
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the conductivity of plasma polymer functionalized electrodes using gold nanoparticles†
Alex
Gheorghiu
 *a,
Daisy
Yang
a,
Tiexin
Li
b,
Essam M.
Dief
b,
Nadim
Darwish
b,
Craig
Priest
ac and
Melanie
MacGregor
*d
*a,
Daisy
Yang
a,
Tiexin
Li
b,
Essam M.
Dief
b,
Nadim
Darwish
b,
Craig
Priest
ac and
Melanie
MacGregor
*d
aUniSA STEM, Future Industries Institute, University of South Australia, Mawson Lakes SA, 5095, Australia. E-mail: alexandru.gheorghiu@mymail.unisa.edu.au
bSchool of Molecular and Life Sciences, Curtin University, Bentley WA 6102, Australia
cAustralian National Fabrication Facility – South Australia Node, University of South Australia, Mawson Lakes SA 5095, Australia
dFlinders Institute for Nanoscale Science & Technology, College of Science and Engineering, Flinders University, Bedford Park SA 5042, Australia. E-mail: melanie.macgregor@flinders.edu.au
First published on 20th November 2023
Abstract
Plasma deposited polyoxazoline thin films (POx) are a promising solution for the rapid, scalable, and substrate-independent immuno-functionalization of electrochemical biosensors. However, a major challenge in using POx thin films in electrochemical sensing is their inherently insulating nature. This work reports the incorporation of gold nanoparticles (AuNPs) between two layers of POx which enhances the conductivity of the films. The size of the AuNPs, their binding density on the POx underlayer, and the POx films’ intrinsic electrical resistance were all factors in improving the overall electrochemical response of the layered construction. Surface bound electrochemical measurements and conductive atomic force microscopy were conducted to uncover a possible mechanism for the observed nanoparticle-mediated electron transport through the insulating matrix. The primary contributor to increasing conductivity in layered constructions is maximising the surface coverage of AuNPs on the surface to provide pathways for current to flow through the insulating matrix. As a proof of concept, POx layered constructions were then used to detect the binding of exosomes to the surface, indicating that these electrodes promise to provide low limits of detection when functionalised with a suitable recognition element.
Introduction
The functionalization of electrodes used in electrochemical biosensors is required to enable the immobilization of a recognition element. Typically, electrode functionalization is achieved using self assembled monolayers (SAM), an approach that requires lengthy incubation steps and surface activation to facilitate ligand binding.1,2 The deposition of plasma polymerized thin films onto electrode surfaces is an alternative method that enables rapid, scalable, and substrate-independent surface functionalization. Previous studies have shown that electrode functionalization with plasma polymerized polyacrylic acid eliminated the need for SAM formation, yet still required a surface activation step to initiate enzyme immobilisation.3 In another study, thin plasma polymerised polyaniline films (PANI) reportedly allowed the adsorption of glucose oxidase without a surface activation step, though the mechanism facilitaing the enzyme immoblisation was not explicitely dicussed and may not extend to other biological recognition elements.4 Our group has reported that plasma polymerized polyoxazoline thin films (POx) are a versatile platform for electrode functionalization.5 The retention of the oxazoline ring in the deposited POx films enables the rapid and irreversible binding of biomolecules to the surface in a click-chemistry type of reaction through–COOH groups.6,7 Electrochemical investigations have shown that the intrinsic structure of the POx films significantly influences the charge transfer resistance of the coating independently of the film thickness.8 Importantly, careful optimization of the deposition conditions was required to balance the POx films’ electrochemical performance and their stability to external processing, such as incubation steps, rinse cycles, and repeated current exposure.The overall performance of single POx layers in electrosensing is somewhat underwhelming because of the inherently insulating nature of plasma polymerised thin films. Their internal structure is a highly branched and crosslinked network consisting primarily of saturated alkyl chains resulting in a high bandgap film which electrons are unable to tunnel through. In addition, this structure restricts the diffusion of redox active species to the electrode and increases the charge transfer resistance.9 As a result, when plain POx films were used to functionalise electrodes for epithelial cell adhesion molecule (EpCAM) detection, the biosensor sensitivity was several orders of magnitude less than similar examples that incorporated a method of signal enhancement such as using conductive nanomaterials.5 In fact, previous work where plasma polymer films were used to functionalize electrodes all integrated conductive nanomaterials into the electrode design in order to obtain good biosensing sensitivity.3,4 Even electrodes functionalized with plasma polymerised polyaniline films required the integration of conductive nanomaterials to achieve good sensitivity, despite PANI exhibiting intrinsic conductivity due to its high degree of conjugation.4 The nanomaterials that were used in these examples were hollow TiO2 spheres3 as well as composites of tin oxide nanoparticles and reduced graphene oxide.4 Gold nanoparticles (AuNPs) are another class of nanomaterial which could be used to improve conductivity.
The unique electrical properties of AuNPs have already been proven useful to enhance charge transport and conductivity in supercapacitors,10 to further the miniaturization of microelectronics,11 and to improve the sensing of biomolecules.12 Adsorbing AuNPs onto gold electrodes passivated with SAMs “switches on” the conductivity independently of the length of alkanethiol used to form the SAM.1,13,14 Similarly, AuNPs mediated efficient electron transport from electroactive proteins such as cytochrome c15 and azurin.16 These results indicated that AuNPs provide efficient pathways for electrons to be conducted to the electrode beneath an insulating layer and that the rate determining step is the charging of the nanoparticle by the redox couple.17 A model by Chazalviel and Allongue18 indeed showed that electron transfer between the metal electrode and nanoparticles in a layered metal-SAM-nanoparticle structure is up to 1012 orders of magnitude more efficient than between a redox molecule in solution and the nanoparticle. More recently, Nichols et al19 also reported improved conductivity in SAM-nanoparticle-SAM constructs due to the unique double monolayer structure of the SAMs, enabling more efficient electron transfer pathways. Thanks to their high surface area-to-volume ratio, bound AuNPs also provide a larger electrode area for the redox process to take place, resulting in enhanced system sensitivity.20
In previous works, AuNPs were bound to POx-coated electrodes with different surface density.5 Both 2-methyl-2-oxazoline (MePOx) and 2-isopropenyl-2-oxazoline (PiPOx) were used a precursor, the latter resulting in a denser,21 more resistive5 film due to its unsaturated sidechain offering more routes for the formation of crosslinks post plasma activation.22 For both MePOx and PiPOx films, increasing the density of AuNPs on the surface resulted in a decrease in the charge transfer resistance. However, this decrease was not proportional to particle density for PiPOx films, which bound a greater density of AuNPs compared to MePOx but showed a greater charge transfer resistance.
In this study, we tested the feasiblity of using AuNPs to enhance the conductivity of POx thin films for biosensing applications through their incorporation into a layered construction between these insulating films. We investigate the electrochemical response of a layered construction where AuNPs are incorporated between two layers of POx using electrochemical impedance spectroscopy (EIS). The addition of a top layer to this system is needed in order to provide a surface to which recognition elements can be attached to facilitate selective biosensing. Furthermore, we probe possible mechanisms through which the AuNPs act to increase the sensitivity of the modified electrode using conductive atomic force microscopy and lastly, investigate the layered construction's performance as an exosome biosensor.
Methods
Plasma polymerization
Plasma polymer deposition was carried out using a custom-built reactor previously described.22 Ignition of the plasma was achieved using a 13.56 MHz radio frequency generator coupled to a matching network (Coaxial Power Systems, UK). Electrode substrates consisted of glass slides coated with a 20 nm chromium adhesion layer and a 200 nm gold layer were used as electrodes. Silicon wafers were used as substrates for thickness measurements. The substrates were primed using an air plasma at 30 W, 2.0 × 10−1 mbar for 3 minutes. The reactor was then evacuated to a base pressure of 2.0 × 10−2 mbar. The pressure in the reactor was monitored with a Pirani gauge (model APG100-XLC, Edwards, UK) and controlled with a needle valve (Chell Instruments, UK) and a ball valve (ANCORP, USA). 2-Methyl-2-oxazoline (MePOx) and 2-isopropenyl-2-oxazoline (PiPOx) were used as the precursors (98%, Sigma-Aldrich, Australia,). Precursors were introduced into the reactor at the required working pressure. The plasma was ignited at the required ignition power for 30 seconds.5Gold nanoparticle synthesis and surface immobilization
AuNPs were synthesis through the reduction of chloroauric acid (HAuCl4) by trisodium citrate (both from Sigma-Aldrich, Australia).5 Briefly, 50 mL of 0.01% HAuCl4 was boiled under reflux with vigorous stirring. To this solution, either 1 mL or 0.3 mL of 1% trisodium citrate was added to synthesize 16 or 68 nm AuNPs respectively. The colloidal suspensions were then boiled for another 20 min under reflux. Nanoparticles were functionalized with –COOH groups by adding 0.02 M sodium hydroxide (Chem-Supply, Australia) and 0.01 M mercaptosuccinic acid (Sigma-Aldrich, Australia) and allowed to stir overnight.AuNP binding is achieved through the unique chemistry of the POx films and the available COOH groups on the AuNP surface.6,7,23 Detailed studies have shown that both the film reactivity (i.e. Nitrogen content) and the diameter of the nanoparticles influence the binding density observed on POx films.24 Here, POx films were incubated with AuNP solutions for 24 h before being rinsed with milliQ water and dried under N2 gas.
