
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational design of carbon nanocomposites with hierarchical porosity: a strategy to improve capacitive energy storage performance†
Agnieszka
Hryniewicka
 *a,
Joanna
Breczko
ab,
Gabriela
Siemiaszko
a,
Krzysztof
Brzezinski
c,
Anna
Ilnicka
d,
Artur P.
Terzyk
d and
Marta E.
Plonska-Brzezinska
*a
*a,
Joanna
Breczko
ab,
Gabriela
Siemiaszko
a,
Krzysztof
Brzezinski
c,
Anna
Ilnicka
d,
Artur P.
Terzyk
d and
Marta E.
Plonska-Brzezinska
*a
aDepartment of Organic Chemistry, Faculty of Pharmacy with the Division of Laboratory Medicine, Medical University of Bialystok, Mickiewicza 2A, 15-222 Bialystok, Poland. E-mail: agnieszka.hryniewicka@umb.edu.pl; marta.plonska-brzezinska@umb.edu.pl
bFaculty of Chemistry, University of Bialystok, Ciolkowskiego 1K, 15-245 Bialystok, Poland
cDepartment of Structural Biology of Prokaryotic Organisms, Institute of Bioorganic Chemistry, Polish Academy of Sciences, Noskowskiego 12/14, 61-074 Poznan, Poland
dFaculty of Chemistry, Nicolaus Copernicus University in Torun, Gagarin Street 7, 87-100 Torun, Poland
First published on 4th December 2023
Abstract
Covalent triazine frameworks (CTFs) constitute an emerging class of high-performance materials due to their porosity and the possibility of structural control at the molecular or atomic level. However, use of CTFs as electrodes in supercapacitors is hampered by their low electrical conductivity and a strong stacking effect between adjacent CTF sheets. Herein, two series of hybrid carbon nano-onion-based CTFs are designed and successfully synthesized using an ionothermal process at 700 °C. The CTFs could undergo framework growth in two or four directions, which was related to a defined number of nitrile groups in the substrate. CTF counterparts without carbon nano-onions were also synthesized as reference materials. The carbon nanocomposites exhibited excellent specific capacitances, with the highest value exceeding 495 F g−1. It should be emphasized that the specific capacitance value of hybrid materials was 1.5–2 times higher than that of the reference CTFs. This study examined the factors responsible for such a significant increase in electrochemical efficiency. This strategy has significantly expanded the scope and application of CTFs as electrode materials for electrochemical energy storage systems.
1. Introduction
The demand for advanced materials used for power devices is constantly growing, and the focus is on the search for safer and more efficient solutions. These include, among others, capacitors that are counted to future technology and devices with unique electrochemical properties that can store a large amount of electric charges. In most cases, the maximum charge is related to the electrode surface area, accessibility to electrolyte ions, surface morphology, and electrical conductivity.1 According to energy storage mechanisms, capacitors can be classified as electric double-layer capacitors (EDLCs) and pseudocapacitors.2 EDLCs are superior to conventional batteries due to their valuable properties, such as high power density, long durability, and fast charging–discharging.3 The search for improving EDLC performance is directly connected to the development of novel materials for electrodes, which will ensure the appropriate operational parameters of the devices. So far, carbon materials focusing on nano-structured and nano-engineered forms have been commonly used in energy storage devices, including supercapacitors (SCs).1,2,4 A sizeable active surface area and high porosity should primarily characterize the electrode material made of nanoscale materials—theoretically, the higher the surface area and electrolyte concentration, the higher the capacitance values.In connection with the above guidelines, we proposed a combination of carbon nanostructures and spherical multilayer fullerenes (called carbon nano-onions, CNOs) with covalent triazine frameworks (CTFs) to create nanocomposites for SCs.5,6 The combination of materials with differential chemical characters increases the capacity and porosity of electrodes, reaching better electrochemical performances.7 Moreover, combining faradaic and non-faradaic processes in SCs can achieve higher energy and power density while maintaining long-term electrochemical stability. Our studies show that using CNOs during the synthesis of macromolecular systems may organize the organic framework of oligomers,8 polymers9,10 or CTFs5 and alter the uniform distribution of pores and pore sizes (micropores (<2 nm), mesopores (2–50 nm) and macropores (>50 nm)) within the materials. Additionally, the combination of two components also guarantees an increase in the total porosity of the material, which is critical when using them as electrodes in SCs.
We are using CNOs obtained via thermal annealing of nanodiamonds, which leads to the formation of nanostructures of small sizes (diameter in the range of 5–6 nm).11 The CNO is a zero-dimensional carbon nanoparticle with a multilayer spherical graphite structure of 1.4–50 nm in diameter.11 The large graphite surface of CNOs (with sp2-hybridized C atoms) enables the formation of supramolecular systems using covalent12–14 and non-covalent functionalization, for example, through electrostatic, van der Waals, and π–π interactions.15,16 These functionalizations allow controlling of the physical and chemical properties of carbon nanostructures, which in turn enable their versatile applications.1,14,17–21
We used nitrogen-rich triazine units that form CTFs to modify the CNO surface.5,6 CTFs are stable and can withstand harsh acidic or alkaline conditions that are destructive to other porous organic polymers. CTFs possess highly conjugated structures and exhibit semiconductive properties, making it possible to use them in SCs. Notably, the high N atom content and the large surface area must be considered to achieve high efficiency in energy storage devices.22 The first reports of the application of CTFs as SCs date back to 2012.23–25 There have been few reports of CTFs with specific capacitance values exceeding 500 F g−1, i.e., anthracene-based CTFs,26 a series of bicarbazole-based CTFs,27 pyrene-functionalized CTFs,28 and polyethynylbenzonitrile-derived frameworks.29 Other CTFs with relatively high capacitance include tetracyanoquinodimethane,30 bipyridine,31 halogen-substituted derivatives,32 and pyridine.33 These materials have suitable N atom contents, hierarchical micro-mesoporous properties, and high specific surface areas with abundant channels for charges, resulting in remarkable cycle stability, high capacity retention, and excellent coulombic efficiencies.
Herein, we design CTFs with different pore diameters and their hybrids with CNOs. Symmetrically substituted moieties with nitrile groups were used to obtain the desired CTFs. One of them, i.e., tetraarylpyrrole[3,2-b]pyrrole, was applied for the first time by our group as a core in a CTF structure.6 It is well known that a small amount of the CNO is enough to markedly increase the surface area of the hybrid material and improve its electrochemical properties.34
2. Results and discussion
2.1. Syntheses of CTF and CTF–CNO materials
Four compounds were selected for syntheses of pristine CTFs and CNO-based CTFs. Two of them, namely 1,4-dicyanobenzene (2CN-1)23,35 and 7,7,8,8-tetracyanoquinodimethane (4CN-1),30 are known as building blocks of organic skeletons, forming two-dimensional (2D) macrostructures with a repeating structural organization (Fig. 1). We used known substrates to study the effects of CNO additives on the structural and chemical properties of the synthesized materials. The other two monomers are pyrrolopyrroles, such as 1,4-bis(6-methylbenzo[d]thiazol-2-yl)-2,5-bis(4-cyanophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole (2CN-2) and 1,2,4,5-tetrakis(4-cyanophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole (4CN-2), which have never been used for these purposes. The new monomers differed in the number of benzonitrile groups substituted in the pyrrolopyrrole core. 2CN-2 and 4CN-2 contained two and four benzonitrile substituents, respectively. Additionally, in the 2CN-2 structure, two benzothiazole substituents are linked to the pyrrolepyrrole core. These substrates synthesized CTFs covalently attached to the CNO surface (CTF–CNO).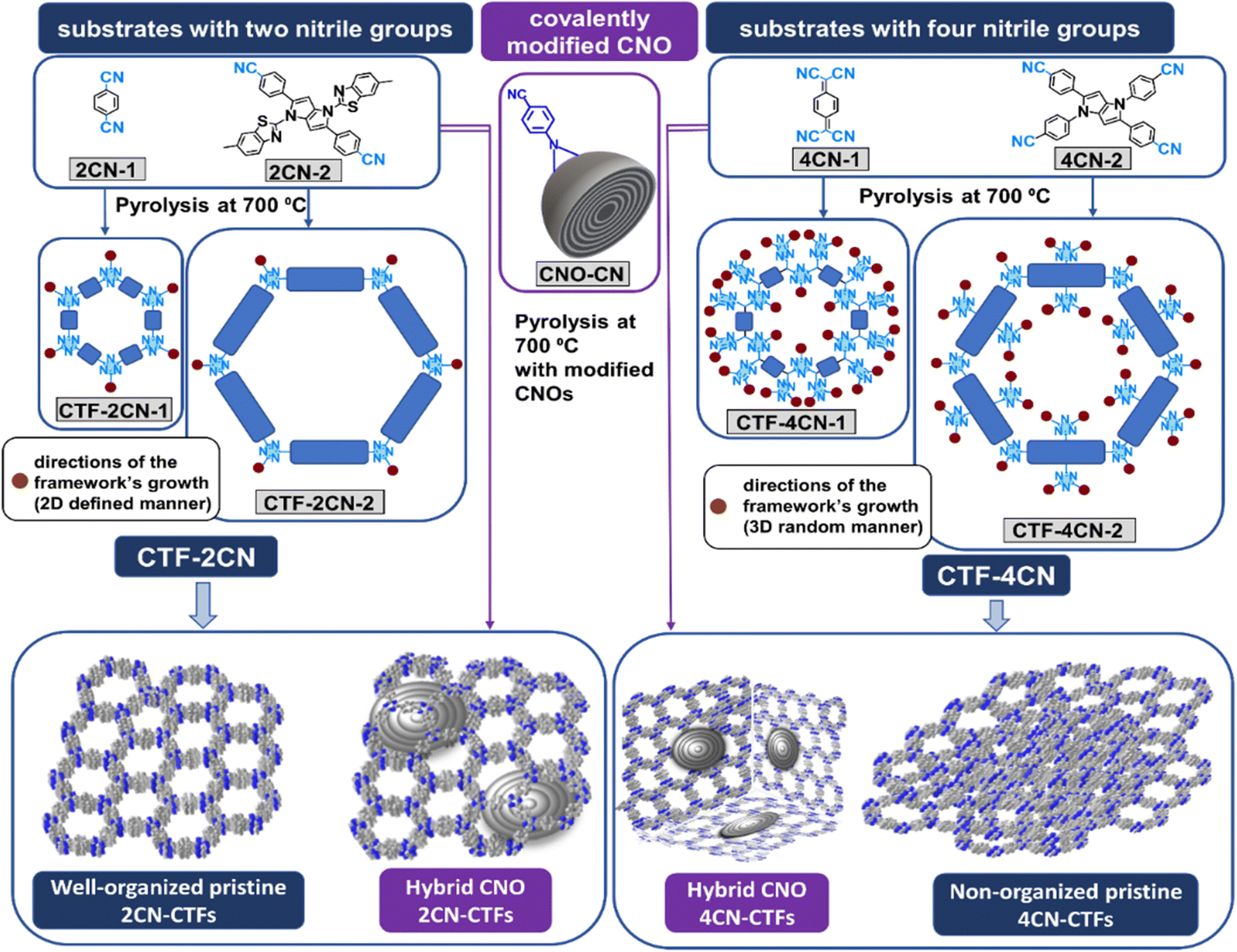 | ||
| Fig. 1 Representative schematic synthesis of CTF and CTF–CNO materials. | ||
Given the experimental determination of the structure, it was expected that tetraaryl-substituted 1,4-dihydropyrrolo[3,2-b]pyrrole monomers with four –CN groups would form a more cross-linked, less ordered framework.36 Conversely, such a skeleton should have a higher N atom content. Adding relatively large CNO structures can result in the framework organization in the three-dimensional (3D) architecture, where the CNO may separate the adjacent CTF sheets. It is very desirable in the case of amorphous frameworks composed of substrates with four –CN groups. Moreover, the nanostructural carbon size may induce a specific orientation of the framework in space.
