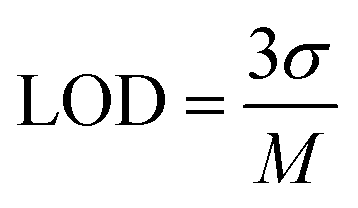DOI:
10.1039/D3MA00561E
(Paper)
Mater. Adv., 2024,
5, 4378-4400
Highly sensitive and selective electrochemical detection of caffeine, theophylline and guaiacol in green tea, green coffee, coffee, and tea extracts using a SnS2 nanoflake-modified electrode†
Received
17th August 2023
, Accepted 24th March 2024
First published on 10th April 2024
Abstract
SnS2 (TS) nanostructures with different morphologies were synthesized in three different solvents, Milli-Q water (W), ethanol (E) and isopropanol (I), using a simple solvothermal method. The phase, morphology and molecular structure of the synthesized TS nanostructures were investigated through powder X-ray diffraction, FE-SEM (field emission scanning electron microscopy), high-resolution TEM and XPS analysis. The synthesized TS nanostructure electrocatalysts were applied for the detection of caffeine (CFN), theophylline (THN) and guaiacol (GCL). During this study, we used methods such as cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). The TS(I)-modified glassy carbon electrode [TS(I)@GCE] showed superior sensitivity towards CFN, THN and GCL. Its electrochemical linearity range was found to be 0.10–0.27, 0.01–0.25 and 0.1–0.20 μM with detection limits of 6.71, 4.0 and 11.2 nM for CFN, THN and GCL, respectively; electrochemical sensitivity was found to be 190.20 μA μM−1 cm−2, 84.72 μA μM−1 cm−2 and 50.32 μA μM−1 cm−2, respectively. The calculated recyclability and long-term storage stability confirmed the robustness of the TS(I)@GCE electrode-based sensor. The detection of CFN, THN and GCL in the presence of dopamine (DPN) as well as in real green coffee, coffee, green tea, and tea samples validated the analytical performance of TS(I)@GCE for the precise and simultaneous detection of biomolecules. To the best of our knowledge, this is the first study on the simultaneous electrochemical detection of CFN, THN and GCL via a TS electrocatalyst. The resultant concentration of the analyte was verified through the spiking method. Consequently, the obtained data varies and represents good contact for the detection of caffeine, theophylline and guaiacol in real samples.
1. Introduction
In their daily routine, most people start their morning with brewed drinks such as tea, coffee, green tea or green coffee. These brewed drinks contain phenolic compounds such as caffeine (CFN), theophylline (THN), and guaiacol (GCL). Although these compounds are beneficial for human beings in some way, their excess consumption can be dangerous. CFN, a xanthine family component, is a stimulant of the central nervous system (CNS). It increases energy metabolism by interfering with active neurons, releasing noradrenaline, dopamine and serotonin.1,2 Consequently, the consumption of CFN in high doses can result in unwanted signs of tremors, headaches, gastric problems, kidney malfunction and anxiety. Caffeine consumption of more than 200 mg per day can result in unconsciousness and possibly death. Asthma, persistent neonatal apnoea disease, and obstructive pulmonary disease can be treated with THN, a phosphodiesterase-inhibiting medication. However, high amounts of THN (>20 μg mL−1) lead to several side effects such as diarrhoea, agitation, cardiac arrhythmias, palpitations, seizures, and insomnia. Besides these side effects, it effectively interferes with the regulation of the neurotransmitters dopamine, gamma-aminobutyric acid (GABA), and histamine.3 Remarkably, the health hazards of theophylline have been noticed when it is administered in the presence of caffeine. GCL is a member of the volatile phenolic compound family, which is widely used in medicine and the food industry to provide characteristic aroma but in a threshold amount.4 It is produced by spore-forming, rod-shaped acidophilic bacteria (Alicyclobacillus acidoterrestris). It grows in acidic pH (2.5–6.0) at temperature values ranging from 25 °C to 60 °C. Since it is found to be unaffected at elevated temperatures and low pH conditions, it has been considered a potential spoilage concern for vegetables and fruit juice. Key spoilage features are odour or phenolic off-flavor produced by guaiacol, 2,6-dichlorophenol, 2,6-dibromophenol.5 Therefore, these three components, namely CFN, THN and GCL, can be dangerous if present in food and medicines simultaneously in excess amounts, and hence the detection of CFN, THN and GCL is essential for the safe use of medicines, food products and beverages. To date, various analytical techniques such as gas chromatography–mass spectrometry,6 gas chromatography,7 fluorescence,8,9 high-performance liquid chromatography,5,10 surface-enhanced Raman spectroscopy,11,12 and colorimetric,13,14 TLC-densitometric,15 kinetic spectrophotometric16 and electrochemical sensing have been used for the sensing of CFN, THN and GCL. However, these approaches, except the electrochemical method involve complicated operation procedures, are time-consuming and have low sensitive. Alternatively, electrochemical methods are rapid, extremely sensitive, less time-consuming, less complicated, and can differentiate various analytes based on their electrochemical oxidation and reduction potentials.55 Nevertheless, despite the above-mentioned merits of the electrochemical method, it is dependent on the modification of the surface of a glassy carbon electrode and size and morphology of nanoparticles.56 Also, the overlapping of the peak potentials of analytes during their simultaneous detection and the electrochemical instability of nanomaterials lead to poor sensitivity and reproducibility. Electrodes modified with noble metals such as gold (Au), platinum (Pt) and palladium (Pd) have been used for the simultaneous detection of samples with stable potentials, but their high cost and low availability limit their electrochemical applications. Recently, square wave voltammetry was employed by B. Mekassa et al. to create poly(L-aspartic acid)/functionalized CNT for the simultaneous detection of CFN and THN.17 It was also reported that [tetra-(5-chloroquinolin-8-yloxy)phthalocyanato] could detect CFN and THN simultaneously using a composite of carbon nanotubes and manganese(III).18 It was reported that E. Murugan et al. simultaneously detected CFN, THN and vanillin using MoS2/PANI@g-C3N4/GEC.19 Qixia Guan et al. developed a covalent metal–organic framework using 1,3,6,8-tetra(4-formyl phenyl)pyrene and 2,6-diaminopyradine for the simultaneous detection of CFN and THN with high-sensitivity.20 To date, there have been numerous studies on the electrochemical-based simultaneous detection of CFN and HQN but there is no report on their detection with GCL. Recently, J. Kalaiyarasi et al. reported the use of graphene nanoflakes for the simultaneous detection of GCL and vanillin.20 Jun-Yong Sun et al. developed γ-Al2O3 hollow spheres functionalized with Pt nanoparticles for the ultra-sensing of GCL.4 Phyllis E. Hayes et al. prepared a Nafion and boron-doped diamond-modified electrode for the electroanalysis of GCL and its analogues. Some researchers prepared gold-nanoparticle-decorated ZrO2–CuO nanocomposites for the electrocatalytic oxidation of formaldehyde.21 The enhanced electrocatalytic oxidation of methanol was reported on an Au–Pt bimetallic particle-modified copper phthalocyanine-carbon nanotube composite electrode.22 The electrochemical determination of levofloxacin using a poly(pyrogallol red)-modified GCE was reported.23 In one study, a nanocomposite created by ultrafine Bi–Sn nanoparticle-decorated carbon aerogels was used for the electrochemical detection of dopamine and clozapine from the serum of rat brain region.24 A ZnO nanorod reduced graphene oxide nanocatalyst was synthesized and a modified GCE was prepared. The modified GCE was utilized for the voltametric detection of dopamine. It was also used for selective detection at a nanomolar concentration.25 An α-Fe2O3 nanoparticle-decorated β-Bi2O3 microsphere-modified electrode was used as a low-cost voltammetric sensor for the robust determination of Cd(II) and Pb(II) in the environment and food such as milk and water.26 Molecularly imprinted polypyrrole film-coated poly(3,4-ethylenedioxythiophene):polystyrene sulfonate-functionalized black phosphorene was employed for the selective and robust detection of norfloxacin in pharmaceutical formulations and milk samples.27 Also, the ultrasensitive, label-free voltammetric determination of norfloxacin based on molecularly imprinted polymers and an Au nanoparticle-functionalized black phosphorus nanosheet nanocomposite in medical, food, and environmental samples was achieved.28 A carbon nitride/graphene oxide composite was facilely synthesized, and a modified glassy carbon electrode was prepared. It was used for the simultaneous electrochemical detection of uric acid and dopamine at a nanomolar concentration.29 However, the complicated procedures for the fabrication of sensors using the above-mentioned materials limit their electrochemical sensing applications. Alternatively, TS nanostructures are being widely used in various fields such as photocatalysis, antioxidant, antimicrobial and battery applications.30–32 They are optimal for electrochemical sensing applications and their high electrochemical redox stability is an added advantage for the simultaneous detection of various analytes.
In this study, we synthesized TS nanostructures via a simple solvothermal method and investigated the influence of different solvents (Milli-Q water, ethanol, and isopropanol). It was observed that the synthesis of TS in Milli-Q water resulted in the formation of nanoparticles with a globular morphology. Alternatively, its synthesis in ethanol and isopropanol resulted in a nanoflake-like morphology. The nanoflakes were intercalated with each other in the case of ethanol, whereas for isopropanol, regular stacking of the TS nanoflakes was observed. Based on the electrochemical analysis, TS synthesised in isopropanol was found to be more electrochemically active than that synthesised in the other solvents, and thus it was used for the separate and simultaneous detection of CFN, THN and GCL. Moreover, the real sample analysis of tea, green tea, coffee and green coffee extracts was performed to validate the results and demonstrate the applicability of the prepared sensor.
2. Resources and procedures
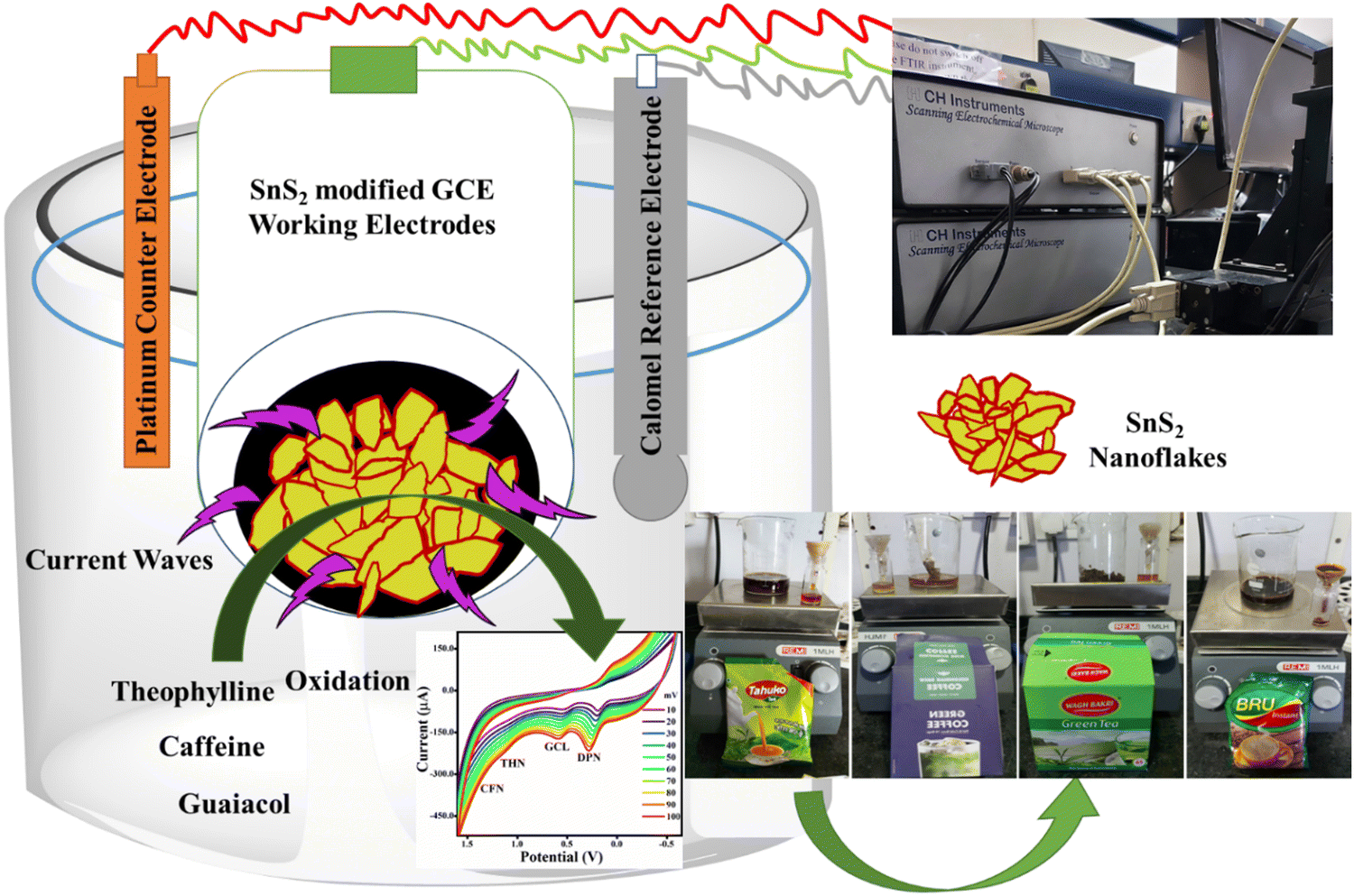
The chemicals and components utilized in this research work were of analytical grade and used without any further purification. Anhydrous tin(IV) chloride solution, thioacetamide, citric acid, isopropanol, caffeine, THN, dopamine, guaiacol and absolute ethanol were purchased from Sigma Aldrich, Bangalore, India. Green tea (Wagh Bakri), tea (Tahuko tea), green coffee (Colombian Brew), and instant coffee (Bru) were purchased from the local Gandhinagar, Gujarat local market for real sample analysis. Milli-Q water was used for the synthesis of the TS nanomaterials and electrochemical sensing experiments.
2.1 Synthesis of TS nanoflakes
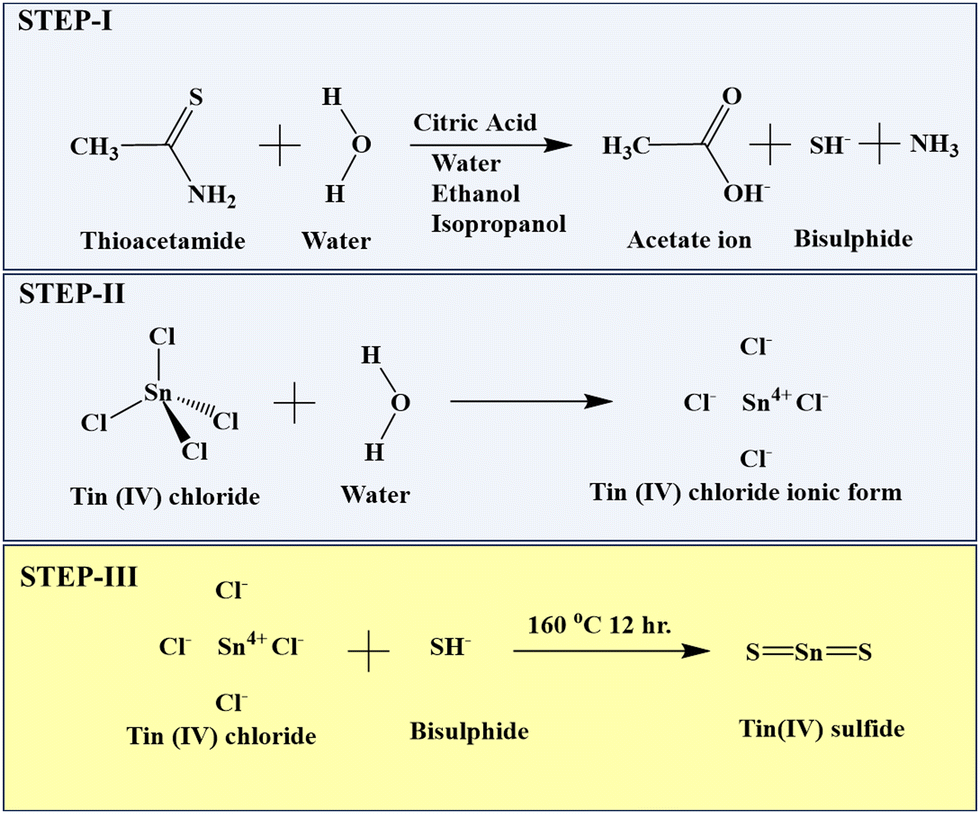
Briefly, 20 mL of solvent was prepared for each of Milli-Q water, ethanol, and isopropanol with 5 mmol citric acid and 10 mmol thioacetamide solution. Then, 20 mL of anhydrous tin chloride was added to each solvent under stirring. The obtained solutions were placed in a Teflon-lined stainless-steel autoclave at 160 °C for 12 h. After cooling to room temperature, the obtained particles were centrifuged, washed in water and ethanol and dried overnight at 60 °C.33 Three different textural changes were obtained at varying volumes of the Milli-Q water, ethanol, and isopropanol solvents. The TS nanoflakes synthesized in Milli-Q water, ethanol and isopropanol were denoted as TS(W), TS(E) and TS(I), respectively. The mechanism for the synthesis of TS is described in Scheme 1.
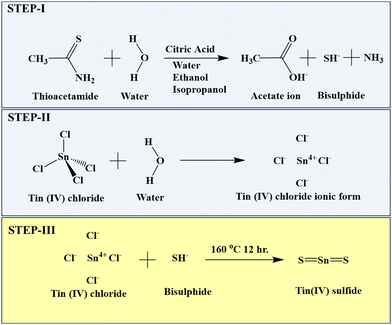 |
| | Scheme 1 Mechanism for the formation of SnS2 nanoflakes. | |
2.2 Electrode preparation and sensing
As required for the electrochemical study, a glassy carbon electrode (GCE 3 mm), platinum wire (Pt electrode), and saturated calomel electrode (SCE, 3 M KCl) were used as the working electrode, reference electrode and counter electrode, respectively. The diameter of the glassy carbon electrode was 3.3 mm, and its geometric surface area was 0.071 cm2. The GCE surface was first activated by rubbing and washing with 1.0 and 0.05 μM alumina slurry, and then a mirror-like finish was achieved by cloth polishing. After sonication (3 min) in Milli-Q water and ethanol mixed solution, the surface of the Milli-Q water GCE was washed and dried overnight in a hot air oven. Finally, 0.2 mg of each TS(W), TS(E) and TS(I) were separately mixed with 5 mL of Milli-Q water and isopropanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and ultrasonicated to form a uniform mixture. For electrochemical studies, different amounts were loaded on the GCE surface to get TS(W)@GCE, TS(E)@GCE and TS(I)@GCE.
:1) and ultrasonicated to form a uniform mixture. For electrochemical studies, different amounts were loaded on the GCE surface to get TS(W)@GCE, TS(E)@GCE and TS(I)@GCE.
2.3 Electrochemical optimization of TS-modified GCE
All three prepared electrodes, i.e., TS(W)@GCE, TS(E)@GCE and TS(I)@GCE, were utilized for voltammetric and impedimetric approaches at room temperature. The comparative optimization studies of the prepared working and bare electrodes were performed by CV and EIS. For optimization, 5 mM of each K3Fe(CN)6 and K4Fe(CN)6 were prepared in 0.1 M KCl (100 mL) and the CV spectra recorded in the scanning potential range of −0.8 to 0.8 V. To obtain the saturated redox behaviour of the TS-modified GCE, 10 consecutive runs of CV were recorded at a scan rate of 100 mV s−1. After that, CV analysis of all the electrodes was executed at varying scan rates (10–100 mV s−1). EIS was carried out at the initially applied potential of 0.1 V and the frequency range of 0.1–1000 Hz.
2.4 Electrochemical detection of CFN, THN and GCL
To calculate CFN, THN, and GCL, CV and differential pulse voltammetry (DPV) were utilized. Following the interpretation of the CV and EIS data, TS(I)@GCE was chosen for sensing because it was proven to be electrochemically more active than TS(W)@GCE and TS(E)@GCE. Therefore, this electrode was considered for further detection herein. The buffer solution (PBS 7.0) was used for all the sensing tests. CV was run in the presence of CFN, THN and GCL (0.25 μM of each). For different concentrations of CFN (0.1–0.27 μM), THN (0.01–0.25 μM) and GCL (0.1–0.27 μM), the DPV technique was executed in the potential window sensing range of 0.2–1.6 V, 0.6–1.3 V, and 0.0–0.8 V, respectively. All experiments were conducted three times to ensure the repeatability of the results. The effect of dopamine (DPN) on CFN, THN and GCL was also checked by introducing it into the reaction system. For the analysis of DPN, 0.1–0.25 μM concentrations were taken followed by DPV analysis in the potential window of −0.1 to 1.5 V. In the presence of CFN, THN, GCL and DPN, their simultaneous detection was performed with the CV and DPV techniques, where the potential window of −0.6 to 1.6 V and 0.0–1.5 V was applied, respectively. For the CV analysis, the amount of CFN, THN, GCL and DPN was fixed at 0.25 μM but in the case of DPV analysis, the amount of these analytes was increased simultaneously. All the trials were performed at room temperature.
