DOI:
10.1039/D4MA00267A
(Paper)
Mater. Adv., 2024,
5, 4818-4831
Preparation of zwitterionic random and block poly(N-α-acrylamide-L-lysine-co-2-methacryloyloxyethyl phosphorylcholine) copolymers and their effect on fibrinolytic activity
Received
15th March 2024
, Accepted 22nd April 2024
First published on 24th April 2024
Abstract
Random copolymers [P(L-r-M)] and block copolymers [P(L-b-M)] composed of N-α-L-lysineacrylamide (LysAA) and 2-(methacryloyloxyethyl phosphorylcholine) (MPC) were prepared by radical polymerization or reversible addition–fragmentation chain transfer (RAFT) polymerization. Poly(LysAA)-immobilized glass substrates (L-g-GL) and poly(LysAA-co-MPC) random copolymer immobilized glass substrates [LM(m:n)-g-GL] were prepared to create biomaterials with good antithrombotic properties. Such materials allow long-term selective binding of serum fibrinolytic factor (plasminogen: Plg; tissue-type plasminogen activator: t-PA), while exhibiting low adsorption of other serum proteins and suppressing their denaturation. The fibrinolytic activity of the LysAA-containing polymers in liquid and solid-phase conditions was evaluated using the chromogenic plasmin substrate H-D-Val-Leu-Lys-p-nitroaniline (S-2251) and Plg/t-PA in the presence of poly(LysAA), L-g-GL or P(L-r,b-M), and LM(m:n)-g-GL. The enzymatic reaction of Plg/t-PA was maximally enhanced under liquid-phase conditions with P(L-r-M) containing 75 mol% of LysAA in the copolymer but not with poly(LysAA). The solid-phase condition was greatly enhanced by LM(8:2)-g-GL containing a mol ratio of LysAA that approximated the liquid-phase condition. Enhancement was also observed for LM(2:8)-g-GL containing 20 mol% LysAA, which was not enhanced in the liquid-phase condition.
Introduction
Plasminogen (Plg) and tissue-type plasminogen activator (t-PA) are serum proteins that play a central role in the fibrinolytic system in human blood.1,2 An anti-thrombotic surface was previously constructed by immobilizing Plg and/or t-PA onto a biomaterial surface.3 We were the first to develop L-serine-bearing polymethacrylate,4–6L-lysine-bearing polymethacrylamide [poly(LysMA)], and polyacrylamide [poly(LysAA)] with zwitterionic structures on the side chains to suppress the adsorption and denaturation of serum proteins as a new class of non-toxic amino acid-derived biomaterials. Furthermore, we reported that poly(LysAA) and poly(LysMA) enhanced fibrinolytic activity based on strong interactions with the lysine binding sites (LBS) of Plg and t-PA viaL-lysine in the polymer side chain.7–10 Poly(2-methacryloyloxyethyl phosphorylcholine) (MPC) is a strong, bio-inert polymer that has been used in a wide range of applications in nanomedicine and bioengineering, in medical devices and sensors, and as an antifouling material.11,12 The zwitterionic structures of LysAA and LysMA resemble that of the side chain of MPC and suppress the adsorption of serum protein and enhance the fibrinolytic activity by Plg and t-PA binding moieties without the need to pre-coat with fibrinolytic factors. The side chains of poly(MPC), poly(LysAA), and poly(LysMA) are terminated with a positive charge, whereas many zwitterionic polymers such as polysulfobetain and polycarbobetain13 have a negative charge. Positively-charged monomers in the side-chain end of copolymers make the copolymers soluble in ethanol, whereas negative charges often make the copolymer insoluble, restricting solvent coating on substrates. Selective binding of blood on poly(LysAA)- or poly(LysMA)-immobilized surfaces should provide high biocompatibility for a long period of time. The binding of Plg to a cell surface stimulates plasmin (Plm) generation by plasminogen activator, in contrast to Plg in solution.14,15 In this study, prior to immobilizing proteins on a material surface and evaluating the biocompatibility of the surface,8,9 we investigated the effect of MPC segments containing a lipid bilayer component on the fibrinolytic promoting effect of poly(LysAA). The materials prepared in this study were intended for practical use in medical applications and were prepared by random radical polymerization to avoid the use of initiators of toxicological concern. Block copolymers were prepared by controlled living/radical polymerization (CLRP) because they could only be prepared by CLRP. We prepared random copolymers [P(L-r-M)] containing various amounts of