Pyrrolepyrroles were synthesized in a condensation reaction using iron(III) perchloride hydrate as a catalyst using a simple, straightforward methodology.37 Pyrrolopyrrole-based and commercially available substrates were pyrolyzed at 700 °C with ZnCl2 in an ionothermal process to obtain pristine CTFs (Fig. 1). The covalently modified CNOs (CNO-CN, 5 wt%)34 were pyrolyzed with the appropriate monomers to obtain the hybrid CTF–CNO materials. ZnCl2 can act as a catalyst, solvent, and porogen.
2.2. Structural characterization of CTF and CTF–CNO
Fourier transform infrared (FTIR) spectroscopy was used to evaluate the synthesis of CTF and CTF–CNO. The trimerization of all the substrates was confirmed by FTIR spectroscopy (Fig. 2). The complete trimerization of the monomers was indicated by the absence of a carbonitrile band at 2230 cm−1 in the spectra of the formed materials (Fig. 2A–C, respectively).35 Furthermore, all CTFs gave rise to a band at approximately 1630 cm−1, the characteristic of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration, indicating the successful formation of triazine graphitic-like domains (Fig. 2B and C).38 This band may also be attributed to aromatic CC skeletal vibrations, indicating the presence of a graphite structure.39 Moreover, the bands between 1550 and 1120 cm−1 correspond to the N-doped graphitic domains, which are related to graphitic CC vibrations, NH stretching, C–N stretching of aromatic amines in benzenoid units, and in-plane distortion vibrations of the C–H bonds in benzene rings.39–41 The broad overlapping bands in the range of 2900–2800 cm−1 were attributed to stretching deformation vibrations of aromatic C–H, suggesting the presence of sp3-hybridized carbon atoms in the CTF and CTF–CNO materials (Fig. 2B and C).42 Moreover, a broad, strong band extending from 3600 to 3000 cm−1 was attributed to the stretching vibrations of N–H and aromatic (C–H) groups and their intra- and intermolecular hydrogen bonding (Fig. 2B and C).
N stretching vibration, indicating the successful formation of triazine graphitic-like domains (Fig. 2B and C).38 This band may also be attributed to aromatic CC skeletal vibrations, indicating the presence of a graphite structure.39 Moreover, the bands between 1550 and 1120 cm−1 correspond to the N-doped graphitic domains, which are related to graphitic CC vibrations, NH stretching, C–N stretching of aromatic amines in benzenoid units, and in-plane distortion vibrations of the C–H bonds in benzene rings.39–41 The broad overlapping bands in the range of 2900–2800 cm−1 were attributed to stretching deformation vibrations of aromatic C–H, suggesting the presence of sp3-hybridized carbon atoms in the CTF and CTF–CNO materials (Fig. 2B and C).42 Moreover, a broad, strong band extending from 3600 to 3000 cm−1 was attributed to the stretching vibrations of N–H and aromatic (C–H) groups and their intra- and intermolecular hydrogen bonding (Fig. 2B and C).
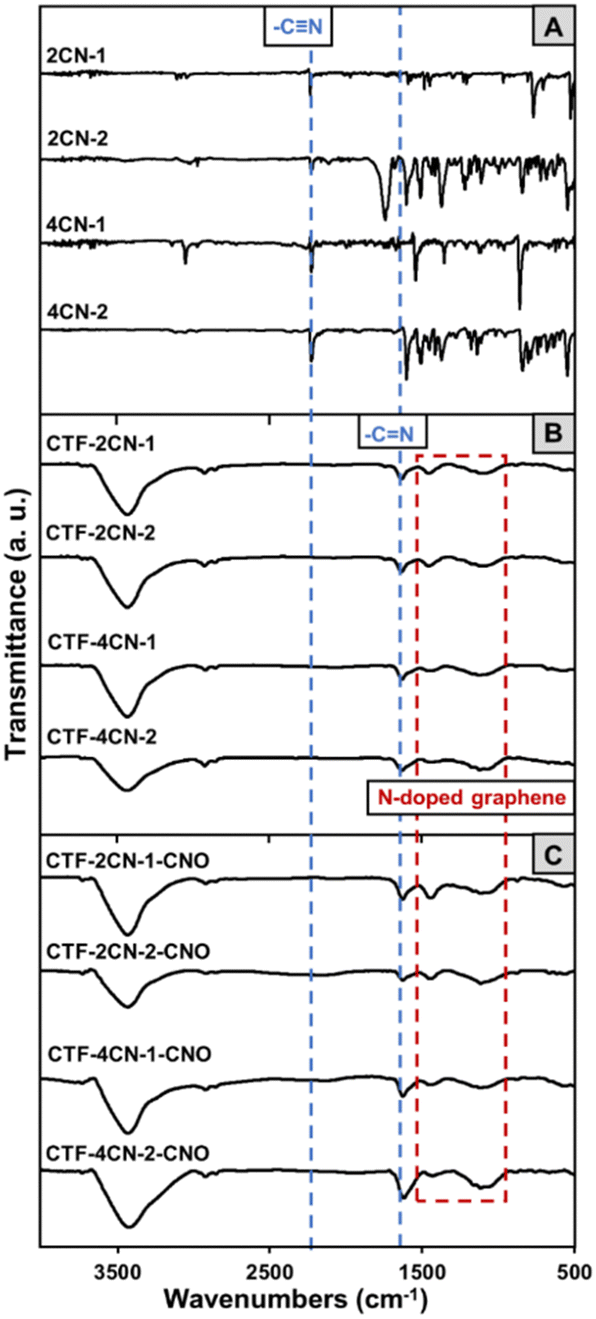 | ||
| Fig. 2 FTIR spectra of (A) monomers, (B) CTF materials and (C) CTF–CNO materials. | ||
The solid-state 13C NMR measurement further confirmed the structures of CTF and CTF–CNO materials. As shown in Fig. S6 (ESI†), aromatic regions are located in the range of 140 to 110 ppm in all spectra of CTFs. Moreover, the chemical shifts at 165–170 ppm are present that can be assigned to the carbon signal from triazine rings.43 Although the relative intensity of this peak is small, its presence indicated the formation of triazine rings. The signal constituting the aromatic region becomes broader for CTF–CNO hybrids (CTF-2CN-1-CNO and CTF-4CN-1-CNO), which may indicate a higher degree of carbonization.44 In the spectra of pyrrolo[3,2-b]pyrrole-based CTFs (pristine and hybrids of CTF-2CN-2 and CTF-4CN-2), there are more aromatic carbon signals, which is related to the presence of pyrrole carbons (see the spectrum of the substrate 2CN-2, Fig. S2, ESI†).
X-ray photoelectron spectroscopy (XPS) was used to determine the elemental compositions of the CTF and CTF–CNO materials. The results indicated that all the studied materials contain C and N atoms with contents between 82 and 91 at% and 4 and 9.5 at%, respectively (Table S1, ESI†). The distribution of elements was defined due to the deconvolution of the high-resolution spectral C 1s (Fig. S5, ESI†) and N 1s (Fig. 3) regions. The C 1s XPS analysis showed that all the materials had a similar distribution pattern of functional groups and defects (Fig. S5 and Tables S2, S3, ESI†). The prominent peak at ∼284.5 eV was assigned to the sp2-hybridized C atom in CC (graphitic/aromatic structure).39,45,46 The peaks at ∼285.0, ∼285.6, and ∼286.4 eV were related to the C–H sp3, C–C sp3, and CN moieties, respectively (Tables S2 and S3, ESI†).46 There were also peaks attributed to C–O–C, CO, and C–N–C groups, with a total content below 10 at%.45,46 It should be noted that CTF-2CN-2 triazine has a significant content of C–N–C groups in its structure (∼288.9 eV),46 compared to other triazines, corresponding to approximately 5 at%.
