
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAu nanoparticle decorated reduced graphene oxide and its electroanalytical characterization for label free dopamine detection†
Chiara
Ingrosso
 ab,
Michela
Corricelli
c,
Anna
Testolin
d,
Valentina
Pifferi
d,
Francesca
Bettazzi
e,
Giuseppe V.
Bianco
f,
Nicoletta
Depalo
a,
Elisabetta
Fanizza
abc,
Marinella
Striccoli
a,
Ilaria
Palchetti
*be,
M. Lucia
Curri
*abc and
Luigi
Falciola
*bd
ab,
Michela
Corricelli
c,
Anna
Testolin
d,
Valentina
Pifferi
d,
Francesca
Bettazzi
e,
Giuseppe V.
Bianco
f,
Nicoletta
Depalo
a,
Elisabetta
Fanizza
abc,
Marinella
Striccoli
a,
Ilaria
Palchetti
*be,
M. Lucia
Curri
*abc and
Luigi
Falciola
*bd
aCNR-IPCF S.S. Bari, c/o Department of Chemistry, Università degli Studi di Bari, via Orabona 4, I-70126 Bari, Italy
bConsorzio Interuniversitario Nazionale per la Scienza e Tecnologia dei Materiali (INSTM), Via G. Giusti, 9 50121 Firenze, Italy
cDepartment of Chemistry, Università degli Studi di Bari, via Orabona 4, I-70126 Bari, Italy. E-mail: marialucia.curri@uniba.it
dDepartment of Chemistry, Università degli Studi di Milano, via Golgi 19, 20133, Milano, Italy. E-mail: luigi.falciola@unimi.it
eDepartment of Chemistry Ugo Schiff, Università degli Studi di Firenze, via della Lastruccia 3-13, 50019 Sesto Fiorentino (Fi), Italy. E-mail: ilaria.palchetti@unifi.it
fCNR-NANOTEC Sez. Bari, c/o Department of Chemistry, Università degli Studi di Bari, Via Orabona 4, I-70126 Bari, Italy
First published on 22nd November 2023
Abstract
A facile and cost-effective in situ colloidal procedure has been optimized for the synthesis of a novel colloidal hybrid nanocomposite based on 1-aminopyrene (AP) functionalized reduced graphene oxide (RGO) sheets, decorated with oleylamine (OLEAM)-coated Au nanoparticles (NPs). The effect of relevant experimental parameters, such as the OLEAM:Au precursor molar ratio and AP–RGO:Au precursor w/w, on the morphological features and spectroscopic characteristics of the nanocomposites has been explored to elucidate their formation mechanism. Au NPs, 9–20 nm in mean size, were grown on the –NH2 groups of the 1-aminopyrene molecules that are anchored at the RGO basal plane via aromatic π–π stacking interactions. The OLEAM ligand coordinating the Au NPs has been found to endow the nanocomposites with dispersibility in organic solvents, thus enabling their solution processability, essential for their fabrication as films onto screen printed carbon electrodes (SPCEs). Cyclic voltammetry and electrochemical impedance spectroscopy were performed to investigate the effect of the AP linker, the OLEAM capping ligand and surface chemistry and morphology of the hybrid nanocomposites on electrical conductivity, heterogeneous charge transfer capability and sensitivity of the nanocomposite modified SPCEs for label-free detection of dopamine (DA), an known biomarker of human neurodegenerative disorders. The AP linker has been found to establish an effective NP-RGO electron coupling, endowing the nanostructured platforms with high electrical conductivity and high electroanalytical activity, which have been found to be dependent on the size of Au NPs. The possibility of easily displacing OLEAM from the Au NP surface by ligand exchange allows modifying the nanoplatform surface chemistry and morphology, which are found to influence the electrochemical reactivity and sensitivity. The hybrid nanoplatforms show superior electroactivity for the determination of DA and viability for the effective detection of H2O2, another relevant biomarker of physiological alterations. The developed platform holds a great promise for innovative point-of-care technological solutions, that, enabling rapid and sensitive monitoring of biomarkers of human diseases, may represent valuable tools in modern medicine.
1. Introduction
Molecular biomarkers are valuable indicators for early diagnosis of human diseases, also enabling the formulation of prognosis and the elaboration of treatment plan. Abnormal levels of dopamine (DA), neurotransmitter of catecholamine involved in regulation of brain functions, are renowned clinical signals of anomalous neural activities1 observed in neurodegenerative disorders such as Parkinson's and Huntington's diseases.2–4 Diagnostic tools capable of monitoring DA levels in biological fluids have attracted interest for diagnosis, prevention, and clinical treatment. Electrochemical (bio)sensors are among the most favoured analytical tools due to their ease of operation, fast response, low LODs, relatively high selectivity, low cost, and portability.5–7Colloidal hybrid nanocomposites based on graphene derivatives and nanoparticles (NPs) merge the interesting properties of graphene8 with the unique size and shape dependent functionalities of colloidal inorganic NPs.9,10 In particular, when graphene derivatives are combined with Au NPs, the resulting nanocomposites synergistically feature the characteristics of the two components, such as large potential window, fast responsivity, high surface area and surface reactivity, high environmental and electrochemical stability, capability of binding biomolecules and high electrocatalytic activity, thus acquiring superior properties promising for many technological applications, including their integration as active elements in electrochemical biosensors.11 Such amenable characteristics have motivated the research towards the development of graphene/Au NP based nanocomposites for the determination of DA.12–17 Sulfonated nitrogen sulphur co-doped graphene (S-NS-GR) flakes, synthesized by thermal annealing, and functionalized via diazotization reaction by phenyl-SO3H groups, with the latter acting as anchor sites for the electrodeposition of bimetallic AuPt NPs, have been synthesized for the determination of DA with a LOD of 0.006 μM in the concentration range of 0.01–50.00 μM.16 RGO and Au NPs, which were electrochemically co-reduced onto ITO, were used for the detection of DA with a LOD of 0.015 μM in the 0.02–200 μM range.12 RGO–MoO2 nanosheets decorated with Au NPs were fabricated by electrochemical deposition onto glassy carbon electrodes for the detection of DA with a LOD of 0.013 μM in the 0.6–260 μM range.14 In general, the most sensitive electroactive systems reported in the literature for DA detection are complex nanostructured platforms, including costly systems based on bimetallic structures.12–17 Moreover, although several types of Au-RGO composites have been already reported,18–21 properties such as the coating density of the Au NPs on the RGO sheets, NP aggregation, electron coupling between the Au NPs and RGO, and surface chemistry of the Au NPs, which are expected to affect the electrochemical activity and sensitivity of the sensor, have not been always fully accounted.
In this work, a facile and cost effective in situ colloidal approach22,23 has been optimized for the synthesis of a novel colloidal hybrid nanocomposite formed of 1-aminopyrene (AP) surface functionalized reduced graphene oxide (RGO), decorated with oleylamine (OLEAM)-coated Au NPs. The suitability of the nanocomposite as an active material for electrode modification, effective in probing the surface interactions with DA, and hence allowing its detection, has been demonstrated. RGO has been selected as it features many of the outstanding properties of graphene,24 including significant chemical reactivity, making it relevant for its functionalization with other nanostructures for preparing multifunctional nanocomposites, promising for a plethora of applications.25,26
The proposed original synthesis route has been suitable conceived to address the preparation of nanocomposites that take advantage of: (i) effective electron coupling between RGO and the Au NPs through the AP linker that is able to anchor by stable aromatic π–π stacking interactions the RGO basal plane22,23,25 and to coordinate, by means of its –NH2 group, the in situ synthesized colloidal Au NPs without detrimentally affecting RGO structural properties,27 leading to a merge of the electron conductivity and electrocatalytic activity of the hybrid nanocomposite components,22,23,25 (ii) the –NH2 groups of AP for avoiding Au NP aggregation and having electrodes with a high surface area, (iii) the stabilizer effect of the oleylamine (OLEAM) ligand, which is used as solvent and as mild reductant of the HAuCl4 × 3H2O precursor,28 and hence it coordinates the growing Au NPs for endowing the nanocomposites with colloidal stability and solution processability for their deposition onto carbon screen-printed electrodes (SPCEs), and (iv) the weak binding of OLEAM at the Au NP surface, to modulate their surface chemistry via simple ligand exchange procedures, and accordingly, the electrochemical reactivity and sensitivity of the nanocomposite modified electrodes.
