
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo1−xS@CNT composite with a three-dimensional skeleton for high-performance magnesium–lithium hybrid batteries†
Changwei
Shan
a,
Wutao
Wei
 *a,
Ximin
Ling
a,
Xuyan
Qin
a,
Zhao
Liu
a,
Mingjie
Song
a and
Liwei
Mi
*ab
*a,
Ximin
Ling
a,
Xuyan
Qin
a,
Zhao
Liu
a,
Mingjie
Song
a and
Liwei
Mi
*ab
aZhongyuan University of Technology, Henan Key Laboratory of Functional Salt Materials, Center for Advanced Materials Research, Longhu Town, Zhengzhou, 450007, China. E-mail: 506952644@qq.com; mlwzzu@163.com
bYaoshan Laboratory, Pingdingshan University, Pingdingshan Henan, 467000, China
First published on 1st February 2024
Abstract
Magnesium–lithium hybrid batteries (MLHB) combine the advantages of magnesium and lithium ions for recharging, thus becoming a very attractive energy storage system. Transitional metal sulfides (TMSs) have attracted attention owing to their large theoretical capacity. However, these materials are accompanied by a huge volume change during charging and discharging processes. This leads to a serious decline in the capacity of their electrode material. In this study, we used low-cost materials to prepare a Co1−xS@CNT composite material and used it as the positive electrode for Mg2+/Li+ hybrid batteries. X-ray diffraction, scanning electron microscopy, and transmitting electron microscopy analyses were employed for the structural and morphological characterisation of the prepared Co1−xS@CNT. Cyclic voltammetry, electrochemical impedance spectroscopy, and X-ray photoelectronic spectroscopy methods were used to determine the mechanism and material reversibility of magnesium ions inserted into the substrate material. The Co1−xS@CNT was assembled as a positive material in a magnesium–lithium hybrid battery and it showed good circulation stability (a capacity retention of 68% after 200 cycles at a current density of 200 mA g−1). This study provides a new idea for the design and synthesis of metal sulfide nanocomposites.
1. Introduction
Based on the demand for renewable energy and the pressure of environmental protection, the demand for high energy density batteries is increasing. Lithium-ion batteries are favored in portable electronic devices and mobile phones because of their high energy density and long cycle life. However, the high cost of lithium and the characteristics of dendrite formation limit their development in the field of electric vehicles.1,2 Rechargeable multivalent batteries (such as Mg, Al, and Ca batteries) have received widespread attention in order to improve safety and reduce costs.3,4 In particular, rechargeable magnesium ion batteries (MIBs) have the advantages of low cost, high theoretical capacity (3833 mA h cm−3 for Mg and 2046 mA h cm−3 for Li), and no dendrite formation.5,6 Despite these advantages, MIBs are unable to provide high specific capacity owing to the complex intercalation chemistry of Mg2+. The strong electrostatic interaction between Mg2+ and a cathode lattice results in its slow diffusion. This ultimately leads to low capacity provided by MIBs, which severely hinders the development of magnesium-ion battery cathodes.7,8 Mg2+/Li+ hybrid batteries have been proposed as a promising candidate to overcome the lack of positive electrode materials. This hybrid battery system has the safety advantage of a dendrite-free Mg anode and the fast storage capability of Li+. Mg2+/Li+ hybrid electrolytes (all-phenyl complex (APC) electrolyte-lithium chloride (LiCl)/tetrahydrofurans (THFs)) are more stable than conventional APC electrolytes.9–11To date, most of the previously reported MLHB cathodes have been traditional lithium embedded cathode materials, such as TiO2,12 Li4Mn5O12,13 Mo6S8,14 Li4Ti5O12,15 and MoO2-C,16 with a maximum reversible capacity of 204 mA h g−1, 155 mA h g−1, 105 mA h g−1, 175 mA h g−1, and 175 mA h g−1, respectively. Evidently, their low capacity results in MLHBs failing to fully unlock their energy density potential; therefore, improvements are still needed. The reduction in charge storage can be attributed to the electrochemical incompatibility of cathodes used with magnesium or lithium salts.17 To increase its capacity, it was necessary to synthesize cathode materials that could accommodate both Mg2+ and Li+ ions without side reactions, and materials based on lithium-driven conversion reactions rather than intercalation processes were selected for MLHBs. For example, Zhang et al. demonstrated that Mg/FeSx has a much greater reversible capacity in MLHB than lithium inserted into the cathode, with FeS and FeS2 having a capacity of 520 mA h g−1 and 600 mA h g−1, respectively, at 0.05C.18
Among the available alternative conversion materials, cobalt sulfides (Co1−xS) have received increasing attention due to their high theoretical capacity (871 mA h g−1) and excellent electrical conductivity (5 × 102 S m−1).19 Unfortunately, in practical applications, the large volume changes inherent in cobalt sulfides cause the electrode to shatter and the polysulfide intermediates to dissolve during the cycle, resulting in a sharp decline in the cycle life and capacity.20 To overcome these shortcomings, researchers modified Co1−xS by changing the morphology of the product, such as nanoparticles. Hollow nanostructured nanocomposites were prepared using carbonaceous materials as a buffer matrix. CNT can improve electronic conductivity, insert/expulse polysulfide intermediates, adapt to the strain caused by volume changes, avoid Co1−xS particle agglomeration, and shorten the ion diffusion length.21
In this study, Co1−xS@CNT composites with three-dimensional network structures and high specific surface area were synthesized by a simple sol–gel method and ice-template-assisted post-freeze drying process. It was used as the battery cathode of Mg2+/Li+ double-salt electrolyte magnesium-based batteries. Among them, Co1−xS nanoparticles have become storage carriers due to their high capacity. Carbon nanotubes are interconnected to form a network of ions and electrons with a high surface area. The network structures can not only facilitate the entry of electrolyte ions into the electrode and ensure the rapid diffusion of electrons, but also prevent the aggregation of Co1−xS. Co1−xS nanoparticles are attached to CNT, which produce strong chemical interactions, reduce volume changes during cycling, and improve the cycle life of the electrode materials. In addition, carbon nanotubes are not chemically treated, maintaining good structural integrity with a high aspect ratio and high conductivity. The network of conductive carbon nanotubes facilitates electron transport, resulting in high-rate capabilities. The instant freezing helps keep the CNT well dispersed without conglomeration and re-stacking. The results showed that compared with the pure Co1−xS electrode material, the Co1−xS@CNT electrode material had outstanding rate performance and long-term cycling performance.