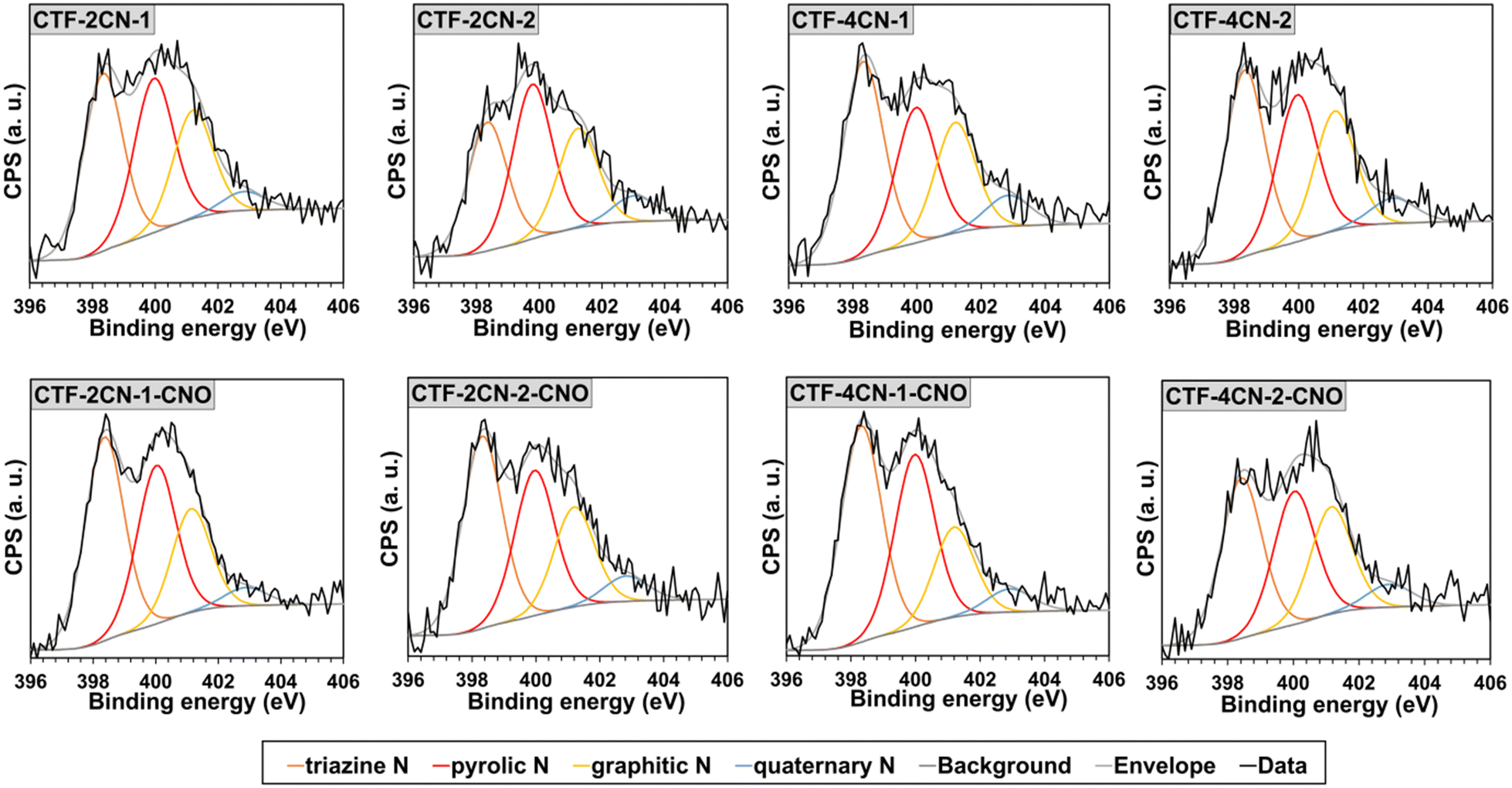 | ||
| Fig. 3 XPS N 1s spectra of the CTF and CTF–CNO spectral regions. | ||
All the materials also containing some defects (∼283.8 eV), which in the CTF, were mainly related to the presence of single and double C vacancies.47,48 After the covalent immobilization of the CTF sheets on the CNO surface, the number of defects in the materials increased, which was mainly attributed to the disruption of the CC bonds with sp2-hybridization in the CNO.
An increase in sp2-hybridized C atoms and a decrease in sp3-hybridized C atoms for the pair of CTF-2CN-2-CNO and CTF-2CN-2 and a couple of CTF-4CN-2-CNO and CTF-4CN-2 (Tables S2 and S3, ESI†), respectively, displayed improvement in graphitic-like domains in the systems. The opposite transformation of C atoms was observed for CTF-2CN-1 after CNO addition (CTF-2CN-1-CNO). Moreover, the elemental compositions of CTF materials were analyzed by elemental analysis, and the results are summarized in Table S1 (ESI†), along with the distribution of elements obtained from XPS. The number of N atoms expressed as a percentage determined by elemental analysis and XPS is almost always consistent. By elemental analysis, we also confirmed the presence of sulfur atoms for CTF-2CN-2 and CTF-2CN-2-CNO at 9.23 and 11.36%, respectively. We observed some discrepancies in the amount of determined carbon. These differences may result from the nature of the measurements of both methods and their accuracy. XPS is the surface analysis of materials. The elemental analysis leads to the complete degradation of the materials and determines the content of individual elements in the entire volume.
The deconvoluted XPS spectra for the N 1s binding state indicated that N was present in four different chemical environments in the CTF and CTF–CNO materials (Fig. 3 and Tables S4, S5, ESI†). Three peaks, namely, pyridinic-N in the covalent triazine skeleton (∼398.3 eV),49,50 pyrrolic-N (∼400.0 eV),51 and tertiary amine in a graphitic network (graphitic-N, ∼401.2 eV), were dominant in the spectra of all the synthesized materials.51,52 The pyridinic-N atoms were the main contribution, with a content of approximately 40 at% for all the materials, excluding CTF-2CN-2, for which this value was approximately 31 at%, followed by pyrrolic-N (about 28–34 at%) and graphitic-N (about 21–25 at%) (Tables S2–S5, ESI†). The graphitic-N atoms were originated from sp2-hybridized N, bonded with three adjacent sp2-hybridized C atoms.53 The N atoms were partially bound in the hexagonal network of graphene. The N atoms appearing in a much smaller amount from 4.1 to 7.1 at% were assigned to the quaternary graphitic N atoms (∼402.8 eV).50,52
High-resolution transmission electron microscopy (HRTEM) studies were performed to analyze the structural organization of the CTF, as well as the effect of adding CNOs to the 2D triazine frameworks. Amorphous phases and crystalline zones were present in all the synthesized materials.54 This heterogeneity in structure may result in higher structural stability and higher conductivity of the hybrid materials. Moreover, the HRTEM images of CTF–CNO hybrids showed the presence of CNOs and graphene ribbons in the structure (Fig. 4F), which increased the crystallinity of the materials. Scanning transmission electron microscopy (STEM) in conjunction with energy-dispersive X-ray (EDX) spectroscopy was used to evaluate the distribution of atoms constituting the synthesized materials and their elemental homogeneity (Fig. 4G and H). The STEM-EDX maps recorded after scanning the CTF-4CN-2-CNO surface show the distribution of the C and N atoms in the material, with a significant predominance of C. The N atoms were not evenly distributed in the material.
 | ||
| Fig. 4 Fig. 4 (A)–(F) HRTEM images of the CTF and CTF–CNO materials; STEM EDS maps of the elemental distribution in CTF-4CN-2-CNO of (G) N atoms and (H) C atoms. | ||
Fig. 4G clearly shows areas with a large amount of N (marked in purple) and darkened areas, which indicate the lack of N. The violet regions, indicating the presence of N atoms, replicated the skeleton of the CTF; therefore, the shaded areas correspond to pores present in the material, which were interconnected. The material's structure with interconnected pores is expected to favor the transport of ions to the electrode surface, which should effectively affect the electrochemical properties of the material.55 In short, in the case of electrodes with many interconnected pores, an even distribution of contact between the electrode and the electrolyte is ensured,56 improving the mass transfer of electrolyte ions into the electrode.55,57 At the same time, in the case of materials with high graphitization and porosity, during repeated charging and discharging of the electrodes, they maintain a 3D structure very well, which ensures their high mechanical stability. Thus, a further advantage of such electrodes is that their porous construction allows the interconnected walls to expand into the open pores, preventing the walls from pulverizing while exhibiting an extremely high level of structural integrity.57,58
Our X-ray powder diffraction (XRD) experiments indicated the transformation of nanodiamond (ND) particles into CNOs at 1650 °C (Fig. 5), at which point we synthesized CNOs. The XRD patterns of the ND and CNO show the presence of a mixture of various phases with different intensities for both materials. For the ND, the most substantial reflection at approximately 2θ = 43.8° corresponded to the diffraction on the (111) basal plane of the diamond structure.59 A broad peak in the 20–27° range was also observed, suggesting a certain amount of a graphite-like phase.60,61 For the CNO nanostructures, these two reflections were also present in similar degrees of theta angles, but their intensity and nature differed. An asymmetric second peak with a maximum of approximately 43.0° was observed, which was attributed to the (002) and (100) graphite planes.62 The broad asymmetric peak centered at approximately 23–27° suggests contributions of some sp2-bonded carbons in the CNO structures. Its asymmetry can be divided into two subpeaks characteristic of turbostratic and graphitic carbon.63,64 For all CTF and CTF–CNO materials, these relatively broad peaks attributed to sp2- and sp3-hybridized C atoms were also predominant. Diffraction broadening originates primarily from the size of the grains. However, a further influence of strains and defects in the semi-crystal material on diffraction broadening cannot be excluded and requires additional studies.