The effect of synthetic parameters, namely the OLEAM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HAuCl4 × 3H2O molar ratio and AP–RGO:HAuCl4 × 3H2O w/w, along with a comprehensive investigation of the spectroscopic and morphological properties of the achieved nanocomposites, has allowed elucidating the NP decoration mechanism of the RGO platform, that has been found to lead to AP–RGO sheets densely coated with uniform multilayers of OLEAM-coordinated Au NPs. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) of the hybrid modified SPCEs were performed before and after exchange of OLEAM. SPCEs were used as disposable electroactive substrates to avoid fouling of their surface and further treatments for their regeneration, and as miniaturised electrochemical cells allowing use of small volumes of samples.
:HAuCl4 × 3H2O molar ratio and AP–RGO:HAuCl4 × 3H2O w/w, along with a comprehensive investigation of the spectroscopic and morphological properties of the achieved nanocomposites, has allowed elucidating the NP decoration mechanism of the RGO platform, that has been found to lead to AP–RGO sheets densely coated with uniform multilayers of OLEAM-coordinated Au NPs. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) of the hybrid modified SPCEs were performed before and after exchange of OLEAM. SPCEs were used as disposable electroactive substrates to avoid fouling of their surface and further treatments for their regeneration, and as miniaturised electrochemical cells allowing use of small volumes of samples.
The easy displacement of OLEAM with acetate ions has been found effective in improving the electrochemical reactivity of the nanocomposite for a successful detection of DA, with a LOD of 0.0025 μM in the 0.013–0.130 μM range, which is among the lowest reported,12–17,29–31 and allows the diagnosis of patho-physiological alterations.32 OLEAM has also been replaced with 6-mercaptohexanol, demonstrating the ability to detect H2O2, which is another interesting biomarker, at a LOD with a clinical relevance for diagnosis of disorders.33–36
The overall results demonstrate the crucial role of the AP linker at the NP/RGO interphase in allowing charge coupling between the materials enhancing electrical conductivity of the electrodes, as well as the effect of the nanocomposite surface morphology and chemistry on accomplishing relatively high electroactivity for the detection of the selected analytes, assessing, as a proof-of-concept, the analytical effectiveness and versatility of the proposed cost effective nanoplatform, indicating its potential for a fast and reliable point-of-care clinical diagnosis of human neurodegenerative disorders.
2. Experimental section
2.1 Chemicals and materials
Commercial reduced graphene oxide (RGO) (1.6 nm flakes) was purchased from Graphene Supermarket. 1-Aminopyrene (AP, 97%), n-methyl-2-pyrrolidone (NMP, 99%), tetrachloroauric(III) acid trihydrate (HAuCl4 × 3H2O, 99.999%), oleylamine (OLEAM, 70%), dopamine (DA) hydrochloride, 6-mercaptohexanol (MCH), acetic acid (AcOH), hydrogen peroxide (H2O2, 30% solution in water), di-sodium hydrogen phosphate dodecahydrate, potassium chloride, potassium dihydrogenphosphate, toluene and methanol were purchased from Sigma Aldrich.2.2 Exfoliation and functionalization of RGO
Commercial RGO was exfoliated and functionalized with AP following the procedure reported in ref. 22, 23 and 25 with minor modifications, which involves sonicating a mixture of RGO and AP powders, prepared in a 1:17 w/w in NMP,22,23,25 followed by purification by four cycles of centrifugation and re-dispersion in methanol, to remove AP in excess.22,23,25
2.3 Synthesis of oleylamine (OLEAM)-coated Au NPs
OLEAM capped Au NPs, 9.1 ± 2.1 nm in size, were synthesized by following the procedure of Hiramatsu et al.28 A reaction flask, filled up with 340 mg of HAuCl4 × 3H2O dispersed in 5 mL of OLEAM and 35 mL of toluene, was stirred under vacuum at 110 °C, and then, left at such a temperature for 5 h under nitrogen atmosphere. The obtained OLEAM-coated Au NPs were purified by three cycles of centrifugation and re-dispersion in methanol, and the isolated pellet was re-dispersed in toluene.2.4 Synthesis of the AP–RGO/Au NP hybrid nanocomposites
The colloidal route of Hiramatsu et al.28 was followed for the synthesis of the AP–RGO/Au NP hybrid nanocomposite, with minor modifications. In a typical experiment, a reaction flask was filled up with 1–5 mg of AP–RGO flakes dispersed by sonication in 15 mL of toluene, 3.5–7.5 mL of OLEAM, and 240–340 mg of HAuCl4 × 3H2O in 35 mL of toluene. At first, the reaction flask was stirred under vacuum at 110 °C, and then, the synthesis was allowed to proceed, at such a temperature, for 5 h under nitrogen atmosphere. The obtained AP–RGO/Au NP hybrid nanocomposites were purified from the excess of OLEAM by three cycles of centrifugation and re-dispersion in methanol. Finally, the isolated AP–RGO/Au NP hybrid pellet was re-dispersed in toluene.To separate the hybrid nanocomposite flakes from the Au NPs homonucleated in the supernatant of the reaction mixture, a post-synthetic precipitation procedure was implemented, inducing, by methanol addition, a gradual flocculation of the larger AP–RGO/Au NP hybrid sample.22,23,25 This pellet was then separated by centrifugation from the supernatant containing the smaller homonucleated OLEAM-capped Au NPs.
2.5 Preparation of the modified screen printed carbon electrodes (SPCEs)
SPCEs were modified by drop-casting 1 μL of a: (i) 0.2 mg mL−1 AP–RGO toluene dispersion, (ii) toluene solution of OLEAM-capped Au NPs synthesized starting from 0.17 M Au precursor, and (iii) AP–RGO/Au NP toluene dispersion synthesized starting from 0.2 mg mL−1 AP–RGO and 0.17 M Au precursor, finally dried in air at room temperature. A layer-by-layer film was prepared by drop casting onto SPCEs, 1 μl of a 0.2 mg mL−1 AP–RGO toluene dispersion followed by drop casting 1 μl of a toluene solution of OLEAM-capped Au NPs synthesized starting from 0.17 M Au precursor.The AP–RGO/Au NP surface chemistry at the electrodes was modified by performing two surface treatments, namely: i. spin-coating (3000 rpm for 40 min) 30 μl of methanol onto the AP–RGO/Au NP modified electrodes, followed by spin-coating (3000 rpm for 40 min) 18 μl of acetic acid, and ii. incubation of the AP–RGO/Au NP modified electrodes, for 1 h, in a 1 mM 6-mercaptohexanol (MCH) aqueous solution under controlled humidity conditions, followed by rinsing with water to remove MCH not specifically chemisorbed.
2.6 Characterization techniques
UV-Vis absorption spectroscopy investigation was performed using a Cary Varian 5000 spectrophotometer. Mid-infrared spectra were acquired using a Varian 670-IR spectrometer equipped with a DTGS (deuterated triglycine sulfate) detector having a spectral resolution of 4 cm−1. For attenuated total reflection (ATR) measurements, a one-bounce 2 mm diameter diamond microprism was used as internal reflection element (IRE). The samples were deposited by drop casting directly onto the upper face of the diamond crystal and the solvent was allowed to evaporate.Transmission electron microscopy (TEM) images were collected using a JEOL JEM-1011 microscope working at an accelerating voltage of 100 kV, equipped with a high-contrast objective lens and a W filament as electron source. Under these conditions, the ultimate point resolution of the microscope was 0.34 nm. The TEM images were recorded with a Gatan SC-1000 Orius Camera, equipped with an optical fiber-coupled 11 Mp CCD. The samples were prepared by dipping a 300 mesh amorphous carbon-coated Cu grid in AP–RGO/Au NP toluene dispersions and by letting then the solvent to evaporate. Size statistical analyses of the NP average size and size distribution were performed using the freeware ImageJ analysis program.
Topography atomic force microscopy (AFM) measurements were performed in air and at room temperature by using a PSIA XE-100 SPM system, operating in tapping mode. A silicon scanning probe microscopy (SPM) sensor for noncontact AFM (Park Systems), having a spring constant of 42 N m−1 and a resonance frequency of 330 kHz, was used. Micrographs were collected on six distinct areas of the sample with a scan size area of 5 μm × 5 μm, by sampling the surface at a scan rate between 1.0 and 0.5 Hz with a resolution of 256 × 256 pixels. Topography AFM images were processed by using the XEI software to obtain statistical data.