MPC and a block copolymer [P(L-b-M)] using LysAA and MPC and studied their utility for regulating anti-thrombogenicity. We also evaluated the interaction between poly(LysAA) and P(L-r-M) with Plg and t-PA using surface plasmon resonance (SPR) measurements. Random graft copolymers with various molar ratios of LysAA and MPC were immobilized on the substrate as an evaluation environment similar to that for medical coatings for practical use, and their utility for anti-thrombogenic modulation was investigated in the same way. Atomic force microscopy (AFM) was used to evaluate the effect of the interaction between the immobilized polymer surface and Plg/t-PA on the fibrinolytic activity.
Results and discussion
Preparation of polymers containing LysAA units
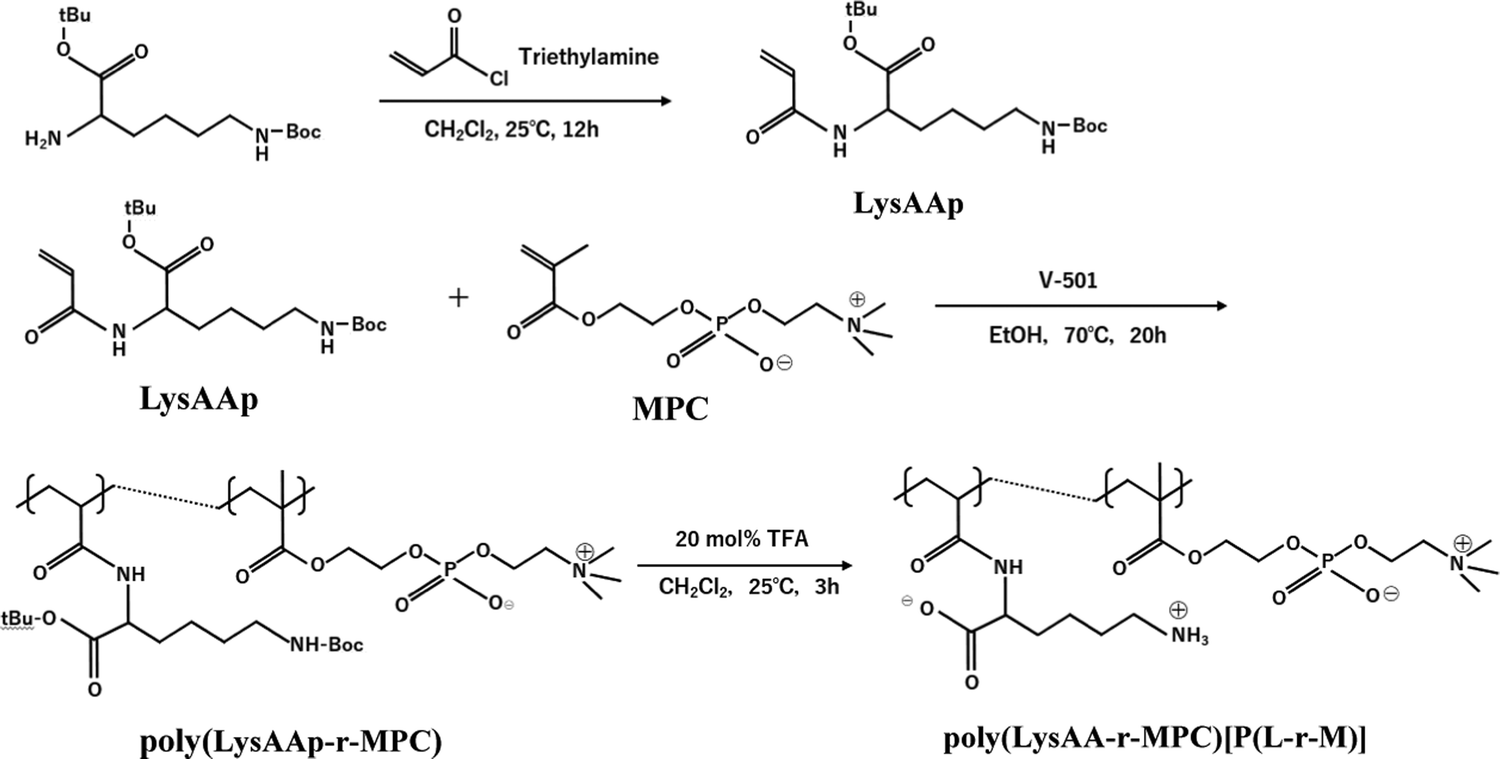
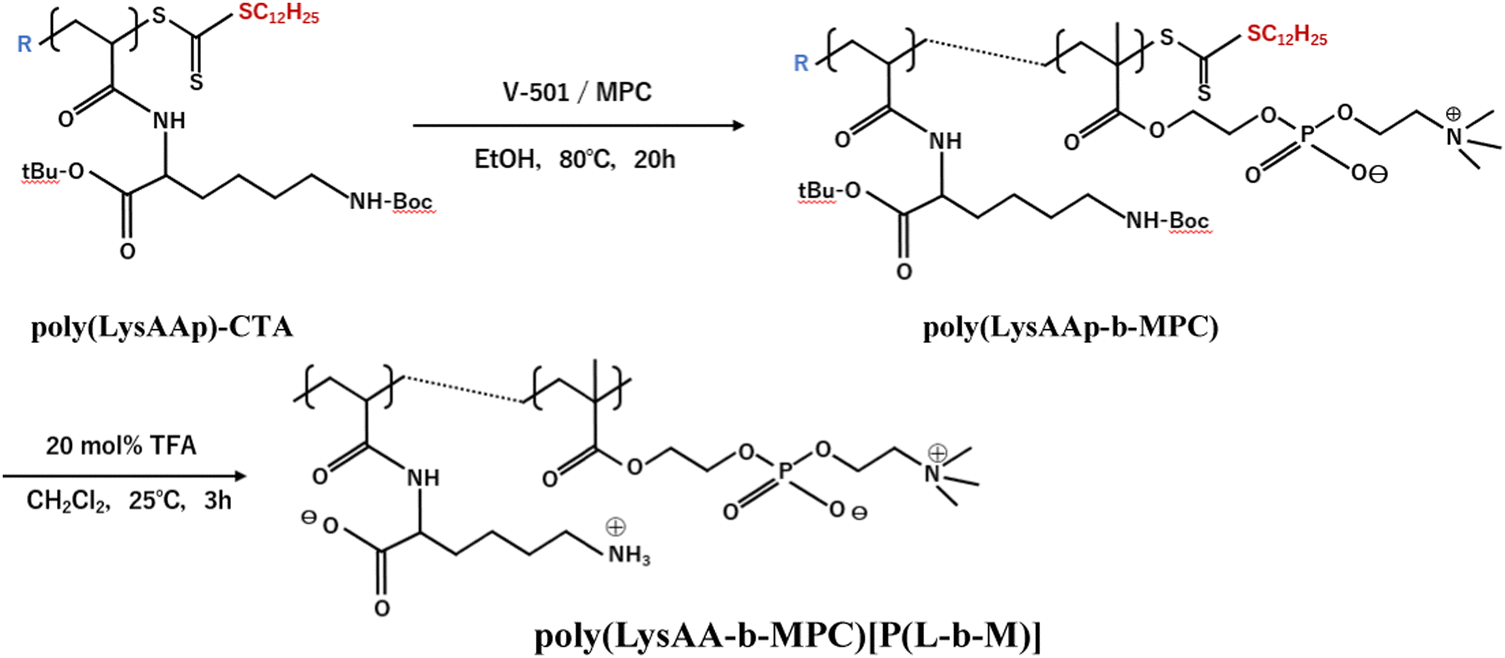
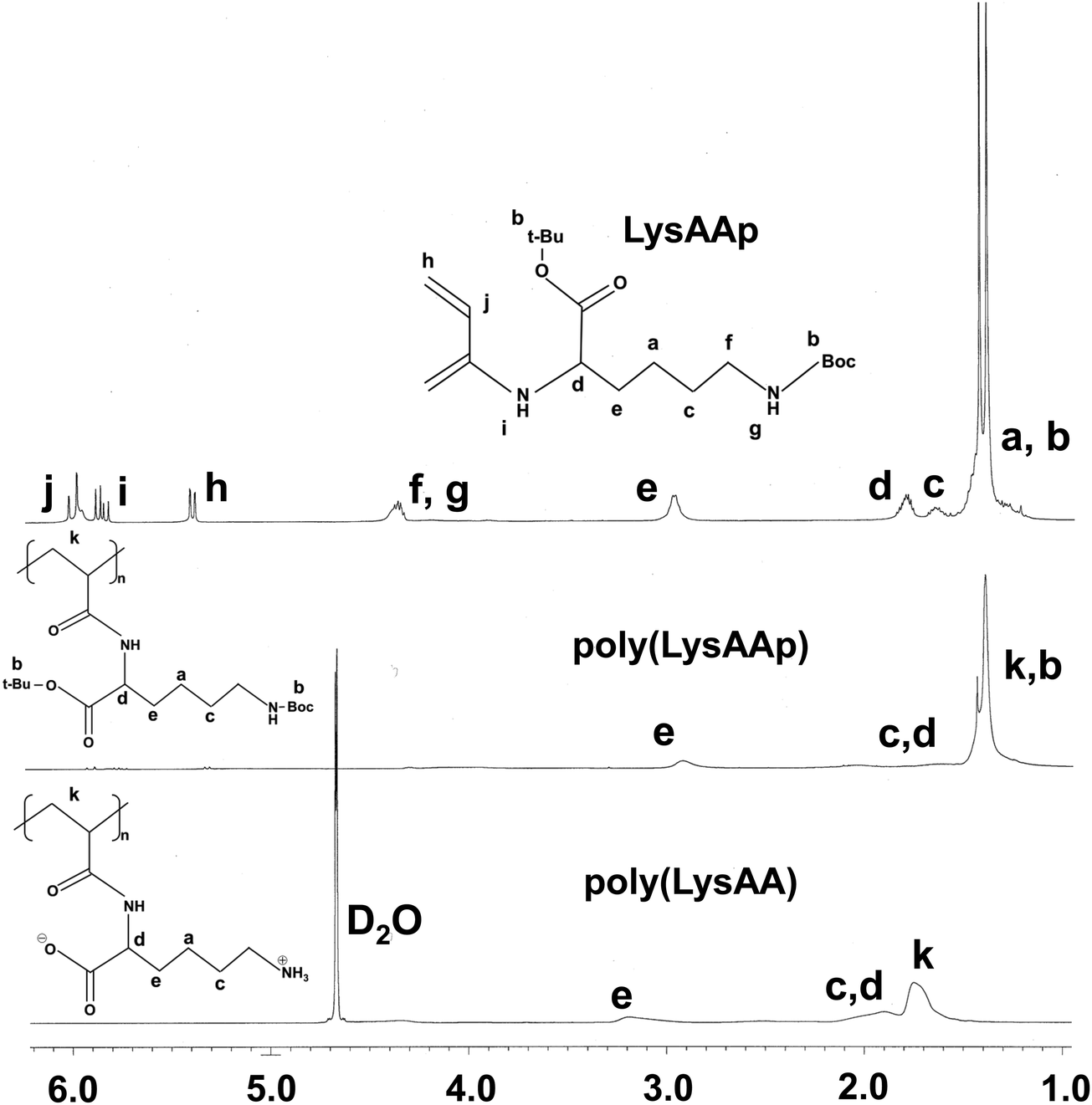
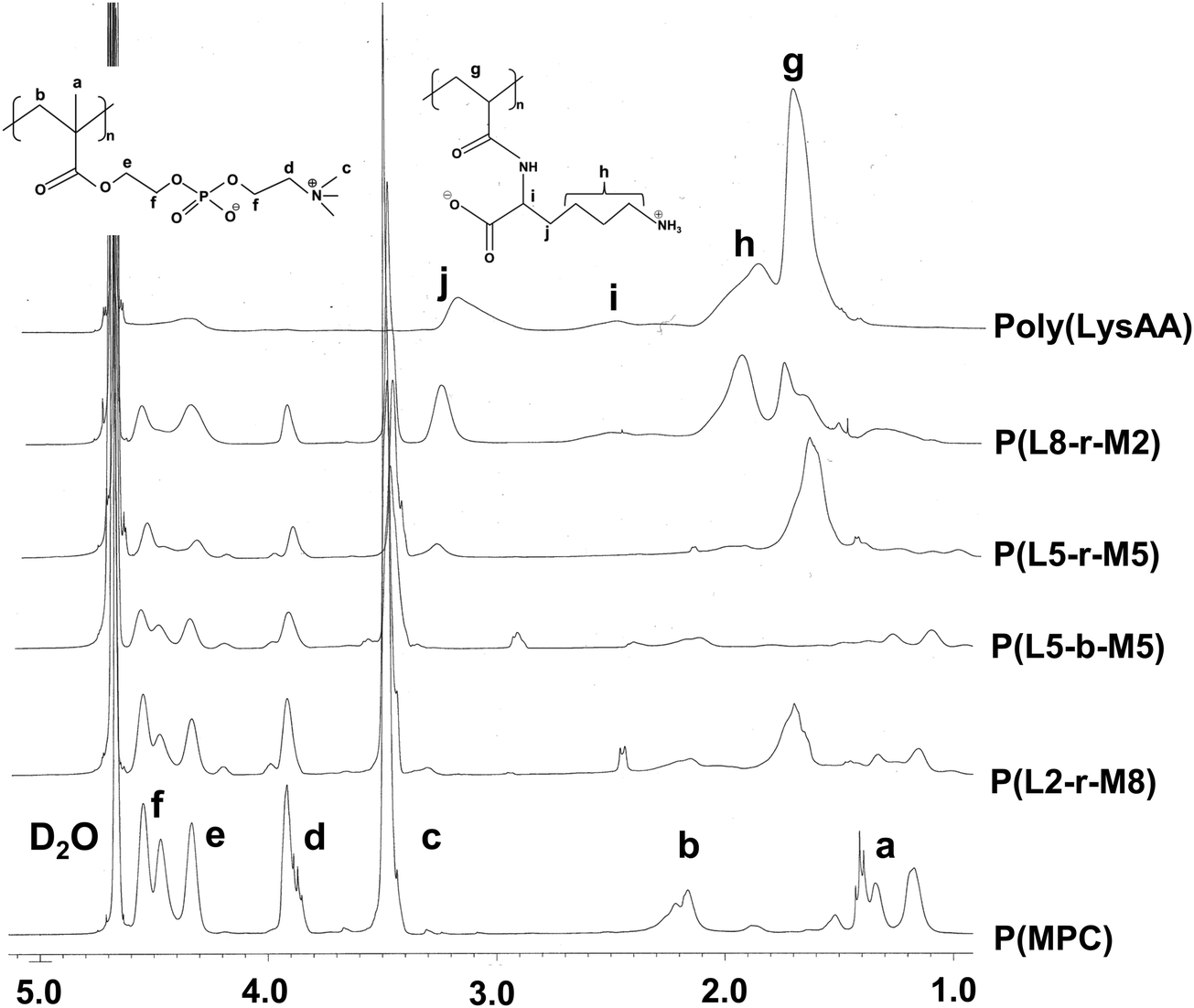
The preparation of P(Lm-r-Mn) and P(Lm-b-Mn) is illustrated in Schemes 1 and 2, respectively, where m and n in the P(Lm-r,b-Mn)s are the comonomer ratio of LysAA and MPC in the feed. 1H-NMR spectra of typical polymers comprising LysAA and MPC are shown in Fig. 1 and 2, respectively. Treatment with trifluoroacetic acid (TFA) at 25 °C for 3 h did not hydrolyze the esters in the MPC units, and quantitative deblocking of the N-tert-butoxycarbonyl (BOC) and tert-butyl (t-Bu) ester groups in LysAAp units was observed. The 1H-NMR spectra of P(L5-r-M5) and P(L5-b-M5) were similar but the peak for P(L5-r-M5) in the methylene region at about 2.8 ppm of LysAA units was broader than that for P(L5-b-M5). The mobility of the discontinuous sequence of LysAA units in the side chain of P(L5-r-M5) was restricted compared with that of the continuous sequence of LysAA in P(L5-b-M5). The polymerization results are summarized in Table 1. The copolymer compositions were estimated by comparison of the integrated intensities of resonance peaks for the methylene group in the side chain of the LysAA unit around 2.8 ppm and the trimethyl ammonium group around 3.2 ppm in the MPC unit (Table 1). Gel permeation chromatography (GPC) elution of P(L5-b-M5) yielded a unimodal curve, confirming that poly(LysAA) and poly(MPC) were not mixed in the obtained P(L5-b-M5). When the copolymers were made under the conditions shown in Table 1, they were randomly arranged. However, 1H-NMR measurements showed polymers with composition ratios different from the preparation ratios. A significant yield reduction was observed for P(L5-r-M5) and P(L8-r-M2) compared to the other copolymers. The lower yields of these polymers were attributed to the fact that a portion of the polymer slipped through the membrane during dialysis due to its low molecular weight. Furthermore, the high yields of the polymers prepared in this study make it difficult to discuss the reactivity ratios. However, Virtanen et al. reported reactivity ratios for copolymers of acrylamide monomer (N-isopropylacrylamide) and methacrylic acid monomer (glycidyl methacrylate).16 Assuming that the monomers reported are the LysAA and MPC monomers we used, the trends are consistent with the copolymer composition ratios reported. Therefore, our copolymer composition ratios were considered reasonable.
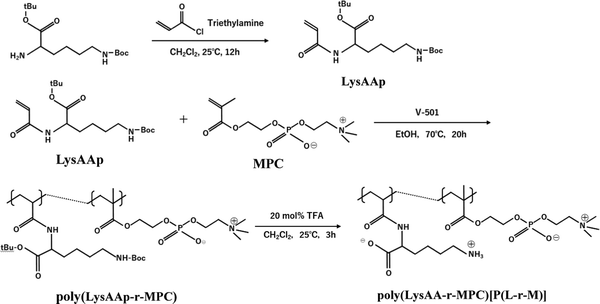 |
| | Scheme 1 Preparation of random copolymer [P(L-r-M)] composed of LysAA and MPC. | |
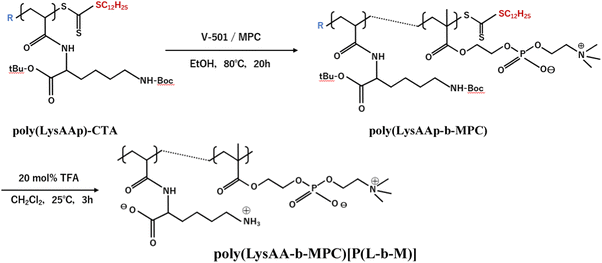 |
| | Scheme 2 Preparation of block copolymer [P(L-b-M)] composed of LysAA and MPC. | |
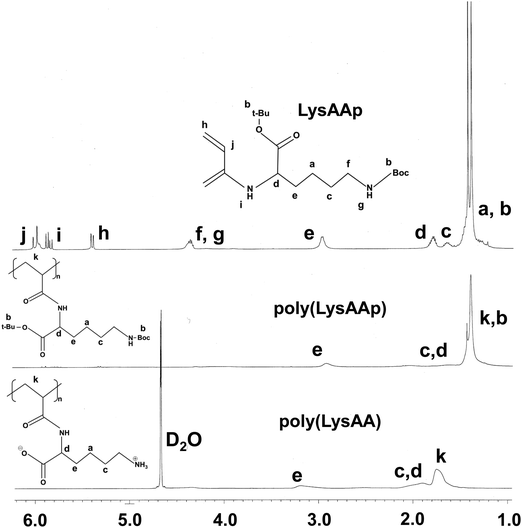 |
| | Fig. 1
1H NMR spectra of LysAAp and poly(LysAAp) in CDCl3, poly(LysAA) in D2O. | |
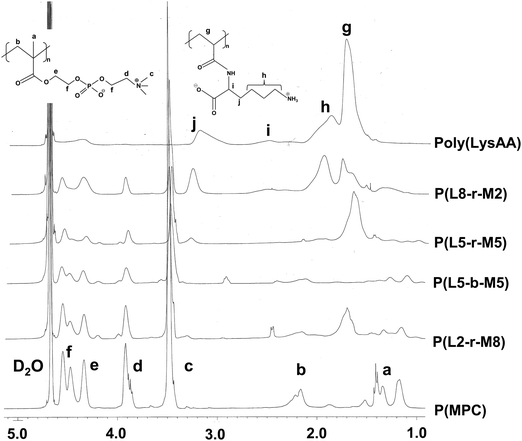 |
| | Fig. 2
1H NMR spectra of LysAA/MPC copolymers and poly(MPC) in D2O. | |
Table 1 Preparation and characterization of zwitterionic polymersa
| Polymer |
Comonomer in feed (mol%) |
Yield [%] |
Molar ratio in copolymerb (mol%) |
M
n × 10−4 (Mw/Mn)c |
| LysAAp |
MPC |
LysAA |
MPC |
|
1 mol% of V-501 as an initiator in total commoner feed (0.186 mol L−1) in ethanol at 80 °C, 20 h.
Determined by 1H NMR in D2O after deblocking procedure.
Determined by GPC in 0.25 mol L−1 NaCl aq. as an eluent with polyoxyethylene standard.
In THF.
Determined by GPC in THF as poly(LysAAp) with protective groups. Mw and Mn represent the weight average molecular weight and the number average molecular weight, respectively.
