
ICP-MS: a tool for detection and quantitation of fosfomycin residues in cleaning samples of finished product by estimation of phosphorous load†
Anirban Roy
Chowdhury
 *,
Sujal
Shah
,
Rahul Y.
Kapse
,
Tushar
Mehta
and
Amit
Mukharya
*,
Sujal
Shah
,
Rahul Y.
Kapse
,
Tushar
Mehta
and
Amit
Mukharya
Amneal Pharmaceuticals Pvt Ltd., Pharmez, Ahmedabad, Gujarat 382213, India. E-mail: anirban.rc16@gmail.com
First published on 3rd November 2023
Abstract
Due to the significance of health risk factors to patients and the requirements imposed by regulatory authorities, the development of a reliable cleaning assessment method holds great importance in the pharmaceutical industry. Reliable, accurate and sensitive analytical techniques are always the need of the hour to establish any cleaning assessment method. Inductively coupled plasma mass spectrometry (ICP-MS) has been acclaimed for its accuracy and sensitivity to detect and analyse trace level elemental toxic metallic impurities. The exploration of using analytical tools to estimate a non-metallic fraction of an organic compound is not very common, especially in cleaning method assessment. This study details using elemental phosphorus in an organic compound as an indirect approach for trace-level analysis in equipment cleaning validation samples. The aim was to determine and quantify trace levels of fosfomycin residue. An analytical method to analyse equipment cleaning samples at a trace level for a fosfomycin tromethamine finished product has not been reported to date. Due to its uniqueness in its structural (non-chromophoric) and physicochemical (acidic) properties, it was a challenging task to develop a sensitive, robust, and rugged cleaning assessment method at a trace level concentration using conventional chromatographic techniques. In order to accomplish this, phosphorus-based quantification using an ICP-MS technique was explored in a rare application. The developed method is benign to the environment, saves time, and is cost effective compared to conventional chromatographic and hyphenated techniques. The developed analytical method was validated as per the principles of ICH guidelines, applicable for the trace-level analysis of equipment cleaning samples. Although the application is specific, the guiding concept can be explored towards analysing other non-chromophoric molecules by optimizing the sample preparation and instrument parameters of the method, e.g., glyphosate (an antibiotic), cisplatin (an anticancer drug), bilanafos (a herbicide), and phosphinothricin (a metabolite of bilanafos).
Introduction
Inductively coupled plasma mass spectrometry (ICP-MS) (Fig. 1) is a sophisticated technique that is generally utilized for the analysis of elemental impurities, such as heavy metals, in the pharmaceutical and other industries.1,2 Apart from this, it also has diverse applications, for instance, in analysing environmental (contaminated water) samples,3 pharmacokinetics studies4 and diagnosis (metal poisoning).5 The detection and quantitation of phosphorus (a non-metallic element) by ICP-MS is a well-established concept. However, the use of ICP-MS in the cleaning assessment method of finished pharmaceutical products has not yet been explored. “Cleaning validation is documented evidence that a proposed equipment cleaning procedure will reproducibly remove the previous product or cleaning agents used in the equipment below the scientifically set maximum allowable carryover level”.6 A cleaning method plays a crucial role in the pharmaceutical industry as cross contamination occurs due to foreign active pharmaceutical ingredients in the finished products that could have adverse effects on patients' health. Due to concerns about drug adulteration, ineffective cleaning processes have resulted in batch disqualification, FDA rejection, and costly fines.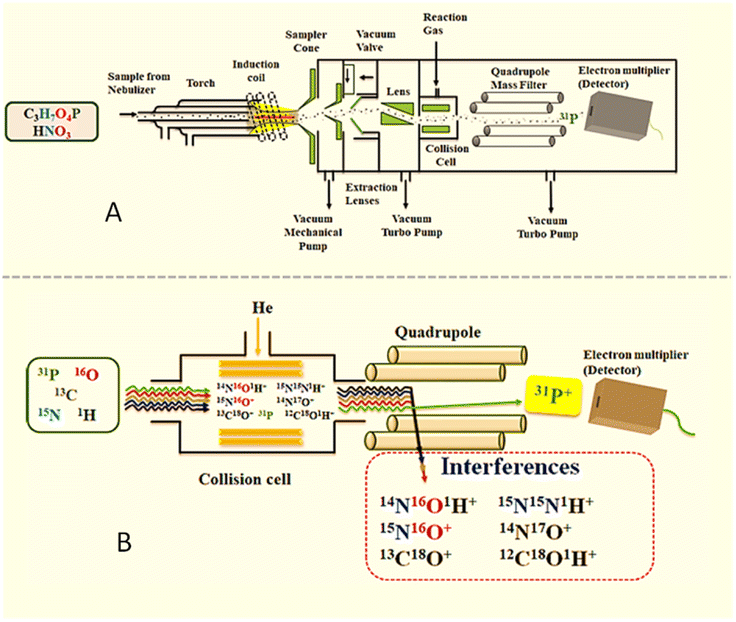 | ||
