
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Food-safe glycidyl-free chain extenders for polylactides†
Hazem M.
Elkholy‡
a,
Mohamed A.
Abdelwahab‡
 a,
Muhammad
Naveed
a,
Khaled
Abdelaziz
bc and
Muhammad
Rabnawaz
*a
a,
Muhammad
Naveed
a,
Khaled
Abdelaziz
bc and
Muhammad
Rabnawaz
*a
aSchool of Packaging, Michigan State University, 448 Wilson Road, East Lansing, Michigan 48824-1223, USA. E-mail: rabnawaz@msu.edu
bDepartment of Animal and Veterinary Sciences, College of Agriculture, Forestry and Life Sciences, Clemson University, Clemson, SC 29634, USA
cClemson University School of Health Research, Clemson, SC 29634, USA
First published on 16th February 2024
Abstract
Glycidyl-based chain extenders are broadly used as processing aids to enhance the physical properties of polyesters, but glycidyl-containing compounds have severe toxicity concerns. Reported herein are glycidyl-free chain extenders produced via the grafting of 9-(oxiran-2-yl)nonyl methacrylate (EAT) and 8-(3-octyloxiran-2-yl)octyl methacrylate (EAI) on biodegradable poly(ε-caprolactone) (PCL). EAI and EAT were grafted onto PCL via free radical grafting using melt extrusion to produce PCL-epoxy-grafted PCL-g-EAI and PCL-g-EAT. The obtained PCL-g-EAI and PCL-g-EAT were characterized by 1H nuclear magnetic resonance (NMR) spectroscopy, intrinsic viscosity measurements, thermogravimetric analysis (TGA), differential scanning calorimetry (DSC) and size exclusion chromatography (SEC). Using reactive melt extrusion, PCL-g-EAI and PCL-g-EAT were tested as potential chain extenders for poly(lactic acid) (PLA). PCL-g-EAI showed comparable or better mechanical properties than those of the commercial chain extender Joncryl ADR. A cytotoxicity assay has been conducted on PLA samples with the new chain extender and showed the absence of cytotoxicity in all samples. The outcomes reported in this paper offer new opportunities for the development of safer chain extenders.
1. Introduction
Poly(lactic acid) (PLA) is a biobased aliphatic polyester that is produced from renewable resources.1 PLA is biocompatible, biodegradable, non-toxic and has high-performance mechanical characteristics. Consequently, it is utilized in various applications, including textiles, packaging, and healthcare.2 However, its lower toughness (impact strength and elongation at break) and high cost limit the application of PLA as a commodity polymer.3 Therefore, various attempts have been made to enhance these properties, such as chemical modification and blending.4,5 PLA was treated with methacryloyloxyethyl isocyanate and trimethyl hexamethylene diisocyanate to obtain a ductile PLA and enhance its mechanical properties.6,7 PLA's rheological, thermal, and mechanical properties and molecular weight were improved after incorporating a chain extender (poly aryl-polymethylene isocyanate).8 Polycarbodiimide and tris(nonyl-phenyl) phosphite were used as chain extenders for PLA systems, which enhanced their linear viscoelastic and thermal properties.9 Moreover, a multifunctional oligomeric epoxy-styrene-acrylic reactive chain extender was used to increase the complex viscosity, elasticity, and elongational viscosity of PLA polymers.10,11 In addition, PLA was chain extended by styrene–acrylonitrile–glycidyl methacrylate (S–ACN–GMA) via the melt-mixing method to enhance its melt viscoelasticity and crystallization behavior.12Joncryl ADR chain extenders containing epoxide groups are some of the most widely used commercial additives for the chemical modification of polyesters to improve their mechanical and thermal behavior.13–15 Yahyaee et al.16 enhanced the poor melt characteristics of PLA with two chain extenders, pyromellitic dianhydride, and Joncryl ADR. The authors indicated that the chain extension reactions increased the molecular weight and elongational rheological characteristics. Benvenuta-Tapia et al.17 used poly(S-co-GMA-b-S) copolymers as chain extenders for the recycling of PLA via melt extrusion. This chemical modification improved the viscosity and elastic modulus of the modified PLA. However, Joncryl ADR chain extenders use GMA as the functional monomer, which upon hydrolysis/degradation forms glycidol. Unfortunately, glycidol derived from GMA is a carcinogen (according to an NTP report),18 which is why glycidyl-free chain extenders are required. Moreover, GMA-based polymers are non-biodegradable and, therefore, biodegradable alternatives can offer environmental benefits.19 This work opens new opportunities for improving the performance of biodegradable PLA polymers without the addition of harmful glycidol-based ADR chain extenders. The new material can be used in food packaging applications.
In this article, we report the development of polycaprolactone (PCL)-based epoxy chain extenders that are glycidyl-free. PCL has been selected as the backbone polymer because it offers numerous beneficial properties such as biodegradability and non-toxicity along with other properties such as excellent rheological and viscoelastic attributes.20,21 While PCL blends with starch and PLA have been reported before,22,23 PCL-epoxy has not been used as a chain extender. Our novel PCL-epoxy was synthesized and tested in PLA in small amounts (1 phr or less). The effects of new PCL-grafted additives on the thermal and mechanical characteristics of PLA were examined.
2. Experimental section
2.1. Materials
11-Hydroxy-1-undecene (98%), cis-9-octadecen-1-ol, 3-chloroperbenzoic acid (≤77%), 4-(dimethylamino)pyridine (DMAP), methacrylic anhydride (94%), dicumyl peroxide (DCP, 98%), benzoyl peroxide (BPO, ≥98%), xylenes (≥98.5%), and polycaprolactone (PCL, average Mn = 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) were purchased from Sigma-Aldrich. Chloroform and ethyl acetate (HPLC grade) were purchased from Fisher Chemical. Poly(lactic acid) (PLA) (Ingeo™ Biopolymer 3001D) was purchased from NatureWorks LLC. PLA has a melting temperature of 200 °C and MFI of 22 g per 10 min (210 °C, 2.16 kg) as indicated in the technical data sheet (TDS). Joncryl ADR 4486 (ADR) was obtained from BASF in flake form with a Tg of 59 °C, Mw of 7250 g mol−1, and an equivalent epoxy weight of 310 g mol−1 as indicated in the TDS.
000 Da) were purchased from Sigma-Aldrich. Chloroform and ethyl acetate (HPLC grade) were purchased from Fisher Chemical. Poly(lactic acid) (PLA) (Ingeo™ Biopolymer 3001D) was purchased from NatureWorks LLC. PLA has a melting temperature of 200 °C and MFI of 22 g per 10 min (210 °C, 2.16 kg) as indicated in the technical data sheet (TDS). Joncryl ADR 4486 (ADR) was obtained from BASF in flake form with a Tg of 59 °C, Mw of 7250 g mol−1, and an equivalent epoxy weight of 310 g mol−1 as indicated in the TDS.
2.2. Characterization
All materials were tested by 500 MHz 1H-NMR spectroscopy (Varian 7600-AS, United States). Around 3 mg of the sample was dissolved in deuterated chloroform (0.7 mL).Attenuated total reflectance Fourier transform infrared (ATR-FTIR) analysis was done using an instrument manufactured by Shimadzu Co., Columbia, MD. Each spectrum was recorded over the range from 4000 to 400 cm−1 with 64 scans and a resolution of 4 cm−1.
The tensile tests were completed according to ASTM D638-14 (type V) at 10 mm min−1 using an Instron model 5565 (MA, USA).
The notched impact samples (obtained using a TMI 2205 notching cutter, USA) were tested according to ASTM D256. A Ray-Ran tester with a hummer pendulum of 5.417 J was used for testing the samples.
The thermal stability of grafted PCL, neat PLA and PLA with the chain extenders was analyzed using a TGA instrument (Thermogravimetric analysis Q50 TA Instrument, USA). Under an N2 flow (40 mL min−1), and at a rate of 10 °C min−1, around 6–10 mg of each sample was heated from room temperature to 600 °C.24
A differential scanning calorimetry Q100 analyzer (TA Instrument) was used to conduct DSC analysis. Around 5–10 mg of copolymer and PLA sample was examined using a heat/cool/heat protocol from −30 °C to 210 °C at a heating rate of 10 °C min−1 under N2 gas. The crystallinity of neat polymer and PLA-chain extender samples was calculated from the 2nd heating curve using the melting enthalpy (ΔHm), cold crystallization enthalpy (ΔHc), and PLA weight fraction (wf) in the sample as illustrated in eqn (1):
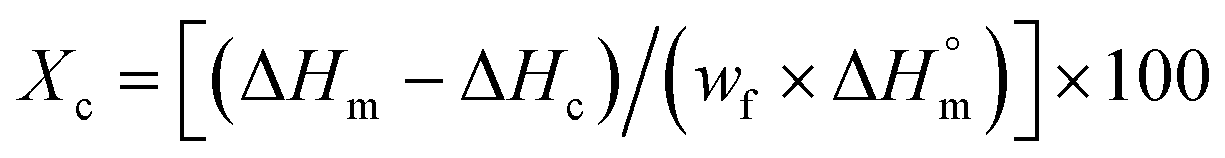 | (1) |
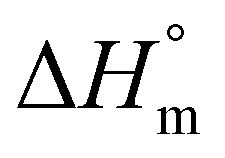 denotes the melting enthalpy values of 100% crystalline PCL and PLA, which are 151.7 and 93.7 J g−1, respectively.25,26
denotes the melting enthalpy values of 100% crystalline PCL and PLA, which are 151.7 and 93.7 J g−1, respectively.25,26
The transparency of the films (∼200 micron film prepared by compression moulding at 230 °C for 2 min and 10 tonnes using a PHI-based press model QL438-C, USA) was investigated using a UV–visible spectrophotometer (Evolution 600, Thermo-Scientific, USA) in Intelli-scan speed mode with a data interval of 0.5 nm in the range 200–800 nm.