[LysAA units in poly(LysAA)] = 0.093 mol L−1 in RAFT agent terminated poly(LysAA), [MPC] = 0.093 mol L−1 at 70 °C.
|
| poly(LysAA)d |
100 |
0 |
95.0 |
100 |
0 |
0.8(1.7)e |
| poly(MPC) |
0 |
100 |
98.0 |
0 |
100 |
2.8(1.6) |
| P(L2-r-M8) |
20 |
80 |
96.0 |
13 |
87 |
0.8(1.7) |
| P(L5-r-M5) |
50 |
50 |
62.7 |
36 |
64 |
0.7(1.6) |
| P(L8-r-M2) |
80 |
20 |
70.0 |
63 |
37 |
0.7(1.6) |
| P(L5-b-M5)f |
50 |
50 |
77.0 |
30 |
70 |
1.8(1.6) |
Kinetic evaluation of fibrinolytic activity of Plg/t-PA in the presence of LysAA-containing polymers
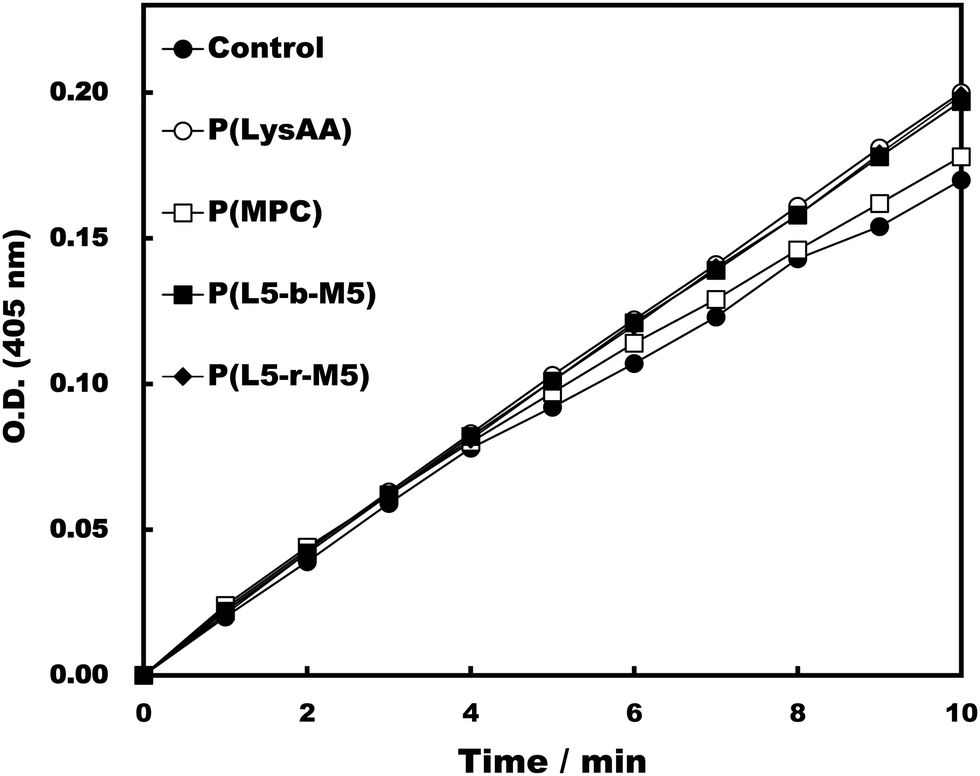
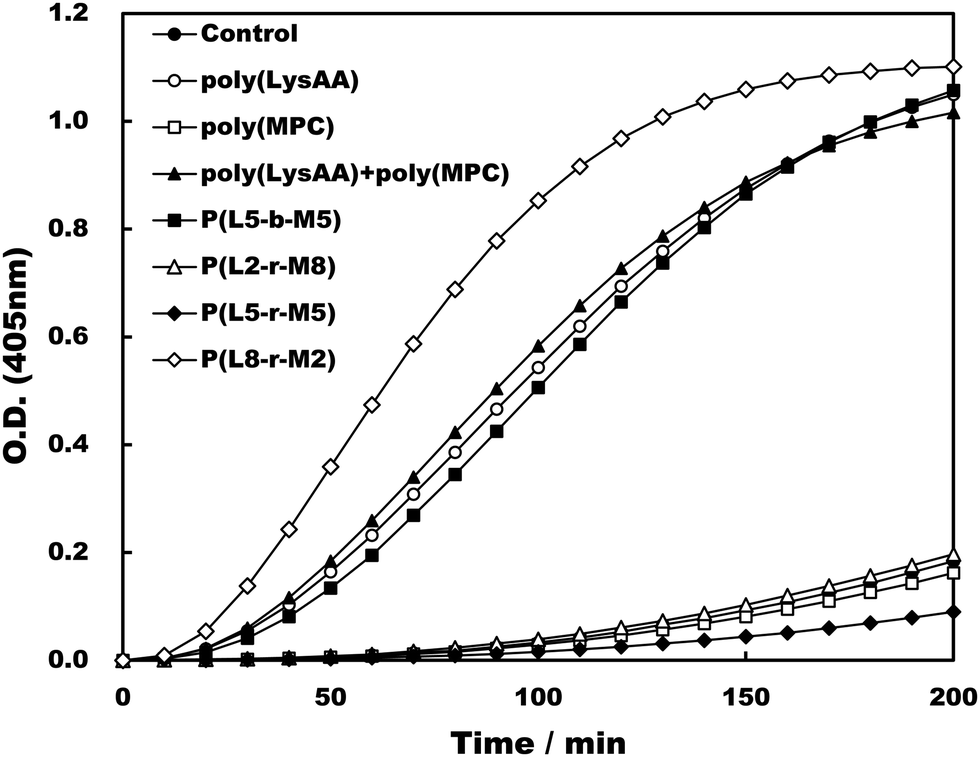
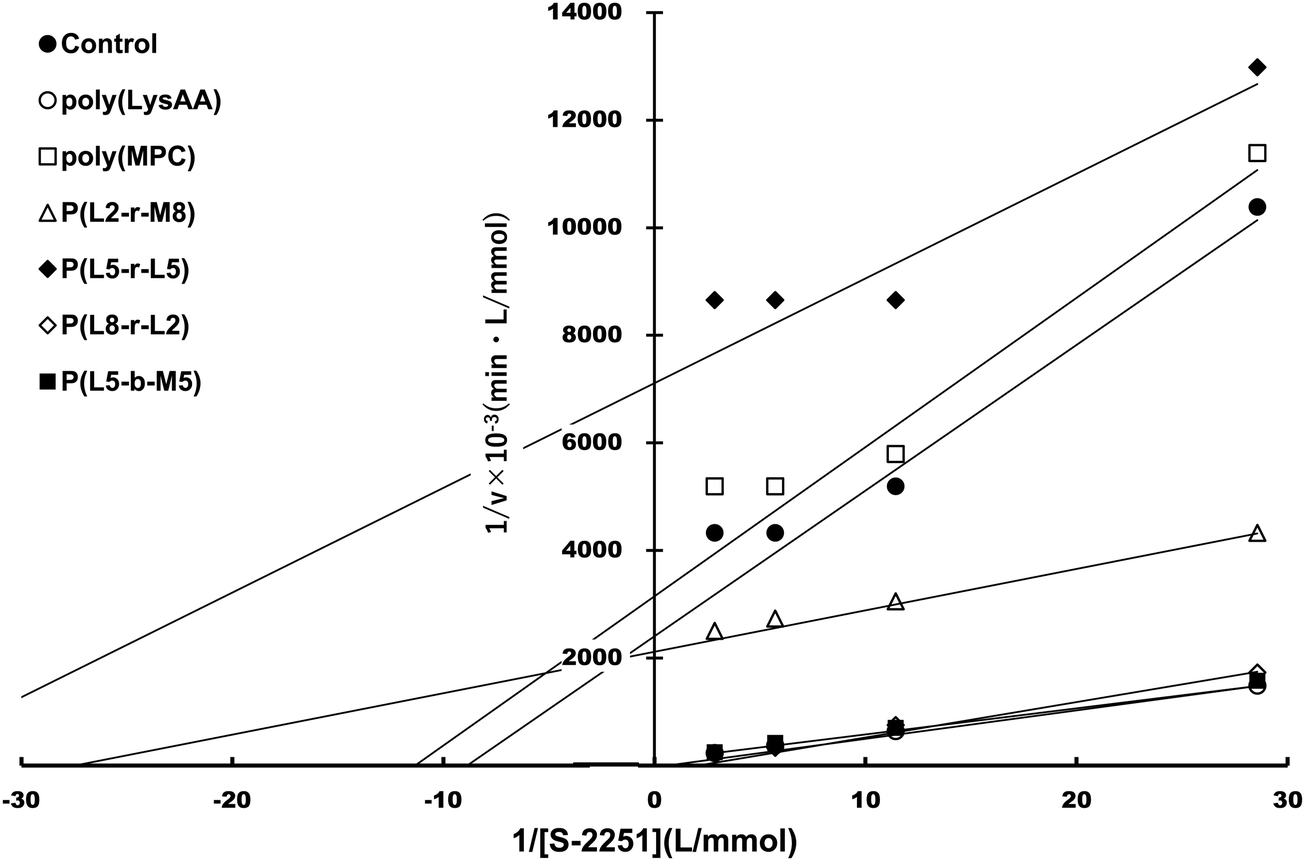
Specific binding of the fibrinolytic proteins Plg and t-PA provides an anti-thrombogenic surface.17 We evaluated enhanced fibrinolytic activity by Plg/t-PA towards poly(LysAA) and poly(LysMA) in the solution phase using the chromogenic Plm substrate S-2251. We concluded that poly(LysAA) is more flexible than poly(LysMA) and readily binds Plg or t-PA to give enhanced fibrinolytic activity.10 The effect of the MPC segment introduced in poly(LysAA) on enhancing fibrinolytic activity by Plg/t-PA was also examined as described previously.7,10 Optimized concentrations of Plg and t-PA in solution were determined by preliminary experiments to obtain the maximum differences in S-2251 hydrolysis between poly(LysAA) and control without poly(LysAA). Fig. 3 shows the Plm activation of S-2251 in the presence of MPC containing poly(LysAA). No significant effect of Plm activity was observed with P(L-r-M), P(L-b-M), poly(LysAA), poly(MPC). These results suggest that the effect of Plm activity on the addition of poly(LysAA), poly(MPC), and P(L-r-M) in the solution phase was similar to that of the control without polymer, indicating that these polymers are inert towards Plm activity. On the other hand, the activity of Plm for S-2251 hydrolysis in the presence of these polymers differs slightly from that of the control, perhaps due to the influence of a reaction field formed by the polymers as described below. Plg and t-PA contain lysine binding sites, allowing them to assemble around LysAA-containing polymers to convert Plm effectively. The activation of S-2251 by P(L5-r-M5), P(L5-b-M5), poly(LysAA), and poly(MPC) was evaluated with Plg/t-PA (Fig. 4). Although S-2251 hydrolysis was not enhanced in the presence of P(L-r-M) containing more than MPC 50 mol% segments, hydrolysis was drastically increased in the presence of P(L8-r-M2) and poly(LysAA). A lag-time was initially observed after adding t-PA to P(L8-r-M2) and poly(LysAA), then S-2251 hydrolysis in the presence of P(L8-r-M2) was faster and the lag-time was shorter than in the presence of poly(LysAA). The addition of either poly(LysAA) or P(L5-b-M5) enhanced fibrinolytic activity by Plg/t-PA, although no enhancement was observed following the addition of poly(MPC) or P(L5-r-M5). Comparison between the addition of P(L5-r-M5) and P(L5-b-M5) with the same concentration of LysAA units in solution indicated that the addition of P(L5-b-M5) produced a fibrinolytic activity enhancement, whereas addition of P(L5-r-M5) did not. These results suggest that the MPC block segment in P(L5-b-M5) does not interfere with binding of the poly(LysAA) segment to Plg and/or t-PA. The lowest S-2251 hydrolysis effects were with P(L2-r-M8) of examined LysAA-containing polymers, poly(MPC), and in the absence of polymer as a control. Binding Plg to P(L2-r-M8) could interfere with the interaction of t-PA. A comprehensive understanding of this phenomenon requires further experimentation. S-2251 hydrolysis in the presence of LysAA-containing copolymers was kinetically analyzed using Lineweaver–Burk plots (Fig. 5). The Michaelis constant (Km) and maximum velocity (Vmax) for S-2251 hydrolysis were determined from the reciprocal concentration of S-2251 and the reciprocal initial velocity after the lag-time, respectively, and are listed in Table 2. S-2251 hydrolysis in the presence of P(L2-r-M8) is very slow for a low concentration of S-2251 ranging from 0.035 mol L−1 to 0.088 mol L−1 due to low conversion of Plm by Plg/t-PA. The fitting plots in the presence of P(L2-r-M8) were drawn at S-2251 concentrations of 0.175 mol L−1 and 0.350 mol L−1 with estimated parameters. P(L8-r-M2) and poly(LysAA) had low affinity for S-2251 and high Vmax compared with that for P(L-r-M)s with 50 mol% or higher MPC content. S-2251 hydrolysis in the presence of mixed poly(LysAA) and poly(MPC) was similar to that at equal concentrations of LysAA units in the polymer, suggesting that the MPC segments act as an inert solvent. The initial stage of S-2251 hydrolysis requires a lag-time to assemble Plg and t-PA around LysAA-containing polymers and subsequent conversion from Plg to Plm by t-PA, followed by the release of Plm to react with S-2251. At a low t-PA concentration (0.04 μg mL−1), S-2251 hydrolysis is slow during the early stage because of the low concentration of Plm converted by Plg/t-PA, and then by interference from the complex formed by Plm/S-2251 around the LysAA-containing polymer. Therefore, the velocity for S-2251 hydrolysis changed. Km was measured as the sum of S-2251 hydrolysis by Plm converted from Plg/t-PA and was found to increase with time. The polymers likely showed a higher apparent Km than without LysAA-containing polymers even if the LysAA-containing polymer enhanced the conversion of Plm from t-PA/Plg. In addition, t-PA is present in human plasma at an extremely low concentration (0.005 μg mL−1) or 1/8th that in the solution used for this experiment.1 These results show that MPC segments with around 20 mol% MPC influenced either Plm conversion and/or fibrinolysis, enhancing fibrinolytic activity compared with poly(LysAA). Thus, an appropriate content of MPC segments in P(L-r-M) is important to regulate the anti-thrombogenicity of this biomaterial, based on optimal fibrinolytic activity enhancement and the suppression of protein adsorption.
