DOI:
10.1039/D4GC05156D
(Paper)
Green Chem., 2025,
27, 133-143
(B,P,Co,Fe)-Ni modified on nanowood for boosting seawater urea electro-oxidation†
Received
14th October 2024
, Accepted 5th November 2024
First published on 6th November 2024
Abstract
Coupling the urea oxidation reaction (UOR) and hydrogen evolution reaction (HER) in seawater is desirable to produce sustainable and green hydrogen due to the reduced energy consumption. However, developing high-performance UOR/HER electrocatalysts in seawater instead of pure water remains a great challenge. Herein, (B,P,Co,Fe)-Ni anchored on Paulownia Wood (PW) is proposed to enhance the overall urea-(sea)water splitting performance. The resulting sample only needs a potential of 1.34 V to deliver a large current density of 100 mA cm−2 for alkaline UOR and features a remarkable durability to maintain 100 mA cm−2 for 100 h. The HER and UOR (HER||UOR) coupled system in alkaline seawater-urea electrolyte for producing H2 demonstrated a more significantly reduced electrolyzer voltage of 1.67 V obtained at 100 mA cm−2 in comparison to that of the HER||OER system (1.98 V). The well-aligned micro-channels and nanopores in wood frameworks not only improve the hydrophilicity and aerophobicity of the whole electrode, which is conducive to the penetration of the electrolyte and release of bubbles, but also shorten the transmission distance of ions and intermediates to accelerate the reaction kinetic process. Density functional theory calculations reveal that Co, Fe, P and B co-doping in Ni effectively adjusts the electronic structure, and the adsorption/desorption behavior of the urea reaction intermediates is regulated by the synergistic effect from multiple components, resulting in an excellent catalytic activity in seawater-urea media. This work promotes a better understanding of the surface electronic structure modulation of nanowood via doping strategy and offers great potential in the design of advanced UOR/HER catalysts for hydrogen production and urea wastewater treatment.
Introduction
Hydrogen energy produced by electrocatalytic water splitting has a high energy density and zero carbon emission, which is crucial to realize hydrogen economy and global carbon neutrality.1–3 Nevertheless, the oxygen evolution reaction (OER, 1.23 V vs. RHE) as a half-reaction of the water splitting process at the anode suffers from slow kinetics and decreases the overall efficiency of water splitting.4,5 Seeking high-efficiency oxidative reactions as a substitute for OER is strongly desirable given the low energy consumption.6,7 The urea oxidation reaction (UOR, CO(NH2)2 + 6OH− → N2 + CO2 + 5H2O + 6e−) has been regarded as a feasible substitution due to the relatively low oxidation potential (0.37 V vs. RHE) and potentially cost-free source from urea-rich wastewater.8–10 Considering the low availability of pure water, utilizing the abundant seawater as an electrolyte has attracted increasing attention.11,12 Since a 6e− transfer is involved in UOR, high-activity electrocatalysts are needed to enhance the reaction kinetics of seawater-urea oxidation. Although noble metal catalysts (e.g., Pt, Rh) have been demonstrated to have excellent catalytic activity for UOR, their scarcity and rising price hinder further development in practical applications.13,14 Consequently, developing efficient and low-cost UOR electrocatalysts is desired to achieve the energy-efficient H2 production and the purification of urea wastewater.15,16 Among them, the alloy-doped nickel-based catalysts show a strong interaction between multiple metals and significantly influence the number of catalytic sites and their intrinsic activities.17–19 This type of nickel-based electrocatalysts promotes the adsorption/desorption of reaction intermediates and achieves the synergistic effect from multiple active centers to improve the bifunctional UOR and HER performances.20–22
The adsorption of urea molecules on the catalyst surface and the subsequent activation/cleavage of the C–N bond are the key steps, affecting the overall UOR rate. The electronic structure of the catalyst surface has a significant effect on the adsorption/activation behaviors of urea molecules, and determines the activity of the catalyst. Heteroatom doping has been considered a promising strategy to optimize the adsorption energy of urea molecules, and improve the UOR activity through adjusting the electronic structures of active centers.23–25 The introduction of heteroatoms achieves an adjustable electron distribution, promotes electron transfer and accelerates the UOR reaction kinetics, resulting in the decline of the potential of urea oxidation. In addition to heteroatom doping, the rational design of a hydrophilic and aerophobic electrode is of great importance for promoting the diffusion of the electrolyte, ions and electrons to enhance the reaction kinetics.26,27 A formed aerophobic electrode is conducive to the overflow of bubbles and exposes more active sites. For instance, assembling open-channels and highly conductive substrates into self-supported electrodes limits the strong adsorption of bubbles on electrode surfaces, allowing bubbles to rupture and overflow readily.28,29 Commercial foam substrates are widely imported and exhibit good structural merits. However, their low abundance, high production cost, and poor stability under high temperature conditions impede further large-scale development and influence the implement of low-carbon economy.30,31 In our previous work, natural porous wood frameworks have been utilized to design self-supported electrodes with excellent OER/HER activities in alkaline seawater.32 The micro-channels and nanopores in wood facilitate the rapid penetration of the electrolyte, and shorten the contact time between active components and electrolyte.33 The wood skeleton accelerates the escape of bubbles and avoids the growth of bubbles, enabling more active sites to be exposed. In addition to the natural aerophobic and hydrophilic properties of wood, the wood cell wall contains rich oxygen-containing functional groups to readily adsorb aqueous metal ions, regulating the generation of a conductive metal layer once processed forward.31,32 Therefore, the use of cost-effective and sustainable wood frameworks as catalytic substrates not only facilitates the optimization of the catalyst structures, but also satisfies the initiative of “carbon peaking” and “carbon neutrality”.
In this study, (B,P,Co,Fe)-Ni anchored on PW (denoted as (B,P,Co,Fe)-Ni@PW) is proposed as a self-supported electrode to enhance UOR and HER activities in an alkaline seawater-urea electrolyte. The resultant (B,P,Co,Fe)-Ni@PW requires only 1.34 V for UOR and 290 mV for HER to achieve a large current density of 100 mA cm−2, and maintains a long cycling stability for 100 h in alkaline urea solution. In a two-electrode seawater-urea electrolyzer ((B,P,Co,Fe)-Ni@PW||(B,P,Co,Fe)-Ni@PW), a voltage of only 1.67 V is required to reach 100 mA cm−2. DFT calculations reveal that the synergistic effect of multiple active centers and the charge redistribution caused by doping resulted in the excellent activity of the (B,P,Co,Fe)-Ni@PW electrode. The micro-channels and nanopores in the sample play a vital role in promoting the infiltration of the electrolyte and the overflow of bubbles, benefiting from the hydrophilic and aerophobic properties of wood. This work proposes a new nanowood-based UOR/HER electrocatalyst applied in seawater medium, facilitating the development of hydrogen energy production and urea pollutant elimination.
Experimental
Materials
The natural Paulownia Wood was cut into pieces with dimensions of 10 mm × 15 mm × 2 mm. NiSO4·6H2O, (NH4)2Fe(SO4)2·6H2O, Co(CH3COO)2·4H2O, Na3C6H5O7·2H2O, CH3COONH4, NaH2PO2·H2O, (NH4)2S2O8, CON2H4, NH3·H2O, KOH and NaBH4 were purchased from Sinopharm Co., Ltd. Dimethylamine borane (DMAB) was purchased from Aladdin Industrial Corporation. Deionized water was used in the experiment, and all reagents were used directly without further purification. The natural seawater was collected from Fushan Bay in Qingdao, Shandong, China.
