
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering the solid-state luminescence of organic crystals and cocrystals†
Aijaz A.
Dar
 *a,
Shaista H.
Lone
a,
Ishtiyaq
Ahmad
a,
Aadil A.
Ahangar
a,
Arshid A.
Ganie
a and
Cherumannil
Femina
b
*a,
Shaista H.
Lone
a,
Ishtiyaq
Ahmad
a,
Aadil A.
Ahangar
a,
Arshid A.
Ganie
a and
Cherumannil
Femina
b
aCrystal Engineering Laboratory, Department of Chemistry, University of Kashmir, Hazratbal, Srinagar-190006, Jammu & Kashmir, India. E-mail: aijazku2015@gmail.com; daraijaz@uok.edu.in
bDepartment of Chemistry, Farook College P. O., Kozhikode 673632, Kerala, India
First published on 23rd November 2023
Abstract
Fine-tuning the solid-state emission of organic materials is a topic of immense commercial significance and academic interest. Of the various approaches employed to achieve emission-tuning, the co-crystal approach is less reported. In this study, we integrate the effect of functionalization and co-crystallization to investigate the optical properties. Three functionalized pyridyl-hydrazone molecules 1a (NO2), 2a (CN) and 3a (Br) have been synthesized and further utilized for cocrystal development with 5-sulfosalicylic acid (5-SSA-2H) to obtain organic salts 1–3, respectively. 1a–3a exhibit varied aggregation-induced emission (AIE) behavior, which is further tuned through co-crystallization. Emission quenching in 1a is attributed to long-range π–π stacking between the organic molecules while no π–π stacking interactions are observed in 2a (464 nm) and 3a (442 nm), resulting in their emissive behavior. Co-crystallization leads to more regulation of the emission wavelengths as 1a undergoes emission turn-on in the organic salt 1 (467 nm), and exhibits brownish luminescence; meanwhile, solid-state emission of 2a and 3a is moderately and significantly red-shifted in 2 (472 nm) and 3 (484 nm), respectively. The emission turn-on in 1 and red-shift in 2 is attributed to J-aggregate formation in their solid state and a significant red-shift of 3 is attributed to the formation of isolated head-to-tail dimers or excimers in the crystal lattice. The results are further supported with powder-X-ray diffraction, AIE and Hirshfeld studies.
Introduction
Organic solid-state emitters have been reported since the 1896 observations of Schmidt; however, this area has garnered attention since the concept of aggregation-induced emission (AIE) was floated by Tang.1,2 This phenomenon expands the application range of these materials as organic light-emitting diodes (OLEDs),3–5 organic field effect transistors (OLETs),6–9 organic photovoltaics (OPVs),10 anti-counterfeiting agents11 and fluorescent sensors.12–14 Organic AIE materials are cost-effective and provide the advantage of molecular design to achieve easy property modulations, good solution processability and facile post-usage disposal over their inorganic counterparts.15 The design and synthesis of novel organic luminescent crystalline materials can be realized through crystal engineering, which represents a greener and more economical alternative to organic synthesis. The resultant crystalline materials provide an opportunity of structure analyses and establishing of the structure-optical property relationship,16 and the photophysical parameters (emission wavelength, quantum yields and life-time) of these materials are a function of intermolecular contacts (hydrogen bonds, charge transfer, π–π interactions, etc.) and the nature of aggregation (H, J, X, I-aggregates).17Co-crystals are an important outcome of crystal engineering, and represent an emergent class of materials with a broad spectrum of applications and prospective opportunities as pharmaceuticals,18–20 devices,21–23 multiple-stimuli responsive materials,24–26 semiconductors,27 lasers,28 optoelectronics,29etc. Emission tuning in single component crystals can be achieved through functionalisation and polymorphism, both of these approaches are well reported. While functionalization involves electronic effects, polymorphism involves the packing effect on the optical properties.29–33 The co-crystal approach is a more facile process through which multi-component crystals can be prepared with better control over design and properties. Charge transfer co-crystals, based on π–π interactions, have been reported and investigated for their electronic and optical properties. Emission tuning in such cocrystals has been reported through coformer selection. As one of the first reports, Jones and co-workers have demonstrated that emission tuning in 1,4-bis-p-cyanostyrylbenzene was realised through co-crystallization with a range of suitable coformers.34 Since then, numerous charge transfer complexes have been developed and reported to exhibit emission tuning.35–42 Chopra and co-workers have demonstrated emission tuning in organic cocrystals utilizing the understanding of intermolecular interactions.43 Oxborrow's group has also utilized π-stacked interactions to develop MASER cocrystals.44 Similarly, arene-perfluorene interactions involving a perfluoro aromatic acceptor and a polyaromatic hydrocarbon do not undergo charge transfer interactions and have been reported to achieve blue-shifted emissions, as compared to red-shifted emissions in charge transfer complexes.45,46 Limited choice of acceptor molecules, however limits the scope of cocrystals developed based on π interactions.
Hydrogen bonding is another significant intermolecular interaction widely used for cocrystal and molecular salt formation, and hydrogen bonded cocrystals/salts with emission or emission tuning, however, are lesser reported. Draper et al. demonstrated the emission switching through hydrogen bonding tendencies of solvent molecules.47 Similarly, Yang and Gazit have demonstrated the use of hydrogen bonds to tune the emission.48 These systems, however, have been reported randomly and there are no established strategies to design emission in hydrogen bonded complexes. Recently we have reported emission tuning of the organic precursor by its cocrystallization, as the pyridyls of the same shape but the different positions of the nitrogen lead to different packing arrangements.49,50 However, advancement in the area remains limited and requires more vigorous endeavours.
In this study, we have attempted to determine the impact of substitution and cocrystallization on the AIE of new solid forms. Planar and linear electronegative substituents were attached to the hydrazone Schiff base of isoniazid to obtain 1a–3a, which were further modified by cocrystallization with 5-sulfosalicylic acid (5-SSA-2H) to synthesize 1–3, Scheme 1. Optical studies of 1a–3a and 1–3 have been performed in detail and the solid-state emission tunning in these materials due to substituent and packing effects has been explained with the help of diffraction and Hirshfeld studies. Furthermore, the AIE studies of both 1a–3a and 1–3 have been reported.