2.3. Synthesis of 9-(oxiran-2-yl)nonyl methacrylate (EAT) and 8-(3-octyloxiran-2-yl)octyl methacrylate (EAI)
EAT and EAI were synthesized in two steps from their unsaturated long-chain alcohol using our recently reported method.27,28 Details about the synthesis and characterization of EAT and EAI are provided in the ESI.†2.4. Synthesis of PCL–EAT and PCL–EAI
PCL and epoxy acrylates (EAT or EAI) were melt processed at 130 °C for 10 min at 100 rpm using a C.W. Brabender Instruments Inc. instrument (Hackensack, NJ, USA) with roller-type blades. After heating the Brabender chamber to 130 °C, PCL was gradually introduced. After the PCL was melted for 2 min, a mixture of epoxy acrylate EAT or EAI and BPO as an initiator was incorporated dropwise over a period of 1 min. The copolymers were mixed for another 7 min to produce PCL-g-EAT and PCL-g-EAI.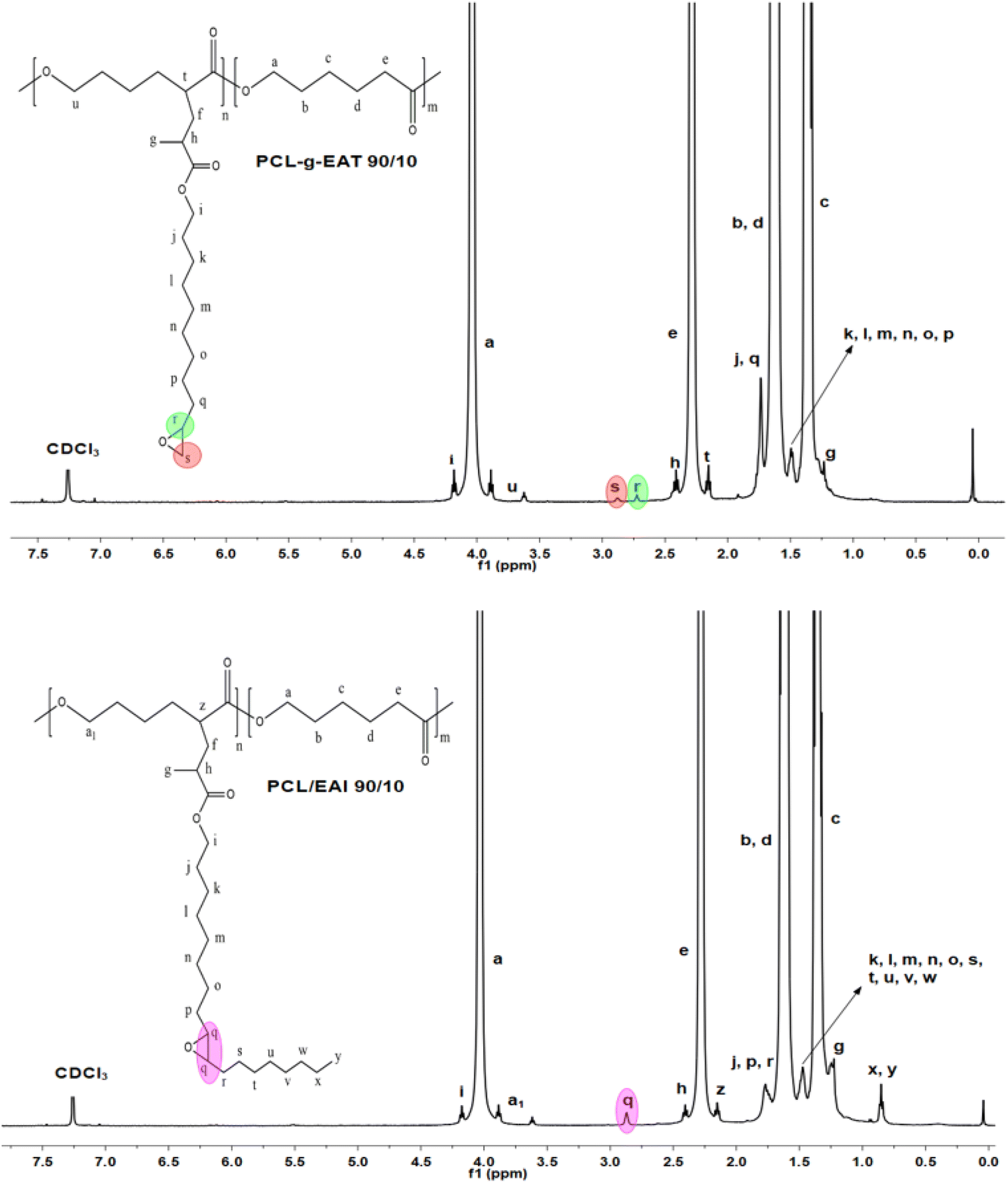 | ||
| Fig. 1 1H-NMR spectra of PCL-g-EAT 90/10 and PCL-g-EAI 90/10. | ||
2.5. General procedure for using PCL–EAI and PCL–EAT as chain extenders in PLA
The moisture of PLA was removed in an oven at 80 °C overnight and mixed with 1 phr of ADR and/or with 1 phr of PCL-g-EAT or PCL-g-EAI. The reactive extrusion of PLA polymer with the chain extender was blended in a DSM Xplore 15cc micro-compounder (The Netherlands) twin-screw extruder at 210 °C, 100 rpm with a 2 min residence time. The extruded sample was conveyed for injection moulding (impact and tensile bars) using DSM micro-injection at 30 °C mold temperature and back pressure of 0.6 MPa.2.6. Cytotoxicity/lactate dehydrogenase (LDH) release assay
The cytotoxic effects of PLA, PLA + ADR, PLA/PCL-g-EAI 90/10, and PLA/PCL-g-EAT 90/10 were determined using the colorimetric CyQUANT™ LDH cytotoxicity assay kit (Thermo Fisher Scientific, Greenville County, SC, USA). Briefly, 2 × 104 cells of the TH-29 cell line (human colorectal adenocarcinoma cells sourced from ATCC) in complete DMEM supplemented with 10% FBS were seeded in triplicate into a 96-well flat-bottom cell culture plate and incubated in a CO2 incubator at 37 °C. Following overnight incubation, the samples were dissolved in chloroform (100 mg ml−1), and then different concentrations (200, 100, 50, 25, 12.5, 6.25, and 3.125 μg mL−1) of PLA, PLA + ADR, PLA/PCL-g-EAI 90/10, and PLA/PCL-g-EAT 90/10 were prepared in DMEM. Subsequently, the cells were exposed to 100 μL of DMEM containing the sample concentrations. Ten microliters of sterile water were added to triplicate wells for the spontaneous release of LDH. The cells were also treated in triplicate with a two-fold serial dilution of chloroform (0.2%, 0.1%, 0.05%, 0.025%, 0.0125%, 0.0065%, and 0.00312%), corresponding to the concentrations carried over with the PLA samples to the treated cells. Additionally, triplicate wells were not treated and served as a negative control. After 24 hours of incubation at 37 °C, 10 μL of 10× lysis buffer was added to triplicate wells for the maximum LDH release. Subsequently, the plate was incubated for 45 min at 37 °C, and supernatants from the untreated cells and PLA sample-, chloroform-, lysis buffer- and water-treated cells were collected.The LDH positive control assay provided in the kit was employed as a positive control in the experiment. The cellular cytotoxicity was measured in the supernatants following the manufacturer's protocol. After measuring the optical density (OD) at 490 nm, the cytotoxicity percentage was calculated using the following formula:
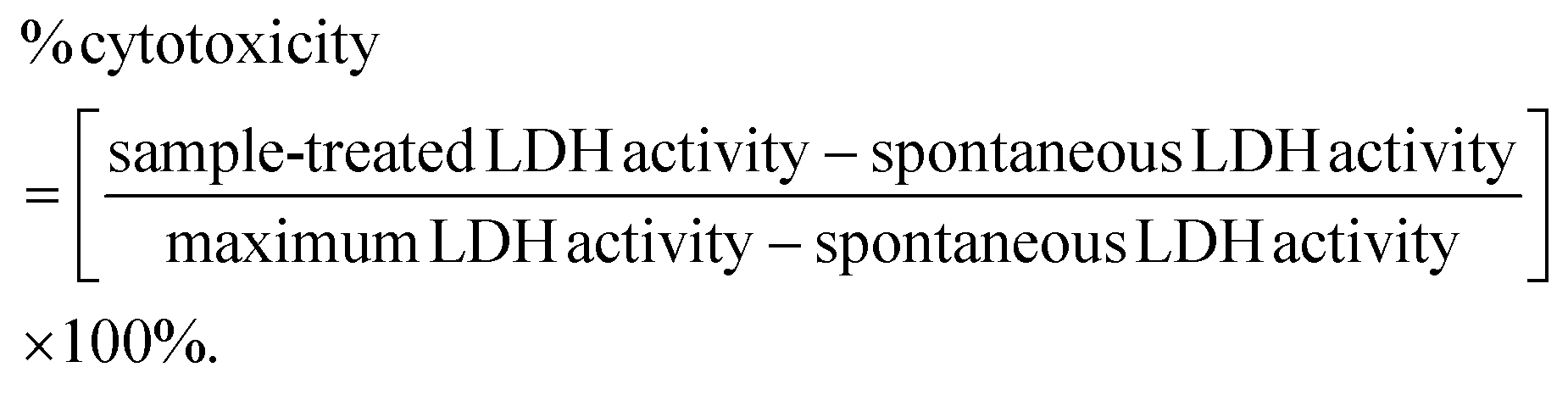
3. Results and discussion
3.1. Synthesis of chain extenders (PCL–EAI and PCL–EAT)