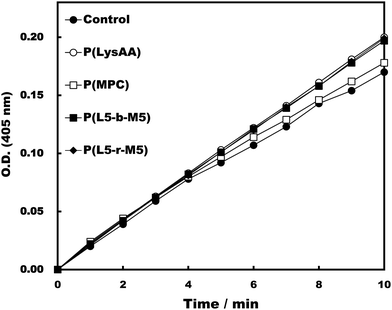 |
| | Fig. 3 Activation test of Plm in the presence of polymers. [LysAA units in polymer] = 0.175 mg mL−1, [Plm] = 0.05 mg mL−1, [S-2251] = 0.35 mmol L−1, [t-PA] = 0.04 μg mL−1. | |
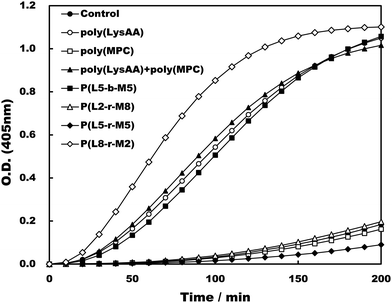 |
| | Fig. 4 The activation of S-2251 by Plg and t-PA in the presence of LysAA polymers and LysAA-containing copolymers. | |
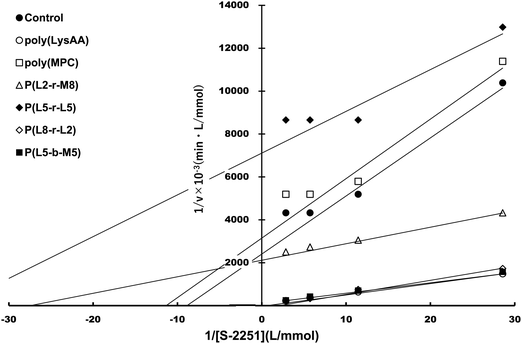 |
| | Fig. 5 Lineweaver–Burk plot of LysAA-containing copolymers. | |
Table 2 Kinetic parameters for the reaction of Plg/t-PA and S-2251 in the presence of polymers in Tris buffer solution (pH = 7.4) at 25 °C
| Polymer |
K
m [mmol L−1] |
V
max [×10−3 mmol L−1 min−1] |
| Control |
0.08 |
0.33 |
| Poly(lysAA) |
0.54 |
10.63 |
| Poly(MPC) |
0.06 |
0.24 |
| P(L2-r-M8) |
0.03 |
0.44 |
| P(L5-r-M5) |
0.04 |
0.17 |
| P(L8-r-M2) |
3.82 |
63.14 |
| P(L5-b-M5) |
2.25 |
8.70 |
Evaluation of the interaction between poly(LysAA) or P(L8-r-M2) and fibrinolytic factors
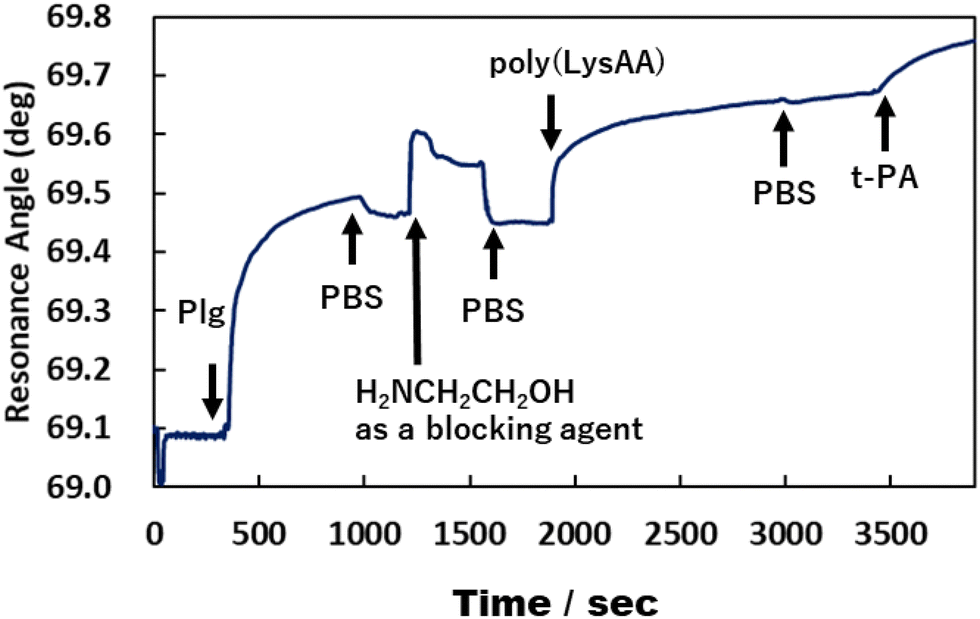
We previously reported that poly(LysAA) shows strong and specific binding to Plg or t-PA, despite low adsorption of other serum proteins such as albumin, γ-globulin, and fibrinogen.10 In addition, the fibrinolytic promoting effect of poly(LysAA) was higher than that of poly(LysMA), and Plg or t-PA bound more specifically to poly(LysAA) than to poly(LysMA). These results suggest that the lysine side chain of poly(LysAA), which has a more flexible main chain than poly(LysMA), more readily orients to interact with the sterically hindered kringle domain of Plg or t-PA than does poly(LysMA). The local density of Plg and t-PA increases around the LysAA-containing polymer due to the lysine binding sites in the protein. Subsequently, the lysine side chain binds to the kringle domain of Plg, inducing a conformational change and enhancing Plg activation by t-PA. Binding between Plg or t-PA and LysAA-containing polymer directly affects the conversion of Plm from Plg/t-PA. We therefore evaluated the interaction between Plg or t-PA and P(L8-r-M2), which has the most enhanced fibrinolytic activity by Plg/t-PA of the LysAA-containing copolymers, using SPR and compared it with the results obtained using poly(LysAA) (Fig. 6 and 7).
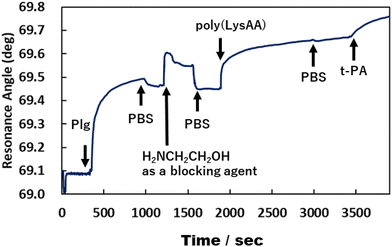 |
| | Fig. 6 Interaction between poly(LysAA), Plg and t-PA by SPR measurement. | |
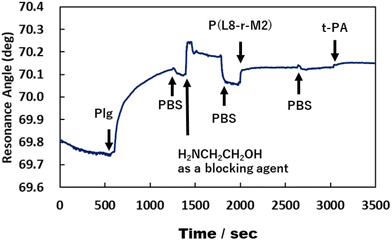 |
| | Fig. 7 Interaction between poly(L8-r-M2), Plg and t-PA by SPR measurement. | |
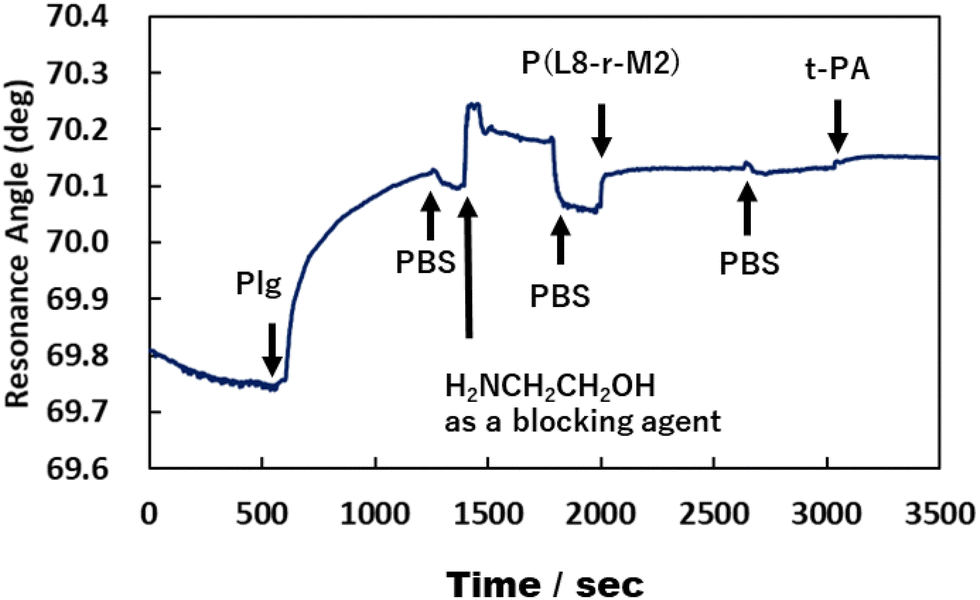
Fig. 6 and 7 show that the ratio of the amount of bound P(L8-r-M2)/immobilized Plg (0.17) on the gold substrate was about 2.5 times lower than that of poly(LysAA) (0.42). The decrease in the binding of P(L8-r-M2) on the immobilized Plg is attributed to the MPC segments in the copolymer restricting the interaction with Plg due to the phosphorylcholine moieties that resist protein adsorption.18,19 For the t-PA-containing analyte, the amount of protein binding to P(L8-r-M2)/immobilized Plg-complex was about 2.1 times lower (15.2 ng cm−2) than that for poly(LysAA) (32.6 ng cm−2). This could result in a release of Plm converted from Plg by t-PA, associated with the LysAA-containing polymer. On the other hand, P(L8-r-M2) likely adsorbs little t-PA based on the MPC segments. We are currently conducting a detailed analysis of Plm conversion on the complexes. The binding of t-PA to the lysine moiety is reportedly lower than that to Plg.10 Moreover, Plg binds to cells with low affinity and high capacity via its lysine binding site, which is associated with its kringle domains, and recognizes the carboxy-terminated lysines of cell surface proteins.20 Therefore, P(L-r-M)s, which exhibit adequate binding to fibrinolytic factors and induce a conformational change of Plg, likely release Plm effectively and increase the fibrinolytic activity of Plm by MPC moieties.
Substrate surface characterization
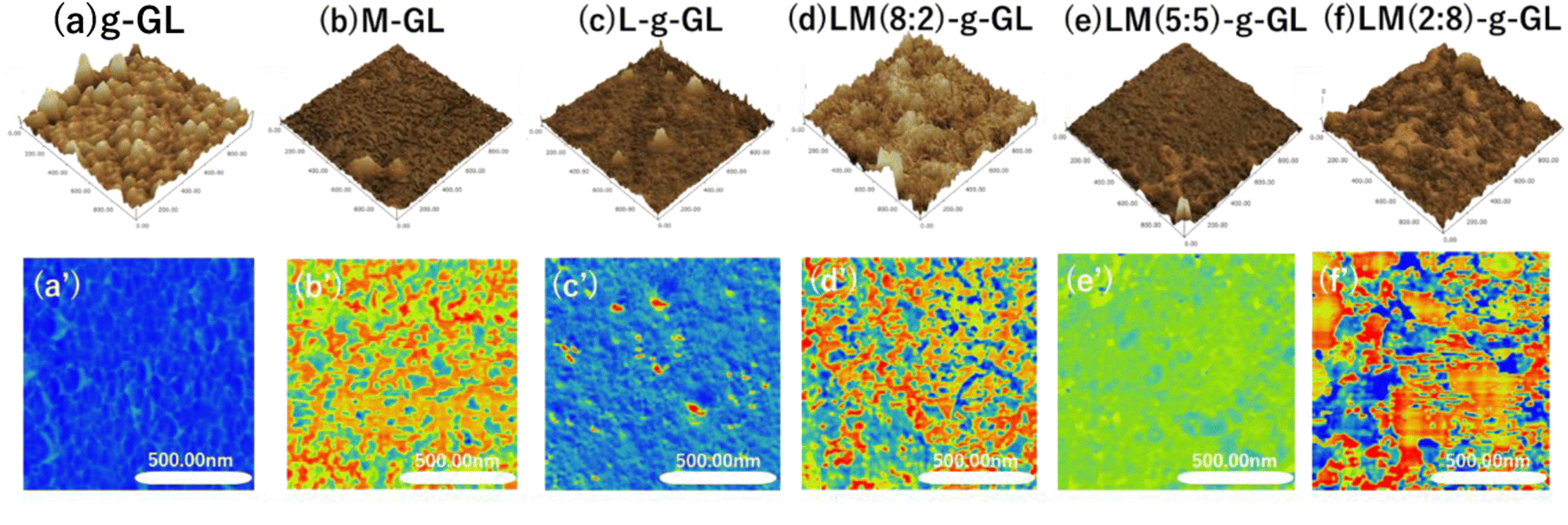
Changes in surface properties due to surface treatment and polymerization of substrates were observed using the phase mode of AFM. Glass substrates were used because of their high chemical resistance when using strong acids during surface treatment and deprotection, and the ease of observing polymer phase separation in AFM viscoelastic images. Fig. 8 shows g-GL, M-GL, L-g-GL, LM(m:n)-g-GL and AFM height measurements (a)–(f), with viscoelastic mapping in phase mode (a′)–(f′).
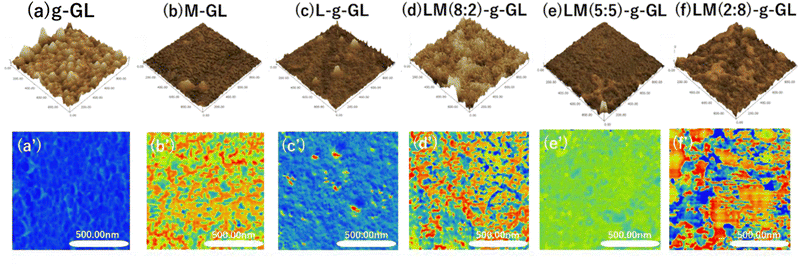 |
| | Fig. 8 Height (a)–(f) and phase (a′)–(f′) images of surface-treated glass substrate, LysAA, and LysAA-co-MPC grafted polymer by AFM. Scanning area (1.0 μm2), cantilever spring constant (1.7 N m−1), height (0–30 nm), phase (0–90 degree), The closer the height image is black, the higher the relative height. Phase images have higher relative viscoelasticity the closer to red they are, and vice versa for blue. Green is in between. | |
Compared to g-GL, the PMD coating and L/M graft polymerization surfaces exhibited a decrease in unevenness, i.e., were smoother. This may be due to the brushed layer surface formed on the g-GL by surface-initiated graft polymerization in the graft-from method. The water contact angle (θ) for the poly(LysAA) grafted surface increased more than that for g-GL, which is likely due to hydrophobization derived from acrylamide main chains and Lys side chains (Table 3). MPC segments were introduced into poly(LysAA) by random copolymerization, and θ decreased with increasing MPC content. We estimated that this was due to an increase in hydrophilicity of the surface of the grafted polymer chain caused by the increase in the MPC segment. Therefore, it is possible to control the surface properties by the introduction of MPC. Phase-separated domains were observed in the PMD coating and on the surface of the L/M grafted polymer chains. Additionally, nano-level phase-separated domains were observed in M-GL, LM(8:2)-g-GL and LM(2:8)-g-GL. These results may be attributed to the fact that one of the monomers has a smaller phase separation than the other when the monomer content is below 30 mol%. The smaller phase separation can homogenize the adhesive force applied to the fibrinolytic factor. Therefore, phase separation has a significant effect on the increase in fibrinolytic activity.