Bulk electrochemical measurements
Bulk electrochemical measurements were conducted using 2 mM potassium hexacyanoferrate(III) (K3[Fe(CN]6), >99%, Sigma-Aldrich, Australia) and potassium hexacyanoferrate(II) (K4[Fe(CN)6]) (99%, Sigma-Aldrich, Australia) dissolved in phosphate buffered saline (PBS) (pH 7.4) water (the redox couple solution). All electrochemical measurements were conducted using glass substrates sputtered with a 20 nm chromium adhesion layer and a 200 nm gold layer as working electrodes.Electrochemical measurements were conducted with an Ivium-n-Stat electrochemical analyzer (Ivium Technologies, The Netherlands) and data were acquired and analyzed using IVIUM software (Ivium Technologies, The Netherlands). The area investigated was delimited using an o-ring with an inner diameter of 7.5 mm in a 3 electrodes Teflon cell (Fig. S1, ESI†). POx coated electrodes were connected as the working electrode, an external coiled Pt wire was used as the counter electrode, and an Ag/AgCl electrode as reference. All potentials are provided as versus Ag/AgCl.
Electrochemical impedance spectroscopy (EIS) measurements were conducted in 750 μL of 2 mM [Fe(CN)6]3/4− in PBS at the open circuit potential. Frequencies were scanned from 100 kHz to 0.1 Hz with an AC amplitude of 5 mV. Data was analyzed using a complex plane plot of the imaginary component vs the real component of the impedance (Nyquist plot). The plot was interpreted by fitting an equivalent Randles circuit model to the data using the complex non-linear least square (CNLS) technique developed by MacDonald and Potter.25
Surface bound electrochemical measurements
To investigate the kinetics of electron transfer at the working electrode, a self assembled monolayer (SAM) of a redox active molecule was formed. A solution of 4 mM 11-(ferrocenyl)-undecanethiol dissolved in ethanol was incubated with electrodes for 16 hours after which, they were removed, rinsed in Milli-Q water, and dried using a stream of nitrogen gas. Electrochemical measurements were conducted in 0.1 M phosphate buffer using a CHI650 electrochemical workstation (CH Instruments, USA). EIS measurements were conducted at the half wave potential (E1/2) obtained in cyclic voltammetry experiments with an AC amplitude of 5 mV between the frequencies of 100 kHz and 0.1 Hz.Biosensing experiments
Experiments using protein analytes and exosomes were conducted to assess the biosensing sensitivity of POx coated electrodes. Anti-prostate specific membrane antigen (anti-PSMA) and PSMA antigen were purchased from Sino Biological (China). Exosome suspensions isolated from cultured LNCap cells were purchased from ATCC (USA).POx coated electrodes to be used in biosensing experiments were functionalized using a previously established protocol.5,26,27 Briefly, 50 μL of 10 μg mL−1 of anti-PSMA antibody (Prostate Specific Membrane Antigen) was incubated on the surfaces for 1 h at room temperature. Next, 50 μL of 0.1 M ethylene diamine (EDM) non-specific block was added for a further 1 hour incubation. Electrodes functionalized in this way are termed antibody functionalized samples. Negative controls were used which were incubated solely in EDM and pristine POx surfaces were used as positive controls. Electrodes were rinsed three times each with 250 μL PBS and the redox couple solution between each incubation step after the blocking step. Increasing concentrations of the analyte (either PSMA antigen or exosome suspension) were conducted for 30 min on separate electrodes for each concentration. PSMA antigen concentrations increased from 1 × 10−4 ng mL−1 to 1 ng mL−1. Exosome concentrations were increased from 1.7 × 100 particles per μL to 1.7 × 105 particles per μL.
X-ray photoelectron spectroscopy (XPS) measurements
XPS analysis was conducted on a Kratos Axis Ultra DLD spectrometer with a monochromatic Al Kα X-ray source at 225 W (1486.6 eV) to assess the chemistry of the plasma polymer films. An area of 0.3 × 0.7 mm with sample charging minimised with an internal flood gun. Survey spectra were collected with a dwell time of 55 ms using 160 eV pass energy and steps of 0.5 eV with three sweeps. Data analysis was conducted using Casa XPS (Version 2.3.24, Casa Software Ltd).28Ellipsometry
Thickness measurements of the POx films were performed using a spectroscopic ellipsometer (V-Vase, J.A. Woolam, MC-200) over a wavelength range of 250–1100 nm at 65°, 70°, and 75°. The plasma polymer layer thickness deposited onto clean silicon wafers was determined using a Cauchy model and fitting procedure.Scanning electron microscopy (SEM)
High resolution imaging of the AuNP functionalised POx surfaces was conducted using a Carl Zeiss Microscopy Crosspy Crossbeam 540 with a GEMINI II column and equipped with a field emission gun to acquire secondary electron images. Images were obtained using an accelerating voltage of 5 kV and a working distance of 3 mm. Acquired SEM images were processed using ImageJ (Version 1.53, US National Institutes of Health).29Conductive atomic force microscopy (cAFM)
A Buker Dimension PF-TUNA atomic force microscope was used to investigate the conductivity of the surfaces in ambient conditions. Current–voltage curves (IV curves) were obtained using solid Pt tips (RMN-25 PT300B, Rocky Mountain Nanotechnology), with a nominal resonant frequency of 20 kHz and a spring constant of 18 N m−1. IV curves were obtained by ramping the potential between −0.1 V and 0.1 V 30 times at each position with a peak force of 2.25 μN and a ramp rate of 1 Hz.Results and discussion
Electrochemical characterization of POx films
Thin films were plasma deposited on sputtered gold substrates from the two oxazoline precursors, MePOx and PiPOx. Both the chemistry (e.g. nitrogen content, Fig. S2, ESI†) and electrochemical responses of the films vary with the deposition condition parameters, namely time, power, pressure and monomer chemistry. The electrochemical properties were studied using electrochemical impedance spectroscopy. The obtained Nyquist Plots (Fig. S3, ESI†) were analysed by fitting a Randles circuit (Fig. S4a, ESI†) to the data and the value of the extracted charge transfer resistance (Rct) are presented in Fig. 1.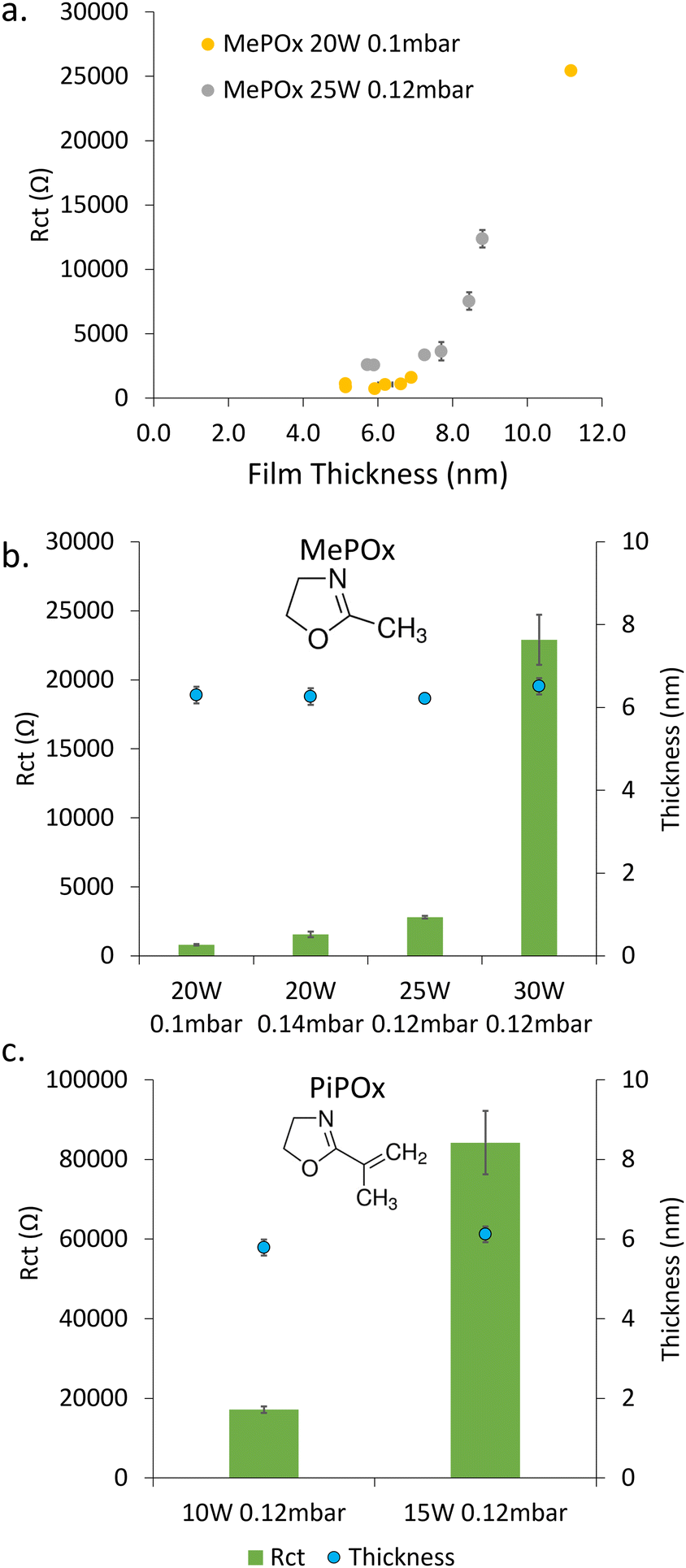 | ||