The synthetic routes leading to GMA-free PCL–EAI and PCL–EAT are shown in Scheme 1. The synthesis of EAT and EAI was performed according to ref. 27–29. The IR spectra of EAT and EAI showed that they exhibited carbonyl group stretching at 1716 cm−1 and double bonds at about 1637 cm−1 (Fig. S1†). The 1H-NMR spectrum shown in Fig. S2 and S3† further confirmed the synthesis of EAT and EAI, respectively. In the next step, EAI and EAT were grafted onto PCL via peroxide (BPO) chemistry to obtain PCL–EAI and PCL–EAT. This grafting onto PCL with different ratios of EAT and EAI was performed by melt reactive extrusion at 130 °C.30,31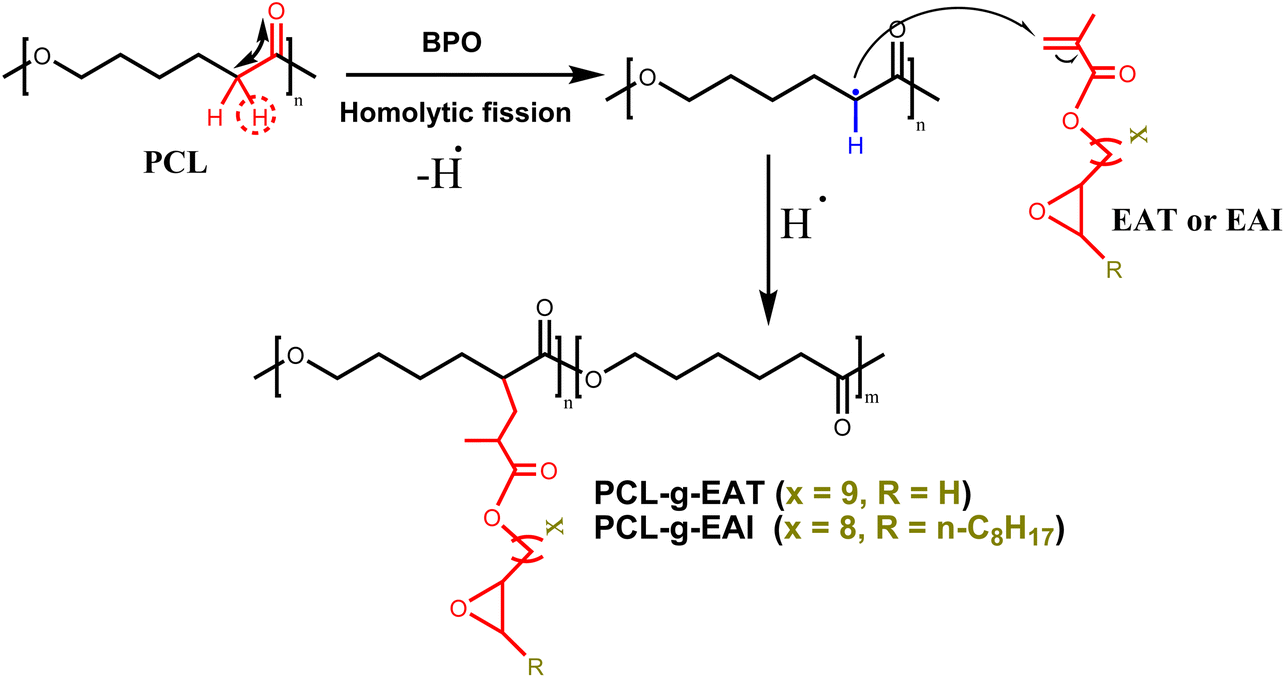 | ||
| Scheme 1 Synthesis of PCL–EAI and PCL–EAT. | ||
The 1H-NMR spectra of PCL-g-EAT and PCL-g-EAI showed the epoxy peaks of grafted EAT at 2.72 and 2.87 ppm, while those of EAI appeared at 2.88 ppm. The grafting degrees (%) were calculated from the 1H-NMR spectra. Based on these calculations, it was found that PCL-g-EAT 95/5 and PCL-g-EAT 90/10 had grafting degrees of 4.87 ± 0.05% and 9.23 ± 0.05%, respectively (Table 1). In addition, the grafting degrees of PCL-g-EAI 95/5 and PCL-g-EAI 90/10 were 4.42 ± 0.04% and 7.45 ± 0.01%, respectively (Table 1). Thus, increasing the amount of epoxy acrylate monomers increases the degree of grafting.
| Polymer | Degree of grafting% from 1H-NMR | η (dl g−1) | M n (kDa) | M w (kDa) | DSC | TGA | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| T c (°C) | T m (°C) | ΔHm (J g−1) | X c (%) | T 10 (°C) | T max (°C) | |||||
| a M n and Mw are the number average molecular weight and weight average molecular weight, respectively. Tc, Tm, ΔHm, and Xc denote the crystallization temperature, melting temperature, melting enthalpy, and crystallization index, respectively, as determined using DSC measurements. T10 is the decomposition temperature at 10 wt% loss and Tmax denotes the maximum derivative peaks, respectively, as determined using TGA measurements. | ||||||||||
| Neat PCL | — | 1.76 ± 0.02 | 53.3 ± 0.02 | 79.7 ± 0.8 | 26.2 ± 0.2 | 56.8 ± 0.2 | 58.2 ± 0.2 | 38.3 ± 0.1 | 380.5 ± 0.4 | 406.6 ± 0.2 |
| PCL-g-EAT 95/5 | 4.87 ± 0.05 | 2.23 ± 0.02 | 66.2 ± 0.5 | 153.0 ± 3.38 | 34.0 ± 1.6 | 58.9 ± 3.5 | 61.1 ± 0.1 | 40.3 ± 0.1 | 371.7 ± 1.7 | 402.4 ± 3.1 |
| PCL-g-EAT 90/10 | 9.23 ± 0.05 | 2.76 ± 0.01 | 75.1 ± 2.5 | 462.0 ± 6.4 | 36.6 ± 0.1 | 59.9 ± 0.1 | 60.6 ± 0.3 | 40.0 ± 0.2 | 369.7 ± 1.6 | 402.8 ± 0.4 |
| PCL-g-EAI 95/5 | 4.42 ± 0.04 | 2.05 ± 0.01 | 60.8 ± 2.7 | 130.4 ± 0.97 | 32.5 ± 0.3 | 59.7 ± 1.5 | 62.2 ± 0.1 | 41.0 ± 0.1 | 367.7 ± 1.8 | 403.8 ± 1.6 |
| PCL-g-EAI 90/10 | 7.45 ± 0.01 | 2.58 ± 0.02 | 66.3 ± 0.6 | 201.2 ± 7.1 | 33.6 ± 0.6 | 60.3 ± 1.6 | 61.2 ± 1.0 | 40.3 ± 0.7 | 358.9 ± 1.0 | 401.3 ± 1.4 |
The reaction of PCL with epoxy acrylates was examined using the torque taken from the melt mixer torque value against time (Fig. 2). The figure shows the feeding of neat PCL and PCL with epoxy acrylate grafted with different ratios. The torque increases at the beginning due to the addition of the polymer pellets, and then the torque decreases over time. The force was increased by adding epoxy acrylate compared to neat PCL. This was due to the increased viscosity that was achieved by adding the chain extender, which suggests that grafting had occurred between PCL and the double bond of EAT and EAI, as illustrated in Scheme 2. Kim et al.31 also observed an increase in force by grafting PCL with the addition of GMA. The torque was increased by increasing the epoxy acrylate (EAT/EAI) content, indicating that the grafting reaction became prominent. The 1H-NMR grafting result supports the torque increase associated with the increase in epoxy acrylate content.