Preparation of (P,Co)-Ni@PW
To activate the non-conducting substrate, Ni doped with Co and P was plated onto the surface of PW and its microchannels via electroless plating. Firstly, PW was dipped in a reducing solution of 5 g L−1 NaBH4 (2 g L−1, NaOH) for 15 min. Then, activated wood was immersed in a plating solution for 20 min at 60 °C. After the reaction was completed, the sample was cleaned multiple times with deionized water, and then dried under vacuum at 60 °C for 24 h (denoted as (P,Co)-Ni@PW). The plating solution includes 30 g L−1 NiSO4·6H2O, 1 g L−1 Co(CH3COO)2·4H2O, 30 g L−1 Na3C6H5O7·2H2O, 12 g L−1 CH3COONH4 and 25 g L−1 NaH2PO2·H2O. The pH of the plating solution was adjusted to 9.5 with NH3·H2O. P-Ni@PW was prepared via the same experimental process (Co(CH3COO)2·4H2O was not added to the plating solution) for comparison.
Preparation of (B,P,Co,Fe)-Ni@PW
The sample was prepared via dissolution of (NH4)2Fe·(SO4)2·6H2O (0.3 g), (NH4)2S2O8 (0.8 g) and DMAB (0.2 g) in 50 mL deionized water. Then, (P,Co)-Ni@PW was immersed in the above solution at room temperature for 2 h, and the solution was replaced every one hour. After the reaction was completed, the sample was cleaned three times with deionized water, and then dried under vacuum at 60 °C for 24 h (denoted as (B,P,Co,Fe)-Ni@PW). The different mass ratios of (NH4)2Fe(SO4)2·6H2O and DMAB (0.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.2, 0.2:0.2, 0.4:0.2, 0.5:0.2, 0.3:0.1, 0.3:0.3 and 0.3:0.4) were prepared, respectively, and reaction times of 4 h and 6 h were used for comparison.
:0.2, 0.2:0.2, 0.4:0.2, 0.5:0.2, 0.3:0.1, 0.3:0.3 and 0.3:0.4) were prepared, respectively, and reaction times of 4 h and 6 h were used for comparison.
Characterizations
Field emission scanning electron microscopy (FESEM, Quanta 250 FEG, FEI, USA) was used to analyze the surface morphologies of the samples. The fine nanostructures and elemental distribution of (B,P,Co,Fe)-Ni@PW were characterized by transmission electron microscopy (TEM, JEM-2800F, Japan) and coupled energy dispersive X-ray spectroscopy (EDS). Powder X-ray diffraction (XRD, DX2700, China) measurements were performed using Cu Kα radiation at a rate (2θ) of 5° min−1 ranging from 10° to 90° to obtain the phase structures of (B,P,Co,Fe)-Ni@PW. The chemical states were revealed by X-ray photoelectron spectroscopy (XPS) on a Thermo Scientific Escalab 250Xi XPS electron spectrometer. The wettability of the samples was tested by a contact angle analyzer (Theta).
Electrochemical performance evaluation
The HER, OER and UOR tests of (B,P,Co,Fe)-Ni@PW were achieved in a standard three-electrode test system from a CHI 760E electrochemical workstation. The (B,P,Co,Fe)-Ni@PW, graphite electrode and Hg/HgO electrode were used as the working electrode, counter electrode and reference electrode, respectively. Solutions of 1 M KOH, 1 M KOH + 0.33 M urea, 1 M KOH + seawater and 1 M KOH + seawater + 0.33 M urea were used as the electrolyte in all electrochemical tests. The linear sweep voltammetry (LSV) polarization curves of HER were acquired at a scan rate of 2 mV s−1, and those of OER and UOR were obtained at a scan rate of 5 mV s−1. All of the potentials involved were based on the reversible hydrogen electrode (RHE): ERHE = EHg/HgO + 0.098 + 0.059 × pH. Electrochemical impedance spectroscopy (EIS) measurements were carried out in the frequency range from 0.1 Hz to 100 kHz with an amplitude of 5 mV. Cyclic voltammetry was performed at different scan rates (20, 40, 60, 80, 100 and 120 mV s−1) for the investigation of the electrochemical double layer capacitance (Cdl). The stability of (B,P,Co,Fe)-Ni@PW in different solutions was tested by chronoamperometric i–t measurement. Tafel slopes were obtained from the Tafel equation (η = blog(j/j0)). The terms η, b, j, and j0 denote the overpotential, Tafel slope, current density, and exchange current density, respectively. The overall water splitting performance of the assembled electrolyzer ((B,P,Co,Fe)-Ni@PW||(B,P,Co,Fe)-Ni@PW) in different solutions was tested. The gas produced from the electrolyzer driven by sunlight was collected by balloon for evaluating the catalytic performances. Inverted centrifuge tubes were used to collect hydrogen for quantitative analysis. The calculation method of the turnover frequency (TOF) is based on the equation: TOF = J/(2F × n), where “J” is the current density under a certain overpotential during the HER process, “F” is the Faraday constant (96 485 C mol−1) and “n” is the mole number of the active metal sites (mol cm−2). The factor 1/2 is presented because two electrons are required to form one hydrogen molecule. The n values were obtained by cyclic voltammetry (CV) in the potential range of 0–0.6 V with a fixed scan rate of 50 mV s−1. The mass activity was obtained by normalizing the potential at 1.40 V vs. RHE to the active mass.
Computational methods
All spin-polarized density functional theory (DFT) methods were performed using the Vienna ab initio simulation package (VASP) code with the projector augmented wave (PAW) method. The generalized gradient approximation (GGA) combined with Perdew–Burke–Ernzerhof (PBE) functional was employed to describe the exchange–correlation term. A 4 × 4 supercell of the Ni–P model contains 4 layers of Ni atoms. A 2 × 2 × 1 grid centered at the gamma (Γ) point was used to sample the Brillouin zone. The projector augmented wave (PAW) pseudo-potentials were used to describe the ionic cores. A vacuum space of 15 Å was added to avoid the interactions between the periodic slabs. The cutoff energy for the plane-wave basis was set to 450 eV. The van der Waals (vdW) interactions were described by using the empirical correction in Grimme's scheme (DFT-D3) in all calculations. The convergence tolerances for energy and force were set to 10−5 eV and 0.05 eV Å−1, respectively. The Gibbs free energy change (ΔG) for each elemental step was defined as:
In this equation, ΔEDFT denotes the electronic energy change that is directly obtained from the DFT calculations, and ΔEZPE and ΔS are the zero-point energy correction and entropy change obtained from frequency calculations at 298.15 K, respectively.