| Fig. 1 (a) Relationship between MePOx film thickness and Rct for two ignition power and monomer flow rate combinations. Fitted Rct values and their corresponding thicknesses for (b) four MePOx deposition conditions and (c) two PiPOx deposition conditions. | ||
The film thickness increases linearly with deposition time (Fig. S5, ESI†), which in turn increases the Rct, as shown in Fig. 1a for MePOx film deposited with two ignition power and flow rate combinations. While greater ignition powers result in films that are inherently more resistive, both deposition conditions tested here show a critical value between 7 and 8 nm above which the resistance increases significantly with small changes in thickness. This is in good agreement with previous works which have shown that films thicker than 8 nm were too insulating for biosensing applications.5 In the following work, the deposition time was therefore set to 30 s in order to generate films 6 nm in thickness.
For any given monomer, the Rct of 6 nm thick films increases significantly with increasing plasma ignition power (Fig. 1b and c). The large increase in resistance between films deposited at 25 W (2800 ± 100 Ω) and 30 W (23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ± 1800 Ω) for MePOx and 10 W (17000 ± 800 Ω) and 15 W (84000 ± 8000 Ω) for PiPOx, indicates that there is a marked change in the films internal structure (i.e. crosslinking) between these ignition powers. When the ignition power is kept constant (20 W), increasing the monomer flow rate from 0.1 mbar to 0.14 mbar also had the effect of increasing the overall resistance of MePOx (from 800 ± 60 Ω to 1600 ± 200 Ω). While the level of crosslinking is unlikely to be increased by providing excess monomer at the same power (no added power provided to break bonds and create additional cross links with the extra monomer material), loosely bound fragments may become trapped within the films, hence adding to the overall resistance.
000 ± 1800 Ω) for MePOx and 10 W (17000 ± 800 Ω) and 15 W (84000 ± 8000 Ω) for PiPOx, indicates that there is a marked change in the films internal structure (i.e. crosslinking) between these ignition powers. When the ignition power is kept constant (20 W), increasing the monomer flow rate from 0.1 mbar to 0.14 mbar also had the effect of increasing the overall resistance of MePOx (from 800 ± 60 Ω to 1600 ± 200 Ω). While the level of crosslinking is unlikely to be increased by providing excess monomer at the same power (no added power provided to break bonds and create additional cross links with the extra monomer material), loosely bound fragments may become trapped within the films, hence adding to the overall resistance.
The difference in chemistry between the two monomers used here induces greater effect on the measured Rct than varying the deposition conditions. This arises from the unsaturated pendant group present in PiPOx, which provides a source of additional crosslinking, resulting in films that typically are denser than MePOx.21 The deposition powers used for PiPOx were reduced to account for this difference in an attempt to limit the degree of crosslinking within the films. Yet, even for films deposited at powers as low as 10 W, the Rct of the PiPOx was 17000 ± 800 Ω – an order of magnitude greater than MePOx films deposited at 20 and 25 W and similar to that obtained using 30 W. When the ignition power for PiPOx deposition is increased to 15 W, the resistance increases further to 84000 ± 8000 Ω – a film that is too resistive to be suitable for biosensing applications. However, PiPOx films deposited at ignition powers below 10 W exhibit instability (Fig. S6, ESI†). Thus, the balance of conductivity and stability obtained by films deposited at 10 W make these films the best suited for biosensing applications.
The two most conductive MePOx films – 20 W, 0.1 mbar and 20 W, 0.14 mbar (MePOx conditions 1 and 2 respectively) and PiPOx 10 W 0.12 mbar, all deposited for 30 s, were therefore chosen for further investigation. These three films are good candidates to evaluate the impact of surface-bound AuNPs on the systems overall electrochemical responses because they have comparable thickness and nitrogen functionality (Fig. S2, ESI†), but different intrinsic Rct.
AuNP immobilisation on POx films
Two nanoparticle sizes were bound to the three most promising POx layers (Table 1) to test the hypothesis that AuNPs can reduce the resistance of films.| Ignition power (W) | Monomer flow rate (mbar) | % Nitrogen | Thickness (nm) | R ct (Ω) | |
|---|---|---|---|---|---|
| PiPOx | 10 | 0.12 | 3.8 ± 0.05 | 5.8 ± 0.2 | 17200 |
| MePOx condition 1 | 20 | 0.14 | 5.0 ± 0.07 | 6.3 ± 0.2 | 1560 |
| MePOx condition 2 | 20 | 0.1 | 4.3 ± 0.08 | 6.3 ± 0.2 | 804 |
The particles binding densities (number of particles per unit area, Fig. 2a) were determined from SEM images analysis (Fig. 2b–g). 16 nm AuNPs bound in higher numbers than 68 nm particles on all three films. Previous work by Visalakshan et al.24 found that smaller particles are able to more densely pack on the modified surfaces than larger ones due to electrostatic repulsion (arising from deprotonated COOH groups on the particle surface) that determines particle–particle separation. A greater surface density of nanoparticles was observed for both particle sizes on PiPOx compared to MePOx, in good agreement with previous studies.5 Of note, particles bound in aggregates on MePOx condition 2 rather than as individual nanoparticles. Previous studies have documented this phenomenon in rather unstable films which elute monomer fragments in solution.6,22 The presence of loosely bound monomer fragments in MePOx condition 2 films, which was suspected in the previous section due to their electrochemical behaviour, is here confirmed as it causes the AuNPs to aggregate. Consequently, although both PiPOx and MePOx condition 2 films have a similar binding density of 16 nm AuNPs, PiPOx features a more evenly distributed surface coverage resulting in a larger increase in surface area than the same size particles on MePOx condition 2. The aggregate structure decreases the effective surface area available to interact with the electrolyte, which could negatively affect the overall conductivity enhancement effect.