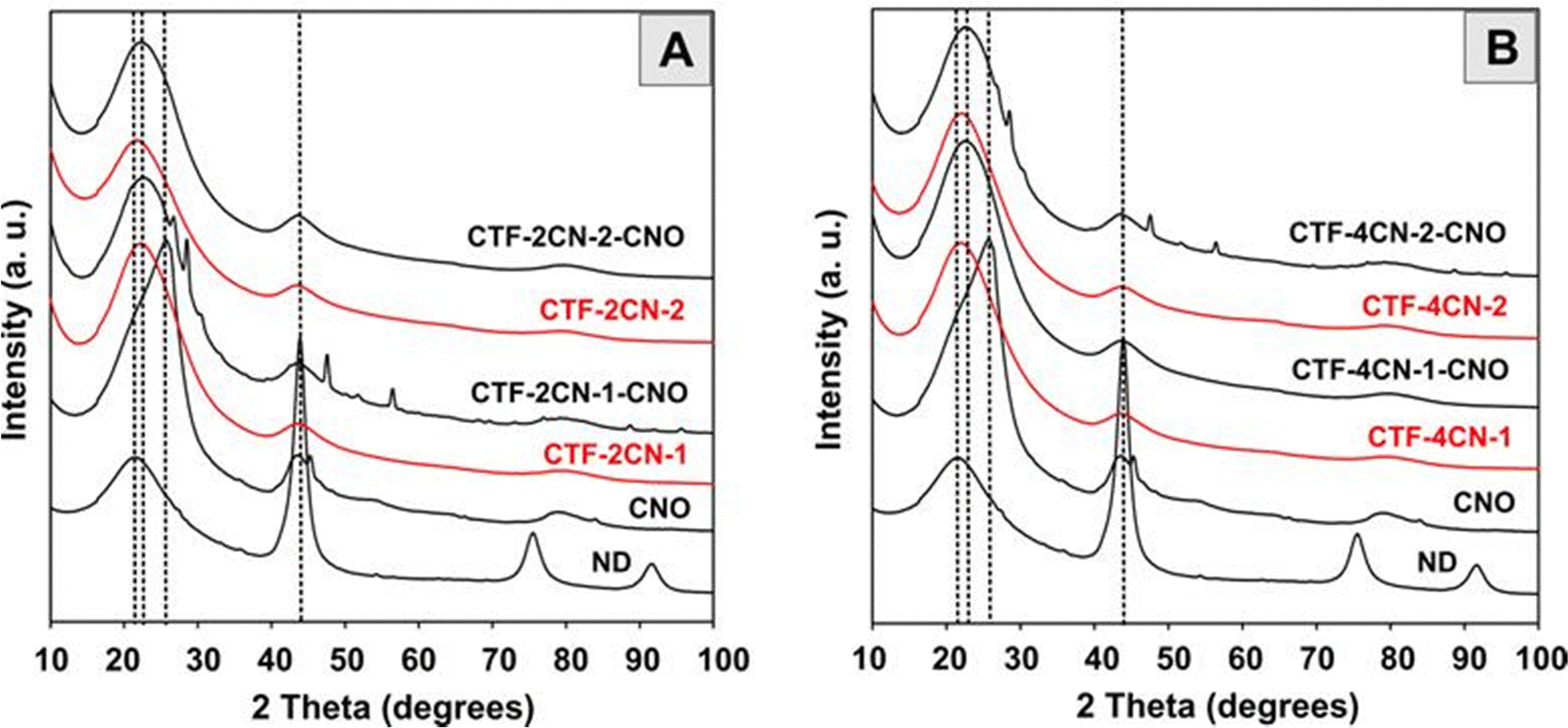 | ||
| Fig. 5 XRD studies of (A) and (B) NDs and CNOs; (A) CTF-2CN-1, CTF-2CN-1-CNO, CTF-2CN-2, and CTF-2CN-2-CNO; and (B) CTF-4CN-1, CTF-4CN-1-CNO, CTF-4CN-2 and CTF-4CN-2-CNO. | ||
Some other reflections, which were sharp with low intensity, were observed for CTF-2CN-1-CNO and CTF-4CN-2-CNO. The peaks at 27.6° and 28.9° were attributed to a layered structural motif, stacked chains of s-triazine and tri-s-triazine units,65–67 which can be attributed to a pseudo-(002) stacking motif of the graphitic carbon nitride (g-C3N4) domains, similar to the turbostratic phase of graphite.68 The interlayer stacking reflection is the characteristic of conjugated aromatic structures – polymerized triazine- and tri-s-triazine-based g-C3N4.69,70 This arrangement is so-called triazine-based g-C3N4.69,71 These motifs can be formed under several conditions, including the appropriate number of N atoms relative to the number of C atoms, the pyrolysis temperature, the pore structure, the planarity of the triazine ring, etc. The other two signals for CTF-2CN-1-CNO and CTF-4CN-2-CNO were the observable characteristics of the graphitic structure. These results reveal the (101) and (004) reflections at 47.4° and 56.29° (CTF-2CN-1-CNO) and 56.09° (CTF-4CN-2-CNO) and the characteristic of g-C3N4.72 These microstructures suggest the presence of highly graphitized domains in the materials.
The CTF and CTF–CNO materials decompose almost totally in an air atmosphere, showing one significant weight loss between 430 and 700 °C with the maximum degradation at 540–610 °C (Fig. S10, ESI†). The second, much smaller degradation of up to 10% of the materials occurs up to 250 °C, with a maximum at about 140 °C. The more significant weight loss is observed for nanocomposites containing CTF-4CN-1 and CTF-4CN-2, e.g., systems containing more heterocyclic rings in the skeleton. It should be emphasized that this stage is observed in both pristine CTFs and nanocomposites and has an analogous course. This proves that the CTF and CNO after the pyrolysis process at high temperatures indicate a high chemical and structural similarity of the obtained carbonaceous materials; the CTF is subject to the graphitization process. Both the CTF and the CNO are made of pentagonal and hexagonal carbon rings. Generally, pyrolyzed materials show high thermal stability up to 600 °C, and their decomposition profiles are similar, degrading continuously over the entire temperature range applied. The CTF and CTF–CNO materials pyrolyzed at high temperatures have a high degree of structural organization and graphitization, resulting in increased thermal stability.
2.3. Textural and electrochemical characteristics of CTF and CTF–CNO
Low-temperature nitrogen adsorption–desorption isotherms were measured at −196 °C. Using Brunauer–Emmett–Teller (BET) theory, the surface area (SBET) was calculated via the approach proposed by Rouquerol et al.73 The pore size distribution (PSD) was calculated by nonlocal density functional theory (DFT) for slit-like carbon nanopores.74–76 Considering the mechanism of these processes, the differential PSDs were analyzed (Fig. 6 and Fig. S7, ESI†), and cumulative nanopore volume distributions (Fig. S7, ESI†) and the micropore (Vmicro) and mesopore (Vmeso) values were calculated (Table 1).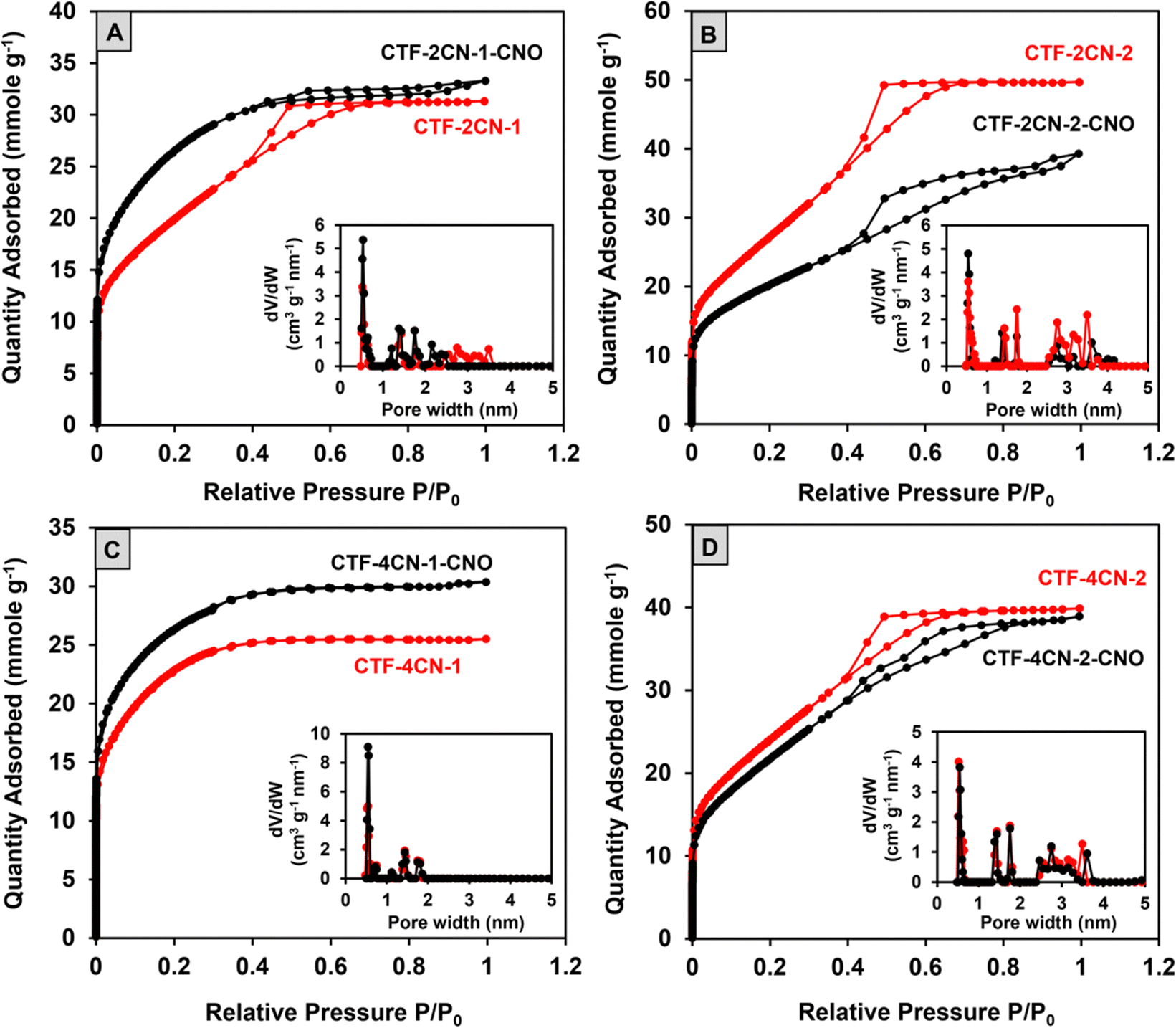 | ||
| Fig. 6 N2 adsorption–desorption isotherms and differential pore size distributions (inset) of the analyzed materials: (A) CTF-2CN-1 and CTF-2CN-1-CNO; (B) CTF-2CN-2 and CTF-2CN-2-CNO; (C) CTF-4CN-1 and CTF-4CN-1-CNO; and (D) CTF-4CN-2 and CTF-4CN-2-CNO. | ||
| Material | S BET (m2 g−1) | V micro (cm3 g−1) | V meso (cm3 g−1) | V total (cm3 g−1) |
|---|---|---|---|---|
| CTF-2CN-1 | 1612 | 0.55 | 0.52 | 1.10 |
| CTF-2CN-1-CNO | 2143 | 0.88 | 0.22 | 1.10 |
| CTF-2CN-2 | 2260 | 0.62 | 1.09 | 1.71 |
| CTF-2CN-2-CNO | 1625 | 0.53 | 0.82 | 1.35 |
| CTF-4CN-1 | 1835 | 0.82 | 0 | 0.82 |
| CTF-4CN-1-CNO | 2126 | 0.96 | 0 | 0.96 |
| CTF-4CN-2 | 1962 | 0.63 | 0.73 | 1.36 |
| CTF-4CN-2-CNO | 1786 | 0.55 | 0.78 | 1.33 |
Most of the nitrogen adsorption–desorption curves shown in Fig. 6 (CTF-2CN-1, CTF-2CN-2, and CTF-4CN-2) are type IVa isotherms according to the IUPAC classification and the characteristic of materials with a predominance of mesopores. The exception is the type I isotherm recorded for CTF-4CN-1 (Fig. 6C), which suggests the microporous nature of this sample. The addition of CNOs impacted the CTF material porosity, and the observed effect strongly depended on the synthesized systems' structural geometry. Namely, the diameter of the CTF ring (see Fig. 1) determined the total pore volume, including the share of micro- and mesopores and SBET (see Table 1). There is an ongoing debate over applying the BET evaluation to evaluate the specific surface area of materials with a relevant proportion of micropores and small mesopores.77,78 Despite these doubts, we decided to use this model because it allows us to compare our results with the literature data (Table S6, ESI†), and the obtained surface area is in fact apparent. In the case of CTF-2CN-1-CNO and CTF-4CN-1-CNO materials containing smaller CTF rings in their structures (Fig. 1), the introduction of CNOs increased SBET (Table 1).