Results and discussion
Synthesis of (B,P,Co,Fe)-Ni@PW
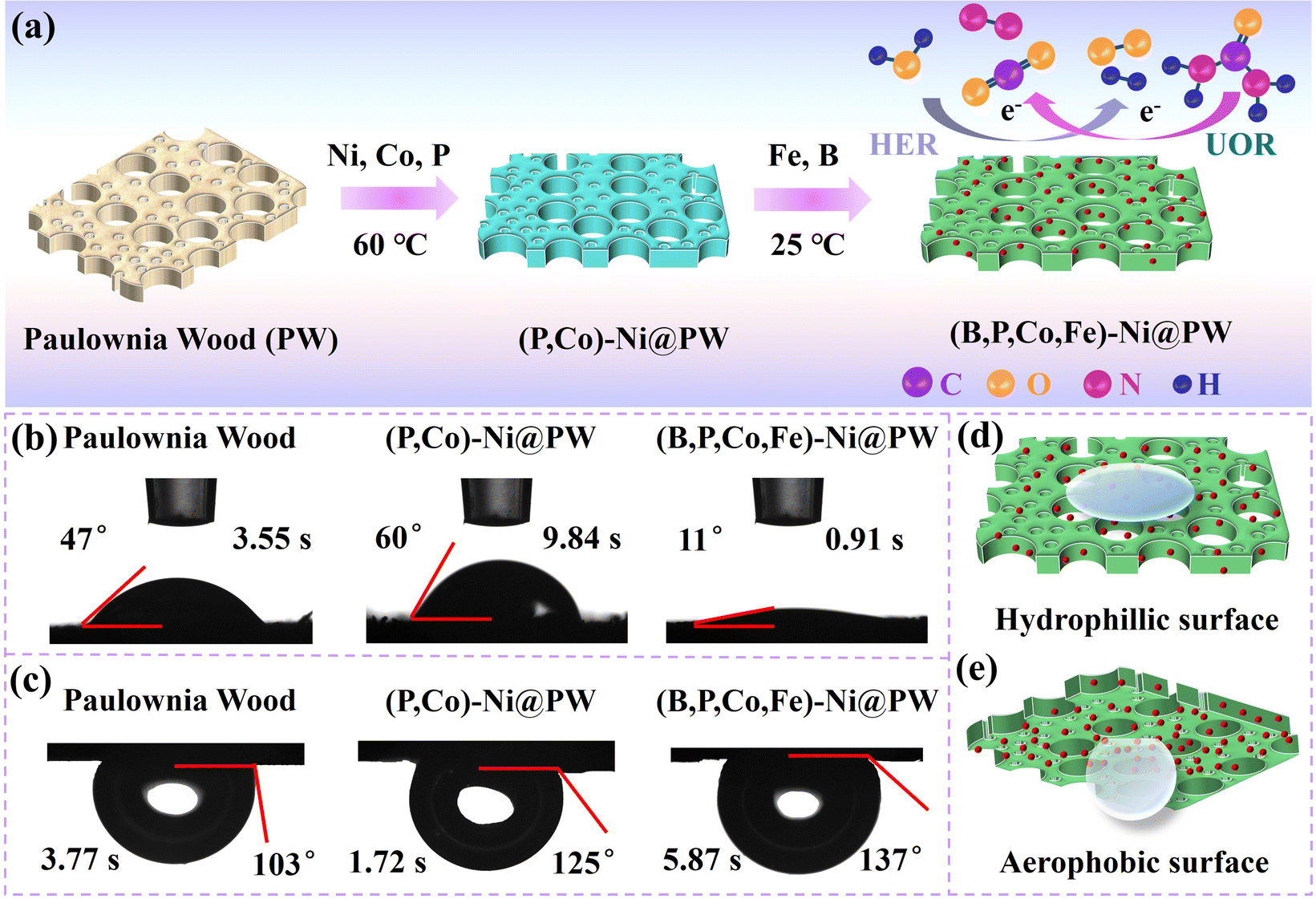
The schematic illustration for the preparation route of (B,P,Co,Fe)-Ni@PW is depicted in Fig. 1a. The natural PW is rich in pore structures and rough surface, with many direct channels along the direction of tree growth. Wood samples with dimensions of 10 mm × 15 mm were cut and immersed into NaBH4 solution. Subsequently, Ni doped with Co and P was grown on the surface of the wood and inside its microchannels via electroless plating. Finally, the Fe and B particles were introduced on (P,Co)-Ni@PW via subsequent electroless plating at room temperature. The sufficient infiltration of electrolyte on the catalyst surface is conducive to the acceleration of the reaction kinetics. During the UOR process, the timely removal of bubbles generated on the catalyst surface can prevent bubble gathering, enabling the adequate exposure of catalytic active sites.17,20 Therefore, constructing a hydrophilic and aerophobic structure is important to improve the UOR performances. As shown in Fig. 1b, (B,P,Co,Fe)-Ni@PW exhibits a smaller water contact angle of 11° than PW (47°) and (P,Co)-Ni@PW (60°), indicating the better hydrophilic behavior of (B,P,Co,Fe)-Ni@PW. This could facilitate sufficient contact between the active sites and reaction species. Fig. 1c demonstrates that (B,P,Co,Fe)-Ni@PW features excellent aerophobic behavior with an underwater bubble contact angle of 137°, higher than that of (P,Co)-Ni@PW (125°) and PW (103°). This is vital for the release of gas products generated on the catalyst surface at high current densities.34 The schematic diagram for water contact angles and bubble contact angle of (B,P,Co,Fe)-Ni@PW are shown in Fig. 1d and e. (B,P,Co,Fe)-Ni@PW has a hydrophilic and aerophobic structure, which may reduce the energy consumption of UOR/HER and improve the activity of the catalyst.
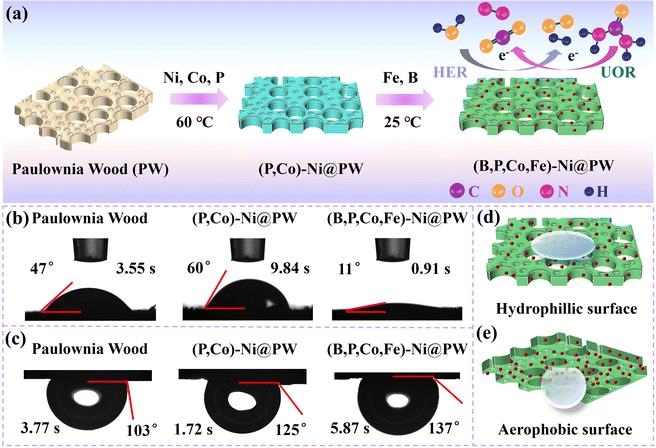 |
| | Fig. 1 (a) Schematic for the preparation process of (B,P,Co,Fe)-Ni@PW. (b) Water contact angle images and (c) bubble contact angles images of PW, (P,Co)-Ni@PW, and (B,P,Co,Fe)-Ni@PW. (d) Schematic for water contact angle and (e) bubble contact angle of (B,P,Co,Fe)-Ni@PW. | |
Morphology of (B,P,Co,Fe)-Ni@PW
The microstructures of PW, (P,Co)-Ni@PW, and (B,P,Co,Fe)-Ni@PW were characterized. As shown in Fig. 2a and Fig. S1,† PW shows interconnected porous structures and has a thin cell wall layer (0.4–0.8 μm), shortening the transmission distance of the electrolytes. Fig. 2b and Fig. S2† show that some microchannels in PW are aligned, which could facilitate the permeation of the electrolyte and the escape of bubbles.32 The natural rich pores and well-arranged microchannels of PW are also demonstrated in Fig. 2c. As shown in the cross and longitudinal sections of wood, the surface of natural PW is rough, which is conducive to anchoring metal ions. The rich pore structures increase the specific surface area, which allows for more active sites to be exposed. The Ni doped with Co, and P is deposited onto the channel walls of wood (Fig. 2d). The natural hierarchical porous structures and inherent toughness of wood are anticipated to serve as good electrode materials. The inset in Fig. 2d and Fig. S3† show that natural leaves are loaded with alloys, demonstrating the universality of the preparation method for various substrates. As shown in Fig. 2e, the cell wall of wood is covered by the (P,Co)-Ni layer, with a total thickness of approximately 3.38 μm. In order to build a strong electric field in the metal and enhance the catalytic performance, further introduction of Fe and B nanoparticles on (P,Co)-Ni@PW is proposed. Fig. 2f and Fig. S4, S5† show that the doping of Fe and B makes the surface of the catalyst rough, and then increases the specific surface area. Several peaks of elements are observed in the EDS spectrum in Fig. 5g. The elemental mapping images in Fig. 2h demonstrate the homogeneous distribution of Ni, Co, Fe, B, P, O and C elements in (B,P,Co,Fe)-Ni@PW.