Raman spectra were collected by using a LabRAM HR Horiba-Jobin Yvon spectrometer with a 532 nm continuous excitation laser source. Measurements were carried out under ambient conditions at a low laser power (1 mW) to avoid laser-induced damage of the sample. The Raman signal from the silicon wafer at 520 cm−1 was used to calibrate the spectrometer, and the accuracy of the spectral measurement was estimated to be 1 cm−1.
Electrochemical investigation was performed using a PGStat30 (Metrohm AutoLab, Utrecht, The Netherlands) equipped with the FRA2 module and the NOVA 2.1 Software (Metrohm AutoLab, Utrecht, The Netherlands). CV and EIS experiments were carried out by means of a three-electrode cell equipped with a saturated calomel electrode (SCE), a Pt wire and carbon screen printed electrodes (SPCEs), as reference, counter and working electrodes, respectively. All the reported potentials were referred to the reference saturated calomel electrode (SCE), and the current values were normalized with respect to the geometrical area of the working electrode (7 mm2). CV and EIS measurements were performed by using a solution of 0.1 M PBS buffer at pH 7.4, to which is also added the 3 mM K4[Fe(CN)6] redox probe, as supporting electrolyte. For EIS, a sinusoidal voltage of 10 mV in amplitude (peak-to-peak), in the frequency range of 100 kHz–10 mHz, was superimposed to the applied bias potential that was set up at a value of +0.25 V.
2.7 Electroanalytical detection of dopamine and H2O2
Electrochemical detection of DA was performed by differential pulse voltammetry (DPV), scanning the potential between −0.2 V and +0.6 V (SCE), with a pulse of 5 mV and an amplitude of 50 mV. DA hydrochloride solutions were prepared in a 0.1 M PBS buffer supporting electrolyte solution at pH 7.4. For H2O2 detection, amperometric measurements were performed under stirring at a potential of +0.70 V and in 0.05 M phosphate buffer at pH 8.5.SPCEs formed with a carbon working electrode (3 mm in diameter), a carbon counter electrode and a silver pseudo-reference electrode, provided by Ecobioservices and Researches (EBSR, Firenze, Italy), were used.
3. Results and discussion
3.1 Decoration of the RGO platform with Au nanoparticles
The synthesis of the hybrid nanocomposite based on 1-amminopyrene (AP) functionalized RGO, decorated with oleylamine (OLEAM)-coated Au NPs, has been carried out by exfoliating and functionalizing RGO with AP, and then, performing the in situ synthesis of the colloidal Au NPs in presence of OLEAM (Scheme 1). | ||
| Scheme 1 Preparation of the AP–RGO hybrid nanocomposite. (The sketches are not drawn to scale). | ||
Commercially available RGO has been exfoliated and functionalized with AP following the approach reported in ref. 22 and 23. Briefly, successive cycles of sonication and stirring of the RGO powder in a solution of AP in N-methyl-2-pyrrolidone (NMP), have been performed, followed by purification of the flakes from AP in excess by centrifugation and re-dispersion in methanol.22,23
AP anchors the RGO basal plane through aromatic π–π stacking interactions that can open small gaps at the graphene sheet edges and penetrate deeper into the graphene multilayers, ultimately generating the AP–RGO complex.22,23 Concomitantly, AP grafts reactive –NH2 groups on RGO, which preserves its C-sp2 structure,27 and enable dispersion of the RGO sheets in solvents.37
TEM micrographs of the AP–RGO flakes show almost transparent sheet-like structures featuring higher image contrast areas, due to wrinkles and folded edges of the RGO sheets (Fig. 1(A)), reasonably originated from mechanical lattice deformations,38 induced by recovery of the conjugated system in GO reduction to RGO,39 and by sheet crumpling, likely occurring upon solvent drying in the deposition onto the TEM grid.
 | ||
| Fig. 1 (A) TEM micrograph and (B) 2-D topography AFM image of AP–RGO and (C) cross-sectional line profile taken along the red line of (B). (D) UV-Vis absorption (left panel) and UV-Vis PL emission spectra (λexc = 300 nm) (right panel) of 1.5 × 10−5 M AP and 0.015 mg mL−1 AP–RGO in NMP. (E) Raman spectra of RGO and AP–RGO onto silicon. | ||
The AFM image shows sheet-like nanostructures (Fig. 1(B)), ca. 4–6 nm high, as assessed from the cross-sectional line profile reported in Fig. 1(C). As the thickness of a single-layer AP–RGO ranges from 0.7 to 1.1 nm with an average value of 0.9 ± 0.3 nm,40 the height profile estimated from the AFM images allows inferring that the exfoliation of the AP–RGO flakes results in few-layer RGO sheets.
The absorption spectrum of the AP–RGO complex shows an increase of the baseline intensity accounted for by scattering phenomena induced by aggregated RGO flakes (Fig. 1(D) left side),41 along with a weak shoulder, in the near UV region, namely centred at ca. 365 nm, which can be accounted for by π–π* transitions of the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– bonds of AP,41 showing intense overlapping signals between 300 and 450 nm, in solution (Fig. 1(D) left side). Also, the line-shape of the PL emission spectrum of AP is significantly modified in the His–RGO complex (Fig. 1(D) right side), assessing its immobilization onto the sheets.42
C– bonds of AP,41 showing intense overlapping signals between 300 and 450 nm, in solution (Fig. 1(D) left side). Also, the line-shape of the PL emission spectrum of AP is significantly modified in the His–RGO complex (Fig. 1(D) right side), assessing its immobilization onto the sheets.42
The Raman spectrum of RGO shows typical G and D peaks of graphene at ca. 1580 cm−1 and 1340 cm−1, respectively, originated from the stretching of the C-sp2 bonds and the breathing modes of C-sp2 atoms in the hexagonal rings, respectively. In particular, the presence of the D peak, derived from a second order Raman process involving the inelastic and elastic scattering of an electron with, respectively, a phonon and a defect, accounts for the existence of defects in the carbon lattice,43 which is also indicated by the very low intensity of the 2D peak at ca. 2700 cm−1, and the presence of the D + D′ peak below 3000 cm−1. After exfoliation and functionalization with AP, no significant changes of the D and G intensity ratio in the RGO Raman spectrum are observed (Fig. 1(E)), assessing the non-covalent nature of the binding of the aromatic ligand at the RGO basal plane.27
At first, to elucidate the role of AP in the synthesis of AP–RGO/Au NPs and the chemical reactivity of the HAuCl4 × 3H2O precursor with RGO, two control experiments were performed, testing the synthesis with: (i) AP–RGO sheets in the absence of OLEAM and in 1-octadecene (ODE), and with (ii) bare RGO in OLEAM.
The former experiment shows that the product is characterized by faceted and partially aggregated, high image contrast nanostructures of 10–250 nm, reasonably accounted for by Au NPs formed by spontaneous galvanic reduction, taking place via electron transfer from RGO to Au(III) (Fig. S1(A) of the ESI†). The latter experiment demonstrates, instead, that the Au NPs do not heteronucleate onto bare RGO flakes, but homonucleate in the reaction solution under control of OLEAM (Fig. S1(B) and (C) of the ESI†). These results thus demonstrate that OLEAM can effectively reduce the HAuCl4 × 3H2O precursor, and that the –NH2 groups of AP act as coordinating sites of the formed NPs, anchoring them onto the RGO basal plane, and show that the AP linker allows transfer of charge between RGO and the Au precursor.22,23,25
An investigation of the effects of the OLEAM:HAuCl4 × 3H2O molar ratio, and of the AP–RGO:HAuCl4 × 3H2O w/w, on the spectroscopy and morphological properties of the hybrid nanocomposite has been performed to elucidate the mechanism underlying the Au NP formation on AP–RGO.
The role of OLEAM in the synthesis was explored by varying the OLEAM:HAuCl4 × 3H2O molar ratio (12:1 and 18:1) keeping constant the AP–RGO:HAuCl4 × 3H2O w/w at 1:340.
