
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe Gore-Tex® effect in externally hydrophobic Metal–Organic Frameworks†
Kaleb L.
Miller
 b,
Rijia
Lin
c,
Jingwei
Hou
c,
Cameron J.
Kepert
*b,
Deanna M.
D’Alessandro
*ab and
Marcello B.
Solomon
*ab
b,
Rijia
Lin
c,
Jingwei
Hou
c,
Cameron J.
Kepert
*b,
Deanna M.
D’Alessandro
*ab and
Marcello B.
Solomon
*ab
aSchool of Chemical and Biomolecular Engineering, The University of Sydney, New South Wales 2006, Australia
bSchool of Chemistry, The University of Sydney, New South Wales 2006, Australia. E-mail: cameron.kepert@sydney.edu.au; deanna.dalessandro@sydney.edu.au; marcello.solomon@sydney.edu.au
cSchool of Chemical Engineering, The University of Queensland, Queensland 4072, Australia
First published on 7th February 2024
Abstract
UiO-66-NH2 ([Zr6O4(OH)4(NH2-bdc)], NH2-bdc = 2-aminoterephthalic acid) was post-synthetically modified using four strategies to improve its external hydrophobicity. The modified MOFs exhibited increased hydrophobicity to water droplets, but their internal hydrophobicity remained unchanged. This highlights a disconnect between hydrophobicity towards vapour and liquid water, referred to as the Gore-Tex® effect.
Introduction
Since their discovery over 30 years ago, Metal–Organic Frameworks (MOFs) have been earmarked as a class of materials capable of revolutionising the CO2 capture, storage and removal industries.1–3 Their potential applications continue to grow as new architectures are discovered. Sustained efforts in MOF syntheses have enabled experimental internal surface areas of up to 6550 m2g−1 to be reached.1,4 Theoretically, the surface areas of MOF materials can reach beyond 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m2g−1.5 High surface areas and accessible pore spaces have enabled the pores of MOFs to play host to a variety of functional chemistries, including fine chemical production,6 sensing7 and gas separations.8 MOFs are well studied on the laboratory bench; however, translating their technology to the industrial scale requires further scrutiny. Of the 90000+ architectures reported to date,9 only a few MOFs have been documented for their commercial use.10,11
000 m2g−1.5 High surface areas and accessible pore spaces have enabled the pores of MOFs to play host to a variety of functional chemistries, including fine chemical production,6 sensing7 and gas separations.8 MOFs are well studied on the laboratory bench; however, translating their technology to the industrial scale requires further scrutiny. Of the 90000+ architectures reported to date,9 only a few MOFs have been documented for their commercial use.10,11
When MOFs are evaluated for their suitability to capture CO2 under laboratory conditions, many industrially relevant conditions are irreplicable or may be overlooked. An important factor in the laboratory is the overall CO2 capacity that a material may possess; however, this is normally determined through thermodynamic measurements under equilibrium conditions by slowly dosing in CO2 to an evacuated sample of material.12 Selectivity – the extent in which a material can favour particular analytes in mixtures without interference from other components13 – is usually determined in the laboratory from a two-component gas mixture under dry conditions,14 or from competitive breakthrough experiments.15,16 In real world conditions, high selectivity for CO2 over multiple gases (N2, O2, H2O, SOx, NOx among others) is often crucial for the performance of a CO2 capture material. The MOF material may also need to operate under non-ideal conditions, such as at elevated temperatures, increased pressures and humid gas streams. A particular industrial challenge is the competitive binding between CO2 and H2O for a solid-state adsorbent. H2O often adsorbs preferentially to CO2 due to its ability to form extensive hydrogen bonding with the MOF, or with itself within the MOF pores, thus reducing the capacity for CO2.17 Furthermore, many MOFs suffer from their sensitivity to moisture and may degrade upon exposure to atmospheric H2O. The archetypal framework MOF-5 ([ZnO4(bdc)3] bdc = 1,4-benzenedicarboxylate) has reported surface areas of 3800 m2g−1 and pore volumes of 1.55 cm3 g−1; however, its hydrolytic instability makes it unsuitable in applications involving humid air as the H2O breaks down the framework.18,19 Improving the hydrophobicity of a MOF is hypothesised to surmount some of these fundamental challenges.
Endowing a MOF with innate hydrophobicity, or introducing a hydrophobic composite material into a MOF may help diminish the detrimental impact of H2O on a pristine framework.20,21 Hydrophobic porous materials are gaining interest for reducing the competition between CO2 and H2O for their deployment into the gas capture and storage field under humid conditions.8,22–25 Hydrophobicity can be achieved via one of three pathways: (1) deliberate pre-synthetic design using ligands with hydrophobic properties, although pre-designed hydrophobicity may not guarantee that the resultant framework will be hydrophobic, since the metal secondary building unit (SBU) of the framework may bind H2O;26 (2) in situ functionalisation of the ligand or SBU to generate an intrinsically hydrophobic MOF;27 or (3) bestowing hydrophobicity through post-synthetic modification (PSM)28via ligand or metal-ion exchange, ligand functionalisation, or guest introduction.29 The benefit of PSM is the ability to produce functional MOFs in a simple, inexpensive and scalable way, which is an enticing pathway towards their industrial deployment.