Table 3 Water contact angles of each substrate
| Substrates |
Water contact angle (θ) |
| Mean (n = 3) |
Error |
| Untreated |
35.8° |
±1.6 |
|
g-GL |
51.9° |
±1.1 |
| M-GL |
67.4° |
±3.0 |
| L-g-GL |
52.8° |
±2.7 |
| LM(8:2)-g-GL |
51.7° |
±3.9 |
| LM(5:5)-g-GL |
45.8° |
±1.6 |
| LM(2:8)-g-GL |
41.1° |
±1.8 |
Evaluation of interactions among L-g-GL, LM(m:n)-g-GL and fibrinolytic proteins Plg/t-PA
In order to determine the hemocompatibility of medical material surfaces, we evaluated the interaction between a substrate material with L or LM (m:n) segments and the plasma proteins Plg or t-PA by measuring the adhesion force by AFM. Table 4 shows the results of thermal frequency calculations after immobilization of Plg and t-PA in PB with COOH-modified probes. After immobilization of Plg and t-PA, the spring constants changed compared to those for the unmodified tips. The change in the spring constant for Plg and t-PA was almost the same, indicating no significant difference in the amount of immobilization of Plg and t-PA.
Table 4 Resonance frequency and spring constant change by fixation of fibrinolytic factor
| COOH group functionalized cantilever |
Treatment |
Resonance frequency (kHz) |
Spring constant (N m−1) |
| SPM-8100FM Tools (Shimadzu Corporation) was used to calculate the resonance frequency and spring constant. The cantilever parameters were set according to CONT-R by Nanoworld (width: 48.000 μm, length: 447.000 μm, spring constant: 0.2 N m−1), and the resonance frequency was calculated from the tuning data of the phase mode measurement. The spring constant was calculated by setting the Q value to 0.007 and the temperature to 300 K and setting the environment to in liquid. |
| Plg |
Untreated |
132.867 |
0.1277 |
| Immobilized |
129.670 |
0.1235 |
| t-PA |
Untreated |
132.970 |
0.1279 |
| Immobilized |
129.654 |
0.1230 |
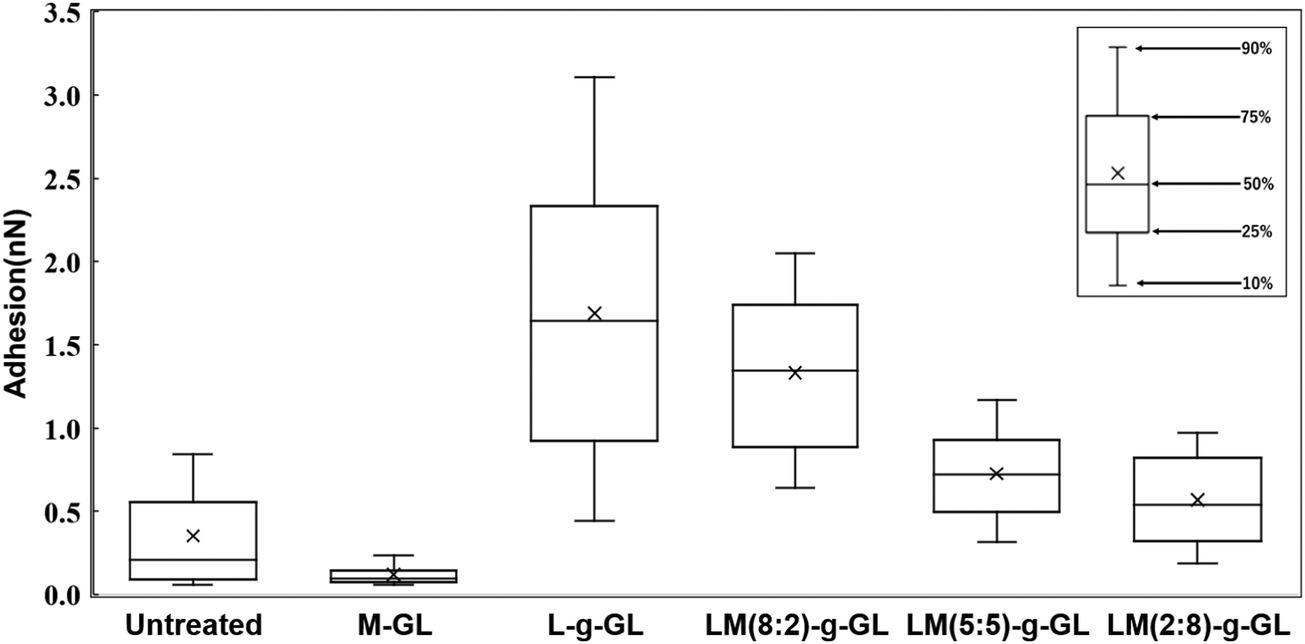
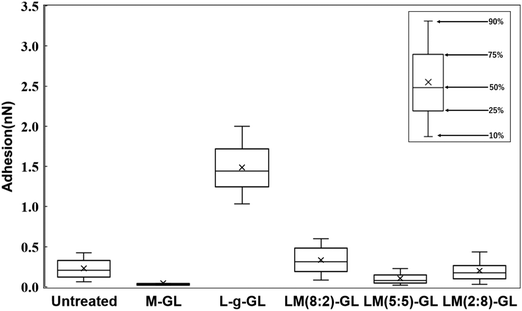
Fig. 9 shows a statistical analysis of the adhesion forces obtained from the AFM force curves for untreated glass, M-GL, L-g-GL, and LM(m:n)-g-GL. The PMD-coated surface (M-GL) showed almost no adhesion force due to the high non-protein adhesion properties of the MPC segment.21 Similarly, the adhesion force for the highly hydrophilic untreated glass (θ = 35.8°) was low. On the other hand, poly(LysAA)-grafted L-g-GL, which has a zwitterionic structure similar to that of MPC, showed a stronger adhesion force than M-GL. These results suggest that poly(LysAA)10 was bound to the LBS of Plg or t-PA. LM(m:n)-g-GL, in which an MPC segment was introduced into poly(LysAA), showed a tendency for the adhesion force to decrease with increasing amount of MPC. We attributed this to the non-protein adhesive MPC segments preventing the LBS-mediated binding of Plg and t-PA. Furthermore, we estimated that the large variation in adhesion force for L-g-GL was due to the nonuniform density and chain length for poly(LysAA) on the L-g-GL immobilized surface, which resulted in inconsistent binding forces with Plg and t-PA.
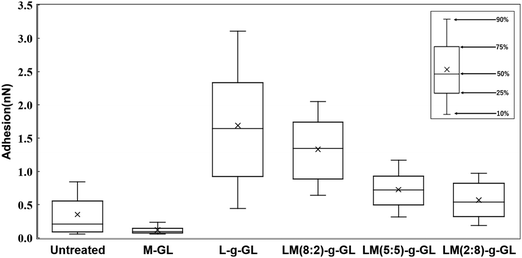 |
| | Fig. 9 Evaluation of interaction force between sample and Plg by AFM. | |
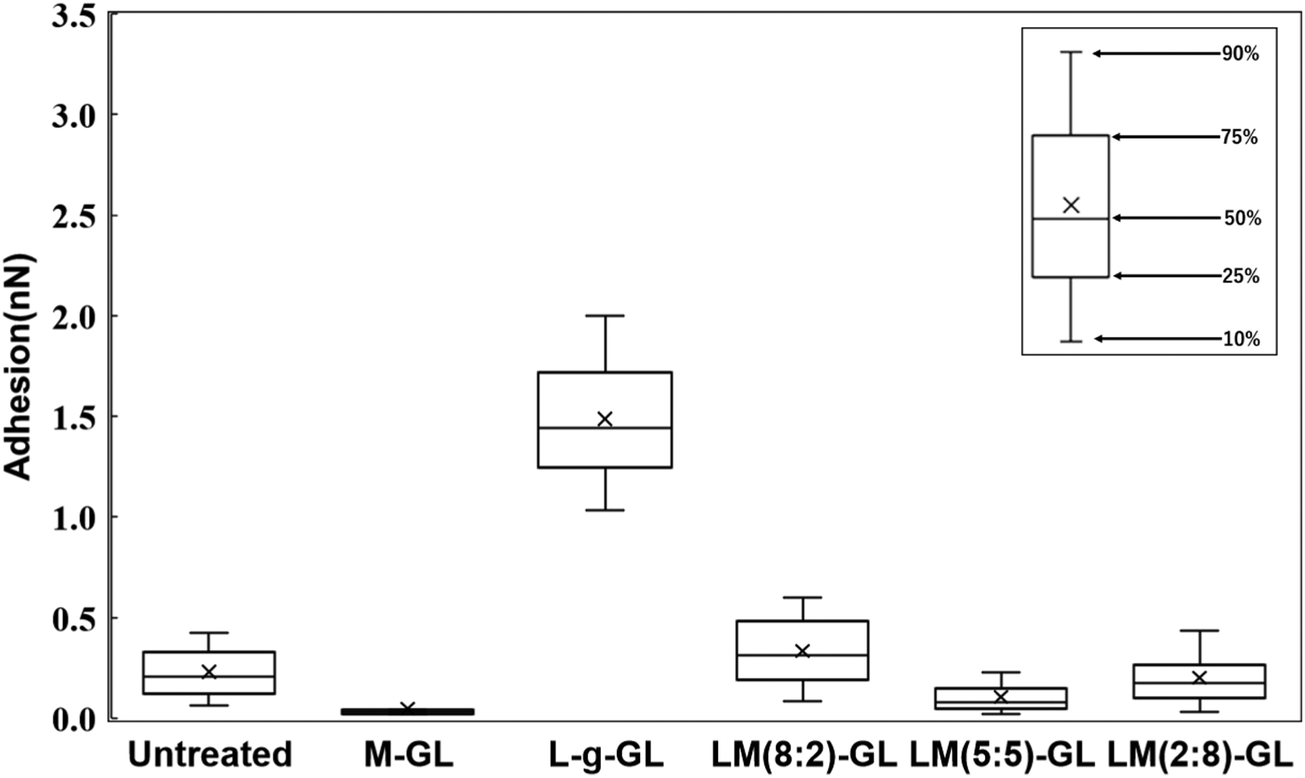
Fig. 10 shows the statistical results for adhesion between t-PA and the substrate. The trend of adhesion force is similar to that for Plg, with the highest adhesion force on the L-g-GL surface. The adhesion force decreased with increasing amount of the MPC segment introduced into the poly(LysAA) chain by copolymerization. The mean adhesion forces for Plg and t-PA on the surface of L-g-GL were approximately the same at around 1.6 nN (Plg: 1.7 nN; t-PA: 1.5 nN). In contrast, the mean adhesion force on LM(8:2)-g-GL was 0.3 nN (t-PA), which was much lower than that for Plg (1.3 nN). LM(5:5)-g-GL and LM(2:8)-g-GL showed mean adhesion forces of 0.7 nN and 0.6 nN for Plg, respectively, while t-PA showed only a low adhesion force of 0.2 nN or less for both surfaces.
 |
| | Fig. 10 Evaluation of interaction force between sample and t-PA by AFM. Same measurement and analysis conditions as in Fig. 9. | |
There are strong effects of the number and affinity of lysine binding sites (LBS) in Plg and t-PA. Plg chains have five kringle (K) domains, with K1, K2, K4, and K5 possessing lysine-binding ability. Several reports have suggested that K4 has a particularly high affinity for lysine (Ka = 24.4 ± 2.5 mM−1).22,23 t-PA has two K domains, with K2 having lysine-binding ability (Ka = 11.9 mM).24 Therefore, we considered that the introduction of a small amount of non-protein adhesive MPC segments greatly decreased the lysine affinity of t-PA compared to that of Plg. Based on these results and existing reports, we believe that the introduction of the MPC segments decreased the adhesion force of Plg and t-PA on the surface of the substrates. Since t-PA has a lower molecular weight than Plg, the effect of the non-protein adhesive domain formed by MPCs due to nano-phase separation is more likely to be stronger in t-PA than in Plg. Accordingly, the decrease in adsorption capacity of t-PA due to MPC is expected to be larger than that of Plg. Furthermore, the introduction of MPC is thought to increase the hydrophilicity of the polymer and promote orientation of the Lys side chain toward the solution. In the LysAA monopolymer, hydrophilicity is reduced by lysine, a hydrophobic amino acid. The change in surface properties with polymer introduction on each substrate is also evident from the water contact angle results (Table 3). In the presence of plasma proteins such as bovine serum protein (BSA), the hydrophobicity of the solution increases and the effect becomes more pronounced. Thus, the presence and action of MPCs may play a very important role under conditions more similar to the in vivo environment.