| Fig. 1 (A) Inductively coupled plasma mass spectrometry (ICP-MS) instrumentation. (B) Removal of interferences. | ||
Pharmaceutical companies meticulously establish equipment-specific cleaning standard operating procedures (SOPs) considering a variety of drug products, thereby upholding good manufacturing practices (GMP). The assurance of the effectiveness of equipment cleaning in the context of change over from one product to another is achieved by testing cleaning validation samples using highly sensitive analytical techniques. This assessment primarily involves the analytical testing of cleaning validation samples, predominantly comprising equipment swab samples and, in certain instances, rinse samples.
Once equipment cleaning procedure is verified, usually after testing at least three batches, the cleaning procedure for a particular product is considered validated. Often, instead of validating every new product, a worst-case compound is chosen as a marker compound to demonstrate equipment cleaning. The choice of this marker compound is based on comprehensive consideration of worst-case scenarios, encompassing attributes such as the compound's solubility in water, dosage strength, and the challenges it poses in terms of propensity to leave residues or colour.
The cleaning assessment method should be capable of measuring at and below the acceptable residue limit that allows change over from one product to another. Hence, a reliable method capable of detecting the lowest concentration of a drug is of a high priority for the pharmaceutical industry. To enable proper evaluation of cleaning validation samples, acceptance criteria for cleaning validation samples need to be defined, usually defined as residue mass per unit equipment surface area, referred to as maximum allowable carry over (MACO). Multiple approaches are being used to determine the MACO value which is usually dependent on the toxicity, solubility, and dose considerations of the drug. Generally, no more than 10 μg g−1 of the previous batch product should be detected in the subsequent batch of finished product or no more than 0.1% of a therapeutic dose of the previous batch active pharmaceutical ingredient (API) should be detected in the next batch of finished product.7 In a modern approach, multiple criteria are applied to determine the MACO value, in conjunction with multiple safety factors to derive a conservative acceptance criterion.
The MACO value is calculated per surface area, usually, in terms of cm−2, from which the swabbing area limit is derived. Generally, after conversion, in terms of analytical solutions, 0.1–1 μg mL−1 level sensitivity is usually attempted. Altogether, “Equipment shall be maintained in a clean and orderly manner”, to prevent adulteration and cross contamination of drug products as mentioned in the FDA's guide on the inspection validation of a cleaning process (7/93).8
Currently, in the pharmaceutical industry, cleaning samples are analysed using analytical techniques such as HPLC, GC, LC-MS/MS, etc. However, it is impractical to use these analytical techniques in the case of fosfomycin tromethamine (a broad-spectrum antibiotic that acts against Gram negative bacteria by inhibiting cell wall biosynthesis)9–17 cleaning samples because of obstacles due to its physicochemical properties. Hence, it is necessary to develop a unique cleaning assessment method for fosfomycin tromethamine. Chemically, fosfomycin is a phosphoenolpyruvate analogue with a framework comprising an epoxide ring and phosphonic group. Fosfomycin tromethamine has a molecular weight of 259.19 g mol−1 and is a highly water-soluble, non-chromophoric drug, with a pKa value of 1.25 and −4.3 for strong basic and strong acidic groups, respectively, and a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value of −0.75.