In contrast, the CTF-2CN-2-CNO and CTF-4CN-2-CNO materials, with a larger diameter of the CTF rings, exhibited lower SBET values than the corresponding CTFs without CNOs (Fig. 1 and Table 1). In addition, it should be emphasized that the increase in porosity observed for the CTF-2CN-1-CNO and CTF-4CN-1-CNO systems originated via a different mechanism because of the diverse nature of the pristine CTF, namely, the nanoporosity (micro- and mesoporosity with pores having diameters smaller than ca. 100 nm). For the CTF-2CN-1-CNO system, the appearance of CNOs led to a reduction in the Vmeso value, indicating a transformation of mesopores into micropores (Table 1, Fig. 6A inset and Fig. S6, ESI†). For the remaining systems in which the decrease in the nanopore volume occurred after CNO addition; for example, in the CTF-4CN-1-CNO system, the result was mainly caused by the decline in the Vmicro and Vmeso values (Table 1, Fig. 6C inset and Fig. S6, ESI†).
The nanopore diameters of all the studied systems were very similar. Namely, three groups of small ultramicropores (width smaller than 0.7 nm) with diameters of approximately 0.5 nm, and supermicropores with diameters of 1.4 nm and 1.8 nm were observed. Except for two microporous systems, the mesopore diameters were about 2.75 nm and 3.5 nm. The only exception was CTF-2CN-2-CNO, where mesopores of approximately 3.5 nm were not observed in the structure.
The cyclic voltammetry (CV) curves of all the synthesized CTF and CTF–CNO materials (Fig. 7A–D) indicated the capacitive nature of these materials, which is promising in the context of their potential use in SCs. On the other hand, the observed deviations from the shape of a perfect rectangle suggest the poor electrical conductivity of the tested material and the pseudocapacitance contribution in the electrode processes.
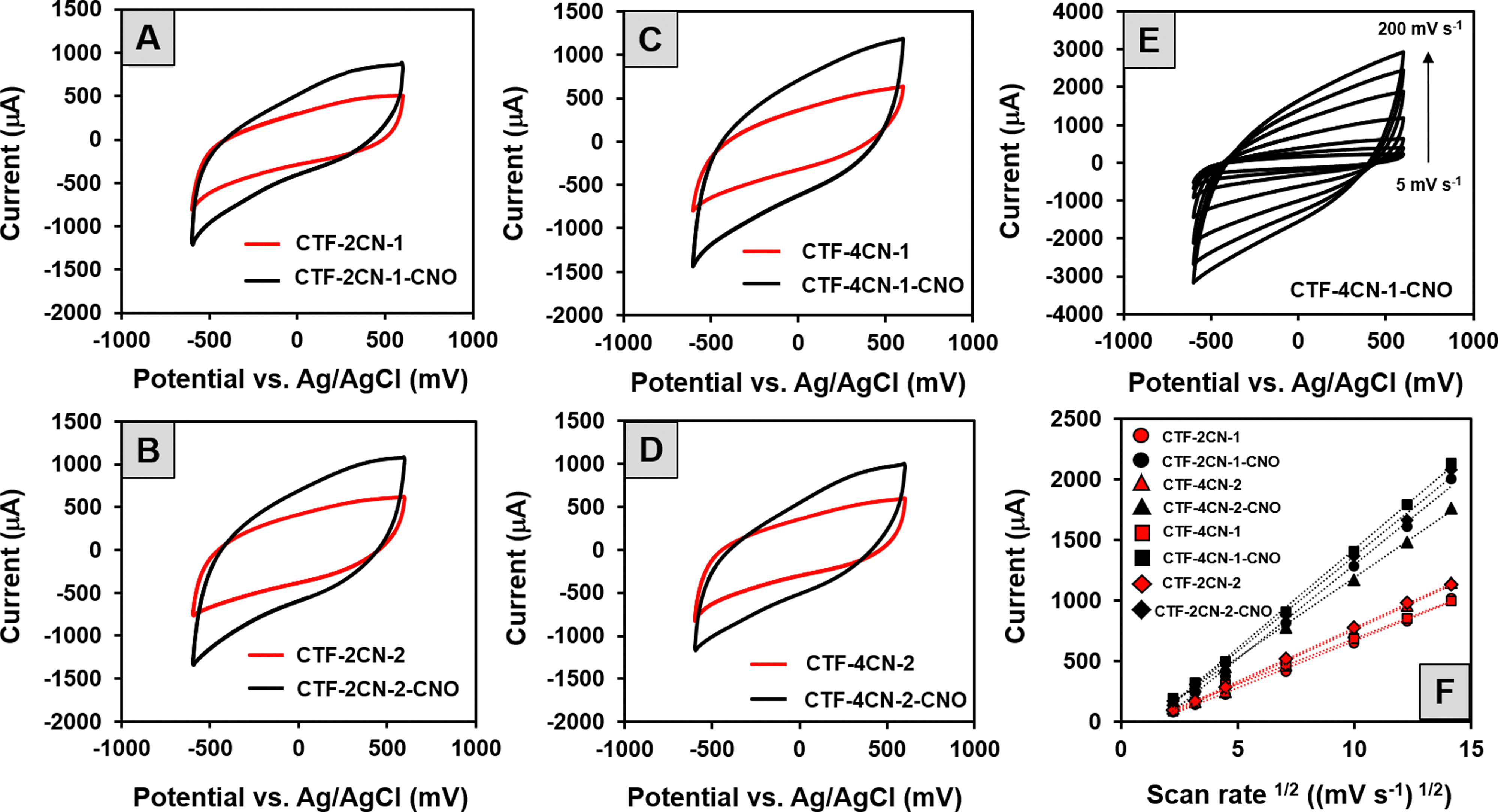 | ||
| Fig. 7 (A)–(D) CVs of the GCE modified with different CTF and CTF–CNO materials recorded in a 1 M H2SO4 solution at a scan rate of 50 mV s−1. (E) CVs of the GCE modified with CTF-4CN-1-CNO materials recorded at different scan rates (from 5 to 200 mV s−1). (F) Dependence of current on the square root of the scan rate at 0.2 V. | ||
The last one is most likely due to the presence of N atoms in the skeleton of the synthesized CTF. Specifically, the CV recorded for the CTF and CTF–CNO in 1 M H2SO4 solution exhibited deviations from a rectangular shape due to the protonation/deprotonation of N atoms with electron lone pairs exposed to an acidic solution.79 It should also be emphasized that the addition of CNOs caused a significant increase in the electrochemical capacitance values of the composites compared to those of the corresponding pristine CTFs. The highest specific capacitances (Cs) calculated from CV measurements are summarized in Table 2, with values of 366 F g−1 (CTF-2CN-1-CNO), 393 F g−1 (CTF-4CN-2-CNO), 486 F g−1 (CTF-2CN-2-CNO) and 495 F g−1 (CTF-4CN-1-CNO). The influence of the scan rate on the current value, as recorded on the electrodes modified with the obtained materials, was also investigated. The CV recorded for the selected CTF-4CN-1-CNO material is shown in Fig. 7E. The analysis showed a linear relationship between the current recorded at 0.2 V vs. Ag/AgCl and the square root of the scan rate (Fig. 7F), which proves that the observed process was diffusion-controlled.
| Material | C S calculated from CV (F g−1) | C S calculated from GCD (F g−1) | E S (W h kg−1) | P S (W kg−1) |
|---|---|---|---|---|
| CTF-2CN-1 | 204 ± 10 | 81 ± 5 (8 A g−1) | 2 | 1712 |
| CTF-2CN-1-CNO | 366 ± 10 | 137 ± 8 (8 A g−1) | 6 | 1996 |
| CTF-2CN-2 | 294 ± 15 | 133 ± 5 (8 A g−1) | 5 | 1842 |
| CTF-2CN-2-CNO | 486 ± 12 | 205 ± 10 (8 A g−1) | 8 | 2007 |
| CTF-4CN-1 | 250 ± 12 | 114 ± 10 (8 A g−1) | 3 | 1674 |
| CTF-4CN-1-CNO | 495 ± 15 | 303 ± 6 (2 A g−1) | 13 | 522 |
| 278 ± 12 (4 A g−1) | 12 | 1033 | ||
| 258 ± 12 (6 A g−1) | 10 | 1500 | ||
| 237 ± 11 (8 A g−1) | 9 | 1970 | ||
| CTF-4CN-2 | 255 ± 10 | 123 ± 10 (8 A g−1) | 4 | 1771 |
| CTF-4CN-2-CNO | 393 ± 10 | 171 ± 5 (8 A g−1) | 6 | 1933 |
Further investigation of the mechanism of the electrode process was performed using electrochemical impedance spectroscopy (EIS). The Nyquist plots obtained for CTF and CTF–CNO materials differed in the spectrum range (Fig. S9, ESI†). The loop corresponding to the contact resistance between the electrode and the porous modifier layer, observed at high frequencies, was more significant in the EIS spectra recorded for pristine CTFs (Fig. S9A, ESI†). In turn, at low frequencies, the behavior of electrodes modified with CTF–CNO materials was strongly controlled by diffusion. It is evidenced by the slope of the linear part of the Nyquist plot close to 45 degrees (Fig. S9B, ESI†). The impedance spectra of CTF-4CN-1-CNO recorded at different potentials (Fig. S9C, ESI†) confirm that this material can be used in the potential window from −0.6 to +0.6 V. The electrical equivalent circuit proposed for the obtained electrodes consists of a constant phase element (CPE) representing the capacitance, solution resistance (RS), charge-transfer resistance (Rct), and the Warburg impedance (ZW) representing the transport of counter-ions through the layer during its charging (Fig. S9C inset, ESI†).