2.5 Real sample (tea, instant coffee, green tea, and green coffee) extract preparation and analysis
In two separate samples of 250 mL of boiling DI water, 1.5 g of tea and instant coffee were combined.34 After 30 min, the mixtures were filtered through Watman filter paper 1. These filtered liquid solutions were then used for further sensing application during this study.
3. Depiction and characterization
The X-ray diffraction patterns of the obtained TS crystal nanostructures were recorded using an XRD diffractometer (Panalytical X pert Pro, Cu Kα, λ = 1.5406 Å mA, 40 kV, Netherlands) with a 2θ step of 0.0300° at a rate of 0.6000° s−1. The morphology of the TS nanostructures was investigated through FE-SEM microscopy (FE-SEM Quanta 200 FEG, Carl Zeiss, Germany) and elemental analysis was performed using the FE-SEM apparatus connected with EDAX (energy-dispersive X-ray analysis). A small portion of the TS nanostructure sample was coated with gold and placed on carbon tape for FE-SEM examination. In Milli-Q water, the TS nanostructures were prepared for HR-TEM examination by dispersing them by ultrasonication for 30 min. After that, each sample was dropped separately onto a carbon-coated copper grid (300 mesh). The TEM and HR-TEM images were recorded using two different HR-TEM instruments, i.e., FEI Talos F 200X and FEI Tecnai F 20 S/TEM. For the functional group analysis, an FT-IR spectrophotometer (XE 70, spectrum 65) was used. For the sample analysis, 198 mg of overnight-dried KBr was combined with 2.0 mg of TS sample, which was pressed into a pellet using a KBr press. The oxidation status of TS was investigated using an Omicron ESCA and X-ray photoelectron spectroscopy (XPS). Before the XPS analysis, the TS sample was placed on carbon tape and a full survey scan as well as high-resolution XPS spectra for each element were recorded. A scanning electrochemical microscope (SECM, CHI920D) workstation was used for electrode optimization and sensing applications.
4. Results and discussion
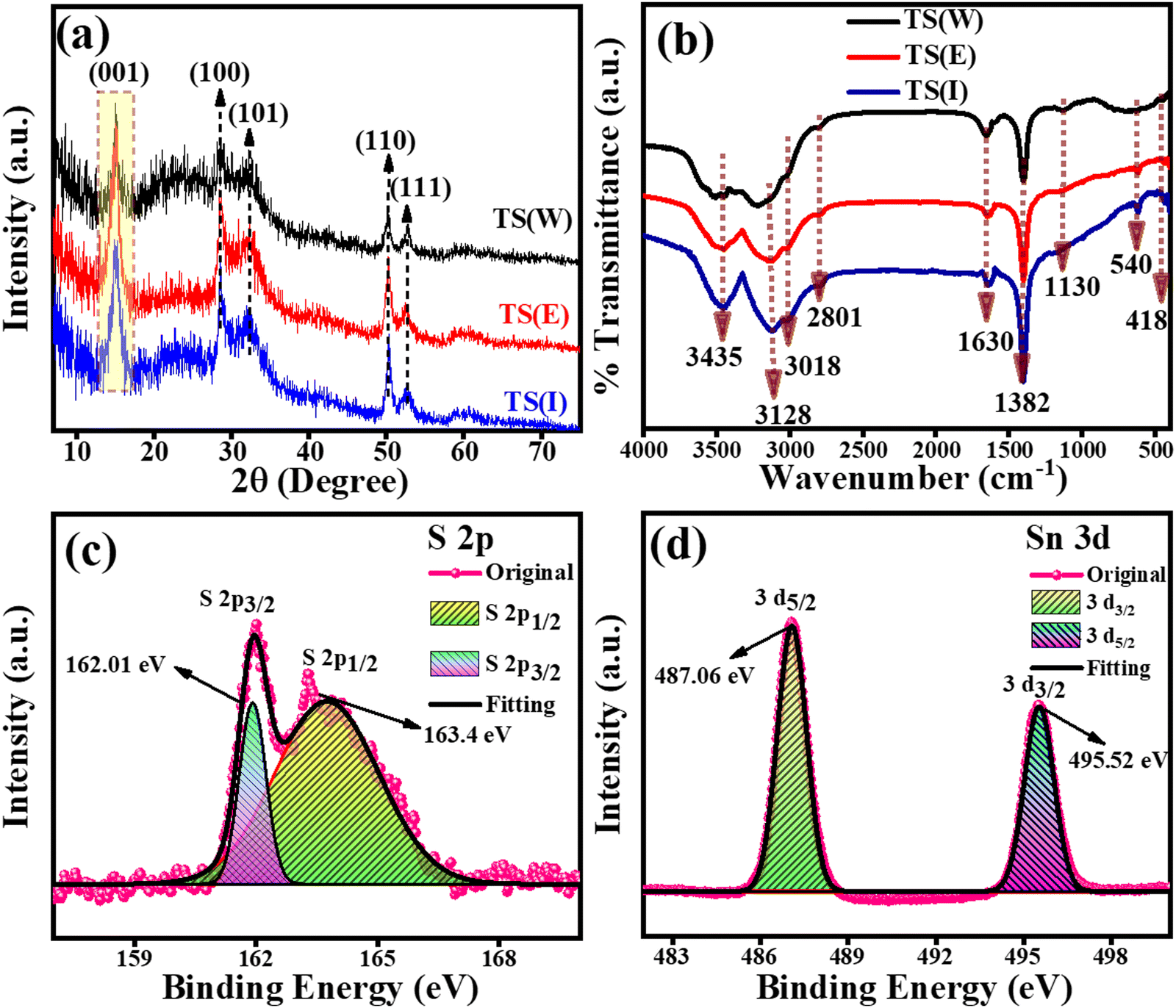
The phase and crystallographic information for the TS(W), TS(E) and TS(I) samples was obtained using XRD.35 The XRD patterns of the TS(W), TS(E) and TS(I) samples are shown in Fig. 1(a). Five major characteristic peaks can be observed at 2θ values of 15.02°, 28.47°, 32.20°, 50.20° and 52.38°, corresponding to the (001), (100), (101), (110) and (111) planes related to the hexagonal crystal phase, respectively. No other impurity peak was detected in the sample. The XRD results well matched the findings by Sungmook Park et al. and were determined to be in good accord with the JCPDS file no. (23-0677).36 According to the XRD analysis, it was concluded that the TS nanostructures were successfully prepared via this simple solvothermal method using non-hazardous solvents such as Milli-Q water (W), ethanol (E) and isopropanol (I).
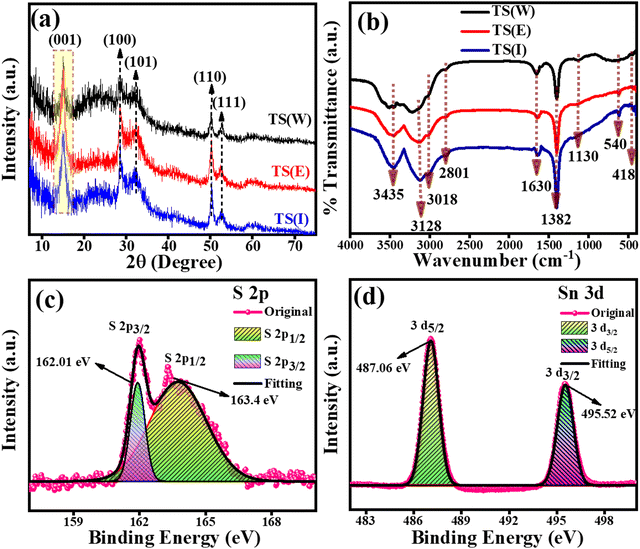 |
| | Fig. 1 PXRD, FT-IR and XPS analysis of TS nanoflakes. (a) PXRD patterns of TS(I), TS(E) and TS(W). (b) FT-IR spectra of TS(I), TS(E) and TS(W). (c) and (d) XPS high magnification spectra of S 2p and Sn 3d for TS(I). | |
The FT-IR spectra of TS(W), TS(E) and TS(I) are presented in Fig. 1(b), in which a total of six characteristic peaks can be observed. No substantial change was observed in all the TS samples. The peak at 540 cm−1 (or broad peak in the case of water) is related to the vibration of the well-matched continuous networks. Any inorganic impurity and the vibrations of the –OH group were indicated by the presence of two peaks at ∼1130 cm−1 and 1382 cm−1. The stretching vibrations associated with the –OH bonds of the hydrated oxides appeared as three peaks at 1630 cm−1, 3128 cm−1 and 3435 cm−1.37,38 The oxidation states of the TS(I) sample were investigated using the full survey scan as well as high-resolution XPS analysis. The high-resolution XPS spectra for the S 2p and Sn 3d spin–orbit core levels are shown in Fig. 1(c) and (d), respectively. In the full survey scan, only the peaks of Sn and S were observed, suggesting the purity of the sample. As indicated in Fig. 1(c), the peaks located at the binding energies of 162.01 eV and 163.4 eV are attributed to the S 2p3/2 and S 2p1/2 spin–orbit core levels, proving the S2− oxidation state of the sulphur element. The binding energy difference between the S 2p3/2 and S 2p1/2 spin–orbit core levels was found to be 1.39 eV and the intensity of S 2p3/2 was found to be higher than that of the S 2p1/2 spin–orbit core levels owing to the degeneracy. The XPS spectra of the Sn 3d spin–orbit core levels are presented by Fig. 1(d), in which the two peaks of Sn3d were observed at the binding energies of 487.06 eV and 495.52 eV, corresponding to the Sn3d5/2 and Sn 3d3/2 spin–orbit core-levels, respectively. The binding energy peak difference was found to be 8.46 eV. According to the XPS results, Sn possesses a +4 oxidation state (Sn4+) in the TS sample and the results were found to match that reported by Hezhang Chen et al.39 The XPS full survey scan profile of TS(I) is shown in Fig. S3 (ESI†). The XPS results of the TS(I) sample were also found to be consistent with the XRD results, revealing the TS molecular structure in the sample.
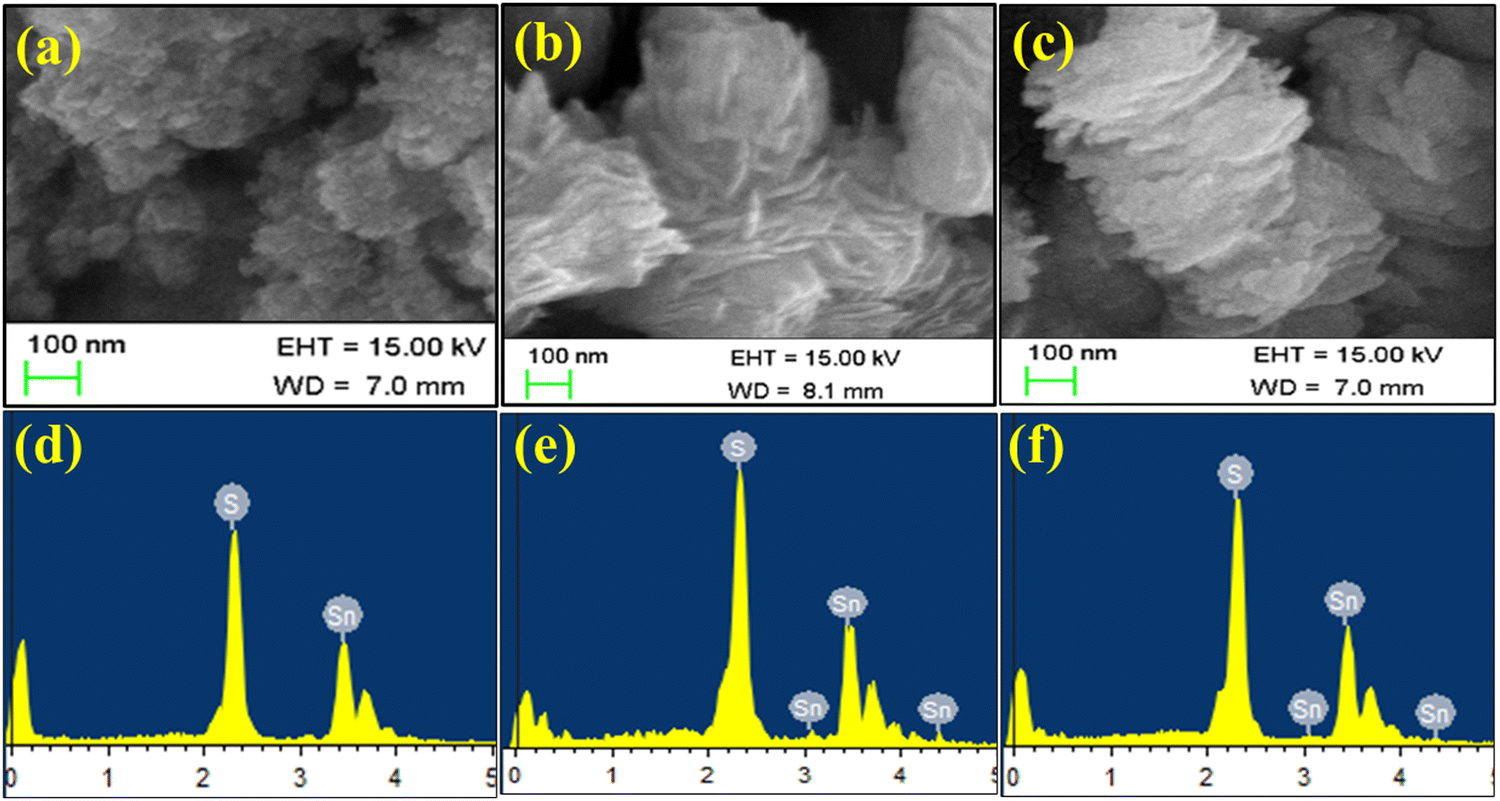
It can be clearly observed in Fig. 2 that in the three solvent-based synthesis processes of (W), (E) and (I), TS(W) showed the smallest particle size. As indicated in Fig. 2(a), TS(W) showed a small globular nanoparticle-like morphology. The TS(E) nanostructures have a disk-shaped morphology, as highlighted in Fig. 2(b). These disks were found to be stacked and interconnected/intercalated with each other. As shown in Fig. 2(c), the TS(I) nanostructures were found to be flake-shaped and stacked but not interconnected. The EDAX patterns of the TS(W), TS I and TS(I) samples are presented in Fig. 2(d)–(f), respectively, depicting that all the samples have Sn and S as their molecular constituents and no other impurity peak was detected. The system-generated EDS composition of the Sn and S elements in TS(W), TS(E), and TS(I) is illustrated in Table S1 (ESI†).
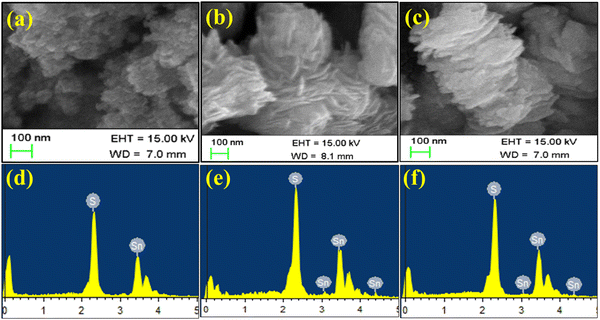 |
| | Fig. 2 FE-SEM micrographs of TS nanoflakes. (a) FE-SEM micrograph of TS(W). (d) EDAX spectra of TS(W). (b) FE-SEM micrograph of TS(E). (e) EDAX spectrum of TS(E). (c) FE-SEM micrograph of TS(I). (f) EDAX spectrum of TS(I). | |
TEM and HR-TEM micrographs of the TS(W), TS(E) and TS(I) samples are displayed in Fig. 3(a)–(c), respectively. As shown in Fig. 3(a), the TS(W) nanostructures appeared as stone pearls with a size of ∼20–30 nm. The TEM image of TS(E) is shown in Fig. 3(b), showing a dendritic appearance, which was taken at the edge of the disk-shaped TS(E) sample. The thickness or the breadth of the edge of the disk-shaped TS(E) was found to be 19.34 nm. Fig. 3(c) reveals the TEM image of the TS(I) sample, showing the flake-like morphology of the TS(I) sample. The HR-TEM images of the TS(W), TS(E) and TS(I) samples are shown in Fig. 3(d)–(f), respectively, suggesting the crystallinity of the samples. There was no significant change in the fringe thickness (d-spacing) of each sample, which was found to grow along the (001) crystallographic plane.
 |
| | Fig. 3 TEM and HR-TEM images of TS nanoflakes. (a)–(c) TEM images of TS(W), TS(E) and TS(I), respectively. (d)–(f) HR-TEM images of TS(W), TS(E) and TS(I). | |
4.1 Optimization of TS(W)-, TS(E)- and TS(I)-modified GCE
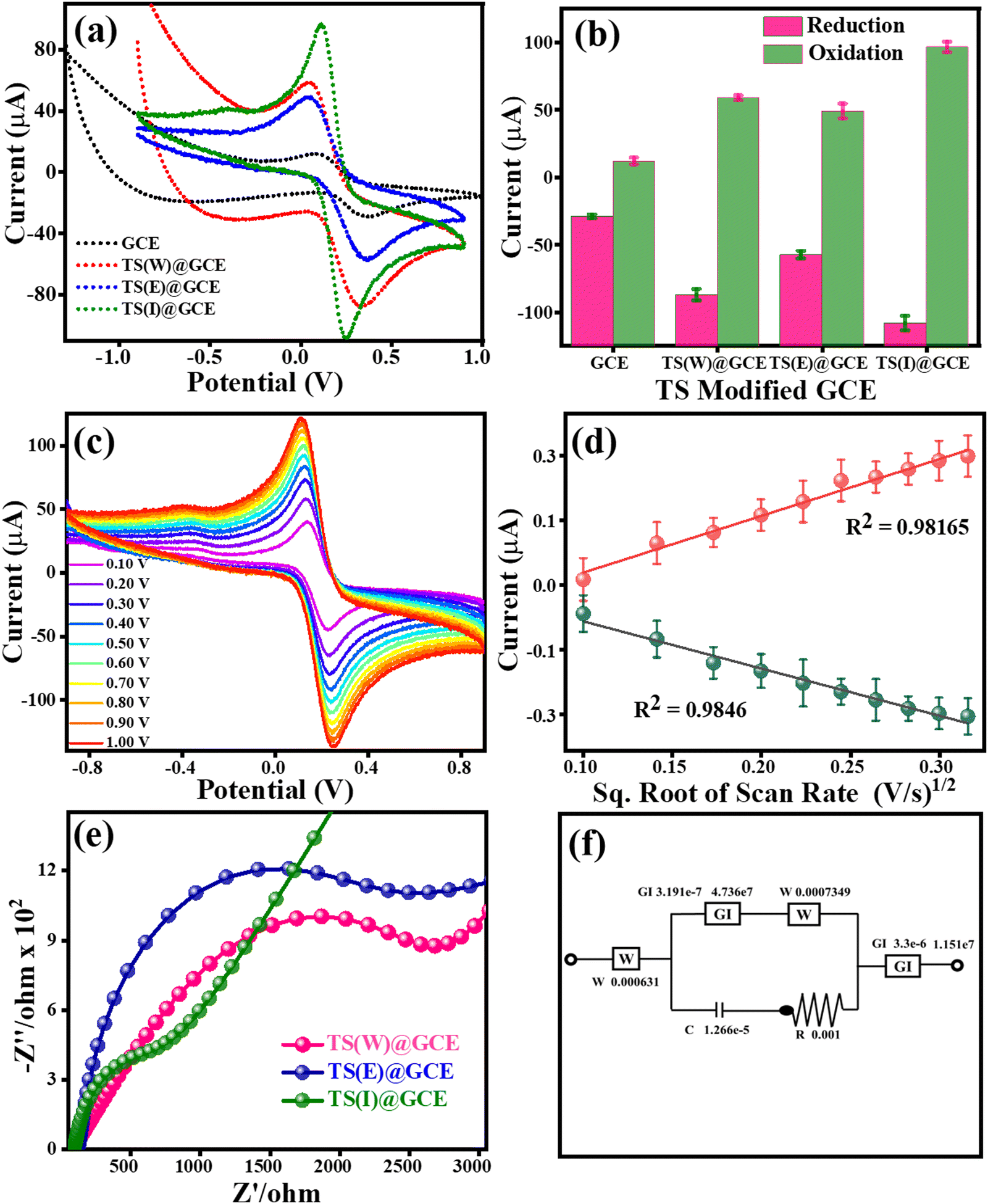
To enhance the performance of the TS(W)@GCE, TS(E)@GCE and TS(I)@GCE electrodes, the electrolyte solution of 0.1 M KCl containing 5 mM of both K3Fe(CN)6 and K4Fe(CN)6 was used. Fig. 4(a) shows the cyclic voltammograms for the GCE, TS(W)@GCE, TS(E)@GCE and TS(I)@GCE electrodes. The redox currents of the TS(I)@GCE electrode were higher than that of the bare GCE and other electrodes, i.e., TS(W)@GCE, TS(E)@GCE. This improved redox behaviour of the TS(I)@GCE electrode denotes an increase in the electrode surface area, which is effective and provides improved active sites for electron transport. Fig. 4(b) represents the dependence of the redox current on the different electrodes, where the maximum redox current was observed for the TS(I)@GCE electrode, and hence this electrode was used for further analysis. As shown in Fig. 4(c), CV analysis was performed to determine the impact of different scan speeds (10 mV s−1 to 100 mV s−1) on the redox behaviour of the TS(I)@GCE electrode. It was discovered that the oxidation and reduction currents increased in tandem with the scan rate. Fig. 4(d) shows the relationship between oxidation current (Ipa) and reduction current (Ipc) and the square root of scan rate (v1/2). We found that Ipa and Ipc rose linearly with the scan rate. Ipa = −90.97 (v1/2) + 2.07 with the regression coefficient R2 = 0.9846 and Ipc = 109.39 (v1/2) − 8.55 with the regression coefficient R2 = 0.9816, which further demonstrated that the linear dependence of these currents can be expressed by the regulated electron transport at the interfacial contact of the TS(I) and GCE surface as well as the strong coupling of the TS(I) and GCE surface. The Randles–Sevcik equation was also used to determine the electrochemical active surface area using the slope of this graph and the electrolyte diffusion coefficient, as follows:
| IP = 269000 × v1/2 × D1/2 × n3/2 × A × C0 |
where v1/2 is the square root of the scan rate, D denotes the diffusion coefficient of the electrolyte, n is the number of electrons involved in the redox reaction, A denotes the electrode surface area, and C0 denotes the molar concentration of redox species. In the case of the TS(I)@GCE electrode, its electrochemically active surface area was determined to be 0.260 cm2. The electrochemically active surface area of the bare GCE is 0.0449 cm2, TS(W)@GCE is 0.0286 cm2 and TS(E)@GCE is 0.0873 cm2. The TS-modified GCE electrode also optimized using electrochemical impedance spectroscopy (EIS), in addition to CV analysis. EIS was used to show the simple electron transfer behaviour of the GCE TS(W)@GCE, TS(E)@GCE, and TS(I)@GCE electrodes in 0.1 M KCl containing 5 mM of both K3Fe(CN)6 and K4Fe(CN)6 electrolyte solutions in the frequency range of 0.001–100000 Hz at a signal amplitude of 5 mV with a potential of 0.1 V. In the obtained Nyquist graph, as shown in Fig. 4(e), at high frequency, the highest altitude semicircle portion indicates the limited ability of charge transfer for the different modified electrodes.