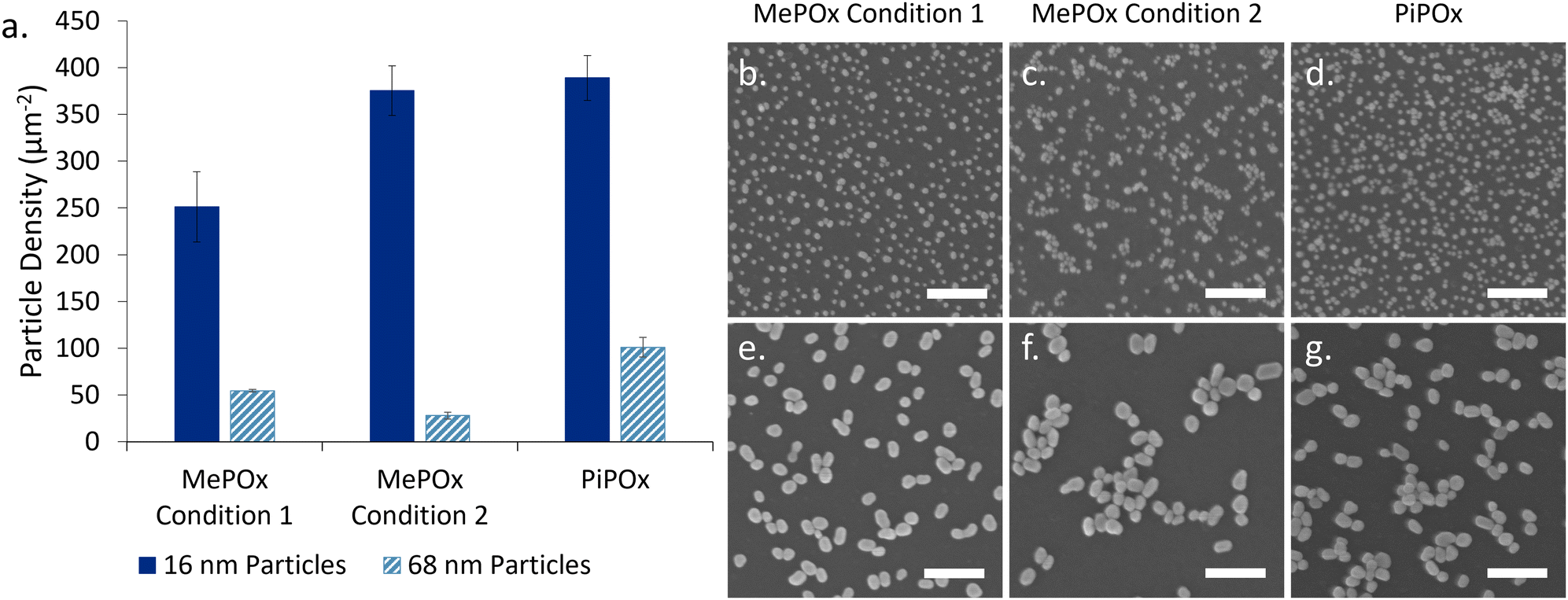 | ||
| Fig. 2 (a) Bound particle density of 16 nm and 68 nm AuNPs on PiPOx, MePOx condition 1, and MePOx condition 2. Representative SEM images of 16 nm AuNPs (b)–(d) and 68 nm AuNPs (e)–(g) bound to each plasma polymer. Scale bar 200 nm. | ||
To harness the conductivity enhancement provided by the AuNPs and simultaneously create a functional surface for biofunctionalization, an additional layer of MePOx is applied over the AuNP-functionalized surfaces. The resistance of this layer-by-layer construction was investigated via EIS (Fig. 3).
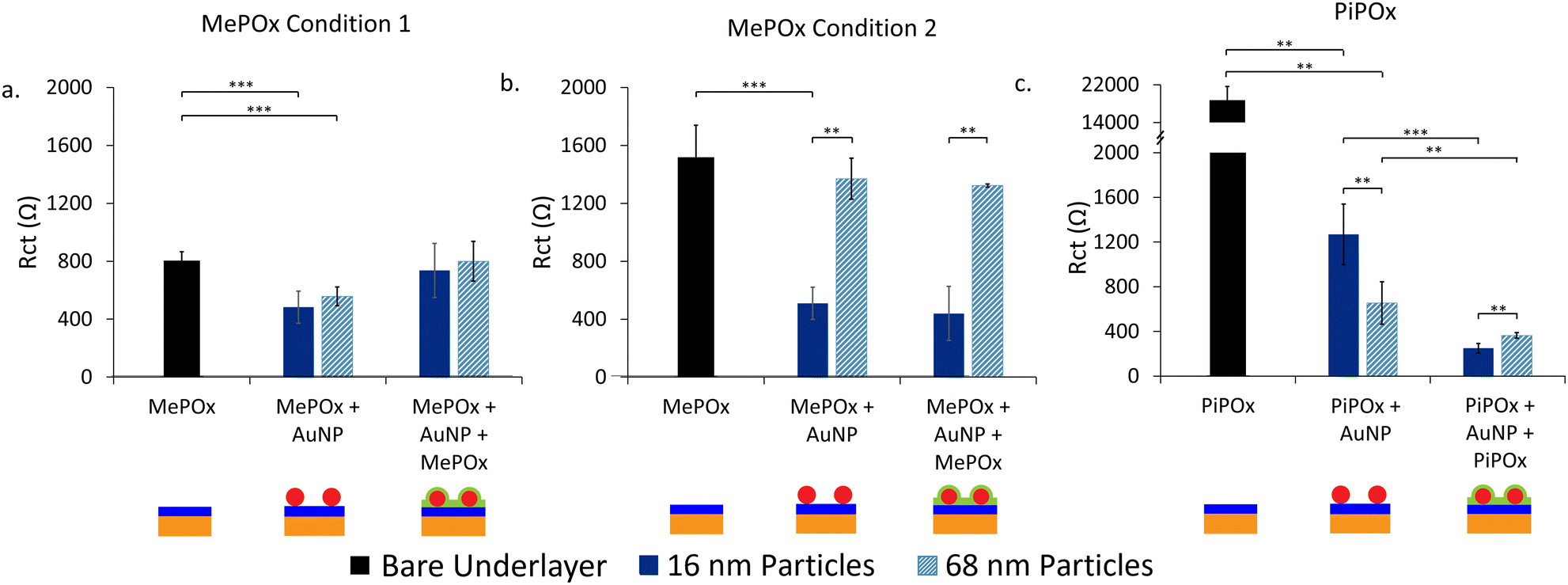 | ||
| Fig. 3 Fitted Rct values for each stage of the sandwich construction with (a) MePOx condition 1, (b) MePOx condition 2, and (c) PiPOx as underlayers. ** represents p < 0.01, *** represents p < 0.001. Schemes beneath give a graphical representation of each stage of layered construction. | ||
Effect of the underlayer on Rct
The effect of the underlayer on the overall system resistance was investigated first while keeping the overlayer constant. MePOx (25 W 0.12 mbar 30 s) was selected as the overlayer for its high nitrogen content, which imparts high bioreactivity that is suitable for biomolecule immobilisation.When MePOx condition 1 is used as the underlayer, a noticeable reduction in Rct is observed upon AuNPs addition, using both 16 and 68 nm AuNPs and an increase in Rct is then observed following the addition of the top layer (Fig. 3a). This is not suprising as the top layer itself is quite resistive (Fig. 1a). What is worth noting, however, is that the overall system does not return to a resistance level equivalent to that of the top layer itself (2800 ± 100 Ω), indicatng that the AuNPs continue to play a role in decreasing the Rct even when incorporated between two POx layers.
When using MePOx condition 2 as the underlayer, the results are not as straight forward. The 16 nm AuNPs do lead to a decrease in Rct, but the 68 nm AuNP do not (Fig. 3b). These results are in keeping with the number of AuNPs bound to the underlayer (Fig. 2a). Indeed, MePOx condition 2 bound fewer 68 nm particles than condition 1 and in the form of aggregates. The aggregation of nanoparticles on the surface led to a lesser increase in effective surface area compared to a surface that bound the same density of individual particles. Hence, the low density of aggregated 68 nm particles on MePOx condition 2 resulted in no appreciable change in Rct. Yet, the Rct does not increase upon addition of the top layer when MePOx condition 2 is used as the underlayer and showed a 71.1% reduction in Rct compared to the bare underlayer. This result – which indicates that AuNPs do improve conductivity even when beneath an insulating layer – is surprising but not isolated.