2 Experimental
2.1 Synthesis of Co1−xS@CNT composites
In this experiment, 5.76 g (0.03 mol) citric acid, and 2.91 g (0.01 mol) cobalt nitrate were mixed in 20 mL water and 5 mL anhydrous ethanol, and heated in a water bath at 90 °C to obtain aerogel. The aerogels were heated to 350 °C at the rate of 5 °C min−1 and kept in a muffle furnace for 12 h to remove excess carbon and obtain Co3O4. Co3O4 was mixed with carbon nanotubes in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 for ultrasonic mixing and frozen with liquid nitrogen. Co3O4@CNT was obtained after freeze-drying. Co3O4@CNT was uniformly mixed with sublimed sulfur in a mass ratio of 1:3. The mixture was then heated to 500 °C at a heating rate of 5 °C min−1 and kept under argon gas in a tube furnace for 2 h to obtain Co1−xS@CNT. The preparation of pure Co1−xS is similar to the above reaction, and Co3O4 was separately freeze-dried after ultrasound.
:2 for ultrasonic mixing and frozen with liquid nitrogen. Co3O4@CNT was obtained after freeze-drying. Co3O4@CNT was uniformly mixed with sublimed sulfur in a mass ratio of 1:3. The mixture was then heated to 500 °C at a heating rate of 5 °C min−1 and kept under argon gas in a tube furnace for 2 h to obtain Co1−xS@CNT. The preparation of pure Co1−xS is similar to the above reaction, and Co3O4 was separately freeze-dried after ultrasound.
2.2 Materials characterization
X-ray diffraction (XRD, Rigaku D/Max-2500, Cu Kα radiation), and X-ray photoelectron spectroscopy (XPS, PHI 5000 Versaprobe, ULVAC PHI) were used to determine the phase composition of the prepared materials. Lattice parameters, Co metal content, and valence states were characterized. The microstructure of the materials was characterized by field emission scanning electron microscopy (FESEM, JEOL JSM-6700) and transmission electron microscopy (TEM). The pore characteristics and pore size distribution of the material were determined using a nitrogen adsorption/desorption instrument (NOVA 2200e).2.3 Electrochemical test
For the MLHBs test, the electrochemical test was performed in a coin-type CR2032 battery. The working electrode consisted of an active material, acetylene black, and polyvinylidene fluoride (PVDF) in a weight ratio of 8:1:1. The mixture was coated on a copper foil and dried for 12 h in a vacuum at 80 °C for use as a working electrode. The mass load of the active material was ∼0.5 mg cm−3. The polished magnesium sheet was used as the negative electrode of the battery, and the glass fiber was used as the diaphragm. The electrolytes were APC and APC-0.8 LiCl derived from 0.8 M LiCl in THF. Cell assembly was performed in an Ar-filled glovebox with concentrations of moisture and oxygen below 0.1 ppm. The charging and discharging performance was performed on the Xinwei battery test system with different current densities, and the voltage range is 0.1–2.0 V. Cyclic Voltammetry (CV) and Electrochemical Impedance Spectral (EIS) were carried out on a CHI 660E electrochemical workstation.
3 Results and discussion
The structure and morphology of Co1−xS@CNT were characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), and transmission electron microscopy (TEM). The complexation of cobalt nitrate hexahydrate and citric acid monohydrate in deionized water and anhydrous ethanol was studied. The ammonia water makes the pH of the solution alkaline, and the H+ in the solution system is reduced, which helps very efficient cross-linking of Co2+ with the organic matter. Then, the water content was further reduced to form an aerogel. The aerogel was annealed to remove volatile components and increase porosity. During the annealing process, metal oxide nanoparticles are formed. In order to obtain lower surface energy, the nano-oxide particles tend to agglomerate. In order to obtain a more uniform composite material, carbon nanotubes were treated ultrasonically. Ultrasonic oscillations disperse CNT and expand the space in the network, facilitating the mixing of materials and carbon nanotubes. The ultrasonic products are rapidly frozen using liquid nitrogen, and the interconnected ice crystals are formed in the CNT network to play a supporting role. Using freeze-drying, rather than traditional high-temperature drying, can make the carbon nanotube network more stable in binding to the nanoparticles.22 The nano-oxide particles coated by the carbon nanotube network were vulcanized by subliming sulfur in an Ar environment and collecting it into Co1−xS@CNT as a black powder. In order to control the experimental variables, the synthesis of pure Co1−xS was performed under ultrasound treatment of the annealed gel directly and then using the same method as above.CNTs have higher electron transport efficiency and better overcome the shortcomings of