
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile methodology for side- and upcycling of HDPE waste via partial creation of unsaturated double bonds†
Wenyu
Wu Klingler
 *a,
Lucie
Perret
a,
Patrick
Rupper
a,
Sandro
Lehner
a,
Xiaoyu
Zhou
b,
Henrik
Eliasson
c,
Rico
Muff
d,
Manfred
Heuberger
ae and
Sabyasachi
Gaan
a
*a,
Lucie
Perret
a,
Patrick
Rupper
a,
Sandro
Lehner
a,
Xiaoyu
Zhou
b,
Henrik
Eliasson
c,
Rico
Muff
d,
Manfred
Heuberger
ae and
Sabyasachi
Gaan
a
aLaboratory for Advanced Fibers, Empa, Swiss Federal Laboratories for Materials Science and Technology, Lerchenfeldstrasse 5, 9014 St Gallen, Switzerland. E-mail: wenyu.wu@empa.ch
bDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir Prelog Weg 1–5, 8093 Zürich, Switzerland
cElectron Microscopy Center, Empa, Swiss Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, 8600 Dübendorf, Switzerland
dTransport at Nanoscale Interfaces Laboratory, Empa, Swiss Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, 8600 Dübendorf, Switzerland
eDepartment of Materials, ETH Zurich, 8092 Zurich, Switzerland
First published on 23rd August 2024
Abstract
Upcycling is emerging as a crucial strategy for enhancing the value of polymers, driving the transition toward a circular material economy. In this study, we present a facile chemical method for converting high-density polyethylene (HDPE) waste into valuable long-chain dicarboxylic acids (LCDCAs) via a key enabling unsaturated double-bond formation step. That is, we propose introducing unsaturation points in HDPE using commercially available heterogeneous catalysts (e.g. Pt/Al2O3). The unsaturation level through C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond formation is around 10%, as confirmed by FTIR, Raman and X-ray photoelectron spectroscopy. A process of microwave-assisted oxidation is demonstrated to break down the dehydrogenated HDPE into a mixture of aliphatic diacids. This approach enhances the recyclability and value of HDPE by the transformation of polymer waste into bifunctional monomers for potentially novel polyester synthesis. This process offers a sustainable and value-added alternative to conventional recycling methods. While further optimizations are needed, initial estimates of the E-factor and sEF provide promising indicators of the potential environmental benefits of this upcycling approach.
C double bond formation is around 10%, as confirmed by FTIR, Raman and X-ray photoelectron spectroscopy. A process of microwave-assisted oxidation is demonstrated to break down the dehydrogenated HDPE into a mixture of aliphatic diacids. This approach enhances the recyclability and value of HDPE by the transformation of polymer waste into bifunctional monomers for potentially novel polyester synthesis. This process offers a sustainable and value-added alternative to conventional recycling methods. While further optimizations are needed, initial estimates of the E-factor and sEF provide promising indicators of the potential environmental benefits of this upcycling approach.
Introduction
In the past century, petroleum-based plastics have undergone remarkable economic growth. However, their beneficial stability turns into a disadvantage once disposed as waste in the environment with alarming consequences.1,2 The growing visibility of climate change emphasizes the urgent necessity for interventions, particularly in waste management and circular material design, two crucial areas with substantial potential to mitigate these impacts.3 For converting polymer waste into valuable new materials on a large scale, complex and energy-intensive procedures are envisioned; along these lines efficient catalytic approaches have been established.4 By 2020 only 14% of common polymers (8.3 billion metric tons have been produced since the 1950s), like polyethylene and polyesters, were mechanically recycled, mainly resulting in lower-value products that end up as depreciated waste after one more life cycle.5,6 Polyolefins in general and, in particular high-density polyethylene (HDPE, 50 million tons per year), are major constituents of solid waste where only about 30% of HDPE is mechanically recycled (Fig. 1a). This general downcycling is limited by the significant degradation of quality after a few cycles. Developing new, high-quality side-/upcycling methods for HDPE is thus a crucial strategy in a sustainable and value-added circular economy. | ||
| Fig. 1 (a) The general post-consumption flow of HDPE, and (b) selected existing side-/upcycling methods for PE treatment, including oxidation,9 catalytic functionalization,10 plasma-assisted cracking,40 tandem hydrogenolysis/aromatization,22 chemical–biological upcycling of mixed polymers,29 fragmentation–oxidation upcycling process,23 and lastly our dehydrogenation and microwave-assisted oxidation approach, for obtaining aliphatic bifunctional monomers. | ||
Catalysts play a significantly important role in side-/upcycling and even in pyrolysis methods (Fig. 1a).7,8 For side-cycling, the creation of certain “functional” points along the inert PE chain is a key step for repurposing the material (Fig. 1b).9,10 Upcycling, which transforms HDPE into lower-molecular-weight products, also offers a promising alternative to mechanical recycling.8,9,11,12 One such widely studied approach is ‘pyrolysis’, in which plastics are decomposed to mixed oil or wax at high temperatures (>400 °C), often assisted by the presence of a catalyst.13 However, efficient and economical chemical processes for HDPE-to-monomer depolymerization are still in their early stages.14 Great efforts have been devoted to energy-efficient catalytic depolymerization methods to recycle HDPE to value-added products (inter alia surfactants, functional molecules).7,15–23 For example, the market price of surfactants ($2.3–$10 per kg) is significantly higher than that of virgin PE ($1.2 per kg).23 The technical capability to upconvert polymeric materials could act as a long-term strategy towards reaching sustainable cycling, where chemical feedstocks are repeatedly polymerized and after their life phase depolymerized, refined and re-polymerized to useful new products. Notably, the possibility to increase the economic value via re-polymerization into new functional materials is increasingly recognized as a central asset allowing evolution in a recycling scenario.24,25
Innovative approaches by introducing and adjusting the HDPE unsaturation points as cleavable linkages showed great potential to introduce functional upgrades into the polymer chemical structure.8,26,27 Further breakdown of the CC bonds by oxidation, metathesis, and other processes could result in telechelic polymers.8,26,28 Recently, a dedicated co-catalytic system, based on tandem dehydrogenation–metathesis reactions at low temperature (∼150 °C), was applied to depolymerize polyethylene (PE).18 The depolymerization relies on the selectivity of one catalyst to partially dehydrogenate PE, forming internal double bonds, while an olefin metathesis catalyst cleaves the polymer into smaller segments at the double bonds. Co(II)/Mn(II)-catalyzed oxidation supported by an N-hydroxyphthalimide initiator, for the production of oxygenated small molecules that are advantaged substrates for biological conversion, has also gained increased prominence more recently.29 A similar method via catalytic oxidation and bioconversion has demostrated the valorization of HDPE waste first to dicarboxylic acids (DCAs) and then to pharmacologically active compounds.30
Long-chain aliphatic diacids, diols, and diamines are essential for creating polymers that fill the niche between polyolefins and traditional polycondensates.28,31–33 Specifically, long-chain dicarboxylic acids (LCDAs) with chain lengths of 10 or more, such as sebacic and dodecanedioic acids, are crucial for synthesizing nylons and other polycondensates, providing benefits like high modulus and heat resistance due to their ability to crystallize.34 These materials, resembling polyethylene, offer potential for degradability in various applications.28,35–37 Currently, LCDAs are produced through chemical synthesis and biotechnological methods, which are often unsustainable or costly.31 There is a significant need for more efficient, economical, and sustainable production methods, potentially through side- and upcycling of polymer waste, to access these monomers more effectively.8,35,38
In accordance with these concepts, we developed a two-step process that enables production of a valuable new class of long-chain aliphatic monomers via catalytic depolymerization of HDPE waste products (Fig. 1b, our approach). By partial creation of unsaturated double bonds, we can enable a range of new possibilities for side- and upcycling. The proposed procedure requires a sequence of dehydrogenation and oxidation steps. Dehydrogenation of HDPE creates reactive points (unsaturated double bonds, 5–10%) along the otherwise inert polymer backbone using industrially available catalysts. A platinum catalyst was employed, inspired by its established use in the dehydrogenation of alkanes, due to its high catalytic activity and stability, e.g. in the Oleflex technologies of UOP.39 In a second step, microwave-assisted oxidation of the unsaturated bonds can directly lead to carboxylic acid-terminated long-chain monomers, which could be re-polymerized to long chain polycondensates. This work has resulted in the development of a new chemical methodology for (partial) depolymerization of HDPE into long-chain aliphatic monomers, which not only has potential to minimize polymer waste but also adds further value supporting the economics and technical evolution of a circular economy. It contributes to providing diverse solutions for chemical recycling; however, some processes within the technology still require further optimization.
Results and discussion
Characterization of catalysts
In order to obtain dehydrogenated HDPE rather than cracking products, selective C–H activation is needed, yet this remains a very challenging process. The cleavage of C–C bonds is more easily activated, since the average bond energy of the C–C bond (347 kJ mol−1) is weaker than that of the C–H bond (414 kJ mol−1).41 Pt is the well-utilized noble metal for the direct dehydrogenation of light alkanes, owing to its facile C–H bond activation over C–C bond scission.41 Al2O3 is a stable carrier of Pt catalysts, in which the coordinatively unsaturated pentacoordinate Al3+ sites on the γ-Al2O3 (100) surface can anchor Pt to make it resistant to migration, such as in the Oleflex process developed by Honeywell UOP.42 Therefore, a commercially available Pt/Al2O3 catalyst was selected deliberately to demonstrate the feasibility and potencial scalability of this work (Fig. 2a). The catalyst was firstly characterized using various methods before its use. The surface area of the Pt/Al2O3 catalyst was determined to be 84 m2 g−1 based on the nitrogen adsorption–desorption isotherm at 77 K using the Brunauer–Emmett–Teller (BET) theory (Fig. S2a and S2b†).43 This is smaller than that of commonly reported alumina matrix, which could be due to the wet impregnation methods used.44 Fig. S2c† gives the measured amounts of total and strongly chemisorbed hydrogen (H2) at 298 K. Strongly chemisorbed H2 was determined from the amount that did not desorb during evacuation at the analysis temperature. It was measured and found to be 0.529 mL g−1, which indicates dispersion of roughly 93% for the supported Pt nanoparticles. The total H/Pt ratios of Pt/Al2O3 at room temperature was close to one, which is often observed for small metal clusters on a supporting matrix.45–47 | ||
| Fig. 2 Upcycling of HDPE via a two-step process: (a) schematic flow with the first dehydrogenation step using 1 wt% Pt/Al2O3 in dry toluene and treated for 36–72 hours at 250 °C under N2; (b) STEM micrograph of the catalyst (Pt/Al2O3) used; (c) GPC traces of the HDPEs with images before and after treatment; and (d) XRD patterns of HDPEs with enlarged area of the spectra between 16° and 26°. | ||
The XRD profiles of the alumina-loaded catalysts showed the characteristic diffraction peaks of the Al2O3 support reported at 2θ = 38.5°, 45.8° and 67.0° (Fig. S3†). Although the introduction of platinum usually induces the appearance of a small peak of metallic platinum, it is not tangible in the present case, indicating the well dispersed Pt on the matrix. It is also a sign that Pt particles should be very small. Consistent with BET and XRD analyses, the TEM and HAADF-STEM images of untreated catalyst show less dense and mesoporous Al2O3 with Pt nanoparticles around 1.0–1.5 nm (Fig. 2b and Fig. S4a–c†). The hydrogen activation of the catalyst introduced certain amounts of aggregation, yet sizes of Pt nanoparticles still remain at around 2 nm (Fig. S4d–f†). The porous nature of the matrix and the low amount of catalyst loading can also be confirmed by SEM and EDX (Fig. S5†). Furthermore, HDPE is a very viscous liquid when molten, which is a critical property that affects its processability for end-of-life treatment. To reduce the viscosity of the reaction mixture and to facilitate the dehydrogenation process by loosening the polymer chains, toluene was used as solvent for improving the homogeneity of the reaction and eventually achieving better conversion. After the reaction, the catalyst was seperated and the solvent was recovered using rotavapor equipment for reuse.
Characterization of the dehydrogenated HDPE
Following the toluene-assisted dehydrogenation process, HDPE was treated using the hydrogen-activated Pt/Al2O3 catalyst. This reaction was conducted at 250 °C for varying durations of 36, 48, and 72 hours. Upon completion of the respective reaction times, the treated HDPEs, designated as HDPE-36, HDPE-48, and HDPE-72, were collected and dried for subsequent characterization. The structural simplicity of HDPE, with its repetition of CH2 units, does not reflect the complexity and properties of the real material. Ideally, the dehydrogenation treatment of HDPE should introduce unsaturation while maintaining the polymer's molecular weight; chain scission is expected to be negligible at this point. This assertion is supported by the analysis of number-averaged molecular weight (Mn), weight-averaged molecular weight (Mw), and the polydispersity (PD = Mw/Mn) values, which were quantified using high-temperature gel permeation chromatography (GPC), with results as depicted in Fig. 2c. Consistency in signal profiles across treated and untreated samples was meticulously noted, indicating no significant molecular weight variation or degradation; Mn values ranged approximately from 13k to 15k, Mw from 50k to 55k, and PD from 3.6 to 4.0, as catalogued in Table S1.† The chromatographic findings suggest that the integrity of the polymer chains remained unaffected by the catalytic treatment, as demonstrated by the alignment of retention times, peak shapes, and intensities with those of the control sample.HDPE exists generally as a mixture of crystalline and amorphous phases. One of the important questions that came up was whether the HDPE morphology changed after relatively harsh dehydrogenation reaction conditions, especially the branching and trans–cis properties.48,49 In principle, melting points of alkenes are similar to those of alkanes. However, since trans-configured alkenes are more symmetrical and hence less polar, they have normally higher melting points than those of cis-alkenes. Additionally, alkenes display weak dipole–dipole interactions due to the electron-attracting sp2 carbon, which might influence their melting points. Differential scanning calorimetry (DSC) analyses (Fig. S6†) further elucidate the polymer's thermal response (i.e. melting and crystallization) to dehydrogenation treatment, using the cooling and second heating curves. HDPE, characterized by minimal to no chain branching, exhibits a highly crystalline structure, reflected in the narrow range of endothermic peaks between 115 and 142 °C during melting. The similarity in melting temperature (Tm2) and crystallization temperature (Tc) values, and the slightly increased crystallinity (Xc) calculated by melting heat of fusion (ΔHm2) values before and after treatment, as documented in Table S1,† imply negligible alterations in the thermal characteristics following the treatment. This preservation of thermal behavior, which is likely attributable to the maintenance of lamellar thickness within the polymer crystals, is indicative of the treatment's efficacy in retaining the polymer's structural integrity.50 Thermogravimetric analysis (TGA, Fig. S7†) corroborates the thermal stability of HDPE, further reinforcing the conclusion that the dehydrogenation process compromises neither the polymer's molecular weight nor its thermal properties, thereby maintaining its intrinsic material complexity.
In addition, XRD is used to confirm the crystallinity of the untreated HDPE blank and its change after dehydrogenation (Fig. S8†). Two sharp crystal diffraction peaks can be observed at ∼21.4° and 23.8°, which correspond to diffraction from the (110) and (200) planes of the orthorhombic crystal structure in the HDPEs, respectively.49 Also, the broad scattering peak lying below the two sharp peaks represents the amorphous portion. The crystalline phase generally increases with increasing molecular weight and decreases with increasing chain branching. There is a subtle decrease in diffraction intensities of the amorphous portion of treated HDPEs (Fig. 2d), which corresponds to the mild change in the crystallinity of HDPE before and after the dehydrogenation treatment, as observed in the thermal analysis.
Importantly, the formation of unsaturated double bonds is observed both in FTIR and Raman spectra (Fig. 3). The FTIR pattern and absorptions are almost identical to those of the blank HDPE (Fig. 3a), except for the appearance of CH– stretching at around 3000 cm−1, which is obvious (Fig. 3c). The CH– bending located at around 890 cm−1 is also observable (Fig. 3d), while the CC stretching peak (i.e. that between 1680 and 1630 cm−1) is too subtle to be observed. This may result from the CC trans-configuration. Since it is symmetrically substituted, the CC stretching is a symmetric stretching of an almost symmetric molecule, similar to the symmetric stretching of CO2. In this case the peak intensity is zero. Furthermore, no observable vibrational peaks near 1100 cm−1 and 1650 cm−1 of the C–O and CO bonds appear, showing no indication of chain oxidation. Meanwhile, HDPE treated at lower temperature (200 °C for 36 and 48 hours) showed no evidence of CH– stretching or CH– bending signals (Fig. S9†). The crystallinity can be estimated from the FTIR spectra by using Zerbi's equation,51 comparing absorbance peaks at 730 cm−1 and 720 cm−1 with that of the internal reference 2019 cm−1 peak. According to Zerbi et al.,51 the bending vibrations at 1474 and 730 cm−1 (crystalline phase) and 1464 and 720 cm−1 (amorphous phase) correspond to the frequency behavior of CH2 symmetric stretching, which is also a function of trans–cis conformation (Fig. 3b). The amorphous phase indicated by the peak at 720 cm−1 slightly decreases.
 | ||
| Fig. 3 (a) FTIR spectra of the HDPE before (blank) and after dehydrogenation (HDPE-36, 48, 72), with (b) enlarged area of the CH2 stretching range, (c) the CH– stretching range, (d) the CH– bending geminal disubstituted alkene range, (e) Raman spectra of the HDPEs before and after dehydrogenation treatment, and (f) the enlarged area at the CC stretching range. | ||
Raman spectra for the untreated HDPE blank and dehydrogenated HDPE in the region between 200 and 3500 cm−1 showed some divergences. For the blank HDPE, the relevant vibrational bands due to skeletal C–C stretching (1050–1150 cm−1), CH– bending (scissoring between 1245 and 1277 cm−1), CH2 and CH3 bending (1400–1500 cm−1), CC stretching (1600–1700 cm−1), and C–H stretching vibrations (2800–3000 cm−1) are shown in Fig. 3e. For the dehydrogenated HDPE, the Raman spectra possess differences in fingerprint (CC stretching) regions;52 therefore, it confirms the existence of double bonds on the HDPE backbones (Fig. 3f). Visually inspecting the spectra offers a fast initial understanding of unsaturation formation. The total unsaturation can be estimated semi-quantitatively by measuring the ratio of the CC stretching (v CC) centered at 1601 cm−1 to the CH2 scissoring deformation (δ CH2) at 1444 cm−1, via Gaussian fitting and integration of the identified band areas at 1540–1670 cm−1 and 1390–1510 cm−1 (Fig. S10†).53 Calculating band intensity ratios is a direct approach to quantifying total unsaturation, and here we obtained roughly 10% unsaturation yield. Additionally, Raman spectroscopy can offer information on the structural characterization of PE. The modified Strobl and Hagedorn method is generally accepted and applied in the investigation of PE morphological structure.54 The normalized transmittance of the 1416 cm−1 band could be used to estimate the crystalline fraction in HDPEs (Fig. S11†), which indicates a slight increase of the crystallinity after treatment (Table S2†).
Fig. 4a depicts the XPS survey scans for HDPE in its native form as well as that for HDPE-72 with the most double bonds as characterized by Raman spectroscopy. As expected, both contain only carbon (C) in significant amounts. In addition, XPS can further confirm double bond formation by distinguishing carbon atoms at different hybridization states.55 Both the C 1s photoelectron peak as well as the X-ray excited C KLL Auger peak can be used to characterize the hybridization fractions of carbon electrons (sp2 and sp3).56 Due to only a small difference of 0.3–1.0 eV in binding energy from C 1s sp2 to sp3, the C 1s peak only gives a semi-quantitative determination of the sp2/sp3 ratio.55,57–59 In the C 1s spectrum fitting, we have fixed the two components of C sp2 and C sp3 to the ranges of literature values of 284.3–284.6 eV and 284.9–285.2 eV, respectively,55,56,58 and restricted the fitted FWHM to be the same in both components.
 | ||
| Fig. 4 Comparison of the XPS spectra of the blank HDPE and HDPE-72. In (a), (c) and (e), the two spectra are offset in the y-direction for better visualization. (a) Survey scans; (b) high-resolution scans for C 1s and (c) curve fitting of the envelope into composite peaks of sp2, sp3 and vibrational fine structure (dashed lines); (d) X-ray excited carbon KLL Auger spectra; (e) first derivative of the carbon KLL Auger spectra obtained by numerical differentiation. | ||
After treatment, HDPE exhibited a slight binding energy shift towards lower energies (see Fig. 4b). This shift results in an increased part of the sp2 component, which is localized at lower binding energies, as is observable from Fig. 4c. The C 1s component towards higher binding energy (at around 285.9 eV) represents vibrational fine structure as described for a reference HDPE spectrum.58 The sp2/sp3 ratio can be more quantitatively evaluated using the shape of the Auger C KLL spectrum (excited by X-ray photons), which represents a fingerprint of the carbon states.60 The method consists of determining the distance (the so-called D parameter) between the most positive maximum and the most negative minimum of the first derivative of the C KLL spectrum (see the ESI†).61 The D parameter is independent of absolute binding energy calibration and hence, the determined sp2 content from it represents a more quantitative value than the one resulting from the fitting of the C 1s peak with components lying close together in binding energy. Fig. 4d depicts the C KLL Auger spectrum for the two investigated samples and Fig. 4e shows the corresponding determination of the D parameter via the first derivative of the C KLL spectrum. The width of the C KLL Auger peak is increased for the HDPE-72 sample. The value of 14.0 eV for the D parameter results in 17.3% of sp2 for HDPE-72, which is consistent with the value obtained from the C 1s peak (see Table S3†). Therefore, successful partial dehydrogenation of the HDPE and formation of desired double bonds are confirmed using various techniques.
Analysis and application of the oxidized products
After achieving the desired partial unsaturation, microwave-assisted oxidation was conducted to facilitate depolymerization towards the target functional monomers. The efficacy of strong oxidation catalysts, specifically diluted nitric acid solution (0.10 g mL−1 in water), was assessed in promoting the oxidative degradation of the dehydrogenated HDPE (1 g) into a mixture of dicarboxylic acids. The reaction was carried out in a Milestone SyntWAVE microwave reactor for 1 hour at 180 °C, and the pressure was kept constant at 60 bar under an N2 atmosphere. The depolymerized solutions of untreated and dehydrogenated HDPEs were directly analyzed using UPLC-MS, which revealed distinct differences between the products derived from dehydrogenated HDPE and those from the blank HDPE. The blank HDPE primarily underwent surface carbonization with a low concentration of detectable organic fragments (Fig. S12†). HDPE-48 was depolymerized into fragments that are completely water-soluble (Fig. 5). This was further confirmed by UPLC-MS analysis, in which the extracted products via total ion chromatography (TIC) and UV spectroscopy channels indicated significant variances in their chemical signatures (Fig. 5b and Fig. S13†). Mass spectrometry (MS) analysis suggested that the primary degradation products were long-chain aliphatic diacids, predominantly those with carbon chain lengths shorter than C13 (Fig. S14†), exhibiting a notable distribution of chain lengths (C9–C13). The distribution of oxidation products is directly influenced by the concentration of double bonds in the treated HDPEs. The oxidation solution of HDPE-36 exhibited a broader range of mass and UV signals compared to HDPE-48 (Fig. S15†), likely due to a lower concentration of double bonds within the HDPE chains. Conversely, the oxidation solution of HDPE-72 demonstrated a narrower product distribution though with relatively lower concentration. Given its relatively narrow product distribution and sufficient concentration, HDPE-48 was selected for further analysis. The resulting diacids were collected, further characterized, and repolymerized.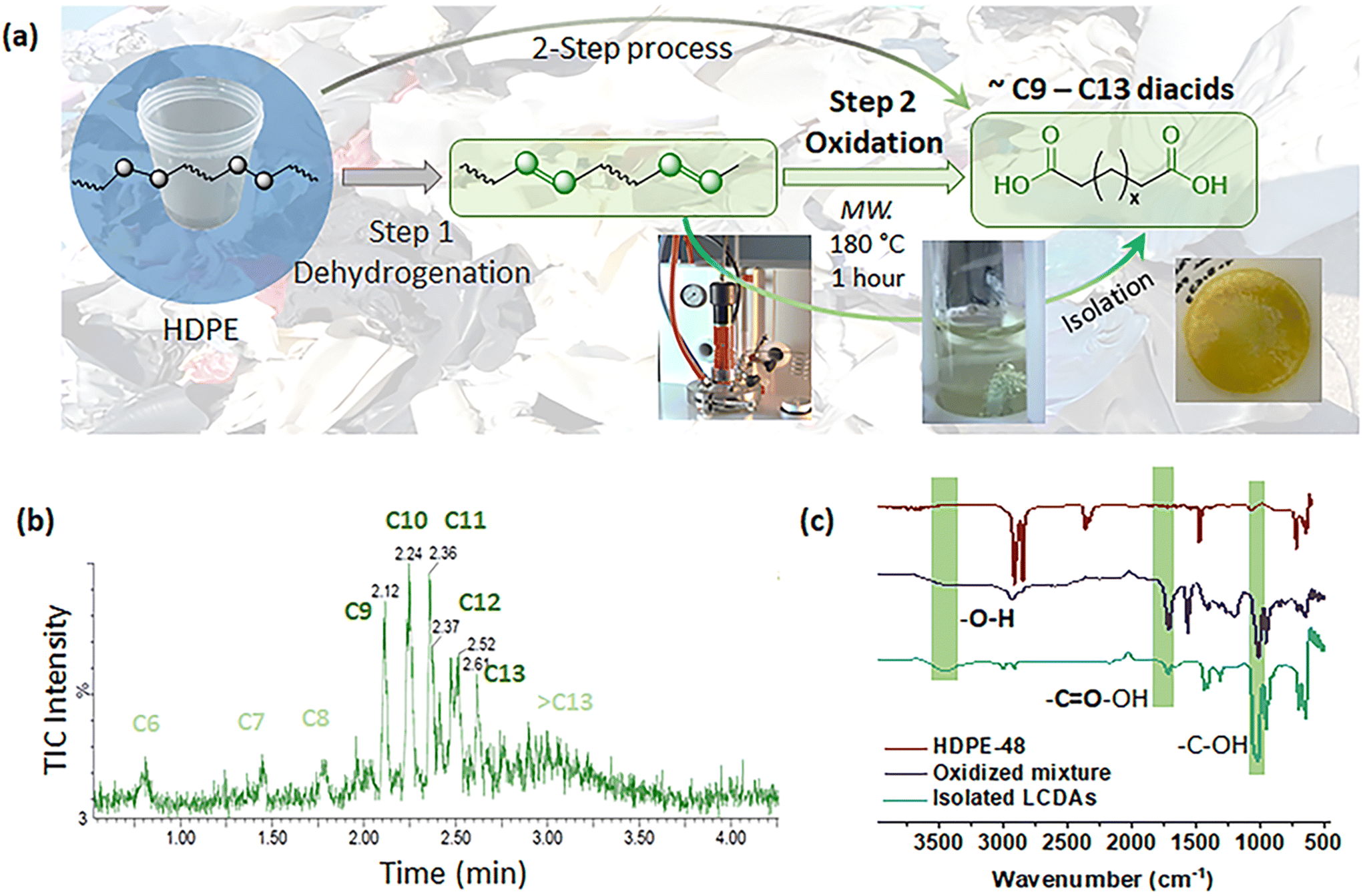 | ||
| Fig. 5 (a) The microwave-assisted oxidation step of dehydrogenated HDPE to LCDA products using nitric acid as the oxidation reagent in a microwave reactor (Shelton, CT) for 1 hour at 180 °C under an N2 atmosphere; (b) UPLC-MS chromatograms of the oxidized fragments detected in the TIC channel, under the ESI-method; and (c) the FTIR spectra comparison of the dehydrogenated HDPE and the oxidative products. | ||
After oxidation of HDPE-48, the crude solution was neutralized using NaOH (1 M) solution for further separation. Chloroform was employed to separate the longer chain DAs from the mixture. Post-separation and drying, 0.84 g of DAs was derived from 1 g of HDPE, resulting in a carbon conversion yield of approximately 58%. In future work, we plan to separate more fragments from the water solution for better recovery of the materials. The confirmation of the isolated LCDAs was further achieved through attenuated total reflection FTIR spectroscopy (Fig. 5c). This analysis highlighted the presence of carboxylic acid groups, as evidenced by characteristic absorption regions indicative of carbonyl (CO) and hydroxyl (–OH) groups. The oxidized products, both pre- and post-isolation, displayed a prominent peak in the range of 3430–3300 cm−1, attributed to O–H stretching vibrations, and an asymmetric stretching vibration of the CO bond at around 1710 cm−1. Additionally, the C–O stretching vibration was identified at approximately 1015 cm−1. Post-extraction spectra indicated the absence of nitric acid-related signals, further confirming the purity of the isolated products. The aliphatic nature of these compounds was corroborated by absorption peaks associated with CH2 stretching modes (2950–2850 cm−1) and scissoring vibrations (1450–1400 cm−1), as well as a C–H bending vibration at 700 cm−1. Nuclear magnetic resonance (NMR) spectroscopy provided further characterization of the diacids, revealing a predominantly linear structure with negligible branching, as inferred from the minimal presence of methyl groups in the 1H and 13C NMR spectra, as shown in Fig. S16.† In summary, the successful formation of LCDAs via this oxidative process has been validated through multiple analytical techniques, with the observed chain-length distribution primarily ranging between C9 and C13. The potential applications, i.e. raw material for polycondensation, and further processing of these diacids are under exploration.
After obtaining functional monomers, and in order to demonstrate an initial application of the isolated LCDAs, they were employed in a molten catalytic condensation process using titanium(IV) butoxide catalyst alongside 1,5-pentanediol, utilizing a two-step polycondensation technique (Fig. 6a).62 The oligomers produced in the first step and the polyester created in the second step appear yellowish and opaque (Fig. 6b). In comparison with HDPE-48, both the oligomer and polyester exhibited notably smaller molecular sizes with results as depicted in Fig. 6c. However, following the two-step polycondensation, the Mn values of polyester increased from 1k to 3k, and Mw from 6k to 10k (Table S4†), quantified using high-temperature GPC. The polyester's thermal properties also presented complex behavior, with noticeable improvements over that of the oligomers (Fig. 6d and e). Despite the polycondensation processes, the FTIR spectra displayed consistent patterns (Fig. 6f). This trial confirms the potential of LCDAs for polyester synthesis, setting the stage for further exploration.
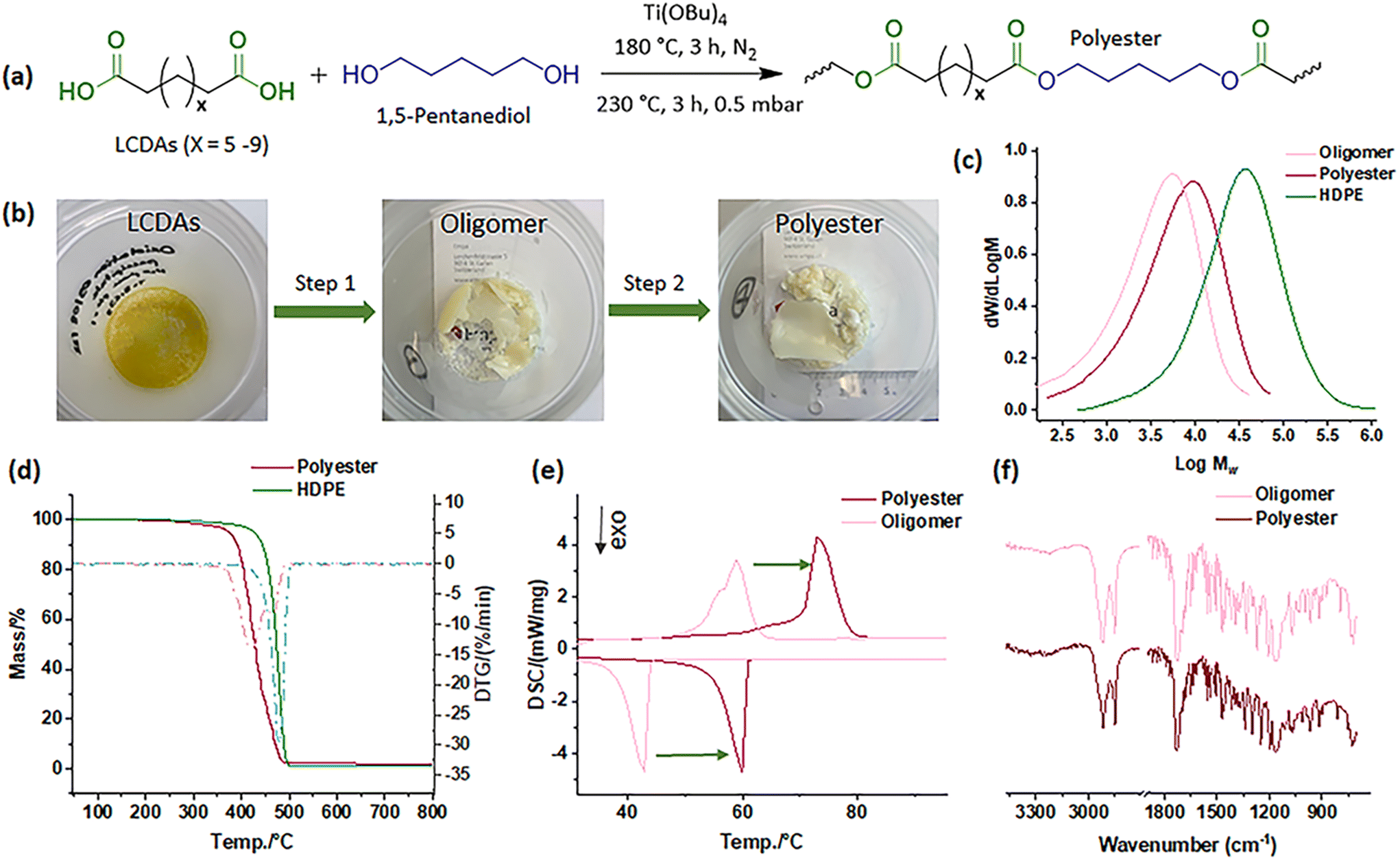 | ||
| Fig. 6 (a) Demonstration of polyester synthesis using a two-step molten polycondensation process of the LCDAs with pentanediol and catalytic amount of Ti(OBu)4 at 180 °C for 3 hours, then with reduced pressure at 230 °C for another 3 hours; (b) the appearances of the oligomer and polyester; (c) comparison between the GPC traces of HDPE-48, oligomer and polyester; (d) TGA and DTG curves of the HDPE-48 and polyester; and (e) the melting and crystallization behaviors and (f) the FTIR spectra comparison of the oligomer and the polyester. | ||
Future studies could systematically explore the effects of diol chain length and the impact of even–odd chain lengths on the chemical, thermal, crystalline, morphological, and mechanical properties of the resultant polyesters. While the recycling approaches for polyesters are more advanced compared to those for polyolefins,19,25 significant hurdles persist, particularly in the separation and decontamination processes. Considerable efforts are essential in order to transition from a linear polymer economy to a more sustainable, circular model.
Environmental consideration
For the developed two-step upcycling process, we have considered specific environmental indicators. Since the E-factor (environmental factor) concept has been instrumental in addressing the issue of waste generation,63 it has driven the development of cleaner processes for sustainable chemical manufacturing. It is defined as the mass ratio of waste to desired product, including reagents, solvent losses, and all process aids, except water (eqn (1)). We reviewed the two-step process and calculated the waste generation accordingly (Fig. S17†) with an estimated 10% solvent loss per step. Depending on the stage of development of the process, the use of simple E-factor (sEF) values for early route scouting activities, without taking solvents and water into account, is also appropriate (eqn (2)):64| E-factor = (mwaste + msolvent loss)/mproduct | (1) |
 | (2) |
The E-factor was calculated and was roughly 9.2, which is in the range of the fine chemical sectors. If solvents are excluded, the sEF was found to be 3.17. Compared with a few selected examples of functional molecule synthesis (Table S4†), this waste-based valorization process has demonstrated similar environmental factors. However, further optimization is essential for upscaling the process in future work. For instance, in the dehydrogenation step, a mechanical process can be envisioned to replace the solvent-assisted solution to address the viscosity issue of polymer melts. Additionally, in the oxidation step, alternative catalytic oxidizing reagents will be screened for effective depolymerization.
Reclaiming of catalyst
In addition to the abovementioned points, the ability to reuse the catalyst holds significant economic relevance. Thus after the dehydrogenation step, the recovered catalyst was collected and underwent regeneration through calcination and hydrogen activation for subsequent utilization, as depicted in Fig. S1.† The TGA and DTG analyses indicated that the degradation of the polymer residue on the catalyst surface requires up to 400 °C in air (Fig. S18†); therefore a relatively harsh regeneration temperature (500 °C) for two hours with a temperature ramp of 3 °C min−1 was applied to reactivate the catalyst. However, TEM and EDX analyses, as presented in Fig. 7, confirmed not only further aggregation of the nanoparticles but also the accrual of a modest amount of carbon residues. The Pt nanoparticles displayed heterogeneity and formed aggregates with dimensions surpassing 10 nm, a significant increase from the approximately 2 nm observed in the freshly activated catalyst prior to use. EDX spectroscopy indicated a notable accumulation of carbon residues after a single catalytic cycle, as illustrated in Fig. 7 and Fig. S19.† | ||
| Fig. 7 (a) TEM image and (b and c) HAADF-STEM images of the recycled catalyst after one dehydrogenation use; (d) and (e) SEM-EDX spectra with a semi-quantitative estimate of the catalyst composition before and after use. | ||
To address nanoparticle aggregation65 and carbon buildup, future investigations should focus on fine-tuning the regeneration conditions, reducing Pt loading on the matrix to less than 0.5%, and incorporating a secondary metal component, such as tin (Sn), gallium (Ga), or indium (In),66,67 into the catalyst composition. This can be achieved using wet impregnation or plasma methods.68 This additional metal is anticipated to form an alloy with Pt under operational conditions, enhancing the catalytic conversion, impeding metal sintering, and crucially, minimizing coke formation, thereby extending the catalyst's operational lifespan. The enhancements observed with platinum alloys are attributed to both geometric effects, which modify the crystalline structure of the catalyst, and electronic effects, influencing the reactivity of platinum's valence electrons and reducing the interaction between olefins and the catalyst.69 Given the superior dehydrogenation efficacy of platinum alloys compared to monometallic platinum, their suitability in the HDPE dehydrogenation process warrants further exploration.
Conclusion
This study has demonstrated a chemical strategy for side- and upcycling of HDPE, addressing the critical need for treatment of plastic waste. By employing a two-step catalytic depolymerization process, we have successfully converted HDPE into value-added functional monomers, specifically long-chain aliphatic diacids, through the introduction of unsaturation imperfections via CC double bond formation within the polymer chains, using a commercially available heterogeneous Pt/Al2O3 catalyst. The use of FTIR, Raman spectroscopy, and XPS for the characterization of the chemically modified HDPE has provided crucial insights, confirming the formation of CC double bonds and enabling a semi-quantitative assessment of unsaturation levels. The successful production of long-chain aliphatic diacids from HDPE-48, with a diverse range of chain lengths (C9–C13), not only underscores the viability of this approach but also highlights the potential of using polymer waste as a chemical feedstock. This represents a step towards the realization of a circular economy by transforming plastic waste into critical resources, thereby mitigating environmental impact and contributing to sustainability. The utilization of these double bonds will be further investigated for HDPE functionalization or repurposed applications.
Meanwhile, this study acknowledges several challenges that require further attention, including the investigation of realistic recycling scenarios with impurities, optimization of the catalyst's lifetime, the purification of diacids, development of cleaner processes, and utilization of the resultant chemicals. The simplified environmental effect of the two-step process is estimated using the E-factor (9.2), which indicates potential to further improve process efficiency and environmental sustainability. Additionally, employing diacids in polycondensation for application development remains valuable for future research. The goals of this research are to refine and expand the applicability of chemical recycling methods for HDPE. By continuing to explore innovative approaches to (partially) depolymerize HDPE more directly, efficiently, and cleanly, we can further contribute to the development of sustainable materials management practices and advance a circular economy.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Swiss Federal Office for the Environment (FOEN), with grant number UTF 666.14.21 and UTF 719.07.23. X. Z. thanks the National Centre of Competence in Research (NCCR) Catalysis (grant 180544) for financial support. We thank Dr. Edith Perret from Empa, St Gallen, Switzerland for assistance during the Raman measurements, and the help of Mr. Robin Pauer from Empa, Dübendorf, Switzerland for the STEM measurements.References
- P. Stegmann, V. Daioglou, M. Londo, D. P. van Vuuren and M. Junginger, Nature, 2022, 612, 272–276 CrossRef CAS PubMed.
- E. MacArthur, Science, 2017, 358, 843 CrossRef PubMed.
- H. Li, H. A. Aguirre-Villegas, R. D. Allen, X. Bai, C. H. Benson, G. T. Beckham, S. L. Bradshaw, J. L. Brown, R. C. Brown, V. S. Cecon, J. B. Curley, G. W. Curtzwiler, S. Dong, S. Gaddameedi, J. E. García, I. Hermans, M. S. Kim, J. Ma, L. O. Mark, M. Mavrikakis, O. O. Olafasakin, T. A. Osswald, K. G. Papanikolaou, H. Radhakrishnan, M. A. Sanchez Castillo, K. L. Sánchez-Rivera, K. N. Tumu, R. C. Van Lehn, K. L. Vorst, M. M. Wright, J. Wu, V. M. Zavala, P. Zhou and G. W. Huber, Green Chem., 2022, 24, 8899–9002 RSC.
- R. Wu, W. Wu Klingler, L. Stieglitz, S. Gaan, B. Rieger and M. Heuberger, Coord. Chem. Rev., 2023, 474, 214844 CrossRef CAS.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- Y. Weng, C.-B. Hong, Y. Zhang and H. Liu, Green Chem., 2024, 26(2), 571–592 RSC.
- K. Faust, P. Denifl and M. Hapke, ChemCatChem, 2023, 15, e202300310 CrossRef CAS.
- A. Arroyave, S. Cui, J. C. Lopez, A. L. Kocen, A. M. LaPointe, M. Delferro and G. W. Coates, J. Am. Chem. Soc., 2022, 144, 23280–23285 CrossRef CAS PubMed.
- J. Y. Q. Teo, C. W. S. Yeung, T. T. Y. Tan, W. W. Loh, X. J. Loh and J. Y. C. Lim, Green Chem., 2022, 24, 6287–6294 RSC.
- L. Chen, K. G. Malollari, A. Uliana, D. Sanchez, P. B. Messersmith and J. F. Hartwig, Chem, 2021, 7, 137–145 CAS.
- M. Chanda, Adv. Ind. Eng. Polym. Res., 2021, 4, 133–150 CAS.
- G. Celik, R. M. Kennedy, R. A. Hackler, M. Ferrandon, A. Tennakoon, S. Patnaik, A. M. LaPointe, S. C. Ammal, A. Heyden, F. A. Perras, M. Pruski, S. L. Scott, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, ACS Cent. Sci., 2019, 5, 1795–1803 CrossRef CAS PubMed.
- I. Barbarias, M. Artetxe, G. Lopez, A. Arregi, L. Santamaria, J. Bilbao and M. Olazar, Catalysts, 2019, 9, 414 CrossRef CAS.
- M. Chu, W. Tu, S. Yang, C. Zhang, Q. Li, Q. Zhang and J. Chen, SusMat, 2022, 2, 161–185 CrossRef CAS.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- X. Zhao and F. You, AIChE J., 2021, 67, e17127 CrossRef CAS.
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC.
- X. Jia, C. Qin, T. Friedberger, Z. Guan and Z. Huang, Sci. Adv., 2016, 2, e1501591 CrossRef PubMed.
- M. Baur, N. K. Mast, J. P. Brahm, R. Habé, T. O. Morgen and S. Mecking, Angew. Chem., Int. Ed., 2023, 62, e202310990 CrossRef CAS PubMed.
- C. Jehanno, M. M. Pérez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2019, 10, 172–186 RSC.
- B. Whajah, N. da Silva Moura, J. Blanchard, S. Wicker, K. Gandar, J. A. Dorman and K. M. Dooley, Ind. Eng. Chem. Res., 2021, 60, 15141–15150 CrossRef CAS PubMed.
- F. Zhang, M. Zeng, R. D. Yappert, J. Sun, Y. H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Science, 2020, 370, 437–441 CrossRef CAS PubMed.
- Z. Xu, N. E. Munyaneza, Q. Zhang, M. Sun, C. Posada, P. Venturo, N. A. Rorrer, J. Miscall, B. G. Sumpter and G. Liu, Science, 2023, 381, 666–671 CrossRef CAS PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- D. Parida, A. Aerts, K. Vanbroekhoven, M. Van Dael, H. Mitta, L. Li, W. Eevers, K. M. Van Geem, E. Feghali and K. Elst, Prog. Polym. Sci., 2023, 101783, DOI:10.1016/j.progpolymsci.2023.101783.
- R. J. Conk, S. Hanna, J. X. Shi, J. Yang, N. R. Ciccia, L. Qi, B. J. Bloomer, S. Heuvel, T. Wills, J. Su, A. T. Bell and J. F. Hartwig, Science, 2022, 377, 1561–1566 CrossRef CAS PubMed.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- M. Häußler, M. Eck, D. Rothauer and S. Mecking, Nature, 2021, 590, 423–427 CrossRef PubMed.
- K. P. Sullivan, A. Z. Werner, K. J. Ramirez, L. D. Ellis, J. R. Bussard, B. A. Black, D. G. Brandner, F. Bratti, B. L. Buss, X. Dong, S. J. Haugen, M. A. Ingraham, M. O. Konev, W. E. Michener, J. Miscall, I. Pardo, S. P. Woodworth, A. M. Guss, Y. Román-Leshkov, S. S. Stahl and G. T. Beckham, Science, 2022, 378, 207–211 CrossRef CAS PubMed.
- C. Rabot, Y. Chen, S. Bijlani, Y.-M. Chiang, C. E. Oakley, B. R. Oakley, T. J. Williams and C. C. C. Wang, Angew. Chem., Int. Ed., 2023, 62, e202214609 CrossRef CAS PubMed.
- F. Stempfle, P. Ortmann and S. Mecking, Chem. Rev., 2016, 116, 4597–4641 CrossRef CAS PubMed.
- M. Piccini, J. Lightfoot, B. C. Dominguez and A. Buchard, ACS Appl. Polym. Mater., 2021, 3, 5870–5881 CrossRef CAS.
- S. Pandey, B. S. Rajput and S. H. Chikkali, Green Chem., 2021, 23, 4255–4295 RSC.
- Z. Wang, M. S. Ganewatta and C. Tang, Prog. Polym. Sci., 2020, 101, 101197 CrossRef CAS.
- Z. Petrovic, Polym. Rev., 2008, 48, 109–155 CrossRef CAS.
- M. Oshinowo, J. R. Runge, M. Piccini, F. Marken and A. Buchard, J. Mater. Chem. A, 2022, 10, 6796–6808 RSC.
- T. F. Nelson, D. Rothauer, M. Sander and S. Mecking, Angew. Chem., Int. Ed., 2023, 62, e202310729 CrossRef CAS PubMed.
- E. Bäckström, K. Odelius and M. Hakkarainen, ACS Sustainable Chem. Eng., 2019, 7, 11004–11013 CrossRef.
- J. J. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- L. S. Diaz-Silvarrey, K. Zhang and A. N. Phan, Green Chem., 2018, 20, 1813–1823 RSC.
- C. Li and G. Wang, Chem. Soc. Rev., 2021, 50, 4359–4381 RSC.
- J. H. Kwak, J. Hu, D. Mei, C.-W. Yi, D. H. Kim, C. H. F. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670–1673 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- H. Jeong, H. B. Bathula, T. W. Kim, G. B. Han, J. H. Jang, B. Jeong and Y.-W. Suh, ACS Sustainable Chem. Eng., 2021, 9, 1193–1202 CrossRef CAS.
- E. Bus and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2007, 9, 2894–2902 RSC.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed.
- G. Bergeret and P. Gallezot, in Handbook of Heterogeneous Catalysis, 2008, pp. 738–765. DOI:10.1002/9783527610044.hetcat0038.
- H. W. Starkweather Jr. and R. H. Boyd, J. Phys. Chem., 1960, 64, 410–414 CrossRef.
- T. Furukawa, H. Sato, Y. Kita, K. Matsukawa, H. Yamaguchi, S. Ochiai, H. W. Siesler and Y. Ozaki, Polym. J., 2006, 38, 1127–1136 CrossRef CAS.
- J. Hoffman, G. Davis and J. Lauritzen, in Treatise on Solid State Chemistry, ed. N.B. Hannay, Plenum, 1976, vol. 3, p. 497 Search PubMed.
- G. Zerbi, G. Gallino, N. Del Fanti and L. Baini, Polymer, 1989, 30, 2324–2327 CrossRef CAS.
- J. R. Beattie, S. E. J. Bell and B. W. Moss, Lipids, 2004, 39, 407–419 CrossRef CAS PubMed.
- H. Wu, J. V. Volponi, A. E. Oliver, A. N. Parikh, B. A. Simmons and S. Singh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3809–3814 CrossRef CAS PubMed.
- G. R. Strobl and W. Hagedorn, J. Polym. Sci., Polym. Phys. Ed., 1978, 16, 1181–1193 CrossRef CAS.
- S. Turgeon and R. W. Paynter, Thin Solid Films, 2001, 394, 43–47 CrossRef.
- B. Lesiak, J. Zemek, J. Houdkova, P. Jiricek and A. Jóźwik, Polym. Degrad. Stab., 2009, 94, 1714–1721 CrossRef CAS.
- S. Kaciulis, Surf. Interface Anal., 2012, 44, 1155–1161 CrossRef CAS.
- G. Beamson and D. Briggs, High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database, Wiley, 1992 Search PubMed.
- B. Lesiak, J. Zemek and J. Houdkova, Polymer, 2008, 49, 4127–4132 CrossRef CAS.
- S. Kaciulis, A. Mezzi, P. Calvani and D. M. Trucchi, Surf. Interface Anal., 2014, 46, 966–969 CrossRef CAS.
- Y. Mizokawa, T. Miyasato, S. Nakamura, K. M. Geib and C. W. Wilmsen, Surf. Sci., 1987, 182, 431–438 CrossRef CAS.
- L. Genovese, M. Gigli, N. Lotti, M. Gazzano, V. Siracusa, A. Munari and M. Dalla Rosa, Ind. Eng. Chem. Res., 2014, 53, 10965–10973 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- W. Zhou, S. R. Docherty, C. Ehinger, X. Zhou and C. Copéret, Chem. Sci., 2023, 14, 5379–5385 RSC.
- M. Abdelgaid, J. Dean and G. Mpourmpakis, Catal. Sci. Technol., 2020, 10, 7194–7202 RSC.
- H. Yan, K. He, I. A. Samek, D. Jing, M. G. Nanda, P. C. Stair and J. M. Notestein, Science, 2021, 371, 1257–1260 CrossRef CAS PubMed.
- D. Hegemann, M. Amberg, A. Ritter and M. Heuberger, Mater. Tech., 2009, 24, 41–45 CrossRef CAS.
- S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc03108c |
| This journal is © The Royal Society of Chemistry 2024 |