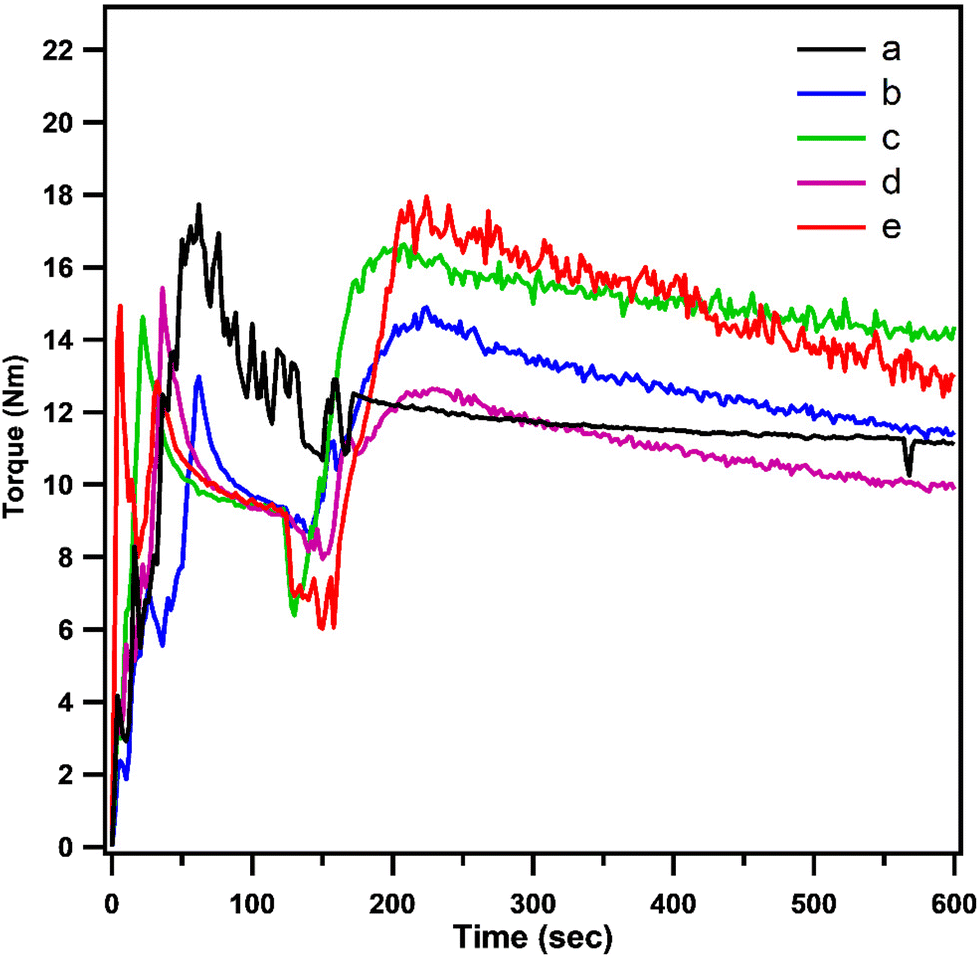 | ||
| Fig. 2 The torque generated during processing in a Brabender system containing neat PCL (a), PCL-g-EAT 95/5 (b), PCL-g-EAT 90/10 (c), PCL-g-EAI 95/5 (d), and PCL-g-EAI 90/10 (e). | ||
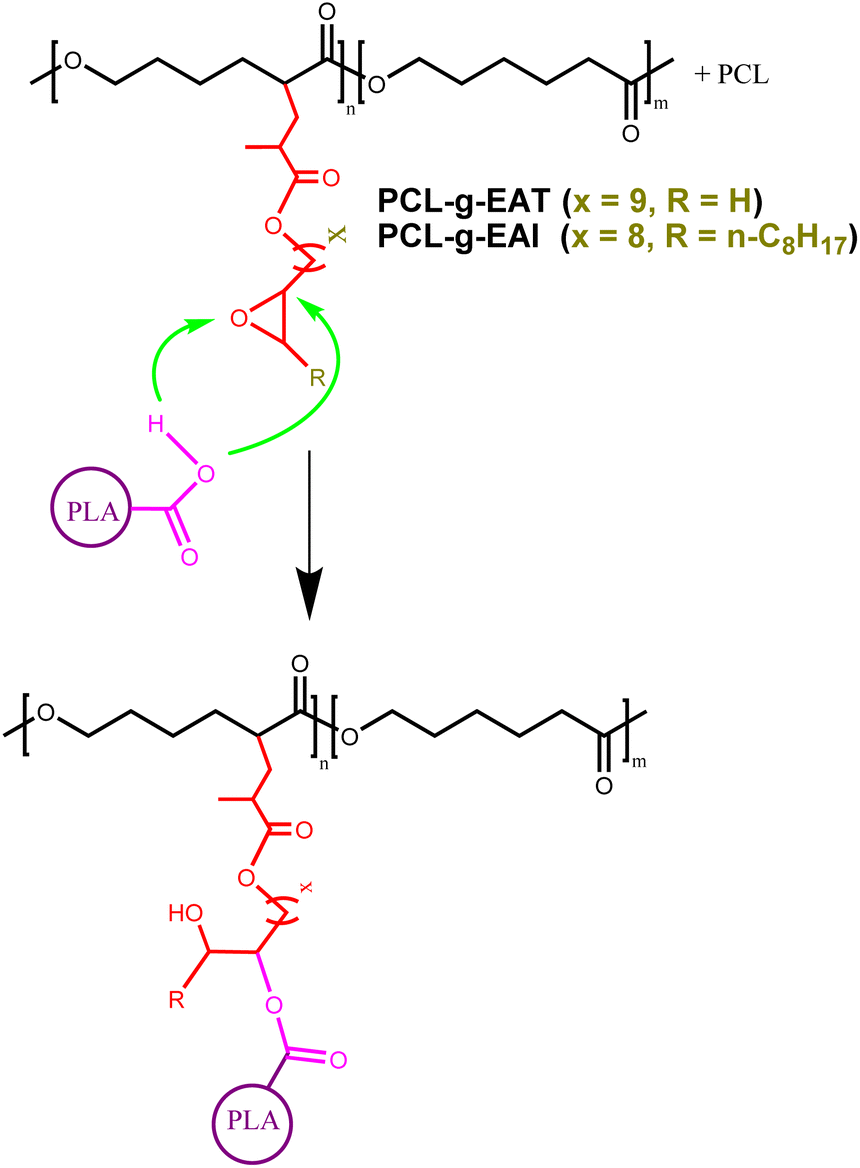 | ||
| Scheme 2 The proposed mechanism for the epoxy reaction with PLA. | ||
The average molecular weights (Mn and Mw) and intrinsic viscosity (IV) of neat PCL and PCL that had been grafted with EAT and EAI (PCL-g-EAT and PCL-g-EAI copolymers, respectively), are shown in Table 1 and Fig. 3. The chemical grafting of PCL with EAT or EAI increased IV, Mn and Mw. Neat PCL exhibited an intrinsic viscosity of 1.75 dl g−1, which increased to 2.22 and 2.78 dl g−1 after grafting with 5 and 10 wt% of EAT, respectively. Similarly, upon grafting with 5 and 10 wt% of EAI, the intrinsic viscosity was raised to 2.05 and 2.58 dl g−1, respectively. SEC analysis illustrated that the melted PCL has Mn and Mw of 53.29 and 79.72 kDa, respectively, which increased to 66.19 and 153.0 kDa after its grafting with 5 wt% of EAT.
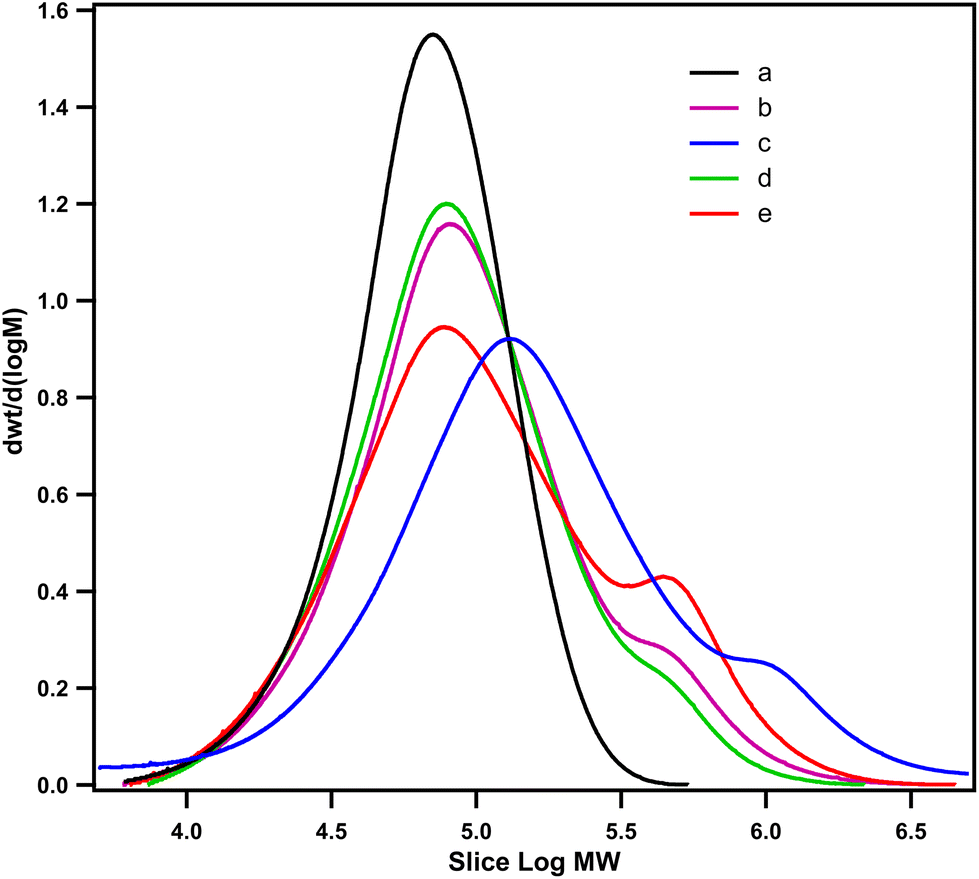 | ||
| Fig. 3 GPC curves of neat PCL (a), PCL-g-EAT 95/5 (b), PCL-g-EAT 90/10 (c), PCL-g-EAI 95/5 (d), and PCL-g-EAI 90/10 (e). | ||
Furthermore, upon the grafting of melted PCL with 5 wt% of EAI, the Mn and Mw are increased to 60.84 and 130.37 kDa, respectively. Additionally, as the amount of EAT and EAI increases, the IV and average molecular weight are also increased because of increases in the grafting degree, as mentioned in Table 1. The degree of grafting and the intrinsic viscosity measurements revealed that EAT showed better grafting with neat PCL than EAI.