Porous materials possess two types of hydrophobicity: internal and external hydrophobicity.30,31 Internal hydrophobicity is defined by the adsorption of water vapour to the internal pore surface of a porous material (as influenced by the competition between weak favourable enthalpic water–surface interactions and unfavourable adsorption entropies), while external hydrophobicity concerns the interaction of liquid H2O at the surface of the material (relating principally to the dominance of water–water over water–surface interaction enthalpies). External hydrophobicity acts on a macroscopic scale compared to internal hydrophobicity and is generally characterised by a water contact angle measurement. Their distinction is essential given that hydrophobic porous materials are used for applications concerning both liquid and gaseous H2O. A challenge arises when measurements for external hydrophobicity are extrapolated to make assumptions about the internal hydrophobicity of a material.30,32 Computational studies performed by Snurr et al. suggest that hydrophobicity indicated by the external water contact angle does not correlate with the hydrophobicity of the internal pore environments, indicated by vapour sorption.30 Porous materials may exhibit external hydrophobicity without exhibiting internal hydrophobicity, similar to Gore-Tex®33 – the material used in hydrophobic clothing.30
External hydrophobicity, as measured by the water contact angle, is frequently assumed to be a reliable metric for characterising the performance of MOFs under humid conditions;23,34–37 however, this is not the case. To the best of our knowledge, we present here the first study which practically demonstrates the differences between internal and external hydrophobicity in MOFs. We explore and apply four hydrophobic PSMs to UiO-66-NH2 – a readily scalable MOF that can be synthesised via green pathways.38 These hydrophobic PSMs include: (1) Enteric coatings – a widespread, cheap class of polymers used in the pharmaceutical industry to protect medications as they pass through the human digestive system,39–41 and have recently found effectiveness in imparting hydrophobicity to MOFs;42 (2) Long-chained alkanes with a terminal group enabling post-synthetic modification to the SBU of a MOF or reaction with a ligand – these have been well studied in their improvement in the stability of MOFs to water;43,44 (3) Organosilicon infiltration – where cheap, commercially available organosilicone-based materials8,24 can create a hydrophobic layer on the exterior surface of the MOF to improve its stability to water; and (4) SBU modification, which may proceed by dative coordination of molecules to unsaturated SBUs.29,45