P value of −0.75.
Even though its structure does not show chromophoric characteristics, it is reported that fosfomycin can be detected in the lowest UV range (below 190–200 nm).18 However, it is a strenuous task to conduct analysis with commonly used solvents and buffers in this UV range. Also, inadequate retention in the reverse phase column due to the presence of a highly acidic, ionizable epoxide ring is observed. Indeed, it can be safely predicted that the application of an RI detector (low sensitivity) would also be futile for low sample concentrations (<1 μg mL−1).19 In mass spectrometry, atmospheric ionization models are widely used in cleaning assessment methods. In this manner, HPLC-MS/MS20–23 (LOQ = 0.75 mg L−1) and GC could be useful to analyse fosfomycin with specific derivatization.24
Nevertheless, the distinctiveness of the structural and physicochemical parameters of fosfomycin directed us to use the ICP-MS technique as a superior choice for cleaning method development. ICP-MS is a convenient tool that can be used to detect metals and a few non-metals in liquid samples at very low concentrations (μg L−1 or ng L−1).25–27 In this study, a cleaning assessment method was developed and validated for fosfomycin tromethamine equipment cleaning samples. The method-based application of ICP-MS was explored as an analytical tool for the analysis of cleaning samples containing a structural backbone featuring metal and inorganic elements. As phosphorus, an inorganic element, is a structural component of fosfomycin, it can be easily detected and sorted from other molecules in a mass spectrum once the analyte progresses through inductively coupled plasma.28–32
It is important to note that the ramifications of cross contamination can be very profound, hence, in terms of practices and limits, a conservative approach is taken into consideration. As an example, hard-to-clean surfaces are targeted for swabbing and the results are extrapolated for the entire equipment to ensure a cautious approach. Similarly, for analytical methods, generic analytical methods are often used, such as the measurement of total organic carbon (TOC), in which TOC is measured regardless of if it is being contributed by excipients or a drug. Likewise, in this research study, total phosphorus accounting is considered as a surrogate for the estimation of fosfomycin as a worst-case scenario. It may be noted that, from a practical aspect, phosphorus-containing compounds are less prominent compared to say, carbon, and in the fosfomycin drug product, no such excipients are used, which provides a reasonable level of specificity of the method.
It is worth noting that equipment cleaning limits derived for the study on the highly conservative side are around 20 μg per swab, which can be sometimes at the 1–5 mg level too, as per industry standards. However, as per stringent requirements implemented at Amneal pharmaceuticals, very conservative and stringent acceptance criteria were applied. The phosphorus-based estimation of compounds has been rarely reported. Bandura et al. reported detection limits of phosphorus of <0.1 ng mL−1 and 0.6 μg g−1 in aqueous and gel biological samples, respectively, using ICP-DRC-MS.29,30 Also, Prange et al. developed a method for the determination of phosphorus-containing pesticides in environmental samples using GC hyphenated with collision-cell ICP-MS.31 In 2008, Webster et al. reported the application of ICP-MS for the selective detection of various non-metals, including phosphorus, in pharmaceuticals.32
Furthermore, when elemental analysis at a trace level is involved, contribution of the sector field ICP-MS (ICP-SFMS) is of prime importance. High resolving power (about 10000) and high sensitivity with a low background (<0.2 ions per sec) give better detection limits up to pg L−1, thus making ICP-SFMS a renowned technique that is used in speciation analysis, especially in biogeochemistry.33 So far as phosphorus determination is concerned, double-focusing sector field ICP-MS (ICP-SFMS) plays an important role among all the alternative techniques, as demonstrated by Becker et al. For estimation of phosphorus in biological as well as aqueous samples, ICP-SFMS is recommended over ICP-QMS (ICP-MS). As ICP-QMS cannot distinguish between background and interference at mass 31 u when a higher concentration of nitric acid (1–2%) is used for sample preparation. However, the separation of disturbing molecular ions (isobaric interferences, 15N16O+; 14N17O+; 14N16O1H+) from atomic ions of an analyte is the key figure of merit in using ICP-SFMS over ICP-QMS. The background in ICP-QMS depends on the concentration of nitrogen in samples, i.e., HNO3 concentration. By preference, both the techniques, i.e., ICP-QMS (μg mL−1) and ICP-SFMS (ng mL−1), are sufficiently sensitive for the determination of phosphorus content in complex matrix samples such as protein.28 However, considering the required application is the analysis of cleaning samples, which involves trace level estimation of a phosphorus-enriched compound in a relatively clean matrix at the μg g−1 level concentration with the usage of nitric acid at a very low concentration (0.0001%) in sample and He collision mode, ICP-QMS has been proven to be the best fit for purpose analysis method in the present application.