Evaluation of fibrinolytic activity by synthetic fibrin chromogenic model substrate S-2251
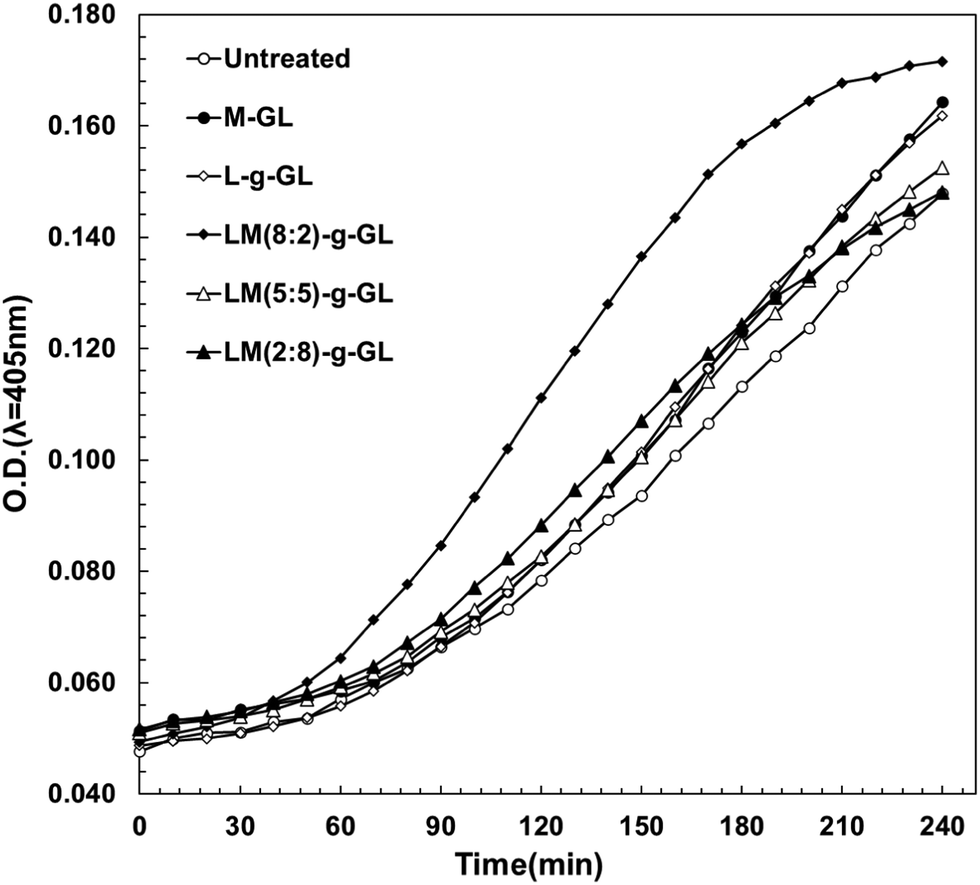
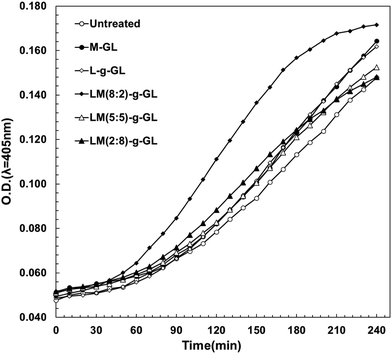
In the evaluation of fibrinolytic activity using S-2251 as a synthetic fibrin chromogenic model substrate in the Plg/t-PA system under liquid-phase conditions, poly(LysAA) and poly(LysAA-co-MPC) (8:2) greatly increased the fibrinolytic activity compared to poly(MPC), poly(LysAA-co-MPC) (5:5), or no additive. We evaluated the fibrinolytic activity of these polymers by immobilizing them on a glass substrate as a surface model for medical materials. The reaction field in the solid-phase system was expected to be reduced compared to that in the liquid-phase system. In order to increase the rate of Plm generation, the amount of Plg and t-PA were twice and 1000 times higher, respectively, compared to the liquid-phase system. We investigated the effect of the presence of LysAA and MPC copolymers and poly(LysAA)-immobilized polymers on the fibrinolytic activity under conditions similar to the environment on the surface of medical materials (Fig. 11).
 |
| | Fig. 11 Evaluation of plasmin generation rate by S-2251. | |
Fig. 11 shows that LM(8:2)-g-GL clearly and strongly enhanced fibrinolytic activity compared to LM(5:5)-, LM(2:8)-g-GL, L-g-GL and M-GL. LM(5:5)- and LM(2:8)-g-GL, which had no activity under liquid-phase conditions, showed slightly enhanced fibrinolytic activity compared to the untreated substrate. The poly(LysAA)-immobilized glass substrate (L-g-GL) showed slightly enhanced fibrinolytic activity compared to the untreated glass substrate. However, it was almost the same as that for the LM(5:5)- and LM(2:8)-g-GL substrates. Plm is generated by the reaction between Plg and t-PA. Therefore, Plm desorption-diffusion from the surface of medical materials is required for the onset of fibrinolytic activity. Clear viscoelastic contrast and nano-phase separation were observed in LM(8:2), LM(5:5), and LM(2:8)-g-GL. Therefore, the phase-separated state, especially nano-phase separation, affected the binding and concentration of Plg and t-PA by LBS, as well as the amount of Plm produced by the Plg/t-PA reaction and the diffusion of Plm in the liquid phase. Compared to L-g-GL, which had almost no phase separation, LM(8:2) and LM(2:8)-g-GL, which formed phase-separated domains, were more easily released from Plg and t-PA after binding. We estimate that the reaction and concentration of Plg and t-PA are significantly enhanced in LM(8:2)-g-GL, which has a significantly lower adhesion force for t-PA compared to L-g-GL, which has a strong adhesion force for both Plg and t-PA.
Experimental
Materials
N-ε-tert-Butoxycarbonyl-α-acrylamide-L-lysine-tert-butyl ester (LysAAp) [99% (1H-NMR)] was synthesized according to a previous report.10 MPC monomer [>96.0%] was kindly donated by Prof. Ishihara (The University of Tokyo). Acryloyl chloride (>98.0%, Tokyo Chemical Industry Co., Ltd, Japan) was purified by distillation before use. Triethylamine (99.0%, Fujifilm Wako, Japan), TFA (98.0%, Fujifilm Wako, Japan), azobis(4-cyanovaleric acid) (V501; Fujifilm Wako, Japan), 2-cyano-4-[(dodecylsulfanylthiocarbonyl sulfanyl)]pentanoic acid (97% (HPLC), CTA; Sigma-Aldrich), dithiobis(succinimidyl undecanoate) [>90.0% (HPLC), Dojindo, Japan], 3-aminopropyltrimethoxysilane (>96.0%, APTMSi; Tokyo Chemical Industry Co., Ltd, Japan), 30% hydrogen peroxide (Fujifilm Wako, Japan), N,N-dicyclohexylcarbodiimide (DCC; Tokyo Chemical Industry Co., Ltd, Japan), N-hydroxysuccinimide (NHS; Tokyo Chemical Industry Co., Ltd, Japan), 1-ethyl-3,3-(dimethylaminopropyl)carbodiimide (EDC; Tokyo Chemical Industry Co., Ltd, Japan), Tris buffered saline (TBS; Nippon Gene, Japan), phosphate buffer saline (PBS; Sigma-Aldrich), phosphate buffer solution (PB; Fujifilm Wako, Japan), synthetic fibrin chromogenic model substrate (S-2251; H-D-Val-Leu-Lys-p-nitroaniline 2HCl, Sekisui Medical Co., Ltd, Japan), plasminogen (Plg; Sigma-Aldrich), plasmin (Plm; Sigma-Aldrich) from human plasma, and tissue-type plasminogen activator (t-PA; Technoclone) from human plasma were purchased and used without further purification. All other reagents were of extra pure grade and were obtained from commercial sources and used without further purification unless otherwise stated.
Synthesis of polymers
The preparation process for P(L5-r-M5) (LysAA/MPC = 50/50 in feed) is described below as a typical example. LysAAp (280 μmol), MPC (280 μmol), and V-501 (1.86 μmol) as a radical initiator were dissolved in 3.0 mL of ethanol solution. After degassing the solution using the freeze–thaw technique, LysAAp and MPC were radically copolymerized at 70 °C for 20 h. The solvent was evaporated, the residue was dissolved in dichloromethane, and then TFA was added as a deblocking agent for N-tert-butoxycarbonyl and tert-butyl groups in the LysAAp units at 25 °C for 3 h. The solvent was evaporated, and the residue was dissolved in water, then neutralized with 0.01 mol L−1 NaOH aqueous solution. Finally, the aqueous solution was dialyzed against water using an acetyl cellulose dialysis membrane (Spectra/PorTM, REPLIGEN, cut-off molecular weight: 3500) for 3 days and then freeze-dried to give a white powder. Yield: 98.0%.
Synthesis of poly(LysAA-b-MPC)
Poly(LysAAp) terminated with chain transfer agent (CTA) [poly(LysAAp)-CTA] was obtained by reversible addition–fragmentation chain transfer polymerization with V-501 (9.35 μmol), LysAAp (280 μmol) and CTA (28.0 μmol) in tetrahydrofuran (THF) solution at 70 °C for 20 h after degassing the solution by freeze-thawing. The solvent was evaporated to give a viscous, yellow-colored solid which was extracted three times with a large amount of hot n-hexane and dried in a vacuum for 24 h. Yield: 99.0%.
Poly(LysAAp)-CTA (280 mmol of LysAA unit), V-501 (1.86 μmol), and MPC (280 mmol) were dissolved in ethanol, degassed by freeze-thawing, and polymerized at 80 °C for 20 h. The solvent was evaporated to give a viscous solid which was dissolved in dichloromethane. The solution was treated with TFA as a deblocking agent at 25 °C for 3 h. The polymer obtained by the evaporation of the solvent was dissolved in water, neutralized with 0.01 mol L−1 NaOH aqueous solution, dialyzed against water, and freeze-dried as described above to give a white powder. Yield: 96.0%.
Evaluation of fibrinolytic activity of Plg/t-PA in the presence of polymers
Plg (136 μg mL−1), polymer ([LysAA units in the polymer] = 175 μg mL−1), S-2251 (0.035–0.350 mmol L−1), and t-PA (0.04 μg mL−1) were dissolved in a TBS buffer solution (pH = 7.4). Plg (50 μL), polymer, and S-2251 were added in that order to the wells of a 96-well microplate (diameter = 6.4 mm) and shaken at 25 °C for 10 min, then 50 μL of t-PA was added and the mixture was shaken at 25 °C for 200 min. The degree of Plm activity from Plg/t-PA in the presence of polymer was evaluated based on the amount of p-nitroaniline, derived from S-2251 using a microplate reader (iMarkTM: Bio-RAD) at a wavelength of 405 nm. Kinetics parameters, Michaelis constant (Km), and maximum velocity (Vmax) for the reaction of Plm and S-2251 in the presence of polymer were calculated from Lineweaver–Burk plots, fitting the plots with a linear least-squares method.
Surface treatment and surface-initiated graft polymerization
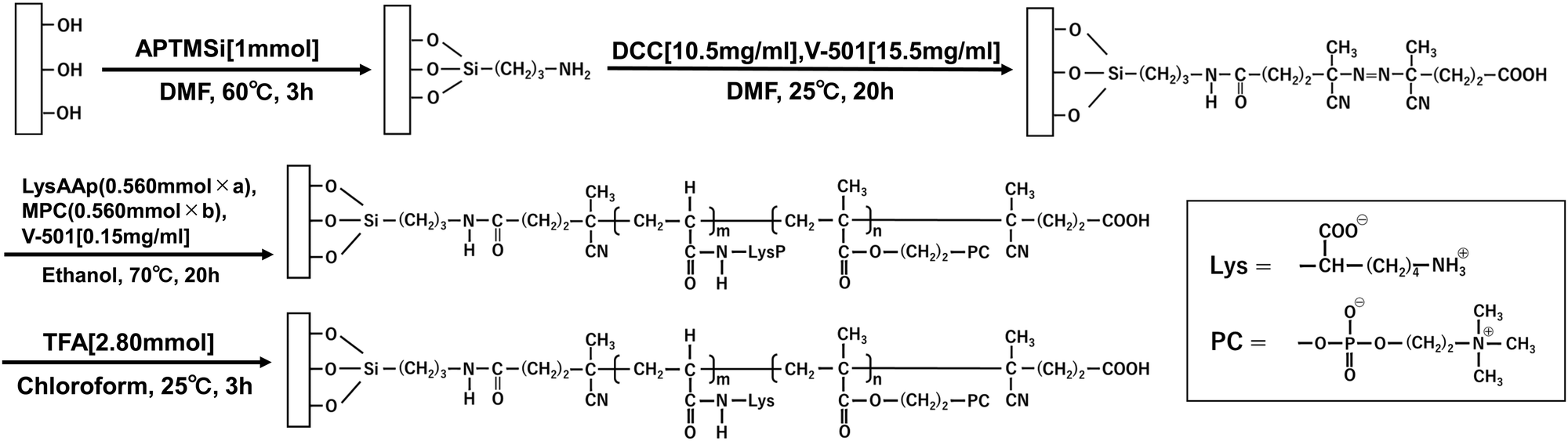
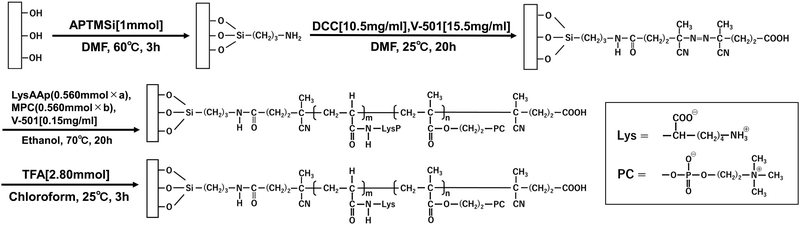
Surface treatment of glass substrates was performed according to a previous report.25Scheme 3 shows the procedure for immobilization and graft polymerization of the initiator onto the glass substrate. Matsunami micro cover glass (thickness = 0.12–0.17 mm) was immersed in Piranha solution [30% H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2SO4 = 1:3 (v/v)] for 1 h at room temperature. The vessel and glass substrate were rinsed at least 3 times with sufficient distilled water, and the water was removed from the vessel and substrate surfaces using an air compressor. The glass substrates were immersed in 2 mL of DMF, and the residual water was dispersed using an ultrasonic cleaner for a few seconds. APTMSi (1 mmol) was added dropwise and allowed to react at 60 °C for 3 h for dehydration condensation with the OH groups on the substrate. The samples were sonicated with DMF and ethanol twice for 5 min each. Baking treatment was performed at 80 °C for 1 h. The samples were cooled and immersed in 2 mL of V-501 [10.5 mg mL−1]/DCC [15.5 mg mL−1] in DMF mixed solution and reacted at 25 °C for 20 h. The samples were sonicated with methanol 3 times for 5 min each and vacuum dried. Sufficiently dried sample (g-GL) was used for graft polymerization. g-GL was immersed in an ethanol solution of LysAAp (0.560 mmol) and V-501 (0.15 mg mL−1). After repeated freezing and nitrogen replacement in a liquid nitrogen bath to remove dissolved oxygen, graft polymerization was carried out at 70 °C for 20 h. After polymerization, the samples were sonicated twice with chloroform and ethanol for 5 min each. After vacuum drying, the Boc and t-Bu protecting groups were deprotected by immersion in TFA (2.80 mmol) chloroform solution at 25 °C for 3 h. The product was sonicated in ethanol, 0.1 M NaOH solution, and distilled water. After vacuum drying, LysAA homopolymer-immobilized substrate L-g-GL was obtained. LysAA/MPC copolymer was polymerized by the same procedure with the L/M ratio adjusted to 0.560 mmol of total monomer.