poor electronic conductivity of Co1−xS electrode material. At the same time, nanoscale particles can effectively shorten the transfer path of electrons and Mg2+, further improving the utilization rate. The crystal structure and phase purity of the prepared Co1−xS@CNT aerogel were determined by XRD. The XRD patterns of Co1−xS and Co1−xS@CNT are shown in Fig. 1a. The XRD curve of Co1−xS@CNT composite showed the diffraction peaks of CNT and Co1−xS, in which the main diffraction peak of Co1−xS was consistent with the standard card (JCPDS No. 42-826), and the diffraction peak was sharp, indicating high crystallinity. The diffraction peaks corresponded to the cubic phase of Co1−xS, which means that the structure of CNT hardly changed even after freeze-drying and high-temperature heat treatment, which is in good agreement with the literature reports. The X-ray diffraction peaks are located at (100), (101), (102), (110), (103), (201), (004), and (202) crystal planes at 30.42°, 35.02°, 46.52°, 54.10°, 61.92°, 65.84°, 72.62°, and 73.90°, respectively. No heterochromatic peaks of Co3O4 and CoS were observed in the XRD images, indicating the high purity of the synthesized Co1−xS. A small peak at 26° is from the CNT and confirms the synthesis of the Co1−xS@CNT composites.23
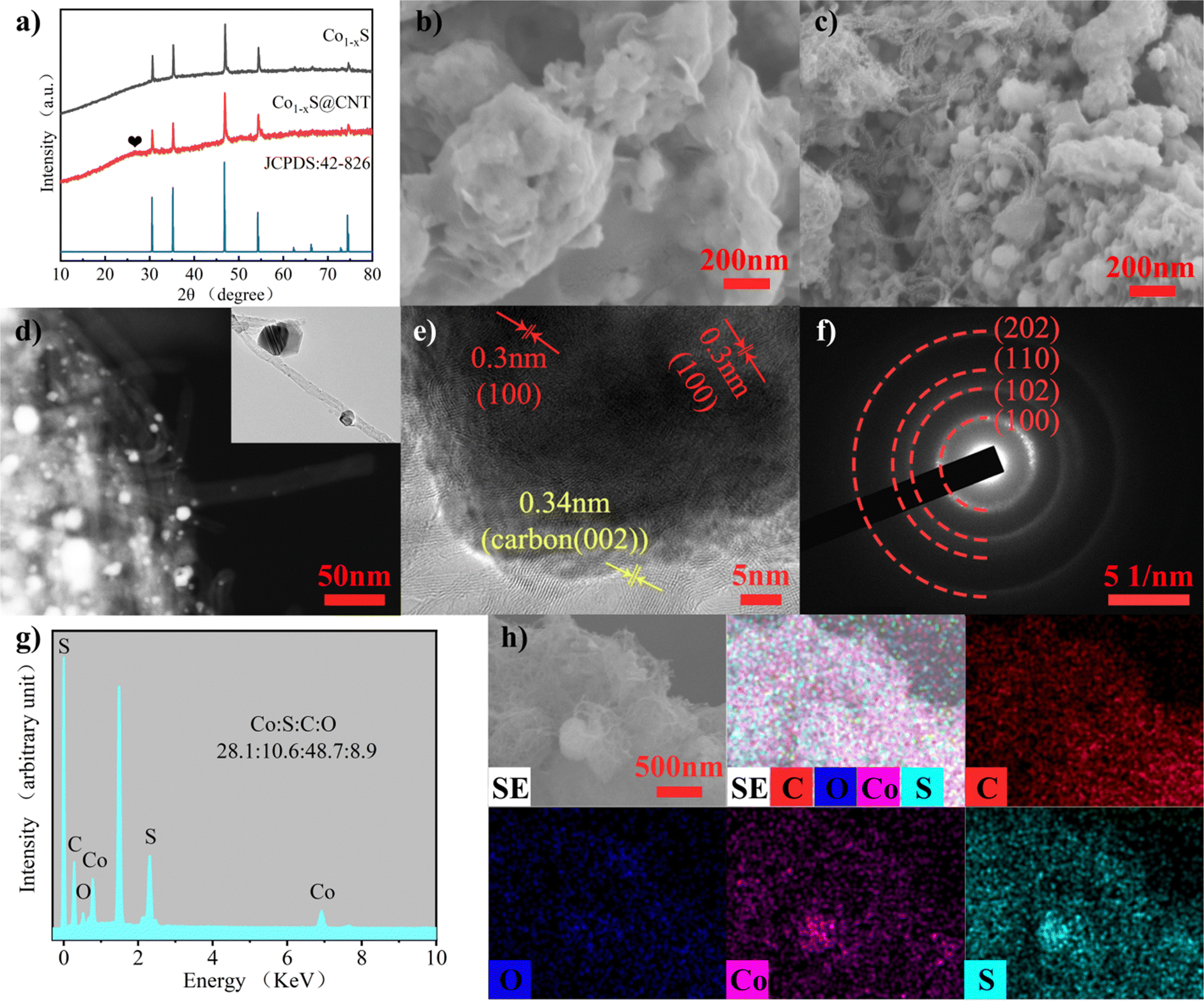 | ||
| Fig. 1 (a) XRD pattern of Co1−xS and Co1−xS@CNTs. (b) SEM images of Co1−xS. (c) SEM images of Co1−xS@CNTs. (d)–(f) TEM images of Co1−xS@CNTs. (g) EDS spectra of Co1−xS@CNTs. (h) Corresponding EDS elemental mapping of Co, S, C, and O elements. | ||
The micromorphology of the samples was characterized using field emission scanning electron microscopy (FESEM). Fig. 1b and c are FESEM images of Co1−xS and Co1−xS@CNT, respectively. The image shows that CNTs form uniform entangled network structures, and Co1−xS appear as clustered braided spheres with a single braided sphere diameter of about 50 nm. Fig. 1b shows Co1−xS nanoparticles clustered together. In the absence of CNT, Co1−xS nanoparticles agglomerate more easily, possibly due to the small size of the nanoparticles, which leads to higher surface energy. In the Co1−xS@CNT composite, Co1−xS is dispersed in a CNT network. This further suggests that there is a strong interaction between CNT and Co1−xS, which can prevent the agglomeration of Co1−xS nanoparticles. After the addition of CNT, the prepared Co1−xS particles are smaller in size, which may be because the oxygen-containing groups in CNT can provide enough nucleation sites for cobalt oxides, and the three-dimensional network structure facilitates the diffusion of cobalt ions.21 Nano-sized particles with large specific surface areas can provide more active sites for electrochemical reactions. The fluffy grid structure of CNT is more conducive to electrolyte penetration and can help the dispersion of Co1−xS, so that the surface area of Co1−xS nanoparticles soaked in the electrolyte is larger, thereby increasing the reactive site and improving its electrochemical performance.23
The microstructure of the sample was further characterized by transmission electron microscopy (TEM) (Fig. 1d–f). It can be seen that Co1−xS@CNT in the TEM image is not filled with a large number of Co1−xS filled with the three-dimensional network structure formed by CNTs as seen in the FESEM image. This phenomenon can be attributed to the separation of some Co1−xS spheroidal particles from CNTs during the TEM test sample preparation due to the ultrasonography performed in Co1−xS@CNT in anhydrous ethanol solution. In addition, high-resolution transmission electron microscopy (HRTEM) images shown in Fig. 1e indicate a 0.30 nm spacing between light and dark lattice fringes, which can be attributed to the (100) lattice plane of Co1−xS. In addition, separated lattice fringes with a spacing of 0.34 nm can be seen, which can be attributed to the (002) lattice plane of CNT, further confirming the good binding of Co1−xS to CNT. According to the selected electron diffraction (SAED) image (Fig. 1f), the clear and bright diffraction ring with lattice characteristics can be attributed to the (100), (102), (110), and (202) planes. The results are consistent with those of XRD analysis. The energy dispersive spectroscopy (EDS) test (Fig. 1g) showed that the atomic ratio of Co:S in Co1−xS@CNT was 28.1:10.6. This is significantly lower than the atomic ratio of Co:S in pure Co1−xS (54.9:28.2) (Fig. S1, ESI†), which is mainly affected by CNT. The element mapping diagram (Fig. 1h) shows that Co1−xS@CNT is mainly composed of Co, S, O, and C elements. Co1−xS nanoparticles are uniformly distributed in the CNT grid, providing more abundant active sites and more stable electrochemical properties for the electrochemical magnesium storage reaction.