 |
| | Fig. 2 (a–c) SEM images of PW. (d and e) SEM images of (P,Co)-Ni@PW. (f) SEM images of (B,P,Co,Fe)-Ni@PW. (g) EDS spectra of the Ni, Co, Fe, B, P, O and C elements. (h) EDS mappings images of the Ni, Co, Fe, B, P, O and C elements in (B,P,Co,Fe)-Ni@PW. | |
Fine structures of (B,P,Co,Fe)-Ni@PW
To interpret the structural information of the as-formed (B,P,Co,Fe)-Ni@PW samples, XRD measurements were conducted. As shown in Fig. S6,† the diffraction peaks belonging to cellulose of PW is located at about 20°. P–Ni@PW, (P,Co)-Ni@PW and (B,P,Co,Fe)-Ni@PW all show the peaks of Ni (PDF#04-0850) in Fig. S7† and Fig. 3a. The wide peaks indicate the existence of the nanocrystalline form in the sample. Compared with the peaks of standard Ni, all of those peaks show a positive shift, indicating that the doping of heteroatoms causes the lattice constant of nickel to decrease. The decrease of the catalyst lattice constant will lead to the increase of lattice defects, the refinement of grains, and the increase of active sites, thus improving the UOR activity.28 The enlarged dotted box in Fig. 3c clearly shows the lattice distance of 0.20 nm, corresponding to the (111) plane for Ni.35 As shown in Fig. S8,† the HRTEM images show that there are many small-size nanoparticles in the catalyst, which is beneficial to promote the activity. To further probe the composition of (P,Co)-Ni@PW and (B,P,Co,Fe)-Ni@PW, XPS measurements were performed to analyze the near-surface properties. Fig. 3d shows the coexistence of (P,Co)-Ni@PW and (B,P,Co,Fe)-Ni@PW. The high-resolution C 1s spectra show the C–C, C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds (Fig. S9†). The peaks of (P,Co)-Ni@PW at 855.9 and 873.7 eV are attributed to Ni 2p3/2 and Ni 2p1/2, respectively, indicating the presence of Ni2+, along with two satellite peaks at 861.6 and 879.8 eV.10 In addition, the signal of Ni0 is at the binding energy of 852.6 eV. Notably, for (B,P,Co,Fe)-Ni@PW, there is a 0.2 eV positive binding energy shift relative to that of (P,Co)-Ni@PW (Fig. 3e). These peak shifts indicate that the co-doping of the nonmetal and metal synergistically modifies the electronic structure, which is conducive to achieving the distribution of charge to enhance the activity of UOR/HER.36,37 Furthermore, the Ni0 peak is not detected in (B,P,Co,Fe)-Ni@PW, indicating that Ni0 was replaced during the doping of Fe and B. The high-resolution O 1s spectra show the metal–O and C–O bond (Fig. S10†).38 The atomic ratios of Ni (88.50%), Co (2.02%) and P (9.48%) in (P,Co)-Ni@PW were calculated by the inductively coupled plasma-optical emission spectrometer (ICP-OES) analysis in Fig. 3f. Furthermore, the atomic ratios of Ni, Co, Fe, P, and B in (B,P,Co,Fe)-Ni@PW are 87.86%, 1.89%, 0.17%, 9.30% and 0.78%, respectively.
O bonds (Fig. S9†). The peaks of (P,Co)-Ni@PW at 855.9 and 873.7 eV are attributed to Ni 2p3/2 and Ni 2p1/2, respectively, indicating the presence of Ni2+, along with two satellite peaks at 861.6 and 879.8 eV.10 In addition, the signal of Ni0 is at the binding energy of 852.6 eV. Notably, for (B,P,Co,Fe)-Ni@PW, there is a 0.2 eV positive binding energy shift relative to that of (P,Co)-Ni@PW (Fig. 3e). These peak shifts indicate that the co-doping of the nonmetal and metal synergistically modifies the electronic structure, which is conducive to achieving the distribution of charge to enhance the activity of UOR/HER.36,37 Furthermore, the Ni0 peak is not detected in (B,P,Co,Fe)-Ni@PW, indicating that Ni0 was replaced during the doping of Fe and B. The high-resolution O 1s spectra show the metal–O and C–O bond (Fig. S10†).38 The atomic ratios of Ni (88.50%), Co (2.02%) and P (9.48%) in (P,Co)-Ni@PW were calculated by the inductively coupled plasma-optical emission spectrometer (ICP-OES) analysis in Fig. 3f. Furthermore, the atomic ratios of Ni, Co, Fe, P, and B in (B,P,Co,Fe)-Ni@PW are 87.86%, 1.89%, 0.17%, 9.30% and 0.78%, respectively.
 |
| | Fig. 3 (a) XRD patterns of (P,Co)-Ni@PW and (B,P,Co,Fe)-Ni@PW. (b and c) HRTEM images of (B,P,Co,Fe)-Ni@PW. (d) XPS survey spectra of (P,Co)-Ni@PW and (B,P,Co,Fe)-Ni@PW. (e) High-resolution XPS spectra of (P,Co)-Ni@PW and (B,P,Co,Fe)-Ni@PW for Ni 2p. (f) Atomic ratios of (P,Co)-Ni@PW and (B,P,Co,Fe)-Ni@PW. | |
UOR, HER and OER performances of (B,P,Co,Fe)-Ni@PW
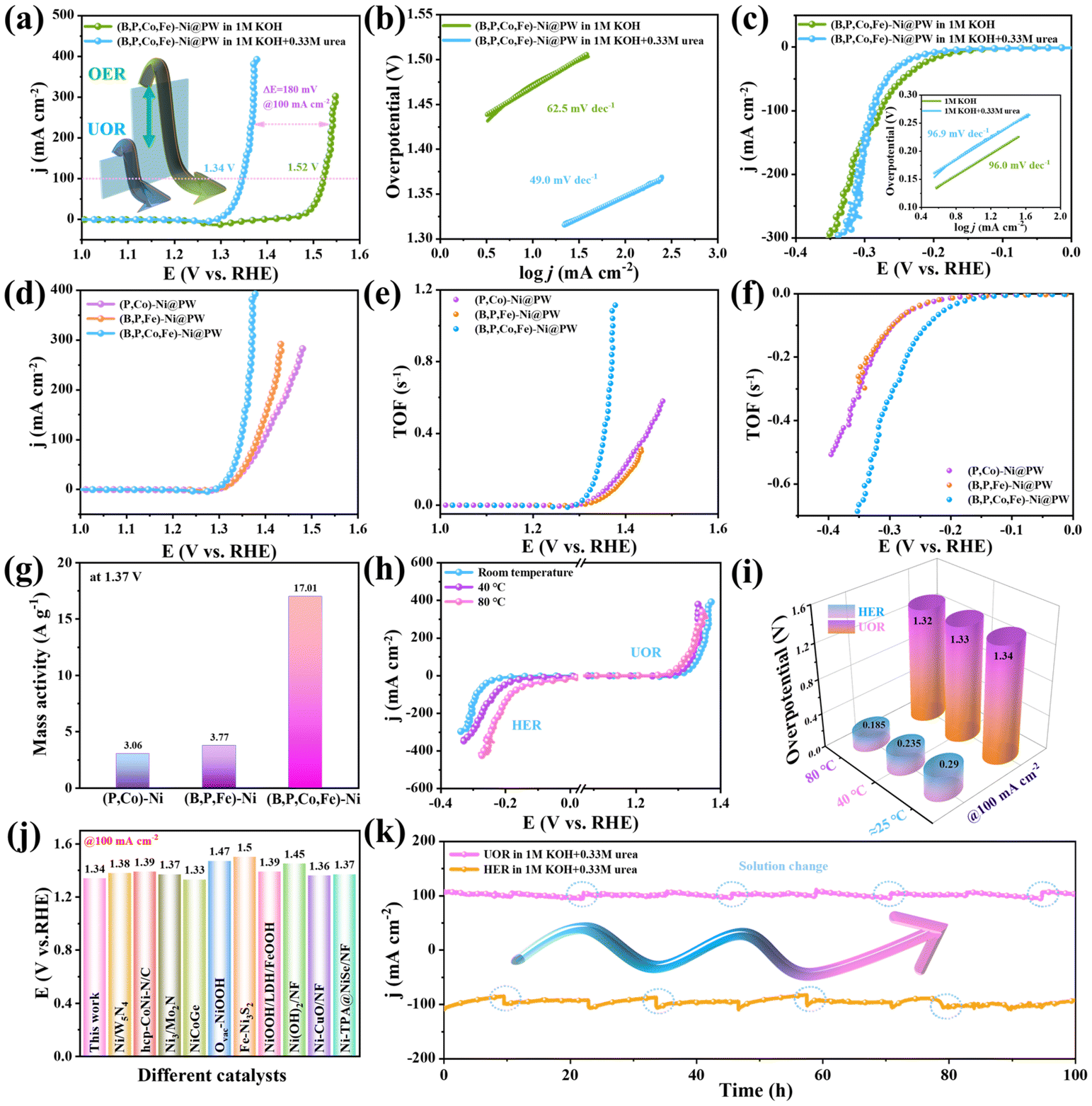