Thermal properties of PCL-grafted epoxy acrylate were examined via DSC and TGA. The cooling and 2nd heating cycles of neat PCL and its PCL-epoxy-grafted copolymers are described by DSC as shown in Table 1 and Fig. S4.† The grafting of neat PCL with EAT and/or EAI led to an increase in the resultant graft copolymer's crystallization temperature (Tc) compared to that of neat PCL. For example, the Tc is shifted to a higher temperature from 26.2 °C for neat PCL to 36.6 and 33.6 °C for PCL-g-EAT 90/10 and PCL-g-EAI 90/10, respectively. It was noticed that PCL-g-EAT 90/10 had a higher Tc than that of PCL-g-EAT 95/5.
Similarly, PCL-g-EAI 90/10 showed a higher Tc than that of PCL-g-EAI 95/5. Thus, as the amount of grafted epoxy acrylate increases, Tc also increases. In addition, the PCL-grafted epoxy acrylates displayed a shift to higher values in terms of Tm, ΔHm, and Xc as compared to the corresponding values observed for neat PCL. The change in these parameters is referred to as the difference in the structural morphology of PCL upon grafting with EAT and EAI. These results agree with the previous study that explained that the grafting of PCL with GMA and diethyl maleate caused a change in the thermal behavior of neat polymer.22
The thermal stabilities of neat PCL and PCL-grafted epoxy acrylate were determined by TGA analysis, as shown in Fig. S5† and Table 1. The DTG curves show that neat PCL and PCL-g-EAT and PCL-g-EAI underwent a single degradation step with a maximum degradation temperature (Tmax) of approximately 407 °C.22 The PCL-g-EAT and PCL-g-EAT graft copolymers decomposed at lower temperatures than neat PCL, as indicated by the values of T10 and Tmax (Table 1). Thus, the chemical grafting of EAT and EAI onto the chains of neat PCL decreased their thermal stabilities compared to that of neat PCL alone. Moreover, the TGA data indicated that PCL-g-EAT was slightly less thermally stable than PCL-g-EAI. By increasing the amount of EAT and EAI that had been grafted onto PCL, the thermal stability was decreased.
3.2. Effect of chain extender on the PLA structure
| Polymer | M n (kDa) | M w (kDa) | M w/Mna | Transmittance% at 660 nm |
|---|---|---|---|---|
| a M w/Mn: the molecular weight distribution and all the data are relative molecular weights with respect to polystyrene. | ||||
| Neat PLA | 39.5 ± 1.4 | 70.4 ± 0.9 | 1.78 ± 0.04 | 74.89 ± 0.79 |
| PLA + ADR | 92.41 ± 0.7 | 241.6 ± 3.8 | 2.61 ± 0.69 | 21.56 ± 1.39 |
| PLA/PCL-g-EAT 95/5 | 38.5 ± 2.7 | 69.9 ± 0.2 | 1.82 ± 0.12 | 54.99 ± 0.79 |
| PLA/PCL-g-EAT 90/10 | 41.0 ± 2.3 | 68.7 ± 1.2 | 1.67 ± 0.06 | 54.61 ± 0.99 |
| PLA/PCL-g-EAI 95/5 | 38.5 ± 2.1 | 66.4 ± 0.8 | 1.72 ± 0.07 | 51.28 ± 1.02 |
| PLA/PCL-g-EAI 90/10 | 37.8 ± 0.4 | 64.2 ± 0.7 | 1.69 ± 0.03 | 52.14 ± 0.01 |
1,2-Epoxy octane was used as a proxy for terminal glycidol-free epoxy, epoxidized plant oil was used as a substitute for glycidol-free internal epoxy, and 1,4-butane diol-diglycidyl was used as a proxy for glycidol epoxy. As PLA reacts with epoxy through its terminal COOH, we therefore used stearic acid as a model compound. After 5 min of heating at 205 °C, we observed new peaks appearing in the NMR spectra for the ring opening of the epoxy with COOH following reaction between the epoxy model compounds and stearic acid (see details in the ESI (Schemes S1–S3 and Fig. S7–S9†)). Our model studies offer conclusive evidence demonstrating that both internal and external epoxy moieties interact with COOH groups. Future research will delve into the kinetics of these reactions, seeking to understand their relative reactivity and any potential by-products that might arise. However, there was no significant molecular weight increase for the combination of PLA and the grafted PCL samples. Given the variation and low level of grafting density noted in Table 1, and that the loading was 1 phr for the grafted PCL with PLA, it could be possible that the loading was too low to statistically ensure that linkages were formed between the PLA and PCL by the chain extenders. However, apparently, it was sufficient for enhancing the mechanical properties of PLA, as discussed in Section 3.2.2.