Results and discussion
Development of PSM strategies and synthesis of UiO-66-NH2
UiO-66-NH2 was successfully synthesised according to the literature methodology, with PXRD and Le Bail data matching that previously reported (Fig. S1 and Table S1, ESI†). SEM images are also consistent with previously reported micrograms (Fig. S2, ESI†).46 Each of the four strategies (Fig. 1) were tested on the same batch of UiO-66-NH2, allowing for their direct comparison.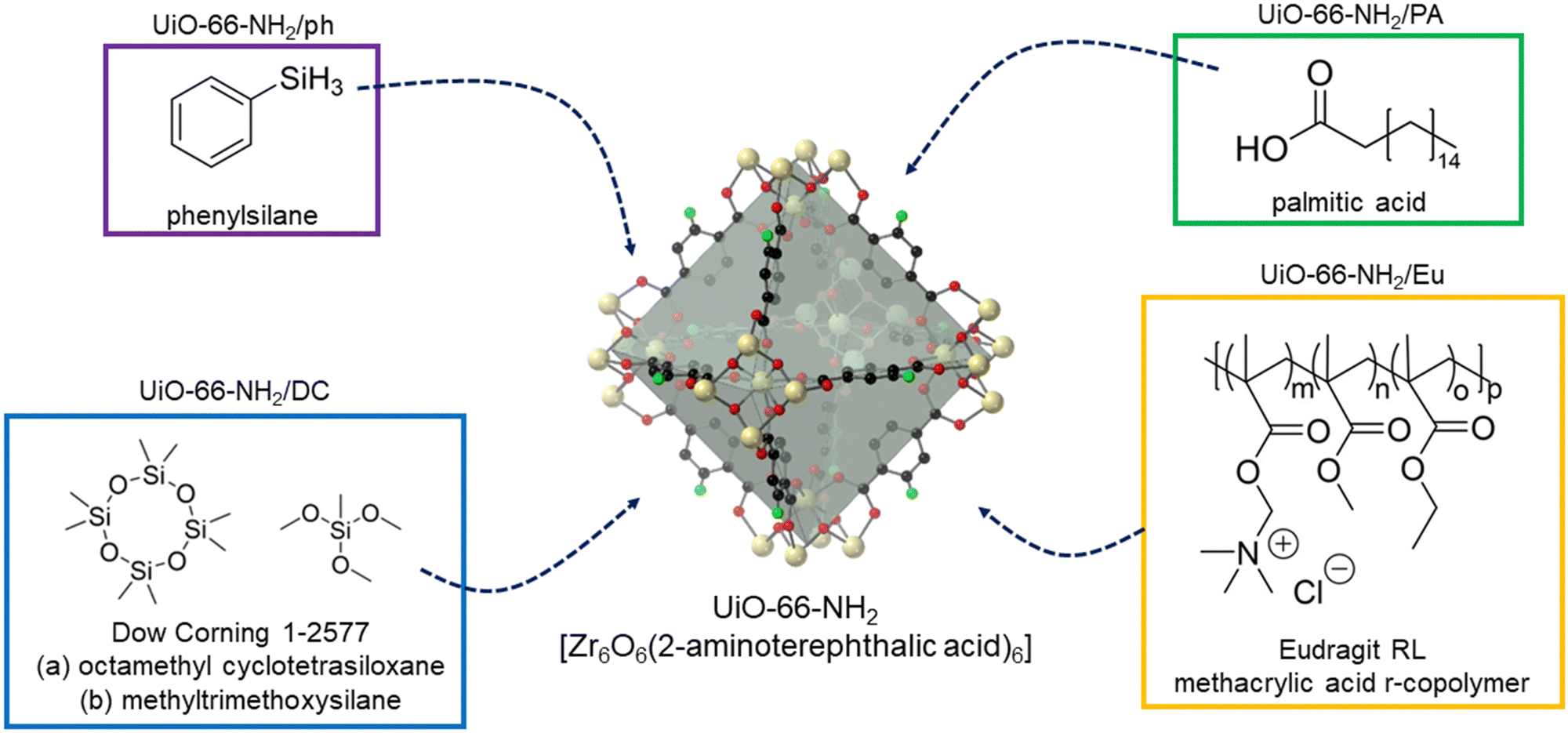 | ||
| Fig. 1 The four post-synthetic modification strategies used to endow UiO-66-NH2 with hydrophobicity. | ||
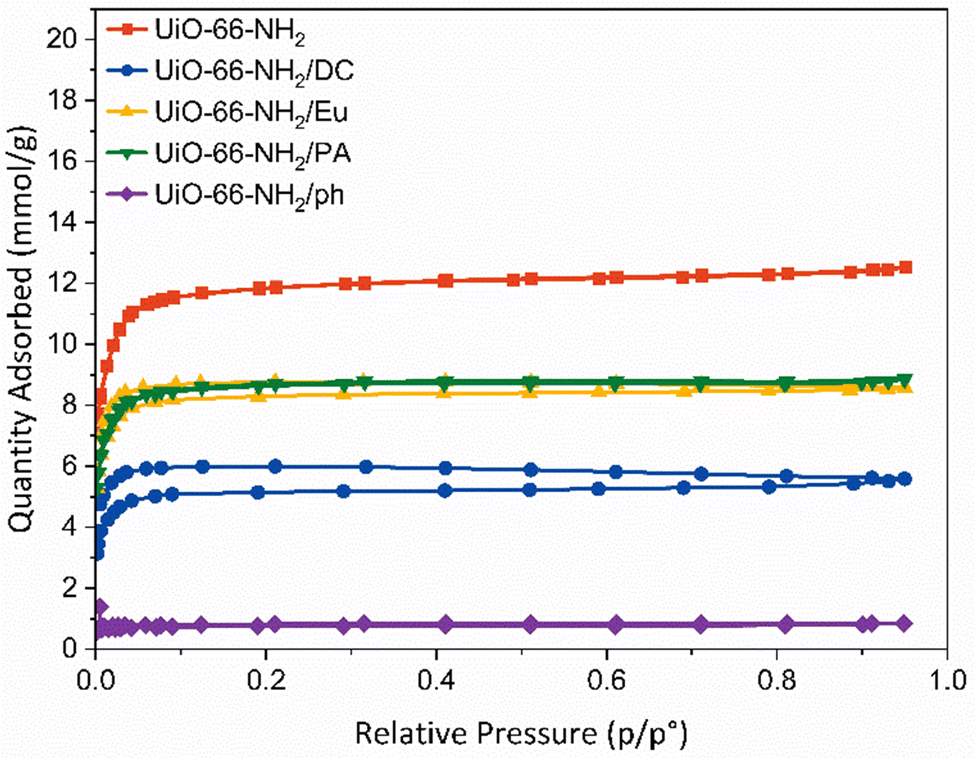 | ||
| Fig. 2 N2 gas sorption measurements performed at 77 K for pristine UiO-66-NH2 (red) and its post-synthetically modified derivatives. UiO-66-NH2/Eu (yellow), UiO-66-NH2/PA (green), UiO-66-NH2/DC (blue) and UiO-66-NH2/ph (purple). Adsorption and desorption isotherms are denoted by filled and hollow symbols, respectively. | ||
Brunauer–Emmett–Teller (BET) surface areas were calculated for each sample.52 Pristine UiO-66-NH2 had a BET surface area of 1054 ± 19 m2g−1, which is consistent with literature reports (Table S4, ESI†).53,54 Post-synthetic modifications of the framework caused the reduction in surface area to varying degrees. The most significant reduction in surface area was to UiO-66-NH2/ph, where a BET surface area of 66 ± 1 m2g−1 was determined. It is likely that the phenylsilane grafting onto the zirconium SBU caused blocking of the framework pore windows from the aromatic ring. This was supported by pore volume measurements, which indicate a reduction from 0.44 to 0.03 cm3 g−1. The DOWSILTM coating resulted in a surface area reduction to 465 ± 6 m2 g−1, which is attributable to pore infiltration of the smaller active hydrophobic agents. The pore volume reduced from 0.44 to 0.19 cm3 g−1, confirming a degree of pore blocking by the organosilicon. The polymeric derivatives, UiO-66-NH2/Eu and UiO-66-NH2/PA, yielded surface areas of 779 ± 9 m2 g−1 and 759 ± 9 m2 g−1, respectively, with respective pore volume reductions to 0.30 and 0.31 cm3 g−1. This suggests that the long-chained nature of the coating agents still allows for adsorption to occur.