Hence, although sensitive methods at the low detection level have been reported, the development of analytical methods for cleaning validation samples imposes unique challenges. One of these challenges is trace-level detection, where obtaining sample recoveries can be challenging, as it is a cumulative challenge of recovery from the swab stick itself as well as the spiked sample on a metal surface. All these conditions should be met as per the technical requirements for an effective cleaning assessment method.
Experimental
Chemicals and laboratory reagents
Nitric acid (ULTREX II ultrapure reagent, J. T. Baker), beryllium standard (ICP-MS grade, Inorganic Ventures), phosphorus standard (ICP-MS grade, Inorganic Ventures), swab [TX714A, Alpha series (Texwipe)], [type-I (Milli-Q)] de-ionized water, and fosfomycin tromethamine API (98.5%, sourced by Interquim S.A. de C.V.) were used in the experiments. Test materials were stored under the conditions as mentioned in the respective supplier's certificate of analysis. All analytical grade chemicals used were ultra-trace metal analysis grade or equivalent. All the volumetric flasks used in sample preparation were PMP (polymethyl pentene), class A with NS (non-sterile) stoppers, PP (polypropylene) or an equivalent quality polymer. Stainless steel, rubber and acrylic coupons (dimensions: 2 × 2 inch2) were used as plausible machine surface materials. All test solutions prepared from the test materials were stored at a room temperature. All sample preparations, quantification and assessments were carried out under a GLP environment.Sample preparation
Instrumentation
A Thermo iCAP RQ ICP-MS instrument was used to determine the concentration of fosfomycin at a trace level during cleaning method development. To attenuate all polyatomic interferences, kinetic energy discrimination mode (helium collision mode) was used. Along with a collision/reaction cell (QCell), a light cell gas (helium) was used that generates effective kinetic energy discrimination (KED) by providing a large number of collisions under high-cell gas pressure. The instrument also features a baffled type cyclonic spray chamber as a secondary particle separator to reduce sample load without compromising detection limits. An auto sampler (ASX-560, CETAC Technologies) was used to deliver the samples to an ICP-MS instrument and to minimize the variability between runs. A quartz nebulizer, standard nickel sampling cones and quartz torch with a 2.5 mm id injector were also used.Methodology
Calculation for critical limit level concentration (CLLC) of phosphorus
Determination of phosphorus load via an indirect strategy was used to assess the concentration of fosfomycin carry over/cross contamination. Contribution of the phosphorus load to the MACO value of fosfomycin was taken into consideration as the central domain of method development. Calculation of the MACO value is a significant and critical step in a cleaning method to ensure patients' safety. The initial assessment of the MACO value allowed us to predetermine the transfer of ineffective drug concentration level and to check for massive cross contamination between two batches. The allowable limit of fosfomycin API residue (the MACO value) based on available toxicity data and therapeutic daily dose was as low as 5 μg inch−2 and 20 μg for a coupon with a 4 inch2 surface area (2 × 2 inch2), as shown in eqn (S2a).† Consequently, the limit of carry over for phosphorus from the fosfomycin tromethamine formulation was 448.7 ng mL−1, as calculated using eqn (S2b).† Conversely, from the obtained phosphorus content, the fosfomycin residue content was calculated as per eqn (S2c).†Method development
Analytical method development involves optimisation of the diluent in sample preparation and the ICP-MS instrument parameters. As the drug product is meant to be administered as an oral solution, the matrix is highly soluble in water, but an optimum level of an acidifying agent such as nitric acid is added to enable the generation of clear solutions and proper ionisation.The developed analytical method was validated as per ICH guidelines. The method validation studies were performed using a spiked sample approach to negate sample variability. After satisfactory completion of method validation, a standard testing procedure was formalised, which was used to test actual equipment cleaning validation samples, which led to validation of the actual equipment cleaning process.
