
Crystalline/amorphous c-NiMo/a-NiMoOx nanoarrays for urea-assisted energy-saving H2 production in alkaline seawater†
Dongxue
Guo
 *a,
Yi
Ping
b,
Chuanjiao
Wang
b,
Changan
Hou
b and
Danhong
Wang
*b
*a,
Yi
Ping
b,
Chuanjiao
Wang
b,
Changan
Hou
b and
Danhong
Wang
*b
aAcademy of National Food and Strategic Reserves Administration, Beijing 100037, China. E-mail: gdx@ags.ac.cn
bTKL of Metal and Molecule Based Material Chemistry, School of Materials Science and Engineering, Nankai University, Tianjin 300350, China. E-mail: dhwang@nankai.edu.cn
First published on 6th November 2024
Abstract
Electrocatalytic seawater splitting is regarded as the most effective method for producing green hydrogen (H2), but it faces issues of high energy consumption and harmful chlorine evolution side reactions. Replacing the sluggish oxygen evolution reaction (OER) with the thermodynamically favorable urea oxidation reaction (UOR) would enable energy-saving and chlorine-free H2 production. Herein, a novel three-dimensional (3D) structured electrocatalyst (c-MoNi/a-NiMoOx) with crystalline MoNi alloy clusters coupled with amorphous NiMoOx nanowires is reported. In the hydrogen evolution reaction (HER) process, the electron redistribution at the crystalline/amorphous interface could effectively regulate the electronic structure, thereby optimizing the Gibbs free energy of water dissociation and hydrogen adsorption. In the UOR process, c-MoNi/a-NiMoOx undergoes surface reconstruction to form highly active β-NiMoOOH. The incorporation of Mo lowers the activation energy barrier of the rate-determining step, thus facilitating the progression of the multi-step UOR process. Excitingly, the urea-assisted seawater electrolysis based on c-MoNi/a-NiMoOx requires an ultralow voltage of 1.68 V to deliver 500 mA cm−2, and displays distinguished long-term stability to keep above 100 mA cm−2 for 300 h. This work may show practical impact on designing efficient electrocatalysts for combing seawater splitting with urea purification.
Introduction
As a secondary energy source with high energy density (142 MJ kg−1) and environmentally friendly nature, hydrogen is a key lever for achieving the goal of carbon peak and carbon neutralization.1–3 Seawater as an unlimited resource accounts for ∼ 96.5% of the Earth's total water supply. In the context of the scarcity of freshwater resources, electrocatalytic seawater splitting using intermittent energy sources (solar, wind, tidal energy, etc.) and alkaline electrolyzers offers a promising avenue for H2 production.4,5 However, such applications have indeed been hampered by the kinetically slow oxygen evolution reaction (OER) and less value-added anodic product (O2).6,7 In addition, the competitive chlorine oxidation reactions caused by the high concentration of chloride ions in seawater and their corrosion of catalysts, and the blockage of the active sites by precipitates such as Ca(OH)2/Mg(OH)2 have brought numerous challenges for achieving efficient alkaline seawater splitting.8,9 Replacing slow water oxidation with thermodynamically favorable small-molecule oxidation reactions (e.g., alcohols, aldehydes, amines, and urea) would enable energy-saving and chlorine-free H2 production.10–15 Among them, the urea oxidation reaction (UOR, CO(NH2)2 + 6OH− → N2 + 5H2O + CO2 + 6e−, E° = 0.37 V vs. RHE) is an appealing one. Its thermodynamic equilibrium potential is much lower than that of the OER (1.23 V vs. RHE).13,16,17 Furthermore, urea decomposes into non-toxic N2 and CO2 during the oxidation process, providing a feasible method for purifying urea-rich wastewater without using extra oxidants. Replacing the OER with the UOR in seawater electrolysis can significantly reduce the overpotential of the anodic reaction.5,18 In this case, the electrolysis cell can operate without chlorine oxidation side reactions even at high current densities, thus addressing the issue of chlorine evolution during seawater electrolysis. Nevertheless, the anodic UOR for electrolytic urea assisted H2 production involves a complex 6e− transfer process and adsorption/desorption of intermediates, resulting in quite sluggish kinetics. The cathodic hydrogen evolution reaction (HER) kinetics are limited by the higher energy barrier of the water dissociation process in alkaline electrolytes.19,20 Therefore, it is of great significance but a big challenge to develop HER/UOR bifunctional catalysts to simultaneously achieve H2 production and urea-rich wastewater purification.Previous studies have found that Ni is an essential metal component for active urease, and Ni-based electrocatalysts have demonstrated remarkable activities for the UOR.21–27 To further enhance their bifunctional activity, doping or alloying Ni with other non-precious metals can modulate the electronic structure of Ni, compensating for the drawbacks of using Ni alone.28,29 Among various Ni-based electrocatalysts, Mo shows a significant synergetic effect with Ni in enhancing the HER performance, as Ni atoms are extensively acknowledged as excellent water dissociation centres, while Mo atoms are conductive to H adsorption.30,31 Furthermore, in comparison to their crystalline counterparts, the inherent disorder of amorphous materials can produce abundant “dangling bonds” and defects in the free-volume region of loosely bound atoms, thus exposing more active sites and altering the electron distribution around the imperfections, thereby improving the catalytic activity.32–35 Besides, their structural flexibility allows them to self-repair and withstand structural disturbances during electrocatalytic processes, which is beneficial to the catalysts’ stability.36–38 Therefore, the establishment of amorphous-crystalline composites would be an effective strategy to construct highly efficient UOR/HER bifunctional electrocatalysts.
Inspired by the aforementioned discussions, a novel 3D structed electrocatalyst c-MoNi/a-NiMoOx constituted of crystalline MoNi alloy clusters and amorphous NiMoOx nanowires was fabricated on Ni foam (NF) through a convenient hydrothermal process followed by thermal annealing in a reducing H2 atmosphere. The obtained c-MoNi/a-NiMoOx showed excellent superhydrophilicity (water contact angle in air ≈ 0°), underwater superaerophobicity (underwater bubble contact ≥ 155°) and ultralow bubble adhesive force. This behavior not only facilitates the exposure of active sites and mass transfer, but also prevents the catalyst from detaching due to the violent release of large bubbles, creating a favorable gas–liquid–solid reaction interface, thus ensuring that the catalyst maintains efficient and stable catalytic performance at high current densities. Importantly, the two-electrode urea-assisted seawater electrolysis based on c-MoNi/a-NiMoOx requires an ultralow voltage of 1.68 V to drive 500 mA cm−2, and displays distinguished long-term stability to maintain 100 mA cm−2 for 300 h. Advanced characterization and density functional theory calculations (DFT) reveal that in the HER process, the electron redistribution at the crystalline/amorphous interface can modulate the electronic structure, thereby optimizing the Gibbs free energy of water dissociation and hydrogen adsorption, facilitating the HER process. In the UOR process, c-MoNi/a-NiMoOx undergoes surface reconstruction to form highly active β-NiMoOOH species. The incorporation of Mo lowers the activation energy barrier of the rate-determining step, which is beneficial for the progression of the multi-step UOR process. This work demonstrates that the crystalline/amorphous configuration greatly optimizes the catalytic activity and stability, which presents a strategy for designing bifunctional catalysts that integrate seawater splitting with urea purification.