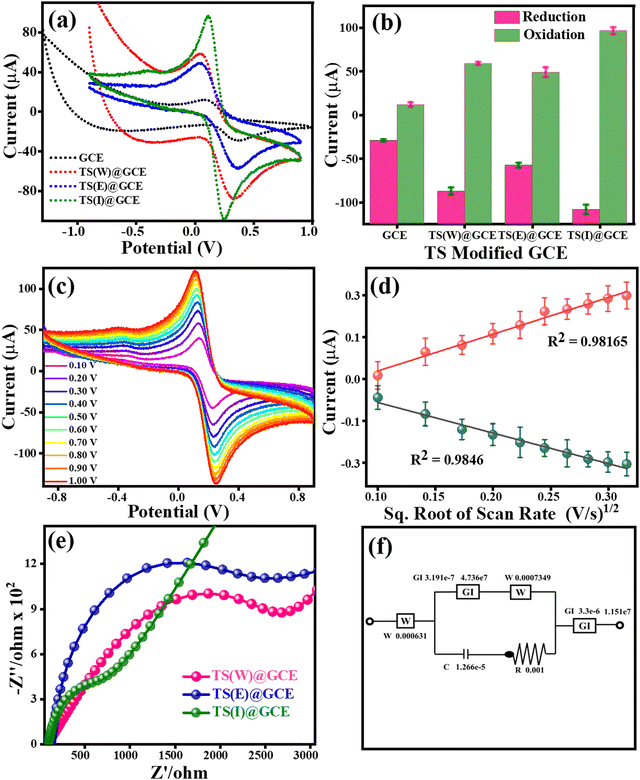 |
| | Fig. 4 Cyclic voltammetry and electrochemical impedance spectroscopy (EIS) analysis of TS(I)@GCE, TS(E)@GCE and TS(W)@GCE. (a) CV analysis of GCE, TS(I)@GCE, TS(E)@GCE and TS(W)@GCE. (b) Dependence of redox currents on the different electrodes. (c) CV analysis of TS(I)@GCE at varying scan rates. (d) Dependence of redox currents (Ipa and Ipc) on scan rate1/2. (e) EIS spectra (Nyquist plots) for GCE, TS(I)@GCE, TS(E)@GCE and TS(W)@GCE. (f) Equivalent circuit for EIS spectra of TS(I)@GCE. | |
The electrode with the lowest hemisphere radius, which indicates less electron transfer resistance and a higher electron transfer rate, is the TS(I)@GCE electrode. Hence, the TS(I)@GCE electrode showed effective charge transportation at the electrode surface and electrolyte. Both the CV and EIS techniques showed that the TS(I)@GCE electrode exhibited the comparative best response among the electrodes. The equivalent circuit for the EIS spectra recorded with the TS(I)@GCE electrode in 0.1 M KCl containing 5 mM of both K3Fe(CN)6 and K4Fe(CN)6 is shown in Fig. 4(f). At the interface of the electrode and electrolyte, an electrical double layer consisting of capacitive characteristics (Cdl) exists. Alternatively, the Warburg impedance describes the created impedance by the TS(I)@GCE electrode diffusion process in one dimension, which is bound by the electrode planar side, for the 45° in the lower frequency region. The layer of the TS(I)@GCE electrode showed a lower diameter than the GCE, indicating less charge transfer resistance (Rct). Thus, the adsorption of the analyte by redox reaction followed by the diffusion of H+ (proton ion) in the low-frequency range results in the Warburg impedance.40 The calculated Rct value for TS(W)@GCE is 2803.25 Ω, TS(E)@GCE is 2513.43 Ω and TS(I)@GCE is 783.42 Ω. The Rct value is inversely proportional to the diffusion rate, which indicates that the diffusion rate for TS(I)@GCE will be much higher than that of the other electrodes used in this study.
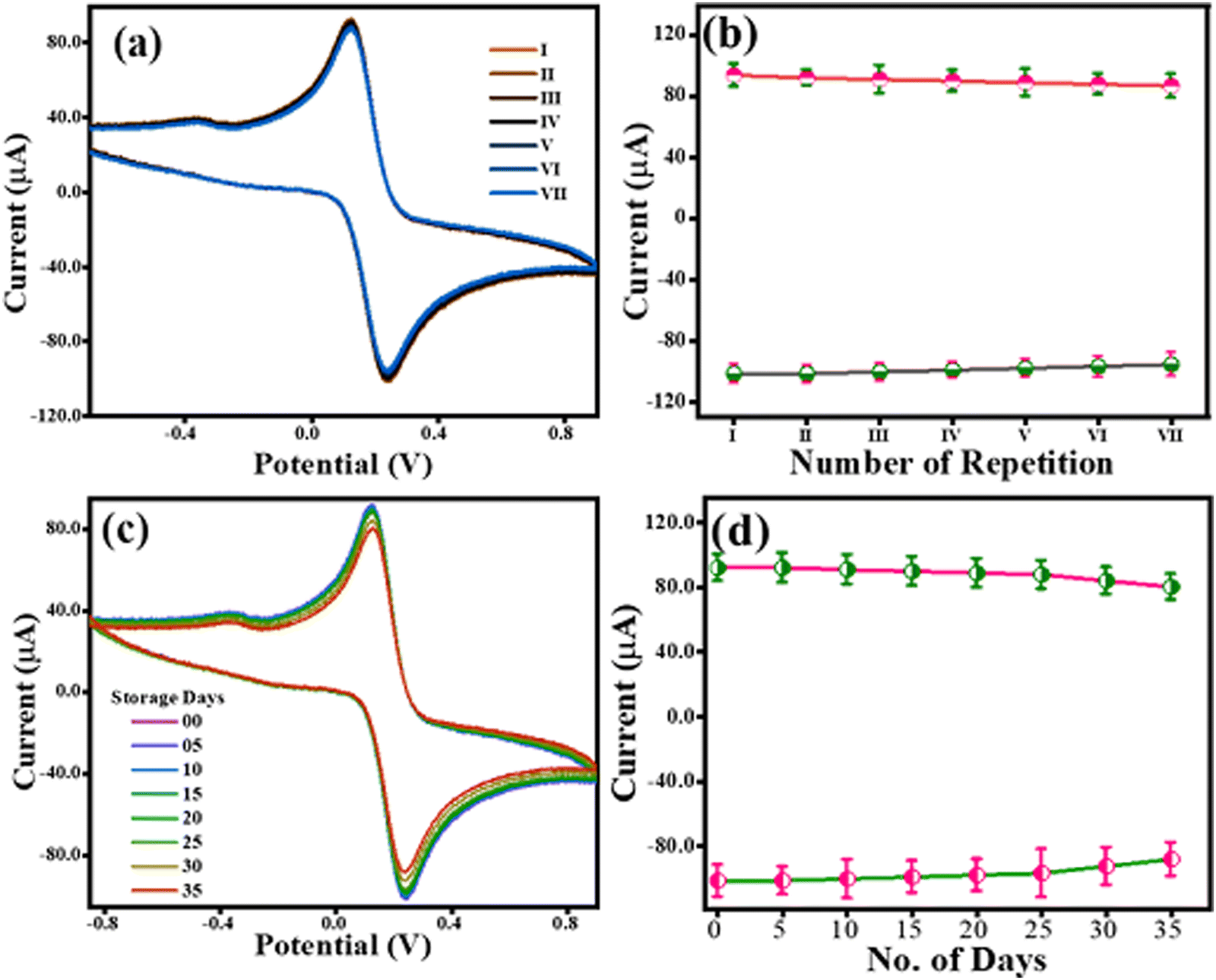
4.2 Stability and reproducibility of the TS(I)@GCE electrode
The reproducibility of the TS(I)@GCE electrode was tested using CV in 0.1 M KCl containing 5 mM of both K3Fe(CN)6 and K4Fe(CN)6. The CV analysis was carried out for the TS(I)@GCE electrode at a constant scan rate of 50 mV s−1. Following the examination, the electrode was carefully taken out of the electrolyte cell and dried in preparation for the subsequent cycle. Fig. 5(a) displays the CV values from seven distinct cycles. The redox current dependence over the seven repetitions is shown in Fig. 5(b). Given that no significant change was recorded even after seven repetitions of CV analysis, the TS(I)@GCE electrode could be used multiple times. Similarly, its stability was tested for up to 35 days with the same electrode (TS(I)@GCE electrode) through CV analysis (Fig. 5(c) and (d)). We found that the TS(I)@GCE electrode worked as normal until day 34 and the change in the current difference was <0.4 μA for both the anodic and cathodic currents. However, on day 35 of storage, the observed anodic and cathodic peak current decreased considerably. That may be due to the mechanical trembling and detachment of the TS(I) nanoflakes from the GCE surface. Hence, the TS(I)@GCE electrode bears good reproducibility and stability and can be effectively utilized for sensing applications. Its reproducibility was also tested in the presence of CFN, THN, and GCL in PBS (pH= 7) buffer. All their dependent linear graphs were drawn within the respective graphs. All the repeatability studies were completed seven times, and all the stability studies were completed on alternating day up to 14 days. Fig. S4(a), (b), and (e) (ESI†) show the differential pulse voltammetry (DPV) graph for the repeatability of CFN, THN, and GCL, respectively. Fig. S4(c), (d), and (f) (ESI†) show the DPV graphs for stability for CFN, THN, and GCL, respectively. No significant change in oxidation peaks was observed. Hence, these electrodes can be used for the stable detection of the analyte.
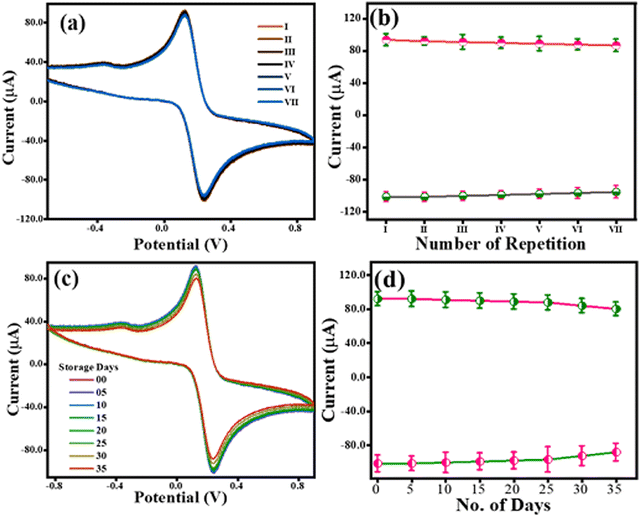 |
| | Fig. 5 Robustness study for the TS(I)@GCE electrode. (a) Repeatability assay for TS(I)@GCE. (b) Dependence of redox currents over different repeatability assays. (c) CV response of TS(I)@GCE after storage for 35 days. (d) Dependence of redox currents over storage time. | |
4.3 Effect of TS(I) loading and buffer pH on CFN, THN, and GCL oxidation
The loading of the catalyst is crucial for the oxidation of THN. To determine how the TS(I) loading affects the GCE, 1–7 μL of 0.2 mg/5 mL TS(I) was loaded and the DPV analysis was performed in the presence of THN. The maximum THN oxidation was noted for 5, 6, and 7 μL TS(I) catalyst, as shown in Fig. 6(a), which also shows the DPV response for each amount of catalyst. The relationship between the THN oxidation current and the amount of TS(I) is shown in Fig. 6(b). Therefore, we opted for a TS(I) catalyst loading of 6 μL for further analysis. Additionally, the DPV approach was used to examine how pH affects the electrochemical behaviour of THN using the TS(I)@GCE electrode. Fig. 6(d) depicts the DPV response to THN oxidation at various buffer pH (3.0 to 11.0). On increasing the pH range from 3.0 to 11.0, the THN oxidation anodic peak potential (Epa) shifted towards the negative side. This suggests the facilitated deprotonation of THN during the oxidation process. This slope shows that the number of electrons transferred throughout the reaction was equivalent to the number of hydrogen ions involved.41 Alternatively, the peak current (Ipa) decreased progressively from pH 9.0 to 11.0 and increased sharply from 3.0 to 7.0. Thus, pH 7 was chosen as the THN oxidation Ipa with the greatest pH. The THN oxidation potential was found to be linearly dependent on the solution pH, as indicated in Fig. 6(c), which can be stated by the regression equation Ep (V) = −0.026 (pH) + 1.22, where Ep (V) is variable, and R2 = 0.9985 indicates the linear dependence of the THN oxidation potential on pH, which can be attributed to the regulated diffusion.
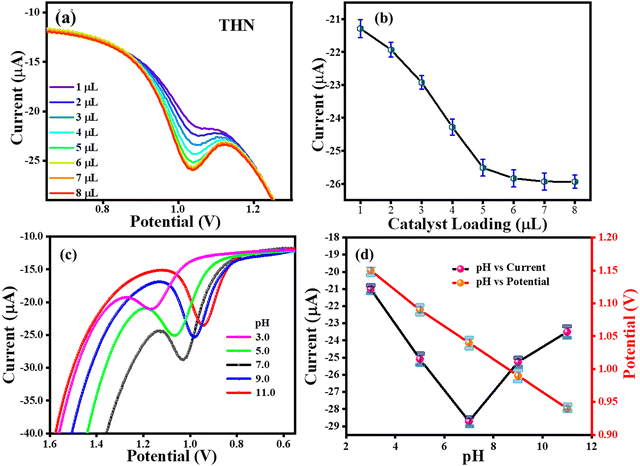 |
| | Fig. 6 Effects of buffer pH and catalyst loading on THN oxidation. (a) DPV response for various loading amounts of THN present at TS(I) GCE. (b) Dependence of THN oxidation current on different amounts of TS(I) loading at GCE. (c) DPV response for THN oxidation at different pH. (d) Dependence of pH on THN oxidation current and potential. | |
However, the loading of the catalyst was also determined to be related to the oxidation of CFN and GCL and repeated with THN. Around 0.2 mg/5 mL TS(I) was loaded on GCE in the volume of 1–8 μL and the DPV analysis was carried out. A higher oxidation was noted at 5 μL, 6 μL, 7 μL and 8 μL, as shown in Fig. S5a, c, and e (ESI†). The relatively linear graphs for the catalyst loading are shown in Fig. S5b, d, and f (ESI†). Therefore, 6 μL of TS(I) catalyst loading was considered for preparing the modified GCE. Moreover, different PBS buffer solutions with pH in the range of 2–8 were prepared, and oxidation of CFN, THN (repeated) and GCL was carried out. The oxidation of CFN is shown in Fig. S6 (ESI†) and its related calibration graph is shown in the inset. The maximum oxidation peaks in DPV were observed in the pH range of 6–8. All the oxidation potential calculations of CFN were carried out at pH 7, which can be considered a good pH for detection. Similarly, the oxidation peak was also observed for THN (repeat), as shown in Fig. S6b (ESI†), and its linear dependent calibration graph at different pH of 2–8 is depicted within the THF oxidation graph. The maximum oxidation of THN was observed at pH 7. As shown in Fig. S6c (ESI†), an increase in the pH range for GCL was observed and its linear relative graph is drawn within that graph. All further experiments for detection were carried out at pH 7, which is the common pH for the detection of CFN, THN, and GCL.