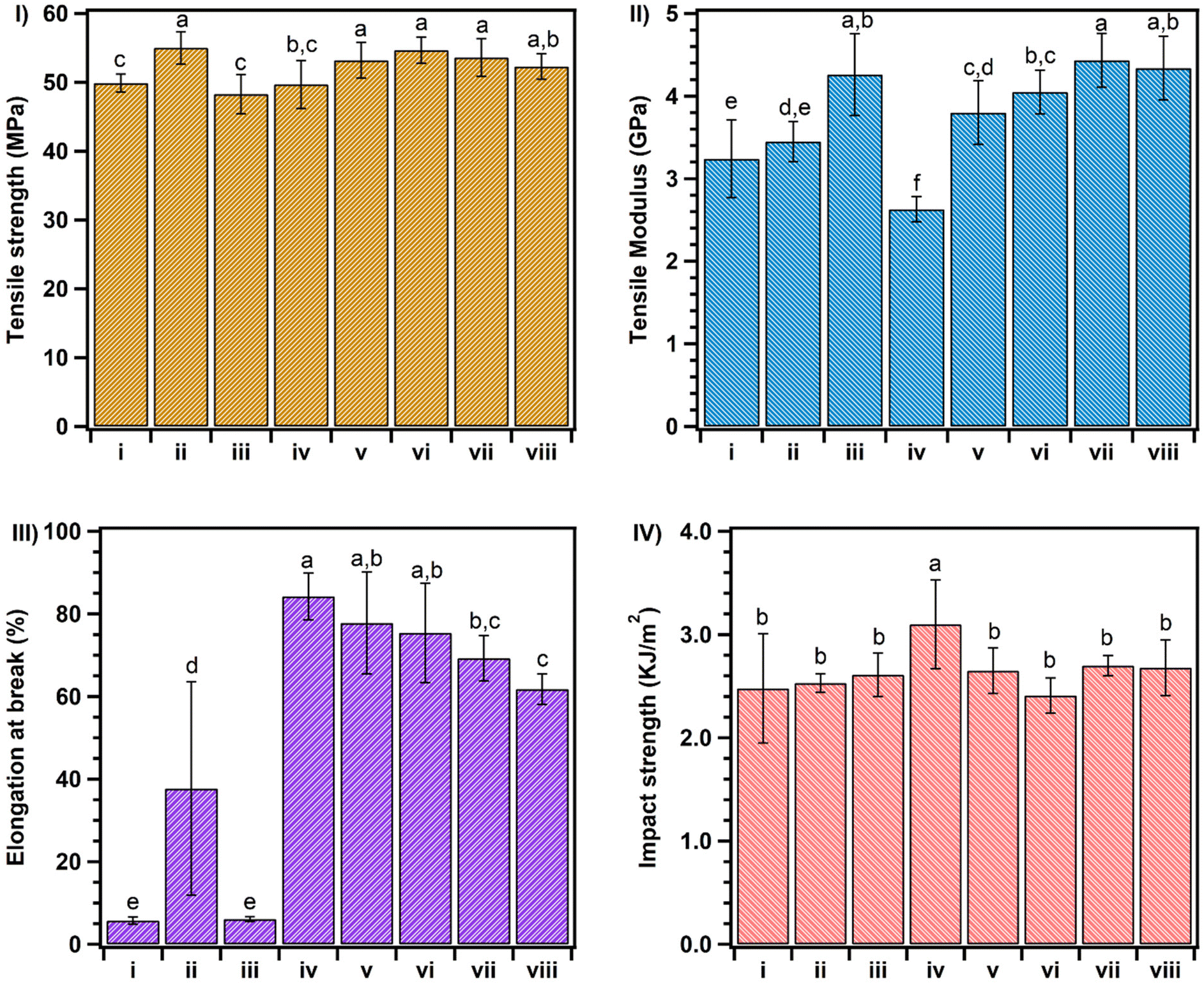 | ||
| Fig. 4 Mechanical properties of PLA samples with different weight ratios of chain extenders. These mechanical properties include (I) the tensile strength, (II) the tensile modulus, (III) elongation at break, and (IV) notched impact strength. The roman numerals for the corresponding samples are as follows: (i) neat PLA, (ii) PLA/ADR, (iii) PLA + 1 phr PCL, (iv) PLA/PCL-g-butyl acrylate, (v) PLA/PCL-g-EAT 95/5, (vi) PLA/PCL-g-EAT 90/10, (vii) PLA/PCL-g-EAI 95/5, and (viii) PLA/PCL-g-EAI 90/10. –Means with the same letter within a graph indicate no significant (P > 0.05) difference using the Duncan multiple range test (calculated with SAS version 9.3 software). | ||
The same result was achieved by incorporating the PCL copolymers (PCL-g-EAT or PCL-g-EAI), which showed comparable or better mechanical properties than those provided by ADR. For example, PLA with 1 phr of PCL-g-EAT 95/5 showed equivalent tensile strength (53.2 MPa) and modulus (3.8 GPa) as well as impact strength (2.65 kJ m−2) compared to PLA with 1 phr of ADR. However, it showed significant enhancement in the elongation at break (77.8%), corresponding to 14- and 2-fold enhancements compared to neat PLA and PLA/ADR, respectively. PLA that had been treated with 1 phr of PCL-g-EAI 95/5 also showed high mechanical strength compared to neat PLA and PLA/ADR. This can be attributed to the interaction between the epoxy groups of the chain extender and the carboxyl groups of PLA.32,35 PLA that had been treated with PCL-g-EAT showed comparable tensile and impact strengths to those of PLA that had been treated with PCL-g-EAI. However, the blend of PLA with PCL-g-EAT showed higher elongation at break and lower tensile modulus than that of PLA and PCL-g-EAI. This may be due to the difference in the reactivity of the epoxy group between PCL-g-EAI (internal epoxy group) and PCL-g-EAT (terminal epoxy group). To confirm the effect of the new chain extender and the reaction of the epoxy groups of the PCL copolymer with the carboxyl groups of PLA, we melt-blended PLA with 1 phr of unmodified PCL, and no enhancement in the mechanical properties was observed except for the tensile modulus (which was increased by ∼31%). This means that no reaction occurred, and the crystallinity of the materials increased, so that the sample became more rigid. However, the addition of 1 phr of PCL-g-butyl acrylate (PCL-g-BA) led to an enhancement of all the mechanical characteristics except for the tensile modulus. This indicates that PCL-g-BA acts as a plasticizer for PLA. Thus, the new chain extenders PCL-g-EAT and PCL-g-EAI worked as chain extenders and enhanced both the toughness and modulus of neat PLA.
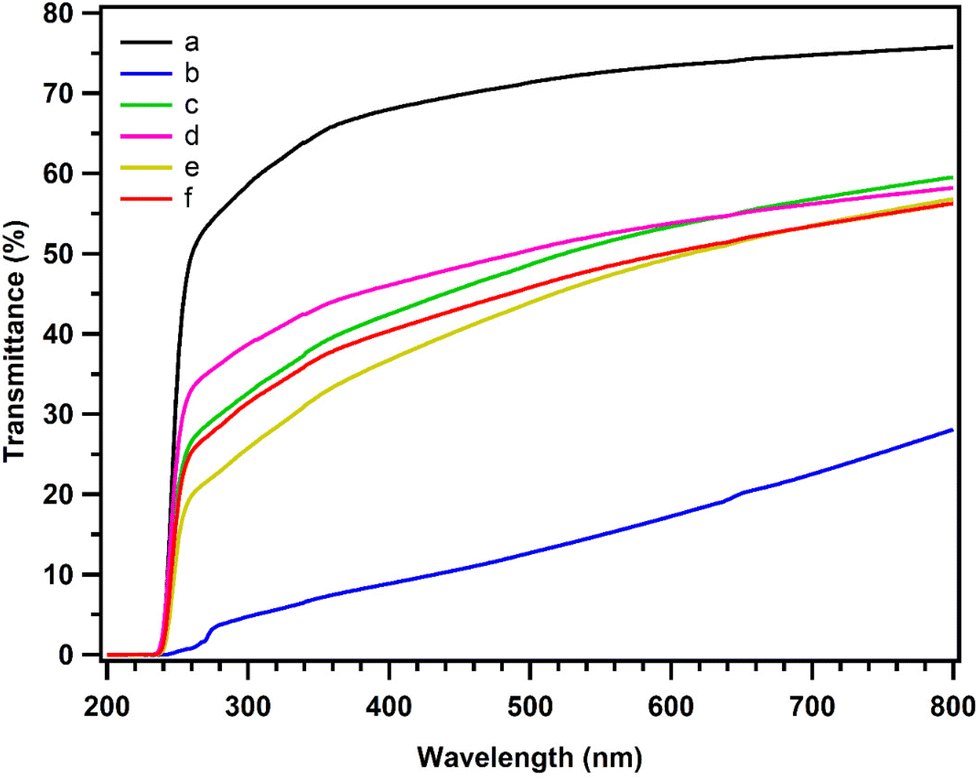 | ||
| Fig. 5 UV spectra of neat PLA (a), PLA/Joncryl ADR (b), PLA/PCL-g-EAT 95/5 (c), PLA/PCL-g-EAT 90/10 (d), PLA/PCL-g-EAI 95/5 (e), and PLA/PCL-g-EAI 90/10 (f). | ||
| Name | TGA data | DSC data | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T 2 (°C) | T max (°C) | T g (°C) | T c (°C) | ΔHc | T m1 (°C) | T m2 (°C) | ΔHm (°C) | X c (%) | |
| a T 2 denotes the decomposition temperature at 2 wt% loss and Tmax is the maximum derivative peak, as determined using TGA measurements. Tm and Tc are the melting and cooling temperatures, respectively. Tg denotes the glass transition temperature, ΔHm is the enthalpy of melting, and Xc is the degree of crystallinity. These data were determined through DSC measurements of the second heating scans. | |||||||||
| Neat PLA | 316.7 ± 1.6 | 362.6 ± 2.0 | 63.7 ± 0.9 | 112.1 ± 0.7 | 33.4 ± 1.0 | 160.7 ± 0.4 | 167.8 ± 0.3 | 36.3 ± 1.2 | 3.1 ± 0.1 |
| PLA/ADR | 323.4 ± 0.5 | 368.6 ± 0.4 | 63.1 ± 1.4 | — | — | — | 164.1 ± 1.0 | — | — |
| PLA/PCL-g-EAT 95/5 | 314.4 ± 0.8 | 363.3 ± 1.6 | 61.4 ± 0.3 | 110.6 ± 0.6 | 31.6 ± 0.3 | 160.1 ± 0.1 | 167.7 ± 0.1 | 34.6 ± 0.3 | 3.3 ± 0.1 |
| PLA/PCL-g-EAT 90/10 | 314.0 ± 0.7 | 364.6 ± 0.7 | 62.6 ± 2.2 | 112.9 ± 1.6 | 33.8 ± 0.5 | 160.9 ± 0.7 | 167.8 ± 0.1 | 37.0 ± 0.4 | 3.5 ± 0.01 |
| PLA/PCL-g-EAI 95/5 | 313.1 ± 1.1 | 363.6 ± 1.1 | 61.7 ± 0.9 | 110.6 ± 2.2 | 32.62 ± 0.21 | 160.2 ± 0.9 | 167.8 ± 0.3 | 35.7 ± 0.1 | 3.3 ± 0.1 |