Results and discussion
Considering analysis is conducted at a trace level, during analysis of a drug substance using the ICP-MS technique, the following critical areas of method development were considered: sample preparation, selection of diluent, selection of a suitable internal standard, selection of instrument parameters, etc.Analysis was performed in both modes (STD and KED) in the ICP-MS measurements; however, more interference was observed in the blank solution in STD mode. Hence, to eliminate the interference, the KED mode was used employing helium as a collision gas. Various instrument parameters were optimized and the parameters were finalized as per Table 1. Milli-Q water and LCMS grade water (Optima grade, Thermo Fisher Scientific) were also screened, but similar interference was observed for phosphorus in both types of water. To mitigate the chances of false positive results and to minimize the polyatomic interference caused by nitric acid (by combining H, N and O, interference may be observed at the mass of P, i.e., 31), nitric acid concentration in diluent was also optimized, and 0.0001% nitric acid was found to be optimum as a diluent in sample preparation. The influences of different internal standards (germanium, yttrium, scandium, and beryllium) were investigated. Beryllium was found to be more compatible for phosphorus and recovery results were found to be acceptable for phosphorus. Furthermore, the internal standard concentration was also optimized to achieve the appropriate response and 1 μg mL−1 concentration of beryllium was found to be appropriate in the final sample. Moreover, a survey run was performed in each run to observe the response of all the elements with a mass in the range of 4.6–245.0 u for observation purposes and to check if any anomalies are observed during routine analysis. However, no such anomalies were observed during method validation and analysis of the swab samples.
| ICP-MS conditions of the developed method | |||||
|---|---|---|---|---|---|
| Parameter | Conditions | Parameter | Conditions | ||
| Acquisition parameters | Dwell time | 0.1 s | Monitor analyte | Minimum delay | |
| Channel | 1 | Uptake | 30 sec | ||
| Spacing | 0.1 u | Wash | 30 sec | ||
| Mode | KED | Maximum delay | |||
| Resolution | Normal | Uptake | 300 sec | ||
| No. of sweep | 10 | Wash | 300 sec | ||
| Survey scan setting | Start mass | 4.6 u | RF plasma power | 1550 W | |
| End mass | 245.0 u | Plasma gas flow | 0.8 L min−1 | ||
| Dwell time | 0.001 sec | Spray chamber temperature | 2.4 °C | ||
| Spacing | 0.2 u | Tune mode | KED | ||
| Resolution | Normal | No. of replicates | 3 | ||
| Mode | KED | — | — | ||
| No. of sweep | 6 | ||||
Method validation
| Surface | Level | No. of set | Accuracy range (%) |
|---|---|---|---|
| Swab stick | 10%, 25%, 150% | 3 | 90.2 to 102.0 |
| SS coupon | 10%, 25%, 150% | 3 | 94.8 to 103.0 |
| Rubber coupon | 10%, 25%, 150% | 3 | 95.9 to 104.6 |
| Acrylic coupon | 10%, 25%, 150% | 3 | 93.0 to 98.3 |
| Swab stick | 100% | 6 | % RSD – 1.1% |
| S – 1.0000 | |||
| % Bias – 0.9–1.0% | |||
| % Drift – 0.6% |
| Set no | Obtained cps (counts per second) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Blank | Coupons | Detergents | Placebo 2 μg mL−1 | Linearity solution – 100% | |||||
| Solution | Swab | SS | Rubber | Acrylic | Alconox | Liquinox | |||
| 1 | 239 | 335 | 293 | 349 | 331 | 18992 |
267 | 281 | 24253 |
| 2 | 251 | 307 | 284 | 335 | 315 | — | — | 279 | — |
| 3 | 248 | 541 | — | — | — | — | — | — | — |
| Mean | 246 | 394 | 289 | 342 | 323 | 18992 |
267 | 280 | 24253 |
| % Interference | 1.0 | 1.6 | 1.2 | 1.4 | 1.3 | 78.3 | 1.1 | 1.2 | — |
| Variable | Condition | System suitability parameters | ||
|---|---|---|---|---|
| Correlation coefficient | % Bias | % Drift | ||
| Plasma RF power | 1519 W (−2%) | 0.9997 | 0.4 & 0.5 | 0.8 |
| 1581 W (+2%) | 0.9996 | 1.1 & 2.5 | 1.3 | |