Indeed, when AuNPs are bound to a PiPOx underlayer (Fig. 3c), we observe the expected decrease in Rct for a PiPOx + AuNP surface. However, when the top layer is added, the Rct decreases further for both 16 nm and 68 nm AuNPs and resulted in a 98.7% and 98.1% reduction in Rct respectively compared to bare PiPOx. A previous study has shown that when AuNPs were bound to PiPOx films in increasing densities,5 a decrease in Rct was observed. However, at the highest bound density, the decrease in Rct was not proportional to the amount of gold detected on the surface. It was hypothesized that this effect was due to electrostatic repulsion between the negatively charged redox couple and the negatively charged AuNPs. The decrease in Rct following deposition of a top layer in Fig. 3c could be attributed to the screening of charges between AuNPs and the redox couple. This enables the AuNPs to provide a path for electrons to tunnel through the insulating layer to the electrode beneath.
Further support for this hypothesis can be observed in the different responses in Rct between the two AuNP sizes. For the intermediate electrodes (PiPOx + AuNP), the 16 nm AuNPs have a greater Rct than the 68 nm ones, but this is reversed for electrodes with the layered construction. These trends correlate with particle densities observed in Fig. 2a and with other studies on the relationship between AuNP size and their binding density.24 Therefore, AuNP coverage of the electrode surface could be a major determinant in the extent to which the Rct is reduced in a layered construction.
Overall, the deposition conditions and chemistry of the underlayer used has been shown to contribute two important factors in the electrochemical response to bound AuNPs. The chemistry of the films dictate the maximum possible binding density of AuNPs and hence, the extent to which the charge transfer resistance through the films is reduced. However, the inherent resistance of the films may also influence the extent to which AuNPs can reduce the resistance.
Effect of the top layer on Rct
Next, the impact of the top layer deposition conditions on the Rct was investigated. PiPOx + 16 nm AuNPs was chosen as the underlayer and nanoparticle combination as this was shown to have the greatest decrease in resistance following deposition of the top layer. MePOx conditions 1 and 2 were deposited on top and compared with the response observed in Fig. 3c (Fig. SI7, ESI†). MePOx condition 1 reduced the resistance to a similar degree to the 25 W film used in Fig. 3c (approx. 260 Ω). The reduction in Rct in the layered construction compared to the intrinsic top layer resistance indicates that the AuNPs enhanced electron transfer to the electrode. However, MePOx condition 1, being the least resistive of the films tested here, resulted in the most conductive layered construction when used in combination with PiPOx and 16 nm AuNPs. This combination of PiPOx-AuNP-MePOx was used for further characterisation.Kinetics of electron transfer
Next, surface bound electrochemical measurements were conducted using a SAM of 11-(ferrocenyl)-undecanethiol to study the rate of electron transfer (ket) as a function of the layered construction structure. Unlike classic electrochemical measurements, where the redox couple is part of the bulk electrolyte, here the redox active species is immobilized on the surface of the electrode. Fig. 4a and b presents a schematic diagram showing the difference between these two type of electrochemical experiments.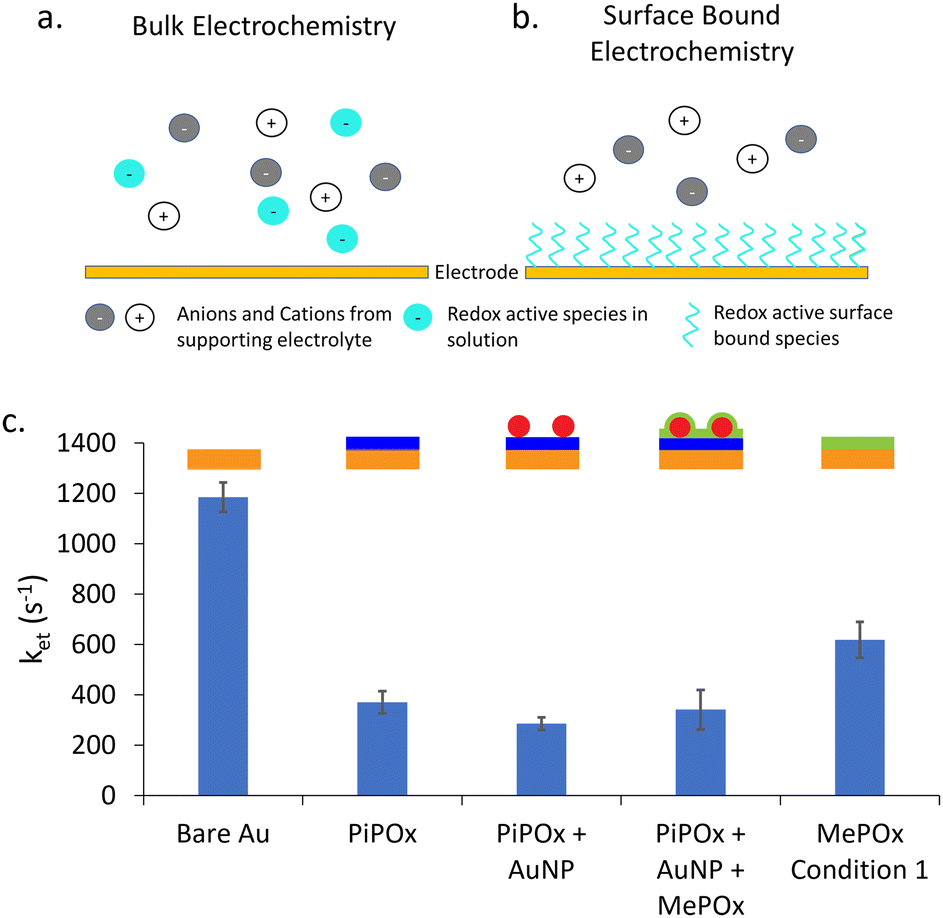 | ||
| Fig. 4 Schematic diagram showing the type of electrochemical experiment being conducted. (a) A bulk electrochemical experiment contains redox active species among supporting ions in solution that need to diffuse to the electrode surface to be oxidised or reduced. (b) A surface bound electrochemical measurement involves the formation of a redox active SAM on the electrode surface, removing the diffusive component of the system. (c) Calculated rate constants of electron transfer (ket) from EIS measurements of 11-(ferrocenyl)-undecanethiol modified electrodes. | ||
A typical bulk electrochemical measurement (Fig. 4a) requires the redox active species to diffuse from the bulk electrolyte to the electrode surface to undergo oxidation or reduction. As such, the Randles circuit that models this system contains a Warburg element to account for the diffusion of ions. In contrast, surface bound electrochemical measurements are not limited by diffusion processes (Fig. 4b) Therefore, the equivalent circuit used to model this kind of experiment replaces the Warburg element with a capacitance of the adsorbed species (Cads) (equivalent circuits are shown in Fig. S4b, ESI†). The rate constant of electron transfer, ket, is then calculated using the extracted Rct and Cad values, following an established method.30 This approach provides complementary information on the systemby enabling the quantitation of ket values, and thus further information on the electron dynamics occurring through the polymer films without the interference of diffusion effects.
The calculated values for ket are presented in Fig. 4c for gold electrodes with SAMs of 11-(ferrocenyl)-undecanethiol formed on them. As expected, bare gold electrodes showed the highest rate constant as there is no insulating layer present to impede the transfer of electrons to the electrode. When a POx coating is added, either MePOx or PiPOx, the ket of the redox process decreases significantly to approximately 400 s−1 and 600 s−1, respectively. The rate constant for PiPOx compared to MePOx is consistent with the previous Rct measurements.
When AuNPs are incorporated into the surface, there is a slight decrease in ket compared to the PiPOx only film, but no significant change is observed when a MePOx top layer is added to the construct. As the PiPOx + AuNP surface has gold surfaces available to the electrolyte, it was expected that a SAM would form on the AuNPs. However, the lack of significant increase in ket seems to invalidate this hypothesis. Suggesting, instead that either the 11-(ferrocenyl)-undecanethiol may not have been able to displace the mercaptosuccinic acid SAM already present on the AuNP surface or that the plasma polymer based layered construction may have little to no effect on the rate determining step of electron transfer – the charging of the nanoparticle by the redox couple.18 Previous work investigating anthraquinone (AQ) based molecular switches showed that this AQ conjugated redox centre improved electron transfer through saturated alkyl groups.31,32 It was proposed that redox active anthraquinone centres surrounded by saturated alkyl groups provided intermediate virtual energy states that facilitate tunneling through saturated norbornylogous bridges, thus reducing the barrier to electron tunelling. It appears that a similar mechanism may be at play here. In the case of PiPOx-AuNP-MePOx layered constructions, this time not at the molecular scale but at the nanoscale, with the AuNPs present within the polymer matrix providing energy states for electron transfer to occur thorugh the saturated polymer.