:H2SO4 = 1:3 (v/v)] for 1 h at room temperature. The vessel and glass substrate were rinsed at least 3 times with sufficient distilled water, and the water was removed from the vessel and substrate surfaces using an air compressor. The glass substrates were immersed in 2 mL of DMF, and the residual water was dispersed using an ultrasonic cleaner for a few seconds. APTMSi (1 mmol) was added dropwise and allowed to react at 60 °C for 3 h for dehydration condensation with the OH groups on the substrate. The samples were sonicated with DMF and ethanol twice for 5 min each. Baking treatment was performed at 80 °C for 1 h. The samples were cooled and immersed in 2 mL of V-501 [10.5 mg mL−1]/DCC [15.5 mg mL−1] in DMF mixed solution and reacted at 25 °C for 20 h. The samples were sonicated with methanol 3 times for 5 min each and vacuum dried. Sufficiently dried sample (g-GL) was used for graft polymerization. g-GL was immersed in an ethanol solution of LysAAp (0.560 mmol) and V-501 (0.15 mg mL−1). After repeated freezing and nitrogen replacement in a liquid nitrogen bath to remove dissolved oxygen, graft polymerization was carried out at 70 °C for 20 h. After polymerization, the samples were sonicated twice with chloroform and ethanol for 5 min each. After vacuum drying, the Boc and t-Bu protecting groups were deprotected by immersion in TFA (2.80 mmol) chloroform solution at 25 °C for 3 h. The product was sonicated in ethanol, 0.1 M NaOH solution, and distilled water. After vacuum drying, LysAA homopolymer-immobilized substrate L-g-GL was obtained. LysAA/MPC copolymer was polymerized by the same procedure with the L/M ratio adjusted to 0.560 mmol of total monomer.
 |
| | Scheme 3 Surface treatment and surface-initiated radical polymerization on glass substrates. | |
Coating
Poly(MPC-co-dodecyl methacrylate) (PMD) [ca. 33 mol% of MPC] was used as previously reported.21,26 Composition, Mw, and Mw/Mn have been reported previously. PMD ethanol solution [1 mg mL−1] was spin-coated on an untreated glass substrate using a spin-coater. The substrate was vacuum dried for 5 min to obtain MPC-coated glass substrates (M-GL).
Surface treatment by AFM probe
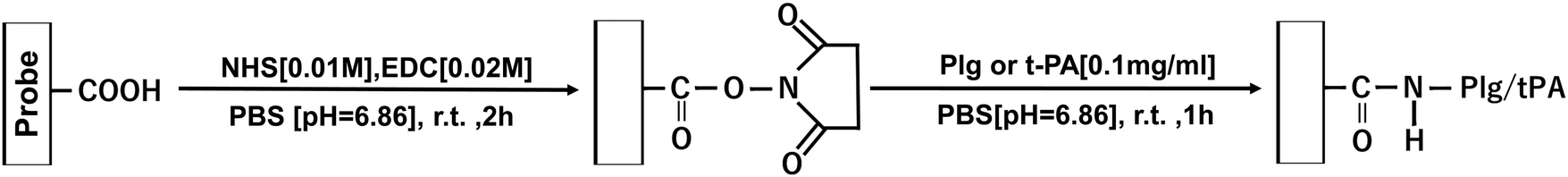
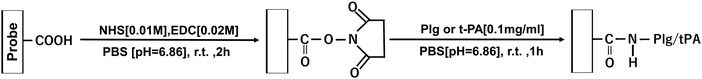
Scheme 4 shows the procedure for immobilizing fibrinolytic factors on carboxyl-modified cantilevers. 450 μL of NHS [0.01 M]/EDC [0.02 M] mixed PB solution was added to an AFM Petri dish type solution cell (Shimadzu), and a COOH group functionalized probe (Novascan, spring constant: 0.12 N m−1) was attached to the AFM system and immersed. The reaction was carried out at room temperature for 2 h to activate the COOH groups. 450 μL of Plg or t-PA PB solution [0.1 mg mL−1] was then added and the mixture was reacted at room temperature for 1 h.
 |
| | Scheme 4 Fibrinolytic factor immobilization on COOH group functionalized tips. | |
Evaluating the rate of plasmin generation over time under substrate immersion conditions
Sample substrates were immersed in PB solution for at least 12 h. The sample substrate was mounted in a 24-well plate (Costar) and 400 μL of PB was added. Plg [0.28 mg mL−1] in 50 μL of PB solution was added and the mixture was incubated at 37 °C for 30 min after pipetting. 50 μL of a PB solution of t-PA [0.04 mg mL−1] and 50 μL of a PB solution of S-2251 [1.4 mM] were added. After pipetting, absorbance (λ = 405 nm) measurements were performed at 37 °C using a plate reader (ARVO-MX: PerkinElmer) every 10 min for 240 min with shaking.
SPR measurements
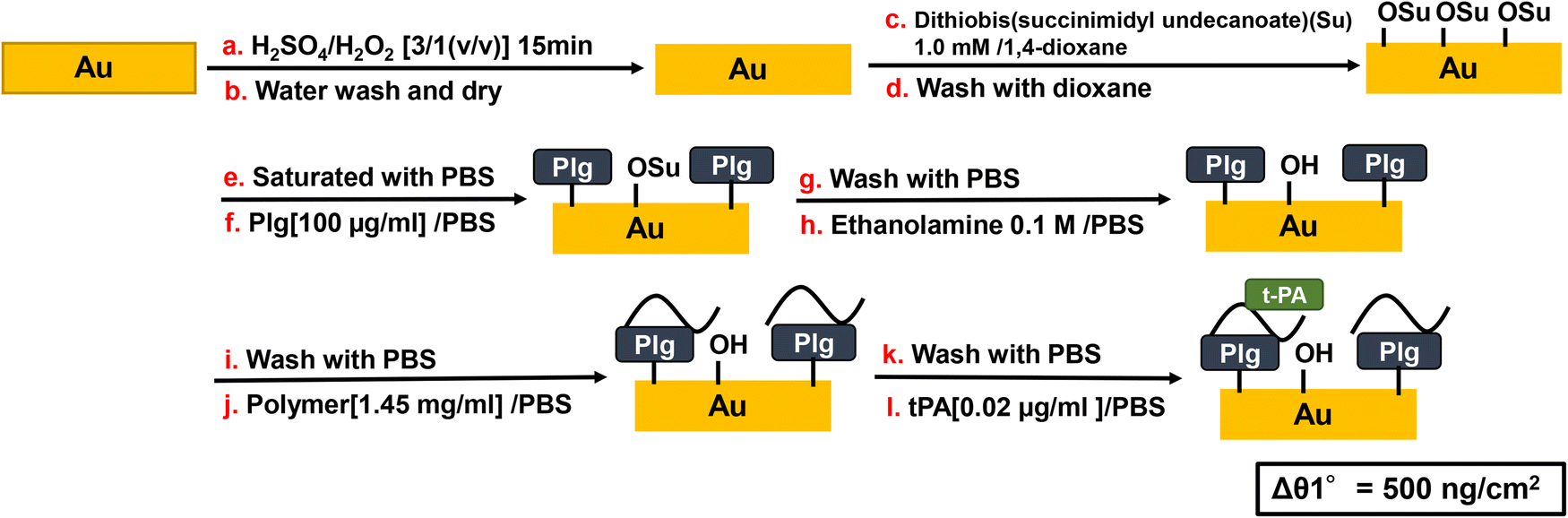
Scheme 5 shows the procedure for SPR measurement and surface treatment of gold sensor chip. A gold sensor chip for SPR (BIAcore 2000, GE Healthcare) was immersed in Piranha solution (H2SO4/H2O2 = 3/1 (v/v)) for 10 min, washed several times with ultrapure water, and dried in air at room temperature. The gold sensor chip was then immediately treated by 1.0 mmol L−1 dithiobis(succinimidyl undecanoate) in 1,4-dioxane at room temperature for 1 h, washed with 1,4-dioxane, and dried under vacuum at room temperature for 2 h. SPR sensorgrams were recorded in the following order. To avoid the direct interaction of Plg and t-PA, Plg was first immobilized on the gold substrate. Plg (100 μg mL−1) in PBS (pH 7.4) was placed on the gold substrate at a flow rate of 10 μL min−1 and washed with PBS. Next, ethanolamine (0.1 mol L−1 in PBS) was added as a blocking agent for the succinimidyl group, the chip was washed with PBS, polymer PBS solution was added (1.45 mg mL−1), the chip was washed with PBS, and then t-PA in PBS solution (0.02 μg mL−1) was added.
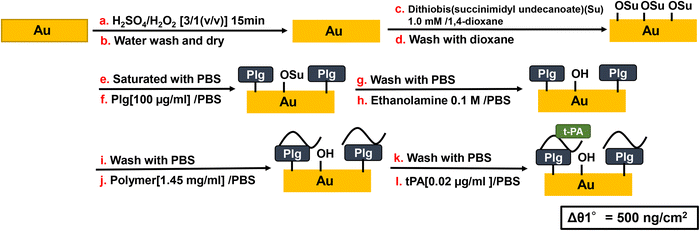 |
| | Scheme 5 Au substrate preparation and SPR measurement procedure. | |
AFM measurements
AFM measurements were performed using a SPM-9700HT (Shimadzu). Viscoelasticity and height images were measured in phase mode for glass substrate characterization. An OMCL-AC240TS [OLYMPUS, spring constant = 1.7 N m−1] was used. Adsorption forces between Plg and t-PA and the sample substrate surface were evaluated from contact mode force curves using a silicon nitride Au-coated COOH chemically functionalized probe [Novascan, Au coating thickness: 30 nm (nominal value); spring constant: 0.12 N m−1]. Fibrinolytic proteins were immobilized on a COOH-functionalized cantilever, and the spring constant was calculated from the resonance frequency change for the probe using thermal frequency calculation software (Shimadzu: SPM-8100FM Tools). Only cantilever parameters that could be handled by the calculation software were evaluated. The preset of CONT-R [Nanoworld, 0.2 N m−1], which has the most similar spring constants and materials, was used in the calculation. Since the Q factor, which indicates the sharpness of the curve at the resonance frequency, is nearly zero in liquid, the error in the spring constant calculation becomes large. The Q factor was fixed such that the spring constant approximated 0.12 N m−1 when calculating the thermal frequency of the cantilever before fixing the fibrinolytic protein, and the change was estimated. Force curve measurements were performed with a scanning area of 1.0 μm2 and 64 pixels. It should be noted that the force curves do not represent the adsorption force over the entire measurement range, since the probe was pushed to the center of the pixel compartments set.
Other measurements
1H-NMR spectra were recorded on a JEOL JNM-ECS400 spectrometer (Japan) at room temperature. Chemical shifts were determined using tetramethylsilane in CDCl3 or sodium trimethylsilylpropanesulfonate-d6 in D2O as an internal standard. GPC was conducted using a Shodex HPLC system (Showa Denko K.K., Japan) equipped with two mixed columns (Shodex OHpak SB-806M HQ × 2) and a differential refractometer (RI) detector in 0.25 mol L−1 NaCl aqueous solution as an eluent at a flow rate of 1.0 mL min−1 at 35 °C for water-soluble polymers. The Mn and Mw/Mn values were determined with polyethylene oxide standards. For THF-soluble polymers, Mn and Mw/Mn values were also measured using a Shimadzu HPLC system (Shimadzu, Japan) equipped with three columns (TOSOH TSKgel G6000HXL, 4000HXL, and 2000HXL) and a RI detector in THF as an eluent at a flow rate of 1.0 mL min−1 at 40 °C. The values were determined using polystyrene standards.