| Wash and uptake time of sample aspiration | 54 s (−10%) | 0.9996 | 1.3 & 1.6 | 0.3 |
| 66 s (+10%) | 0.9996 | 0.7 & 1.3 | 0.6 | |
| Sample name | Obtained concentration (ng mL−1) | ||
|---|---|---|---|
| Initial (0 hours) | 143 hours | % Difference | |
| Spiked sample (100%) | 480.324 | 484.205 | 0.8 |
| Linearity solution (100%) | 442.021 | 412.578 | 6.7 |
Sample analysis
Based on the successful completion of the method validation, actual testing of equipment cleaning validation samples was also performed, and the results were found to be below detection levels. Hence, this proved the effectiveness of the actual equipment cleaning procedures. The results of the swab sample analysis are provided in Table S2.†Conclusion
The study details the development of a method that enables the trace level detection of equipment cleaning samples using ICP-MS for the non-chromophoric molecule fosfomycin. Using a conventional analytical technique, this would not be possible, e.g., for a HPLC-based technique with either UV or a refractive index detector. The study utilized a unique aspect of fosfomycin, i.e., the presence of phosphorus in the molecule. The development of an analytical method is critical to ensure equipment cleaning procedures remain for new products introduced into premises. This adheres to the regulatory compliance requirements for the pharmaceutical industry as well as ethical considerations for patient safety concerns. The method is totally eco-friendly, green and economical as the use of organic solvents is totally omitted. Also, the method is time saving, as the run time is short. The developed method was validated in terms of its specificity, linearity, accuracy, precision and robustness. The method was also demonstrated as specific with a commonly used detergent (liquinox) and linear (LOD 0.1 μg mL−1 and LOQ 0.2 μg mL−1 of fosfomycin), with a coefficient of determination (R) of 1.00. Furthermore, the study showed that the method is accurate and precise. The results indicate that the method is robust with respect to ICP-MS. Robustness data indicate that the operating range of the method in terms of RF power (±2%) was found to be between 1519 W and 1581 W. In addition, variation in sample uptake and wash time (±10%) was found to be acceptable at between 54 s and 66 s. From the solution stability results, it can be concluded that the standard solution of phosphorus (at the 100% level) and spiked sample of fosfomycin (at the 100% level) are stable for up to 143 h at room temperature. For all the studied parameters, the method was found suitable for the detection of fosfomycin at a trace level concentration of 0.1 μg mL−1. The developed analytical method was used to analyse actual equipment cleaning samples and an equipment cleaning procedure was validated for fosfomycin.Apart from fosfomycin, there are future prospects that ICP-MS can be utilized as an analytical tool for the analysis of cleaning samples of various non-chromophoric drugs containing metals or inorganic elements, such as glyphosate (an antibiotic), cisplatin (an anticancer drug), Li2CO3 (used in the treatment of manic disorders), bismuth subsalicylate (an antacid), sodium zirconium cyclosilicate (for the treatment of chronic kidney disorders), arsenic trioxide (for the treatment of acute leukemia), bilanafos (a herbicide), phosphinothricin (a metabolite of bilanafos), etc. However, optimization of the method needs to be done for each of the elements separately, which solely depends on the matrix composition of the finished pharmaceutical product as well as the drug itself. Compared to fosfomycin, which contains only phosphorus as a non-metal, the molecules in which metallic components are present might be more straightforward considering the better ionization potential of metals, enabling the development of more sensitive analytical methods.