To further elucidate a possible mechanism for how AuNPs contribute to reducing the Rct of POx films, conductive AFM was carried out on the electrode surfaces at each stage of the layered construction.
Large area conductive AFM measurements
Current–voltage (I–V) curves were acquired across several spots on the electrode surfaces and the current response is presented in Fig. 5a. Rather than a specific shape or trend in the trace for each type of sample, three distinct categories of traces were observed across all samples tested. Traces where a steep switch in current occurs between −2 mV and 2 mV (Fig. 5a, left) are conductive traces and the plateaus at −600 nA and 600 nA indicate saturation of the current detector. Another group of traces showed no current response to ramping the potential (Fig. 5a, right), and these are referred to as insulating traces. Lastly, traces that showed some current in response to the ramping potential but did not saturate the detector (Fig. 5a, middle) correspond to intermediate conductivity profiles. Fig. 5b presents a schematic of hypothesized AFM tip locations in relation to AuNPs on the surface. Conductive traces are hypothesized to result from having the tip touching the AuNP and there being the smallest distance possible between the platinum tip and the AuNP, giving electrons a direct path to the nanoparticles. Traces that show intermediate conductivity are hypothesized to correspond to the tip touching the plasma polymer film in the proximity of a nanoparticle, enabling lateral conduction of electrons from the AFM tip to the AuNP. Lastly, insulating films showing no response to current are likely to be in areas where there are no AuNPs present in close proximity and therefore no paths for electrons to tunnel to the electrode.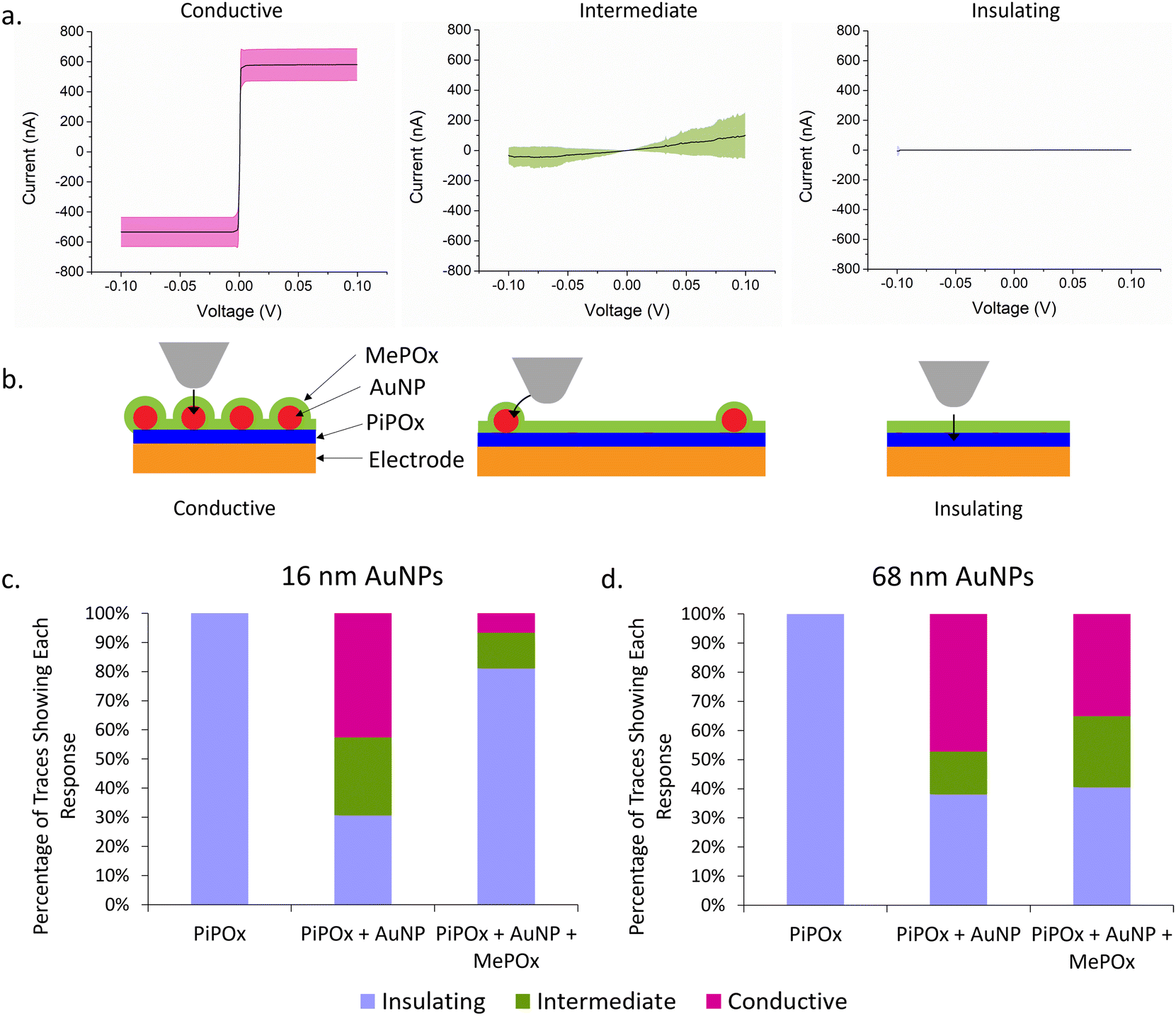 | ||
| Fig. 5 (a) Representative I–V curves of characteristic responses observed when probing the conductivity of the surface. Solid line represents average, shaded area represents standard deviation. (b) Schematic diagram showing hypothesised location of conductive AFM tip in relation to AuNPs on the surface of the electrode. Arrows represent proposed path of electrons. (c) Proportion of IV traces showing each response type in (a) on layered constructions with 16 nm AuNPs. (d) Proportion of IV traces showing each response type in (a) on layered constructions with 68 nm AuNPs. | ||
The percentage of each type of current transfer occuring at different stages of the layered construction are repesented in Fig. 5c and d for 16 and 68 nm AuNPs, respectively. A lone layer of PiPOx only returns insulating traces, confirming the high resistance of the film as observed previously. When 16 nm AuNPs are added to the PiPOx layer (Fig. 5c), 42.7% of the traces are conductive with an extra 26.7% showing intermediate conductivity – a total of 69.3% of traces showing conductivity. On the other hand, when 68 nm AuNPs are used (Fig. 5d), 61.8% of the traces show conductivity (47.3% conductive traces and 14.5% intermediate conductivity traces). The fewer intermediate traces observed with 68 nm particles are consistent with the nanoparticle binding density trends observed in Fig. 2, where this particle size bound in small aggregates, leaving a greater distance between nanoparticle aggregates. This greater distance between aggregates would decrease the probability of tunelling from the tip and therefore, less probability of observing an intermediate trace in these spaces. More even surface coverage – resulting from fewer nanoparticle aggregates – reduces the average interparticle distance and increases the probability of the tip interacting with the surface in a region where conductivity cany be measured. Therefore, the greater proportion of conductive traces in samples with 16 nm AuNPs suggests that more even surface coverage of AuNPs is the primary determinant of reducing the overall Rct.
When the top layer is deposited on samples with 68 nm AuNPs, we see no significant change in the total number of conductive traces, but these are made up of a greater number of intermediate conductivity traces (24.3% versus 14.5%). This result provides support for the ability of AuNPs to provide a path for electrons to tunnel through the POx top layer and reach the electrode beneath. However, when a layer of MePOx is deposited on top of the smaller AuNPs, the percentage of insulating responses increases significantly from 30.7% to 81.1% of all traces. Li et al.33 investigated the effect of insulating SiO2 shells on AuNPs of different sizes and found that the tunnelling current detected decreased with decreasing particle diameter when the shell thickness is kept constant. They attributed this effect to weaker electromagnetic fields produced by smaller nanoparticles and proposed that this is evidence of surface plasmon coupling contributing to the conductivity. Surface plasmons on particle surfaces induce magnetic fields, the intensity of which are related to the particle size. Coupling between the magnetic fields from nanoparticles and the magnetic field of the cAFM tip may accelerate electrons and contribute to the enhanced electron conduction through insulating films. Other work based on the development of conductive protein films using AuNPs reached a similar conclusion.34 This is consistent with the observed effects here, where the larger AuNPs maintained a detectable tunnelling current through the insulating MePOx top layer whereas, the smaller 16 nm particles saw a greater proportion of insulating traces when the same top layer is added. Furthermore, due to the high surface coverage of 16 nm AuNPs, the changes in nanotopography following deposition of the top layer may reduce the accessibility of the platinum tip to the areas between nanoparticles (Fig. 5b). This in turn could prevent the tip from reaching close enough to the nanoparticle to record a conductive or intermediate trace, thereby inflating the number of insulating traces recorded.