4.4 Effect of scan rate on the detection of CFN, THN, GCL and DPN
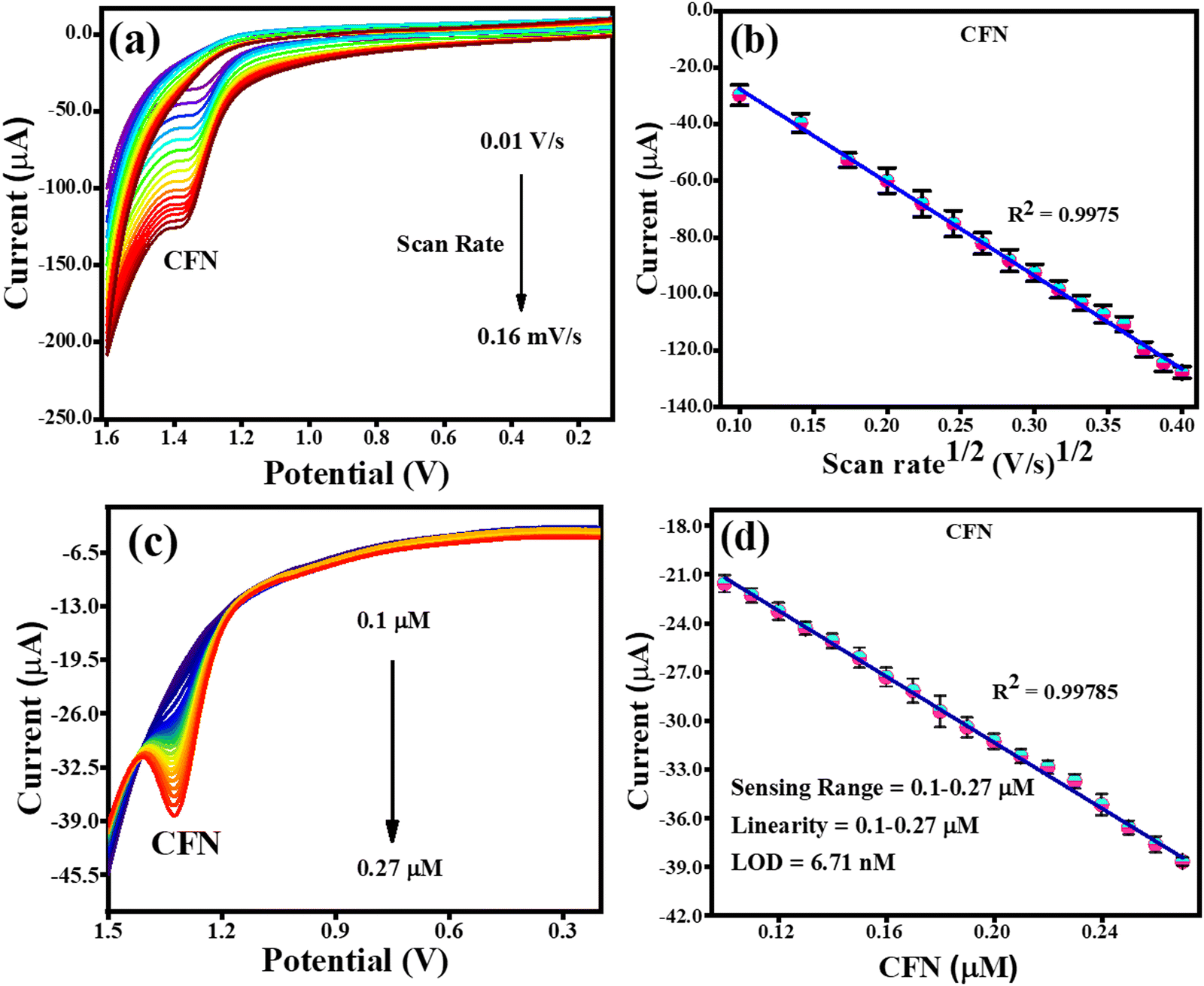
The effect of scan rate was investigated by CV analysis with fixed concentrations of CFN, THN, GCL and DPN at pH 7.0. Fig. 7(a) illustrates the effect of the scan rate on the oxidation of CFN, causing the oxidation potential of CFN to shift towards the higher potential end. Alternatively, Fig. 7(b) shows the relation between the CFN oxidation current and the square root of the scan rate. The regulated diffusion process is satisfied by the linear connection of CFN oxidation. The regression equation Ipa (μA) = −0.00033 (scan rate1/2) + 5.4 × 10−6 with the regression coefficient R2 = 0.9975 could be used to express this linearity dependence. Additionally, the differential pulse voltammetry (DPV) method was used to assess the analytical performance of the TS(I)@GCE electrode for the detection of CFN in a pH 7.0 buffer solution.
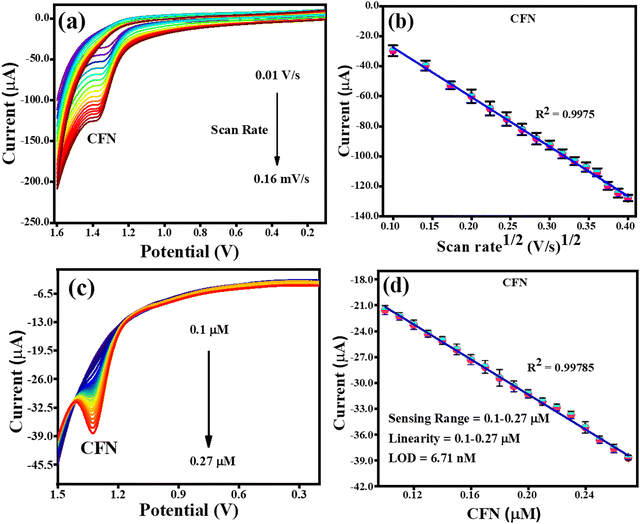 |
| | Fig. 7 CV and DPV response for the oxidation of CFN. (a) Effect of CV scan rate on the oxidation of CFN. (b) Dependence of CFN oxidation current on scan rate1/2. (c) DPV response for CFN oxidation with increasing concentration. (d) Dependence of oxidation current on CFN concentration. | |
This method was found to be more sensitive to produce a better electrochemical performance than the CV. Therefore, DPV was carried out in the range of −0.2 V to 1.6 V with the amplitude and frequency of 0.025 V and 5 Hz, respectively, to obtain favourable observations. The DPV response to CFN in the concentration range of 0.1–0.27 μM is shown in Fig. 7(c), where it can be seen that there was no change in the oxidation potential of CFN after increasing its concentration. Additionally, Fig. 7(d) shows the relationship between the CFN oxidation current (Ipa) and its concentration, from which it can be deduced that the current increased linearly with an increase in its concentration. This linearity dependence can be expressed by the following equation: Ipa (μA) = −1.01 × 10−4 (CFN, μM) − 1.1 × 10−5 with the regression coefficient R2 = 0.9978. Additionally, the following equation was used to compute the limit of detection (LOD) of CFN (S/N = 3):
where
σ is the leftover standard deviation at a small concentration and
M denotes the calibration of the straight-line slope. The linearity range and calculated LOD of CFN were determined to be 0.1 to 0.27 μM and 6.71 nM, respectively.
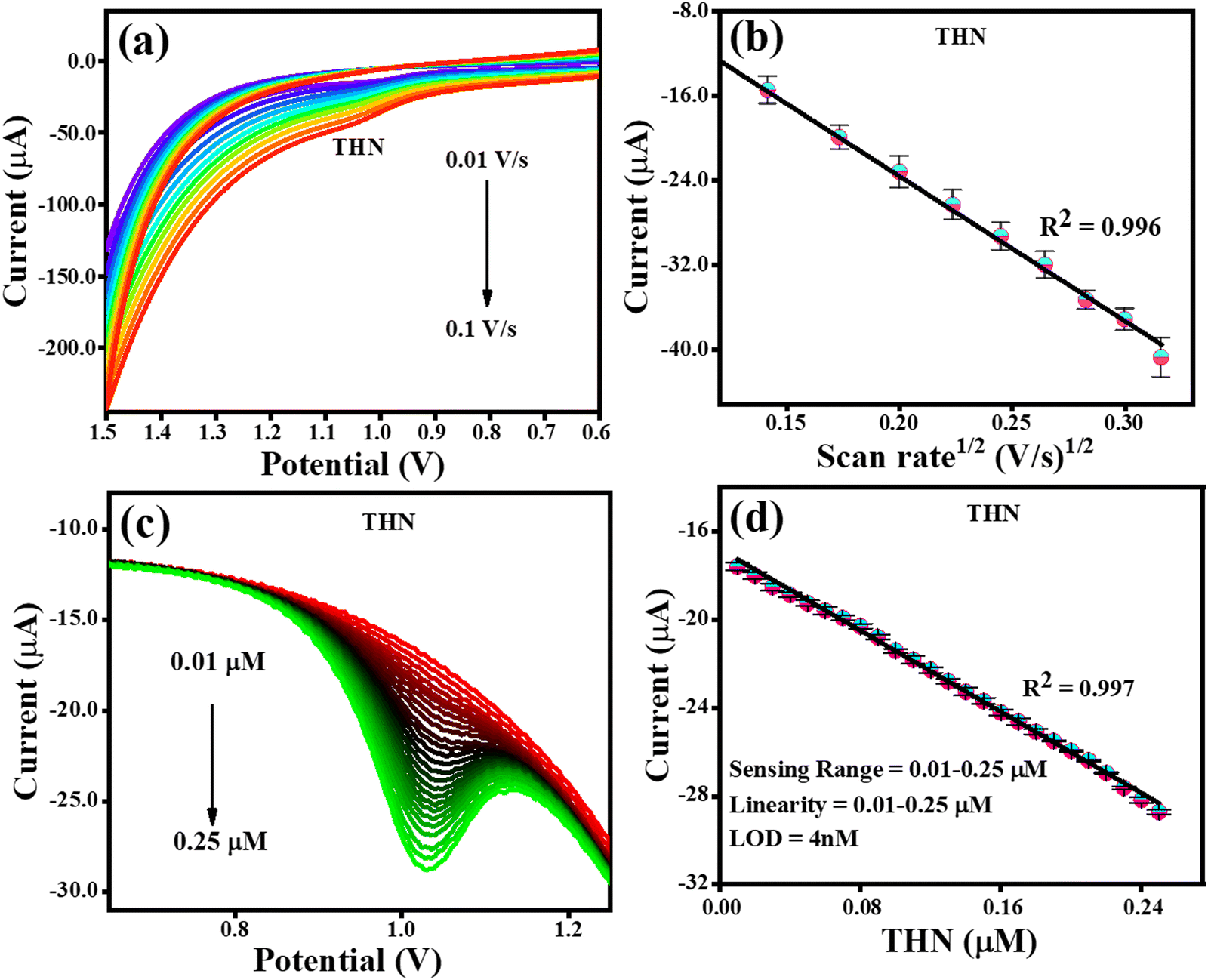
The electrochemical detection of THN was carried out similar to the electrochemical detection of CFN employing the CV and DPV techniques in PBS buffer at pH 7. The effect of scan rate on the oxidation of THN was examined using CV analysis. Fig. 8(a) depicts the CV analysis at a variable scan rate (0.01 V s−1 to 0.1 V s−1) and a constant THN concentration. The link between the change in scan rate and the oxidation potential of THN was demonstrated to have steadily increase with the scan rate. Fig. 8(b) shows the relationship between the square root of the scan rate and the THN oxidation current. The finding that the oxidation of THN was linearly dependent on the square root of scan rate, satisfying the regression equation Ipa = −0.0013 (scan rate1/2) + 3.7 × 10−6 with the regression coefficient R2 = 0.996, provided additional evidence for the controlled diffusion of THN at the TS(I)@GCE electrode surface. The DPV curve for the analysis of THN at the TS(I)@GCE electrode at different concentrations was obtained after refining the parameters for determining THN. In a wide range of THN concentrations, it was found that the oxidation peak currents are proportional to the concentrations (Fig. 8(c)). Ipa(μA) = −45.99 × 10−6 (THN, μA) − 16.81 × 10−6 was used as the linear regression equation, and the regression coefficient was 0.997. THN was deposited at various potentials in the range of 0.9 to 1.1 V and studied.
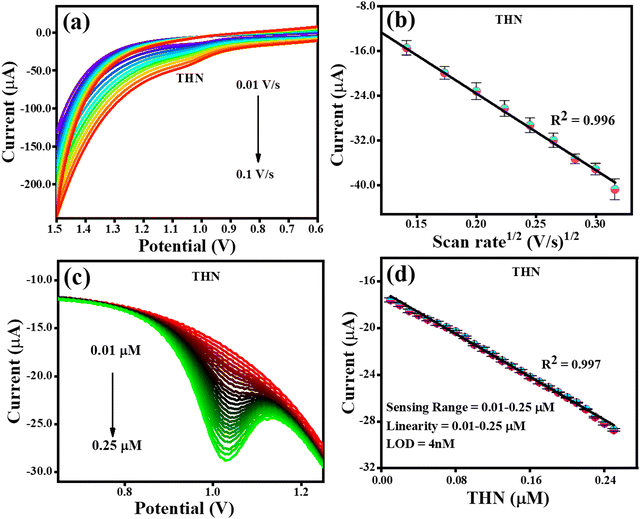 |
| | Fig. 8 CV and DPV responses for THN oxidation. (a) Effect of CV scan rate on the oxidation of THN. (b) Dependence of THN oxidation current on scan rate1/2. (c) DPV response for THN oxidation reaction with an increase in concentration. (d) Dependence of oxidation current on THN concentration. | |
As a result, it was determined that the projected limit of detection (LOD) for THN from the calibration plot is 4.0 nM, with a wide linearity range of 0.01–0.25 μM. The methodology utilised in this study was compared to that in related works in the literature, as indicated in Table 1, and the results were found to be acceptable when compared to recent studies (Fig. 8(d)).
Table 1 Analysis of the analytical performance of the TS(I)@GCE electrode in comparison to that of other electrodes for the detection of CFN, THN, GCL, and DPN
| Catalyst |
Analyte |
Linear range (μM) |
LOD (nM) |
Ref. |
| MIP/CNPs-SO3H/GCE: molecularly imprinted polymer/carbon nanoparticles-SO3H modified GCE, P(L-Asp)f-MWCNTs: poly(L-aspartic acid)/functionalized multiwalled carbon nanotubes, βH-MnO2-NF: βH-manganese oxide-nanoflower, GNP-CHIT-IL-/rGO/GCE: gold nanoparticle-chitosan-ionic liquid/graphene/glassy carbon electrode, RGO/GCE: reduced graphene oxide-modified GCE, GNF: graphene nanoflakes, p(L-Arg)ERGO: poly(L-arginine) reduced graphene oxide, PANI: polyaniline, GO/P(ANI-co-THI): poly(aniline-co-thionine) modified graphene oxide. |
| MIP/CNPs-SO3H/GCE |
THN |
0.005–30 |
1.4 |
48
|
| P(L-Asp)/f-MWCNTs/GCE |
CFN THN |
1–150 |
280 |
17
|
| 0.1–50 |
20 |
| βH-MnO2-NF/GCE |
CFN THN |
0.01–320 |
10.1 |
49
|
| 0.1–320 |
5.9 |
| Anisotropic GNP-CHIT-IL-/rGO/GCE |
CFN THN |
0.025–2.49 |
4.42 |
50
|
| 0.025–2.5 |
1.32 |
| RGO/GCE |
GCL |
0.5–500 |
200 |
51
|
| GNF modified GCE |
GCL |
0.03–46 |
9.8 |
20
|
| Pt-γ-Al2O3/GCE |
GCL |
0.05–30 |
17.9 |
4
|
| PANI/Pt–Au–FeOOH |
DPN |
0.001–10 |
1.0 |
52
|
| GO/P(ANI-co-THI) |
DPN |
2–500 |
2000 |
53
|
| rGO/Bi2S3-1/GCE |
DPN |
0.01–40 |
12.3 |
54
|
|
TS(I)@GCE
|
CFN
|
0.10–0.27 |
6.71
|
This work
|
|
TS(I)@GCE
|
THN
|
0.01–0.25 |
4.0
|
This work
|
|
TS(I)@GCE
|
GCL
|
0.10–0.20 |
11.2
|
This work
|
|
TS(I)@GCE
|
DPN
|
0.10–0.27 |
5.19
|
This work
|
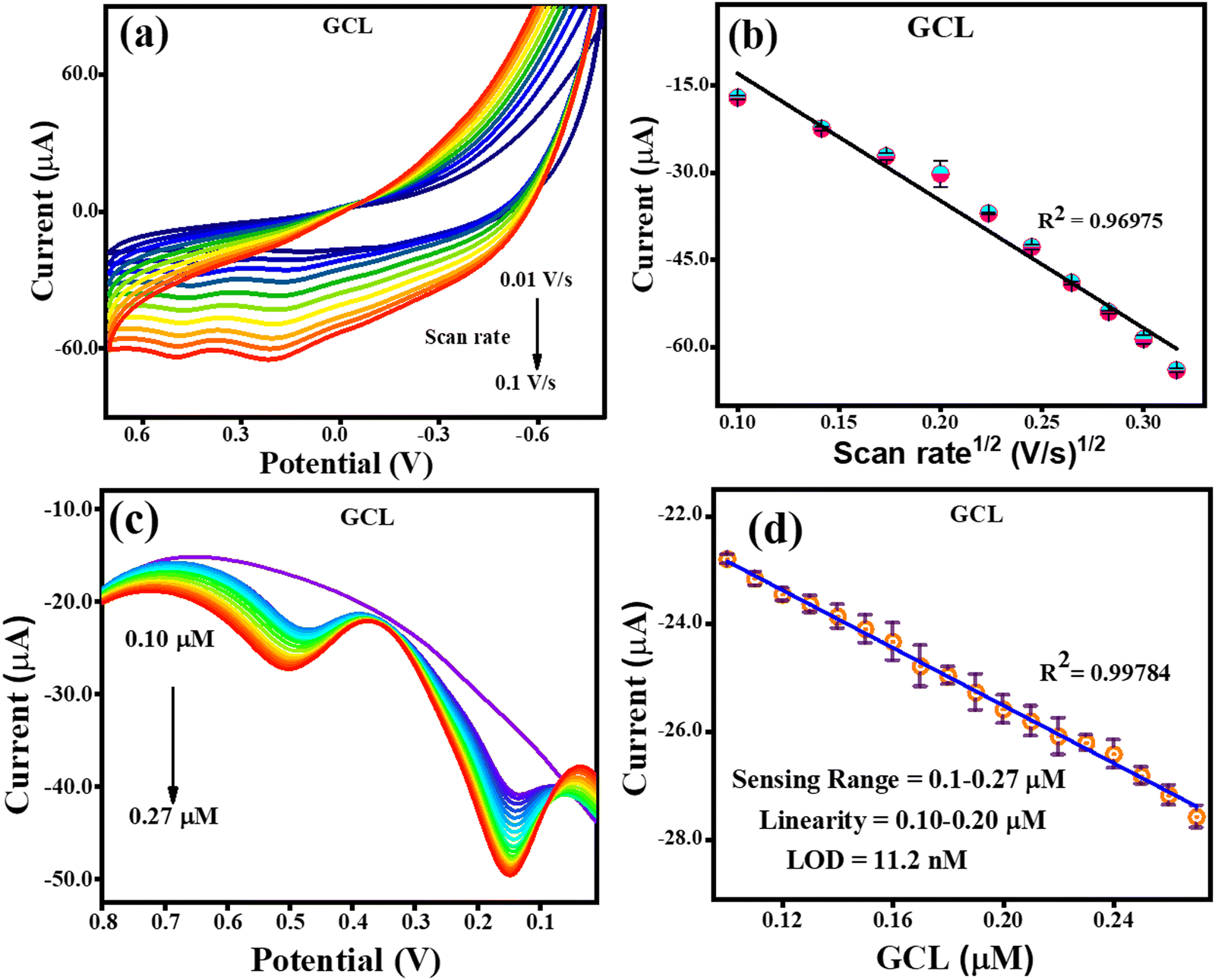
Given that the electrochemical process increases the sensitivity, lowers the limit of detection, and increases the amount of THN absorbed on the electron surface, the impact of its accumulation was investigated. The methods used to detect GCL were the same as that used to identify CFN and THN, changing the scan rate (10–100 mV s−1), while maintaining a constant concentration of GCL. According to Fig. 9(a), the oxidation potential of GCL (Epa) was similarly discovered to be pushed towards the higher potential end for the scan rate. Fig. 9(b) shows how the square root of the scan rate affects the GCL oxidation current. The oxidation of GCL was discovered to be linearly dependent on scan rate1/2, satisfying the regression equation Ipa = −0.0021 (scan rate1/2) + 8.9 × 10−6 with the regression coefficient R2 = 0.9697, further confirming the controlled diffusion of GCL at the TS(I)@GCE electrode surface. However, during CV, only oxidation peaks were observed, and therefore the electrochemical reaction was found to be irreversible. Fig. 9(c) shows the oxidation peak current for GCL (Ipa, μA) by using DPV. It increased remarkably in a linear manner with an increase in concentration from 0.1 to 0.27 μM as the sensing range.