Pore size distribution data from gas sorption measurements demonstrate the impact of PSM on the parent UiO-66-NH2 framework (Fig. S24, ESI†). The pristine UiO-66-NH2 exhibits two different sized micropores (16 Å and 18.5 Å). UiO-66-NH2/Eu and UiO-66-NH2/DC both reveal a reduction in the larger 18.5 Å pore size, implying that it is filled with fragments of the enteric and organosilicon coatings. A broader Gaussian peak shape in the pore size at 16 Å indicates less gas entering the pores. The two micropores are preserved upon coating with a long-chain alkane, indicating that the coating of the alkane mostly occurs on the surface of the material. Unlike in the case of Eudragit RL® or DOWSIL™ coating, there are no bulky moieties in the PA, which may only result in a partial blockage of the larger pores. UiO-66-NH2/ph demonstrates the greatest variation in pore size, with micropores of 13 Å, 16 Å, 18.5 Å and 21–24 Å determined. π–π stacking from the phenylsilane moieties may be creating additional pores within the framework material.
The interaction of the frameworks with CO2 was examined using CO2 adsorption experiments at 288, 298 and 308 K (Fig. 3 and Fig. S25, ESI†). At atmospheric pressure (1013.2 mbar), the adsorption of CO2 was 1.99 mmol g−1, which is comparable to that observed in the literature.55,56 The isosteric heat of adsorption for pristine UiO-66-NH2 was found to be 27.47 kJ mol−1. Of greatest interest is the improvement of CO2 uptake in the UiO-66-NH2/Eu sample. The adsorption of CO2 at 1000 mbar increased to 2.19 mmol g−1 and the isosteric heat of adsorption increased to 29.12 kJ mol−1, which indicates moderate physisorption of CO2. It is likely that the trimethylammonium moiety within the Eudragit RL® structure provides an additional site of interaction for CO2. Although the highest heat of adsorption was recorded for the phenylsilane grafted derivative (31.57 kJ mol−1), the significant reduction in CO2 uptake to 0.61 mmol g−1 precludes its use as an enhanced CO2 capture material.
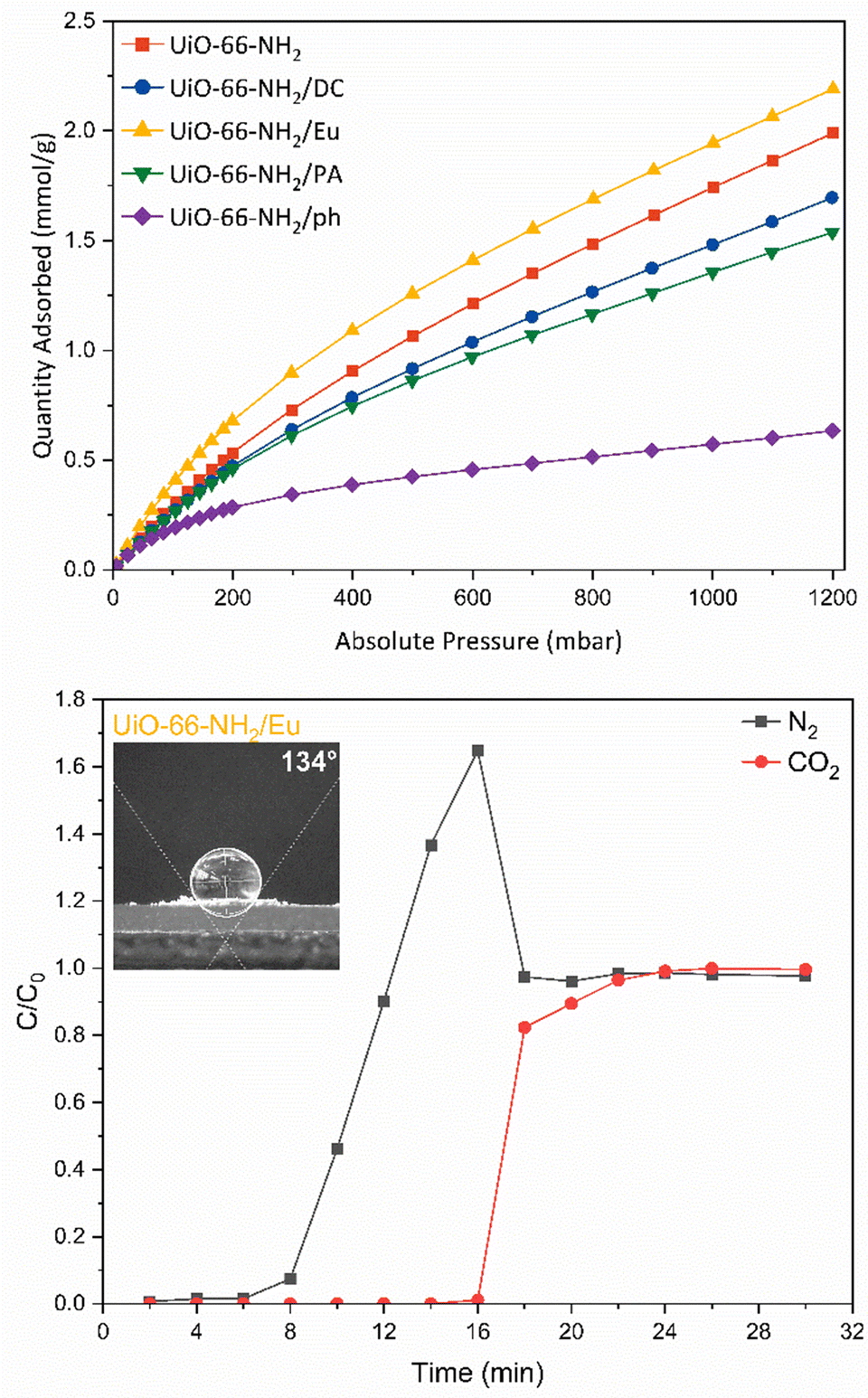 | ||
| Fig. 3 CO2 adsorption isotherms at 298 K of UiO-66-NH2 (red), UiO-66-NH2/DC (blue), UiO-66-NH2/Eu (yellow), UiO-66-NH2/PA (green) and UiO-66-NH2/ph (purple) (above). Breakthrough calculations for UiO-66-NH2/Eu for N2 (black) and CO2 (red). Inset: Water contact angle for UiO-66-NH2/Eu (below). | ||
Real time CO2 separation from N2 for each composite material was examined via fixed-bed column breakthrough experiments in a custom-made breakthrough rig, using a binary CO2/N2 mixture (50:50) (Fig. S26, ESI†). The GC was calibrated for the N2 and CO2 signal (Fig. S27, ESI†). N2 breakthrough was first observed, prior to CO2 (Fig. 3 and Fig. S28, ESI†). For pristine UiO-66-NH2, the breakthrough point of N2 was 6–8 min, with saturation reached at 10 min (Fig. S28a, ESI†). For CO2 the breakthrough point was 8–10 min, while CO2 saturation was reached at 26 min. UiO-66-NH2/DC (Fig. S28b, ESI†), UiO-66-NH2/Eu (Fig. 3), and UiO-66-NH2/PA ((Fig. S28c, ESI†) all demonstrated improved separation between N2 and CO2 compared to the parent material. For UiO-66-NH2/Eu, N2 took longer to pass through the column than in the parent material, breaking through at 8–10 min and reaching saturation at 18 min. In comparison, the breakthrough point of CO2 was 16–18 min, and saturation was reached at 22 min. The increased separation in the UiO-66-NH2/Eu sample is consistent with the behaviour observed in the CO2 isotherm. UiO-66-NH2/Eu consistently demonstrated a higher uptake of CO2 in the thermodynamic adsorption isotherm compared to the parent material, while functional quaternary amine groups in the enteric coating promoted improved interactions with CO2. A practical CO2 capacity of 0.98 mmol g−1 was calculated from the breakthrough curve. For UiO-66-NH2/DC, the breakthrough point of N2 was at 6–8 min, while saturation was reached at 14 min. The CO2 breakthrough point was at 10–12 min while saturation was slowly reached at 32 min. A practical CO2 capacity of 0.59 mmol g−1 was calculated from the breakthrough curve. The silyl functional groups improved the interaction of CO2 with the material and did not greatly impact the CO2 capacity of the composite at low partial pressures. UiO-66-NH2/PA only demonstrated a small improvement in separation compared to the parent material. A practical CO2 capacity of 0.54 mmol g−1 was calculated from the breakthrough curve. Each modified sample measurement displayed a roll-up feature,57 indicating the concentration of the weaker sorbent –N2 – briefly exceeds that of the feed. This is a common phenomenon in breakthrough curves for pure sorbent beds.57
Internal hydrophobicity was initially tested through vapour sorption experiments (Fig. 4). Of all the materials, UiO-66-NH2/ph demonstrated the lowest vapour uptake of 4.85 mmol g−1 at 76% relative humidity; however, this may be due to the reduced porosity calculated by the BET surface area and pore volume. Furthermore, there is a very gradual water uptake at low humidities, with typical S-shaped behaviour not observed.58 The other candidate materials all showed similar H2O vapour uptake performances as the pristine UiO-66-NH2, which implied that the external hydrophobic coating does not improve the performance of the MOF under humid conditions. In some cases, the water vapour uptake increased from 20.4 mmol g−1 at 35% relative humidity in the pristine UiO-66-NH2 to 23.5 mmol g−1 and 25.7 mmol g−1 in UiO-66-NH2/Eu and UiO-66-NH2/DC at 35% relative humidity, respectively. UiO-66-NH2/PA demonstrated slight improvements in hydrophobicity at higher relative humidities. The water vapour uptake was reduced to 18.3 mmol g−1 at 35% relative humidity. There is little variation in hydrophobicity at low humidities. Interestingly, the increased uptake in vapour for Eu and DC may be due to the interactions displayed by the pristine polymers. Vapour sorption of these materials indicate H2O uptake of up to 2.5 mmol g−1 at 80% relative humidity (Fig. S30, ESI†).