Layered constructions in biosensing
Lastly, the optimised layered construction was used in a biosensing experiment to assess any improvement in sensitivity gained by the lowered baseline resistance of the AuNP-doped plasma polymer bilayer configuration.To functionalise electrodes for biosensing applications, POx coated electrodes were incubated first with anti-PSMA antibody, then blocked by incubating with EDM. EDM binds to the POx through electrostatic interactions between its positive dipole and negatively charged sites on the surface, hence, a reduction in Rct is observed compared to unfunctionalised POx surfaces.35,36
Firstly, detection of the PSMA antigen was attempted using single layer POx-functionalised electrodes (Fig. 6a). No significant trend was observed between the concentration of PSMA antigen and measured Rct, indicating that there may be an incompatibility between the antibody and the PSMA antigen or that the sensitivity of this system is not sufficient for this type of analyte in a concentration range comparable to other examples in the literature.37,38 The inherent resistance of the MePOx films is likely too great for small changes in resistance caused by a protein binding to be detected. Therefore, exosomes isolated from LNCap cells (Fig. 6b) were used instead as they are physically larger and would induce a greater change in resistance upon binding.
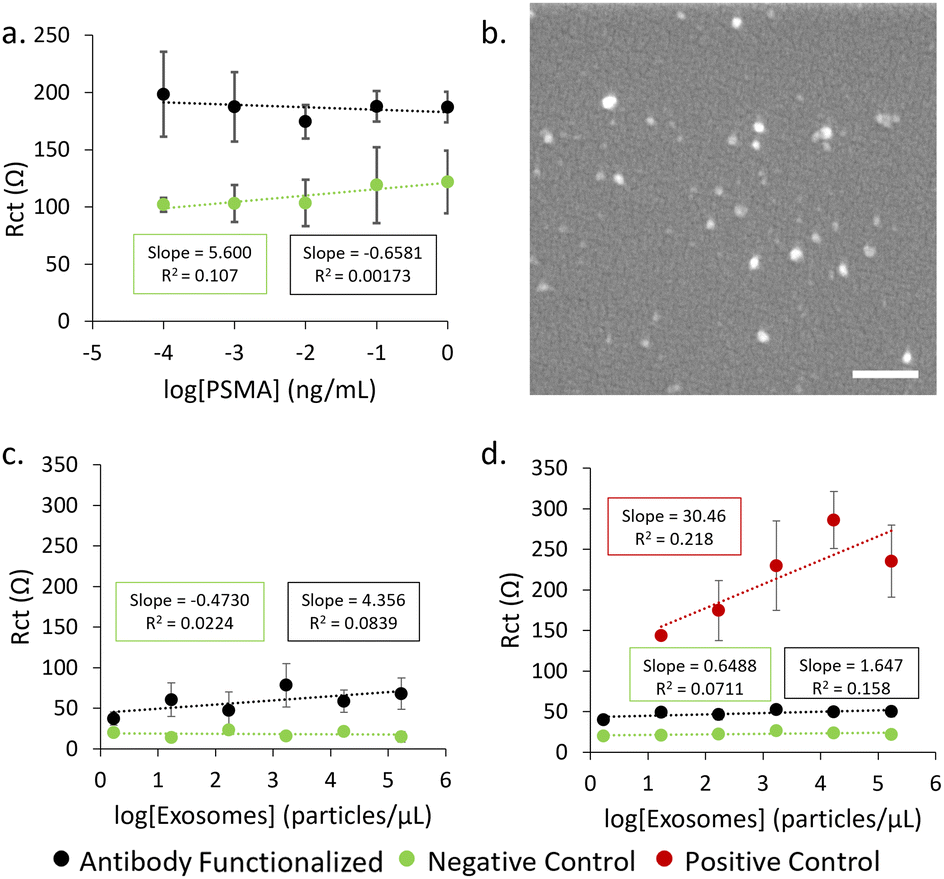 | ||
| Fig. 6 (a) Response in Rct for PSMA antigen detection when MePOx condition 1 is used to functionalize the electrode. (b) SEM image of LNCaP derived exosomes immobilised on MePOx condition 1 coated Si. Scale bar represents 200 nm. Response in Rct for LNCap derived exosome detection when MePOx condition 1 is used to functionalize the electrode (c) and when the full layered construction is used to functionalize the electrode (d). | ||
When exosomes are incubated on electrodes functionalized with a single layer of MePOx condition 1 (Fig. 6c), there is some response in Rct to increasing exosomes concentrations binding on the antibody functionalised electrode compared to the blocked negative control. A limit of detection (LOD) was calculated to be 96.1 particles per μL using the method 3SD/(slope) where SD is the standard deviation of the regression line.39 However, if the optimised PiPOx-AuNP-MePOx layered construction is used (Fig. 6d) to functionalize the electrodes, we observe a correlation between Rct and exosome concentration leading to an LOD of 35.2 particles per μL. This enhancement in sensitivity compared to MePOx only arises primarily due to a lower standard deviation calculated for the slope and highlights that the layered construction provides more reproducible data. The small correlation between Rct and exosome concentration that was observed for antibody functionalised electrodes here also indicates that there is an incompatibility between the selected antibody and the exosomes despite LNCap cells being reported to express PSMA.40,41 However, the LOD calculated for the positive control – where exosomes are captured non-specifically onto the surface – was 12.9 particles per μL. This is comparable to other reported examples in the literature of exosome biosensors where LODs were determined to be 96 particles per μL,42 70 particles per μL43 and low as 20 particles per μL.44,45 The calculated LOD for the positive control indicates that a biosensor using the POx layered construction has promise to achieve a low LOD comparable to other literature when a suitable recognition element is used.
Conclusions
We have demonstrated that there is a complex link between the deposition conditions of POx thin films and their chemical and electrochemical response. Increases in the plasma ignition power and monomer flow rate led to elevated film resistances at a given thickness. Furthermore, another major influence on the films’ resistivity was the chemistry of the monomer, with unsaturated monomers (PiPOx) providing additional pathways for crosslinking during the plasma polymerization. To reduce the charge transfer resistance of the films, in turn making them more suitable to biosensing, a layered construction of AuNPs incorporated between two layers of POx was optimized. A complex relationship between the inherent resistance of the underlayer and the bound nanoparticles was observed. In situations where particle density was kept constant but particle distribution on the surface differed, more even surface coverage combined with higher resistance underlayers resulted in a greater decrease in Rct. No significant relationship between the kinetics of electron transfer (ket) and the presence of AuNPs was observed. Tunnelling currents detected through cAFM show that 68 nm AuNPs maintain the ability to promote the tunnelling of electrons through the insulating top layer. Yet, further work is required to elucidate the mechanism that enables the significant reduction in Rct in the layered construction when 16 nm AuNPs are used. Lastly, a biosensor based on this method of functionalization promises to be sensitive and suitable for the detection of exosomes.Author contributions
AG: conceptualization, data curation, formal analysis, investigation, methodology, project administration, validation, visualization, writing – original draft. DY: project administration, supervision, writing – review & editing. TL: investigation, methodology. ED: investigation, methodology, writing – review & editing. ND: conceptualization, methodology, resources, software, writing – review & editing. CP: methodology, resources, supervision, writing – review & editing. MM: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the Australian Research Council Future Fellowship [grant number: FT200100301]. The authors also acknowledge the Australian Academy of Science, on behalf of the Department of Industry, Innovation, and Science. The Regional Collaborations Program is supported by the Australian Government under the National Innovation and Science Agenda. The authors also acknowledge the instruments and scientific and technical assistance of Microscopy Australia at the Future Industries Institute, University of South Australia, a facility that is funded by the University, and State and Federal Governments. This work used the NCRIS and Government of South Australia enabled Australian National Fabrication Facility - South Australian Node (ANFF-SA).References
- J. B. Shein, L. M. H. Lai, P. K. Eggers, M. N. Paddon-Row and J. J. Gooding, Langmuir, 2009, 25, 11121–11128 CrossRef CAS.