The TEM investigation of the sample obtained at the 12:1 OLEAM:HAuCl4 × 3H2O molar ratio shows ca. 200 nm in size irregular structures, with a high contrast image, that can be reasonably ascribed to the AP–RGO flakes fragmented during the sonication, densely coated with a multilayer of spherical nanostructures compatible with Au NPs (Fig. S2 of ESI†). In addition, the TEM image shows also spherical Au NPs that homonucleate in the reaction mixture concomitantly to the AP–RGO/Au NP hybrid flakes (Fig. S2(A), ESI†).22,23 The separation of the hybrid flakes from the homonucleated Au NPs was achieved by means of post-synthesis precipitation and the recovered nanostructures, depicted in the TEM images of Fig. S2(B), ESI† and of Fig. 2(A)–(D), collected at lower and higher magnification, respectively, are compatible with AP–RGO flakes still densely coated with layers of Au NPs, supporting the effectiveness of the NPs anchoring onto the sheets.
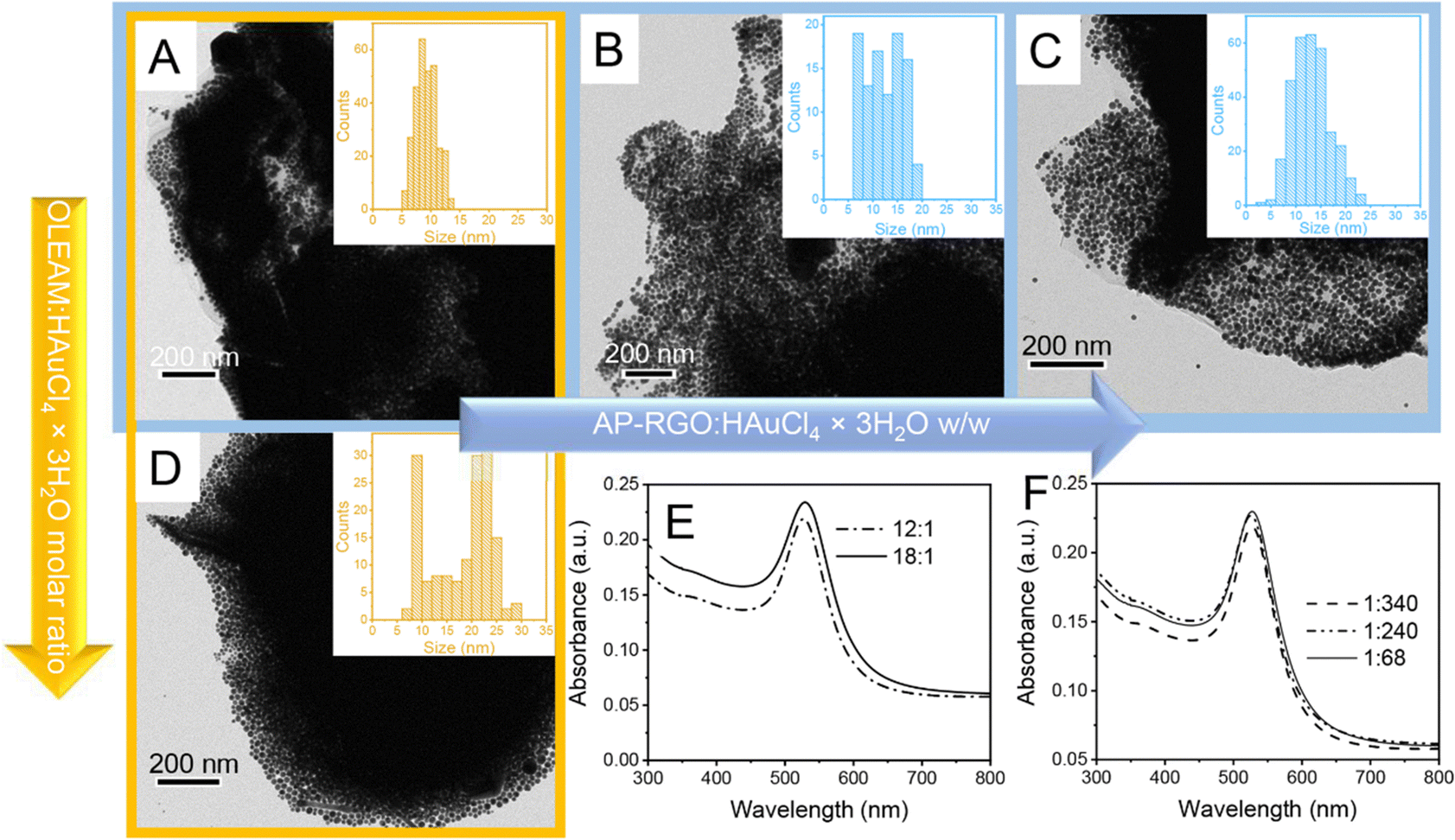 | ||
| Fig. 2 (A)–(D) TEM images and (E) and (F) UV-Vis absorption spectra of AP–RGO/Au NPs synthesized with the 12:1 (A)–(C) and 18:1 (D) OLEAM:HAuCl4 × 3H2O molar ratio, at the 1:340 (A) and (D), 1:240 (B) and 1:68 (C) AP–RGO:HAuCl4 × 3H2O w/w. Absorption spectra were obtained from 1:120 diluted nanocomposite toluene dispersions synthesized in (E) starting from 0.17 M Au precursor and 0.2 mg mL−1 AP–RGO and in (F) from 0.12 M Au precursor and 0.2 mg mL−1 AP–RGO for the 1:240 w/w, 0.17 M Au precursor and 0.2 mg mL−1 AP–RGO for the 1:340 w/w, and 0.17 M Au precursor and 1 mg mL−1 AP–RGO for the 1:68 w/w. | ||
The mean sizes of the NPs estimated from the statistics performed on the TEM images are 9.2 ± 1.8 nm for sample in panel A (Fig. 2) and 19.1 ± 6.5 nm for sample in panel D (Fig. 2), with the Au NP size distribution narrower in sample in panel A than in that in panel D, obtained, instead at a higher OLEAM:HAuCl4 × 3H2O molar ratio. The UV-Vis absorption spectra of both samples show a narrow and intense localized surface plasmon resonance (LSPR) band at 536 nm (Fig. 2(E)), which originates from the typical collective excitation of the free conduction band electrons in spherical Au NPs.22,28
Another set of experiments were performed by changing the AP–RGO:HAuCl4 × 3H2O w/w to investigate the effect of the –NH2 coordinating sites of AP anchored onto the RGO basal plane on the synthesis mechanism.
Fig. 2(B) and (C) report the TEM images of the hybrid nanocomposites synthesized with the AP–RGO:HAuCl4 × 3H2O w/w of 1:240 and 1:68, respectively, at the fixed OLEAM:HAuCl4 × 3H2O molar ratio of 12:1, which show a dense and uniform coating of Au NPs, organized in a multilayer onto the RGO sheets. The presence of homonucleated Au NPs has also been observed under these experimental conditions (data not shown).
It is worth noting that when the AP–RGO:HAuCl4 × 3H2O w/w increases passing from 1:340 in sample in panel A up to 1:240 in sample in panel B and to 1:68 in sample of panel C (Fig. 2), the NP size distribution is still monomodal, but larger than in sample in panel A, with also a higher average NP size, namely 12.3 ± 3.6 nm and 12.7 ± 3.7 nm for samples in panels B and C, respectively.
The Raman spectrum of the AP–RGO/Au NPs (Fig. 2(A)) shows a decrease of the D and G peak intensity ratio with respect to that of the AP–RGO complex after decoration with the Au NPs (Fig. 3(A)), passing from 1.11, for the AP–RGO sample, to 0.9 for AP–RGO/Au NPs, being such a modification likely due to a healing effect on the pristine defects of the RGO basal plane, occurring during the formation of the NPs at 110 °C assisted by OLEAM.45 Finally, the broad background signal in the Raman spectrum of the AP–RGO/Au NP can be explained by the intrinsic PL emission of AP.41
 | ||
| Fig. 3 (A) Raman spectrum of AP–RGO and AP–RGO/Au NPs. (B) FTIR-ATR spectra of neat OLEAM (a) and AP–RGO/Au NPs (b). | ||
In Fig. 3(B) the FTIR-ATR spectra of AP–RGO/Au NPs and of the neat OLEAM are compared, pointing out that, in the high wavenumber region, the spectrum of the hybrid nanocomposite shows the stretching vibrations of the OLEAM alkyl groups, namely at 2962 cm−1 the –CH3 asymmetric stretching and at 2922 cm−1 and 2852 cm−1, the asymmetric and symmetric stretching of –CH2, respectively, whereas the two –NH2 asymmetric and symmetric stretching vibrations of the neat OLEAM, pointing at 3370 cm−1 and 3293 cm−1, respectively, cannot be detected. In the finger print region, the hybrid nanocomposite shows intense signals at 1259 cm−1, 1084 cm−1 and 1015 cm−1, which can be ascribed to the –C–N– stretching, along with the strong signal due to the –N–H bending in the OLEAM molecules at 795 cm−1. The coordination of OLEAM to the Au NP surface can be thus safely confirmed, accounting for the observed colloidal stability of the nanocomposite in organic solvents. In addition, the presence of such coordinating molecules can be considered responsible for the Au NP assembly in multistacks onto the RGO sheets (Fig. 2), due to hydrophobic interactions among the interdigitating OLEAM alkyl chains.