The pore structure of Co1−xS@CNT was studied using N2 adsorption/desorption experiments (Fig. 2a). The calculated surface area of Co1−xS@CNT via Brunauer–Emmett–Teller (BET) analysis was 225 m2 g−1 and the pore diameter was 3.7–40 nm, which is consistent with the SEM observation. It has a typical mesoporous structure, which facilitates the penetration of the electrolyte into the solid material to a certain extent. The abundance of mesoporous nanostructures may provide more active sites in the Co1−xS@CNT layer and may be a favorable low-resistance pathway for ion transport during the charge–discharge processes.24 The mesoporous structure not only provides a transport channel for the electrolyte to enter the electrode but also acts as a buffer during the insertion/expulsion of ions, delaying the crushing and exfoliation of the active substance.25 Overall, the prepared Co1−xS@CNT has a large specific surface area and mesoporous nanostructure, which eliminates kinetic limitations and provides a long cycle life.26
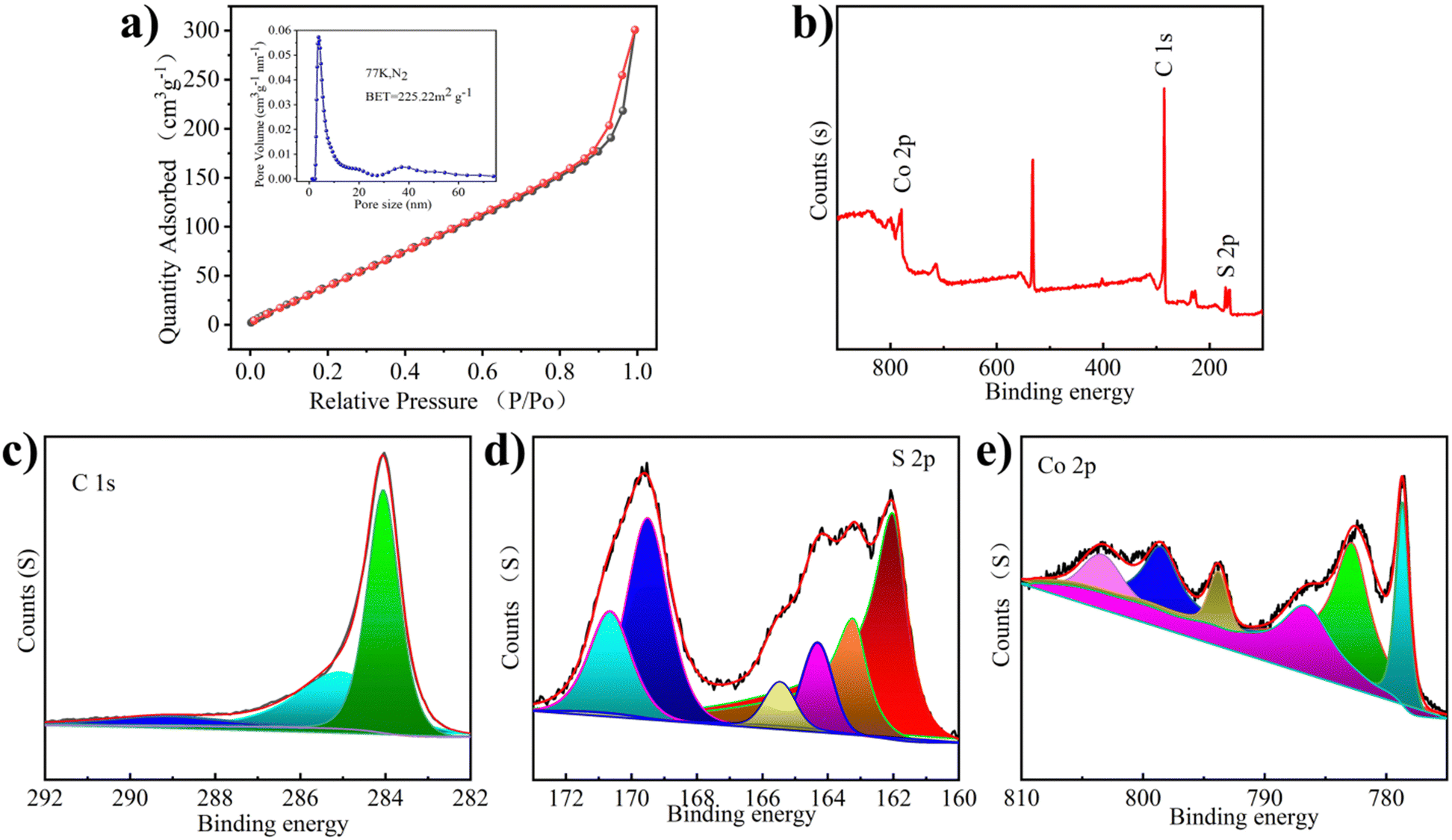 | ||
| Fig. 2 (a) N2 adsorption–desorption isotherms, inset: corresponding pore size distribution. (b) Full XPS spectrum of Co1−xS@CNT. (c) High resolution C 1s XPS spectrum. (d) High resolution S 2p XPS spectrum. (e) High resolution Co 2p XPS spectrum. | ||
The chemical composition and valence states of the prepared Co1−xS@CNT were analyzed by X-ray photoelectron spectroscopy (XPS). The presence of Co, S, and C elements was confirmed from the XPS full-scan spectrum of the Co1−xS@CNT composite (Fig. 2b). This is consistent with the results of the element mapping and EDS spectra. As shown in Fig. 2c, the high-resolution C 1s spectrum of the composite shows three peaks at 284.5, 286.0, and 289.2 eV, attributed to the C–C, C–O–C, and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively.22 Interestingly, in the S 2p spectrum of Fig. 2d, the formation of S–C (163.4 and 165.3 eV) directly demonstrates the close connection between the carbon matrix and the nanoparticles, which can serve as an effective bridge for the electron transfer between graphene and active Co1−xS nanoparticles. The two peaks at 162.3 eV and 163.4 eV can be attributed to the spin-coupled orbitals of the metal sulphides, indicating the successful formation of cobalt sulfide.27 The peaks at 168.8 and 170.2 eV correspond to S–O (SOx−). The S–C structure is very efficient in the oxygen reduction process, while the oxidized S is always inactive.28 The Co 2p spectrum shows peaks at 786.0 and 803.3eV, Fig. 2e, belonging to the satellite peak, while the peaks at 778.7 and 782.7 eV, and 793.7 and 799.2 eV belong to Co 2p3/2 and Co 2p1/2, respectively.29 These chemical bonds could keep the whole structure stable when Mg2+ is detached and embedded.