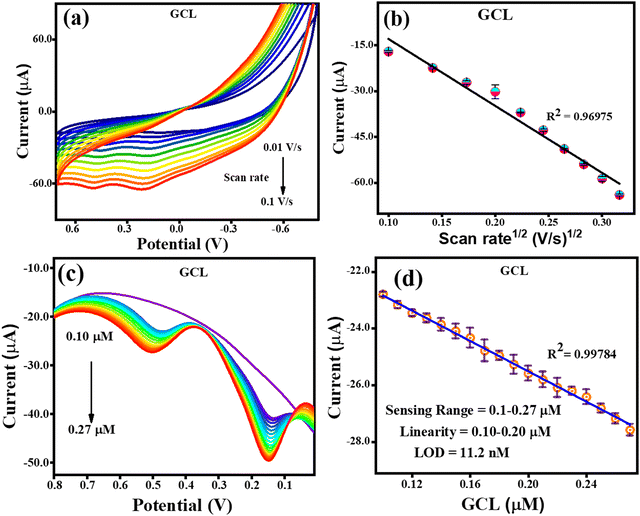 |
| | Fig. 9 CV and DPV response for GCL oxidation. (a) Effect of CV scan rate on the oxidation of GCL. (b) Related GCL oxidation current dependence on scan rate1/2. (c) GCL oxidation DPV response with an increase in concentration. (d) Dependence of oxidation current on GCL concentration. | |
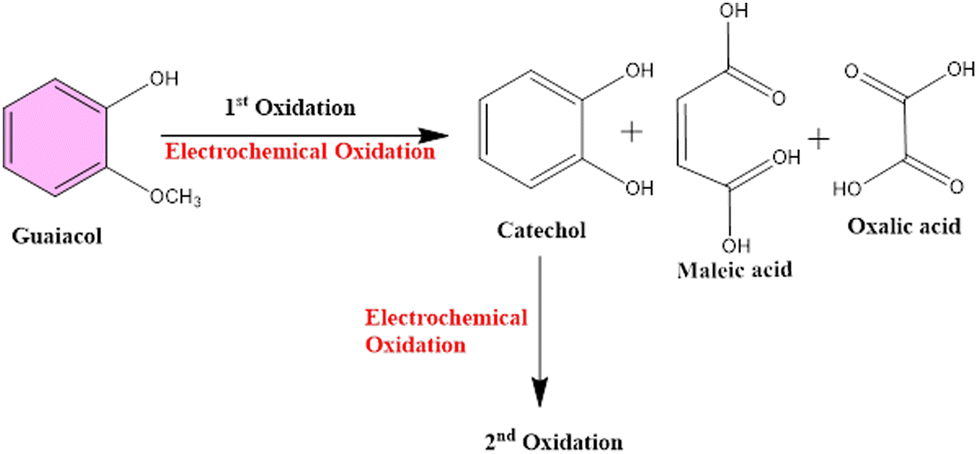
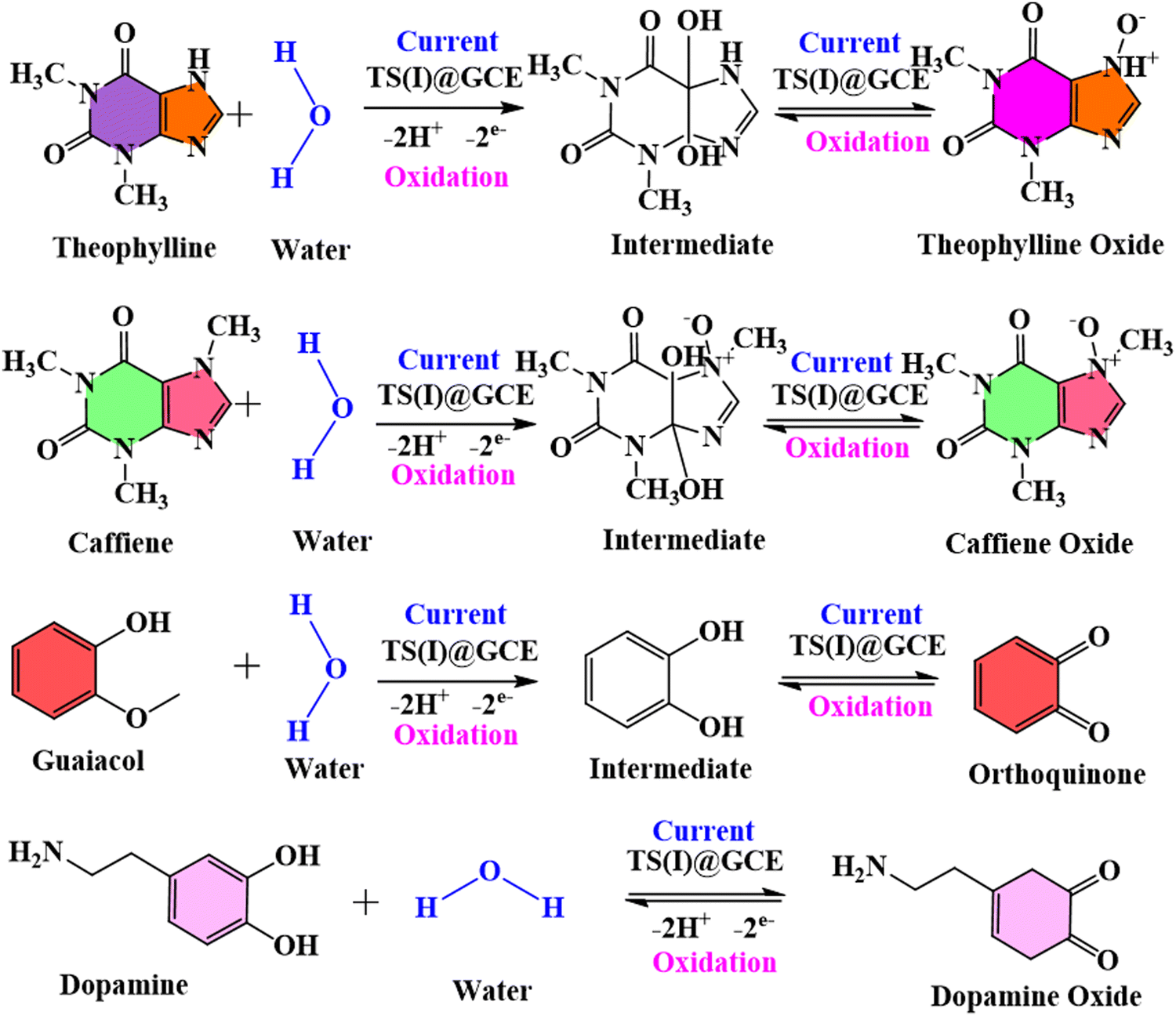
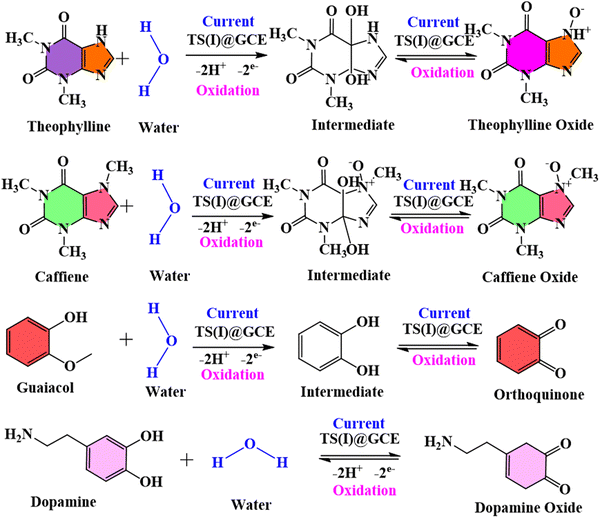
There was no change recorded in the GCL oxidation potential even after increasing the concentration, suggesting the stability of the TS(I)@GCE electrode. The dependence of the GCL oxidation current on its concentration is shown in Fig. 9(d), which was also used for the LOD calculations. The linear regression equation was expressed as Ipa (μA) = −26.72 × 10−6 (GCL, μA) − 20.16 × 10−6 with the regression coefficient R2 = 0.9978. At various potentials in the range of 0.0 to 0.8 V, which correspond to CFN and THN, GCL deposition was observed. As a result, based on the calibration plot at ∼0.5 V the limit of detection (LOD) for GCL was determined to be 11.2 nM, with a large linearity range of 0.1–0.2 μM. According to Milczarek, the irreversible oxidation is associated with the second oxidation peak at a lower potential (0.19 V).42 Therefore, one explanation for this peak can be that oxygenates such as guaiacol and phenols cause the pyrolysis of lignin to produce coke. Guaiacol is produced via the pyrolysis of lignin in low-boiling-point oxygen with a polar-protic solvent. The produced guaiacol can be hydrolysed into catechol, and subsequently to phenol as the reaction temperature and duration increase.43–45Scheme 2 shows the mechanism for the two oxidation peaks of guaiacol. The peak in the range of 0.4–0.5 V was used for the further detection and quantification of GCL. The second oxidation peak coincided with the oxidation peak of the catechol group. In a previous study, the hydrothermal oxidation of GCL in sub-and super-critical water was catechol, phenol and o-cresol.46 It was unselectively oxidized to low-molecular-weight oxalic acid and carboxylic acids such as acetic acid and formic acid, which can be considered the first oxidation potential. In the second oxidation, the catechol group gets oxidized, which is found in dopamine as well. Hence, in this work, the oxidation potential peaks at 0.5 V and 0.19 ± 0.01 V are related to the oxidation of guaiacol. It is obvious that DPN is a neurotransmitter, which can be found in beverages; however, it is also detected in real samples. Focusing on the molecular structure of DPN, it consists of a catechol group bearing ethylamine or derivative side chain. Indeed, the DPV response of GCL (as indicated in Fig. 9(c)) shows two oxidation peaks. Between them, the oxidation peak at 0.1 V is attributed to the oxidation of catechol, but the DPN molecular structure consists of catechol.47 Thus, to compare the catechol groups in GCL and DPN, the detection of DPN was carried out in the same manner as the other analytes to confirm its influence. To detect standard DPN, the CV and DPV electrochemical methods were also employed. The peculiar DPN oxidation at about 0.18 V can be used for its detection, which is equivalent to the catechol oxidation of guaiacol at 0.19 V. In the CV analysis, 20 μM of DPN, scan rate in the range of 10–100 mV s−1 and pH 7.0 PBS buffer electrolyte were utilized for sensing and achieving the maximum oxidation peak.
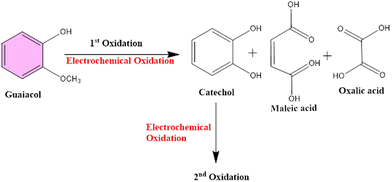 |
| | Scheme 2 Represents the mechanism of oxidation of two oxidation peaks of guaiacol. | |
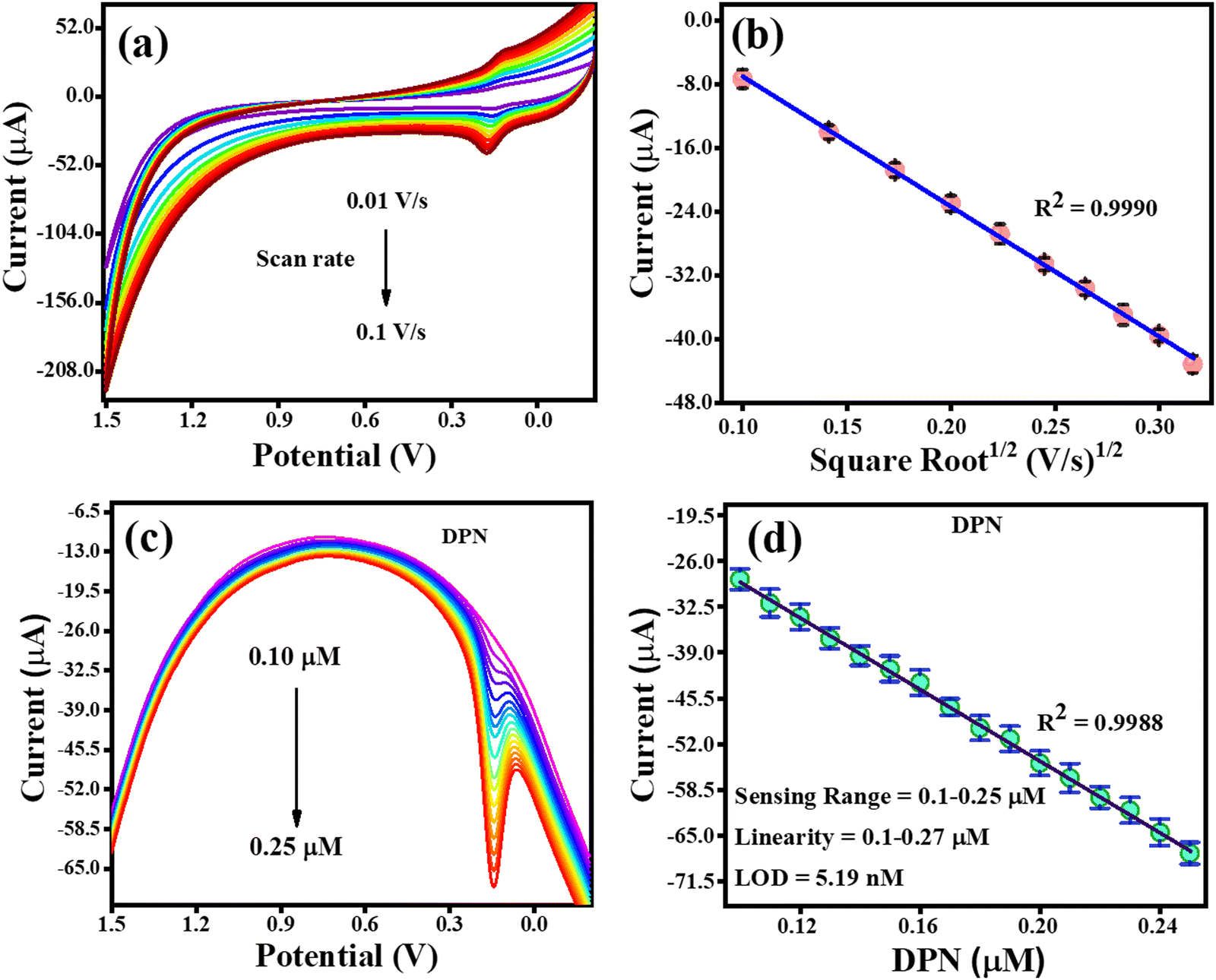
The CV analysis with an increasing scan rate (0.01–0.1 V s−1) and a constant DPN concentration is shown in Fig. 10(a). The variation in scan rate was found to have caused the oxidation potential of DPN to steadily shift towards the higher potential end. The association between the DPN oxidation current and the square root of the scan rate is depicted in Fig. 10(b). The regulated diffusion of DPN at the TS(I)@GCE electrode surface was further confirmed by the discovery that the oxidation of DPN is linearly dependent on the square root of the scan rate, satisfying the regression equation Ipa = −0.0016 (scan rate1/2) + 9.3 × 10−6 with the regression coefficient R2 = 0.99. The analysis of DPN at the TS(I)@GCE electrode at various concentrations was performed when the necessary conditions were attained. It was discovered that the oxidation peak current is proportional to the concentration of DPN (Fig. 10(c)). Fig. 10(d), which was utilised for the LOD calculations, depicts the relation between the DPN oxidation current and concentration. Ipa(μA) = −256.46 × 10−6 (DPN, μA) − 3.54 × 10−6 is the linear regression equation, and the regression coefficient R2 = 0.9988. With a vast linearity range of 0.1 to 0.25 μM, the established limit of detection (LOD) for DNP was 5.19 nM based on the calibration plot. The results from the literature included in Table 1 were compared to the approach proposed in this work for the detection of DPN, and the results obtained were determined to be reliable compared to previous studies.
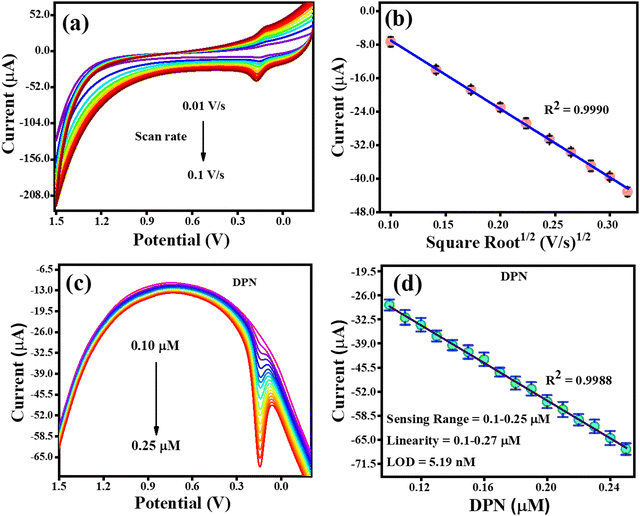 |
| | Fig. 10 CV and DPV response for DPN oxidation. (a) Effect of CV scan rate on the oxidation of DPN. (b) Dependence of DPN oxidation current on scan rate1/2. (c) DPV response with an increase in the concentration of DPN. (d) Relay oxidation current dependence on DPN concentration. | |
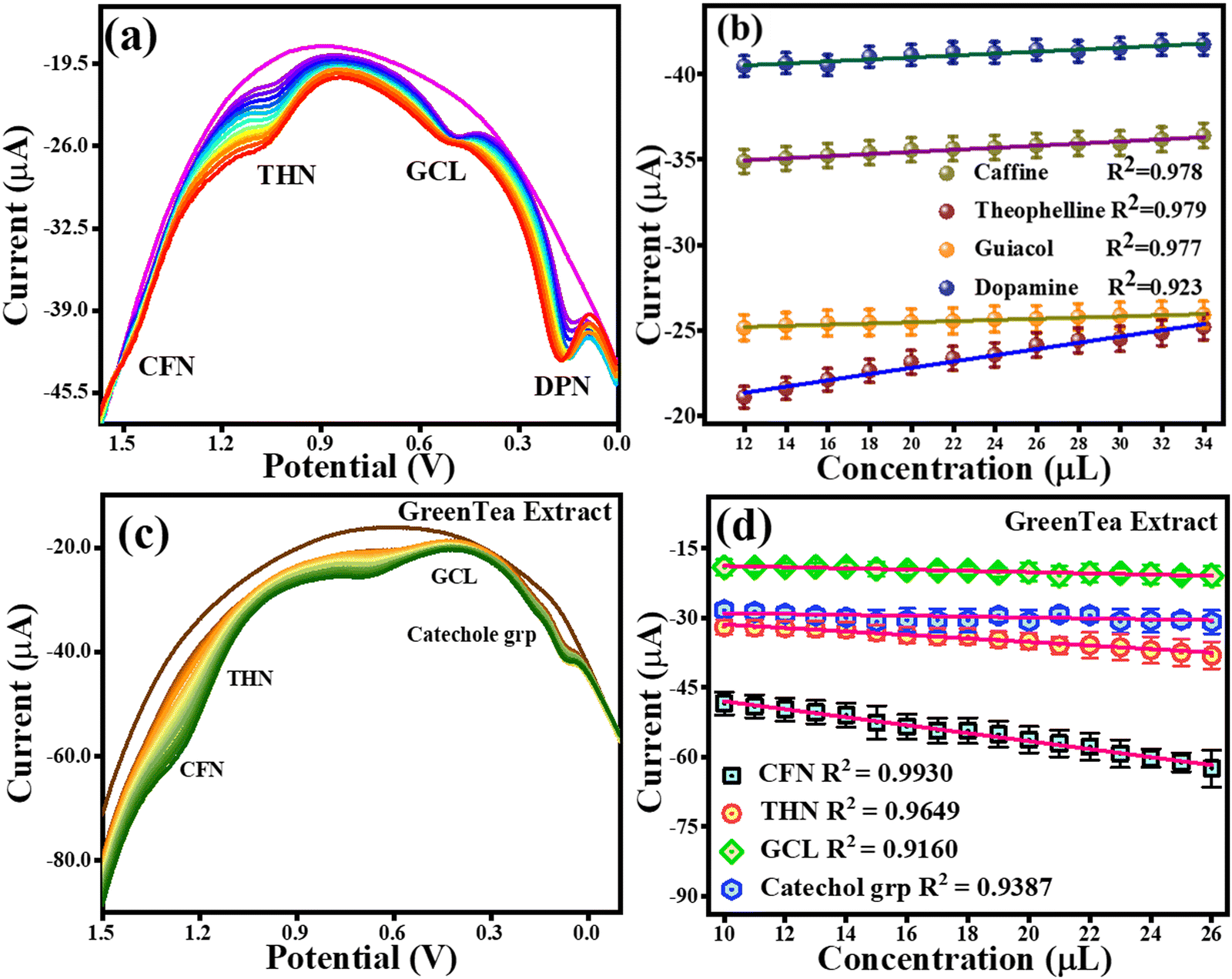
4.5 Simultaneous detection of CFN, THN, GCL and DPN as a proof-of-concept experiment for their selectivity
A CV analysis was performed with a fixed analyte concentration for the simultaneous detection and a proof-of-concept experiment for the selectivity of CFN, THN, GCL, and DPN. The CV curves with the varying scan rates are shown in Fig. S2a (ESI†). The anodic peak potentials based on CV analysis for 20 μM concentration of each CFN, THN, GCL, and DPN were found at 1.3 V, 1.0 V, 0.5 V and 0.18 ± 0.01 V, respectively. It was found that the oxidation potentials of each CFN, THN, GCL, and DPN shifted towards the higher potential end using different scan rates (0.01 V s−1 to 0.1 V s−1). Further, a DPV analysis was also performed to investigate their hindrance to each other with an increase in their concentration. The DPV responses for CFN, THN, GCL, and DPN an increase in their concentrations are presented in Fig. 11(a), showing that there was no change in the oxidation potential peaks of CFN, THN, GCL, and DPN. Fig. 11(b) shows the calibration graph in the related figure with an increase in concentration in the range of 12–34 μL. As a result, CFN, THN, GCL, and DPN can all be determined simultaneously and selectively using the TS(I)@GCE electrode.