The study of the synthesis of the hybrid nanocomposite leads to interesting considerations, such as the increase of the size of the Au NPs in the nanocomposite with the increase of the OLEAM:HAuCl4 × 3H2O molar ratio from 12:1 in sample of panel A to 18:1 in sample of panel D (Fig. 2). This evidence can be explained by considering that, after the injection of HAuCl4 × 3H2O in the reaction solution, Au(III), in the form of AuCl4− ions, is thermally reduced to Au(I), in the form of AuCl2− ions. Such AuCl2− ions yield stable complexes with OLEAM as [AuCl(RNH2)],46 that reduce to Au(0) with oxidation of OLEAM. The latter forms the aminium radical cation that reacts with another molecule of OLEAM forming the secondary ammine R-CH2-NH-R-CH2-NH3+.47 Thus, it is likely that the increase of concentration of OLEAM can lead to a more reduction of the Au precursor, including also that unreacted in sample in panel A of Fig. 2, resulting in the increase of the Au NP size observed in sample in panel D of the same figure.
The synthesis process also shows that higher is the AP–RGO:HAuCl4 × 3H2O w/w, larger is the size of the achieved Au NPs, which increases passing from the sample of panel A to that in panel B and panel C (Fig. 2). This evidence is likely ascribed to the enhancement of concentration of the –NH2 groups of AP–RGO, that, replacing OLEAM in the coordination of the Au clusters, increases the OLEAM concentration available in solution for the further reduction of the unreacted HAuCl4 × 3H2O precursor in sample of panel A (Fig. 2), resulting in an enhancement of the NP size.
The broadening of the size distribution of the Au NPs in samples B–D can be explained considering that the Au NPs likely form according to the classical diffusion limited growth mechanism, starting with a burst of heteronucleation events at the AP sites anchored onto the RGO basal plane,28 which then proceed with the rapid growth of Au NPs occurring with the deposition of the Au atoms diffusing from the bulk of the reaction solution onto the Au nuclei pre-formed onto RGO. It is likely that the mild reducing behavior of OLEAM prolongs the Au precursor reduction, leading to multiple heteronucleation events of new Au NPs. This takes place concomitantly with the growth of the preformed ones,28 due to the energetically favoured heteronucleation process,48 resulting in an uneven growth rate over differently sized Au nuclei.49 This evidence confirms that the –NH2 groups of the AP linker act as coordinating sites.22,23,25
3.2 AP–RGO/Au NP hybrid modified screen-printed carbon electrodes (SPCEs)
:1 and 18:1 OLEAM:HAuCl4 × 3H2O molar ratio, and at the 1:340 AP–RGO:HAuCl4 × 3H2O w/w, were deposited by drop casting from toluene solutions onto disposable screen-printed carbon electrodes (SPCEs) for their application in label free electrochemical detection of dopamine (DA). Such electrodes are hereafter indicated as A_OLEAM and D_OLEAM, respectively.
The surface morphology of the A_OLEAM electrodes, characterized by scanning electron microscopy (SEM), points out the nanoporous surface morphology of the SPCEs modified with the nanocomposite sample, as bright contrast and spherical nanostructures can be detected (Fig. 4(A)), reasonably ascribed to the OLEAM-capped Au NPs anchored onto the AP–RGO sheets (Fig. 2(A)).
 | ||
| Fig. 4 SEM micrographs of SPCEs modified with A_OLEAM (A), A_CH3OH/AcOH (B) and A_MCH (C), respectively. | ||
The long alkyl chain OLEAM ligand is known to act as a dielectric coating on NP films hampering NP–NP charge transfers therein,50 hence, in view of the application of the hybrid nanocomposite as active material for the electrochemical detection of analytes, it has been replaced with shorter in length ligand molecules. For this purpose, two distinct treatments were performed; the former involves spin-coating methanol first, and then, acetic acid onto the hybrid electrodes, and the latter involves incubating the hybrid electrodes in an aqueous solution of 6-mercaptohexanol (MCH).
In the former treatment, as methanol is a poor solvent for the OLEAM molecules coordinating the Au surface in the as-synthesized nanocomposite, it is used to only wash away the weakly bound OLEAM molecules,51 and then acetic acid displaces OLEAM, protonating the –NH2 groups of the bound OLEAM molecules, allowing coordination of acetate ions to the Au NP surface.52
The latter treatment uses, instead, MCH, a molecule that is reported to lead, when applied to Au NP based electrodes, to electron transfer kinetics faster than those of electrodes treated with other thiols able to chemisorb at the Au(0) NP surface,53 thanks to the semi-covalent bond of energy of ca. 188 kJ mol−1,51 thus resulting in a more effective displacement of OLEAM that binds the NPs with a lower energy of ca. 44 kJ mol−1.52
The treatment of the hybrid modified electrodes by incubation in MCH leads to smooth nanostructures with larger and irregular shape, as shown in the SEM image of Fig. 4(C), likely due to nanowelding phenomena caused by desorption of OLEAM, which is responsible for an increase of the NP surface energy, that, destabilizing the NPs, induces their diffusion and coalescence onto the AP–RGO basal plane.54
The SEM image of Fig. 4(B) shows that the surface morphology of the A_OLEAM electrodes is mainly retained upon treatment with methanol and acetic acid. Such electrodes, hereafter indicated as A_CH3OH/AcOH, present bright contrast Au NPs with no evident aggregation phenomena, and show a more densely packed NP layout, likely due to the removal of the long alkyl chain OLEAM molecules and the concomitant decreases of the interparticle distance. On the other hand, the MCH treated SPCEs, hereafter indicated as A_MCH electrodes, show smooth nanostructures with larger and irregular shape (Fig. 4(C)). This evidence is likely due to nanowelding phenomena caused by desorption of the insulating OLEAM layer, which is responsible for an increase of the NP surface energy, that, destabilizing the NPs, induces their diffusion and coalescence onto the AP–RGO basal plane (Fig. 4(C)).54
 | ||
| Fig. 5 (A) and (C) CV curves collected at the scan rate of 0.1 V s−1 and step potential of 0.005 V and (B) and (D) Bode plots at +0.25 V in 0.1 M PBS (pH = 7.4) of SPCEs modified by (A) and (B) AP–RGO, OLEAM-Au NPs and A_OLEAM, (C) and (D) A_OLEAM, D_OLEAM, A_CH3OH/AcOH, D_CH3OH/AcOH and (inset of C) A_MCH and D_MCH. | ||
Fig. 5(A) shows that the AP–RGO/Au NPs and the A_OLEAM electrodes exhibit a similar electrochemical behaviour, presenting an anodic plateau and a cathodic peak in the 0.2–0.6 V range, that the bare AP–RGO electrodes instead do not show. The cathodic peak can be ascribed to the reduction of the electrochemically active Au NPs that are at the surface of the AP–RGO sheets.
The Bode plots of Fig. 5(B) show a pure-like capacitive behaviour for the AP–RGO electrodes, whereas the OLEAM–Au NP modified electrodes show a deviation from such a behaviour due to the presence of the metal NPs, thus suggesting electron transport properties intermediate between the two components of the nanocomposite.
The effect of the Au NP size and surface chemistry on the current intensities and red/ox potentials of the hybrid modified electrodes was investigated by means of CV and EIS measurements of A_OLEAM and D_OLEAM electrodes, as deposited and after treatment with CH3OH/AcOH and MCH. The D_OLEAM electrodes are hereafter indicated as D_CH3OH/AcOH and D_MCH after the treatment.