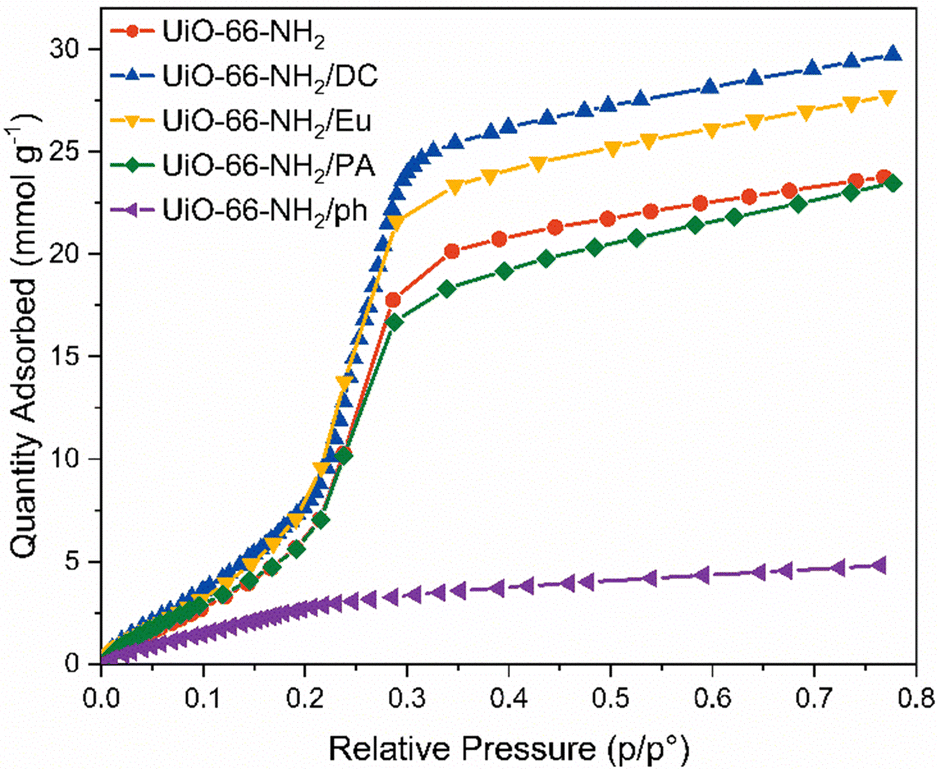 | ||
| Fig. 4 Water vapour adsorption measurements performed at 298 K on pristine UiO-66-NH2 (red), UiO-66-NH2/DC (blue), UiO-66-NH2/Eu (yellow), UiO-66-NH2/PA (green) and UiO-66-NH2/ph (purple). | ||
Conclusions
This project sought to investigate the impact of post-synthetically modifying UiO-66-NH2 with hydrophobic functionalities to reduce the competition between CO2 and H2O in more industrially relevant conditions. Four broad, facile strategies (enteric coatings, long-chained alkane, organosilicon infiltration and phenylsilane grafting) were chosen, which led to four representative candidate materials of interest (UiO-66-NH2/Eu, UiO-66-NH2/PA, UiO-66-NH2/DC and UiO-66-NH2/ph). The post-synthetic modifications occurred under mild conditions and with short reaction times, which are conducive to a scalable process implementable in an industrial scenario. The post-synthetic modification was confirmed not to interfere with the structure of the parent material, as determined by PXRD, FT-IR, TGA and SEM characterisation.Given that hydrophobicity in MOFs is generally characterised using water contact angles, alternative methods need to be developed to characterise hydrophobicity to vapour. Although these post-synthetically modified materials demonstrated external hydrophobicity, vapour sorption measurements revealed minimal impact on hydrophobicity when compared to the parent framework. Despite endowing the parent framework with an external hydrophobicity to liquid water, the modified frameworks were not sufficiently hydrophobic in the presence of humid air. The materials studied in this work demonstrate behaviour akin to that seen in Gore-Tex®; therefore, we propose the Gore-Tex® effect as a descriptor of this phenomenon.59 Of the studied materials, the best coating performance was achieved using UiO-66-NH2/Eu. Until now, MOF literature tends to use the water contact angle as a sufficient predictor of overall hydrophobicity.60 This study indicates that such an assumption is not valid and that alternative methods are required to correctly characterise MOF hydrophobicity in an industrial setting. Nevertheless, we have demonstrated that we can improve the hydrophobicity of a MOF to liquid water, with efforts underway to expand the candidate MOFs that can be tested. There is also further interest in studying these materials which possess varied hydrophobicity for membranes where vapour transport is essential, e.g., particular aquaporins.
Author contributions
DD developed the idea for this paper. DD, CK and MS supervised the experimental work. MS developed the initial optimisation strategies for each composite. KM, supported by MS, synthesised, and physically characterised bulk composite samples, including their gas and vapour sorption. Breakthrough experiments were performed by RL and JWH. KM and MS drafted the final paper, with input received from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the funding provided by the Australian Research Council (JH and DD – DP230101901, CK – DP220103690, JH – FT210100589, RL – DE230100147). This project is also funded The University of Queensland Foundation Research Excellence Awards. We gratefully acknowledge the technical and scientific assistance of Sydney Microscopy & Microanalysis, the University of Sydney node of Microscopy Australia. We wish to thank Mr Rohan Gillespie from Southern Green Gas Ltd. for discussions on hydrophobic porous materials. We would like to thank Prof. Chiara Neto for providing access to the goniometer for contact angle measurements, and Dr. Isaac Gresham for facilitating the measurements. We would like to thank Prof. Kim Chan for providing insights into enteric coatings. Finally, we gratefully acknowledge the support of Dr. David Dharma for setting up vapour sorption experiments.Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- D. M. D’Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- T. C. Wang, W. Bury, D. A. Gomez-Gualdron, N. A. Vermeulen, J. E. Mondloch, P. Deria, K. Zhang, P. Z. Moghadam, A. A. Sarjeant, R. Q. Snurr, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 3585–3591 CrossRef CAS PubMed.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. O. Yazaydin and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS PubMed.