To confirm the electrochemical properties of the materials, they were also subjected to galvanostatic charge–discharge (GCD) tests with two-electrode configurations (Fig. 8A and B). In a wide positive potential window (0 to +0.6 V vs. Ag/AgCl), the GCD cycles showed a triangular shape with a slight deviation from a perfect profile observed in the part of the curve corresponding to the discharge process. The conclusions drawn from the GCD tests are similar to those obtained by the CV analysis. Namely, the studied materials showed a capacitive nature, but the course of the charging and discharging processes also indicated the participation of some pseudocapacitive currents.
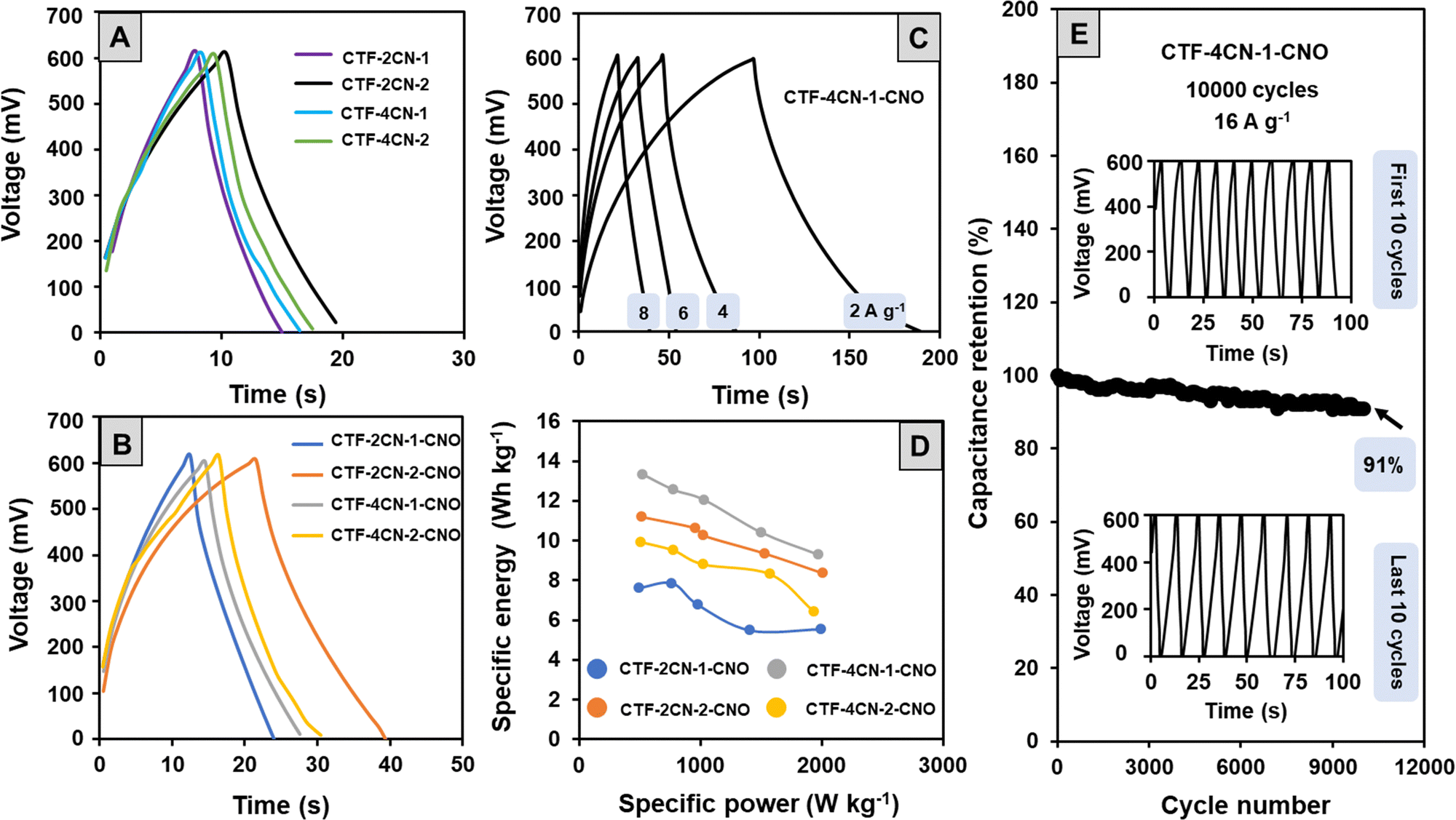 | ||
| Fig. 8 (A) and (B) GCD curves of different materials recorded in 1 M H2SO4 at a specific current of 8 A g−1. (C) GCD curves of CTF-4CN-1-CNO materials at different specific currents. (D) Ragone plots of the specific energy and power of CTF–CNO materials. (E) Capacitance retention as a function of cycle number for CTF-4CN-1-CNO materials at a specific current of 16 A g−1 (inset: first and last 10 GCD cycles). | ||
The CS values determined from the GCD measurements at a current density of 8 A g−1 were lower than the CS values calculated from the CV curves (Table 2). This discrepancy results from relatively high current density and using a two-electrode configuration in the GCD electrochemical studies (CV measurements were carried out using a three-electrode system). The CS values were also calculated for CTF-4CN-1-CNO at different current densities (Fig. 8C). The CS value was 303 F g−1 at 2 A g−1, 66% of this value (237 F g−1). It indicates that the interconnected micropore structure provides an efficient pathway for the electrolyte ion movement through the porous carbon materials.
Analysis of the GCD cycles recorded for the CTF–CNO materials at different current densities, an example of which is shown in Fig. 8C, also allowed us to determine the values of energy density (Edensity) and power density (Pdensity) (Table 2). The high active surface area of the porous electrodes increased Edensity, while the charge storage mechanism of EDL led to a high Pdensity.79 The relationship between these parameters is presented in Ragone plots (Fig. 8D). High values of Edensity indicate the excellent prospects of using the obtained structures to form SCs. For the material showing the best electrochemical properties (CTF-4CN-1-CNO), we also tested the capacitive retention after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at a current density of 16 A g−1 (Fig. 8E). The capacitive retention was maintained at 91% of its initial value (Fig. 8E), and the shape of the recorded curves remained unchanged.
000 cycles at a current density of 16 A g−1 (Fig. 8E). The capacitive retention was maintained at 91% of its initial value (Fig. 8E), and the shape of the recorded curves remained unchanged.
The CS values calculated for the obtained materials in an aqueous acidic solution were much higher than those typically reported for carbon-based SCs (50–200 F g−1).80 In theESI,† we also showed a summary of the specific capacitance for other triazine systems in correlation with the materials obtained by our group (Table S6, ESI†). The CS values of CTF and composite materials (CNO–CTF) are in the upper limit of the specific capacitance value, and a value of 303 (2 A g−1) recorded in the two-electrode system for CTF-4CN-1-CNO is the highest value recorded in the literature. The CS values usually correlate with the specific surface area ranging from approximately 500 m2 g−1 to 1000–2000 m2 g−1. For our materials, with SBET values ranging from 1800 to 2200 m2 g−1, we achieved higher CS values within the 200–500 F g−1 range for a three-electrode system and up to 300 F g−1 for a two-electrode system. The highest CS value was obtained for the CTF-4CN-1-CNO material, for which SBET was 2126 m2 g−1, indicating the substantial contribution of microporosity.
3. Experimental
3.1. Materials
4-Cyanobenzaldehyde (98%), 4-cyanoaniline (98%), 2-amino-6-methylbenzothiazole (98%), 1,4-dicyanobenzene (98%), 7,7,8,8-tetracyanoquinodimethane (98%), iron(III) perchlorate hydrate Fe(ClO4)3·H2O (crystalline), diacetyl (pure), and anhydrous zinc chloride (98%) were purchased from Sigma-Aldrich, USA. Glacial acetic acid (99.5–99.9%), methanol (99.8%), THF (99.5%), ethanol (96%), and concentrated hydrochloric acid (HCl) were purchased from Avantor Performance Materials Poland S.A. Toluene (99.8%), acetonitrile (99.8%), and dichlorobenzene (99%) were purchased from Sigma-Aldrich, USA. All reagents and solvents were used as received. Nanodiamond (ND, powder, 97 wt%, Carbodeon Diamond®Molto) with a crystal size of 4.2 ± 0.5 nm was used for the preparation of CNOs (annealing treatment under an inert atmosphere and reduced pressure of ultradispersed ND particles).81The CTFs were labeled according to the number of nitrile groups (-CN). The CTFs with two –CN groups were denoted as 2CN-X (1,4-dicyanobenzene and 1,4-bis(6-methylbenzo[d]thiazol-2-yl)-2,5-bis(4-cyanophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole), and those with four –CN groups were denoted as 4CN-X (7,7,8,8-tetracyanoquinodimethane and 1,2,4,5-tetrakis(4-cyanophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole), where X is 1 for known materials and 2 for new materials (pyrrolopyrrole-based).
All CTFs were synthesized by grinding the monomer with ZnCl2 under inert conditions and placing the mixture in a quartz test tube. A significant excess of ZnCl2 (5 weight, 5–23 molar equivalents, depending on the monomer's molar mass) was necessary to achieve complete dissolution. A Carbolite Gero STF 16/180 tube furnace was used for performing pyrolysis (with 3216 Controller).
3.2. Synthetic procedures
2CN-2. 1,4-bis(6-methylbenzo[d]thiazole-2-yl)-2,5-bis(4-cyanophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole. General procedure A was followed with 2-amino-6-methylbenzothiazole (1312 mg, 8 mmol). After the reaction, the solvents were evaporated, and the product was purified by crystallization from acetonitrile. After drying under vacuum, a yellow solid was obtained with a yield of 22% (1053 mg). 1H NMR (500 MHz, CDCl3) δ: 9.27 (s, 2H), 8.25 (d, J = 8.3 Hz, 4H), 8.05 (d, J = 8.3 Hz, 4H), 7.87 (m, 4H), 7.37 (d, J = 8.3 Hz, 2H), 2.46 (s, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ = 170.0, 166.0, 149.8, 138.8, 136.0, 134.9, 133.5, 130.8, 128.8, 123.0, 122.5, 118.8, 115.3, 21.6 ppm; HRMS (TOF AP+): m/z calcd for C36H23N6S2: 603.1426 [M + H]+; found: 603.1434.
4CN-2. 1,2,4,5-tetrakis(4-cyanophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole. General procedure A was followed with 4-cyanoaniline (944 mg, 8 mmol). After the reaction, the precipitate was filtered off and washed with water and acetonitrile. The product was purified by crystallization from acetonitrile. After drying under vacuum, a yellow solid was obtained with a yield of 39% (792 mg). The 1H NMR and 13C NMR spectra were consistent with those previously reported.37 HRMS (ESI+): m/z calcd for C34H18N6: 501.1593 [M]+; found: 501.1596.