Author contributions
Anirban Roy Chowdhury: supervision and conceptualisation of method development, validation, review, and interpretation of data. Sujal Shah: practical execution, method development, validation, and data analysis. Rahul Y. Kapse: data analysis, manuscript draft writing and editing of intellectual contents. Tushar Mehta and Amit Mukharya: supervision, resources, data interpretation, and review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by Amneal pharmaceuticals Pvt. Ltd. We thank the administration of Amneal pharmaceuticals for making available the ICP-MS instrument and all the requirements for this study.Notes and references
- W. Chen, Y. Yang, K. Fu, D. Zhang and Z. Wang, Front. Pharmacol., 2022, 13, 891273, DOI:10.3389/fphar.2022.891273.
- J. S. Barin, P. A. Mello, M. F. Mesko, F. A. Duarte and E. M. M. Flores, Anal. Bioanal. Chem., 2016, 408, 4547–4566, DOI:10.1007/s00216-016-9471-6.
- V. Balaram, L. Copia, U. Saravana Kumar, J. Miller and S. Chidambaram, Geosyst. Geoenviron., 2023, 100210, DOI:10.1016/j.geogeo.2023.100210.
- P. Songvut, N. Pholphana, T. Suriyo, N. Rangkadilok, D. Panomvana, P. Puranajoti and J. Satayavivad, Sci. Rep., 2023, 13, 2534, DOI:10.1038/s41598-023-28612-1.
- J.-P. Goullé, E. Saussereau, L. Mahieu and M. Guerbet, Bioanalysis, 2014, 6, 2245–2259, DOI:10.4155/bio.14.190.
- Australian Government, Department of Health and Aged Care, Therapeutic Goods Administration, https://www.tga.gov.au/, accessed 17 February 2023 Search PubMed.
- R. Abhishek, J. At. Mol., 2014, 4, 779–783 Search PubMed.
- U.S. Food and Drug administration, Validation of Cleaning Processes (7/93), FDA, accessed 17 February 2023 Search PubMed.
- S. S. Patel, J. A. Balfour and H. M. Bryson, Drugs, 1997, 53, 637–656, DOI:10.2165/00003495-199753040-00007.
- F. M. Kahan, J. S. Kahan, P. J. Cassidy and H. Kropp, Ann. N. Y. Acad. Sci., 1974, 235, 364–386, DOI:10.1111/j.1749-6632.1974.tb43277.x.
- M. Aghamali, M. Sedighi, A. Zahedi bialvaei, N. Mohammadzadeh, S. Abbasian, Z. Ghafouri and E. Kouhsari, J. Med. Microbiol., 2019, 68, 11–25, DOI:10.1099/jmm.0.000874.
- A. Dinh, J. Salomon, J. P. Bru and L. Bernard, Scand. J. Infect. Dis., 2011, 44, 182–189, DOI:10.3109/00365548.2011.616221.
- M. E. Falagas, A. C. Kastoris, A. M. Kapaskelis and D. E. Karageorgopoulos, Lancet Infect. Dis., 2010, 10, 43–50, DOI:10.1016/S1473-3099(09)70325-1.
- A. Castañeda-García, J. Blázquez and A. Rodríguez-Rojas, Antibiotics, 2013, 2, 217–236, DOI:10.3390/antibiotics2020217.
- S. E. D. Brown, E. I. Vivas, C. T. Walsh and R. Kolter, J. Bacteriol., 1995, 177, 4194–4197, DOI:10.1128/jb.177.14.4194-4197.1995.
- S. Eschenburg, M. Priestman and E. Schönbrunn, J. Biol. Chem., 2005, 280, 3757–3763, DOI:10.1074/jbc.M411325200.
- S. Sastry and Y. Doi, J. Infect. Chemother., 2016, 22, 273–280, DOI:10.1016/j.jiac.2016.01.010.
- M. Piponski, T. B. Stoimenova, T. Melnyk, S. Kovalenko, E. L. Todevska, M. Velkovski, S. E. Deeb, Y. Mysula and L. Logoyda, Sci. Pharm., 2022, 90, 35, DOI:10.3390/scipharm90020035.
- H. Liu, H. Wang and V. Bruce Sunderland, J. Liq. Chromatogr. Relat. Technol., 2006, 29, 15–24, DOI:10.1080/10826070500358209.
- R. A. Wijma, S. Bahmany, E. B. Wilms, T. van Gelder, J. W. Mouton and B. C. P. Koch, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1061, 263–269, DOI:10.1016/j.jchromb.2017.07.036.
- T. Shopova, T. Hüppe, B. Wolf, D. I. Sessler, T. Volk, H. V. Groesdonk, S. Kreuer and F. Maurer, J. Chromatogr. Sci., 2021, 59, 165–174, DOI:10.1093/chromsci/bmaa092.
- J. Martens-Lobenhoffer and S. M. Bode-Böger, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 990, 164–168, DOI:10.1016/j.jchromb.2015.03.029.
- S. Baldelli, M. Cerea, D. Mangioni, L. Alagna, A. Muscatello, A. Bandera and D. Cattaneo, J. Chemother., 2022, 34, 25–34, DOI:10.1080/1120009X.2021.1963617.
- S. Y. Kim, K.-S. Ju, W. W. Metcalf, B. S. Evans, T. Kuzuyama and W. A. van der Donk, Antimicrob. Agents Chemother., 2012, 56, 4175–4183, DOI:10.1128/AAC.06478-11.
- S. Thiab, M. Wainwright and P. Riby, Br. J. Pharm., 2017, 2, S2–S4, DOI:10.5920/bjpharm.2017.12.
- United States Pharmacopeia, General Chapter, 〈233〉 Elemental Impurities—Procedures, United States Pharmacopeia, Rockville, MD, 2023, DOI:10.31003/USPNF_M5193_02_01.
- International council for harmonisation of technical requirements for pharmaceuticals for human use, ICH harmonised guideline, guideline for elemental impurities Q3D(R2), Final version Adopted on 26 April 2022, Microsoft Word – Q3D-R2_Guideline_Step3_2022_0308_topublish (ich.org), https://database.ich.org/, accessed 17 February 2023 Search PubMed.
- J. A. Becker, S. F. Boulyga, C. Pickhardt, J. A. Becker, S. Buddrus and M. Przybylski, Anal. Bioanal. Chem., 2003, 375, 561–566, DOI:10.1007/s00216-002-1737-5.
- D. R. Bandura, V. I. Baranov and S. D. Tanner, Anal. Chem., 2002, 74, 1497–1502, DOI:10.1021/ac011031v.
- D. R. Bandura, O. I. Ornatsky and L. Liao, J. Anal. At. Spectrom., 2004, 19, 96, 10.1039/B308901K.
- Y. Zhang, J. Qu, Y. Chang and Y. Li, J. Anal. At. Spectrom., 2021, 36, 429–438, 10.1039/d0ja00462f.
- D. Pröfrock, P. Leonhard, S. Wilbur and A. Prange, J. Anal. At. Spectrom., 2004, 19, 623–631, 10.1039/B310530J.
- M. Moldovan, E. M. Krupp, A. E. Holliday and O. F. X. Donard, J. Anal. At. Spectrom., 2004, 19, 815–822, 10.1039/b403128h.
Footnote |
| † Electronic supplementary information (ESI) available: eqn (S1)–(S3); Tables S1 and S2; Fig. S1. See DOI: https://doi.org/10.1039/d3ja00247k |
| This journal is © The Royal Society of Chemistry 2024 |