Conclusions
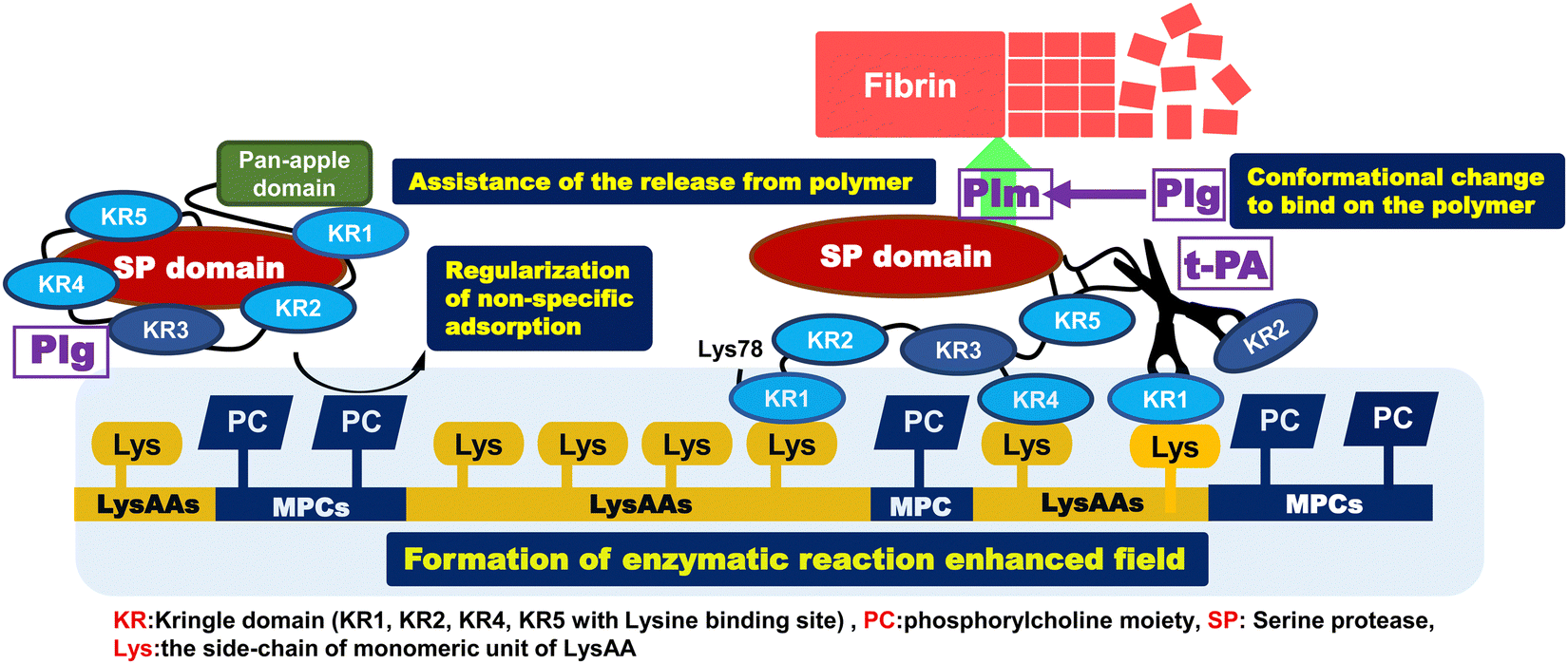
Our aim was to prepare a highly anti-thrombogenic biomaterial that suppresses protein adsorption, prevents denaturation of these proteins, and selectively binds fibrinolytic factors (Plg, t-PA) as a serum protein. Here, we report the enhancement or regulation of fibrinolytic activity, the preparation and characterization of copolymers composed of LysAA and MPC, and enzymatic fibrinolytic activity using a synthetic chromogenic Plm substrate (S-2251). The results are summarized below and a brief mechanism for assembly and activation of Plg around P(L8-r-M2) and LM(8:2)-g-GL is illustrated in Scheme 6.
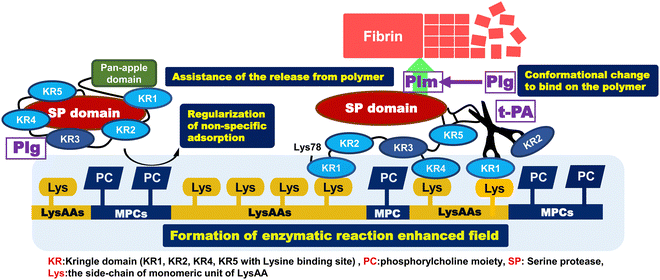 |
| | Scheme 6 Mechanism for assembly and activation of Plg around P(L8-r-M2) and LM(8:2)-g-GL. | |
[1] Addition of about 20 mol% MPC segment to poly(LysAAp) provided poly(LysAA) bearing L-lysine in the side-chain [P(L8-r-M2)]. L-Lysine selectively bound to Plg and t-PA via kringle domains and showed the highest fibrinolytic activity enhancement of the examined LysAA-containing polymers.
[2] Fibrinolytic activity enhancement was compared between P(L5-r-M5) and P(L5-b-M5). The activity in the presence of P(L5-b-M5) was similar to that of poly(LysAA), and the activity in the presence of P(L5-r-M5) was lower than that of P(L5-b-M5) and poly(LysAA). This indicated that increasing the amount of introduced MPC segments in the P(L-r-M) restricted the specific binding of Plg and t-PA to LysAA segments because the phosphorylcholine moieties restricted protein adsorption.
[3] We observed clearly different phase separation in the LysAA/MPC copolymer-immobilized substrate compared to the homopolymerized LysAA-immobilized substrate. In addition, nano-phase separation was observed in the copolymer substrate to which a small amount of monomer was added. The surface characteristics such as water contact angle also changed significantly.
[4] We showed selective adhesion between Plg/t-PA and the polymer-immobilized substrate with lysine sidechains. The selective adhesion of fibrinolytic factors is expected to be strongly influenced by the number and lysine affinity of LBS in Plg and t-PA. Furthermore, AFM measurements suggested that the interaction between Plg/t-PA and the material surface may be related to nano-phase separation and the size of the conformations.
[5] The fibrinolytic activity enhancement for the copolymers, which were not active in the liquid-phase condition, was the same or slightly higher compared to that for the poly(LysAA) homopolymer-immobilized substrate. The LM(8:2) copolymer, which showed a large increase in fibrinolytic activity in the liquid-phase condition, also showed a clear enhancement of fibrinolytic activity in the solid-phase condition. We estimated that the addition of a slight amount of MPC caused nano-phase separation at the material interface, which increased the concentration and reaction of Plg-tPA.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The authors gratefully acknowledge Prof. Kazuhiko Ishihara (The University of Tokyo) for donating MPC monomer, use of the SPR apparatus and helpful advice for SPR measurements and analysis.
References
- D. C. Rijken and H. R. Lijnen, New insights into the molecular mechanisms of the fibrinolytic system, J. Thromb. Haemostasis, 2008, 7, 4–13, DOI:10.1111/j.1538-7836.2008.03220.x.
- K. Okada, S. Ueshima, H. Matsuo, N. Nagai, N. Kawao, M. Tanaka and O. Matsuo, A synthetic peptide derived from staphylokinase enhances plasminogen activation by tissue-type plasminogen activator, J. Thromb. Haemost., 2011, 9, 997–1006, DOI:10.1111/j.1538-7836.2011.04257.x.
- M. Ashcraft, M. Douglass, Y. Chen and H. Handa, Combination strategies for antithrombotic biomaterials: an emerging trend towards hemocompatibility, Biomater. Sci., 2021, 9, 2413–2423, 10.1039/d0bm02154g.
- K. Shiraishi, T. Ohnishi, K. Sugiyama, K. Okada and O. Matsuo, Surface modified poly(methyl methacrylate) microspheres with O-methacryloyl-L-serine moiety, Chem. Lett., 1997, 863–864, DOI:10.1246/cl.1997.863.
- K. Shiraishi, T. Ohnishi and K. Sugiyama, Preparation of poly(methyl methacrylate) microspheres modified with amino acid moieties, Macromol. Chem. Phys., 1998, 199(9), 2023–2028, DOI:10.1002/(SICI)1521-3935(19980901)199:9<2023::AID-MACP2023>3.0.CO;2-V.
- K. Shiraishi, K. Miura, G. Asami, M. Kohta and K. Sugiyama, Analysis of pH response for amphoteric poly(O-methacryloyl-L-serine) and interaction with serum protein by fluorescence spectroscopy, Kobunshi Ronbunshu, 2003, 60(1), 30–37, DOI:10.1295/koron.60.30.
- K. Shiraishi, M. Kohta and K. Sugiyama, Preparation of zwitterionic polymethacrylamide modified with L-lysine and its effect on fibrinolytic activity, Chem. Lett., 2004, 33(6), 646–647, DOI:10.1246/cl.2004.646.
- N. Tanigawa, K. Shiraishi and K. Sugiyama, Blood compatibility of self-assembled poly(N-α-methacrylamide-L-Lysine-b-dimethylsiloxiane) copolymers, Kobunshi Ronbunshu, 2008, 65(2), 150–156, DOI:10.1295/koron.65.150.
- R. Hamawaki, T. Ishihara, A. Tominaga, K. Shiraishi, K. Sugiyama, Y. Nitta, T. Nakatani and K. Okamoto, Grafting of biocompatible polymers on DLC thin films by plasma irradiation-post polymerization technique for application on biomedical devices and cell microarrays, J. Photopolym. Sci. Tech., 2011, 24(4), 447–452, DOI:10.2494/photopolymer.24.447.
- K. Shiraishi, S. Wakisaka, J. Satozaki and K. Sugiyama, Preparation of poly[(meth)-acrylamide] having L-lysine moiety and its effect on fibrinolytic activity, Kobunshi Ronbunshu, 2012, 69(1), 39–46, DOI:10.1295/koron.69.39.
- Y. Iwasaki and K. Ishihara, Cell membrance-inspired phospholipid polymers for developing medical devices with excellent biointerfaces, Technol. Adv. Mater. Sci., 2012, 13, 064101, DOI:10.1088/1468-6996/13/6/064101.
- T. Goda, K. Ishihara and Y. Miyahara, Critical update on 2-methacryloyloxyethyl phosphorylchline (MPC) polymer science, J. Appl. Polym. Sci., 2015, 132, 41766–41776, DOI:10.1002/app.41766.
- S. Jiang and Z. Cao, Ultralow fouling, functionalizable, and hydrolyzable zwitterionic materials and their derivatives for biological applications, Adv. Mater., 2010, 22, 920–932, DOI:10.1002/adma.200901407.
- Y. Gong, S. Kim, J. Felez, D. K. Grella, F. J. Castellino and L. A. Miles, Conversion of Glu-plasminogen to Lys-plasminogen is necessary for optimal stimulation of plasminogen activation on the endothelial cell surface, J. Biol. Chem., 2001, 276(22), 19078–19083, DOI:10.1074/jbc.M101387200.
- L. A. Miles, F. J. Castellino and Y. Gong, Critical role for conversion of Glu-plasminogen to Lys-plasminogen for optimal stimulation of plasminogen activation on cell surface, Trends Cardiovasc. Med., 2003, 13(1), 21–30, DOI:10.1016/S1050-1738(02)00190-1.
- J. Virtanen and H. Tenhu, Studies on Copolymerization of N-Isopropylacrylamide and Glycidyl Methacrylate, J. Polym. Sci., Part A: Polym. Chem., 2001, 39(21), 3716–3725, DOI:10.1002/pola.10017.
- K. Yu, Y. Mei, N. Hadjesfandiari and J. N. Kizhakkedathu, Engineering biomaterials surfaces to modulate the host response, Colloids Surf., B, 2014, 124, 69–79, DOI:10.1016/j.colsurfb.2014.08.009.
- Y. Dang, M. Quan, C.-M. Xing, Y.-B. Wang and Y.-K. Gong, Biocompatible and antifouling coating of cell membrane phosphorylcholine and mussel catechol modified multi-arm PEGs, J. Mater. Chem. B, 2015, 3, 2350–2361, 10.1039/c4tb02140a.
- C. Kojima, R. Katayama, T.-L. Nguyen, Y. Oki, A. Tsujimoto, S. Yusa, K. Shiraishi and A. Matsumoto, Different antifouling effect of random and block copolymers comprising 2-methacryloyloxyethyl phosphorylcholine and dodecyl methacxrylate, Eur. Polym. J., 2020, 135, 109932, DOI:10.1016/j.eurpolymj.2020.109932.
- E. F. Plow, T. Herren, A. Redlitz, L. A. Miles and J. L. Hoover-Plow, The cell biology of the plasminogen system, FASEB J., 1995, 9, 939–945, DOI:10.1096/fasebj.9.10.7615163.
- R. Katayama, M. Ikeda, K. Shiraishi, A. Matsumoto and C. Kojima, Formation of hydrophobic domains on the poly(MPC-co-dodecyl methacrylate)-coated surface recognized by macrophage-like cells, Langmuir, 2019, 35, 12229–12235, DOI:10.1021/acs.langmuir.9b00178.
- S. S. An, C. Carreño, D. N. Martin, J. Schaller, F. Albericio and M. Llinás, Lysine-50 is a likely site for anchoring the plasminogen N-terminal peptide to lysine-binding kringles, Protein Sci., 1998, 7, 1960–1969, DOI:10.1002/pro.5560070911.
- A. M. Petros, V. Ramesh and M. Lilnás,
1H NMR Studies of Aliphatic Ligand Binding to Human Plasminogen Kringle 4, Biochemistry, 1989, 28, 1368–1376, DOI:10.1021/bi00429a064.
- I. J. Byeon, R. F. Kelly and M. Llinás,
1H NMR Structural Characterization of a Recombinant Kringle 2 Domain from Human Tissue-Type Plasminogen Activator, Biochemistry, 2019, 28(24), 9350–9360, DOI:10.1021/bi00450a016.
- T. Nousou, S. Hirao, T. Ogawa and K. Shiraishi, Preparation of poly(acrylamide-co-acrylnitrile) grafted glass and thermal stimulated detachment of surface-attached human immortalized mesenchymal stem cells, J. Appl. Polym. Sci., 2022, 139(22), 52257, DOI:10.1002/app.52257.
- T. Ueda, H. Oshida, K. Kurata, K. Ishihara and N. Nakabayashi, Preparation of 2-Methacryloyloxyethyl Phosphorylcholine Copolymers with Alkyl Methacrylates and Their Blood Compatibility, Polym. J., 1992, 24(11), 1259–1269, DOI:10.1295/POLYMJ.24.1259.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Yuto
Oki
a,
Tatsuki
Nousou
a,
Yuto
Oki
a,
Tatsuki
Nousou