Fig. 5(C) shows that both the A_OLEAM and D_OLEAM electrodes have the same CV curves, specifically a cathodic peak ascribed to the background reduction at the potential of +0.4 V (SCE), while D_OLEAM shows a higher current, likely due to an increase of the electroactive surface area.
The evidence of the cathodic peak at +0.4 V for both A_OLEAM and D_OLEAM electrodes can be explained by considering that the dielectric properties of OLEAM limit both the interparticle electron transfer and the heterogeneous charge transfers at the electrode/electrolyte interface, thus mitigating the differences between the electrochemical properties of the two modified SPCEs. Indeed, A_OLEAM and D_OLEAM also show the same frequencies in the Bode plots of Fig. 5(D).
After treatment with CH3OH/AcOH, both A_CH3OH/AcOH and D_CH3OH/AcOH exhibit an increase of the current, which can be ascribed to the displacement of OLEAM with the shorter alkyl moiety acetate ions.55 In addition, A_CH3OH/AcOH shows the reduction of Au at lower potentials than D_CH3OH/AcOH, that is at +0.36 V vs. +0.40 V, as a more energetically favourable process. This finding can be explained by a higher catalytic activity of the smaller Au NPs and a higher charge transfer capability at the electrode/electrolyte interface, due to a larger surface area and a higher NP surface free energy.56 Similarly, the Bode plots show a frequency much higher for A_CH3OH/AcOH than for the D_CH3OH/AcOH, thus confirming its higher electrochemical activity.
Finally, the hybrid modified SPCEs treated with MCH present a current line-shape significantly different with respect to A_CH3OH/AcOH and D_CH3OH/AcOH (inset of Fig. 5(C)). In fact, both A_MCH and D_MCH show a total pseudocapacitive current significantly higher, and present enhanced anodic and cathodic currents, with two reduction peaks at low potentials, +0.22 V and −0.014 V, in agreement with the lowest frequencies they show in the Bode plots (Fig. 5(D)).
The observed pseudocapacitive behaviour can be likely ascribed to nanowelding phenomena occurring among Au NPs after treatment with MCH (Fig. 4(C)), which can account for the formation of electron percolation pathways among interconnected Au NPs57 and enhancement of the electrode conductivity. In addition, the pseudocapacitive currents are much more enhanced in D_MCH than in A_MCH (Fig. 5C), likely due to the lower electrochemical reactivity of larger Au NPs.56
CV and EIS curves were recorded also in the presence of the inner-sphere redox probe K4[Fe(CN)6] (Fig. S4, ESI†) in the buffer PBS solution. The selected probe shows electrochemical behavior that is more sensitive to the chemistry (i.e. oxygen moieties, impurities, and adsorption sites) and to the structure of the electrode surface, rather than to the electrode electron density of states (DOS).58
The CV curves and Bode plots collected at the SPCEs modified by AP–RGO and OLEAM-coated Au NPs, and at the A_OLEAM electrodes, in PBS solution and in the presence of the redox probe (Fig. S4, ESI†), confirm the higher catalytic activity of the nanocomposite with respect to the neat hybrid components, and the higher catalytic activity and electrochemical surface reactivity of the A_CH3OH/AcOH and A_MCH electrodes accountable for the smaller Au NPs.56
The effect of AP on the electrochemical properties of hybrid nanocomposite modified electrodes and the charge transfers at their surface were further investigated by comparing them with those of SPCEs modified by a layer-by-layer junction, prepared by drop casting, first, a dispersion of AP–RGO in toluene, and then, a dispersion of previously prepared OLEAM-Au NPs. CV and EIS measurements recorded with and without the redox K4[Fe(CN)6] probe (Fig. S5, ESI†) have demonstrated a significant enhancement of the electrochemical activity of A_OLEAM with respect to the layer-by-layer junction, thus remarking the effectiveness of the AP linker in electronically coupling the hybrid components.
3.3 Electrochemical detection of dopamine
The investigation and assessment of the electroanalytical potential of the hybrid nanocomposites conveniently designed for the SPCE modification, to address the detection of the DA, were performed by differential pulse voltammetry (DPV).DPV curves show that all the investigated SPCEs can detect DA (Fig. 6(A)), likely due to aromatic π–π stacking interactions between DA and the AP–RGO platform59 and to the coordination of its –OH and –NH2 groups to the Au NP surface (Fig. 6(D)).60 Besides, the oxidation current of DA at A_OLEAM is higher than that at the layer-by-layer junction and at the OLEAM-Au NPs alone, also reasonably accounted for the electron coupling function of AP. Finally, the current at the A_OLEAM is comparable to that at the AP–RGO modified SPCEs (Fig. 6(A)) likely due to the dielectric behaviour of OLEAM at the surface of the nanocomposite. After displacement of the insulating layer of OLEAM with CH3OH/AcOH, the oxidation currents of DA at A_CH3OH/AcOH and D_CH3OH/AcOH show respectively a ca. 8-fold and 2-fold increase with respect to that at AP–RGO, thanks to the synergistic combination of the electrochemical reactivity and electrical conductivity of the Au NPs and RGO, which is granted by the AP linker coupling agent, and to the easier access of DA molecules to the surface of the NPs, which penetrate through a thinner surface coating layer of acetate ions to reach the NP surface. Besides, the oxidation current at the A_CH3OH/AcOH is almost 4-fold higher than that at the D_CH3OH/AcOH (Fig. 6(B)).
 | ||
| Fig. 6 DPV curves at (A) A_OLEAM, OLEAM–Au NPs, AP–RGO and layer-by-layer junction, and (B) A_OLEAM and D_OLEAM, before and after treatment with CH3OH/AcOH, after addition of 10−5 M DA, between −0.2 V and +0.6 V (SCE), with pulse of 5 mV and amplitude of 50 mV in 0.1 M PBS (pH 7.4). (C) DPV recorded at A_CH3OH/AcOH after consecutive additions of DA, from 0.013 to 0.130 μM, and the corresponding calibration plot (inset). (D) Sketch of the detection of DA at the CH3OH/AcOH treated hybrid electrodes. (The sketches are not drawn to scale). | ||
This result may be explained by the higher electrochemical activity of the smaller Au NPs56 and of their higher curvature, which results in a less closely packed layer of coordinating species at the NP surface, favouring the DA intercalation between such ligands and, accordingly more effective chemical interactions at the NP surface.
It is worth noting that the oxidation current of DA at both the A_MCH and D_MCH electrodes shows an irregular line-shape without any oxidation peak (data not reported), likely due to the MCH coverage of the modified electrode surface, which may somewhat hinder the aromatic π–π stacking interactions of DA with the AP–RGO surface, as well as its coordination by the –OH and –NH2 groups to the Au nanostructures, with the semi-covalent Au–thiol bonds stronger than these interactions.55,61
A calibration curve for DA in the 0.013–0.130 μM concentration range was obtained by recording the DPV current at A_CH3OH/AcOH electrodes (Fig. 6(D)), and two trends are observed, probably due to the fouling action of DA with a LOD of 0.0025 μM (RSD < 1%). The two equations of the calibration linear ranges, and the corresponding squared values of R, are y = 41x + 1.236 × 10−5 with R2 = 0.998 and y = 22x + 1.310 × 10−5 with R2 = 0.987, respectively. It is worth noting that the estimated LOD is among the lowest reported for electroanalytical devices integrating graphene/Au NP nanocomposites presented in recent literature12–17,29–31 (Table S1, ESI†) and it is suitable for diagnosis of human disorders.32
3.4 Effect of the electrode surface chemistry on the detection of H2O2
The two treated electrodes were also tested for possible detection of H2O2, one of the intracellular reactive oxygen species, which is typically generated under normal physiological conditions, at rather low concentrations,36 in enzymatic and non-enzymatic reactions resulting from conversion of intracellular reactive oxygen species,62 and hence, when unregulated because of pathological conditions, it may increase up to ∼10−3 M36 producing oxidative stress and leading to a decline of cognitive functions, also causing Alzheimer's and Parkinson's disease.33–36CV scans and amperometry curves were collected to investigate the current response of H2O2 at the A_OLEAM and D_OLEAM electrodes, before and after treatment with MCH and CH3OH/AcOH. These measurements were performed at pH 8, which is the optimal pH at which enzymes such as superoxide dismutase (SOD) and glutathione peroxide rapidly convert superoxide to H2O2.63
Panel A of Fig. 7 shows the high current density at the hybrid modified SPCEs after treatment with MCH (Fig. 7(A)). On the other hand, D_OLEAM electrodes have a higher capacitive current (Panel A) and a higher current at the Au oxidation peak with respect to A_OLEAM (Panel B), in agreement with the results of Fig. 5(C) and Fig. S4(C), ESI.†
 | ||
| Fig. 7 (A) and (B) CV curves of bare SPCEs, A_OLEAM, D_OLEAM, A_MCH and D_MCH, collected at the scan rate of 50 mV s−1 in 0.1 M PBS buffer (pH 8.0), for neat (A) and added with 50 mM H2O2 (B). (C) Amperometric response of H2O2 at SPCEs, as bare and modified with A_OLEAM, D_OLEAM, A_MCH and D_MCH, collected in 0.05 M PBS (pH 8.0) at +0.70 V (SCE). (D) Sketch of the detection of H2O2 using the MCH treated hybrid electrodes. (The sketches are not drawn to scale). | ||
Amperometric measurements at the bare SPCEs display only a slight increase of the current density to successive additions of H2O2, a detectable increase at A_OLEAM and D_OLEAM, which is, as expected, higher in A_OLEAM56 (Fig. 7(C)), and a significant enhancement at A_MCH, which reaches the steady-state value within few seconds.