O bonds, respectively.22 Interestingly, in the S 2p spectrum of Fig. 2d, the formation of S–C (163.4 and 165.3 eV) directly demonstrates the close connection between the carbon matrix and the nanoparticles, which can serve as an effective bridge for the electron transfer between graphene and active Co1−xS nanoparticles. The two peaks at 162.3 eV and 163.4 eV can be attributed to the spin-coupled orbitals of the metal sulphides, indicating the successful formation of cobalt sulfide.27 The peaks at 168.8 and 170.2 eV correspond to S–O (SOx−). The S–C structure is very efficient in the oxygen reduction process, while the oxidized S is always inactive.28 The Co 2p spectrum shows peaks at 786.0 and 803.3eV, Fig. 2e, belonging to the satellite peak, while the peaks at 778.7 and 782.7 eV, and 793.7 and 799.2 eV belong to Co 2p3/2 and Co 2p1/2, respectively.29 These chemical bonds could keep the whole structure stable when Mg2+ is detached and embedded.
Fig. 3a shows the CV curve of Co1−xS@CNT‖APC-LiCl‖Mg half battery for the first five cycles at 0.2 mV s−1. The potential window was set to 0–2.0 V. To obtain the true capacity of the electrode material, the battery first runs 10 cycles at 200 mA g−1 current density, also known as the activation process. In the cycle, the oxidation peak and the reduction peak have a slight displacement. Moreover, the intensity of the peaks increases with the number of cycles, which is very consistent with the charge–discharge curves shown in Fig. 3b–e. CV curve can confirm the electrode reaction process of Co1−xS@CNT in MLHB. During the discharge process, reduction peaks appeared at 0.47 V and 0.98 V. The reduction peak of 0.98 V is attributed to the formation of Li+-embedded Co1−xS and LixCo1−xS. Another reduction peak at 0.47 V is attributed to the conversion of LixCo1−xS to the metal Co and Li2S. During charging, the oxidation peaks at 1.38 V and 1.80 V are attributed to the oxidation of the above products (Co + Li2S → LixCo1−xS and LixCo1−xS → Li+ + Co1−xS).22,26 The test results show that the CV curve repeats well in the first five laps of the test, indicating that the Co1−xS@CNT cathode has excellent reversibility and stability.
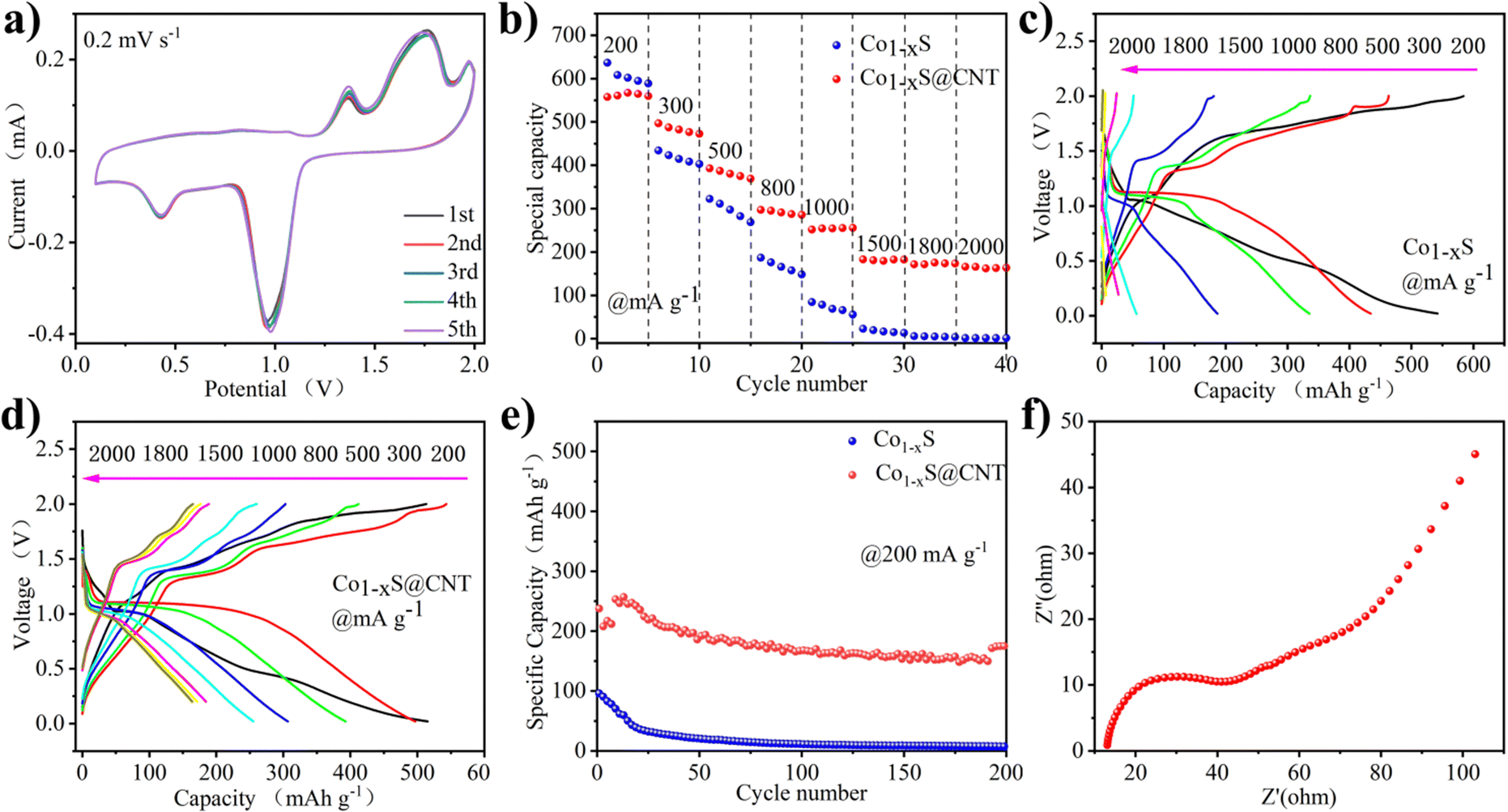 | ||
| Fig. 3 (a) CV curves at 0.2 mV s−1 of Co1−xS@CNTs. (b) Rate performance of Co1−xS and Co1−xS@CNTs. (c) Charge–discharge profiles under different current densities of Co1−xS. (d) Charge–discharge profiles under different current densities of Co1−xS@CNTs. (e) Long-term cycling of Co1−xS and Co1−xS@CNT electrodes at 200 mA g−1. (f) Nyquist plots for Co1−xS@CNTs. | ||
In order to illustrate the potential of the synthesized original Co1−xS and Co1−xS@CNT for battery performance, the specific capacity, rate performance, and cycle performance were studied in the APC-LiCl hybrid electrolyte. For half-cell rate performance, the applied current varies from 200 to 2000 mA g−1. As shown in Fig. 3b–e, at 200, 300, 500, 800, 1000, 1500, and 2000 mA g−1, the discharge capacities of Co1−xS were 636.85, 434.13, 322.58, 186.86, 84.54, 22.94, 6 and 1.67 mA h g−1, respectively. The discharge capacities of Co1−xS@CNT were 557.73, 496.72, 392.85, 297.18, 251.82, 183.06, 171.52 and 165.89 mA h g−1, respectively. The above results show that pure Co1−xS as electrode material has poor rate performance and fast capacity decay at high current density. After doping with CNT, the material rate performance was significantly improved.