Experimental section
Preparation of NiMoO4 precursors on NF
First, a piece of 2 × 2.5 cm2 NF was sequentially sonicated in acetone and 0.5 M HCl solution for 10 minutes. Then, it was rinsed with deionized water and ethanol several times and vacuum dried at 50 °C for 12 hours. NiMoO4 precursors were synthesized on NF through a simple hydrothermal method. 594 mg Ni(NO3)2·6H2O, 312 mg (NH4)6Mo7O24·4H2O and 180 mg CO(NH2)2 were dissolved in 20 mL deionized water. After vigorous stirring for 30 min, the obtained transparent green solution was transferred to a 50 mL Teflon-lined stainless-steel autoclave with the pretreated NF, then maintained at 90 °C for 8 h. After cooling to room temperature, the NF with yellow-green materials was taken out and washed with a mixture of ethanol and deionized water several times, followed by drying at 60 °C for 12 h. For brevity, all the catalysts on NF are named without mentioning NF.Preparation of c-NiMo/a-NiMoOx nanowire arrays and other contrast catalysts
c-NiMo/a-NiMoOx was obtained by the controlled H2 reduction annealing method. In detail, the prepared NiMoO4 precursor was placed in a crucible and then placed in the center of a tube furnace. Under pure H2 atmosphere, the furnace was heated to 450 °C at a heating rate of 5 °C min−1 and maintained for 2 h. After cooling to room temperature, c-MoNi/a-NiMoOx nanowire arrays were obtained. For contrast, the NiMoO4 catalyst was prepared by only annealing the NiMoO4 precursor using the same method in an Ar atmosphere. The synthesis process of c-Ni/a-NiO was the same as that of c-MoNi/a-NiMoOx except that (NH4)6Mo7O24·4H2O was not added during the synthesis process.Results and discussion
Structural morphology and chemical composition
The self-supported c-MoNi/a-NiMoOx bifunctional catalyst was synthesized by a two-step process as illustrated in Scheme 1. Commercial Ni foam, in spite of its high electrical conductivity and 3D topology, was used as a conductive support (Fig. S1†). First, the NiMoO4 precursor was formed in situ on the NF skeleton by a convenient hydrothermal process at 90 °C for 8 h. Then, it was annealed at 450 °C for 2 h under a pure H2 atmosphere to obtain the c-MoNi/a-NiMoOx. Optical photographs confirm that the prepared catalysts exhibit significant color variation and uniformity throughout the synthesis process (Fig. S2†). As the diffraction intensity of the NF substrate is too strong to cover the other peak signals, the powder was scraped from the 3D NF skeleton for X-ray diffraction (XRD) testing. As revealed in Fig. S3,† the diffraction peaks in the XRD pattern of the NiMoO4 precursor are indexed to the triclinic NiMoO4·xH2O phase.39 After being reduced under a pure H2 atmosphere, the original peaks disappeared completely, and the peaks at s of 37.5°, 43.9° and 51.1° are correlated to metallic MoNi (PDF #65-6903) (Fig. 1a).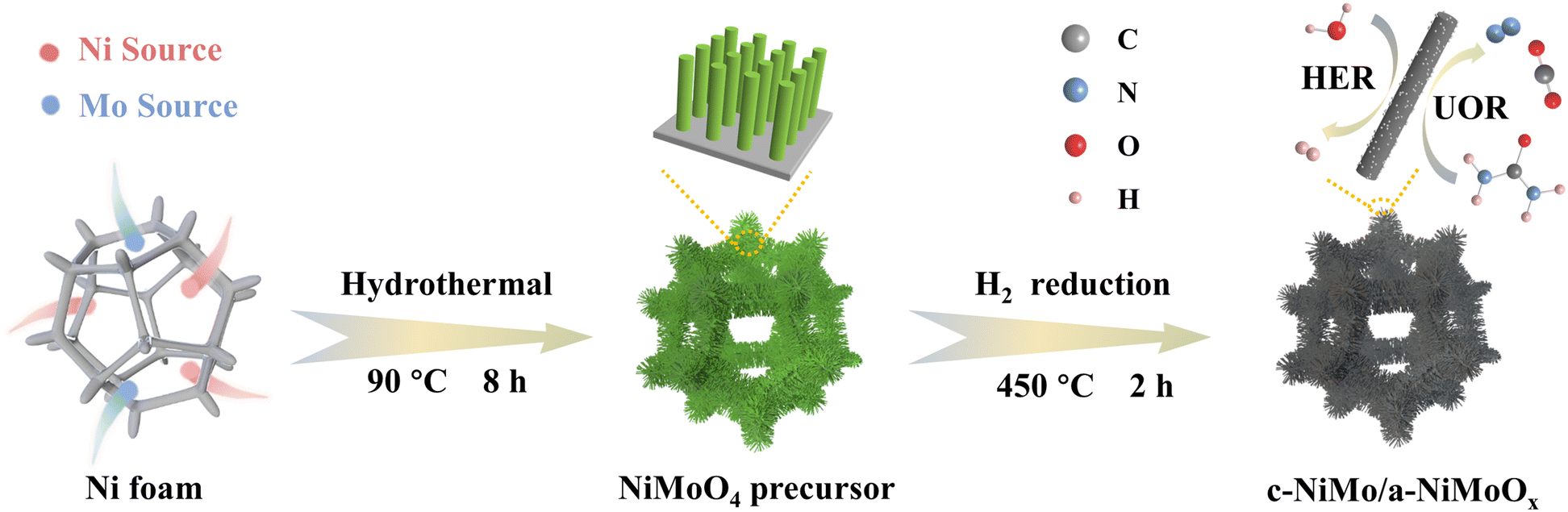 | ||
| Scheme 1 Schematic synthetic procedure for c-MoNi/a-NiMoOx. | ||
 | ||
| Fig. 1 (a) XRD pattern. (b and c) SEM images at different magnifications. (d) TEM image. (e) HRTEM image; insets show the SAED pattern. (f) HAADF-STEM image and corresponding elemental mappings of the c-MoNi/a-NiMoOx catalyst. Air-bubble contact angles under water, static water-droplet contact angles and gas-bubble adhesive force measurements (insets 1–3 show the bubble states during the measurement process) of (g) Ni foam and (h) c-MoNi/a-NiMoOx. | ||
Fig. 1b–e and Fig. S4, S5† show the typical morphology and structure of the as-prepared catalysts. First, the uniform 3D NiMoO4 nanowire arrays are directly grown on NF via the facile hydrothermal reaction of Ni(NO3)2·6H2O, (NH4)6Mo7O24·4H2O and CO(NH2)2 (Fig. S4†). TEM images also reveal the typical nanowire morphology of the NiMoO4 precursor, while the high-resolution TEM (HRTEM) image shows characteristic lattice fringes with an interplanar spacing of 0.89 nm, which can be easily indexed to the (100) plane of the NiMoO4·xH2O phase (Fig. S5†). The selected-area electron diffraction (SAED) pattern confirms that the NiMoO4 precursor has a single crystal structure. Additionally, the energy-dispersive X-ray spectroscopy (EDX) elemental mapping performed in STEM mode shows the presence of Ni, Mo, and O and their even distribution within the sample. After the controlled H2 reduction annealing process, the obtained c-MoNi/a-NiMoOx successfully inherits the nanowire array structure of the NiMoO4 precursor (Fig. 1b and c); the average diameter of the c-MoNi/a-NiMoOx nanowire is approximately 100 nm. The TEM image depicted in Fig. 1d manifests the presence of ultrasmall clusters tightly anchored to the nanowires in c-MoNi/a-NiMoOx, which is in stark contrast to the smooth-surfaced NiMoO4 precursor. As shown in Fig. 1e, the HRTEM images suggest that the c-MoNi/a-NiMoOx exhibits mixed-crystalline characteristics with a significant number of heterointerfaces between the crystalline and amorphous materials (marked with yellow dashed lines). The well-resolved lattice fringes with an interplanar spacing of 0.206 nm can be assigned to the (133) plane of the MoNi alloy. There are no obvious lattice fringes in the surrounding region of the MoNi alloy, indicating poor crystallinity of the NiMoOx. The SAED pattern of c-MoNi/a-NiMoOx in Fig. 1e shows weak crystallinity or an amorphous structure, consistent with the HRTEM results. HAADF-STEM images and corresponding elemental mappings further confirm a uniform distribution of Ni, Mo, and O elements throughout the entire nanowires (Fig. 1f). Furthermore, inductively coupled plasma-optical emission spectroscopy (ICP-OES) was conducted to quantify the atomic ratio of the c-MoNi/a-NiMoOx sample, showing Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mo = 1:1 (Table S1†). The aforementioned results demonstrate that the c-MoNi/a-NiMoOx composite catalyst containing crystalline MoNi clusters and amorphous NiMoOx has been successfully synthesized. In order to elucidate the contributions of each component in the prepared samples, a series of comparative samples were fabricated. As can be seen from the XRD pattern, when the NiMoO4 precursor was treated under a pure Ar atmosphere, the obtained NiMoO4 shows obvious diffraction peaks corresponding to the monoclinic NiMoO4 phase (PDF #86-0361), and no other impurity peaks appear (Fig. S6a†). In addition, c-Ni/a-NiO was prepared by controlled addition of molybdenum sources and the diffraction peaks of the obtained samples are indexed to the metallic Ni phase (PDF #04-0850) (Fig. S6b†). Combined with TEM characterization, it was confirmed that c-Ni/a-NiO consists of Ni nanocrystals uniformly distributed on amorphous NiO (Fig. S7†). The SEM images show that NiMoO4 still inherits the nanowire-array morphology of the original precursor, while c-Ni/a-NiO appears as aggregates of irregular particles (Fig. S8†).
:Mo = 1:1 (Table S1†). The aforementioned results demonstrate that the c-MoNi/a-NiMoOx composite catalyst containing crystalline MoNi clusters and amorphous NiMoOx has been successfully synthesized. In order to elucidate the contributions of each component in the prepared samples, a series of comparative samples were fabricated. As can be seen from the XRD pattern, when the NiMoO4 precursor was treated under a pure Ar atmosphere, the obtained NiMoO4 shows obvious diffraction peaks corresponding to the monoclinic NiMoO4 phase (PDF #86-0361), and no other impurity peaks appear (Fig. S6a†). In addition, c-Ni/a-NiO was prepared by controlled addition of molybdenum sources and the diffraction peaks of the obtained samples are indexed to the metallic Ni phase (PDF #04-0850) (Fig. S6b†). Combined with TEM characterization, it was confirmed that c-Ni/a-NiO consists of Ni nanocrystals uniformly distributed on amorphous NiO (Fig. S7†). The SEM images show that NiMoO4 still inherits the nanowire-array morphology of the original precursor, while c-Ni/a-NiO appears as aggregates of irregular particles (Fig. S8†).
It is evident that the wettability of the electrode surface is essential for the efficiency of gas-involved electrocatalytic reactions, specifically at high current densities.40–42 Contact-angle measurements were evaluated to examine the wettability characteristics of the obtained samples. As shown in Fig. 1g, the c-MoNi/a-NiMoOx surface is superhydrophilic, as demonstrated by its zero contact angles. In contrast, the bare Ni foam exhibits a water contact angle of 131.8° (Fig. 1h). The underwater gas-bubble contact angle for c-MoNi/a-NiMoOx is close to 155.1°, whereas that for the Ni foam is 136.2°, implying superaerophobic properties of the c-MoNi/a-NiMoOx surface. The superhydrophilic/superaerophobic properties of the c-MoNi/a-NiMoOx results in rapid diffusion of electrolyte and fast bubble release from the electrode surface, thus enhancing mass transfer and improving its catalytic performance. Meanwhile, there is no adhesive force between the gas bubbles and electrode surface measured for c-MoNi/a-NiMoOx, whereas Ni foam has a large bubble adhesive force (57.9 μN). This fast bubble release ability not only facilitates the exposure of active sites but also prevents the catalyst from detaching due to the violent release of large bubbles, creating a favorable gas–liquid–solid reaction environment.
The surface chemical composition and valence states of the elements were further examined utilizing X-ray photoelectron spectroscopy (XPS) measurement. As shown in Fig. S9a,† the XPS survey spectra of c-MoNi/a-NiMoOx reveal the coexistence of Ni, Mo, and O elements. In the Ni 2p high-resolution XPS spectra of c-MoNi/a-NiMoOx, the peaks at 852.5 and 869.6 eV can be ascribed to Ni0 2p, and the peaks at 856.0 and 873.8 eV can be assigned to Ni2+ 2p, accompanied by a pair of satellite peaks (Fig. 2a). The Mo 3d spectrum displayed in Fig. 2b is deconvoluted into Mo0 (227.7 and 230.9 eV), Mo4+ (228.7 and 232.0 eV), Mo5+ (229.8 and 233.1 eV) and Mo6+ (232.0 and 235.2 eV), further confirming the partial reduction of NiMoO4 to NiMoOx and the formation of the MoNi alloys. Notably, the binding energy of Ni in c-MoNi/a-NiMoOx shifts towards higher energy compared with NiMoO4 and c-Ni/a-NiO; on the contrary, the binding energy of Mo in c-MoNi/a-NiMoOx shifts towards lower energy compared with NiMoO4, implying the strong electronic interactions caused by the establishment of crystalline/amorphous heterointerfaces. This modified electronic structure can optimize the adsorption/desorption properties of the intermediate, resulting in better electrocatalytic performance.35,43 Regarding the O 1s spectrum, the analysis of NiMoO4 presents a very limited number of oxygen vacancies at 531.7 eV (Fig. S9b†). After the H2 reduction process, the peak intensity of the oxygen vacancies in c-MoNi/a-NiMoOx significantly increased, demonstrating the presence of abundant oxygen vacancies. Electron paramagnetic resonance (EPR) spectroscopy further provides strong evidence for the existence of oxygen vacancies in c-MoNi/a-NiMoOx, where the symmetric signal at g = 2.002 is derived from unpaired electrons captured at the oxygen vacancies (Fig. S10†). It has been reported that the presence of oxygen vacancies could increase the carrier concentration and lead to faster electron transfer of the catalyst, thus enhancing the electrocatalytic performance.44 Further information on the phase composition was investigated by Raman spectra (Fig. S11†), where the characteristic peaks of c-MoNi/a-NiMoOx located at 946, 891, 820 and 360 cm−1 are typical MoO4 tetrahedra for β-NiMoO4.45 The broad peak at 720 cm−1 can be ascribed to the modification of the Mo2–O bond for MoO3−x.46 The Raman peaks also confirm the existence of low-valence Mo. Furthermore, the Ni K-edge X-ray absorption near-edge structure (XANES) spectrum of c-MoNi/a-NiMoOx and the corresponding reference samples disclose that the near-edge absorption energy of Ni is higher than that of Ni but lower than that of NiO (Fig. 2c). The k2-weighted extended X-ray absorption fine structure (EXAFS) spectra of c-MoNi/a-NiMoOx were also obtained after background subtraction and edge-step normalization at the Ni K-edge, and both Ni–Ni (≈ 2.0 Å) and N–O bonds (≈ 2.5 Å) were detected (Fig. 2d). The Mo K-edge XANES spectrum of c-MoNi/a-NiMoOx reveals the existence of the MoO3−x phase (Fig. 2e), and the Mo–O (≈ 1.5 Å) and Mo-Mo (≈ 2.3 Å) bonds could be observed in the k2-weighted EXAFS spectra (Fig. 2f). Altogether, it can be clearly confirmed that the NiMoO4 precursor transforms into amorphous NiMoOx and crystalline MoNi alloy clusters after the H2 reduction process.
 | ||
| Fig. 2 The high-resolution XPS spectra of (a) Ni and (b) Mo of the prepared catalysts. (c) Ni K-edge XANES and (d) EXAFS spectra of c-MoNi/a-NiMoOx, Ni and NiO. (e) Mo K-edge XANES and (f) EXAFS spectra of c-MoNi/a-NiMoOx, Mo and MoO3. | ||
Evaluation of the electrocatalytic HER performance
The electrocatalytic performance towards the HER was investigated by a standard three-electrode configuration in 1.0 M KOH electrolyte. The optimal annealing temperature was determined to be 450 °C, at which the prepared catalyst exhibits the best HER activity (Fig. S12†). Fig. 3a shows the linear sweep voltammetry (LSV) curves of c-MoNi/a-NiMoOx, NiMoO4, c-Ni/a-NiO and commercial Pt/C in alkaline electrolyte with a scan rate of 2 mV s−1 for the HER. Impressively, the c-MoNi/a-NiMoOx electrode needs ultralow overpotentials of 20, 82 and 185 mV at −10, −100 and −500 mA cm−2, respectively, which is much lower than those of c-Ni/a-NiO (208, 380 and 525 mV at −10, −100 and −500 mA cm−2) and NiMoO4 (203, 352 and 497 mV at −10, −100 and −500 mA cm−2), and even outperforms the Pt/C catalyst (30, 113 and 329 mV at −10, −100 and −500 mA cm−2) (Fig. 3b). Notably, the HER performance of the c-MoNi/a-NiMoOx catalyst is superior to other reported non-noble-metal HER electrocatalysts (Table S2†). To study the HER kinetics in alkaline water, Tafel slopes obtained from LSV curves are presented in Fig. 3c. As it is evident, c-MoNi/a-NiMoOx exhibits a low Tafel slope of 59.8 mV dec−1, smaller than that of c-Ni/a-NiO (156.7 mV dec−1), NiMoO4 (135.3 mV dec−1) and Pt/C catalysts (85.9 mV dec−1), indicating rapid reaction kinetics. Mass transfer is another key factor in determining HER performance, especially at high current density. The proportion of overpotential to current density (Δη/Δlog|j|) is considered be an important indicator for evaluating the HER performance at high current densities, as it takes into account the effects of mass transport. In order to meet the prerequisites required for practical applications, the ratio of Δη/Δlog|j| at high current density must be sufficiently low. As displayed in Fig. 3d, the HER performance of the Pt/C catalyst is strongly affected and possesses a much higher Δη/Δlog|j| ratio than that of c-MoNi/a-NiMoOx, which indicates that the high current density performance of c-MoNi/a-NiMoOx is better than that of the Pt/C catalyst. For a more profound investigation of the HER kinetics and interface behaviors, electrochemical impedance spectroscopy (EIS) was employed. As shown in Fig. 3e, c-MoNi/a-NiMoOx exhibits the smallest semicircle and the lowest charge transfer resistance (Rct) among all of the catalysts, which means that faster charge transfer is achieved during the HER process. To estimate the number of active sites, the electrochemical double layer capacitance (Cdl) was obtained and calculated from the cyclic voltammetry (CV) curves in the non-Faraday region at different scan rates (Fig. S13†). c-MoNi/a-NiMoOx reveals a Cdl value of 412.9 mF cm−2, which is larger than that of c-Ni/a-NiO (1.7 mF cm−2) and NiMoO4 (2.2 mF cm−2) from Fig. 3f, indicating its larger electrochemical surface area (ECSA) and increased active sites. | ||
| Fig. 3 HER performance of the fabricated catalysts in 1.0 M KOH. (a) HER polarization curves. (b) Overpotentials of the catalysts at −10, −100 and −500 mA cm−2. (c) Tafel slopes. (d) Ratios of Δη/Δlog|j| in different current density ranges. (e) Nyquist plots (inset: equivalent circuit diagram). (f) Cdl values. | ||
Considering that c-MoNi/a-NiMoOx will be employed as an anode for urea-assisted water electrolysis, the HER performance of the catalyst was also investigated in 1.0 M KOH + 0.33 M Urea electrolyte. As shown in Fig. S14a,† the nearly coincident LSV curves demonstrate that the presence of urea in the electrolyte does not negatively affect the catalyst's HER activity. Furthermore, the long-term catalytic stability was appraised using chronopotentiometry (CP); as depicted in Fig. S14b,† c-MoNi/a-NiMoOx was able to remain stable at 100 mA cm−2 for 20 h, indicating its highly stable performance in urea-containing alkaline electrolytes. Meanwhile, the structure, morphology and surface chemical state of the c-MoNi/a-NiMoOx catalyst after the HER test were analyzed by XRD, Raman, SEM, TEM and XPS. The results demonstrate that the material does not change after the HER test (Fig. S15–17†). The comprehensive post-mortem examination confirms the excellent structural and morphological stability of the c-MoNi/a-NiMoOx catalyst, making it a promising candidate for the alkaline HER.
Evaluation of electrocatalytic UOR performance
The UOR performances of the as-prepared catalysts were evaluated in 1.0 M KOH with a concentration of 0.33 M urea. As shown in Fig. S18,† the c-MoNi/a-NiMoOx catalyst annealed at 450 °C under a H2 atmosphere exhibits the most outstanding UOR activity, which is in accordance with the HER results, signifying the excellent dual-functionality of the catalyst. Fig. 4a shows the LSV curves of the c-MoNi/a-NiMoOx catalyst in 1.0 M KOH electrolyte with or without 0.33 M urea. The well-designed c-MoNi/a-NiMoOx catalyst exhibits a distinguished UOR activity, compared with the OER, and the voltage required to reach the current density of 100 mA cm−2 is significantly reduced by 215 mV, demonstrating that the UOR is more energy-efficient than the OER. The UOR/OER potential histogram plots of c-MoNi/a-NiMoOx at different current densities are intuitively shown in Fig. 4b. To deliver the current densities of 10, 100 and 500 mA cm−2, the required potentials for the UOR are 1.311, 1.361 and 1.466 V (vs. RHE), while the required potentials for the OER are 1.479, 1.576 and 1.702 V (vs. RHE). Notably, the remarkable UOR activity of the c-MoNi/a-NiMoOx catalyst also surpasses those of most of the recently reported catalysts (Table S3†). As revealed in Fig. 4d, the c-MoNi/a-NiMoOx exhibits the smallest Tafel slope value of 30.4 mV dec−1, against the c-Ni/a-NiO (39.2 mV dec−1), NiMoO4 (35.6 mV dec−1) and RuO2 (77.5 mV dec−1), confirming that the c-MoNi/a-NiMoOx possesses the fastest reaction kinetics for the UOR. In addition, the Nyquist plots of the c-MoNi/a-NiMoOx and control samples were also collected. The smallest Rct of c-MoNi/a-NiMoOx among all the prepared catalysts (Fig. 4e) indicates that c-MoNi/a-NiMoOx has a favorable charge-transfer kinetics during the UOR process. Furthermore, the ECSA for the UOR was assessed by measuring the Cdl of the as-obtained catalysts from the CV curves (Fig. S19†). The Cdl value of c-MoNi/a-NiMoOx is determined to be 4.6 mF cm−2, which is significantly higher than that of c-Ni/a-NiO (3.0 mF cm−2), NiMoO4 (3.0 mF cm−2) and RuO2 (1.2 mF cm−2), indicating that c-MoNi/a-NiMoOx provides more exposure of active sites for the UOR (Fig. 4f). Aside from the efficient UOR catalytic activity, the electrochemical stability of the catalyst is also an essential factor. As shown in Fig. S20a,† the voltage has no apparent attenuation after chronoamperometric measurement at a current density of 100 mA cm−2 for a long period of 20 h (Fig. S20a†), verifying the excellent stability of the as-prepared c-MoNi/a-NiMoOx catalyst. Furthermore, the effect of urea concentration on the performance of the UOR was investigated. As depicted in Fig. S20b,† after increasing the urea concentration from 0 to 0.3 M, the OER current density was completely replaced by the UOR current density, implying that the reaction switches from the OER to the UOR.47,48 | ||
| Fig. 4 (a) Comparison of the OER and UOR polarization curves of c-MoNi/a-NiMoOx. (b) Voltages of the catalysts at 10, 100 and 500 mA cm−2. (c) UOR polarization curves, (d) Tafel plots, (e) EIS spectra and (f) Cdl curves of the prepared catalysts. | ||
It is well-known that transition metal-based catalysts may undergo surface reconstruction process to form the corresponding oxides/oxyhydroxides as actual active sites in the OER/UOR. To determine the active phase and reveal the reason why the as-prepared c-MoNi/a-NiMoOx catalyst possesses extraordinary electrocatalytic UOR activity, the morphology, composition and valence state of the catalyst after the UOR tests were studied. SEM analyses suggest that the morphology of the original nanowire arrays is well maintained after the UOR test (Fig. S21†), which would guarantee outstanding stability during the electrolysis process. XRD analysis shows that the diffraction peak of MoNi was significantly weaker than that of the initial catalyst, indicating that partial phase evolution occurred during the UOR (Fig. S22a†). Raman spectroscopy was conducted to illustrate the surface structure of the c-MoNi/a-NiMoOx catalyst under urea oxidation conditions. Apparently, two new peaks appeared at ∼474 and 554 cm−1, which are attributed to the bending and stretching vibrations of β-NiOOH (Fig. S22b†). These results indicate that the MoNi phase could be partially transformed into amorphous (oxy)hydroxides after the UOR. Accordingly, TEM images demonstrated that there is no obvious lattice fringe on the catalyst surface after the UOR, suggesting the generation of potential reconstruction-derived amorphous (oxy)hydroxides on the catalyst surface (Fig. S23a and b†). The HAADF-STEM and corresponding element mapping images show the uniform elemental distribution of Ni, Mo and O elements in the post-c-MoNi/a-NiMoOx sample (Fig. S23c†). In addition, EDS results show a decrease in the signal intensities of the Mo elements, confirming the partial dissolution of Mo species (Fig. S23d†). This phenomenon has also been widely observed in previous reports.49,50 Under harsh oxidation conditions, the metal elements are oxidized to a higher valence state. When Mo is oxidized to MoO42−, it can dissolve into KOH electrolyte. The surface chemical state of the c-MoNi/a-NiMoOx catalyst after the UOR test was further analyzed using XPS measurements (Fig. S24†). For the Ni 2p3/2 spectrum, the peaks of Ni0 disappeared while peaks of Ni3+ appeared, implying the formation of β-NiOOH (Fig. S24b†). For the Mo 3d spectrum, after the UOR test, the peaks of Mo0 disappeared, consistent with the TEM results (Fig. S24c†). In summary, the c-MoNi/a-NiMoOx undergoes surface reconstruction during UOR testing, and the resulting β-NiMoOOH species are believed to be the real highly catalytically active species.
Mechanism analysis for the HER and UOR
Density functional theory (DFT) calculations were performed to further explore the UOR/HER mechanism on c-MoNi/a-NiMoOx. Since amorphous materials have a high degree of short-range order, the MoNi/NiMoO4 composite structure is constructed by using the NiMoO4 crystal structure instead of amorphous NiMoOx. The differential charge density distribution of the MoNi/NiMoO4 structure is presented in Fig. 5a, in which the yellow and blue regions indicate charge accumulation and depletion, respectively. The electron transfer occurs from MoNi to NiMoO4 in the MoNi/NiMoO4 structure, indicating that the formation of a composite structure effectively promotes charge redistribution at the interface. The Bader charge analysis shows a net transfer of 1.97 e− from MoNi to NiMoO4, which is consistent with the XPS results. Such a pronounced electron redistribution is expected to accelerate the HER process in the alkaline electrolyte and effectively enhance the catalytic performance. Generally, the typical process of the HER in alkaline conditions is a two-step electrochemical process including electrochemical hydrogen adsorption (Volmer step) and electrochemical desorption (Heyrovsky step) or chemical desorption (Tafel step). Due to the lack of hydrogen ions in alkaline media, the adsorbed hydrogen (H*) can only be formed by water dissociation. Therefore, the Volmer step is critical for the HER. As presented in Fig. 5b, MoNi/NiMoO4 has a lower water dissociation energy than NiMoO4, indicating that the composite structure can accelerate the Volmer step of the HER and increase hydrogen sources for the next step. It is well-known that the Gibbs free energy of hydrogen adsorption (ΔGH*) is extensively acknowledged to be a key parameter for evaluating catalyst activity. For an efficient HER process, the ΔGH* value onto a catalyst should be close to zero (not too strong nor too weak). Fig. 5c shows that the ΔGH* value significantly decreases after the MoNi/NiMoO4 composite is constructed, implying that the interfacial interaction is favorable for optimizing the adsorption of the H* intermediates. Taken together, the results indicate that the MoNi/NiMoO4 composite can optimize the electronic structure through the strong electron interaction at the interface, so as to realize the cooperative optimization of the water dissociation and the hydrogen adsorption step, and effectively improve the alkaline hydrogen evolution reaction kinetics. | ||
| Fig. 5 (a) The charge density difference analysis of NiMo/NiMoO4; yellow and blue refer to electron-rich and electron-deficient areas, respectively. (b) Gibbs free energy (ΔG) diagram of the water dissociation path. (c) Gibbs free energy diagram for H adsorption. (d) Diagram of the crystal structure model of β-NiOOH and β-NiMoOOH. (e) Urea adsorption energy at β-NiOOH and β-NiMoOOH. The Gibbs free energy profiles of the proposed UOR pathway at (f) β-NiOOH and (g) β-NiMoOOH; the insert images are 6e− steps of urea oxidation on the catalysts. | ||
For the UOR, due to the surface reconstruction behaviors of the catalysts under oxidation conditions, the models of β-NiOOH and β-NiMoOOH were constructed in combination with the XRD/Raman/HRTEM/XPS results (Fig. 5d). As presented in Fig. 5d, the calculation results show that β-NiMoOOH (−1.93 eV) exhibits stronger urea adsorption energy compared with β-NiOOH (−1.73 eV), making it more favorable for decomposing urea (Fig. 5e). The Gibbs free energy of the UOR pathway was subsequently calculated to reveal the superiority of β-NiMoOOH. As shown in Fig. 5f, the rate-determining step (RDS) for β-NiOOH involves the initial conversion of oxidized and dehydrogenated CO(NH2)2* into CONH2NH*, with a thermodynamic barrier of 1.83 eV. The RDS for β-NiMoOOH involves the dehydrogenation of CONHN* into CONN* with a thermodynamic barrier of 1.72 eV. These results indicate that the incorporation of Mo leads to a decrease of the thermodynamic barrier, which is conducive to accelerating the multi-step UOR process.
Evaluation of the electrocatalytic activity for HSE
Inspired by the remarkable electrocatalytic HER and UOR performances of c-MoNi/a-NiMoOx, a two-electrode urea-assisted water electrolyzer was constructed, in which the as-prepared c-MoNi/a-NiMoOx was used as both the anode and cathode catalyst (Fig. 6a). Impressively, compared with the conventional water electrolysis, this hybrid electrolyzer shows excellent overall urea electrolysis activity (Fig. 6a), which only requires a lower voltage of 1.327 V to achieve 10 mA cm−2. Moreover, electrolytic water requires 1.676 and 1.935 V to reach 100 and 500 mA cm−2, while electrolytic urea requires only 1.436 and 1.661 V (Fig. 6b). Such excellent catalytic performance is better than most recently reported electrocatalysts for urea-assisted water electrolysis (Table S4†). In addition, the amounts of evolved H2 on the cathode were quantified by the drainage method. The calculation results show that c-MoNi/a-NiMoOx can achieve nearly 100% faradaic efficiency (FE) for H2, indicating that the cathode currents mainly result from the generation of H2 (Fig. 6c). Remarkably, the hybrid urea electrolyzer can be efficiently driven by a 1.5 V commercial dry battery, and continuous bubbles were rapidly released from the electrode surface during electrolysis (Fig. S25 and Video S1†). Furthermore, the solar (Fig. 6d and Video S2†) and wind (Fig. S26 and Video S3†) powered urea electrolyzers are capable of continuous production of H2, confirming the possibility of applying c-MoNi/a-NiMoOx in H2 generation by renewable energy sources. Encouraged by the excellent performance, the UOR activity and stability of the c-MoNi/a-NiMoOx in alkaline seawater were further studied. It is noteworthy that the HER/UOR activity of c-MoNi/a-NiMoOx is almost unchanged in the 1.0 M KOH + 0.33 M Urea + seawater electrolyte, confirming its promising potential in seawater electrolysis (Fig. S26†). Then, c-MoNi/a-NiMoOx was used to assemble a two-electrode hybrid seawater electrolyzer (HSE). The comparison results show that the c-MoNi/a-NiMoOx couple maintained good activity, and a low potential of only 1.676 V (vs. RHE) was required to reach a high current density of 500 mA cm−2 (Fig. 6e). As depicted in (Fig. 6f), energy-saving and chlorine-free H2 production might be realized by feeding valueless seawater and industrial urea sewage into a renewables-powered HSE. In addition, the stability test of the hybrid seawater electrolyzer was carried out by a chronoamperometric method at a current density of 100 mA cm−2. As a result, the potential shows negligible change within 300 h, validating the preeminent stability of c-MoNi/a-NiMoOx. The above results indicate that the present c-MoNi/a-NiMoOx has good application prospects in energy-saving H2 production and urea–rich wastewater purification.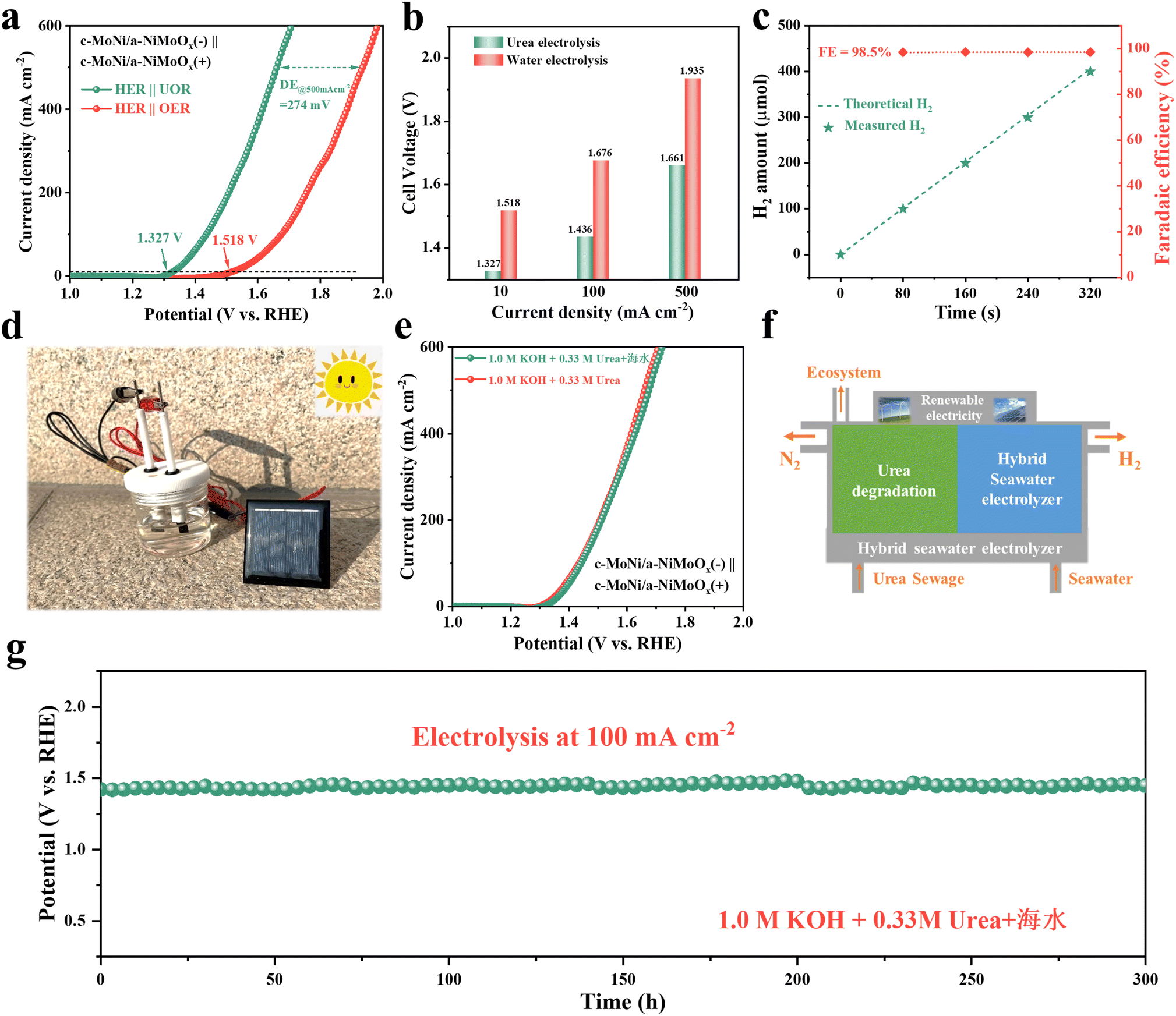 | ||
| Fig. 6 (a) The polarization curves of c-MoNi/a-NiMoOx for overall water splitting and urea electrolysis. (b) Comparison of the voltages at different current densities. (c) Faraday efficiency of c-MoNi/a-NiMoOx in 1.0 M KOH + 0.33 M urea. (d) Diagram of urea-assisted water splitting driven by solar panels. (e) The polarization curves of c-MoNi/a-NiMoOx for urea electrolysis in 1.0 M KOH + 0.33 M urea + seawater electrolyte. (f) Schematic diagram of the hybrid seawater electrolyzer. (g) Chronopotentiometry test of c-MoNi/a-NiMoOx in 1.0 M KOH + 0.33 M urea + seawater electrolyte. | ||
Conclusions
In summary, a crystalline/amorphous composite catalyst of c-MoNi/a-NiMoOx has been successfully fabricated by the combination of hydrothermal and subsequent controlled H2 reduction annealing methods. According to thorough characterization experiments and DFT calculations, the c-MoNi/a-NiMoOx composite structure could effectively regulate the electronic structure through the strong electron interaction at the interface, realizing the cooperative optimization of the water dissociation and the hydrogen adsorption step, which was beneficial for boosting the alkaline HER kinetics. Excitingly, the c-MoNi/a-NiMoOx achieves an overpotential of 185 mV at a current density of 500 mA cm−2. In the UOR process, c-MoNi/a-NiMoOx undergoes surface reconstruction to form highly active β-NiMoOOH species. The incorporation of Mo leads to a decrease of the thermodynamic barrier, which is conducive to accelerating the multi-step UOR process. As a result, it needs a small voltage of 1.466 V to achieve a current density of 500 mA cm−2. Moreover, in a urea-assisted overall seawater splitting system, the c-MoNi/a-NiMoOx||c-MoNi/a-NiMoOx electrodes display a low cell voltage of 1.676 V at 500 mA cm−2, and demonstrate remarkable long-term stability at 100 mA cm−2 for 300 h, implying the potential for practical applications. Overall, the present work provides valuable insights into the rational design of bifunctional electrocatalysts for urea-assisted energy-efficient hydrogen production and environment-related applications.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22372082) and the 111 project (B18030) from China.References
- Y. Yang, R. Zou, J. Gan, Y. Wei, Z. Chen, X. Li, S. Admassie, Y. Liu and X. Peng, Green Chem., 2023, 25, 4104–4112 Search PubMed.
- K. Xu, L. Liang, T. Li, M. Bao, Z. Yu, J. Wang, S. M. Thalluri, F. Lin, Q. Liu, Z. Cui, S. Song and L. Liu, Adv. Mater., 2024, 36, 2403792 CrossRef CAS PubMed.
- Z. Y. Yu, Y. Duan, X. Y. Feng, X. Yu, M. R. Gao and S. H. Yu, Adv. Mater., 2021, 33, 2007100 CrossRef CAS PubMed.
- L. Chen, C. Yu, J. Dong, Y. Han, H. Huang, W. Li, Y. Zhang, X. Tan and J. Qiu, Chem. Soc. Rev., 2024, 53, 7455–7488 RSC.
- L. Guo, J. Chi, J. Zhu, T. Cui, J. Lai and L. Wang, Appl. Catal., B, 2023, 320, 121977 CrossRef CAS.
- H. Dotan, A. Landman, S. W. Sheehan, K. D. Malviya, G. E. Shter, D. A. Grave, Z. Arzi, N. Yehudai, M. Halabi, N. Gal, N. Hadari, C. Cohen, A. Rothschild and G. S. Grader, Nat. Energy, 2019, 4, 786–795 CrossRef CAS.
- Z. Yu and L. Liu, Adv. Mater., 2024, 36, 2308647 Search PubMed.
- D. Liu, Y. Cai, X. Wang, Y. Zhuo, X. Sui, H. Pan and Z. Wang, Energy Environ. Sci., 2024, 17, 6897–6942 RSC.
- W. He, X. Li, C. Tang, S. Zhou, X. Lu, W. Li, X. Li, X. Zeng, P. Dong, Y. Zhang and Q. Zhang, ACS Nano, 2023, 17, 22227–22239 CrossRef CAS.
- X. Wang, J. Wang, X. Sun, S. Wei, L. Cui, W. Yang and J. Liu, Nano Res., 2018, 11, 988–996 CrossRef CAS.
- Y. Qi, Y. Zhang, L. Yang, Y. Zhao, Y. Zhu, H. Jiang and C. Li, Nat. Commun., 2022, 13, 4602 CrossRef CAS PubMed.
- Y. Zhao, N. Jia, X. R. Wu, F. M. Li, P. Chen, P. J. Jin, S. Yin and Y. Chen, Appl. Catal., B, 2020, 270, 118880 CrossRef CAS.
- T. Wang, X. Cao and L. Jiao, Angew. Chem., Int. Ed., 2022, 61, e202213328 CrossRef CAS PubMed.
- F. Sun, J. Qin, Z. Wang, M. Yu, X. Wu, X. Sun and J. Qiu, Nat. Commun., 2021, 12, 4182 CrossRef CAS PubMed.
- T. Li, B. Wang, Y. Cao, Z. Liu, S. Wang, Q. Zhang, J. Sun and G. Zhou, Nat. Commun., 2024, 15, 6173 CrossRef CAS PubMed.
- H. Sun, W. Zhang, J. G. Li, Z. Li, X. Ao, K.-H. Xue, K. K. Ostrikov, J. Tang and C. Wang, Appl. Catal., B, 2021, 284, 119740 CrossRef CAS.
- K. Ye, G. Wang, D. Cao and G. Wang, Top. Curr. Chem., 2018, 376, 42 CrossRef PubMed.
- Z. Yu, Y. Li, V. Martin-Diaconescu, L. Simonelli, J. Ruiz Esquius, I. Amorim, A. Araujo, L. Meng, J. L. Faria and L. Liu, Adv. Funct. Mater., 2022, 32, 2206138 CrossRef CAS.
- E. T. Sayed, T. Eisa, H. O. Mohamed, M. A. Abdelkareem, A. Allagui, H. Alawadhi and K. J. Chae, J. Power Sources, 2019, 417, 159–175 CrossRef CAS.
- S. K. Geng, Y. Zheng, S. Q. Li, H. Su, X. Zhao, J. Hu, H. B. Shu, M. Jaroniec, P. Chen, Q. H. Liu and S. Z. Qiao, Nat. Energy, 2021, 6, 904–912 CrossRef CAS.
- T. Wang, L. Miao, S. Zheng, H. Qin, X. Cao, L. Yang and L. Jiao, ACS Catal., 2023, 13, 4091–4100 CrossRef CAS.
- S. J. Patil, N. R. Chodankar, S. K. Hwang, G. S. Rama Raju, Y. S. Huh and Y. K. Han, Small, 2022, 18, 2103326 CrossRef CAS PubMed.
- Z. Liu, C. Zhang, H. Liu and L. Feng, Appl. Catal., 2020, 276, 119165 CrossRef CAS.
- S. Wang, L. Zhao, J. Li, X. Tian, X. Wu and L. Feng, J. Energy Chem., 2022, 66, 483–492 CrossRef CAS.
- J. Wang, F. Mo, J. Fei, W. Ling, M. Cui, H. Lei, L. Jiang and Y. Huang, Carbon Neutralization, 2022, 1, 267–276 CrossRef.
- H. Qin, Y. Ye, J. Li, W. Jia, S. Zheng, X. Cao, G. Lin and L. Jiao, Adv. Funct. Mater., 2023, 33, 2209698 CrossRef CAS.
- X. Zheng, J. Yang, P. Li, Z. Jiang, P. Zhu, Q. Wang, J. Wu, E. Zhang, W. Sun, S. Dou, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202217449 CrossRef CAS PubMed.
- Y. Li, X. Tan, R. K. Hocking, X. Bo, H. Ren, B. Johannessen, S. C. Smith and C. Zhao, Nat. Commun., 2020, 11, 2720 CrossRef CAS PubMed.
- Y. Jin, X. Yue, C. Shu, S. Huang and P. K. Shen, J. Mater. Chem. A, 2017, 5, 2508–2513 RSC.
- J. Zhang, T. Wang, P. Liu, Z. Liao, S. Liu, X. Zhuang, M. Chen, E. Zschech and X. Feng, Nat. Commun., 2017, 8, 15437 CrossRef CAS.
- A. Nairan, P. Zou, C. Liang, J. Liu, D. Wu, P. Liu and C. Yang, Adv. Funct. Mater., 2019, 29, 1903747 CrossRef CAS.
- Y. Wang, X. Li, Z. Huang, H. Wang, Z. Chen, J. Zhang, X. Zheng, Y. Deng and W. Hu, Angew. Chem., Int. Ed., 2023, 62, e202215256 CrossRef CAS PubMed.
- H. Han, H. Choi, S. Mhin, Y. R. Hong, K. M. Kim, J. Kwon, G. Ali, K. Y. Chung, M. Je, H. N. Umh, D. H. Lim, K. Davey, S. Z. Qiao, U. Paik and T. Song, Energy Environ. Sci., 2019, 12, 2443–2454 RSC.
- J. Wang, L. Han, B. Huang, Q. Shao, H. L. Xin and X. Huang, Nat. Commun., 2019, 10, 5692 CrossRef CAS.
- J. T. Ren, L. Chen, H. Y. Wang, W. W. Tian, X. L. Song, Q. H. Kong and Z. Y. Yuan, ACS Catal., 2023, 13, 9792–9805 CrossRef CAS.
- L. Li, X. Zhang, M. Humayun, X. Xu, Z. Shang, Z. Li, M. F. Yuen, C. Hong, Z. Chen, J. Zeng, M. Bououdina, K. Temst, X. Wang and C. Wang, ACS Nano, 2024, 18, 1214–1225 CrossRef CAS.
- L. Qiao, A. Zhu, D. Liu, J. Feng, Y. Chen, M. Chen, P. Zhou, L. Yin, R. Wu, K. W. Ng and H. Pan, Chem. Eng. J., 2023, 454, 140380 CrossRef CAS.
- L. Li, H. Sun, X. Xu, M. Humayun, X. Ao, M. F. Yuen, X. Xue, Y. Wu, Y. Yang and C. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 50783–50793 CrossRef CAS.
- K. Eda, Y. Kato, Y. Ohshiro, T. Sugitani and M. S. Whittingham, J. Solid State Chem., 2010, 183, 1334–1339 CrossRef CAS.
- H. Shi, T. Wang, J. Liu, W. Chen, S. Li, J. Liang, S. Liu, X. Liu, Z. Cai, C. Wang, D. Su, Y. Huang, L. Elbaz and Q. Li, Nat. Commun., 2023, 14, 3934 CrossRef CAS.
- Q. Ren, L. Feng, C. Ye, X. Xue, D. Lin, S. Eisenberg, T. Kou, E. B. Duoss, C. Zhu and Y. Li, Adv. Energy Mater., 2023, 13, 2302073 CrossRef CAS.
- Y. Zhou, N. Jin, Y. Ma, Y. Cui, L. Wang, Y. Kwon, W.-K. Lee, W. Zhang, H. Ge and J. Zhang, Adv. Mater., 2023, 35, 2209500 CrossRef CAS.
- Z. Dong, F. Lin, Y. Yao and L. Jiao, Adv. Energy Mater., 2019, 9, 1902703 CrossRef CAS.
- H. S. Kim, J. B. Cook, H. Lin, J. S. Ko, S. H. Tolbert, V. Ozolins and B. Dunn, Nat. Mater., 2017, 16, 454–460 CrossRef CAS PubMed.
- Z. Zhang, L. Li, Y. Li, Y. Zheng, Q. Wu, L. Xie, B. Luo, J. Hao and W. Shi, Chem. Eng. J., 2023, 469, 143846 CrossRef CAS.
- V. R. Jothi, K. Karuppasamy, T. Maiyalagan, H. Rajan, C. Y. Jung and S. C. Yi, Adv. Energy Mater., 2020, 10, 1904020 CrossRef CAS.
- Q. Zheng, Y. Yan, J. Zhong, S. Yan and Z. Zou, Energy Environ. Sci., 2024, 17, 748–759 RSC.
- Z. Zhu, K. Ge, Z. Li, J. Hu, P. Chen and H. Bi, Small, 2023, 19, 2205234 CrossRef CAS PubMed.
- D. Guo, Z. Zhao, M.-Y. Zong, C. Fan, W. Zheng and D. Wang, ACS Sustainable Chem. Eng., 2023, 11, 8362–8373 CrossRef CAS.
- X. Xu, H. Liao, L. Huang, S. Chen, R. Wang, S. Wu, Y. Wu, Z. Sun and H. Huang, Appl. Catal., B, 2024, 341, 123312 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05042h |
| This journal is © The Royal Society of Chemistry 2025 |