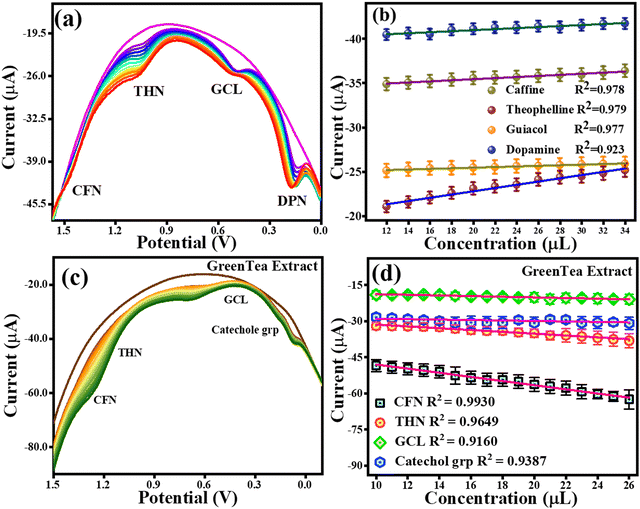 |
| | Fig. 11 DPV and CV responses for the oxidation of CFN, THN, GCL and DPN. (a) Effect of DPV scan rate on CFN, THN, GCL and DPN oxidation. (b) Related DPV calibration graph for CFN, THN, GCL and DPN oxidation. (c) DPV response for CFN, THN, GCL and catechol group oxidation with increasing concentration of green tea extract. (d) Dependence of oxidation currents on CFN, THN, GCL and the catechol group. | |
4.6 Amperometric detection of CFN, THN and GCL and selectivity
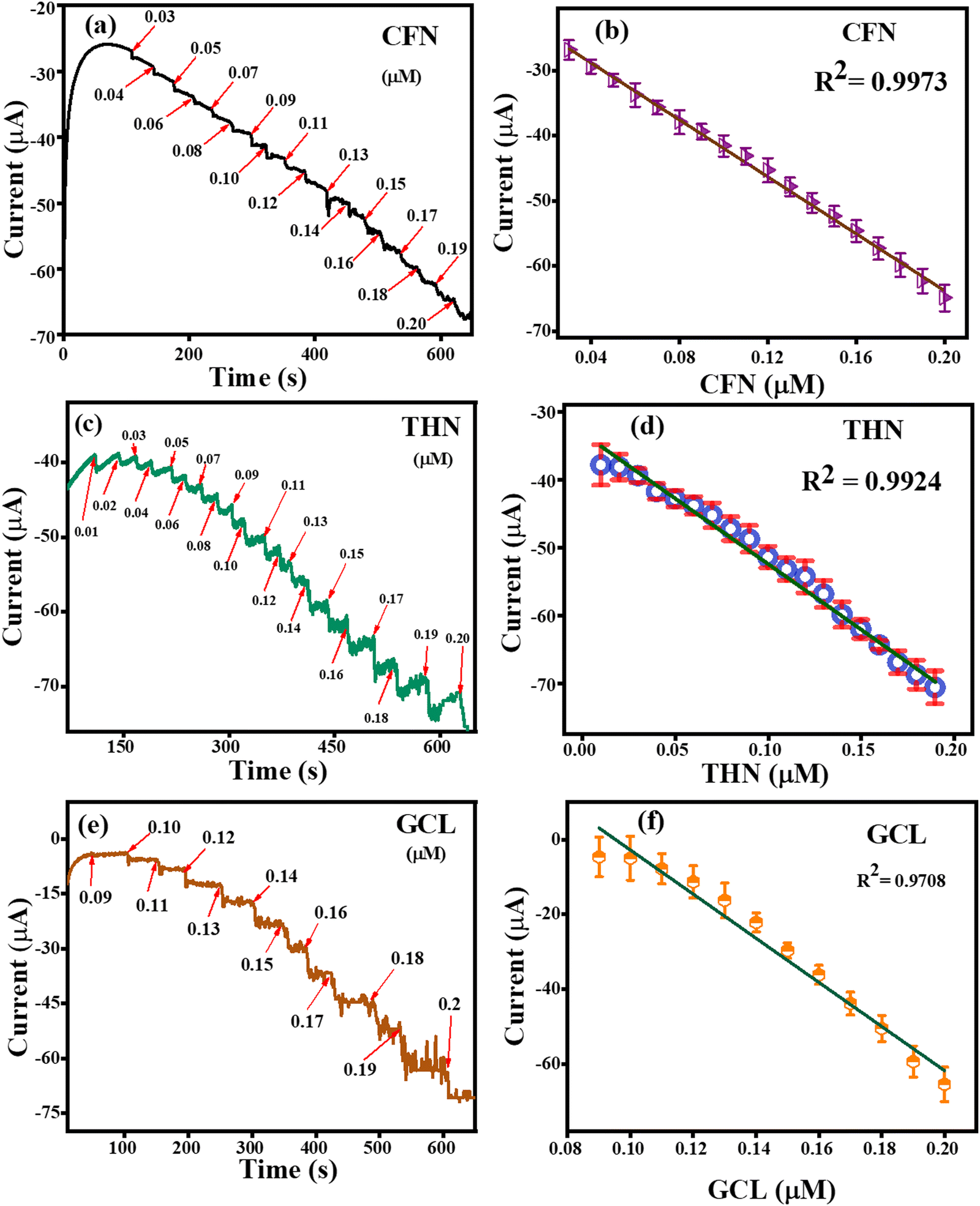
The amperometric detection of CFN, THN and GCL was carried out under all the pre-optimized conditions using the amperometric (i–t) technique. Fig. 12(a) represents the amperometric response of successively added caffeine at a fixed potential of 1.34 V. For the detection of CFN, the amperometric technique was performed for 600 s, and CFN was added after getting the saturated current. The response for CNF oxidation took less than 3 s, confirming the fast electron transportation between the electrolyte and electrode surface, which can be attributed to the adsorption of CFN molecules on the surface of the TS(I)-modified electrode. Fig. 12(b) represents the dependence of oxidation current on the CFN concentration added. The linear response of the current satisfied the regression equation: IP (μA) = − 21.8156 (CFN, μM) − 2.829 with regression coefficient, R2 = 0.9973. Similarly, the amperometric detection of THN was obtained in the same way as for CFN at a different applied potential (1.1 V). Fig. 12(c) represents the amperometric current response for the successive addition of THN between 0 and 600 s. Fig. 12(d) indicates the dependence of oxidation current on THN concentration with the regression equation: IP (μA) = − 19.2806 (THN, μM) − 3.95134 with the regression coefficient, R2 = 0.9924. Similarly, the amperometric detection of GCL was obtained in the same way as for CFN but at a different applied potential (0.45 V). Fig. 12(e) represents the amperometric current response for the successive addition of GCL between 0 and 600 s. Fig. 12(f) indicates the dependence of oxidation current on GCL concentration with the regression equation: IP (μA) = − 11.79177 (GCL, μM) − 0.64623 with the regression coefficient, R2 = 0.9708. Thus, the amperometric detection of CFN, THN and GCL confirmed the efficiency of the TS(I)-modified GCE electrode because of its high sensitivity and fast response to the added analytes.
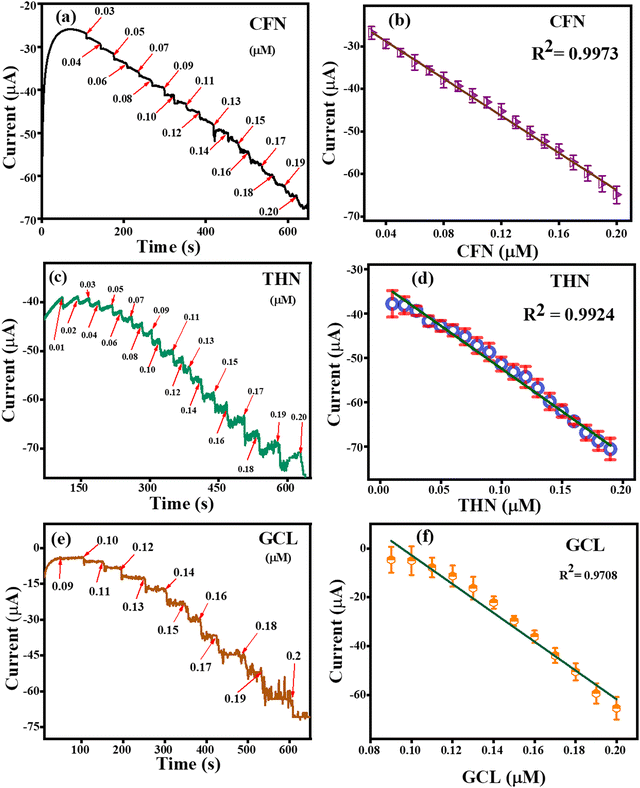 |
| | Fig. 12 (a) Amperometric detection of CFN. (b) Related linear range calibration graph for CFN. (c) Amperometric detection of THF. (d) Related linear range calibration graph for THF. (e) Amperometric detection of GCL. (f) Related linear range calibration graph for GCL. | |
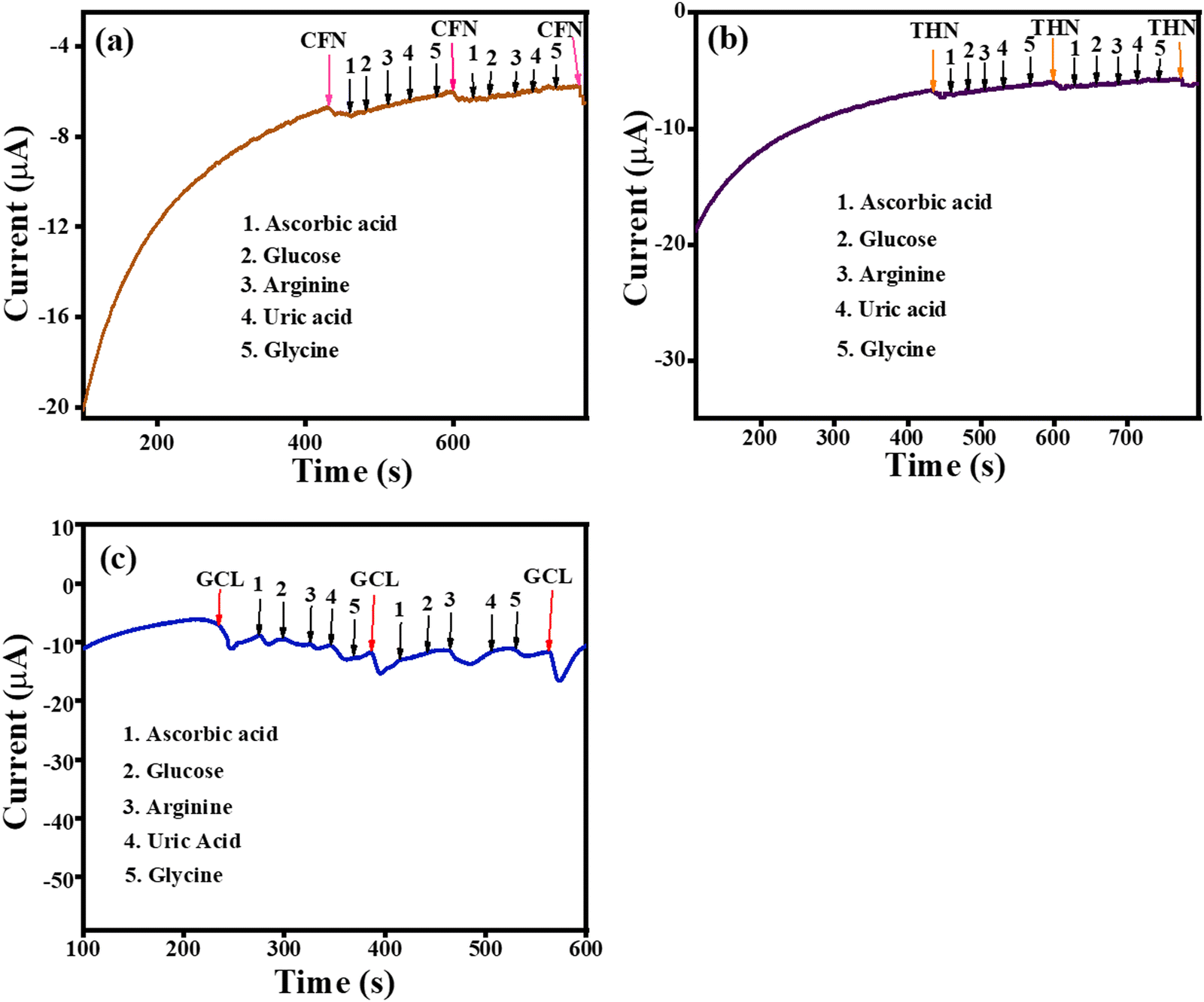
The amperometric selectivity for CFN, THN and GCL was investigated by the amperometric (i– t) technique against ascorbic acid, glucose, arginine, and uric acid interferences alternately added to the reaction system. As shown in Fig. 13(a), the amperometric response was observed only for CFN at the applied potential of 1.34 V and no disturbance in the amperometric current was observed for any of the interferences. Similarly, the oxidation current was only observed for THN at an applied potential of 1.1 V (Fig. 13(b)) and the oxidation current was only observed for GCL at an applied potential of 0.45 V (Fig. 13(c)). There was no disturbance recorded for any of the interferences. Given that all the interferences have different oxidation potentials to that of CFN, THN and GCL, the amperometric response was only for CFN, THN and GCL even after the addition of the different interferences. The selectivity analysis confirmed that the sensor based on the TS(I)-modified electrode is highly selective for CFN THN and GCL and could be applied for detection in the presence of various analytes.
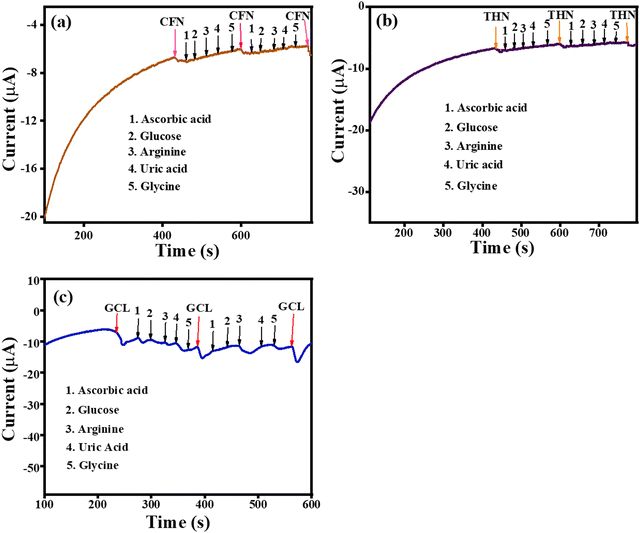 |
| | Fig. 13 (a) Amperometric selectivity graph for CFN. (b) Amperometric selectivity graph for THN. (c) Amperometric selectivity graph for GCL. | |
4.7 Detection of CFN, THN, GCL and catechol in real sample extracts and their quantification
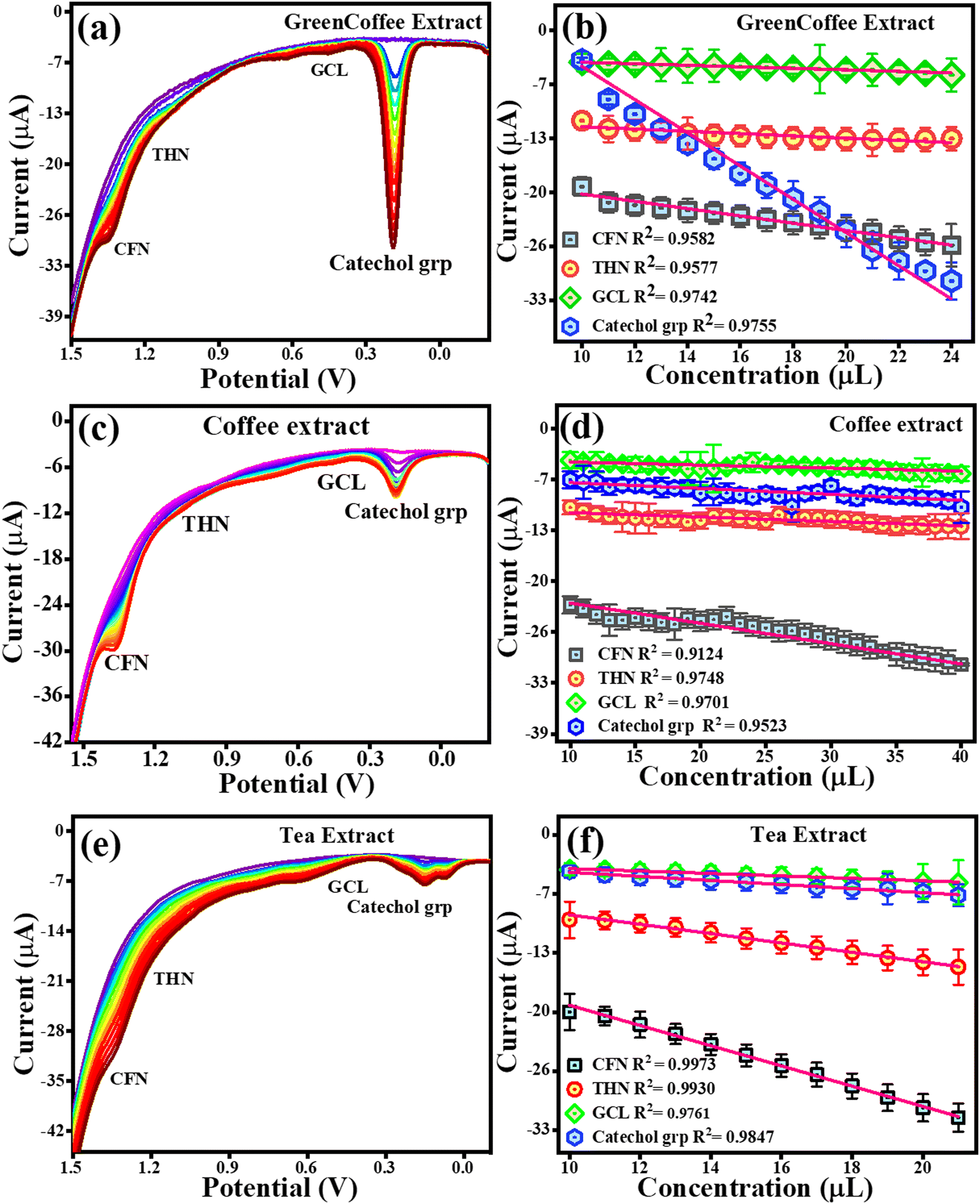
To estimate CFN, THN, GCL and catechol group in real samples, the TS(I)@GCE electrode was used under all the pre-optimized conditions. 1.5 g of all the real samples (green tea, green coffee, coffee, and tea) was added to Milli-Q water (250 mL) to obtain their extract. The results are shown in Fig. 11(c) and (d) for green tea extract and Fig. 14 depicts for other extracts. DPV analysis was done with increasing concentrations of each real sample ranging from 10–26 μL for their quantification. Fig. 14(c) depicts the DPV response upon the addition of green tea extract. The peaks for the oxidation potentials of CFN, THN, GCL and the catechol group in the DPV spectra of green tea extract were observed, further suggesting the presence of these analytes. The dependence of CFN, THN, GCL and the catechol group oxidation currents on the green tea extract concentration is shown in Fig. 14(d). We observed a linear dependence of the oxidation current on the green tea extract concentration. The regression equations can be used to express the linear dependence of the oxidation currents for CFN, THN, GCL, and the catechol group: Ipa (μA) = −0.85 × 10−6 (green tea extract, μL) − 39.42 × 10−6, Ipa (μA) = −0.37 × 10−6 (green tea extract, μL) − 27.64 × 10−6, Ipa (μA) = −0.13 × 10−6 (green tea extract, μL) − 17.47 × 10−6 and Ipa (μA) = −0.095 × 10−6 (green tea extract, μL) − 28.02 × 10−6 with the regression coefficient of R2 = 0.9930, R2 = 0.9649, R2 = 0.9160, and R2 = 0.9387, respectively. The DPV behaviour upon the addition of green coffee extract is shown in Fig. 14(a). The peaks for the oxidation potentials of CFN, THN, GCL and the catechol group in the DPV spectra of green coffee extract were observed, suggesting the presence of these analytes. The dependence of the oxidation currents of CFN, THN, GCL and the catechol group on the green coffee extract concentration is shown in Fig. 14(b). We found a linear dependence of the oxidation currents on the green coffee extract concentration. The regression equations can be used to express the linear dependence of the oxidation current of the catechol group for CFN, THN, GCL, and other variables: Ipa (μA) = −0.44 × 10−6 (green coffee extract, μL) − 15.34 × 10−6, Ipa (μA) = −0.13 × 10−6 (green coffee extract, μL) – 10.27 × 10−6, Ipa (μA) = −0.091 × 10−6 (green coffee extract, μL) − 2.96 × 10−6 and Ipa (μA) = −2.0 × 10−6 (green coffee extract, μL) − 15.67 × 10−6 with the regression coefficient of R2 = 0.9582, R2 = 0.9577, R2 = 0.9742, and R2 = 0.9755, respectively. The DPV behaviour upon the addition of coffee extract is shown in Fig. 14(c). The peaks for the oxidation potentials of CFN, THN, GCL and the catechol group in the DPV spectra of coffee extract were observed, suggesting the presence of these analytes. The dependence of the CFN, THN, GCL and the catechol group oxidation currents on the coffee extract concentration is shown in Fig. 14(d). Again, we observed the linear dependence of the oxidation current on the coffee extract concentration. The linear dependence of the oxidation currents for CFN, THN, GCL and the catechol group can be expressed by the regression equations: Ipa (μA) = −0.26 × 10−6 (coffee extract, μL) − 19.66 × 10−6, Ipa (μA) = −0.056 × 10−6 (coffee extract, μL) − 10.14 × 10−6, Ipa (μA) = −0.038 × 10−6 (coffee extract, μL) − 3.86 × 10−6 and Ipa (μA) = −0.075 × 10−6 (coffee extract, μL) − 6.10 × 10−6 with the regression coefficient of R2 = 0.124, R2 = 0.9748, R2 = 0.9701, and R2 = 0.9523, respectively.