CTF-2CN-1 . General procedure B was followed with dicyanobenzene (2CN-1, 200 mg, 1.56 mmol) and ZnCl2 (1062 mg, 7.81 mmol, 5 eq.) to afford 154 mg of the product (77% yield) after purification and drying.
CTF-4CN-1 . General procedure B was followed with 7,7,8,8-tetracyanoquinodimethane (4CN-1, 200 mg, 0.98 mmol) and ZnCl2 (1067 mg, 7.84 mmol, 8 eq.) to afford 196 mg of the product (98% yield) after purification and drying.
CTF-2CN-2 . General procedure B was followed with 1,4-bis(6-methylbenzo[d]thiazole-2-yl)-2,5-bis(4-cyanophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole (2CN-2, 200 mg, 0.33 mmol) and ZnCl2 (1032 mg, 7.59 mmol, 23 eq.) to afford 182 mg of the product (91% yield) after purification and drying.
CTF-4CN-2 . General procedure B was followed with 1,2,4,5-tetrakis(4-cyanophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole (4CN-2, 200 mg, 0.39 mmol) and ZnCl2 (1066 mg, 7.84 mmol, 20 eq.) to afford 190 mg of the product (95% yield) after purification and drying.
CTF-2CN-1-CNO . General procedure C was followed with dicyanobenzene (2CN-1, 100 mg, 0.78 mmol), CNO-CN (5 mg), and ZnCl2 (531 mg, 3.91 mmol, 5 eq.) to afford 72 mg of the product (69% yield) after purification and drying.
CTF-4CN-1-CNO . General procedure C was followed with 7,7,8,8-tetracyanoquinodimethane (4CN-1, 100 mg, 0.49 mmol), CNO-CN (5 mg), and ZnCl2 (533 mg, 3.92 mmol, 8 eq.) to afford 100 mg of the product (95% yield) after purification and drying.
CTF-2CN-2-CNO . General procedure C was followed with 1,4-bis(6-methylbenzo[d]thiazole-2-yl)-2,5-bis(4-cyanophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole (2CN-2, 100 mg, 0.17 mmol), CNO-CN (5 mg), and ZnCl2 (520 mg, 3.82 mmol, 23 eq.) to afford 90 mg of the product (86% yield) after purification and drying.
CTF-4CN-2-CNO . General procedure C was followed with 1,2,4,5-tetrakis(4-cyanophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole (4CN-2, 100 mg, 0.2 mmol), CNO-CN (5 mg) and ZnCl2 (533 mg, 3.92 mmol, 20 eq.) to afford 95 mg of the product (90% yield) after purification and drying.
4. Conclusions
In conclusion, we successfully synthesized pristine CTFs using 1,4-bis(6-methylbenzo[d]thiazol-2-yl)-2,5-bis(4-cyanophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole (2CN-2) and 1,2,4,5-tetrakis(4-cyanophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole (4CN-2) forming macrostructures with a repeating structural organization. This synthesized CTF was organized in a 3D manner on the CNO surface, creating CTF–CNO systems with the organized hierarchical porosity. This was achieved by adding only 5% by the weight of the CNO. A high porosity, about 2000 m2 g−1, and interconnections between micropores and mesopores significantly increased the electrochemical performance of the obtained composites. The CS value was increased 1.5–2 times compared to the reference CTF value, reaching 495 F g−1 in the three-electrode system in 1 M H2SO4 (CTF-4CN-1-CNO). Using the GCD method in a two-electrode system, the CS value of the same system was 303 (2 A g−1), reaching the highest value from the literature data for triazine systems. For the material showing the best electrochemical properties (CTF-4CN-1-CNO), we also tested the capacitive retention at a current density of 16 A g−1 that was at 91% of the initial value after 10000 cycles, and the shape of the recorded curves remained unchanged.
Apart from high porosity, several factors may be responsible for such a significant increase in electrochemical performance. The presence of highly graphitized domains characterized the CNO–CTF materials we obtained, the interconnected pores preventing the walls of the pores from pulverizing while exhibiting an extremely high level of structural integrity, morphological heterogeneity resulting from the simultaneous presence of amorphous phases and crystalline zones that may result in higher structural stability and higher conductivity of the hybrid materials, a high percentage of the pyridinic-N, pyrrolic-N and graphitic-N atoms, which are responsible for the pseudocapacitance in materials. The efficient combination of all these factors made composites excellent electrode materials. Due to these features, the CTF–CNO materials showed attractive properties for practical applications in electrochemistry.
Author contributions
Conceptualization, methodology, supervision, project administration, visualization, data curation and funding acquisition: M. E. P.-B. methodology, visualization, validation, formal analysis and investigation: A. H., G. S., J. B., K. B., A. I. and A. P. T. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 4th December 2023, which contained errors in Fig. 7 and 8.Acknowledgements
We gratefully acknowledge the financial support from the National Science Centre, Poland, grants #2017/25/B/ST5/01414 and #2019/35/B/ST5/00572 to M. E. P.-B. We gratefully acknowledge Prof. Luis Echegoyen from the University of Texas at El Paso for providing the CNOs. The research was carried out with the equipment purchased thanks to the financial support of the European Regional Development Fund (POIG.02.01.00-06024/09, Maria Curie-Sklodowska University, Poland).References
- M. E. Plonska-Brzezinska and L. Echegoyen, J. Mater. Chem. A, 2013, 1, 13703 RSC.
- R. Kumar, E. Joanni, S. Sahoo, J.-J. Shim, W. K. Tan, A. Matsuda and R. K. Singh, Carbon, 2022, 193, 298–338 CrossRef CAS.
- Y. Soneda, Handbook of Advanced Ceramics, Elsevier, 2013, pp. 211–222 Search PubMed.
- H. Aghajani, A. T. Tabrizi, R. Ghorbani, S. Behrangi, M. Stupavska and N. Abdian, J. Alloys Compd., 2022, 906, 164284 CrossRef CAS.
- A. Hryniewicka, J. Breczko, G. Siemiaszko, A. N. Papathanassiou, K. Góra-Marek, K. A. Tarach, K. Brzezinski, A. Ilnicka, A. P. Terzyk, K. H. Markiewicz, L. Echegoyen and M. E. Plonska-Brzezinska, Sci. Rep., 2023, 13, 10737 CrossRef CAS.
- A. Hryniewicka, J. Breczko, G. Siemiaszko, K. Brzezinski, A. Ilnicka, A. Terzyk and M. Plonska-Brzezinska, ChemRXiv, 2022, preprint DOI:10.26434/chemrxiv-2022-qpgjj.
- M. Zhong, M. Zhang and X. Li, Carbon Energy, 2022, 4, 950–985 CrossRef CAS.
- G. Siemiaszko, J. Breczko, A. Hryniewicka, A. Ilnicka, K. H. Markiewicz, A. P. Terzyk and M. E. Plonska-Brzezinska, Sci. Rep., 2023, 13, 6606 CrossRef CAS.
- G. Siemiaszko, A. Hryniewicka, J. Breczko, O. F. Delgado, K. H. Markiewicz, L. Echegoyen and M. E. Plonska-Brzezinska, ACS Appl. Polym. Mater., 2022, 4, 2442–2458 CrossRef CAS PubMed.
- G. Siemiaszko, A. Hryniewicka, J. Breczko, K. Brzezinski and M. E. Plonska-Brzezinska, Chem. Commun., 2022, 58, 6829–6832 RSC.
- M. E. Plonska-Brzezinska, ChemNanoMat, 2019, 5, 568–580 CrossRef CAS.
- L. Zhou, C. Gao, D. Zhu, W. Xu, F. F. Chen, A. Palkar, L. Echegoyen and E. S.-W. Kong, Chem. – Eur. J., 2009, 15, 1389–1396 CrossRef CAS.
- O. Mykhailiv, M. Imierska, M. Petelczyc, L. Echegoyen and M. E. Plonska-Brzezinska, Chem. – Eur. J., 2015, 21, 5783–5793 CrossRef CAS PubMed.
- S. Sek, J. Breczko, M. E. Plonska-Brzezinska, A. Z. Wilczewska and L. Echegoyen, ChemPhysChem, 2013, 14, 96–100 CrossRef CAS.
- M. Bartkowski and S. Giordani, Nanoscale, 2020, 12, 9352–9358 RSC.
- M. E. Plonska-Brzezinska, A. Molina-Ontoria and L. Echegoyen, Carbon, 2014, 67, 304–317 CrossRef CAS.
- J. K. McDonough, A. I. Frolov, V. Presser, J. Niu, C. H. Miller, T. Ubieto, M. V. Fedorov and Y. Gogotsi, Carbon, 2012, 50, 3298–3309 CrossRef CAS.
- F.-D. Han, B. Yao and Y.-J. Bai, J. Phys. Chem. C, 2011, 115, 8923–8927 CrossRef CAS.
- N. Keller, N. I. Maksimova, V. V. Roddatis, M. Schur, G. Mestl, Y. V. Butenko, V. L. Kuznetsov and R. Schlögl, Angew. Chem., Int. Ed., 2002, 41, 1885–1888 CrossRef CAS.
- C. Portet, G. Yushin and Y. Gogotsi, Carbon, 2007, 45, 2511–2518 CrossRef CAS.
- C. Zhu, F. Xu, J. Chen, H. Min, H. Dong, L. Tong, K. Qasim, S. Li and L. Sun, J. Power Sources, 2016, 303, 159–167 CrossRef CAS.
- Y. Zhang and S. Jin, Polymers, 2019, 11, 31 CrossRef.
- L. Hao, B. Luo, X. Li, M. Jin, Y. Fang, Z. Tang, Y. Jia, M. Liang, A. Thomas, J. Yang and L. Zhi, Energy Environ. Sci., 2012, 5, 9747–9751 RSC.
- L. Hao, J. Ning, B. Luo, B. Wang, Y. Zhang, Z. Tang, J. Yang, A. Thomas and L. Zhi, J. Am. Chem. Soc., 2015, 137, 219–225 CrossRef CAS PubMed.
- Y. Su, Y. Liu, P. Liu, D. Wu, X. Zhuang, F. Zhang and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 1812–1816 CrossRef CAS PubMed.
- M. G. Mohamed, S. U. Sharma, N.-Y. Liu, T. H. Mansoure, M. M. Samy, S. V. Chaganti, Y.-L. Chang, J.-T. Lee and S.-W. Kuo, Int. J. Mol. Sci., 2022, 23, 3174 CrossRef CAS.
- M. G. Mohamed, A. F. M. EL-Mahdy, M. M. M. Ahmed and S. Kuo, ChemPlusChem, 2019, 84, 1767–1774 CrossRef CAS PubMed.
- M. G. Mohamed, A. F. M. EL-Mahdy, Y. Takashi and S.-W. Kuo, New J. Chem., 2020, 44, 8241–8253 RSC.
- M. M. Vadiyar, X. Liu and Z. Ye, ACS Appl. Mater. Interfaces, 2019, 11, 45805–45817 CrossRef CAS.
- Y. Li, S. Zheng, X. Liu, P. Li, L. Sun, R. Yang, S. Wang, Z.-S. Wu, X. Bao and W.-Q. Deng, Angew. Chem., Int. Ed., 2018, 57, 7992–7996 CrossRef CAS.
- Y. Zhang, B. Zhang, L. Chen, T. Wang, M. Di, F. Jiang, X. Xu and S. Qiao, J. Colloid Interface Sci., 2022, 606, 1534–1542 CrossRef CAS PubMed.
- Y. Gao, C. Zhi, P. Cui, K. A. I. Zhang, L.-P. Lv and Y. Wang, Chem. Eng. J., 2020, 400, 125967 CrossRef CAS.
- C. Wu, H. Zhang, M. Hu, G. Shan, J. Gao, J. Liu, X. Zhou and J. Yang, Adv. Electron. Mater., 2020, 6, 2000253 CrossRef CAS.
- A. Hryniewicka, J. Breczko, G. Siemiaszko, A. Papathanassiou, K. Góra-Marek, K. A. Tarach, K. Brzezinski, A. Ilnicka, A. P. Terzyk, K. H. Markiewicz, L. Echegoyen and M. E. Plonska-Brzezinska, ChemRXiv, 2022, preprint DOI:10.26434/chemrxiv-2022-h0n3l.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- D. H. Friese, A. Mikhaylov, M. Krzeszewski, Y. M. Poronik, A. Rebane, K. Ruud and D. T. Gryko, Chem. – Eur. J., 2015, 21, 18364–18374 CrossRef CAS.
- M. Tasior, O. Vakuliuk, D. Koga, B. Koszarna, K. Górski, M. Grzybowski, L. Kielesinski, M. Krzeszewski and D. T. Gryko, J. Org. Chem., 2020, 85, 13529–13543 CrossRef CAS.
- N. A. Elessawy, J. El Nady, W. Wazeer and A. B. Kashyout, Sci. Rep., 2019, 9, 1129 CrossRef PubMed.
- C. Xia, X. Hai, X.-W. Chen and J.-H. Wang, Talanta, 2017, 168, 269–278 CrossRef CAS PubMed.
- N. A. Kumar, H. Nolan, N. McEvoy, E. Rezvani, R. L. Doyle, M. E. G. Lyons and G. S. Duesberg, J. Mater. Chem. A, 2013, 1, 4431–4435 RSC.
- A. Kellenberger, E. Dmitrieva and L. Dunsch, Phys. Chem. Chem. Phys., 2011, 13, 3411 RSC.
- C. M. Masemola, N. Moloto, Z. N. Tetana, S. S. Gqoba, P. K. Mubiayi and E. C. Linganiso, Mater. Chem. Phys., 2022, 287, 126229 CrossRef CAS.
- K. Wang, L.-M. Yang, X. Wang, L. Guo, G. Cheng, C. Zhang, S. Jin, B. Tan and A. Cooper, Angew. Chem., Int. Ed., 2017, 56, 14149–14153 CrossRef CAS.
- S. Mukherjee, M. Das, A. Manna, R. Krishna and S. Das, J. Mater. Chem. A, 2019, 7, 1055–1068 RSC.
- M. Koinuma, H. Tateishi, K. Hatakeyama, S. Miyamoto, C. Ogata, A. Funatsu, T. Taniguchi and Y. Matsumoto, Chem. Lett., 2013, 42, 924–926 CrossRef CAS.
- M. K. Rabchinskii, S. A. Ryzhkov, D. A. Kirilenko, N. V. Ulin, M. V. Baidakova, V. V. Shnitov, S. I. Pavlov, R. G. Chumakov, D. Y. Stolyarova, N. A. Besedina, A. V. Shvidchenko, D. V. Potorochin, F. Roth, D. A. Smirnov, M. V. Gudkov, M. Brzhezinskaya, O. I. Lebedev, V. P. Melnikov and P. N. Brunkov, Sci. Rep., 2020, 10, 6902 CrossRef CAS PubMed.
- Y.-C. Chiang, Y.-J. Chen and C.-Y. Wu, Materials, 2017, 10, 1296 CrossRef PubMed.
- A. Barinov, O. B. Malcioglu, S. Fabris, T. Sun, L. Gregoratti, M. Dalmiglio and M. Kiskinova, J. Phys. Chem. C, 2009, 113, 9009–9013 CrossRef CAS.
- D. Y. Osadchii, A. I. Olivos-Suarez, A. V. Bavykina and J. Gascon, Langmuir, 2017, 33, 14278–14285 CrossRef CAS PubMed.
- T. Wei, Q. Zhang, X. Wei, Y. Gao and H. Li, Sci. Rep., 2016, 6, 22646 CrossRef CAS PubMed.
- N. K. Gupta, B. Peng, G. L. Haller, E. E. Ember and J. A. Lercher, ACS Catal., 2016, 6, 5843–5855 CrossRef CAS.
- R. Arrigo, M. Hävecker, S. Wrabetz, R. Blume, M. Lerch, J. McGregor, E. P. J. Parrott, J. A. Zeitler, L. F. Gladden, A. Knop-Gericke, R. Schlögl and D. S. Su, J. Am. Chem. Soc., 2010, 132, 9616–9630 CrossRef CAS PubMed.
- S. Chen, X. Hai, C. Xia, X.-W. Chen and J.-H. Wang, Chem. – Eur. J., 2013, 19, 15918–15923 CrossRef CAS.
- Y. Yang, Z. Lu, J. Xia, Y. Liu, K. Wang and X. Wang, Chem. Eng. Sci., 2021, 229, 116054 CrossRef CAS.
- Z. Liu, X. Yuan, S. Zhang, J. Wang, Q. Huang, N. Yu, Y. Zhu, L. Fu, F. Wang, Y. Chen and Y. Wu, NPG Asia Mater., 2019, 11, 12 CrossRef CAS.
- G. Lui, G. Li, X. Wang, G. Jiang, E. Lin, M. Fowler, A. Yu and Z. Chen, Nano Energy, 2016, 24, 72–77 CrossRef CAS.
- D. McNulty, E. Carroll and C. O’Dwyer, Adv. Energy Mater., 2017, 7, 1602291 CrossRef.
- L. J. Fu, T. Zhang, Q. Cao, H. P. Zhang and Y. P. Wu, Electrochem. Commun., 2007, 9, 2140–2144 CrossRef CAS.
- S. Tomita, M. Fujii and S. Hayashi, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 245424 CrossRef.
- S. Tomita, A. Burian, J. C. Dore, D. LeBolloch, M. Fujii and S. Hayashi, Carbon, 2002, 40, 1469–1474 CrossRef CAS.
- A. E. Aleksenskii, M. V. Baidakova, A. Y. Vul’, V. Y. Davydov and Y. A. Pevtsova, Phys. Solid State, 1997, 39, 1007–1015 CrossRef.
- Q.-H. Yang, P.-X. Hou, M. Unno, S. Yamauchi, R. Saito and T. Kyotani, Nano Lett., 2005, 5, 2465–2469 CrossRef CAS PubMed.
- S.-W. Bian, Z. Ma and W.-G. Song, J. Phys. Chem. C, 2009, 113, 8668–8672 CrossRef CAS.
- A. Vinu, P. Srinivasu, D. P. Sawant, T. Mori, K. Ariga, J.-S. Chang, S.-H. Jhung, V. V. Balasubramanian and Y. K. Hwang, Chem. Mater., 2007, 19, 4367–4372 CrossRef CAS.
- T. Komatsu, Macromol. Chem. Phys., 2001, 202, 19–25 CrossRef CAS.
- B. V. Lotsch, M. Döblinger, J. Sehnert, L. Seyfarth, J. Senker, O. Oeckler and W. Schnick, Chem. – Eur. J., 2007, 13, 4969–4980 CrossRef CAS.
- A. Schwarzer, T. Saplinova and E. Kroke, Coord. Chem. Rev., 2013, 257, 2032–2062 CrossRef CAS.
- W. Ruland and B. Smarsly, J. Appl. Crystallogr., 2002, 35, 624–633 CrossRef CAS.
- F. Fina, S. K. Callear, G. M. Carins and J. T. S. Irvine, Chem. Mater., 2015, 27, 2612–2618 CrossRef CAS.
- D. M. Teter and R. J. Hemley, Science, 1996, 271, 53–55 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- M. Kim, S. Hwang and J.-S. Yu, J. Mater. Chem., 2007, 17, 1656–1659 RSC.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Studies in Surface Science and Catalysis, Elsevier, 2007, vol. 160, pp. 49–56 Search PubMed.
- C. Lastoskie, K. E. Gubbins and N. Quirke, J. Phys. Chem., 1993, 97, 4786–4796 CrossRef CAS.
- P. Tarazona, Mol. Phys., 1984, 52, 81–96 CrossRef CAS.
- J. P. Olivier, J. Porous Mater., 1995, 2, 9–17 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- B. Lobato, L. Suárez, L. Guardia and T. A. Centeno, Carbon, 2017, 122, 434–445 CrossRef CAS.
- M. Zhong, E. K. Kim, J. P. McGann, S.-E. Chun, J. F. Whitacre, M. Jaroniec, K. Matyjaszewski and T. Kowalewski, J. Am. Chem. Soc., 2012, 134, 14846–14857 CrossRef CAS PubMed.
- C. Liu, F. Li, L.-P. Ma and H.-M. Cheng, Adv. Mater., 2010, 22, E28–E62 CAS.
- V. L. Kuznetsov and Yu. V. Butenko, in Synthesis, Properties and Applications of Ultrananocrystalline Diamond, ed. D. M. Gruen, O. A. Shenderova and A. Y. Vul’, Springer-Verlag, Berlin/Heidelberg, 2005, vol. 192, pp. 199–216 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma01003a |
| This journal is © The Royal Society of Chemistry 2024 |