The UOR activity of (B,P,Co,Fe)-Ni@PW was primarily evaluated in 1.0 M KOH with 0.33 M urea. As presented in Fig. 4a, (B,P,Co,Fe)-Ni@PW shows a UOR onset potential of 1.30 V, which is significantly lower than that of OER (1.47 V). To achieve a large current density of 100 mA cm−2, the UOR potential for (B,P,Co,Fe)-Ni@PW is only 1.34 V, while the OER requires a potential of 1.52 V. This result proves that the potential is greatly reduced by replacing OER with UOR, thereby reducing the energy consumption. To explore the reaction kinetics of the catalysts, Fig. 4b displays the Tafel slopes based on the UOR and OER polarization curves. (B,P,Co,Fe)-Ni@PW shows a Tafel slope of 49.0 mV dec−1 for UOR, which is lower than that for OER (62.5 mV dec−1), suggesting the superior catalytic kinetics of UOR. The charge transfer resistance (Rct) obtained from the Nyquist plot is related to the UOR kinetics at the catalyst/electrolyte interface.39 (B,P,Co,Fe)-Ni@PW shows a charge transfer resistance of 0.4 Ω for UOR, which is lower than that for OER with 1.4 Ω (Fig. S11†). As shown in Fig. 4c, (B,P,Co,Fe)-Ni@PW holds a HER overpotential of 290 mV at 100 mA cm−2 in 1.0 M KOH + 0.33 M urea, which is slightly higher than that in 1.0 M KOH (278 mV). To attain a higher current density of over 140 mA cm−2, a smaller overpotential in 1.0 M KOH + 0.33 M urea is required in comparison with that in only 1.0 M KOH, demonstrating the good HER activity of (B,P,Co,Fe)-Ni@PW in a wide potential range. (B,P,Co,Fe)-Ni@PW exhibits similar kinetics for HER in 1.0 M KOH + 0.33 M urea (96.9 mV dec−1) and 1.0 M KOH (96.0 mV dec−1), respectively. In addition, the semicircle diameter of EIS in Fig. S12† is similar. Therefore, the concomitant urea contained in the electrolyte shows a negligible impact on the HER activity, suggesting a good prospect of (B,P,Co,Fe)-Ni@PW to promote H2 production in the HER||UOR coupled system. The reaction mass ratio of Fe to B is 0.3:0.2 and the reaction time is set to 2 h as the optimal reaction conditions, thus preparing an efficient electrode with excellent bifunctional UOR and HER activities (Fig. S13–S18†).
 |
| | Fig. 4 (a) Polarization curves for OER and UOR of (B,P,Co,Fe)-Ni@PW with and without 0.33 M urea. (b) The corresponding Tafel slopes. (c) HER polarization curves of (B,P,Co,Fe)-Ni@PW with and without 0.33 M urea. (d) TOF values for UOR. (e) TOF values for HER. (f) Mass activity at 1.37 V (vs. RHE). (g) UOR polarization curves of (P,Co)-Ni@PW, (B,P,Fe)-Ni@PW and (B,P,Co,Fe)-Ni@PW. (h) HER and UOR performance of (B,P,Co,Fe)-Ni@PW at different temperatures. (i) Comparison of the HER and UOR performance. (j) Comparison of the potentials needed at 100 mA cm−2 for (B,P,Co,Fe)-Ni@PW and recently reported UOR catalysts. (k) Chronoamperometry curves of (B,P,Co,Fe)-Ni@PW for HER and UOR. | |
Activity analysis of (P,Co)-Ni@PW, (B,P,Fe)-Ni@PW and (B,P,Co,Fe)-Ni@PW
The performance comparisons of (P,Co)-Ni@PW, (B,P,Fe)-Ni@PW, and (B,P,Co,Fe)-Ni@PW are displayed in Fig. 4d and Fig. S19.† The performances of (B,P,Fe)-Ni@PW and (B,P,Co,Fe)-Ni@PW are significantly better than that of (P,Co)-Ni@PW, indicating that the double doping of Fe and B significantly enhances the catalytic activities of UOR and HER.40 Moreover, Co doped in Ni further improves the catalytic efficiency. The Tafel slopes also match the HER and UOR activities of (P,Co)-Ni@PW, (B,P,Fe)-Ni@PW and (B,P,Co,Fe)-Ni@PW in Fig. S20 and S21,† demonstrating that (B,P,Co,Fe)-Ni@PW can accelerate electron transfer and thus boost the reaction kinetics.17 Additionally, the double-layer capacitance (Cdl), which is proportional to the electrochemically active surface area (ECSA) of the samples, was measured by cyclic voltammetry (Fig. S22–S25†). The Cdl value of (B,P,Co,Fe)-Ni@PW (5.18 mF cm−2) is larger than that of (B, P, Fe)-Ni@PW (3.60 mF cm−2) and (P,Co)-Ni@PW (3.56 mF cm−2), which demonstrates that (B,P,Co,Fe)-Ni@PW could expose more active sites. The turnover frequency (TOF) was used to evaluate the intrinsic catalytic activity.22 As shown in Fig. 4e and f, the TOF values of (B,P,Co,Fe)-Ni@PW at different potentials are higher than those of (B,P,Fe)-Ni@PW and (P,Co)-Ni@PW, respectively, revealing the better intrinsic catalytic activity. The mass activity at a potential of 1.37 V was calculated in Fig. 4g. (B,P,Co,Fe)-Ni@PW shows superior intrinsic activity compared to that of (P,Co)-Ni@PW and (B,P,Fe)-Ni@PW. The co-doping of metal and nonmetal can not only increase the accessible active sites, but also synergistically optimize the electronic structure of the catalyst.13 During the UOR process, the surface electronic structure has a significant impact on the adsorption/activation behaviour of the reaction species, consequently determining the inherent catalytic activity. The external environmental effects on the performance of the catalysts were evaluated by changing the electrolyte temperature. Three electrochemical systems were designed with different electrolyte temperatures (Room temperature = 25, 40 and 80 °C) to investigate the HER and UOR activities of (B,P,Co,Fe)-Ni@PW. As shown in Fig. 4h, the η value gradually decreased as the electrolyte temperature increased. Fig. 4i shows that the overpotential is as low as 185 mV for HER and the potential is 1.32 V for UOR to achieve 100 mA cm−2, smaller than those at 25 °C (290 mV and 1.34 V), and 40 °C (235 mV and 1.33 V), respectively. Compared with recently reported transition metal-based electrocatalysts (Fig. 4j and Table S1†), (B,P,Co,Fe)-Ni@PW exhibits superior UOR activity in alkaline urea solution.8,10,19,21,41–46
Stability analysis of (B,P,Co,Fe)-Ni@PW