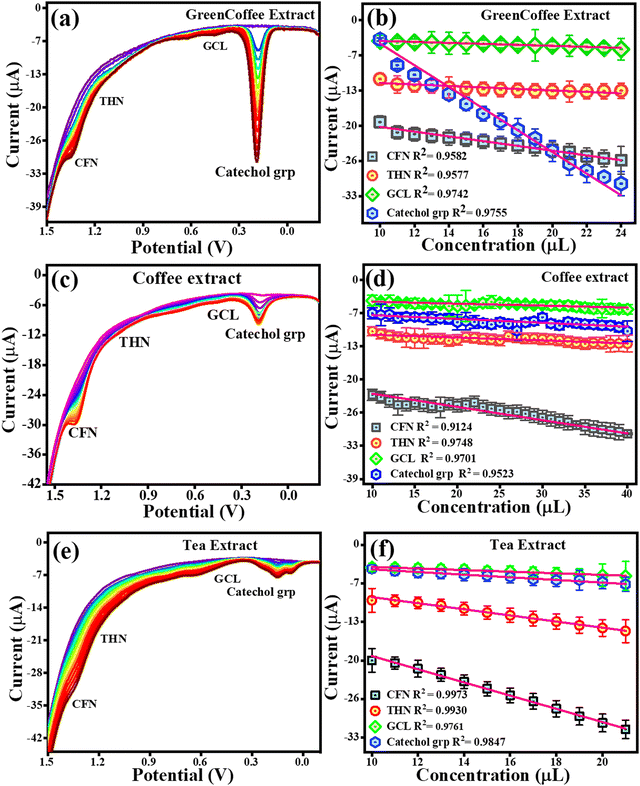 |
| | Fig. 14 DPV response for the oxidation of CFN, THN, GCL and the catechol group. (a) DPV response for CFN, THN, GCL and catechol group oxidation in green coffee extract. (b) Dependence of oxidation current on the concentration of the green coffee extract. (c) DPV response for CFN, THN, GCL and catechol group oxidation in the coffee extract. (d) Dependence of oxidation current on concentration of the coffee extract. (e) DPV response for CFN, THN, GCL and catechol group oxidation in the tea extract. (f) Dependence of oxidation current on concentration of the tea extract. | |
Fig. 14(e) displays the DPV response upon the addition of tea extract. The peak of oxidation potentials of CFN, THN, GCL and the catechol group in the DPV spectra of tea extract were observed, further suggesting the presence of these analytes. The dependence of the oxidation currents of CFN, THN, GCL and the catechol group on the tea extract concentration is shown in Fig. 14(f). Again, we found the linear dependence of the oxidation current on the tea extract concentration. The regression equations can be used to express the linear dependence of the oxidation currents for CFN, THN, GCL, and the catechol group: Ipa (μA) = −1.11 × 10−6 (tea extract, μL) − 7.61 × 10−6, Ipa (μA) = −0.51 × 10−6 (tea extract, μL) − 3.64 × 10−6, Ipa (μA) = −0.13 × 10−6 (tea extract, μL) − 2.37 × 10−6 and Ipa (μA) = −0.22 × 10−6 (tea extract, μL) − 1.88 × 10−6 with the regression coefficient of R2 = 0.9973, R2 = 0.9930, R2 = 0.9761, and R2 = 0.9847, respectively. For the quantification analysis, the DPV results for the standard and real samples were compared and we found that 0.106 μM of CFN, 0.0534 μM of THN, 0.158 μM of GGL and 0.158 μM of catechol group were present in 20 μL of green coffee extract. Similarly, 0.161 μM CFN, 0.0717 μM THN, 0.0711 μM GCL and 0.0186 μM catechol group were found in 20 μL of green tea extract. In the case of coffee extract, 0.044 μM CFN, 0.0403 μM THN, 0.0769 μM GCL, and 0.0134 μM catechol group were detected. Similarly, 0.217 μM CFN, 0.112 μM THN, 0.0953 μM GCL, and 0.0174 μM catechol group were found in the tea extract. These results are also tabulated in Table 2. A graphical representation of the electrochemical detection of CFN, THN, GCL and DPN is shown in Scheme 3.
Table 2 Electrochemical quantification of CFN, THN and GCL in real samples
| Real sample |
CFN detected (μM) |
THN detected (μM) |
GCL detected (μM) |
Catechol detected (μM) |
| Green tea |
0.161 |
0.0717 |
0.0711 |
0.0186 |
| Green coffee |
0.106 |
0.534 |
0.0626 |
0.158 |
| Coffee |
0.0440 |
0.0403 |
0.0769 |
0.0134 |
| Tea |
0.217 |
0.112 |
0.0953 |
0.0174 |
 |
| | Scheme 3 Oxidation mechanism of THN, CFN, GCL and DPN on the TS(I)@GCE electrode. | |
4.8 Use of spiking method for the validation of detected CFN, THN, and GCL in real sample extracts
After the preparation of the green tea, green coffee, coffee and tea extracts, the DPV response of the TS(I)-modified GCE was observed with spiked CFN, THN and GCL. All the analyte spiking conditions were the same, with an increase in the concentration of the analytes of 0 μM, 0.16 μM, 0.17 μM, 0.18 μM, 0.19 μM, and 0.20 μM. Where, each 0 μM condition was the without spiked extracts of green tea, green coffee, coffee and tea. Therefore, DPV characteristic peaks were found at the potentials of 1.34 V, 1.1 V, and 1.45 V, respectively.
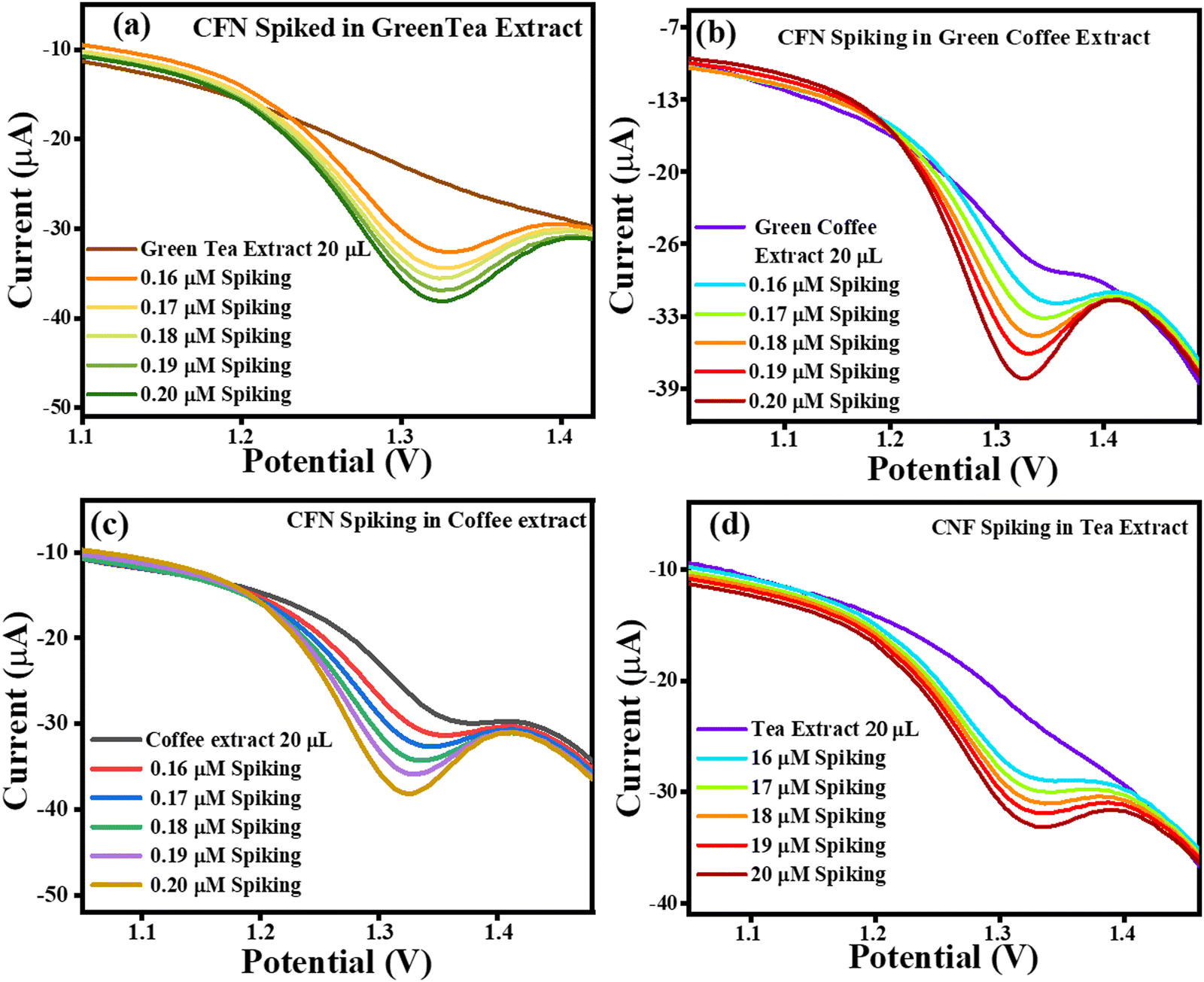
The current vs. potential graph for CFN spiking is shown in Fig. 15(a) for green tea extract, Fig. 15(b) for green coffee extract, Fig. 15(c) for coffee extract, and Fig. 15(d) for tea extract. Subsequently, the accuracy of the sensing system was also investigated through recovery analysis using 20 μL, without spiked green tea extract consisting of 0.161 μM, 0.106 μM, 0.0440 μM and 0.217 μM of CFN. This calculation was done based on the correlation between the standard CFN concentration vs. current and in extract vs. current. After, spiking was continued, and the correlated recoveries are shown in Table 3, where the percentage recoveries were found in the approximate range of 124–237%, signifying the accuracy of the TS(I)-modified electrode. Thus, the present sensing system is reliable for the effective selective detection of CFN in real samples of green tea, green coffee, coffee and tea.
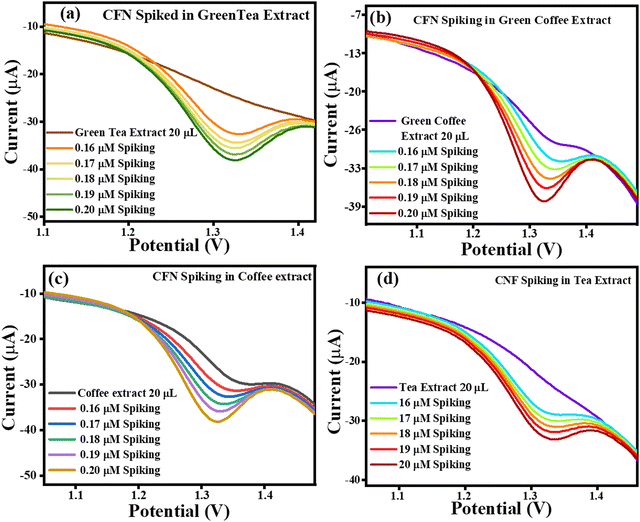 |
| | Fig. 15 Current vs. potential graph for (a) CFN spiked in the green tea extract, (b) CFN spiked in the green coffee extract, (c) CFN spiked in the coffee extract and (d) CFN spiked in the tea extract. | |
Table 3 CFN spiking results in the green tea extract, green coffee extract, coffee extract and tea extract
| Extract |
CFN spiking (μM) |
Recovery |
Recovery (%) |
RSD (%) (n = 3) |
| Green tea |
0 |
0.161 |
|
|
| 0.16 |
0.32742 |
204.6375 |
2.305906786 |
| 0.17 |
0.334641 |
196.8476471 |
1.999157306 |
| 0.18 |
0.344751 |
191.5283333 |
2.291508944 |
| 0.19 |
0.354861 |
186.7689474 |
2.000783405 |
| 0.20 |
0.364971 |
182.4855 |
2.000158917 |
| Green coffee |
0 |
|
|
|
| 0.16 |
0.26999 |
168.74375 |
2.000074077 |
| 0.17 |
0.280416 |
164.9505882 |
2.713825174 |
| 0.18 |
0.29172 |
162.0666667 |
2.03277115 |
| 0.19 |
0.301328 |
158.5936842 |
2.067514469 |
| 0.20 |
0.3213 |
160.65 |
2.063492063 |
| Coffee |
0 |
|
|
|
| 0.16 |
0.206244 |
128.9025 |
2.530982719 |
| 0.17 |
0.21721 |
127.7705882 |
2.002670227 |
| 0.18 |
0.22848 |
126.9333333 |
2.00017507 |
| 0.19 |
0.23634 |
124.3894737 |
2.001353982 |
| 0.20 |
0.24888 |
124.44 |
2.00096432 |
| Tea |
0 |
|
|
|
| 0.16 |
0.38077 |
237.98125 |
2.001208078 |
| 0.17 |
0.391257 |
230.1511765 |
2.00124215 |
| 0.18 |
0.400573 |
222.5405556 |
1.999635522 |
| 0.19 |
0.41107 |
216.3526316 |
1.999659425 |
| 0.20 |
0.42117 |
210.585 |
1.999192725 |
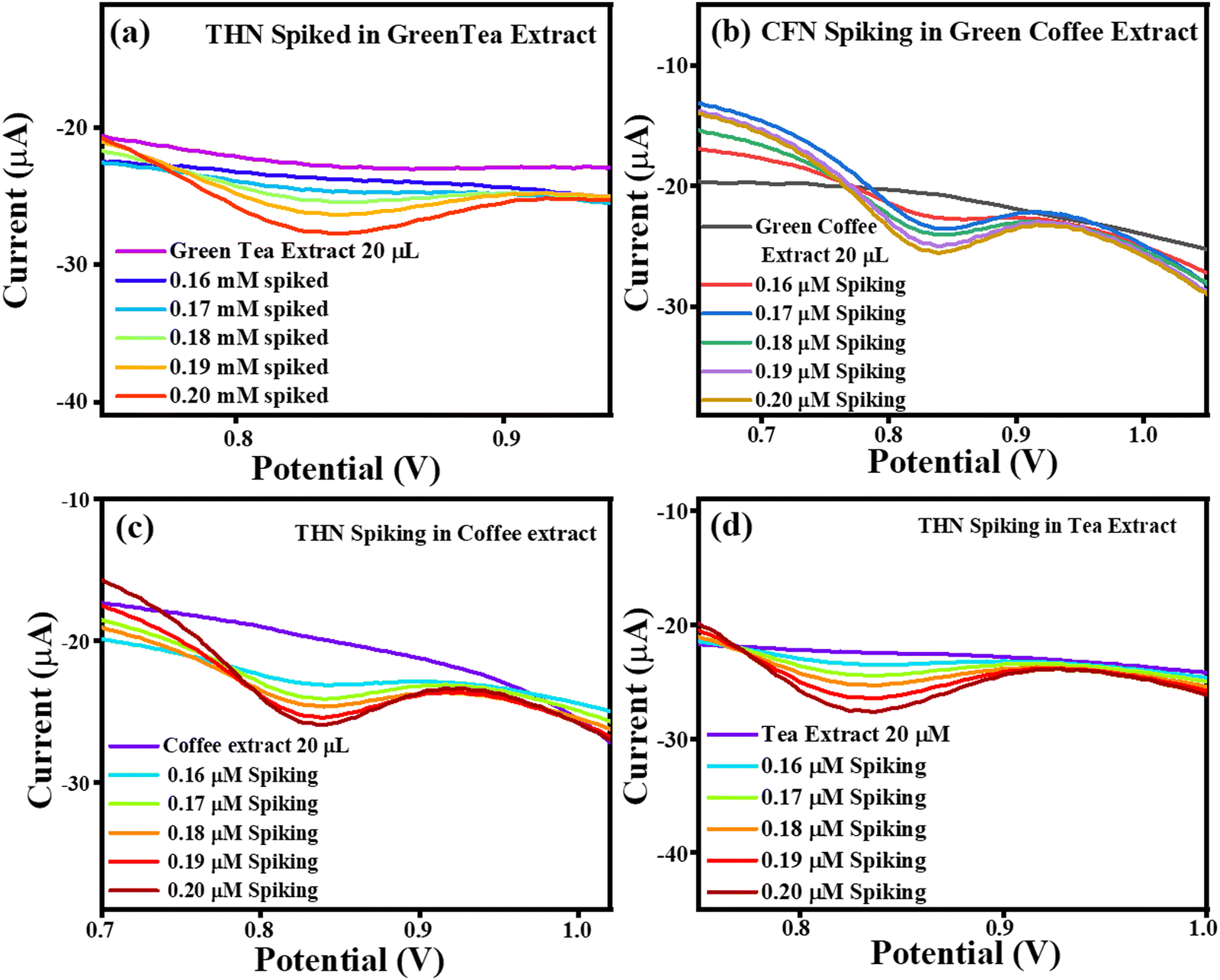
The current vs. potential graph for THN spiking is shown in Fig. 16(a) for green tea extract, Fig. 16(b) for green coffee extract, Fig. 16(c) for coffee extract, and Fig. 16(d) for tea extract. Subsequently, the accuracy of the sensing system was also investigated through recovery analysis using 20 μL, without spiked green tea extract consisting of 0.0717 μM, 0.534 μM, 0.0403 μM and 0.112 μM of THN. This calculation was done based on the correlation between the standard THN concentration vs. current and in extract vs. current. After, spiking was continued, with the correlated recoveries shown in Table 4, and the percentage recoveries were found to be in the approximate range of ∼132–173%, signifying the accuracy of the TS(I)-modified electrode. Thus, the present sensing system is reliable for the effective selective detection of THN in real samples of green tea, green coffee, coffee and tea.