| PLA/PCL-g-EAI 90/10 | 313.5 ± 1.4 | 364.2 ± 0.3 | 62.6 ± 0.1 | 112.4 ± 1.6 | 33.6 ± 1.2 | 160.6 ± 0.9 | 167.7 ± 0.3 | 36.8 ± 1.4 | 3.4 ± 0.1 |
The second heating cycle of neat PLA and its counterparts that had been chain-extended with ADR and the graft copolymers PCL-g-EAT and PCL-g-EAI were recorded via DSC, as shown in Fig. 6 and Table 3. The DSC thermograms illustrated that there were no significant differences in the Tc, Tm1, and Tm2 of neat PLA and PLA that had been treated with the chain extenders. However, the reaction of PLA with the PCL-epoxy-grafted copolymers resulted in a slight decrease in Tg. After incorporating ADR into neat PLA, the Tc disappeared, Tm decreased slightly, and the double peaks disappeared. These changes were observed because the polymer crystallization was hindered as the molecular weight increased, leading to the production of only one type of crystal (α-ordered phase).37,41
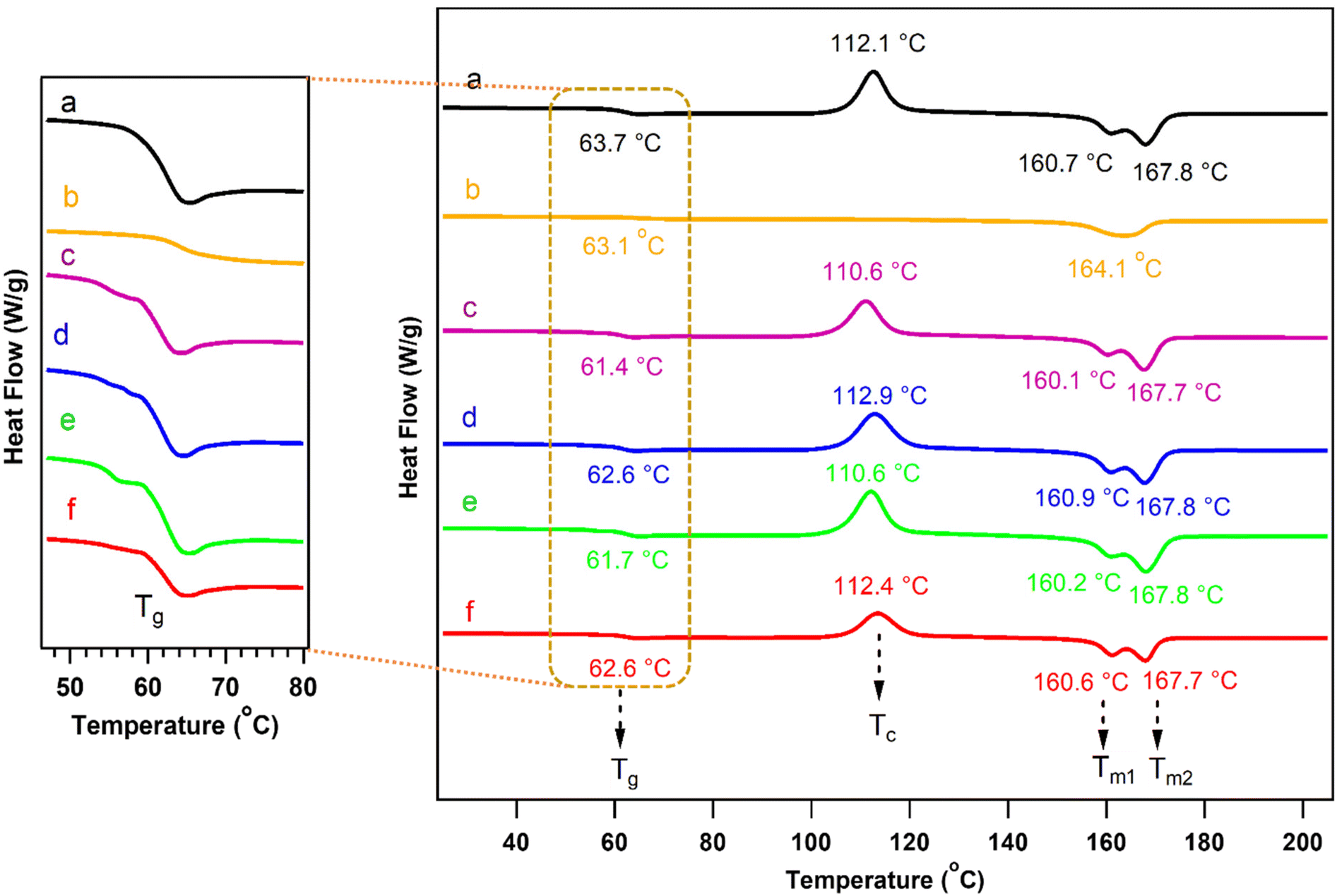 | ||
| Fig. 6 DSC thermograms (recorded during the second heating cycle) for neat PLA (a), PLA/Joncryl ADR (b), PLA/PCL-g-EAT 95/5 (c), PLA/PCL-g-EAT 90/10 (d), PLA/PCL-g-EAI 95/5 (e), and PLA/PCL-g-EAI 90/10 (f). Magnifications of the DSC graphs are provided at the left to show the Tg temperature. | ||
Neat PLA and PLA with lower grafting amounts of the PCL copolymers (PCL-g-EAT 95/5 and PCL-g-EAI 95/5) showed equivalent crystallinity values. However, the PLA samples with higher PCL copolymer grafting (PCL-g-EAT 90/10 and PCL-g-EAI 90/10) showed a shift in the % of crystallinity to a higher value than that of neat PLA. Therefore, increasing the epoxy content of PCL-epoxy-grafted copolymers from 5 to 10%, led to an enhancement of the % crystallinity.
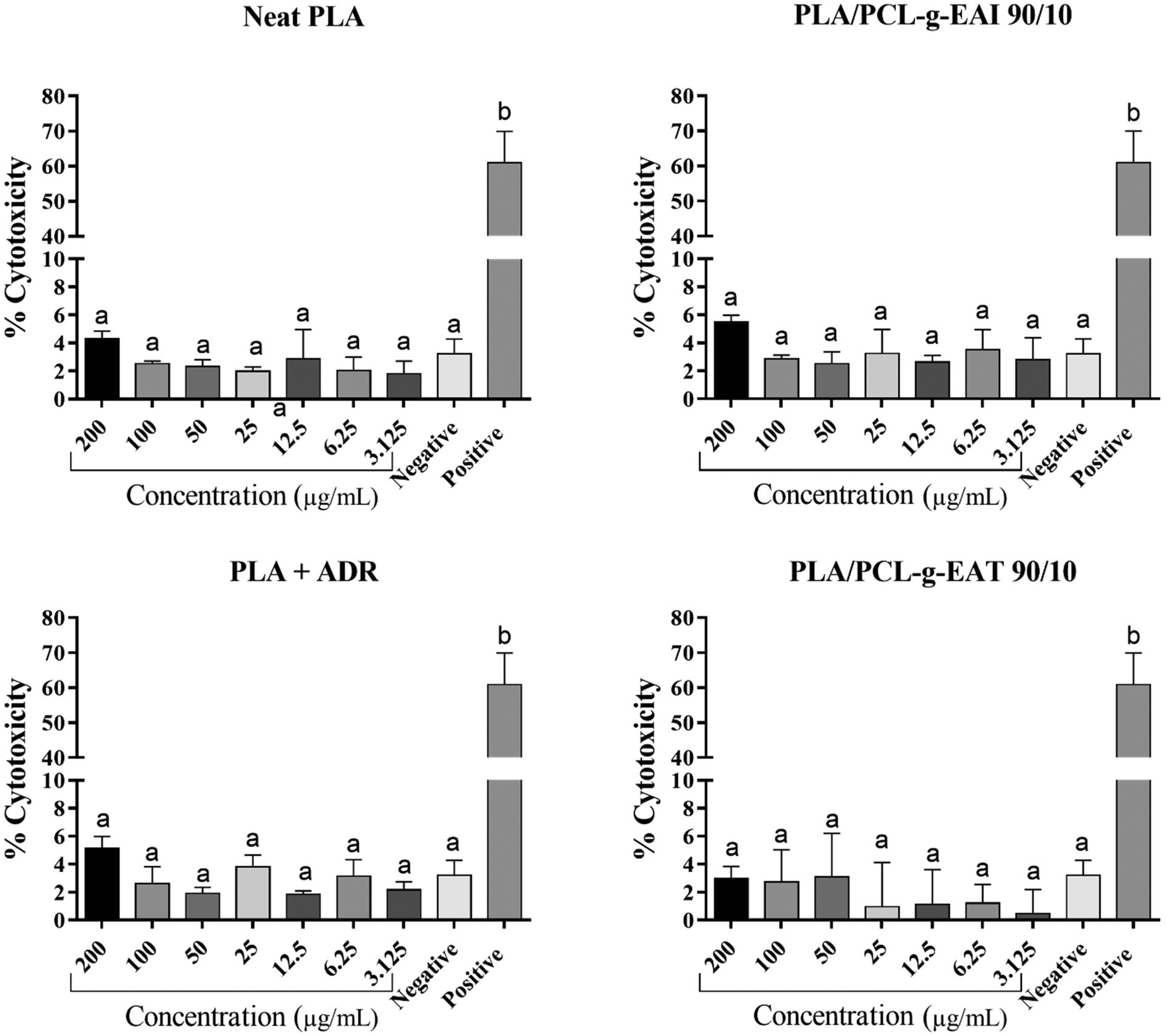 | ||
| Fig. 7 Cytotoxicity test for neat PLA, PLA/Joncryl ADR, PLA/PCL-g-EAI 90/10, PLA/PCL-g-EAT 90/10. Means with the same letter within a graph indicate no significant (P > 0.05) difference using the Duncan multiple range test. | ||
4. Conclusion
In summary, for the first time, PCL-functionalized internal and external reactive epoxy groups were successfully synthesized as evident from NMR spectroscopy, SEC, and intrinsic viscosity analysis. The obtained PCL-epoxy polymers were used as chain extenders for the polyester in PLA. Even though no increase in the average molecular weight of PLA was observed upon the use of PCL-g-EAT or PCL-g-EAI, the chain extension of PLA with these graft copolymers (1 phr) yielded excellent mechanical properties, which were better than those achieved with the commercial chain extender ADR. PLA with PCL-g-EAT 95/5 (1 phr) exhibited comparable tensile strength (53.2 MPa), modulus (3.8 GPa), and impact strength (2.65 kJ m−2) compared to those of PLA with 1 phr ADR. Furthermore, it showed a significant increase in elongation at break (77.8%), which is 14 and 2 times greater than those of neat PLA and PLA + ADR, respectively. The treatment of PLA with 1 phr of PCL-g-EAT led to a higher elongation at break compared to that observed for PLA that had been treated with PCL-g-EAI. TGA and DSC data demonstrated that the thermal characteristics of the PLA systems that had been treated with the graft copolymers have no significant difference relative to those of neat PLA. No cytotoxic effects were observed in the cells at any tested concentrations of the PLA samples with the new chain extenders. This work opens new opportunities for improving the performance of biodegradable PLA polymers without using harmful glycidol-based ADR chain extenders, which may provide uses in areas such as food packaging.Conflicts of interest
The authors declare no competing financial interest. H.M.E and M. A. A. are on leave from the Department of Chemistry, Faculty of Science, Tanta University, Tanta, 31527, Egypt.Acknowledgements
The authors thank the Environmental Research and Education Foundation for partially supporting this project.References
- T. Singh, P. Pattnaik, A. Aherwar, L. Ranakoti, G. Dogossy and L. Lendvai, Polymers, 2022, 14, 2603 CrossRef CAS PubMed.