- H. Konnerth, B. M. Matsagar, S. S. Chen, M. H. G. Prechtl, F. K. Shieh and K. C. W. Wu, Coord. Chem. Rev., 2020, 416, 213319 CrossRef CAS.
- S. Wu, H. Min, W. Shi and P. Cheng, Adv. Mater., 2020, 32, e1805871 CrossRef PubMed.
- W. Zhang, Y. Hu, J. Ge, H. L. Jiang and S. H. Yu, J. Am. Chem. Soc., 2014, 136, 16978–16981 CrossRef CAS PubMed.
- S. M. Moosavi, A. Nandy, K. M. Jablonka, D. Ongari, J. P. Janet, P. G. Boyd, Y. Lee, B. Smit and H. J. Kulik, Nat. Commun., 2020, 11, 4068 CrossRef CAS PubMed.
- L. L. Huang, L. Yu, B. Li, B. B. Li, H. Wang and J. Li, ACS Mater. Lett., 2022, 4, 1053–1057 CrossRef CAS.
- M. Rubio-Martinez, C. Avci-Camur, A. W. Thornton, I. Imaz, D. Maspoch and M. R. Hill, Chem. Soc. Rev., 2017, 46, 3453–3480 RSC.
- O. P. Murphy, M. Vashishtha, P. Palanisamy and K. V. Kumar, ACS Omega, 2023, 8, 17407–17430 CrossRef CAS PubMed.
- N. Kumar, S. Mukherjee, N. C. Harvey-Reid, A. A. Bezrukov, K. Tan, V. Martins, M. Vandichel, T. Pham, L. M. van Wyk, K. Oyekan, A. Kumar, K. A. Forrest, K. M. Patil, L. J. Barbour, B. Space, Y. Huang, P. E. Kruger and M. J. Zaworotko, Chemistry, 2021, 7, 3085–3098 CrossRef CAS PubMed.
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030–3040 RSC.
- S. D. Kenarsari, D. L. Yang, G. D. Jiang, S. J. Zhang, J. J. Wang, A. G. Russell, Q. Wei and M. H. Fan, RSC Adv., 2013, 3, 22739–22773 RSC.
- R. Krishna, ACS Omega, 2020, 5, 16987–17004 CrossRef CAS PubMed.
- P. G. Boyd, A. Chidambaram, E. Garcia-Diez, C. P. Ireland, T. D. Daff, R. Bounds, A. Gladysiak, P. Schouwink, S. M. Moosavi, M. M. Maroto-Valer, J. A. Reimer, J. A. R. Navarro, T. K. Woo, S. Garcia, K. C. Stylianou and B. Smit, Nature, 2019, 576, 253–256 CrossRef CAS PubMed.
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS PubMed.
- S. Hausdorf, F. Baitalow, T. Böhle, D. Rafaja and F. O. R. L. Mertens, J. Am. Chem. Soc., 2010, 132, 10978–10981 CrossRef CAS PubMed.
- L. H. Xie, M. M. Xu, X. M. Liu, M. J. Zhao and J. R. Li, Adv. Sci., 2020, 7, 1901758 CrossRef CAS PubMed.
- B. Wang, X.-L. Lv, D. Feng, L.-H. Xie, J. Zhang, M. Li, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 6204–6216 CrossRef CAS PubMed.
- Y. S. Bae, J. Liu, C. E. Wilmer, H. Sun, A. N. Dickey, M. B. Kim, A. I. Benin, R. R. Willis, D. Barpaga, M. D. LeVan and R. Q. Snurr, Chem. Commun., 2014, 50, 3296–3298 RSC.
- N. Ding, H. Li, X. Feng, Q. Wang, S. Wang, L. Ma, J. Zhou and B. Wang, J. Am. Chem. Soc., 2016, 138, 10100–10103 CrossRef CAS PubMed.
- X. Qian, F. Sun, J. Sun, H. Wu, F. Xiao, X. Wu and G. Zhu, Nanoscale, 2017, 9, 2003–2008 RSC.
- C. A. Fernandez, S. K. Nune, H. V. Annapureddy, L. X. Dang, B. P. McGrail, F. Zheng, E. Polikarpov, D. L. King, C. Freeman and K. P. Brooks, Dalton Trans., 2015, 44, 13490–13497 RSC.
- A. X. Zhu, R. B. Lin, X. L. Qi, Y. Liu, Y. Y. Lin, J. P. Zhang and X. M. Chen, Microporous Mesoporous Mater., 2012, 157, 42–49 CrossRef CAS.
- D. D. Zu, L. Lu, X. Q. Liu, D. Y. Zhang and L. B. Sun, J. Phys. Chem. C, 2014, 118, 19910–19917 CrossRef CAS.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- D. Sun, P. R. Adiyala, S. J. Yim and D. P. Kim, Angew. Chem., Int. Ed., 2019, 58, 7405–7409 CrossRef CAS PubMed.
- A. von Wedelstedt, H. Chen, G. Kalies and R. Q. Snurr, Langmuir, 2020, 36, 13070–13078 CrossRef CAS PubMed.
- M. De Toni, R. Jonchiere, P. Pullumbi, F. X. Coudert and A. H. Fuchs, Chem. Phys. Chem., 2012, 13, 3497–3503 CrossRef CAS PubMed.