- Z. Alhalili, D. Figueroa, M. R. Johnston, J. Shapter and B. Sanderson, Aust. J. Chem., 2016, 69, 1402–1412 CrossRef CAS.
- Z. Zhang, S. Zhang, L. He, D. Peng, F. Yan, M. Wang, J. Zhao, H. Zhang and S. Fang, Biosens. Bioelectron., 2015, 74, 384–390 CrossRef CAS.
- S. Wu, F. Su, X. Dong, C. Ma, L. Pang, D. Peng, M. Wang, L. He and Z. Zhang, Appl. Surf. Sci., 2017, 401, 262–270 CrossRef CAS.
- E. Alvarez de Eulate, A. Gheorghiu, C. Amoura, A. Whiteley, C. Priest and M. N. MacGregor, Adv. Mater. Technol., 2021, 6, 2001292 CrossRef CAS.
- M. N. Macgregor-Ramiasa, A. A. Cavallaro and K. Vasilev, J. Mater. Chem. B, 2015, 3, 6327–6337 RSC.
- M. MacGregor, U. Sinha, R. M. Visalakshan, A. Cavallaro and K. Vasilev, Plasma Processes Polym., 2019, 16, 1800130 CrossRef.
- A. Gheorghiu, D. Yang, I. Delcheva, C. Priest and M. MacGregor, Plasma Processes Polym., 2023, e2200233 CrossRef.
- J. Wang, K. G. Neoh, L. Zhao and E. T. Kang, J. Colloid Interface Sci., 2002, 251, 214–224 CrossRef CAS.
- H. Liu, Z. Guo, S. Wang, X. Xun, D. Chen and J. Lian, J. Alloys Compd., 2020, 846, 156504 CrossRef CAS.
- M. Homberger and U. Simon, Philos. Trans. R. Soc., A, 2010, 368, 1405–1453 CrossRef CAS.
- G. Liu, S. G. Iyengar and J. J. Gooding, Electroanalysis, 2012, 24, 1509–1516 CrossRef CAS.
- J. Dyne, Y.-S. Lin, L. M. H. Lai, J. Z. Ginges, E. Luais, J. R. Peterson, I. Y. Goon, R. Amal and J. J. Gooding, ChemPhysChem, 2010, 11, 2807–2813 CrossRef CAS PubMed.
- T. Albrecht, S. F. L. Mertens and J. Ulstrup, J. Am. Chem. Soc., 2007, 129, 9162–9167 CrossRef CAS.
- C. Engelbrekt, K. H. Sørensen, J. Zhang, A. C. Welinder, P. S. Jensen and J. Ulstrup, J. Mater. Chem., 2009, 19, 7839–7847 RSC.
- P. S. Jensen, Q. Chi, J. Zhang and J. Ulstrup, J. Phys. Chem. C, 2009, 113, 13993–14000 CrossRef CAS.
- C. R. Bradbury, J. Zhao and D. J. Fermín, J. Phys. Chem. C, 2008, 112, 10153–10160 CrossRef CAS.
- J.-N. Chazalviel and P. Allongue, J. Am. Chem. Soc., 2011, 133, 762–764 CrossRef CAS.
- P. Zhou, X. Qiao, D. C. Milan, S. J. Higgins, A. Vezzoli and R. J. Nichols, Phys. Chem. Chem. Phys., 2023, 25, 7176–7183 RSC.
- A. Azadbakht, A. R. Abbasi, Z. Derikvand and Z. Karimi, J. Electroanal. Chem., 2015, 757, 277–287 CrossRef CAS.
- A. Michelmore, D. A. Steele, D. E. Robinson, J. D. Whittle and R. D. Short, Soft Matter, 2013, 9, 6167–6175 RSC.
- K. M. Chan, C. Amoura, A. Whiteley, J. Rouget, H. Safizadeh Shirazi, A. Cavallaro, K. Vasilev and M. MacGregor, Biointerphases, 2020, 15, 051005 CrossRef CAS PubMed.
- M. N. Macgregor, A. Michelmore, H. Safizadeh Shirazi, J. Whittle and K. Vasilev, Chem. Mater., 2017, 29, 8047–8051 CrossRef CAS.
- R. M. Visalakshan, M. N. MacGregor, A. A. Cavallaro, S. Sasidharan, A. Bachhuka, A. M. Mierczynska-Vasilev, J. D. Hayball and K. Vasilev, ACS Appl. Nano Mater., 2018, 1, 2796–2807 CrossRef CAS.
- J. R. Macdonald and L. D. Potter, Solid State Ionics, 1987, 24, 61–79 CrossRef.
- M. Macgregor-Ramiasa, K. McNicholas, K. Ostrikov, J. Li, M. Michael, J. M. Gleadle and K. Vasilev, Biosens. Bioelectron., 2017, 96, 373–380 CrossRef CAS PubMed.
- M. MacGregor, H. Safizadeh Shirazi, K. M. Chan, K. Ostrikov, K. McNicholas, A. Jay, M. Chong, A. H. Staudacher, T. D. Michl, A. Zhalgasbaikyzy, M. P. Brown, M. N. Kashani, A. Di Fiore, A. Grochowski, S. Robb, S. Belcher, J. Li, J. M. Gleadle and K. Vasilev, Biosens. Bioelectron., 2021, 171, 112699 CrossRef CAS.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- T. Li, C. Peiris, E. M. Dief, M. MacGregor, S. Ciampi and N. Darwish, Langmuir, 2022, 38, 2986–2992 CrossRef CAS.
- N. Darwish, M. N. Paddon-Row and J. J. Gooding, Acc. Chem. Res., 2014, 47, 385–395 CrossRef CAS PubMed.
- N. Darwish, I. Díez-Pérez, P. Da Silva, N. Tao, J. J. Gooding and M. N. Paddon-Row, Angew. Chem., Int. Ed., 2012, 51, 3203–3206 CrossRef CAS PubMed.
- C. Li, C. Xu, D. Cahen and Y. Jin, Sci. Rep., 2019, 9, 18336 CrossRef CAS.
- S. H. Mejias, E. López-Martínez, M. Fernandez, P. Couleaud, A. Martin-Lasanta, D. Romera, A. Sanchez-Iglesias, S. Casado, M. R. Osorio, J. M. Abad, M. T. González and A. L. Cortajarena, Nanoscale, 2021, 13, 6772–6779 RSC.
- D. E. P. Souto, J. V. Silva, H. R. Martins, A. B. Reis, R. C. S. Luz, L. T. Kubota and F. S. Damos, Biosens. Bioelectron., 2013, 46, 22–29 CrossRef CAS.
- K. Kimura, Y. Toshiyasu and R. Fujishiro, Bull. Chem. Soc. Jpn., 1966, 39, 1681–1683 CrossRef CAS.
- O. Jalil, C. M. Pandey and D. Kumar, Bioelectrochemistry, 2021, 138, 107733 CrossRef CAS PubMed.
- K. Bravo, F. G. Ortega, G. A. Messina, M. I. Sanz, M. A. Fernández-Baldo and J. Raba, Clin. Chim. Acta, 2017, 464, 64–71 CrossRef CAS.
- J. Miller and J. Miller, Statistics and Chemometrics for Analytical Chemistry, Pearson Education, London, 6th edn, 2010 Search PubMed.
- F. Lee, A. Yu, A. Anderson, D. Marrinucci, W. W. Magargal, V. A. DiPippo and W. C. Olson, J. Clin. Oncol., 2014, 32, 266 Search PubMed.
- P. Tsui, M. Rubenstein and P. Guinan, J. Biomed. Biotechnol., 2005, 2005, 328927 Search PubMed.
- Y. An, T. Jin, Y. Zhu, F. Zhang and P. He, Biosens. Bioelectron., 2019, 142, 111503 CrossRef CAS PubMed.
- H. Dong, H. Chen, J. Jiang, H. Zhang, C. Cai and Q. Shen, Anal. Chem., 2018, 90, 4507–4513 CrossRef CAS.
- Y. Cao, L. Li, B. Han, Y. Wang, Y. Dai and J. Zhao, Biosens. Bioelectron., 2019, 141, 111397 CrossRef CAS.
- S. Wang, L. Zhang, S. Wan, S. Cansiz, C. Cui, Y. Liu, R. Cai, C. Hong, I. T. Teng, M. Shi, Y. Wu, Y. Dong and W. Tan, ACS Nano, 2017, 11, 3943–3949 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00641g |
| This journal is © The Royal Society of Chemistry 2024 |