Kawaguchi, K. Ohara, K. Li, D. Yu, R. Mole, T. Hattori, T. Kikuchi, S.-I. Yano, Z. Zhang, Z. Zhang, W. Ren, S. Lin, O. Sakata, K. Nakajima and Z. Zhang, Nature, 2019, 567, 506–510 CrossRef CAS PubMed.
- J. M. Bermúdez-García, M. Sánchez-Andújar and M. A. Señarís-Rodríguez, J. Phys. Chem. Lett., 2017, 8, 4419–4423 CrossRef.
- J.-C. Tan, P. Jain and A. K. Cheetham, Dalton Trans., 2012, 41, 3949–3952 RSC.
- S. Sobczak, A. Chitnis, M. Andrzejewski, M. Mączka, S. Gohil, N. Garg and A. Katrusiak, CrystEngComm, 2018, 20, 5348–5355 RSC.
- G. Feng, D. Gui and W. Li, Cryst. Growth Des., 2018, 18, 4890–4895 CrossRef CAS.
- L.-J. Ji, S.-J. Sun, Y. Qin, K. Li and W. Li, Coord. Chem. Rev., 2019, 391, 15–29 CrossRef CAS.
- S. Grover, S. Burger, K. T. Butler, K. Hemmer, P. Vervoorts, G. Kieslich and R. Grau-Crespo, CrystEngComm, 2023, 25, 3439–3444 RSC.
- K. L. Svane, A. C. Forse, C. P. Grey, G. Kieslich, A. K. Cheetham, A. Walsh and K. T. Butler, J. Phys. Chem. Lett., 2017, 8, 6154–6159 CrossRef CAS PubMed.
- G. Kieslich, S. Kumagai, A. C. Forse, S. Sun, S. Henke, M. Yamashita, C. P. Grey and A. K. Cheetham, Chem. Sci., 2016, 7, 5108–5112 RSC.
- W. Li, A. Thirumurugan, P. T. Barton, Z. Lin, S. Henke, H. H.-M. Yeung, M. T. Wharmby, E. G. Bithell, C. J. Howard and A. K. Cheetham, J. Am. Chem. Soc., 2014, 136, 7801–7804 CrossRef CAS.
- G. Kieslich, A. C. Forse, S. Sun, K. T. Butler, S. Kumagai, Y. Wu, M. R. Warren, A. Walsh, C. P. Grey and A. K. Cheetham, Chem. Mater., 2016, 28, 312–317 CrossRef CAS.
- L. He, P.-P. Shi, M.-M. Zhao, C.-M. Liu, W. Zhang and Q. Ye, Chem. Mater., 2021, 33, 799–805 CrossRef CAS.
- I. E. Collings, J. A. Hill, A. B. Cairns, R. I. Cooper, A. L. Thompson, J. E. Parker, C. C. Tang and A. L. Goodwin, Dalton Trans., 2016, 45, 4169–4178 RSC.
- M. Mączka, S. Sobczak, M. Kryś, F. F. Leite, W. Paraguassu and A. Katrusiak, J. Phys. Chem. C, 2021, 125, 10121–10129 CrossRef.
- A. Sieradzki, A. Nowok, S. Sobczak, K. Roszak, A. Szeremeta, M. Maczka, A. Katrusiak and S. Pawlus, Research Square, 2023 DOI:10.21203/rs.3.rs-3657532/v1.
- J. Y. Lee, S. Ling, S. P. Argent, M. S. Senn, L. Cañadillas-Delgado and M. J. Cliffe, Chem. Sci., 2021, 12, 3516–3525 RSC.
- H. T. Stokes, D. M. Hatch and B. J. Campbell, ISODISTORT, ISOTROPY Software Suite, https://iso.byu.edu Search PubMed.
- B. J. Campbell, H. T. Stokes, D. E. Tanner and D. M. Hatch, J. Appl. Crystallogr., 2006, 39, 607–614 CrossRef CAS.
- H. L. B. Boström, CrystEngComm, 2020, 22, 961–968 RSC.
- J. García-Ben, A. García-Fernández, P. Dafonte-Rodríguez, I. Delgado-Ferreiro, U. B. Cappel, S. Castro-García, M. Sánchez-Andújar, J. M. Bermúdez-García and M. A. Señarís-Rodríguez, J. Solid State Chem., 2022, 316, 123635 CrossRef.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, Gaussian 16 Rev. B.01, Wallingford, CT, 2016 Search PubMed.
- J. García-Ben, L. N. McHugh, T. D. Bennett and J. M. Bermúdez-García, Coord. Chem. Rev., 2022, 455, 214337 CrossRef.
- L. Zhou, X. Zheng, P.-P. Shi, Z. Zafar, H.-Y. Ye, D.-W. Fu and Q. Ye, Inorg. Chem., 2017, 56, 3238–3244 CrossRef CAS PubMed.
- C. Ye, L. N. McHugh, C. Chen, S. E. Dutton and T. D. Bennett, Angew. Chem., Int. Ed., 2023, 62, e202302406 CrossRef CAS.
- Y. Wang and W. J. Thomson, Thermochim. Acta, 1995, 255, 383–390 CrossRef CAS.
- N. Khan, D. Dollimore, K. Alexander and F. Wilburn, Thermochim. Acta, 2001, 367–368, 321–333 CrossRef CAS.
- N. J. Brooks, B. L. L. E. Gauthe, N. J. Terrill, S. E. Rogers, R. H. Templer, O. Ces and J. M. Seddon, Rev. Sci. Instrum., 2010, 81, 64103 CrossRef.
- G. S. Pawley, J. Appl. Crystallogr., 1981, 14, 357–361 CrossRef CAS.
- J. Gonzalez-Platas, M. Alvaro, F. Nestola and R. Angel, J. Appl. Crystallogr., 2016, 49, 1377–1382 CrossRef CAS.
- M. J. Cliffe and A. L. Goodwin, J. Appl. Crystallogr., 2012, 45, 1321–1329 CrossRef CAS.
- I. E. Collings, M. Bykov, E. Bykova, M. Hanfland, S. van Smaalen, L. Dubrovinsky and N. Dubrovinskaia, CrystEngComm, 2018, 20, 3512–3521 RSC.
- Z. Yang, G. Cai, C. L. Bull, M. G. Tucker, M. T. Dove, A. Friedrich and A. E. Phillips, Philos. Trans. R. Soc., A, 2019, 377, 20180227 CrossRef CAS.
- H. Gao, C. Li, L. Li, W. Wei, Y. Tan and Y. Tang, Dalton Trans., 2020, 49, 7228–7233 RSC.
- K. T. Jacob, S. Raj and L. Rannesh, Int. J. Mater. Res, 2007, 98, 776–779 CrossRef CAS.
- N. L. Evans, P. M. M. Thygesen, H. L. B. Boström, E. M. Reynolds, I. E. Collings, A. E. Phillips and A. L. Goodwin, J. Am. Chem. Soc., 2016, 138, 9393–9396 CrossRef CAS PubMed.
- A. M. Mroz, L. Turcani and K. E. Jelfs, Electron. Struct., 2023, 5, 45004 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details to material synthesis and characterisation (SCXRD, PXRD, NMR, TGA, DSC, HPPXRD and DFT). CCDC 2309035, 2309036, 2309038, 2309039, 2309042, 2309043, 2309049 and 2309050. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ma00587b |
| This journal is © The Royal Society of Chemistry 2024 |