Stability is an important factor for evaluating the performance of a catalyst. As shown in Fig. 4k, (B,P,Co,Fe)-Ni@PW demonstrates long stability for 100 h during UOR and HER at about 100 mA cm−2. The SEM analyses show that (B,P,Co,Fe)-Ni@PW maintains the initial structure after a long-term cycle (Fig. S26 and 27†). After being stable for 100 h, the CV curves were tested to determine the activity change of the catalyst. As shown in Fig. S28,† the areas of the CV curves did not exhibit any obvious change (only a slight decline) in comparison with the initial ones (Fig. S24†), indicating that the long-term cycling retains the activity of the catalyst, further demonstrating that the structure of the catalyst was not destroyed. The good stability of the self-supported electrode at a high current density is not only due to the stability of the elaborate structure, but also a result of the hierarchical porous structures and three-dimensional well-aligned micro-channel of wood. The natural organic framework of wood shortens the transport path of the electrolyte, ions and active intermediates, maintaining the high activity. Importantly, the porous structure of wood limits the growth of bubbles adsorbed on the electrode, accelerates the rupture and release of bubbles, and avoids the reduction of catalytic efficiency resulting from the blocking of active sites during the long-term catalytic process.
Performances of the overall urea-water splitting system
Taking into account the excellent electrochemical performances of (B,P,Co,Fe)-Ni@PW toward UOR in an alkaline environment, the overall urea–water splitting system was implemented in a 1.0 M KOH + 0.33 M urea aqueous solution with a two-electrode configuration using the same bifunctional electrode as both cathode and anode (Fig. 5a). The charge-polarized surface is generated by the charge redistribution caused by multi-component doping, which can adjust the surface electronic structure of the host catalyst. The optimization of the electronic structure can improve the adsorption capacity of urea molecules and induce decomposition to produce CO2 and N2. Notably, the hydrophilic and aerophobic wood-based electrode facilitates the permeation of the electrolyte and the overflow of gas generated on the surface, improving the catalytic efficiency and reducing energy consumption. As shown in Fig. 5b, the entire (B,P,Co,Fe)-Ni@PW||(B,P,Co,Fe)-Ni@PW system demonstrates the superiority of the anodic UOR compared to conventional water splitting. To attain 10 and 100 mA cm−2, the voltages of (B,P,Co,Fe)-Ni@PW||(B,P,Co,Fe)-Ni@PW are 1.48 and 1.66 V, respectively. This signifies that (B,P,Co,Fe)-Ni@PW||(B,P,Co,Fe)-Ni@PW is a promising candidate for overall urea–water electrolysis in alkaline conditions. The influences of seawater and urea on the electrolyzer performances were evaluated in electrolytes including 1 M KOH, 1 M KOH + seawater, and 1 M KOH + 0.33 M urea + seawater. The voltages to attain 100 mA cm−2 are 1.89 V (1 M KOH), 1.98 V (1 M KOH + seawater), and 1.67 V (1 M KOH + 0.33 M urea + seawater), respectively. The composition of seawater is complex, including an abundance of Mg2+, Ca2+ and various microorganisms. In particular, the formed Mg(OH)2 and Ca(OH)2 in the alkaline electrolyte may partially cover the surface of (B,P,Co,Fe)-Ni@PW, resulting in the reduced activity. The voltage differences between the HER||UOR and HER||OER are gradually expanded with increasing current densities. Due to the abundance of seawater and the shortage of freshwater on the earth, the ability to use seawater instead of freshwater as an electrolyte is significant. The performance comparison results show that the UOR-assisted seawater splitting improve the efficiency of hydrogen production. As shown in Fig. 5c, the Tafel slopes of 244.7, 192.8, 263.6, and 208.6 mV dec−1 are attained in the electrolytes containing 1 M KOH, 1 M KOH + 0.33 M urea, 1 M KOH + seawater, and 1 M KOH + 0.33 M urea + seawater, respectively. The two-electrode HER||UOR electrolyzer in 1 M KOH + 0.33 M urea + seawater exhibits a faster electron transfer rate and superior catalytic kinetics. Impressively, the as-assembled system shows high stability for 200 h at a large current density of 100 mA cm−2 (Fig. 5d). By comparing the volume of the final balloons in different solutions at the same time, the amount of gas collected by the balloon for HER||UOR is larger than that of HER||OER (Fig. 5e and g). Fig. S29 and S30† display the initial state of the balloon in the device. After several hours of sunlight, the energy-efficient hydrogen production can be achieved with the assistance of UOR. As depicted in Fig. 5f, the electrode surface of HER||UOR has more bubbles than that of HER||OER. In addition, the H-type electrolyzer is used to separate the anode and cathode gas, and a simple overall (urea-)water splitting device was assembled and driven by two batteries (each is 1.5 V). The gases of the cathode and anode were collected at the same time for comparison in Fig. S31.† After 20 min, the HER||OER and HER||UOR electrolyzers collected 8.5 and 11.5 mL of hydrogen, respectively, demonstrating that UOR improved the efficiency of the overall water splitting process.