Such evidence accounts for the higher sensitivity of the hybrid electrodes treated with MCH for the detection of H2O2, due to the enhancement of the film conductivity, that is provided by formation of percolation pathways among the Au NPs, resulting interconnected for the MCH induced nanowelding phenomena.57
The LOD estimated for the A_OLEAM electrodes is ca. 2.9 μM, while the A_CH3OH/AcOH modified counterparts show a lower sensitivity (Fig. S6, ESI†) with a LOD of 40 μM. Conversely, in the case of the A_MCH, it decreases down to ca. 0.3 μM with a satisfactory reproducibility of the electrodes, namely at a concentration of 100 μM (1008 ± 45 nA) the %RSD is 5% (n = 5), and with the equation of the calibration linear range y = 10.22x + 7.76 and R2 = 0.995, demonstrating the effectiveness of the electrode in the detection of H2O2 for the monitoring of abnormal physiological processes.33–36
4. Conclusions
A novel hybrid nanocomposite formed of 1-amminopyrene (AP) modified RGO sheets decorated with Au NPs has been prepared by a facile, in situ and cost-effective colloidal route. Au NPs, ranging from 9–20 nm as mean size, have been found to heteronucleate and grow on the –NH2 groups of the AP–RGO sheets in OLEAM. The role of OLEAM solvent as mild reductant of the HAuCl4 × 3H2O precursor and surface coordinating agent for the grown NPs has been demonstrated. On the other hand, AP has been found to firmly anchor the NPs via aromatic π–π stacking interactions onto the sheets and to as coordinating site for the formed NPs. The size distribution of the NPs decorating the flakes has been found to be affected by an interplay of OLEAM and AP concentrations.A suitable tuning of the AP–RGO:HAuCl4 × 3H2O w/w and of the OLEAM:HAuCl4 × 3H2O molar ratio has led to hybrid nanocomposites formed by AP–RGO flakes, densely and uniformly decorated with a multilayer layout of monodisperse Au NPs coordinated by OLEAM, featuring intense absorption signals in the visible spectral range.
The dispersibility of the nanocomposite in organic solvent, ensured by the coordination of the OLEAM ligand to the Au NP surface, has enabled the prompt modification of disposable screen printed electrodes (SPCEs) for the label free detection of dopamine (DA), biomarker of human neurodegenerative diseases.
An enhanced conductivity and charge transfer capability of the AP–RGO/Au NP modified SPCEs have been found due to the effective electron coupling of the nanocomposite components. The novel manufactured electroanalytical nanoplatform has shown high electrochemical activity, higher for the nanocomposite featuring smaller Au NPs, and with a sensitivity to the tested analytes, tuneable by modifying the Au NP surface chemistry. Indeed, the LOD for the detection of DA has been determined, and to the best of our knowledge, it is among the lowest reported for this class of graphene/Au nanocomposite based electrochemical sensors, thus envisioning their potential for innovative point-of-care technology for the clinical diagnosis of DA.
The electroactivity, spectroscopy properties and processability from solution of the manufactured nanocomposites make them interesting for applications in optical (bio)sensors, (photo)electrocatalysis, (photo)catalysis, photodetectors, and photovoltaic cells. Finally, the synthetic strategy developed here can be extended to the manufacturing of hybrid nanocomposites formed of RGO, as well as of other carbon nanostructured derivatives, and NPs of different chemical compositions, for targeting other technology purposes.
Author contributions
Conceptualization: C. I., V. P., A. T., F. B., I. P., M. L. C. and L. F.; investigation; C. I., M. C., G. V. B., V. P.,A. T. and F. B.; methodology: C. I., V. P., A. T., F. B., I. P. and L. F.; supervision: I. P., M. L. C. and L. F.; writing – original draft preparation: C. I., V. P., A. T.; writing – review and editing: I. P., L. F., M. S., E. F., N. D. and M. L. C.; funding acquisition: C.I., I. P. and L. F. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge TOPSIS bilateral CNR-BAS (n. prot. 0089480/2018) and TOOL bilateral CNR-MHESR Projects and the Academic Horizon Europe Seeds Project BIOMAD funded by University of Bari (Italy).Notes and references
- A. A. Grace, Neuroscience, 1991, 41, 1 CrossRef CAS.
- A. Baskerville and A. J. Douglas, CNS Neurosci. Ther., 2010, 16, 92 Search PubMed.
- D. Chaudhury, J. J. Walsh, A. K. Friedman, B. Juarez, S. M. Ku, J. W. Koo, D. Ferguson, H. C. Tsai, L. Pomeranz, D. J. Christoffel, A. R. Nectow, M. Ekstrand, A. Domingos, M. S. Mazei-Robison, E. Mouzon, M. K. Lobo, R. L. Neve, J. M. Friedman, S. J. Russo, K. Deisseroth, E. J. Nestler and M. H. Han, Nature, 2013, 493, 532 CrossRef CAS.
- Q. J. Huys, P. N. Tobler, G. Hasler and S. B. Flagel, Prog. Neurobiol., 2014, 211, 31 CAS.
- S. Chakraborty and C. R. Raj, Biosens. Bioelectron., 2009, 24, 3264 CrossRef CAS PubMed.
- G. Li, J. Wu, X. Qi, X. Wan, Y. Liu, Y. Chen and L. Xu, Mater. Today Chem., 2022, 26, 101043 CrossRef CAS.
- G. Li, X. Qi, J. Wu, L. Xu, X. Wan, Y. Liu, Y. Chen and Q. Li, J. Hazard. Mater., 2022, 436, 129107 CrossRef CAS PubMed.
- G. V. Bianco, A. Sacchetti, C. Ingrosso, M. M. Giangregorio, M. Losurdo, P. Capezzuto and G. Bruno, Carbon, 2018, 129, 869 CrossRef CAS.
- C. Ingrosso, A. Petrella, M. L. Curri, M. Striccoli, P. Cosma, P. D. Cozzoli and A. Agostiano, Appl. Surf. Sci., 2005, 246, 367 CrossRef CAS.
- M. L. Curri, R. Comparelli, M. Striccoli and A. Agostiano, Phys. Chem. Chem. Phys., 2010, 12, 11197 RSC.
- D. Quesada-González and A. Merkoçi, Chem. Soc. Rev., 2018, 47, 4697 RSC.
- X. Huang, W. Shi, N. Bao, C. Yu and H. Gu, Microchim. Acta, 2019, 186, 310 CrossRef.
- C. Tan, J. Zhao, P. Sun, W. Zheng and G. Cui, New J. Chem., 2020, 44, 4916 RSC.
- J. Gu, Q. An, J. Chen, Y. He and W. Huang, Inorg. Chem. Commun., 2023, 147, 110282 CrossRef CAS.
- X. Yan, Y. Gu, C. Li, B. Zheng, Y. Li, T. Zhang, Z. Zhang and M. Yang, Sens. Actuators, B, 2018, 257, 936 CrossRef CAS.