The rate properties of pure Co1−xS and Co1−xS@CNT are similar. Because CNT does not provide a capacity contribution, the capacity of Co1−xS at a small current is higher than that of Co1−xS@CNT. The advantages of CNT are mainly reflected in the capacity preservation and the improvement of coulomb efficiency. Fig. 3e shows the cyclic performance of Co1−xS and Co1−xS@CNT. The maximum capacity of pure Co1−xS at 200 mA g−1 is 96.04 mA h g−1. However, after 200 cycles, the capacity decays to 7.66 mA h g−1, with a capacity retention rate of only 7.9%. The pure Co1−xS electrode material decays too quickly during the cycle, which may be due to the accumulation of the electrode material. However, the Co1−xS@CNT electrode exhibits excellent cyclic stability at 200 mA g−1 current density. After 200 cycles, the reversible capacity of the Co1−xS@CNT electrode remains at 57 mA h g−1, with a capacity retention rate of 68%. The improved performance of Co1−xS@CNT batteries is primarily attributed to the superior conductivity of the carbon nanotube network. The three-dimensional interconnected porous structure not only accommodates the volume expansion due to crushing but also accelerates the absorption of electrolytes, thereby increasing the transport rate of metal ions to the electrons.30,31 Furthermore, the Co1−xS nanoparticles surrounded by carbon nanotubes are smaller in size, which effectively increases the specific surface area, guarantees more active sites, and relieves stress in ion insertion/expulsion reactions. This ultimately improves the performance of the nanocomposites and also demonstrates the advantages of CNTs as doping materials.
The charge transfer on the electrode surface was analyzed by EIS, and the Nyquist diagram of the material was obtained. As shown in Fig. 3f, in the high-frequency region, only a slight curvature was observed, and no significant semicircle was observed. This indicated that the charge transfer resistance between the active material and electrolyte interface is negligible. In the low-frequency region, the Nyquist diagram is almost a vertical line, which indicates that the electrode performance is not limited by the electrolyte diffusion process, which is a very ideal capacitive behavior.32
The mechanism of the electrode reaction was studied by using in situ XPS. Fig. 4a–d shows XPS spectra under different charging and discharging states. The results of XPS can explain the mechanism of the electrochemical reaction. Fig. 4a shows the XPS peak of Mg 1s in charge and discharge states. During discharge, the peak binding energy shifts slightly towards higher binding energy. After charging to 2 V, the peak binding energy returns to the initial state, indicating that the process of Mg insertion/expulsion has good reversibility. In Fig. 4b, the peak of S 2p consists of the Co–S bond, S–C bond, and S 2p3/2 bond. It can be seen that the position of the S–O bond energy changes little, only in the intensity of the peak. The S–C bond is slightly offset, the initial state is restored after charging, and the peak strength change is also small. The stability of the S–C bond provides a good basis for the reversible circulation of materials. The binding energy of 161.4 eV responds to the 2p3/2 orbital of S2− binding to metal ions, and the apparent change may be due to the influence of the S2− self-acting force during the insertion of the double salt metal ions.23Fig. 4c shows the XPS peaks of Co 2p under different charging and discharging states. Co 2p3/2 at 778.7 eV disappeared during discharge, indicating a reduction reaction. The peak value of Co 2p3/2 appears gradually with charge, indicating that the accompanying reaction during charge and discharge is reversible. Fig. 4d shows the XPS peak of Li 1s in charge and discharge states, which is accompanied only by the change of peak intensity during charge and discharge. The peak binding energy recovered after full charge, indicating that the reaction involving Li+ has excellent reversibility.