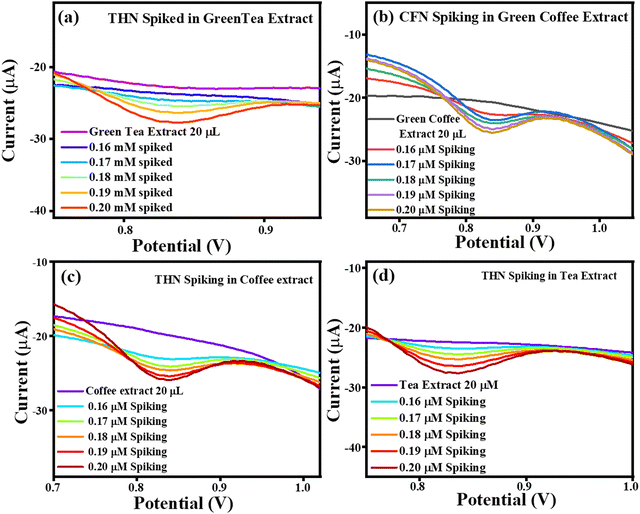 |
| | Fig. 16 Current vs. potential graph for (a) THN spiked in the green tea extract, (b) THN spiked in the green coffee extract, (c) THN spiked in the coffee extract and (d) THN spiked in the tea extract. | |
Table 4 THN spiking results in the green tea extract, green coffee extract, coffee extract and tea extract
| Extract |
THN spiking (μM) |
Recovery |
Recovery (%) |
RSD (n = 3) (%) |
| Green tea |
0 |
|
|
|
| 0.16 |
0.2361023 |
147.5639375 |
1.999133426 |
| 0.17 |
0.2448421 |
144.0247647 |
2.001289811 |
| 0.18 |
0.256734 |
142.63 |
1.998177102 |
| 0.19 |
0.2653638 |
139.6651579 |
2.001026515 |
| 0.20 |
0.2776774 |
138.8387 |
1.998722258 |
| Green coffee |
0 |
|
|
|
| 0.16 |
0.216601 |
135.375625 |
1.99906741 |
| 0.17 |
0.226751 |
133.3829412 |
2.002196242 |
| 0.18 |
0.238068 |
132.26 |
1.999428735 |
| 0.19 |
0.2460774 |
129.5144211 |
1.99937093 |
| 0.20 |
0.258468 |
129.234 |
2.000247613 |
| Coffee |
0 |
|
|
|
| 0.16 |
0.2039054 |
127.440875 |
2.049970231 |
| 0.17 |
0.214506 |
126.18 |
1.999944058 |
| 0.18 |
0.2249263 |
124.9590556 |
1.996209425 |
| 0.19 |
0.2339848 |
123.1498947 |
1.957392104 |
| 0.20 |
0.2429433 |
121.47165 |
2.041628643 |
| Tea |
0 |
|
|
|
| 0.16 |
0.277168 |
173.23 |
1.998787739 |
| 0.17 |
0.28764 |
169.2 |
1.999026561 |
| 0.18 |
0.296088 |
164.4933333 |
1.999405582 |
| 0.19 |
0.305926 |
161.0136842 |
2.000483777 |
| 0.20 |
0.31824 |
159.12 |
1.998491704 |
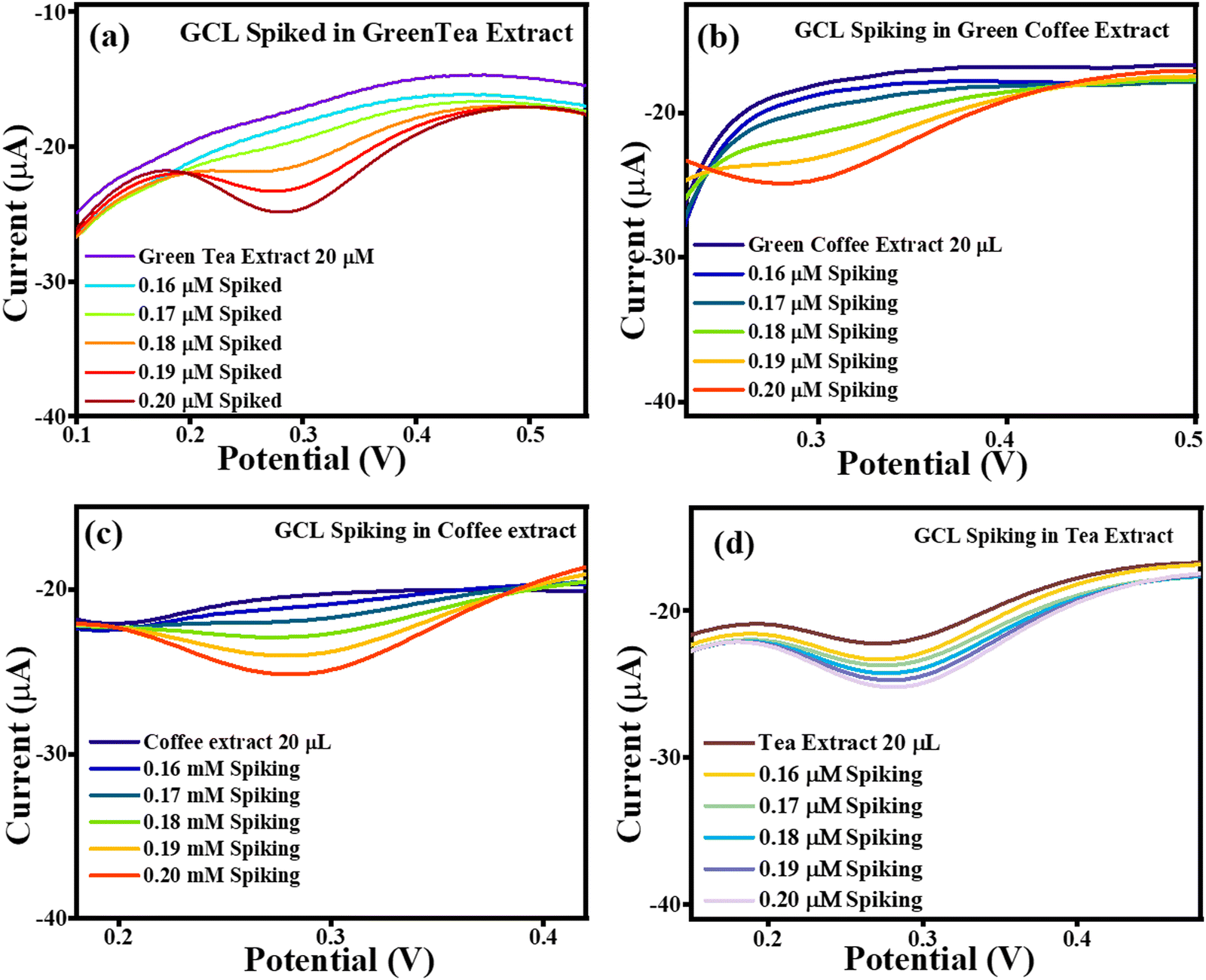
The current vs. potential graph for GCL spiking is shown in Fig. 17(a) for green tea extract, Fig. 17(b) for green coffee extract, Fig. 17(c) for coffee extract, and Fig. 17(d) for tea extract. Subsequently, the accuracy of the sensing system was also investigated through recovery analysis using 20 μL, without spiked green tea extract consisting of 0.0711 μM, 0.0626 μM, 0.0769 μM and 0.0953 μM of GCL. This calculation was done based on the correlation between the standard GCL concentration vs. current and in extract vs. current. After, spiking was continued, with the obtained correlated recoveries shown in Table 5, and the percentage recoveries were found to be in the approximate range of ∼133–182%, signifying the accuracy of the TS(I)-modified electrode. Thus, the present sensing system is reliable for the effective selective detection of GCL in real samples of green tea, green coffee, coffee and tea (Scheme 4).
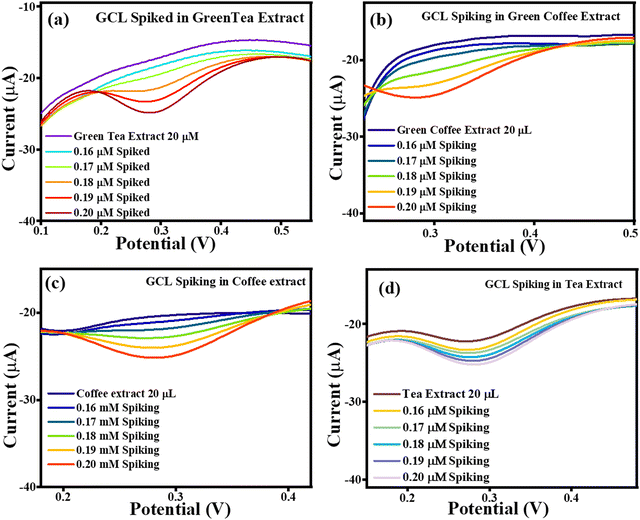 |
| | Fig. 17 Current vs. potential graph for (a) GCL spiked in the green tea extract, (b) GCL spiked in the green coffee extract, (c) GCL spiked in the coffee extract and (d) GCL spiked in the tea extract. | |
Table 5 GCL spiking results in the green tea extract, green coffee extract, coffee extract and tea extract
| Extract |
GCL spiking (μM) |
Recovery |
Recovery (%) |
RSD (n = 3) (%) |
| Green tea |
0 |
|
|
|
| 0.16 |
0.235722 |
147.32625 |
1.998116425 |
| 0.17 |
0.2447165 |
143.9508824 |
1.998230606 |
| 0.18 |
0.2551176 |
141.732 |
1.999078072 |
| 0.19 |
0.266322 |
140.1694737 |
2.001336728 |
| 0.20 |
0.276522 |
138.261 |
1.999840881 |
| Green coffee |
0 |
|
|
|
| 0.16 |
0.2266068 |
141.62925 |
1.999057398 |
| 0.17 |
0.236089 |
138.8758824 |
1.999246047 |
| 0.18 |
0.2472094 |
137.3385556 |
1.99830589 |
| 0.19 |
0.257652 |
135.6063158 |
1.998820114 |
| 0.20 |
0.2673268 |
133.6634 |
2.001295792 |
| Coffee |
0 |
|
|
|
| 0.16 |
0.2395059 |
149.6911875 |
1.999950732 |
| 0.17 |
0.2506035 |
147.4138235 |
1.999173994 |
| 0.18 |
0.2597259 |
144.2921667 |
1.998260474 |
| 0.19 |
0.2719711 |
143.1426842 |
2.000212523 |
| 0.20 |
0.2827149 |
141.35745 |
1.998479741 |
| Tea |
0 |
|
|
|
| 0.16 |
0.2581083 |
161.3176875 |
1.999160817 |
| 0.17 |
0.2695448 |
158.5557647 |
1.999667588 |
| 0.18 |
0.327607 |
182.0038889 |
1.999346778 |
| 0.19 |
0.2907207 |
153.0108947 |
1.998481704 |
| 0.20 |
0.3015013 |
150.75065 |
1.999991376 |
 |
| | Scheme 4 Electrochemical detection of CFN, THN, GCL, and DPN. | |
5. Conclusion
In this work, the synthesis of tin sulphide (TS) nanostructures in different solvents (Milli-Q water, ethanol and isopropanol) via the solvothermal method with a unique morphology was reported. There was no change recorded in the molecular structure and crystallographic phase of TS, but its morphology and size were found to change depending on the solvent. The as-synthesized TS(W), TS(E) and TS(I) were used for the modification of a GCE. The TS(I)@GCE electrode was found to be more electrochemically active than the TS(W)@GCE and TS(E)@GCE electrodes, and therefore it was used for the electrochemical detection of CFN, THN, GCL and DPN. The voltammetric limit of detection (LOD) for CFN, THN, GCL and DPN using the TS(I)@GCE electrode was found to be 6.71, 4.0, 11.2, and 5.19 nM with the linear range of 0.1–0.27, 0.01–0.25, 0.1–0.2 and 0.1–0.27 μM, respectively. The TS(I)@GCE electrode was further employed for the quantification of these analytes in green coffee, green tea, coffee and tea real samples. Interestingly, catechol group-containing species was also detected in the real samples. The excellent robustness and capability of the TS(I)@GCE electrode for real sample quantification make it appropriate for practical use. Further, to validate the resultant concentration of CNF, THF and GCL, the spiking method was also employed for a comparison of the results to utilize this system for commercial-level applications.
Conflicts of interest
There is no conflict of interest to declare.
Acknowledgements
The authors warmly acknowledge the Central University of Gujarat for providing the laboratory as well as a central instrumental facility for all experimental works. Amisha Kushwaha, especially, acknowledges the Council of Scientific & industrial research (CSIR) for providing the research fellowship. Amisha Kushwaha cordially acknowledges All India Institute of Medical Sciences (AIIMS, New Delhi) and Punjab University, Chandigarh for HR-TEM and XRD analysis of all samples.
References
- A. Nehlig, J. L. Daval and G. Debry, Brain Res. Brain Res. Rev., 1992, 17, 139–170 CrossRef CAS PubMed.
- G. Singh, A. Kushwaha and M. Sharma, Mater. Chem. Phys., 2022, 279, 125782 CrossRef CAS.
- P. Forsythe, L. P. A. McGarvey, L. G. Heaney, J. MacMahon and M. Ennis, Clin. Sci., 1999, 96, 349–355 CrossRef CAS.
- J. Y. Sun, T. Gan, Y. P. Deng, Z. X. Shi and Z. Lv, Sens. Actuators, B, 2015, 211, 339–345 CrossRef CAS.
- K. S. Bahçeci and J. Acar, Eur. Food Res. Technol., 2007, 225, 873–878 CrossRef.
- T. Arinobu, H. Hattori, T. Kumazawa, X. P. Lee, Y. Mizutani, T. Katase, S. Kojima, T. Omori, R. Kaneko, A. Ishii and H. Seno, Forensic Toxicol., 2009, 27, 1–6 CrossRef CAS.
- C. Zambakjian and A. A. Sakur, Futur. J. Pharm. Sci., 2020, 6, 1–8 CrossRef.
- C. Du, C. Ma, J. Gu, L. Li and G. Chen, Sensors, 2020, 20, 819 CrossRef PubMed.
- J. F. Wu, X. Gao, L. Ge, G. C. Zhao and G. F. Wang, RSC Adv., 2019, 9, 19813–19818 RSC.
- B. Srdjenovic, V. Djordjevic-Milic, N. Grujic, R. Injac and Z. Lepojevic, J. Chromatogr. Sci., 2008, 46, 144–149 CAS.
- P. Liu, R. Liu, G. Guan, C. Jiang, S. Wang and Z. Zhang, Analyst, 2011, 136, 4152–4158 RSC.
- O. Alharbi, Y. Xu and R. Goodacre, Anal. Bioanal. Chem., 2015, 407, 8253–8261 CrossRef CAS PubMed.
- H. Deng, B. Wang, M. Wu, B. Deng, L. Xie and Y. Guo, Int. J. Food Sci. Technol., 2019, 54, 202–211 CrossRef CAS.
- X. Ma, Z. Guo, Z. Mao, Y. Tang and P. Miao, Mikrochim. Acta, 2018, 33 CrossRef PubMed.
- N. Abdelwahab and E. Abdelaleem, J. Planar Chromatogr.--Mod. TLC, 2013, 26, 73–77 CrossRef CAS.
- Z. Xia, Y. Ni and S. Kokot, Food Chem., 2013, 141, 4087–4093 CrossRef CAS PubMed.
- B. Mekassa, M. Tessema and B. S. Chandravanshi, Sens. Bio-Sensing Res., 2017, 16, 46–54 CrossRef.
- Ç. C. Koçak, A. Nas, H. Kantekin and Z. Dursun, Talanta, 2018, 184, 452–460 CrossRef PubMed.
- E. Murugan and A. Dhamodharan, Diamond Relat. Mater., 2021, 120, 108684 CrossRef CAS.
- J. Kalaiyarasi, S. Meenakshi, K. Pandian and S. C. B. Gopinath, Microchim. Acta, 2017, 184, 2131–2140 CrossRef CAS.
- K. V. Özdokur, Ç. C. Koçak, Ç. Eden, Z. Demir, Ç. Çirak, E. Yavuz and B. Çağlar, ChemistrySelect, 2022, 7, e202201411 CrossRef.
- Ç. C. Koçak and Z. Dursun, Mater. Sci. Eng., B, 2022, 275, 115485 CrossRef.
- Ç. C. Koçak, B. Aslışen, Ş. Karabiberoğlu, K. V. Özdokur, A. Aslan and S. Koçak, ChemistrySelect, 2022, 7, e202201864 CrossRef.
- P. Veerakumar, S. Manavalan, S. M. Chen, A. Pandikumar and K. C. Lin, Nanoscale, 2020, 12, 22217–22233 RSC.
- F. Li, B. Ni, Y. Zheng, Y. Huang and G. Li, Surf. Interfaces, 2021, 26, 101375 CrossRef CAS.
- G. Li, X. Qi, G. Zhang, S. Wang, K. Li, J. Wu, X. Wan, Y. Liu and Q. Li, Microchem. J., 2022, 179, 107515 CrossRef CAS.
- G. Li, J. Wu, X. Qi, X. Wan, Y. Liu, Y. Chen and L. Xu, Mater. Today Chem., 2022, 26, 101043 CrossRef CAS.
- G. Li, X. Qi, J. Wu, L. Xu, X. Wan, Y. Liu, Y. Chen and Q. Li, J. Hazard. Mater., 2022, 436, 129107 CrossRef CAS PubMed.
- Y. Xia, G. Li, Y. Zhu, Q. He and C. Hu, Microchem. J., 2023, 190, 108726 CrossRef CAS.
- J. Johny, S. S. Guzman, B. Krishnan, J. A. A. Martinez, D. Avellaneda Avellaneda and S. Shaji, Appl. Surf. Sci., 2019, 470, 276–288 CrossRef CAS.
- A. J. Khimani, S. H. Chaki, S. M. Chauhan, A. V. Mangrola, R. R. Meena and M. P. Deshpande, Nano-Struct. Nano-Objects, 2019, 18, 100286 CrossRef CAS.
- J. Zheng, X. Xiong, G. Wang, Z. Lin, X. Ou, C. Yang and M. Liu, Chem. Eng. J., 2018, 339, 78–84 CrossRef CAS.
- Y. C. Zhang, J. Li, M. Zhang and D. D. Dionysiou, Environ. Sci. Technol., 2011, 45, 9324–9331 CrossRef CAS PubMed.
- A. Kushwaha, G. Singh, U. K. Gaur and M. Sharma, New J. Chem., 2023, 47, 3276–3289 RSC.
- A. Kushwaha, G. Singh and M. Sharma, RSC Adv., 2020, 10, 13050–13065 RSC.
- S. Park, J. Park, R. Selvaraj and Y. Kim, J. Ind. Eng. Chem., 2015, 31, 269–275 CrossRef CAS.
- J. Srivind, S. Balamurugan, K. Usharani, D. Prabha, M. Suganya, V. S. Nagarethinam and A. R. Balu, J. Mater. Sci.: Mater. Electron., 2018, 29, 9016–9024 CrossRef CAS.
-
A. Zhu, L. Qiao, Z. Jia, P. Tan, Y. Liu, Y. Ma and J. Pan, C–S bond induced ultrafine SnS2 dot/porous g-C3N4 sheet 0D/2D heterojunction: Synthesis and photocatalytic mechanism investigation, 2017, vol. 46 Search PubMed.
- H. Chen, B. Zhang, J. Zhang, W. Yu, J. Zheng, Z. Ding, H. Li, L. Ming, D. A. M. Bengono, S. Chen and H. Tong, Front. Chem., 2018, 6, 1–9 CrossRef CAS PubMed.
- R. K. Shervedani and H. A. Alinajafi-Najafabadi, Res. Int. J. Electrochem., 2011, 2011, 11 Search PubMed.
- A. Karthika, C. Sudhakar, A. Suganthi and M. Rajarajan, J. Sci. Adv. Mater. Devices, 2019, 4, 554–560 CrossRef.
- N. Di Fidio, J. W. Timmermans, C. Antonetti, A. Maria, R. Galletti, R. J. A. Gosselink, R. J. M. Bisselink and T. M. Slaghek, New J. Chem., 2021, 45, 9647 RSC.
- H. Zhang, Y. Wang, S. Shao and R. Xiao, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- I. Graça, J. M. Lopes, M. F. Ribeiro, F. Ramôa Ribeiro, H. S. Cerqueira and M. B. B. de Almeida, Appl. Catal., B, 2011, 101, 613–621 CrossRef.
- P. Feng, H. Wang, H. Lin and Y. Zheng, Carbon Resour. Convers., 2019, 2, 1–12 CrossRef CAS.
- J. Su, L. Yang, R. N. Liu and H. Lin, Chin. J. Catal., 2014, 35, 622–630 CrossRef CAS.
- A. Kushwaha, G. Singh and M. Sharma, Microchem. J., 2021, 166, 106224 CrossRef CAS.
- S. Güney and F. Cebeci, Sens. Actuators, B, 2015, 208, 307–314 CrossRef.
- N. A. Nia, M. M. Foroughi and S. Jahani, Talanta, 2021, 222, 121563 CrossRef CAS PubMed.
- G. Yang, F. Zhao and B. Zeng, Talanta, 2014, 127, 116–122 CrossRef CAS PubMed.
- Y. Wu, M. Huang, N. Song and W. Hu, Anal. Methods, 2014, 6, 2729–2735 RSC.
- L. Lin, C. Miao, S. Weng, S. Ying, F. Chen, C. Ye, H. Zhuang and D. You, J. Electroanal. Chem., 2020, 877, 114519 CrossRef CAS.
- N. Nan Song, Y. Zhu Wang, X. Yun Yang, H. Long Zong, Y. Xian Chen, Z. Ma and C. Xiang Chen, J. Electroanal. Chem., 2020, 873, 114352 CrossRef.
- X. Yan, Y. Gu, C. Li, B. Zheng, Y. Li, T. Zhang, Z. Zhang and M. Yang, Sens. Actuators, B, 2018, 257, 936–943 CrossRef CAS.
- K. Harikrishnan, G. Singh, A. Kushwaha, G. Singh, V. P. Singh, U. K. Gaur and M. Sharma, J. Environ. Chem. Eng., 2022, 10, 108717 CrossRef CAS.
- V. P. Singh, G. Singh, R. Patel, U. K. Gaur and M. Sharma, J. Environ. Chem. Eng., 2023, 6, 111208 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a