 |
| | Fig. 5 (a) Schematic of the urea-assisted water electrolyzer using (B,P,Co,Fe)-Ni@PW as both anode and cathode catalysts. (b) Polarization curves of the (B,P,Co,Fe)-Ni@PW electrodes in HER||UOR and HER||OER coupled systems in different electrolytes. (c) The corresponding Tafel slopes. (d) Chronoamperometry curves of the (B,P,Co,Fe)-Ni@PW||(B,P,Co,Fe)-Ni@PW. Photographs showing the electrolyzer driven by solar panels illuminated by sunlight of (e) 1 M KOH + 0.33 M urea, (f) gas generated, and (g) 1 M KOH. | |
DFT calculations
Density functional theory (DFT) calculations were employed to confirm the optimization of the electronic structure caused by doping and the synergistic effect between multiple active components to enhance the UOR performance. In the design and synthesis of (B,P,Co,Fe)-Ni@PW, (P,Co)-Ni@PW was formed by electroless plating in the first step. Co and P were incorporated into the lattice of Ni through the oxidization/reduction reaction, which was consistent with the synthesis of P-Ni@PW. Fig. 6a and Fig. S33† show the built models of P–Ni, (P,Co)-Ni and (B,P,Co,Fe)-Ni (111). The charge density differences of P–Ni, (P,Co)-Ni and (B,P,Co,Fe)-Ni are displayed in Fig. 6b and Fig. S34.† There are obvious charge accumulation regions and charge depletion regions around the urea molecule. The change of the region reveals the redistribution of charge density caused by doping, adjusting the adsorption energy of the oxygen-containing intermediates.43 As shown in Fig. 6c, the adsorption energy of the urea molecule on P–Ni, (P,Co)-Ni and (B,P,Co,Fe)-Ni is −1.15, −1.20 and −1.25 eV, respectively. The introduction of Co improves the adsorption of the urea molecule on P–Ni, and the further introduction of Fe and B achieves the synergistic effect of multiple components, thereby optimizing the adsorption energy. The improvement of the adsorption capacity of urea molecules promotes the UOR process and inhibits the OER reaction.47 The Gibbs free energy was calculated to reveal the UOR mechanism. As displayed in Fig. 6d and 6e, the rate-determining step (RDS) for P–Ni, (P,Co)-Ni and (B,P,Co,Fe)-Ni is the dehydrogenation step from *CO(NH·N) to *CO(N·N). The incorporation of Co regulates the adsorption/desorption behavior of the urea reaction intermediates and reduce the free energy (3.28 eV → 2.68 eV) of the rate-determining step (RDS). In the second step of electroless plating, the incorporation of Fe and B further improves the adsorption/desorption capacity and reduces the energy barrier of RDS (2.68 eV → 2.09 eV). Co, Fe, P, and B co-doping in Ni effectively optimizes the electronic structure, and the adsorption/desorption behavior of the urea reaction intermediates is regulated by the synergistic effect from multiple components, resulting in the excellent catalytic activity in seawater-urea media. The typical UOR pathways involve the listed steps: *CO(NH2)2 → *CO(NH2·NH) → *CO(NH2·N) → *CO(NH·N) → *CO(N·N) → *CO + N2 → *CO·OH → *CO·O → CO2.9 Firstly, urea molecules are adsorbed on the surface of the catalyst, and then four H atoms are removed to form *CO(N·N) in Fig. 6f and Fig. S34.† Secondly, the strong C–N bond is broken, and the N–N bond is coupled to form N2 under the action of the applied voltage and then escapes. Finally, the residues of C–O are penetrated by OH-, forming the *CO·OH structure and then forming CO2.48 The above results demonstrate that doping can effectively adjust the electronic structure, and the synergistic effect between multiple components optimizes the adsorption/desorption of the urea reaction intermediates, thereby improving the intrinsic activity of UOR.
 |
| | Fig. 6 (a) DFT-optimized structure model of (B,P,Co,Fe)-Ni@PW. (b) Charge density difference for (B,P,Co,Fe)-Ni@PW. (c) Adsorption energy of CO(NH2)2*. (d) Free energy diagram of the UOR on the P–Ni, (P,Co)-Ni@PW and (B,P,Co,Fe)-Ni@PW. (e) ΔG diagram of P–Ni, (P,Co)-Ni@PW and (B,P,Co,Fe)-Ni@PW. (f) Schematic of the proposed UOR mechanism. | |
Conclusions
Heteroatom doping in (B,P,Co,Fe)-Ni regulates the surface electronic structure of the catalyst to promote the adsorption/desorption of the reaction intermediates. The electron density of the active sites is optimized via doping, accelerating electron transfer. Benefiting from the directional channels and nanopores in wood frameworks, the diffusion of the electrolyte, ions, and electrons is accelerated and the overflow of bubbles is promoted. Due to the optimized electronic structures, the synergistic effect of multiple active centers, well-aligned channels, good hydrophilic and aerophobic properties, (B,P,Co,Fe)-Ni@PW exhibits outstanding electrocatalytic activities for UOR, HER and urea-assisted overall seawater splitting in alkaline medium. The (B,P,Co,Fe)-Ni@PW electrode shows excellent activities for UOR and HER in 1 M KOH + 0.33 M urea, with a potential of 1.34 V and an overpotential of 290 mV at 100 mA cm−2, respectively. The assembled seawater-urea electrolyzer ((B,P,Co,Fe)-Ni@PW||(B,P,Co,Fe)-Ni@PW) exhibits a low cell voltage of 1.67 V to obtain 100 mA cm−2, providing a cost-effective and high-efficiency strategy for energy-saving hydrogen production. This work proposes a high-performance and low-cost wood-based UOR/HER electrocatalyst through the regulation of the electronic structure of catalytic sites for producing green hydrogen and realizing hydrogen economy.
Author contributions
Hongjiao Chen: writing original draft, data curation, investigation, formal analysis. Kewei Zhang: data curation, resources. Yanzhi Xia: investigation, resources. Jian Li: data curation, methodology. Bin Hui: supervision, conceptualization, methodology, formal analysis, resources, funding acquisition, writing – review & editing.
Data availability
All the data supporting the findings of this work are available within the article and its ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Excellent Youth Foundation of Shandong Province (No. ZR2022YQ22), National Natural Science Foundation of China (No. 32101451), and Youth Innovation Team Project of Shandong Province (No. 2022KJ303).
References
- D. R. Paudel, U. N. Pan, R. B. Ghising, P. P. Dhakal, V. A. Dinh, H. Wang, N. H. Kim and J. H. Lee, Nano Energy, 2022, 102, 107712 CrossRef CAS.
- C. Rong, X. Shen, Y. Wang, L. Thomsen, T. Zhao, Y. Li, X. Lu, R. Amal and C. Zhao, Adv. Mater., 2022, 34, 2110103 CrossRef CAS PubMed.
- K. Shah, R. Dai, M. Mateen, Z. Hassan, Z. Zhuang, C. Liu, M. Israr, W. C. Cheong, B. Hu, R. Tu, C. Zhang, X. Chen, Q. Peng, C. Chen and Y. Li, Angew. Chem., Int. Ed., 2021, 61, e202114951 CrossRef PubMed.
- H. Jiang, M. Sun, S. Wu, B. Huang, C. S. Lee and W. Zhang, Adv. Funct. Mater., 2021, 31, 2104951 CrossRef CAS.
- T. Wang, P. Wang, W. Zang, X. Li, D. Chen, Z. Kou, S. Mu and J. Wang, Adv. Funct. Mater., 2021, 32, 2107382 CrossRef.
- F. N. Indah Sari, S. Marsaor Sihotang, S.-Y. Li, Y.-H. Shen and J.-M. Ting, ACS Sustainable Chem. Eng., 2023, 11, 1207–1220 CrossRef CAS.
- D. Khalafallah, W. Huang, M. Zhi and Z. Hong, Energy Environ. Mater., 2023, 2, e12528 Search PubMed.
- H. Sun, J. Liu, H. Kim, S. Song, L. Fei, Z. Hu, H. J. Lin, C. T. Chen, F. Ciucci and W. Jung, Adv. Sci., 2022, 9, 2204800 CrossRef CAS PubMed.
- X. Zheng, J. Yang, P. Li, Z. Jiang, P. Zhu, Q. Wang, J. Wu, E. Zhang, W. Sun, S. Dou, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202217449 CrossRef CAS.
- Y. Zhou, B. Chu, Z. Sun, L. Dong, F. Wang, B. Li, M. Fan and Z. Chen, Appl. Catal., B, 2023, 323, 122168 CrossRef CAS.
- L. Guo, J. Chi, J. Zhu, T. Cui, J. Lai and L. Wang, Appl. Catal., B, 2023, 320, 121977 CrossRef CAS.