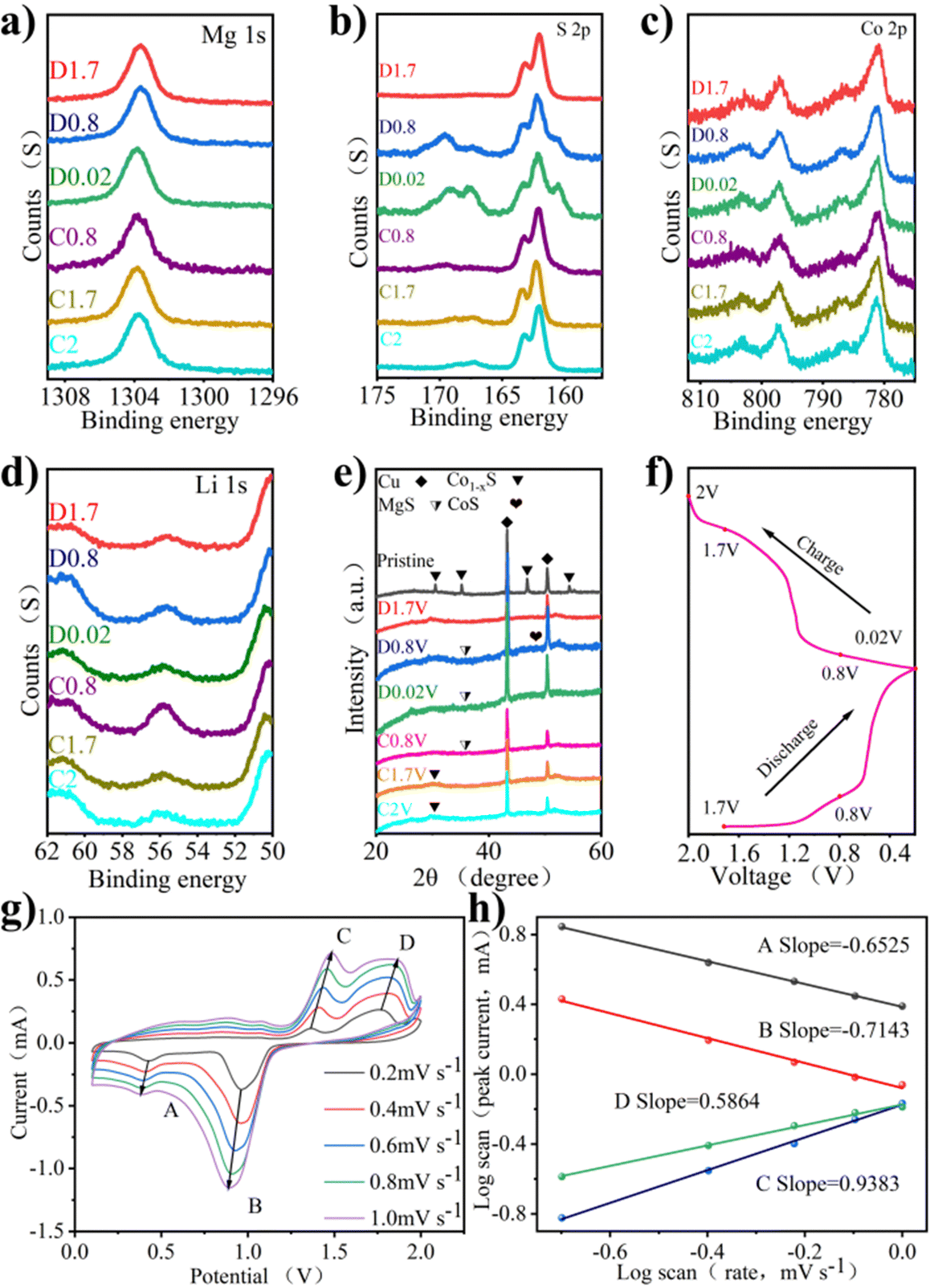 | ||
| Fig. 4 (a) Ex situ XPS spectra of Mg 1s. (b) Ex situ XPS spectra of S 2p. (c) Ex situ XPS spectra of Co 2p. (d) Ex situ XPS spectra of Li 1s. (e) Ex situ XRD patterns of the Co1−xS@CNT microspheres under different electrochemical stages. (f) Corresponding charge–discharge profiles. (g) Corresponding charge–discharge profiles. (h) CV curves at different scan rates of Co1−xS@CNT. (i) logi–logv curves of Co1−xS@CNT. | ||
The mechanism of the electrode reaction was studied using in situ XRD. Fig. 4e shows the XRD patterns of Co1−xS@CNT at different charging and discharging points after 5 cycles. The peak at 26.5° is due to CNT, peaks at 43.3° and 50.5° are attributed to copper foil acting as a fluid collector, and peaks at 30.5°, 35.2°, 36.8°, and 54.3° are due to the original Co1−xS. The peak of Co1−xS decreased significantly at 0.02 V, indicating that Co1−xS participated in the electrode reaction. As the discharge progresses, the characteristic peaks of the original material become wider and weaker, indicating that there are other ions embedded, and the preliminary judgment is Li+. At the same time, CoS(JCPDS No. 1-1279) shows weak peaks, which can be attributed to low crystallinity. As the charge progresses, the diffraction peak of Co1−xS@CNT reappears, indicating that the reaction is reversible.
The CV curve shows the oxidation properties and reduction properties of the material in the range of 0–2 V to understand the Co1−xS@CNT rate properties. The REDOX peak in Fig. 4g is related to the intercalation of ions. It is obvious that the CV curves at different scanning speeds show similar reduction and oxidation peaks, and the enclosed area of CV curves increases with the increase in the scanning speed. When the scanning rate increases from 0.2 mV s−1 to 1.0 mV s−1, the positions of the reduction peak and oxidation peak shift to low potential and high potential, respectively. The reason for this phenomenon may be that the material as a whole has sufficient time to reach the equilibrium potential of the oxidation and reduction reactions at a low scanning rate. With the increase of the scanning rate, the diffusion rate of ions to the bulk phase makes it difficult to reach a balance with the charge transfer rate, forcing the surface reaction to increase, resulting in the overpotential of the oxidation peaks and reduction peaks. In other words, the ion diffusion rate generated by the electrode reaction cannot meet the ion consumption rate, resulting in the peak moving in the direction of the increase of overpotential. In order to confirm the above statement, the relationship between the scanning rate and the peak current was further analyzed. It is generally believed that the value of the scanning rate and peak current follows a power law relationship:
| I = a·vb | (1) |
| logi = loga + blogv | (2) |
In eqn (1), i is the current and v is the scan rate; a and b in the equation represent adjustable values. The value of b is evaluated from the slope in eqn (2). The b value represents the electrochemical reaction under the control of different behaviors, b = 0.5 indicates the typical diffusion control behavior, and b = 1 indicates that the contribution of the current is completely controlled by the surface.32 As shown in Fig. 4h, the log(v)–log(i) figure shows that the peak b values of the cathode and anode are −0.6525, −0.7143, 0.5864, and 0.9383, respectively. Most of the b values are close to 0.5, indicating that diffusion and surface control behavior exist simultaneously, but the diffusion control current is the main one.
In summary, the addition of CNTs can enhance the uniform dispersion of Co1−xS nanoparticles, thus increasing active sites in Co1−xS nanoparticles, which is more conducive to contact with the electrolyte. The network structure of CNT has a rich buffer region that can be used as a local memory to hold the electrolyte. In addition, CNT can reduce the volume change of electrode material during the cycle, accelerating the electrochemical reaction and improving the cycle stability.