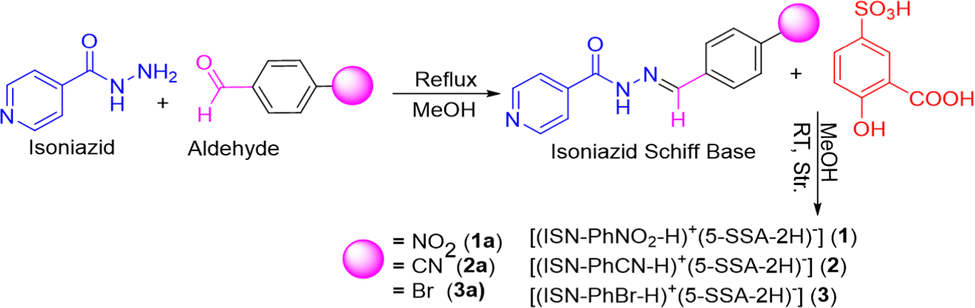 | ||
| Scheme 1 Schematic representation of the reaction pathway. | ||
Experimental section
Methods and materials
Isoniazid (99%, Sigma Aldrich), 4-nitrobenzaldehyde (99%, Sigma Aldrich), 4-formylbenzonitrite (99%, Sigma Aldrich), p-bromobenzaldehyde (97%, Sigma Aldrich) and 5-sulphosalicylic acid (5-SSA-3H) (99%, Sigma Aldrich) were purchased from Sigma Aldrich and used without further purification. The Schiff bases 1a–3a have been synthesized by refluxing the methanolic solution of isoniazid and aldehydes for 2–3 hours. Methanol (SD fine), DMF (SRL), and distilled water have been used as solvents for crystallization and/or for photo-physical studies. Melting points have been determined on the MP70 melting point system capillary apparatus (Mettler Toledo) in closed-end capillaries. Infrared spectroscopic data for molecular salts has been obtained using a 630 FT-IR (4000–650 cm−1) in ATR mode. Ground crystals of the products are placed on the crystal plate of the infrared instrument, to record the spectrum. Diffuse reflectance measurements of the products have been recorded on a Shimadzu-2600 spectrometer on BaSO4 discs and converted to Kubelka–Munk function and Tauc plots in origin using an appropriate conversion formula.51 The absorption studies have been carried out on a Shimadzu-2600 Spectrophotometer.SC-XRD studies
Single-crystal data were collected on a Rigaku Saturn CCD diffractometer using a graphite monochromator (Mo Kα, λ = 0.71073 Å). The selected crystals were mounted on the tip of a glass pin using mineral oil and placed in the cold flow produced with a Cryo-cooling device. Complete hemispheres of data were collected using ω and φ scans (0.3°, 16 s perframe). Integrated intensities were obtained with Rigaku Crystal Clear-SM Expert 2.1 software, and they were corrected for absorption correction. Structure solution and refinement were performed with the SHELX package. The structures were solved by direct methods and completed by iterative cycles of ΔF syntheses and full-matrix least-squares refinement against F. Crystal refinement parameters of 1a–3a are given in Table S1, ESI.†Fluorescence measurements
Solid state emission was recorded on a Shimadzu RF-5301PC Spectrofluorometer by making use of BaSO4 discs on which compounds 1a–3a, 1–3, and 5-SSA-2H were placed and emission was recorded at room temperature in normal mode by exciting them at 320 nm. Emission in the solution state was recorded on the same instrument by taking 10−3 M solution in the cuvettes and each was excited at 300 nm.Powder X-ray studies
Powder (PXRD) data of compounds 1–3 was recorded with a BRUKER-AXS-D8-ADVANCE diffractometer (Cu kα, λ = 1.5406) at room temperature with theta ranging from 10 to 40.Hirshfeld studies
The Hirshfeld analysis was carried out through CrystalExplorer 17.5 software. Color coding mapped on the dnorm surface represents the contacts that indicate short (red color), intermediate (white color), and long contacts (blue color) compared with the sum of van der Waals interactions. The details are provided in the ESI.† fileResults
To investigate the combined effect of the substituents and crystal packing on the properties, three hydrazone Schiff bases of isoniazid with nitro (1a), cyano (1b) and bromo (1c) substituents at the para-position of the aldehydic end were synthesized and utilized for the preparation of organic salts 1–3. The formation of the products is validated by Fourier-transform Infrared (FT-IR) spectroscopy. The characteristic absorptions in the spectrum of 1–3 at 1670–1680 cm−1 and 1640–1575 cm−1 correspond to the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN, respectively. The absorption bands at 1320–1170 cm−1 and 1150–1030 cm−1 correspond to the asymmetrical and symmetrical stretching of the sulfonic group, respectively, and the stretching frequency around 2872, 2887 and 2976 cm−1 in 1, 2 and 3, respectively, corresponds to the protonation of the pyridinium end of the Schiff bases (N+–H bond), indicating proton transfer or salt formation. Furthermore, the diagnostic peaks for different functional groups are observed at 1576 cm−1 and 1391 cm−1 for –NO2, 2219 cm−1 for –CN and 696 cm−1 for –Br in 1–3, respectively (Fig. S1–S3, ESI†).
O and CN, respectively. The absorption bands at 1320–1170 cm−1 and 1150–1030 cm−1 correspond to the asymmetrical and symmetrical stretching of the sulfonic group, respectively, and the stretching frequency around 2872, 2887 and 2976 cm−1 in 1, 2 and 3, respectively, corresponds to the protonation of the pyridinium end of the Schiff bases (N+–H bond), indicating proton transfer or salt formation. Furthermore, the diagnostic peaks for different functional groups are observed at 1576 cm−1 and 1391 cm−1 for –NO2, 2219 cm−1 for –CN and 696 cm−1 for –Br in 1–3, respectively (Fig. S1–S3, ESI†).
Thermal studies indicate significant stability of the products and the salts 1–3 form melts at temperatures greater than 230 °C. The Thermogravimetric analyses (TGA) and differential scanning analyses (DSA) of the products validate augmented thermal stability as they start to decompose after 230 °C and show endothermic dips in the DTA curves at 267, 281 and 266 °C for 1–3, respectively, which can be attributed to their melting points. Augmented thermal stabilities of these organic salts may be attributed to the ionic interactions between crystal components, due to proton transfer. The TGA and DSC curves of 1–3 are provided in Fig. 1a and b, and the early irregularity in the DSC curve of 1c plausibly arises due to the scattering of the product on heating. Powder X-ray diffraction studies of 1–3 have been carried out to understand the phase changes on the size reduction of the crystals. A comparison of the experimental and the simulated data confirms that there is no significant phase change on size reduction from single crystals to microcrystalline phase. Moreover, the appearance of sharp peaks in the powder X-ray diffraction indicates retention of the polycrystalline nature even on size reduction. Experimental and simulated powder X-ray data of product 1 are given in Fig. 1c and for 2 and 3 are provided in Fig. S4 and S5, ESI.†
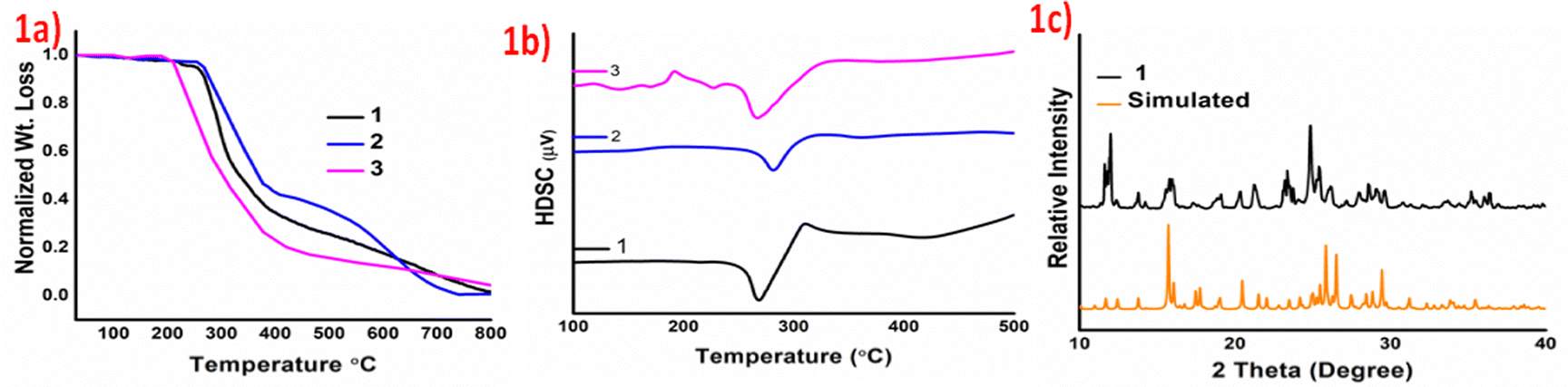 | ||
| Fig. 1 (a) TGA, (b) DSC of cocrystals 1–3, and (c) PXRD of cocrystal-1 with its simulated data. | ||
Optical properties
Solid-state luminescence of materials is an intriguing phenomenon with significant prospective consequences. Besides commercial implications, the process is of great academic interest. The organic salts 1–3, obtained through cocrystallization of 1a–3a with 5-SSA-2H, exhibit intriguing colour modulation on formation and the chromic changes are also accompanied by changes in the photo-luminescence. The optical images of 1–3 and their precursors 1a–3a, recorded under visible and long ultra-violet exposure, are provided in Fig. 2. The solid-state modulation of the photo-physical parameters in 1a–3a is a consequence of the functionalization and the Schiff base 1a with nitro substitution is non-emissive, while 2a and 3a exhibit blue and cyan emission, respectively. Co-crystallization of 1a–3a with blue emitter 5-SSA-2H, further modulates the luminescence of organic salts 1–3, which undergo red-shift in their emission. The results stipulate the integration of functionalization and crystal packing to achieve fine-tuned AIE in the products. | ||
| Fig. 2 (Left) Optical images of 1a–3a and 1–3 recorded under visible and UV exposure and crystal morphologies of 1–3, at 10× magnification and (Right) optical images of 10−3 M aqueous solutions of 1a–3a and 1–3, recorded under visible and UV exposure. | ||
To understand the role of intra and intermolecular charge transfer interactions, optical studies have been reported in both solution and the solid state. Solution phase studies of 1a–3a and 1–3, have been carried out for their 10−3 M aqueous solutions, obtained by heating the resultant solutions up to 80 °C until clear solutions were obtained (Fig. 3a). The solid-state studies have been reported by loading the samples on BaSO4 discs. The absorption bands corresponding to the intermolecular charge transfer interactions observed beyond 400 nm are absent in the solution phase spectra of these compounds, implying that the solution phase optical response in the materials arises from the independent molecules, which do not undergo intramolecular charge transfer. Meanwhile the products exhibit broad absorptions beyond 400 nm in the solid state, indicating aggregation-induced intermolecular charge transfer interactions (Fig. 3b). Interestingly, the aqueous solutions of the Schiff base precursors 1a–3a are weakly emissive, while the corresponding solutions of 1–3 exhibit blue emission with different intensities with the maximum emission centered at 420, 425 and 418 nm for 1–3, respectively (Fig. 3c). The solid-state luminescence spectra of these compounds exhibit intriguing modulations. The Schiff base 1a remains non-emissive, while as 2a (ϕ 0.10%) and 3a (ϕ 0.07%) emit in the blue region with emission maxima at 464 and 442 nm, respectively. Emission of 1a on crystallization is switched on in 1 (ϕ 0.14%) and the organic complex exhibits emission with maximum activity at 467 nm. The luminescence of 2 (ϕ 0.13%) and 3 (ϕ 0.12%) is red-shifted vis-à-vis their precursors 2a and 3a as well as their solution phase luminescence, with λmax values of 472 and 484 nm, respectively. The un-normalized solid-state emission spectra of 1a–3a and 1–3 are provided in Fig. 3d, and their optical images are given in Fig. 2. The KM plots, Tauc plots, and solid-state emission of 1a–3a, and 1–3 are provided in Fig. S6–S18, ESI.† Comparative solution and solid state emission parameters of the products are provided in Table S2, ESI.†
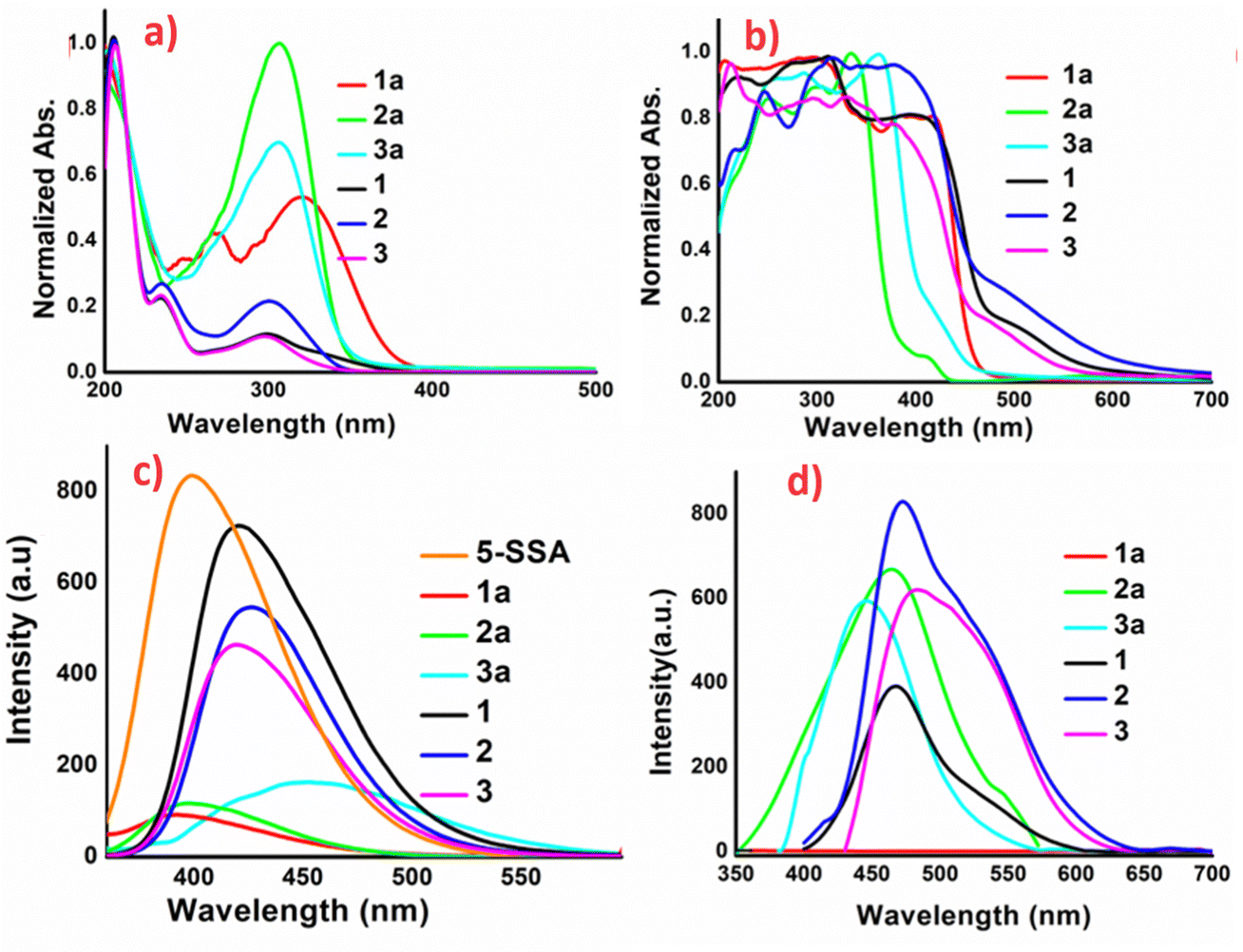 | ||
| Fig. 3 Photophysical studies: (a) solution phase absorption spectra of 10−3 M aqueous solutions, (b) solid phase diffuse-reflectance spectra reported as BaSO4 diluted discs, (c) solution phase emission spectra of 10−3 M aqueous solutions and (d) solid-state emission spectra reported as BaSO4 diluted discs, of 1a–3a and 1–3. | ||
Solution phase AIE studies, involving incremental addition of a bad solvent to the non-emissive solutions, have been carried out for 1a–3a and 1–3, to gain further insights into the emission behavior. The AIE studies have been carried out by dissolution of 1a–3a and 1–3 in DMF to prepare 10−3 M solutions, to obtain non-emissive solutions. Water was further added as a bad solvent to make them emissive; in this case, 1a–3a do not exhibit any significant emission turn-on at slit-width values from 3 to 10, while 1–3 exhibit gradual increases in the emission intensity up to a fw (mole fraction of water) value of 80% and a sharp augmentation in intensity at fw 90%. The λmax values for AIEgen solutions of 1–3 are nearly identical and located at 400 nm; however, the behavior is different vis-à-vis aggregation as 1 undergoes a gradual change in emission turn-on while 2 and 3 undergo abrupt emission turn on with the addition of water. As the AIEgen emission and solid-state emission of 1–3 are not the same, we believe that the AIEgen emission arises from the coformer 5-SSA-2H, which was also confirmed by the AIE studies of the organo-sulfonate. Quantum yields corresponding to the maximum emission values i.e., fw 90% for AIEgens 1–3 are 72.0%, 45.0%, and 45.2%, respectively. Dynamic light scattering (DLS) studies of these systems validate that the emission turn-on arises due to increase in the particle size. The AIE, DLS plots and optical images under daylight and UV light, of 1–3 are provided in Fig. S19–S25, ESI.†
Solid-state structure analyses
To understand the effect of substituents on the crystal packing and underlying reasons for the variable solid-state emission, single crystal structure analyses of 1–3 have been carried out and compared with previously reported crystal structures of 1a–3a.52 The diffraction quality crystals have been obtained directly from the reaction mixture through the slow evaporation method.1 crystallizes in orthorhombic chiral space group P212121, with two-fold screw symmetry. As anticipated from our previous results,53–59 the precursors undergo proton transfer to form a robust charge-assisted sulfonate-pyridinium synthon: N1–H1N⋯O1 [D–H⋯A: 145.30(1)° D⋯A: 2.281(1) Å]. Proton transfer is substantiated by nearly equal S–O bond distances in the acid former and relaxed C–N–C bond angle in the base former [122.43(2)°] The isoniazid Schiff base is non-planar and forms convergent trifurcated hydrogen bonds with sulfonate oxygen: O2⋯H17–C17[150.33°, 2.340 Å], O2⋯H2N–N2 [133.71(1)°, 2.153(3) Å] and O2⋯H14–C14 [122.65(2)°, 2.607(1) Å] interactions (Fig. 4a). The free Schiff-base 1a crystallizes in a nearly planar configuration, and is curved in 1 due to lateral interactions of the NO2 group with the carbonyl oxygen of the isoniazid residue [O7⋯N4: 3.028(1) Å]. The nitro group oxygen O8 forms a second hydrogen bond with the pyridinium end of the isoniazid: O8⋯H1N–N1 [2.929(2) Å], while the carboxylic OH group forms bifurcated interaction with carbonyl oxygen and azo nitrogen: O6–H6A⋯O7 [166.39(1)°, 1.925(1) Å] and O6–H6A⋯N3 [117.05(2)°, 2.693(2) Å] to form 2-dimensional hydrogen bonded tapes with cylindrical voids (Fig. S26, ESI†). The tapes further aggregate with weak interactions, including hydrogen bonds: O9⋯H10–C10 [122.21(1)°, 2.627(2) Å], O5⋯H6–C6 [157.27(1)°,2.370(1) Å], O3⋯H20–C20[150.21(2)°, 2.404(1) Å], O5⋯H6–C6 [157.27(1)°, 2.370(2) Å], O5⋯H18–C18 [117.63(2)°, 2.636(1) Å], O4⋯H20–C20 [126.76(1)°, 2.605(1) Å], π–π interactions, and interaction between the sulfonate oxygen O2 and C21 [3.065(1) Å], to form an intriguing 3-dimensional network of hydrogen-bonded tapes (Fig. S27, ESI†).
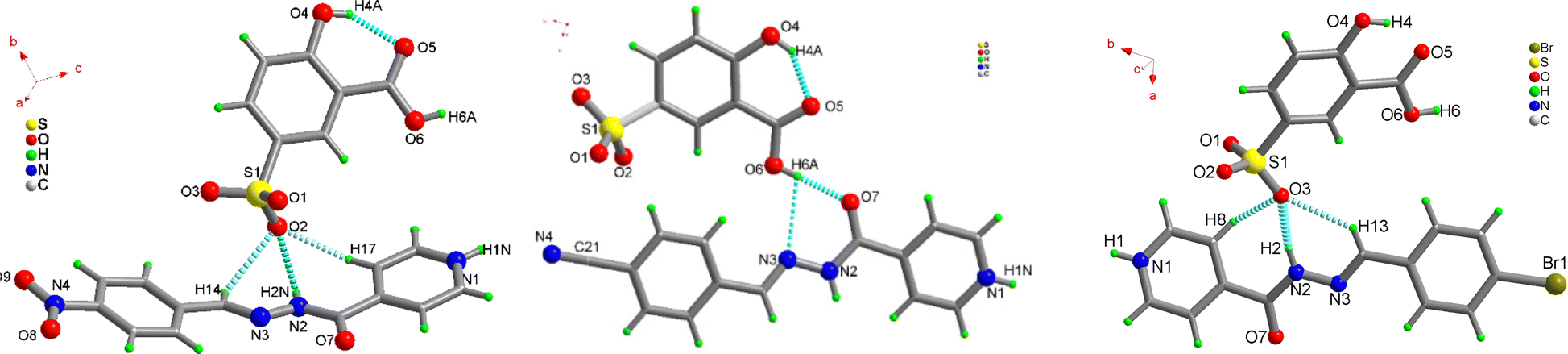 | ||
| Fig. 4 Molecular structure diagrams of 1, 2, and 3. | ||
For 2, the molecular complex crystallizes in a monoclinic P21/n symmetry system. The ionic sulfonate-pyridinium synthon is the major intermolecular interaction between the crystal components. The substitution of the nitro functional group in 1 by the nitrile group in the Schiff base in 2, does not significantly impact the interactions between the co-formers. The trifurcated hydrogen bonding interaction about the sulfonate oxygen and the –CN–NH– linker of the Schiff base, involving interactions: O2⋯H14–C14 [130.40(1)°; 2.582(2) Å], O2⋯H2N–N2 [144.89(1)°; 2.075(2) Å] and O2⋯H11–C11 [164.45(1)°; 2.371(1) Å], remains nearly unchanged but unlike in the 1 Schiff base, 2 is nearly planar and not curved (Fig. 4b). The Schiff base 2a establishes O6–H6A⋯O7 [174.72(1)°; 2.693(3) Å] and O6–H6A⋯N3 [108.80(1)°; 3.044(2) Å] contacts on the other side with the hydroxyl of the carboxylate group. The nitrile group does not establish any strong intermolecular contacts.
The organo-sulfonate hydrogen-bonded chains, stabilized by O4–H4A⋯O3 [116.55(2)°; 2.609(1) Å] hydrogen bonds, with further help from weak intermolecular contacts: C6–H6⋯O5 [152.25(1)°; 2.414(1) Å], C8–H8⋯O4 [107.64(1)°; 3.093(2) Å] and C12–H12⋯N14 [132.58(1)°; 2.447(1) Å], and π–π stacking between Schiff base cations, grow further. The final 3-dimensional hetero-aggregated lattice of 2 is stabilized by a centrosymmetric R22(10) synthon stabilized by C17–H17⋯N4 [148.82(2)°; 2.604(1) Å] interactions (Fig. S28, ESI†).
3 crystallizes in a monoclinic P21/n crystal system with a molecule of each component in the asymmetric unit. The Schiff base is nearly planar and protonated about the pyridyl center and the C–N–C bond angle is relaxed [122.69(2)°] (Fig. 4c). The ionic sulfonate-pyridinium synthon is masked by the Schiff base: O3⋯H2–N2 [165.74(1)°,1.915(2) Å] and O7⋯H1–N1 [169.0(2)°,1.874(3) Å]. As observed in 1 and 2, 3 forms trifurcated hydrogen bonds on one side: O3⋯H13–C13 [137.40(1)°, 2.661(3) Å] and O3⋯H8–C8 [129.53(1)°,2.549(3) Å] and bifurcated interactions: O7⋯H1–N1 [169.0(1)°,1.874(2) Å] and N3⋯H1–N1 [113.71(3) °, 3.288(2) Å] on the other side of the –CN–NH– linker. With the aid of a chalcogen bond: O3⋯Br1 [3.554(1) Å] and O3⋯N1 [3.135(1) Å], 3 grows into a 3-dimensional solid (Fig. S29, ESI†). The hydrogen-bonded lattice is further stabilized by inverted π-interactions between cationic base formers.
Hirshfeld studies
To further understand the impact of functionalization and cocrystallization on intermolecular interactions and crystal packing Hirshfeld analyses of 1a–3a and 1–3 have been carried out on Crystal Explorer 17.5 software.60,61 The Hirshfeld surface generated within a radius of 3.8 Å and calculated over dnorm provides a three-dimensional picture of close contacts in a crystal, which can be summarized in their fingerprint plots. Hirshfeld surface properties as well as the fingerprint plots provide significant details of the intermolecular contacts as well as packing of compounds 1a–3a and 1–3. Also, the curvedness and shape index provide further information about molecular packing, particularly about π-stacking in the compounds. Fingerprint analyses have been carried out to understand the effect of changing functionality on intermolecular contacts, which are responsible for the crystal packing of the materials. The fingerprint analysis of 1–3 indicates that the main interactions responsible for the formation of these co-crystals are O–H/H–O, H–H and C–H/H–C contributions. The interactions, however, exhibited significant variation with change in the functionality as the O–H/H–O contribution decreases from 41.8% in 1 to 33.4% in 2, and 29.3% in 3, which is due to more oxygen centers in 1 and 2 than 3. The contribution of π–π or C/C is much less in all three solids, but relatively higher in 2 [13.5%] than 1 [8.1%] and 3 [7.5%]. The contributions of other interactions in the precursors 1a–3a and 1–3 are provided in Fig. 4. The Hirshfeld and fingerprint plots of 1–3 are provided in Fig. S30–S35, ESI.†Discussion
Aroyl hydrazone was substituted with electron-withdrawing substituents, 1a–3a, and subsequently co-crystallized with 5-SSA-2H to understand the combined effect of substitution and cocrystallization on the crystal packing and optical properties. The nitro aroyl hydrazone 1a undergoes aggregation-caused quenching due to close-packed π–π interactions, while the π-stacking interactions in nitrile 2a and bromo 3a aroyl hydrazones are insignificant, leading to aggregation-induced emission (Fig. 5a–c). These Schiff bases undergo remarkable emission tuning on cocrystallization as the resultant organic salts exhibit improved red-shifted luminescence. 1a undergoes emission turn-on in 1 which in addition to the protonation of its pyridyl functionality can be attributed to the packing modulations on co-crystallization. Instead of complete head-to-tail π-overlap the 1a molecules in 1 undergo slipped π-interactions forming J-type aggregates (Fig. 5a). The 2a molecules undergo a moderate red-shift of 8 nm on co-crystallization in 2 which is accompanied by augmented intensity. Though the molecules in 2a are stacked, they are twisted enough to prevent π-overlap, while the 2a molecules in 2 aggregate as J-type aggregates with slipped head-to-tail overlap, leading to the red-shifted emission (Fig. 5b). 3a molecules in 3 undergo a significant red shift of 26 nm. Crystal packing comparison of pure 3a and its organic complex 3 reveals that the molecules aggregate orthogonally in the pure form, while the 3a molecules form isolated head-to-tail dimers in the crystal lattice of 3. The red shift can be plausibly attributed to better π-overlap behavior with the 3a dimers, and subsequent excimer formation (Fig. 5f).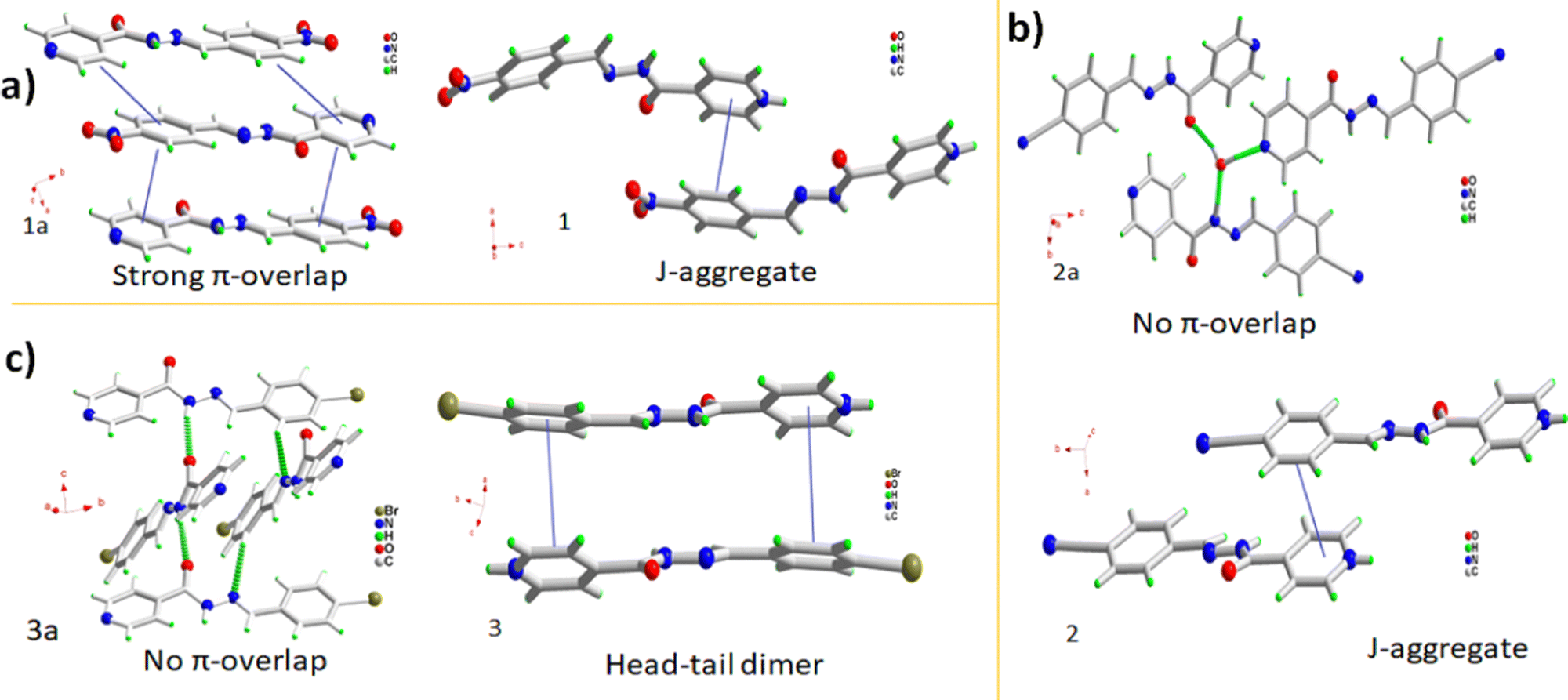 | ||
| Fig. 5 Packing arrangements of 1–3. (a) Long range π-overlap in 1a and J-aggregate formation in 1, (b) molecular aggregation in 2a and J-aggregate in 2 and (c) molecular aggregation in 3a and head-to-tail dimer in 3. | ||
The solution phase optical studies of the products provide further insights into their optical behavior. The solution phase absorption studies of 1a–3a and 1–3 do not show any charge transfer interactions indicating their existence as isolated molecular species. The charge transfer interactions emerge in their solid state and the interactions get stronger on cocrystallization. Similarly, the precursors 1a–3a do not exhibit emission in their aqueous solutions, possibly due to free molecular rotations, while 1–3 exhibit nearly similar emission behavior, which arises due to different contributions from frontier orbitals due to substitution effects. The concentration-dependent solution phase optical studies for the products do not indicate the possibility of excimer emission on aggregation.
The solid-state emission tuning achieved through the combined effect of functionalization and crystallization indicates enough scope for property engineering. Currently, we are working on more variations vis-a-vis functional groups and their steric bulk and electronic effects on pyridyl hydrazones with a follow-up of conformer variation. The studies can help to establish the structure–property relationship for the development of desired solid-state emitters for practical applications.
Conclusions
Using the principles of crystal engineering we demonstrate the combined effect of functionalization and crystallization on the structural and optical properties of the organic solids and their co-crystals. The solid-state emission has been engineered and explained vis-à-vis crystal structure–propertystructure–property relationship. The emission quenching in 1a is attributed to strong π–π interactions, which are absent in emissive solids 2a and 3a. The emission turn-on of 1a on co-crystallization in 1 and moderate redshift of 2a in 2 is attributed to the formation of J-type aggregates in their solid state, while the significant red shift in 3 plausibly arises due to excimer emission of isolated head-to-tail hydrazone dimers. The results represent one of the first attempts to integrate the impact of functional group and co-crystallization on the AIE of organic materials and indicate enough scope for the development of functional solid-state emitters.Author contributions
D. A. A. has perceived the problem, carried out the crystallographic studies, interpreted the data and prepared the manuscript. S. H. L. has synthesised and characterized the products and generated the data. I. A. and A. A. G. have helped with data plotting/improvisation and manuscript preparation. C. F. has carried out computational studies and helped during the manuscript prepration. A. A. A. has helped during the revision, which involved the reproduction of data and products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. A. D. acknowledges DST-SERB, New Delhi for funding under the Core Research Grant (CRG/2022/003693) and S. H. L. is thankful to DST-KIRAN for Women Scientist-A fellowship DST/WOS-A/CS-73/2019(G) WISE KIRAN. All the authors thank the Department of Chemistry and the University of Kashmir for the support and facilities.Notes and references
- F. Würthner, Aggregation-Induced Emission (AIE): A Historical Perspective, Angew. Chem., Int. Ed., 2020, 59, 14192–14196 CrossRef.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhuc and B. Z. Tang, Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole, Chem. Commun., 2001, 1740–1741 RSC.
- T. Tsujimura, OLED Display Fundamentals and Applications, John Wiley & Sons, Inc., 2017 Search PubMed.
- S. K. Park, J. H. Kim, T. Ohto, T. Yamada, A. O. F. Jones, D. R. Whang, I. Cho, S. Oh, S. H. Hong, J. E. Kwon, J. H. Kim, Y. Olivier, R. Fischer, R. Resel, J. Gierschner, H. Tada and S. Y. Park, Highly Luminescent 2D-Type Slab Crystals Based on a Molecular Charge-Transfer Complex as Promising Organic Light-Emitting Transistor Materials, Adv. Mater., 2017, 29, 1701346 CrossRef.
- X. Y. Shen, J. W. Y. Lam, P. Lu, Y. Lu, Z. Wang, R. Hu, N. Xie, H. S. Kwok, Y. Zhang, J. Z. Sun and B. Z. Tang, Efficient solid emitters with aggregation-induced emission and intramolecular charge transfer characteristics: molecular design, synthesis, photophysical behaviors, and OLED application, Chem. Mater., 2012, 24, 1518–1528 CrossRef.
- Y. Z. Yuan, P. Lu, S. Chen, J. W. Y. Lam, Z. Wang, Y. Lu, H. S. Kwok, Y. Ma and B. Z. Tang, Changing the behavior of chromophores from aggregation-caused quenching to aggregation induced emission: development of highly efficient light emitters in the solid state, Adv. Mater., 2010, 22, 2159–2163 CrossRef.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Semiconducting π- conjugated systems in field-effect transistors: a material odyssey of organic electronics, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS.
- J. Zhang, H. Geng, H. Virk, Y. Zhao, J. Tan, C. Di, W. Xu, K. Singh, W. Hu, Z. Shuai, Y. Liu and D. Zhu, Sulfur-bridged annulene-TCNQ Cocrystal: A self-assembled ‘‘molecular level heterojunction’’ with air stable ambipolar charge transport behaviour, Adv. Mater., 2012, 24, 2603–2607 CrossRef CAS.
- H. D. Wu, F. X. Wang, Y. Xiao and B. G. Pan, Preparation and ambipolar transistor characteristics of co-crystal microrods of dibenzotetrathiafulvalene and tetracyanoquinodimethane, J. Mater. Chem. C, 2013, 1, 2286–2289 RSC.
- Z. Qin, H. Gao, H. Dong and W. Hu, Organic Light-Emitting Transistors Entering a New Development Stage, Adv. Mater., 2021, 33, 2007149 CrossRef CAS.
- Q. Qi, C. Li, X. Liu, S. Jiang, Z. Xu, R. Lee, M. Zhu, B. Xu and W. Tian, Solid-State Photoinduced Luminescence Switch for Advanced Anticounterfeiting and Super-Resolution Imaging Applications, J. Am. Chem. Soc., 2017, 139, 16036–16039 CrossRef CAS.
- K. Li, T.-B. Ren, S. Huan, L. Yuan and X.-B. Zhang, Progress and Perspective of Solid-State Organic Fluorophores for Biomedical Applications, J. Am. Chem. Soc., 2021, 143, 21143–21160 CrossRef CAS PubMed.
- D. Dai, Z. Li, J. Yang, C. Wang, J. R. Wu, Y. Wang, D. Zhang and Y. W. Yang, Supramolecular Assembly-Induced Emission Enhancement for Efficient Mercury(II) Detection and Removal, J. Am. Chem. Soc., 2019, 141, 4756–4763 CrossRef CAS PubMed.
- P. Kumari, S. K. Verma and S. M. Mobin, Water soluble two-photon fluorescent organic probe for long-term imaging of lysosomes in live cells and tumor spheroids, Chem. Commun., 2018, 54, 539–542 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Aggregation-induced emission: the whole is more brilliant than the parts, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS.
- S. Feng, S. Gonga and G. Feng, Aggregation-induced emission and solid fluorescence of fluorescein derivatives, Chem. Commun., 2020, 56, 2511–2513 RSC.
- M. P. Lijina, Alfy Benny, Ebin Sebastian and Mahesh Hariharan, Keeping the chromophores crossed: evidence for null exciton splitting, Chem. Soc. Rev., 2023, 52, 6664–6679 RSC.
- A. A. Ahangar, H. Qadri, A. A. Malik, M. A. Mir, P. A. H. Shah and A. A. Dar, Physicochemical and Anti-fungal Studies of Pharmaceutical Co-crystal/Salt of Fluconazole, Manuscript ID: mp-2023-000879.R1.
- O. N. Kavanagh, D. M. Croker, G. M. Walker and M. J. Zaworotko, Pharmaceutical cocrystals: from serendipity to design to application, Drug Discovery Today, 2019, 24, 796–804 CrossRef CAS.
- P. Grobely, A. Mukherjee and G. R. Desiraju, Drug-drug co-crystals: Temperature-dependent proton mobility in the molecular complex of isoniazid with 4-aminosalicylic acid, CrystEngComm, 2011, 13, 4358–4364 RSC.
- Y. Liu, A. Li, S. Xu, S. Xu, Y. Liu, W. Tian and B. Xu, Reversible Luminescent Switching in an Organic Cocrystal: Multi-Stimuli-Induced Crystal-to-Crystal Phase Transformation, Angew. Chem., Int. Ed., 2020, 59, 15098–15103 CrossRef CAS PubMed.
- Q. Liu, L. Zhao, W. Wu, Y. He, K. Song, J. Qi, H. Li and Z. Chen, Reversible photo/thermal stimuli-responsive electrical bistability performance in supramolecular co-crystals accompanied by crystalline-to-amorphous transformations, J. Mater. Chem. C, 2020, 8, 3258–3267 RSC.
- Y. Lei, W. Dai, J. Guan, S. Guo, F. Ren, Y. Zhou, J. Shi, B. Tong, Z. Cai and J. Zheng, et al., Wide-Range Color-Tunable Organic Phosphorescence Materials for Printable and Writable Security Inks, Angew. Chem., Int. Ed., 2020, 59, 16054–16060 CrossRef CAS.
- I. Ahmad, A. A. Malik and A. A. Dar, Multi-Stimuli- Responsive Organo-Sulfonated Anil and Its Organic Complex, Cryst. Growth Des., 2022, 22, 6483–6492 CrossRef CAS.
- I. Ahmad, A. A. Ganie, S. Ahmad, A. A. Ahangar, C. M. Reddy and A. A. Dar, A high Z' structure of an organic salt with unusual high phase stability, mechano and vapo-fluorochromism, CrystEngComm, 2023, 25, 3164–3170 RSC.
- W. Wang, L. Luo, P. Sheng, J. Zhang and Q. Zhang, Multi-Functional Features of Organic Charge-Transfer Complexes: Advances and Perspectives.
- C. Wang, H. Dong, L. Jiang and W. Hu, Organic semiconductor crystals, Chem. Soc. Rev., 2018, 47, 422–500 RSC.
- X. Wang, Z. Z. Li, M. P. Zhuo, Y. Wu, S. Chen, J. Yao and H. Fu, Tunable Near-Infrared Organic Nanowire Nanolasers, Adv. Funct. Mater., 2017, 27, 1703470 CrossRef.
- S. K. Park, S. Varghese, J. H. Kim, S. J. Yoon, O. K. Kwon, B. K. An, J. Gierschner and S. Y. Park, Tailor-made highly luminescent and ambipolar transporting organic mixed stacked charge-transfer crystals: an isometric donor-acceptor approach, J. Am. Chem. Soc., 2013, 135, 4757–4764 CrossRef CAS PubMed.
- N. Genevaz, P. Chávez, V. Untilova, A. Boeglin, C. Bailly, L. Karmazin and L. Biniek, Tuning crystallochromism in diketopyrrolopyrrole-co-thieno[3,2-b]thiophene derivatives by the architecture of their alkyl side chains, J. Mater. Chem. C, 2018, 6, 9140–9151 RSC.
- X. Zhang, J. Wang, Y. Liu, Y. Hao, F. Yu, D. Li, X. Huang, L. Yu, T. Wang and H. Hao, Tunable Emission of Organic Fluorescent Crystals through Polymorphic Manipulation, J. Phys. Chem. C, 2021, 125, 6189–6199 CrossRef CAS.
- R. Li, S. Xiao, Y. Li, Q. Lin, R. Zhang, J. Zhao, C. Yang, K. Zou, L. Dongsheng and Y. Tao, Polymorphism-dependent and piezochromic luminescence based on molecular packing of a conjugated molecule, Chem. Sci., 2014, 5, 3922–3928 RSC.
- R. Misra, T. Jadhav, B. Dhokale and S. M. Mobin, Reversible mechanochromism and enhanced AIE in tetraphenylethene substituted phenanthroimidazoles, Chem. Commun., 2014, 50, 9076–9078 RSC.
- D. Yan, A. Delori, G. O. Lloyd, T. Friščić, G. M. Day, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, A Cocrystal Strategy to Tune the Luminescent Properties of Stilbene-Type Organic Solid-State Materials, Angew. Chem., Int. Ed., 2011, 50, 12483–12486 CrossRef CAS PubMed.
- J. D. Wuest, Co-crystals give light a tune-up, Nat. Chem., 2012, 4, 74–75 CrossRef CAS PubMed.
- D. Yan and D. G. Evans, Molecular crystalline materials with tunable luminescent properties: from polymorphs to multi-component solids, Mater. Horiz., 2014, 1, 46–57 RSC.
- Y. Wang, W. Zhu, H. Dong, X. Zhang, R. Li and W. Hu, Organic Cocrystals: New Strategy for Molecular Collaborative Innovation, Top. Curr. Chem., 2016, 83, 374 Search PubMed.
- Y. Huang, Z. Wang, Z. Chen and Q. Zhang, Organic Cocrystals: Beyond Electrical Conductivities and Field-Effect Transistors (FETs), Angew. Chem., Int. Ed., 2019, 58, 9696 CrossRef CAS.
- Y. Huang, Z. Wang, Z. Chen and Q. Zhang, Organic Cocrystals: Beyond Electrical Conductivities and Field-Effect Transistors (FETs), Angew. Chem., Int. Ed., 2019, 58, 9696 CrossRef CAS.
- W. Wang, L. Luo, P. Sheng, J. Zhang and Q. Zhang, Multifunctional Features of Organic Charge-Transfer Complexes: Advances and Perspectives, Chem. – Eur. J., 2020, 27, 464 CrossRef PubMed.
- J. Gierschner, J. Shi, D. Roca-Sanjuán, B. Milián-Medina, S. Varghese and S. Y. Park, Luminescence in Crystalline Organic Materials: From Molecules to Molecular Solids, Adv. Opt. Mater., 2021, 9, 2002251 CrossRef CAS.
- H.-Y. Liu, Y.-C. Li and X.-D. Wang, Recent advances in organic donor–acceptor cocrystals: design, synthetic approaches, and optical applications, CrystEngComm, 2023, 25, 3126 RSC.
- R. Bhowal, S. Biswas, A. Thumbarathil, A. L. Koner and D. Chopra, Exploring the Relationship between Intermolecular Interactions and Solid-State Photophysical Properties of Organic Co-Crystals, J. Phys. Chem. C, 2019, 123, 9311–9322 CrossRef CAS.
- W. Ng, S. Zhang, H. Wu, I. Nevjestic, A. J. P. White and M. Oxborrow, Exploring the Triplet Spin Dynamics of the Charge-Transfer Co-crystal Phenazine/1,2,4,5-Tetracyanobenzene for Potential Use in Organic Maser Gain Media, J. Phys. Chem. C, 2021, 125, 14718–14728 CrossRef CAS.
- Y. Zhang, H. Wu, Y. Wang, L. Sun, S. Li, Y. Ren, Y. Sun, F. Yang, X. Zhang and W. Hu, Cocrystal engineering for constructing two-photon absorption materials by controllable intermolecular interactions, J. Mater. Chem. C, 2022, 10, 2562–2568 RSC.
- Y. Sun, Y. Lei, L. Liao and W. Hu, Competition between Arene–Perfluoroarene and Charge-Transfer Interactions in Organic Light-Harvesting Systems, Angew. Chem., Int. Ed., 2017, 56, 10352–10356 CrossRef CAS.
- S. P. Anthony, S. Varughese and S. M. Draper, Switching and tuning organic solid-state luminescence via a supramolecular approach, Chem. Commun., 2009, 7500–7502 RSC.
- B. Dong, M. Wang, C. Xu, Q. Feng and Y. Wang, Tuning Solid-State Fluorescence of a Twisted π-Conjugated Molecule by Regulating the Arrangement of Anthracene Fluorophores, Cryst. Growth Des., 2012, 12, 5986–5993 CrossRef CAS.
- A. A. Ganie, A. A. Ahangar, A. Dhir, A. K. Gupta and A. A. Dar, Hetero-Aggregation-Induced Tunable Emission in Multicomponent Crystals, J. Phys. Chem. C, 2023, 127(19), 9257–9267 CrossRef CAS.
- I. Ahmad and Aijaz A. Dar, Switching the Solid-State Emission of Organic Crystals through Coformer Choice and Vapochromism, J. Phys. Chem. C, 2023, 127(37), 18684–18693 CrossRef CAS.
- A. A. Ganie, S. Rashid, A. A. Ahangar, T. M. Ismail, P. K. Sajith and A. A. Dar, Expanding the Scope of Hydroxyl-pyridine Supramolecular Synthon to Design Molecular Solids, Cryst. Growth Des., 2022, 22(3), 1972–1983 CrossRef CAS.
- M. V. N. de Souza, S. M. S. V. Wardell, J. L. Wardell, J. N. Low and C. Glidewell, Acta Crystallogr., Sect. C: Struct. Chem., 2007, 63, o166–o168 CrossRef CAS.
- A. A. Dar and A. A. Ganie, Irreversible Thermochromism in organic salts of sulfonated Anils, Cryst. Growth Des., 2020, 20, 3888–3897 CrossRef CAS.
- A. A. Ganie and A. A. Dar, Water Switched Reversible Thermochromism in Organic Salts of Sulfonated Anil, Cryst. Growth Des., 2021, 21, 3014–3023 CrossRef CAS.
- A. A. Ganie, P. Vishnoi and A. A. Dar, Utility of Bis-4-pyridines as Supramolecular Linkers for 5-Sulfosalicylic Acid Centers: Structural and Optical Investigations, Cryst. Growth Des., 2019, 19, 2289–2297 CrossRef CAS.
- I. Ahmad, A. A. Ganie and A. A. Dar, Achievement of enhanced solubility and improved optics in molecular complexes based on a sulfonate−pyridinium supramolecular synthon, CrystEngComm, 2020, 22, 3933–3942 RSC.
- A. A. Ganie, T. M. Ismail, P. K. Sajith and A. A. Dar, Validation of the supramolecular synthon preference through DFT and physicochemical property investigations of pyridyl salts of organosulfonates, New J. Chem., 2021, 45, 4780–4790 RSC.
- A. A. Ganie, A. A. Ahangar and A. A. Dar, Sulfonate···Pyridinium Supramolecular Synthon: A Robust Interaction Utilized to Design Molecular Assemblies, Cryst. Growth Des., 2019, 19, 4650–4660 CrossRef CAS.
- A. A. Ganie, R. Marimuthu, S. T. Islam and A. A. Dar, Molecular salts of the isoniazid derivatives. Expanding the scope of sulfonate-pyridinium synthon to design materials, J. Solid State Chem., 2022, 307, 1227622 CrossRef.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, 2017 CrystalExplorer17.5.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR, powder X-ray diffraction, Hirshfeld studies, structure diagrams, absorption and emission spectra, AIE and DLS studies and crystallographic table. CCDC 2255995–2255997. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ma00853c |
| This journal is © The Royal Society of Chemistry 2024 |