- K. Zhang, X. Chen, Z. Li, Y. Wang, S. Sun, L. Wang, T. Guo, D. Zhang, Z. Xue, X. Zhou and X. Lu, Talanta, 2018, 178, 315 CrossRef CAS.
- Y. Hou, K. Sheng, Y. Lu, C. Ma, W. Liu, X. Men, L. Xu, S. Yin, B. Dong, X. Bai and H. Song, Microchim. Acta, 2018, 185, 1 CrossRef CAS.
- X. Xia, S. Guo, W. Zhao, P. Xu, H. Yu, T. Xu and X. Li, Sens. Actuators, B, 2014, 202, 846 CrossRef CAS.
- L. Liu, J. Liu, Y. Wang, X. Yan and D. D. Sun, New J. Chem., 2011, 35, 1418 RSC.
- X. Xu, Q. Zhenga, G. Baia, L. Song, Y. Yao, X. Cao, S. Liu and C. Yao, Electrochim. Acta, 2017, 242, 56 CrossRef CAS.
- R. Muszynski, B. Seger and P. V. Kamat, J. Phys. Chem. C, 2008, 112, 5263 CrossRef CAS.
- C. Ingrosso, M. Corricelli, A. Disha, E. Fanizza, G. V. Bianco, N. Depalo, A. Agostiano, M. Striccoli and M. L. Curri, Carbon, 2019, 152, 777 CrossRef CAS.
- C. Ingrosso, V. Valenzano, M. Corricelli, A. Testolin, V. Pifferi, G. V. Bianco, R. Comparelli, N. Depalo, E. Fanizza, M. Striccoli, A. Agostiano, I. Palchetti, L. Falciola and M. L. Curri, Carbon, 2021, 182, 57 CrossRef CAS.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270 CrossRef CAS.
- C. Ingrosso, G. V. Bianco, V. Pifferi, P. Guffanti, F. Petronella, R. Comparelli, A. Agostiano, M. Striccoli, I. Palchetti, L. Falciola, M. L. Curri and G. Bruno, J. Mater. Chem. A, 2017, 5, 9307 RSC.
- G. Li, X. Wan, Y. Xia, D. Tuo, X. Qi, T. Wang, M. Mehmandoust, N. Erk, Q. He and Q. Li, ACS Appl. Nano Mater., 2023, 6, 17040 CrossRef CAS.
- V. M. Georgakilas, A. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156 CrossRef CAS PubMed.
- H. Hiramatsu and F. E. Osterloh, Chem. Mater., 2004, 16, 2509 CrossRef CAS.
- Y. Xia, G. Li, Y. Zhu, Q. He and C. Hu, Microchem. J., 2023, 190, 108726 CrossRef CAS.
- F. Li, B. Ni, Y. Zheng, Y. Huang and G. Li, Surf. Interfaces, 2021, 26, 101375 CrossRef CAS.
- Q. Li, Y. Xia, X. Wan, S. Yang, Z. Cai, Y. Ye and G. Li, Mater. Sci. Eng., 2020, 109, 110615 CrossRef CAS.
- S. M. Matt and P. J. Gaskill, J. Neuroimmune Pharmacol., 2020, 15, 114 CrossRef CAS PubMed.
- J. Yang, J. Yang, S. H. Liang, Y. Xu, A. Moore and C. Ran, Sci. Rep., 2016, 6, 35613 CrossRef CAS PubMed.
- D. Praticò, Trends Pharmacol. Sci., 2008, 29, 609 CrossRef.
- P. Jenner, Ann. Neurol., 2003, 53, S26 CrossRef CAS.
- Y. Luo, H. Liu, Q. Rui and Y. Tian, Anal. Chem., 2009, 81, 3035 CrossRef CAS.
- D. Konios, M. M. Stylianakis and E. Kymakis, J. Colloid Interface Sci., 2014, 430, 108 CrossRef CAS.
- M. Ahmad, H. An, Y. S. Kim, J.-H. Lee, J. Jung, S.-H. Chun and Y. Seo, Nanotechnology, 2012, 23, 285705 CrossRef.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291 RSC.
- M. Zhang, R. R. Parajuli, D. Mastrogiovanni, B. Dai, P. Lo, W. Cheung, R. Brukh, P. L. Chiu, T. Zhou, Z. Liu, E. Garfunkel and H. He, Small, 2010, 6, 1100 CrossRef CAS PubMed.
- L. Li, X. Zheng, J. Wang, Q. Sun and Q. Xu, ACS Sustainable Chem. Eng., 2013, 1, 527 CrossRef CAS.
- Y.-Y. Ou and M. H. Huang, J. Phys. Chem. B, 2006, 110, 2031 CrossRef CAS.
- A. Ferrari, Solid State Commun., 2007, 143, 47 CrossRef CAS.
- S. Mourdikoudis and L. M. Liz-Marzań, Chem. Mater., 2013, 25, 1465 CrossRef CAS.
- D. Thangaraju, R. Karthikeyan, N. Prakash, S. M. Babu and Y. Hayakawa, Dalton Trans., 2015, 44, 15031 RSC.
- A. Kumar, S. Mandal, P. R. Selvakannan, R. Pasricha, A. B. Mandale and M. Sastry, Langmuir, 2003, 19, 6277 CrossRef CAS PubMed.
- A. Kisner, S. Lenk, D. Mayer, Y. Mourzina and A. Offenhausser, J. Phys. Chem. C, 2009, 113, 20143 CrossRef CAS.
- X. Dong, X. Ji, J. Jing, M. Li, J. Li and W. Yang, J. Phys. Chem. C, 2010, 114, 2070 CrossRef CAS.
- V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588 CrossRef CAS.
- S. Pud, A. Kisner, M. Heggen, D. Belaineh, R. Temirov, U. Simon, A. Offenhausser, Y. Mourzina and S. Vitusevich, Small, 2012, 9, 846 CrossRef.
- L. H. Dubois and R. G. Nuzzo, Annu. Rev. Phys. Chem., 1992, 43, 437 CrossRef CAS.
- A. Halder and N. Ravishankar, Adv. Mater., 2007, 19, 1854 CrossRef CAS.
- E. Koposova, X. Liu, A. Kisner, Y. Ermolenko, G. Shumilova, A. Offenhäusser and Y. Mourzina, Biosens. Bioelectron., 2014, 57, 54 CrossRef CAS PubMed.
- M. Ding, D. C. Sorescu, G. P. Kotchey and A. Star, J. Am. Chem. Soc., 2012, 134, 3472 CrossRef CAS PubMed.
- Z. Yang, M. Wang, Y. Shi, X. Song, Z. Lin, Z. Ren and J. Bai, J. Mater. Chem., 2012, 22, 21009 RSC.
- W. Plieth, J. Phys. Chem., 1982, 86, 3166 CrossRef CAS.
- M. Han, F. Chen, M. Li, R. Yu, Y. Xu, Y. Jiang, C. Liu and J. Hu, Electrochim. Acta, 2020, 334, 135626 CrossRef CAS.
- L. Liu, S. Ryu, M. R. Tomasik, E. Stolyarova, N. Jung, M. S. Hybertsen, M. L. Steigerwald, L. E. Brus and G. W. Flynn, Nano Lett., 2008, 8, 1965 CrossRef CAS.
- F. Seven, T. Gölcez and M. Şen, J. Electroanal. Chem., 2020, 864, 114104 CrossRef CAS.
- D. S. Kim, E. S. Kang, S. Baek, S. S. Choo, Y. H. Chung, D. Lee, J. Min and T. H. Kim, Sci. Rep., 2018, 8, 1 Search PubMed.
- H. Schlicke, J. H. Schroder, M. Trebbin, A. Petrov, M. Ijeh, H. Weller and T. Vossmeyer, Nanotechnology, 2011, 22, 305303 CrossRef.
- P. Newsholme, E. Rebelato, F. Abdulkader, M. Krause, A. Carpinelli and R. Curi, J. Endocrinol., 2012, 214, 11 CAS.
- Y. C. Awasthi, E. Beutler and S. K. Srivastava, J. Biol. Chem., 1975, 250, 5144 CrossRef CAS; B. B. Keele Jr., J. M. McCord and I. Fridovich, J. Biol. Chem., 1971, 246, 2875 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00785e |
| This journal is © The Royal Society of Chemistry 2024 |