- Z. Yu, Y. Li, V. Martin-Diaconescu, L. Simonelli, J. R. Esquius, I. Amorim, A. Araujo, L. Meng, J. L. Faria and L. Liu, Adv. Funct. Mater., 2022, 32, 2206138 CrossRef CAS.
- W. K. Han, J. X. Wei, K. Xiao, T. Ouyang, X. Peng, S. Zhao and Z. Q. Liu, Angew. Chem., Int. Ed., 2022, 61, e202206050 CrossRef CAS.
- H. Sun, J. Liu, G. Chen, H. Kim, S. Kim, Z. Hu, J.-M. Chen, S.-C. Haw, F. Ciucci and W. Jung, Small Methods, 2022, 6, 2101017 CrossRef CAS PubMed.
- C. Pei, S. Chen, T. Zhao, M. Li, Z. Cui, B. Sun, S. Hu, S. Lan, H. Hahn and T. Feng, Adv. Mater., 2022, 34, 2200850 CrossRef CAS.
- L. Wang, S. Zhu, Y. Wang, Z. Liu, Y. Liu, Q. Wang, M. Gu, K. Li, X. Sun, L. Yang and M. Shao, Chem. Eng. J., 2023, 460, 141826 CrossRef CAS.
- P. Li, W. Li, Y. Huang, Q. Huang and S. Tian, Small, 2023, 19, 2300725 CrossRef CAS PubMed.
- Y. Wang, Y. Liu, M. Zhang, B. Liu, Z. Zhao and K. Yan, Sci. China Mater., 2022, 65, 1805–1813 CrossRef CAS.
- P. Wang, X. Bai, H. Jin, X. Gao, K. Davey, Y. Zheng, Y. Jiao and S. Z. Qiao, Adv. Funct. Mater., 2023, 33, 2300687 CrossRef CAS.
- J. Li, X. Xu, X. Hou, S. Zhang, G. Su, W. Tian, H. Wang, M. Huang and A. Toghan, Nano Res., 2023, 16, 8853–8862 CrossRef CAS.
- T. Wang, L. Miao, S. Zheng, H. Qin, X. Cao, L. Yang and L. Jiao, ACS Catal., 2023, 13, 4091–4100 CrossRef CAS.
- X. Xu, C. Zhang, J. Li, H. Liu, G. Su, Z. Shi and M. Huang, Chem. Eng. J., 2023, 452, 139362 CrossRef CAS.
- Z. Li, X. Zhang, Y. Kang, C. C. Yu, Y. Wen, M. Hu, D. Meng, W. Song and Y. Yang, Adv. Sci., 2021, 8, 2002631 CrossRef CAS PubMed.
- M. Liu, K.-A. Min, B. Han and L. Y. S. Lee, Adv. Energy Mater., 2021, 11, 2101281 CrossRef CAS.
- T. Wang, L. Chen, C. Chen, M. Huang, Y. Huang, S. Liu and B. Li, ACS Nano, 2022, 16, 2306–2318 CrossRef CAS PubMed.
- X. Qin, B. Yan, D. Kim, Z. Teng, T. Chen, J. Choi, L. Xu and Y. Piao, Appl. Catal., B, 2022, 304, 120923 CrossRef CAS.
- Y. Wang, Y. Xue and C. Zhang, Small, 2021, 17, 2006587 CrossRef CAS PubMed.
- H. Cheng, Y. Pan, X. Wang, C. Liu, C. Shen, D. W. Schubert, Z. Guo and X. Liu, Nano-Micro Lett., 2022, 14, 63 CrossRef CAS.
- H. Zhang, G. Zhang, Q. Gao, M. Tang, Z. Ma, J. Qin, M. Wang and J.-K. Kim, Chem. Eng. J., 2020, 379, 122304 CrossRef.
- B. Hui, J. Li, Y. Lu, K. Zhang, H. Chen, D. Yang, L. Cai and Z. Huang, J. Eng. Chem., 2021, 56, 23–33 CAS.
- J. Kang, F. Yang, C. Sheng, H. Xu, J. Wang, Y. Qing, Y. Wu and X. Lu, Small, 2022, 18, 2200950 CrossRef CAS.
- H. Chen, Y. Zou, J. Li, K. Zhang, Y. Xia, B. Hui and D. Yang, Appl. Catal., B, 2021, 293, 120215 CrossRef CAS.
- B. Hui, K. Zhang, Y. Xia and C. Zhou, Electrochim. Acta, 2020, 330, 135274 CrossRef CAS.
- Y. Liao, S. Deng, Y. Qing, H. Xu, C. Tian and Y. Wu, J. Eng. Chem., 2023, 76, 566–575 CAS.
- C. Fu, J. Fan, Y. Zhang, H. Lv, D. Ji and W. Hao, J. Colloid Interface Sci., 2023, 634, 804–816 CrossRef CAS PubMed.
- J. Wu, Z. Zhai, S. Yin and S. Wang, Adv. Funct. Mater., 2023, 202308198, DOI:10.1002/adfm.202308198.
- M. Pan, G. Qian, T. Yu, J. Chen, L. Luo, Y. Zou and S. Yin, Chem. Eng. J., 2022, 435, 134986 CrossRef CAS.
- H. Chen and B. Hui, Int. J. Hydrogen Energy, 2023, 8, 279 Search PubMed.
- R. Lin, L. Kang, T. Zhao, J. Feng, V. Celorrio, G. Zhang, G. Cibin, A. Kucernak, D. J. L. Brett, F. Cora, I. P. Parkin and G. He, Energy Environ. Sci., 2022, 15, 2386–2396 RSC.
- X. Zheng, J. Yang, P. Li, Z. Jiang, P. Zhu, Q. Wang, J. Wu, E. Zhang, W. Sun, S. Dou, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202217449 CrossRef CAS PubMed.
- L. Jin, R. Ji, H. Wan, J. He, P. Gu, H. Lin, Q. Xu and J. Lu, ACS Catal., 2023, 13, 837–847 CrossRef CAS.
- M. Cai, Q. Zhu, X. Wang, Z. Shao, L. Yao, H. Zeng, X. Wu, J. Chen, K. Huang and S. Feng, Adv. Mater., 2022, 35, 2209338 CrossRef.
- D. Li, W. Wan, Z. Wang, H. Wu, S. Wu, T. Jiang, G. Cai, C. Jiang and F. Ren, Adv. Energy Mater., 2022, 12, 2201913 CrossRef CAS.
- P. Li, Y. Huang, X. Ouyang, W. Li, F. Li and S. Tian, Chem. Eng. J., 2023, 464, 142570 CrossRef CAS.
- H. Qin, Y. Ye, J. Li, W. Jia, S. Zheng, X. Cao, G. Lin and L. Jiao, Adv. Funct. Mater., 2022, 33, 2209698 CrossRef.
- S. W. Tatarchuk, J. J. Medvedev, F. Li, Y. Tobolovskaya and A. Klinkova, Angew. Chem., Int. Ed., 2022, 61, e202209839 CrossRef CAS PubMed.
- P. Guo, S. Cao, W. Huang, X. Lu, W. Chen, Y. Zhang, Y. Wang, X. Xin, R. Zou, S. Liu and X. Li, Adv. Mater., 2024, 36, 2311766 CrossRef CAS.
- M. Liu, W. Zou, S. Qiu, N. Su, J. Cong and L. Hou, Adv. Funct. Mater., 2023, 34, 2310155 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Yanzhi
Xia
a,
Jian
Li
a,
Yanzhi
Xia
a,
Jian
Li