- K. Jayaramulu, F. Geyer, A. Schneemann, Š. Kment, M. Otyepka, R. Zboril, D. Vollmer and R. A. Fischer, Adv. Mater., 2019, 31, 1900820 CrossRef PubMed.
- P. Möder, H. Vierheilig, M. Alt and A. Wiemken, Plant Soil, 1993, 152, 201–206 CrossRef.
- J. H. Choe, J. R. Park, Y. S. Chae, D. W. Kim, D. S. Choi, H. Kim, M. Kang, H. Seo, Y.-K. Park and C. S. Hong, Commun. Mater., 2021, 2, 3 CrossRef CAS.
- J. G. Nguyen and S. M. Cohen, J. Am. Chem. Soc., 2010, 132, 4560–4561 CrossRef CAS PubMed.
- Y. Wan, D. Kong, F. Xiong, T. Qiu, S. Gao, Q. Zhang, Y. Miao, M. Qin, S. Wu, Y. Wang, R. Zhong and R. Zou, Chin. J. Chem. Eng., 2023, 61, 82–89 CrossRef.
- Y. Wang, Z. Hu, T. Kundu, Y. Cheng, J. Dong, Y. Qian, L. Zhai and D. Zhao, ACS Sustainable Chem. Eng., 2018, 6, 11904–11912 CrossRef CAS.
- S. R. Wenger, E. R. Kearns, K. L. Miller and D. M. D'Alessandro, ACS Appl. Energy Mater., 2022 Search PubMed.
- G. Kaur, J. Grewal, K. Jyoti, U. K. Jain, R. Chandra and J. Madan, in Drug Targeting and Stimuli Sensitive Drug Delivery Systems, ed. A. M. Grumezescu, William Andrew Publishing, 2018, pp. 567–626 Search PubMed.
- W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 149–173 CrossRef CAS PubMed.
- X. Lu, C. Huang, M. B. Lowinger, F. Yang, W. Xu, C. D. Brown, D. Hesk, A. Koynov, L. Schenck and Y. Su, Mol. Pharm., 2019, 16, 2579–2589 CrossRef CAS PubMed.
- D. G. Madden, A. B. Albadarin, D. O’Nolan, P. Cronin, J. J. T. Perry, S. Solomon, T. Curtin, M. Khraisheh, M. J. Zaworotko and G. M. Walker, ACS Appl. Mater. Interfaces, 2020, 12, 33759–33764 CrossRef CAS PubMed.
- L. Feng, S. H. Lo, K. Tan, B. H. Li, S. Yuan, Y. F. Lin, C. H. Lin, S. L. Wang, K. L. Lu and H. C. Zhou, Matter, 2020, 2, 988–999 CrossRef.
- H. F. Zhang, M. Li, X. Z. Wang, D. Luo, Y. F. Zhao, X. P. Zhou and D. Li, J. Mater. Chem. A, 2018, 6, 4260–4265 RSC.
- P. Deria, J. E. Mondloch, E. Tylianakis, P. Ghosh, W. Bury, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2013, 135, 16801–16804 CrossRef CAS PubMed.
- H. X. Luo, F. W. Cheng, L. Huelsenbeck and N. Smith, J. Environ. Chem. Eng., 2021, 9, 105159 CrossRef CAS.
- Y. H. Cai, D. Y. Chen, N. J. Li, Q. F. Xu, H. Li, J. H. He and J. M. Lu, ACS Sustainable Chem. Eng., 2019, 7, 2709–2717 CrossRef CAS.
- J. Gu, J. Li and Q. Ma, Nanomaterials, 2022, 12, 1134 CrossRef CAS PubMed.
- B. Yuan, G. Gou, T. Fan, M. Liu, Y. Ma, R. Matsuda and L. Li, Angew. Chem., Int. Ed., 2022, 61, e202204568 CrossRef CAS PubMed.
- C. Fang, C. L. Lu, M. H. Liu, Y. L. Zhu, Y. Fu and B. L. Lin, ACS Catal., 2016, 6, 7876–7881 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- S. Y. Xu, Q. H. Gao, C. Zhou, J. X. Li, L. G. Shen and H. J. Lin, Mater. Chem. Phys., 2021, 274, 125182 CrossRef CAS.
- C. Gomes Silva, I. Luz, F. X. Llabres i Xamena, A. Corma and H. Garcia, Chemistry, 2010, 16, 11133–11138 CrossRef PubMed.
- X. Jiang, S. W. Li, S. S. He, Y. P. Bai and L. Shao, J. Mater. Chem. A, 2018, 6, 15064–15073 RSC.
- J. Winarta, B. H. Shan, S. M. Mcintyre, L. Ye, C. Wang, J. C. Liu and B. Mu, Cryst. Growth Des., 2020, 20, 1347–1362 CrossRef CAS.
- A. Kapoor and R. J. Yang, AIChE J., 1987, 33, 1215–1217 CrossRef CAS.
- M. R. Samarghandi, M. Hadi and G. McKay, J Adsorption Sci., 2014, 32, 791–806 CrossRef.
- L. Hunter and J. Fan, Textiles and fashion, Elsevier, 2015, pp. 739–761 Search PubMed.
- Y. Li, H. T. Wang, Y. L. Zhao, J. Lv, X. Zhang, Q. Chen and J. R. Li, Inorg. Chem. Commun., 2021, 130, 108741 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00716b |
| This journal is © The Royal Society of Chemistry 2024 |