4 Conclusion
A high-performance magnesium–lithium hybrid battery was constructed by synthesizing nanometer-size Co1−xS@CNT composite material with cheap materials and simple and efficient technology. This hybrid battery exhibited a high energy density and a long cycle life. Compared with pure magnesium-based electrolytes, Mg2+/Li+ hybrid electrolyte has higher dynamic performance, lower charge transfer resistance, and a faster ion-diffusion rate. The highly conductive, high-porosity CNT allows the electrolyte to diffuse easily and quickly collecting electrons from the external circuit, which provides more active sites. Co1−xS nanoparticles maintain a diameter as low as 50 nm and a uniform dispersion in a carbon nanotube network. Co1−xS nanoparticles uniformly distributed in the carbon nanotube network can improve cyclic stability and provide short paths for ion transport to facilitate ion diffusion dynamics. As a result, Co1−xS@CNT enables high reversible specific capacity (greater than 500 mA h g−1 at 0.2 A g−1) and excellent cycling performance (68% capacity retention after 200 cycles). These excellent electrochemical properties confirm that Co1−xS-based materials have broad application prospects in MLHB, paving the way for the development of more electrode materials with high capacity and long-cycle stability. In addition, the carbon nanotubes used in this work have inherent interconnect structure, low cost, and easy operation characteristics, which show obvious advantages over graphene materials. We believe that carbon nanotubes can be an excellent alternative to graphene materials, and this mixture of carbon nanotubes based on high-performance materials can offer great promise in large-scale energy storage device applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21671205), the Henan Province Key Research and Development and Promotion Project (Scientific and Technological Breakthroughs) (Grant No. 232102240081).References
- J. Liu, Y. Zhong, X. Li, T. Ying, T. Han and J. Li, Nanoscale Adv., 2021, 3, 5576 RSC.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 RSC.
- J. Muldoon, C. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683 CrossRef CAS.
- H. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265 RSC.
- M. Mao, T. Gao, S. Hou and C. Wang, Chem. Soc. Rev., 2018, 47, 8804 RSC.
- Q. Zhang, J. Du, A. He, Z. Liu and C. Tao, J. CO2 Util, 2019, 34, 635 CrossRef CAS.
- R. Dubey, T. Colijn, M. Aebli, E. Hanson, R. Widmer, K. Kravchyk, M. Kovalenko and N. Stadie, ACS Appl. Mater. Interfaces, 2019, 11, 39902 CrossRef CAS PubMed.
- T. Arthur, R. Zhang, C. Ling, P. Glans, X. Fan, J. Guo and F. Mizuno, ACS Appl. Mater. Interfaces, 2014, 6, 7004 CrossRef CAS PubMed.
- C. Liu, G. Zhao, L. Zhang, P. Lyu, X. Yu, H. Huang, G. Maurin, K. Sun and N. Zhang, J. Mater. Chem. A, 2020, 8, 22712 RSC.
- C. Pei, F. Xiong, J. Sheng, Y. Yin, S. Tan, D. Wang, C. Han, Q. An and L. Mai, ACS. Appl. Mater. Interfaces, 2017, 9, 17060 CrossRef CAS PubMed.
- J. Cho, J. Ha, J. Oh, S. Lee, K. Kim, K. Lee and J. Lee, J. Phys. Chem. C, 2020, 124, 25738 CrossRef CAS.
- Q. Fu, R. Azmi, A. Sarapulova, D. Mikhailova, S. Dsoke, A. Missiul, V. Trouillet, M. Knapp, N. Bramnik and H. Ehrenberg, Electro. Acta, 2018, 277, 20 CrossRef CAS.
- Y. Cen, Y. Liu, Y. Zhou, P. Jiang, J. Hu, Q. Xiang, B. Hu, C. Xu, D. Yu and C. Chen, ChemElectroChem, 2020, 7, 1115 CrossRef CAS.
- Y. Cheng, Y. Shao, J. Zhang, V. Sprenkle, J. Liu and G. Li, Chem. Commun., 2014, 50, 9644 RSC.
- N. Wu, Z. Yang, H. Yao, Y. Yin, L. Gu and Y. Guo, Angew. Chem., 2015, 127, 5849 CrossRef.
- C. Wu, J. Hu, J. Tian, F. Chu, Z. Yao, Y. Zheng, D. Yin and C. Li, ACS Appl. Mater. Interfaces, 2019, 11, 5966 CrossRef CAS PubMed.
- E. Levi, Y. Gofer and D. Aurbach, Chem. Mater., 2010, 22, 860 CrossRef CAS.
- Y. Zhang, J. Xie, Y. Han and C. Li, Adv. Funct. Mater., 2015, 25, 7300 CrossRef CAS.
- G. Liu, B. Wang, L. Wang, Y. Yuan and D. Wang, RSC. Adv., 2016, 6, 7129 RSC.
- H. Wang, N. Mao, J. Shi, Q. Wang, W. Yu and X. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 2882 CrossRef CAS PubMed.
- S. Zeng, H. Chen, H. Wang, X. Tong, M. Chen, J. Di and Q. Li, Small, 2017, 13, 1700518 CrossRef PubMed.
- M. Asif, M. Rashad and Z. Ali, Nanoscale, 2020, 12, 14267 RSC.
- Z. Ye, P. Li, W. Wei, C. Huang, L. Mi, J. Zhang and J. Zhang, Adv. Sci., 2022, 9, 2200067 CrossRef CAS.
- F. Han, C. Zhang, B. Sun, W. Tang, J. Yang and X. Li, Carbon, 2017, 118, 731 CrossRef CAS.
- Y. Meng, D. Wang, Y. Wei, K. Zhu, Y. Zhao, X. Bian, F. Du, B. Liu, Y. Gao and G. Chen, J. Power Sources, 2017, 346, 134 CrossRef CAS.
- C. Liu, G. Zhao, L. Zhang, X. Yu, H. Huang, K. Sun and N. Zhang, Chem. – Eng. J., 2021, 405, 126726 CrossRef CAS.
- Y. Xu, Y. Hao, G. Zhang, X. Jin, L. Wang, Z. Lu and X. Sun, Part. Part. Syst. Charact., 2016, 33, 569 CrossRef CAS.
- J. Ren and Z. Yuan, ACS Sustainable Chem. Eng., 2019, 7, 10121 CrossRef CAS.
- Y. Liu, W. Jiang, M. Liu, L. Zhang, C. Qiang and Z. Fang, Langmuir, 2019, 35, 16487 CrossRef CAS PubMed.
- J. He, Y. Chen, W. Lv, K. Wen, C. Xu, W. Zhang, W. Qin and W. He, ACS Energy Lett., 2016, 4, 820–826 CrossRef.
- D. Luo, F. Lin, W. Xiao, C. Luo, Z. Li and X. Li, Trans. Indian Ceram. Soc., 2016, 3, 161–165 CrossRef.
- Z. Guo, W. Wei, J. Shi, P. Wang, Z. Ye and L. Mi, Nanoscale, 2023, 15, 1702 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma01089a |
| This journal is © The Royal Society of Chemistry 2024 |