- B. L. C Cunha, J. O. Bahú, L. F. Xavier, S. Crivellin, S. D. A. de Souza, L. Lodi, A. L. Jardini, M. I. R. B. Filho, M. Schiavon, V. O. C. Concha, P. Severino and E. B. Souto, Bioengineering, 2022, 9, 164 CrossRef CAS PubMed.
- A. N. Frone, M. Ghiurea, C. A. Nicolae, A. R. Gabor, S. Badila and D. M. Panaitescu, Molecules, 2022, 27, 2390 CrossRef CAS PubMed.
- A. A. Pérez-Fonseca, V. S. Herrera-Carmona, Y. Gonzalez-García, A. S. Martin del Campo, M. E. González-López, D. E. Ramírez-Arreola and J. R. Robledo-Ortíz, Polym. Adv. Technol., 2021, 32, 3483–3494 CrossRef.
- M. A. Abdelwahab, S. Jacob, M. Misra and A. K. Mohanty, Polymer, 2021, 212, 123153 CrossRef CAS.
- B.-K. Chen, C.-H. Shen, S.-C. Chen and A. F. Chen, Polymer, 2010, 51, 4667–4672 CrossRef CAS.
- S. Lin, W. N. Guo, C. Y. Chen, J. L. Ma and B. B. Wang, Mater. Des., 2012, 36, 604–608 CrossRef CAS.
- Y. Hao, Y. Li, Z. Liu, X. Yan, Y. Tong and H. Zhang, Fibers Polym., 2019, 20, 1766–1773 CrossRef CAS.
- N. Najafi, M. Heuzey, P. Carreau and P. M. Wood-Adams, Polym. Degrad. Stab., 2012, 97, 554–565 CrossRef CAS.
- M. Mihai, M. A. Huneault and B. D. Favis, Polym. Eng. Sci., 2010, 50, 629–642 CrossRef CAS.
- M. A. Abdelwahab, S. Taylor, M. Misra and A. K. Mohanty, Macromol. Mater. Eng., 2015, 300, 299–311 CrossRef CAS.
- Y. Li, J. Mi, H. Fu, H. Zhou and X. Wang, ACS Omega, 2019, 4, 12512–12523 CrossRef CAS PubMed.
- Y. Kahraman, Y. Alkan Goksu, B. Özdemir, B. Eker Gümüş and M. Nofar, J. Appl. Polym. Sci., 2022, 139, 51833 CrossRef CAS.
- M. A. Abdelwahab, A. Khan, L. M. Matuana, R. Auras and M. Rabnawaz, ACS Appl. Polym. Mater., 2022, 4, 9272–9283 CrossRef CAS.
- M. A. Abdelwahab, B. P. Chang, A. K. Mohanty and M. Misra, Polym. Test., 2022, 114, 107681 CrossRef CAS.
- N. Yahyaee, A. Javadi, H. Garmabi and A. Khaki, Macromol. Mater. Eng., 2020, 305, 1900423 CrossRef CAS.
- J. J. Benvenuta-Tapia, P. Champagne, J. A. Tenorio-López, E. Vivaldo-Lima and R. Guerrero-Santos, Polymers, 2021, 13, 2791 CrossRef CAS PubMed.
- J. Aasa, F. Granath and M. Törnqvist, Food Chem. Toxicol., 2019, 128, 54–60 CrossRef CAS PubMed.
- A. Samir, F. H. Ashour, A. A. A. Hakim and M. Bassyouni, npj Mater. Degrad., 2022, 6, 68 CrossRef CAS.
- R. Ilyas, M. Zuhri, M. N. F. Norrrahim, M. S. M. Misenan, M. A. Jenol, S. A. Samsudin, N. Nurazzi, M. Asyraf, A. Supian and S. P. Bangar, Polymers, 2022, 14, 182 CrossRef CAS PubMed.
- Z. U. Arif, M. Y. Khalid, R. Noroozi, A. Sadeghianmaryan, M. Jalalvand and M. Hossain, Int. J. Biol. Macromol., 2022, 218, 930–968 CrossRef CAS PubMed.
- A. K. Sugih, J. P. Drijfhout, F. Picchioni, L. P. Janssen and H. J. Heeres, J. Appl. Polym. Sci., 2009, 114, 2315–2326 CrossRef CAS.
- M. P. Guaras, V. A. Alvarez and L. N. Ludueña, J. Polym. Res., 2015, 22, 1–12 CrossRef CAS.
- The new plastics economy: Rethinking the future of plastics, Ellen MacArthur Foundation, 2016, https://www.ellenmacarthurfoundation.org/publications/the-new-plastics-economy-rethinking-the-future-of-plastics.
- A. C. De Figueiredo, J. M. Anaya-Mancipe, A. O. d. S. De Barros, R. Santos-Oliveira, M. L. Dias and R. M. d. S. M. Thiré, Molecules, 2022, 27, 5351 CrossRef CAS PubMed.
- D. Nakayama, F. Wu, A. K. Mohanty, S. Hirai and M. Misra, ACS Omega, 2018, 3, 12412–12421 CrossRef CAS PubMed.
- M. Feigel, J. M. Breitsameter, K. Lechner, B. Rieger and O. Hinrichsen, Ind. Eng. Chem. Res., 2023, 62, 13389–13400 CrossRef CAS.
- J. Mestry, M. A. Abdelwahab, H. M. Elkholy and M. J. R. Rabnawaz, Resour. Conserv. Recycl., 2024, 203, 107420 CrossRef CAS.
- N. Neumann, S. Thinius, G. Abels, A. Hartwig, K. Koschek and L. Boskamp, Polymer, 2023, 276, 125886 CrossRef CAS.
- J. John, J. Tang, Z. Yang and M. Bhattacharya, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1139–1148 CrossRef CAS.
- C.-H. Kim, K. Cho and J.-K. Park, Polymer, 2001, 42, 5135–5142 CrossRef CAS.
- M. A. Ghalia and Y. Dahman, Int. J. Biol. Macromol., 2017, 95, 494–504 CrossRef PubMed.
- A. Jaszkiewicz, A. K. Bledzki, A. Duda, A. Galeski and P. Franciszczak, Macromol. Mater. Eng., 2014, 299, 307–318 CrossRef CAS.
- P. Giri, C. Tambe and R. Narayan, Biomass Extrusion and Reaction Technologies: Principles to Practices and Future Potential, American Chemical Society, 2018, ch. 1, vol. 1304, pp. 1–15 Search PubMed.
- S. R. Rathi, E. B. Coughlin, S. L. Hsu, C. S. Golub, G. H. Ling and M. J. Tzivanis, Polymers, 2014, 6, 1232–1250 CrossRef.
- J.-W. Rhim, S.-I. Hong and C.-S. Ha, LWT – Food Sci. Technol., 2009, 42, 612–617 CrossRef CAS.
- M.-E. Grigora, Z. Terzopoulou, K. Tsongas, P. Klonos, N. Kalafatakis, D. N. Bikiaris, A. Kyritsis and D. Tzetzis, Polymers, 2021, 13, 1381 CrossRef CAS PubMed.
- A. Hambardzumyan, L. Foulon, B. Chabbert and V. Aguie-Beghin, Biomacromolecules, 2012, 13, 4081–4088 CrossRef CAS PubMed.
- S. Duangphet, D. Szegda, J. Song and K. Tarverdi, J. Polym. Environ., 2014, 22, 1–8 CrossRef CAS.
- Y.-M. Corre, J. Duchet, J. Reignier and A. Maazouz, Rheol. Acta, 2011, 50, 613–629 CrossRef CAS.
- S. S. Karkhanis and L. M. Matuana, Polym. Eng. Sci., 2019, 59, 2211–2219 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04200f |
| ‡ Both these authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |