
Metal- and additive-free TfOH catalyzed chemoselective O- and S-trifluoroethylation of oxindoles, isoindolines and thio-oxindoles†
Manisha
Lamba
 ,
Prasoon Raj
Singh
,
Shubham
Bhatt
and
Avijit
Goswami
*
,
Prasoon Raj
Singh
,
Shubham
Bhatt
and
Avijit
Goswami
*
Department of Chemistry, Indian Institute of Technology Ropar, Rupnagar, Punjab-140001, India. E-mail: agoswami@iitrpr.ac.in
First published on 23rd November 2023
Abstract
Fluorine-containing moieties are of great interest in the development of new synthetic methods due to their considerable medicinal value. Thus, researchers have explored direct trifluoroethylation using CF3CH2-containing precursors; however, the high cost, prolonged synthesis time, toxic nature, and instability towards transition metal catalysis of these precursors pose challenges. As an alternative, conducting trifluoroethylation by in situ production of 2,2,2-trifluorodiazoethane (CF3CHN2) from CF3CH2NH2·HCl in solution is a comparatively easy, safe, and greener approach. However, previous reports using 2,2,2-trifluorodiazoethane in solution also demonstrate the use of transition metal-catalyzed pathways under harsh reaction conditions and hazardous solvents. Herein, we have developed a triflic acid (a commercially available and cheap Brønsted acid) catalyzed protocol for O-trifluoroethylation of 3,3-disubstituted, monosubstituted, unsubstituted, and chalcone-based oxindoles and isoindolines, and S-trifluoroethylation of thio-oxindoles in the presence of in situ generated 2,2,2-trifluorodiazoethane in solution. This highly efficient metal- and additive-free strategy offers a mild and feasible approach to achieve chemoselective trifluoroethylation with good to excellent yields in a very short reaction time (10 minutes). This methodology has a broad substrate scope, making it a versatile approach that can be effectively employed for all types of cyclic amides. Additionally, the method is scalable for larger operations. The calculation of various green chemistry metrics, such as high atom economy (93%), high carbon efficiency (100%), high reaction mass efficiency (74%), and low E-factor(0.40 kg waste per kg product), confirms the eco-friendliness of this approach. The EcoScale evaluation showcases the simplicity, effectiveness, and eco-friendliness of this approach.
Introduction
The trifluoromethyl moiety (–CF3) is a prevalent bioisostere in a number of natural and agrochemical products and plays multifarious roles. In particular, the introduction of fluorine into a compound has the capability of influencing its physiochemical properties and bioactivity such as metabolic stability, acidity or basicity, and lipophilicity.1 Many CF3-containing drugs are under clinical trials,2 while many more have already been approved by the Food and Drug Administration (FDA).3 In particular, interest in these functionalities has greatly enhanced after recognizing the extensive involvement of isotopically labeled CF3 groups in biomolecules for imaging purposes.4 Besides their roles in active pharmaceutical ingredients (APIs) and radiolabeling, these moieties are widely accepted as solvents or additives [e.g., hexfluoroisopropanol (HFIP), trifluoroethanol (TFE) and trifluoroethanedithiol (TFET)] used extensively in various chemical and biological transformations.5In this regard, protocols for trifluoromethylation are extensively explored.6 In sharp contrast, the corresponding homologous trifluoroethylation is relatively less reported. Furthermore, chemoselective trifluoroethylation is more challenging for amides, thioamides, enolates, oximes, nitrites, etc. due to the presence of more than one reaction centre. Despite various potential utilities of the trifluorethyl group in many biologically active compounds (Fig. 1(i)),7 strategies for introducing it are rare. In order to enhance the success in the ongoing drug discovery studies, there is high demand to synthesize a library of molecules that can imitate the structural and functional characteristic features found in many pharmaceutical and natural products. Oxindoles are one of these classes of cyclic amides which are naturally occurring alkaloids and have potential for various bioactivities (Fig. 1(ii)).8 These amide frameworks offer both opportunities and challenges for late-stage functionalization as they have intrinsic directing groups and show amide–iminol tautomerism.
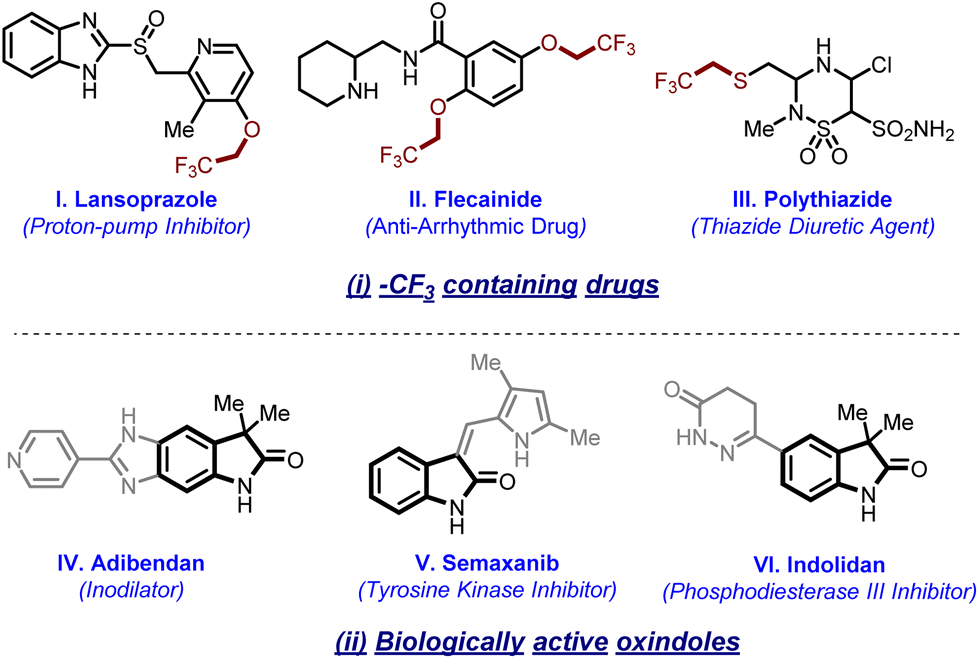 | ||
| Fig. 1 Representative examples of (i) –CF3-containing drugs and biomarkers and (ii) biologically active 2-oxindoles. | ||
In the context of fluorine-containing moieties and developing new modes of synthesizing them, direct trifluoroethylation using CF3CH2-containing precursors, viz., CF3CHCl2, CF3CH2I, [ArI(CH2CF3)]+(OTf)−, etc., has been mainly explored, but either these motifs are expensive or they require days for their synthesis.9 Moreover, the instability of these alkyl electrophiles towards transition metal-catalyzed pathways (CF3CH2-M) leads to hydrodehalogenation (CF3CH3) or β-F elimination (CF2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) in coupling reactions.10 On the other hand, trifluoroethylation can easily be achieved by in situ generation of 2,2,2-trifluorodiazoethane (CF3CHN2) in solution from CF3CH2NH2·HCl.11 Although safety studies specific to CF3CHN2 in solution have not been carried out to our knowledge, Pavel K. Mykhailiuk suggests, “it is safer to use its diluted solutions in organic solvents generated either in situ or in flow.”12 CF3CHN2 shows a variety of reactivity modes and tends to react as an N-electrophile and a C-nucleophile. In addition, it also exhibits reactivity via CF3CH carbene and undergoes cycloadditions.12 To date, only one report illustrates the trifluoroethylation of acyclic amides using CF3CHN2 in the presence of a silver(I) catalyst (Scheme 1a).13 Moreover, chemoselective installation of the trifluoroethyl moiety in cyclic amides and thioamides is still unexplored. As a part of our ongoing research on exploring the reactivity of oxindoles and thio-oxindoles,14 we have unfolded the metal- and additive-free TfOH catalyzed chemoselective trifluoroethylation of oxindoles, isoindolines, and thio-oxindoles (Scheme 1b).
CH2) in coupling reactions.10 On the other hand, trifluoroethylation can easily be achieved by in situ generation of 2,2,2-trifluorodiazoethane (CF3CHN2) in solution from CF3CH2NH2·HCl.11 Although safety studies specific to CF3CHN2 in solution have not been carried out to our knowledge, Pavel K. Mykhailiuk suggests, “it is safer to use its diluted solutions in organic solvents generated either in situ or in flow.”12 CF3CHN2 shows a variety of reactivity modes and tends to react as an N-electrophile and a C-nucleophile. In addition, it also exhibits reactivity via CF3CH carbene and undergoes cycloadditions.12 To date, only one report illustrates the trifluoroethylation of acyclic amides using CF3CHN2 in the presence of a silver(I) catalyst (Scheme 1a).13 Moreover, chemoselective installation of the trifluoroethyl moiety in cyclic amides and thioamides is still unexplored. As a part of our ongoing research on exploring the reactivity of oxindoles and thio-oxindoles,14 we have unfolded the metal- and additive-free TfOH catalyzed chemoselective trifluoroethylation of oxindoles, isoindolines, and thio-oxindoles (Scheme 1b).
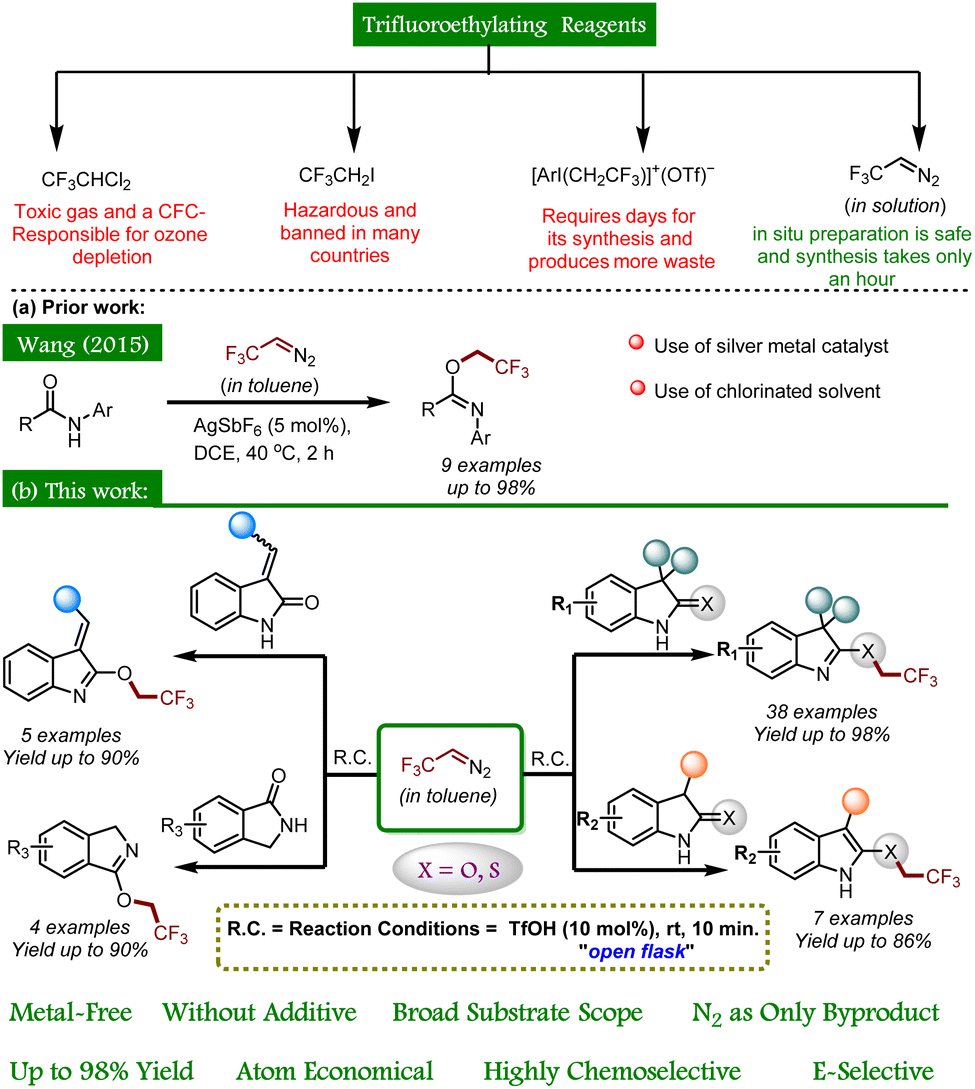 | ||
| Scheme 1 Synthetic approaches for chemoselective O- and S-trifluoroethylation of amides and thioamides using CF3CHN2 in toluene. | ||
Results and discussion
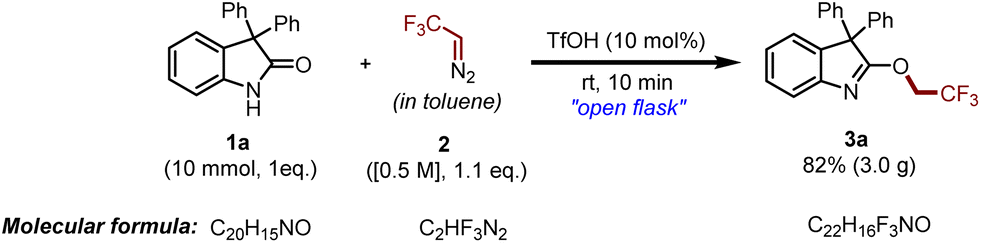
In the course of obtaining the optimized reaction conditions for the O-trifluoroethylation of oxindoles, we started a reaction by taking 3,3-diphenyl indoline-2-one 1a as the model substrate for screening various initiators in the presence of in situ generated 2,2,2-trifluorodiazoethane 2 in toluene (Table 1). At the onset, the reaction showed good results under transition metal catalysts through carbene formation (CuI and CuO), but we wanted to introduce the trifluoroethyl group under metal-free conditions as it is the need of the hour to advance not only selective, efficient, and high-yielding approaches but also environment-friendly, green, and safe approaches for organic reactions (not shown in Table 1). Initially, nitrogenous bases like 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), pyridine, 2,6-di-tert-butyl-pyridine (DTBPy), 4-(dimethylamino)pyridine (DMAP) and quinolone were employed as initiators in this survey; however, in all these cases, either the reaction became sluggish or a complex mixture was observed (Table 1, entries 1–5). Interestingly, when DIPEA was utilized as a base, the desired product, 3a, was observed in 70% yield (Table 1, entry 6). The reaction was further explored with tertiary amines like NEt3, NPh3, and NBn3; however, further improvement in the yield was not observed (Table 1, entries 7–9). Moreover, an array of inorganic bases like NaH, t-BuOK, K2CO3, and Cs2CO3 was also investigated; unfortunately, none of these could offer us the desired product (Table 1, entries 10–13). Afterward, we turned our attention toward Lewis and Brønsted acids as reaction promoters for the mentioned reaction. Lewis acids like AlCl3 and BF3·OEt2 were found to be completely ineffective for the said reaction (Table 1, entries 14 and 15). At this point, we enquired about the proton source of trifluoroethylation in the case of a base and performed a reaction in the presence of a drop of D2O and observed the formation of O-CHDCF3 (see the ESI, section IV†). In order to provide a better proton source, we performed the reaction under acidic conditions, and to our delight, the use of Brønsted acids like p-TSA and trifluoroacetic acid (TFA) furnished the product, 3a, with moderate yields (Table 1, entries 16 and 17). In the process of enhancing the yield, the reaction was further performed using TfOH, and to our delight, targeted product 3a was obtained in excellent yield within 10 min at ambient temperature (Table 1, entries 18 and 19). Further screening of other reaction parameters like varying solvents from toluene to benzene, DCM, DCE, and CHCl3 could not prove to be effective (not shown in Table 1).|
|
||||
|---|---|---|---|---|
| Entry | Initiator | Temp. | Time | Yield of 3ab |
| n.r. = no reaction, c.m. = complex mixture.a Reaction conditions: 1a (0.2 mmol, 1 eq.), 2 ([0.5 M], 1.1 eq.), initiator = (2 eq.), N2 atmosphere, neat.b Isolated yield.c Initiator used 10 mol%.d Initiator used 20 mol%.e Reaction performed in an open flask. | ||||
| 1 | DBU | 80 °C | 24 h | n.r. |
| 2 | Pyridine | 80 °C | 10 h | c.m. |
| 3 | DTBPy | 80 °C | 24 h | n.r. |
| 4 | DMAP | 80 °C | 24 h | n.r. |
| 5 | Quinoline | 80 °C | 20 h | n.r. |
| 6 | DIPEA | 80 °C | 5 h | 70% |
| 7 | NEt3 | 80 °C | 10 h | 60% |
| 8 | NPh3 | 80 °C | 8 h | 30% |
| 9 | NBn3 | 80 °C | 8 h | 47% |
| 10 | NaH | rt | 8 h | c.m. |
| 11 | KOt-Bu | 80 °C | 8 h | n.r. |
| 12 | K2CO3 | 80 °C | 10 h | c.m. |
| 13 | Cs2CO3 | 80 °C | 10 h | c.m. |
| 14c | AlCl3 | 80 °C | 10 h | n.r. |
| 15c | BF3·OEt2 | 80 °C | 24 h | n.r. |
| 16c | p-TSA | rt | 2 h | 67% |
| 17d | TFA | rt | 2 h | 42% |
| 18c | TfOH | rt | 10 min | 89% |
| 19 , | TfOH | rt | 10 min | 90% |

Having optimal reaction conditions in hand, we further explored several oxindoles 1 and thio-oxindoles 4 with in situ generated 2,2,2-trifluorodiazoethane (in toluene) 2 to determine the substrate scope and generality of the proposed protocol as shown in Scheme 2. With this context, we began our study with 3,3-diphenyl substituted oxindole 1a that afforded the desired product 3a in 90% yield. Afterward, different halide (–F, –Cl, –Br, and –I) substituted oxindole derivatives (1b–1m) were investigated, and it was found that all of them furnished the product (3b–3m) in good to excellent yields. Next, oxindoles with electron-donating groups (–Me and –OMe) were investigated and the desired compounds 3n–3r were obtained in good yields. Analogously, the reaction worked well with 5-NO2 substituted oxindole 1s and furnished the corresponding desired product 3s in 82% yield, which indicates the incredible functional group tolerance of the developed protocol. Further, the 3,3-substituted dibenzyl oxindole 1t also delivered the chemoselective product 3t in good yield. Meanwhile, substituents at the C3 position having p-EDG and p-EWG attached to the aryl group on the oxindoles were also exploited under optimized reaction conditions and achieved the targeted products 3u–3x with high yields. Remarkably, 2-oxindole having deuterated diphenyl groups at the C3 position delivered O-trifluoroethylated derivative 3y in 90% yield. The structure of compound 3y was unambiguously determined using single crystal XRD analysis. Next, we have explored the scope of 3,3-dialkyl substituted oxindoles under the standard reaction conditions, and product 3z was obtained in 82% yield. Similarly, spiro derivatives also performed well in the reaction and furnished the products 3aa and 3ab with excellent yields.
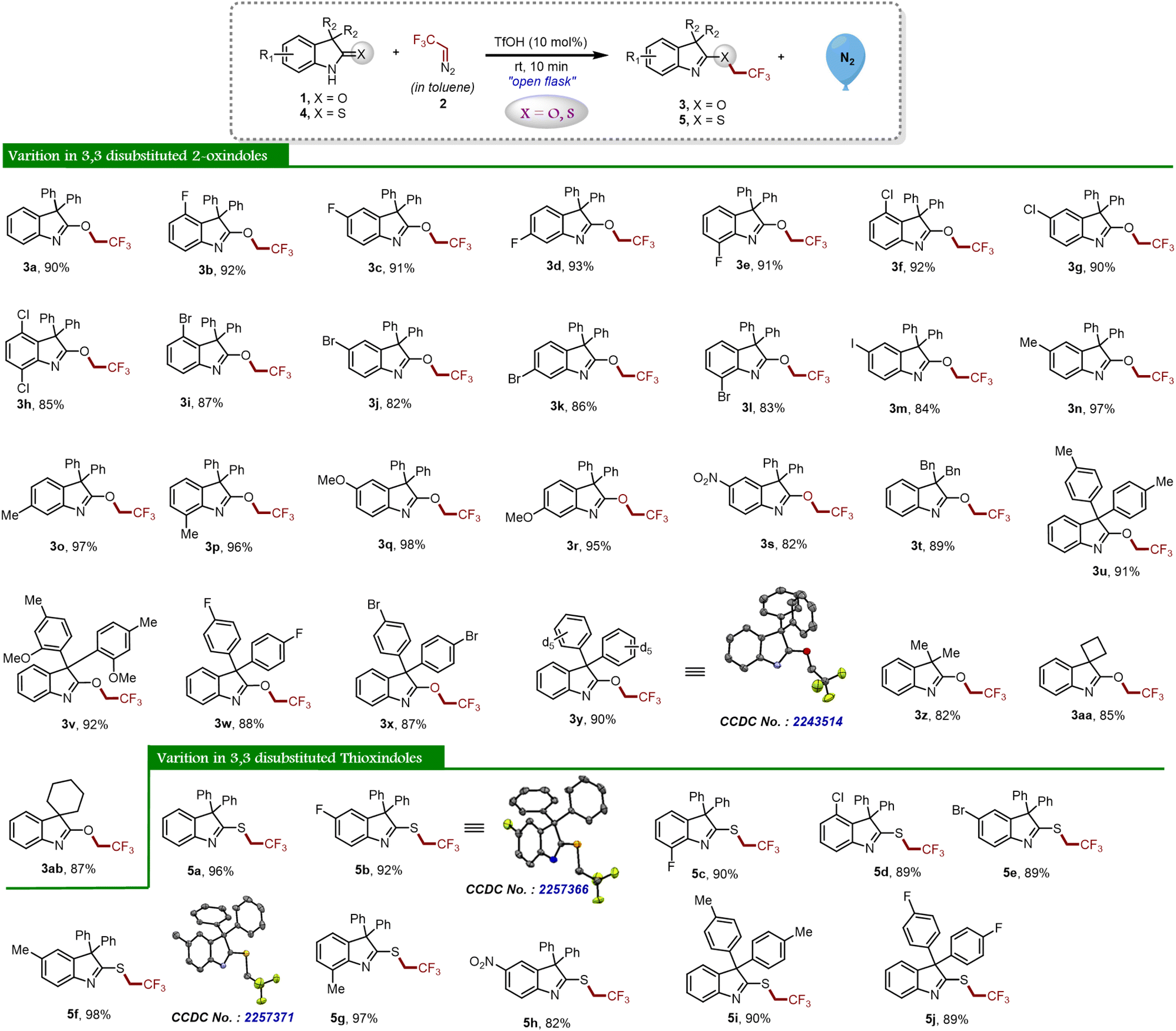 | ||
| Scheme 2 Synthesis of O-trifluoroethylated oxindole and S-trifluoroethylated thio-oxindoles derivatives. Reaction conditions: 1 or 4 (0.2 mmol, 1 eq.), 2 ([0.5 M], 1.1 eq.), TfOH (10 mol%). Isolated yields. | ||
To establish the potential of the newly developed methodology, we applied the same reaction protocol for thio-oxindoles, and as expected, we observed the chemoselective S-trifluoroethylation product 5a in almost quantitative yield (Scheme 2). Initially, we explored halide substituted thio-oxindoles derivatives and it is worth noting that the compounds 5b–5e were successfully obtained in good to excellent yields. The reaction went well with methyl-substituted thio-oxindoles derivatives 4f and 4g and afforded the corresponding S-trifluoroethyl decorated oxindole derivatives 5f and 5g, respectively. In addition, the formation of compound 5h demonstrated that the reaction was not limited to electron-donating derivatives, but also worked well for electron-withdrawing substituents. In a similar manner, introducing the –Me and –F moieties on the phenyl ring at the third position of the thio-oxindoles furnished the products 5i and 5j in good yields.
Encouraged by the efficiency of this protocol, we further explored the possibility of O-trifluoroethylation on several oxindole-based chalcones 6a–e, which are known to inhibit melanogenesis (production of extra pigment melanin).15 Besides this, 6e is also known to exhibit biological activities as a potent anti-oxidative agent and a tyrosinase inhibitor.16 A mixture of cis and trans isomers of the compounds 6 were subjected to this approach to furnish the desired products 7a–e with the E-isomer as the major product, as shown in Scheme 3. It is noteworthy that, even in the presence of a hydroxyl substituent in 6e, we were able to achieve O-trifluoroethylation of amide exclusively.
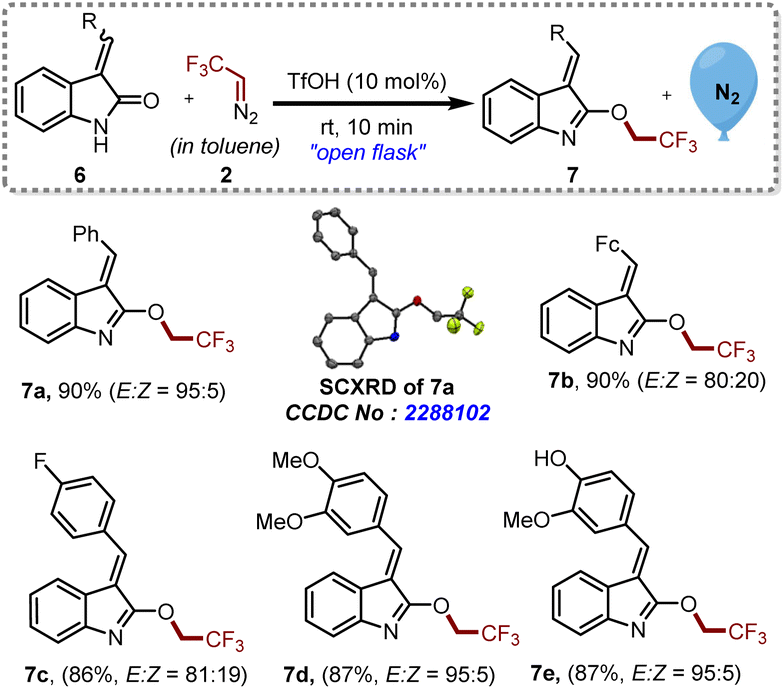 | ||
| Scheme 3 Synthesis of trifluoroethylated oxindole-based chalcone derivatives. Reaction conditions: 6 (0.2 mmol, 1 eq.), 2 ([0.5 M], 1.1 eq.), TfOH (10 mol%). Isolated yields. E:Z was determined through crude NMR. | ||
In order to investigate the effect of substituents at the C3 position of oxindoles and thio-oxindoles, monosubstituted and unsubstituted oxindoles and thio-oxindoles were tested under the established reaction conditions (Scheme 4). By utilizing the optimized reaction conditions, 2-trifluoroethyoxy indole derivatives 9a–e were obtained due to the chemoselective proton abstraction from the C3 position of oxindoles over the NH proton present on 8a–e. On the other hand, C3 unsubstituted 2-oxindole 8f offered the product 9f only in trace amounts, which might be due to identical pKa values of C3-H and NH as represented in Scheme 4.17 Luckily, with C3 unsubstituted thio-oxindoles 10, we were able to obtain the corresponding trifluoroethylated derivative 11 in good yield.
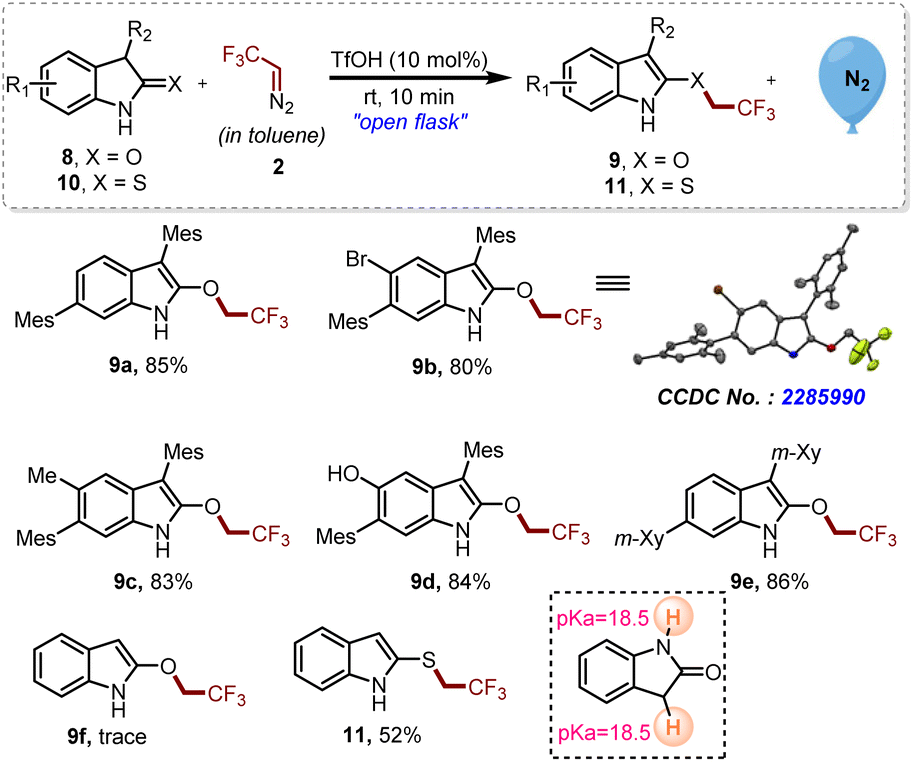 | ||
| Scheme 4 Exploration of mono- and unsubstituted oxindoles and thio-oxindoles for chemoselective trifluoroethylation. Reaction conditions: 8 or 10 (0.2 mmol, 1 eq.), 2 ([0.5 M], 1.1 eq.), TfOH (10 mol%). Isolated yields. | ||
In the course of in-depth studies, we focused our attention on analyzing the conjugation effect of the aromatic ring on the carbonyl part of compound 12 and checked the practicality of the established strategy. To our surprise, exclusive chemoselective O-trifluoroethylation over N-trifluoroethylation was observed, and as a result, a library of substituted isoindoline derivatives 13a–d was synthesized in good to excellent yields as shown in Scheme 5.
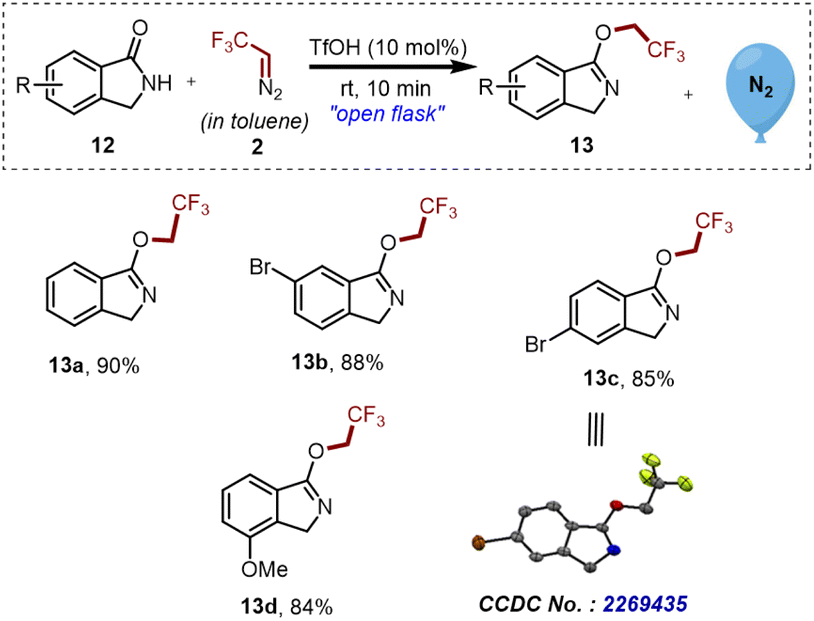 | ||
| Scheme 5 Synthesis of 3-(2,2,2-trifluoroethoxy)-1H-isoindole derivatives 13a–d. Reaction conditions: 12 (0.3 mmol, 1 eq.), 2 ([0.5 M], 1.1 eq.), TfOH (10 mol%). Isolated yields. | ||
To gain insight into the reaction, we carried out two competitive experiments which are summarized in Scheme 6. Initially, we performed an experiment with a mixture of electron-rich oxindole 1n and electron-deficient oxindole 1r under the standard reaction conditions and achieved compound 3n in 87% yield and compound 3r in 69% yield. This result suggests that 1n is more reactive than 1r in the reaction medium. Similarly, another experiment performed between oxindole 1a and thio-oxindoles 4a demonstrated that the synthesis of S-trifluoroethylated product 5a was predominant as compared to that of O-trifluoroethylated product 3a.
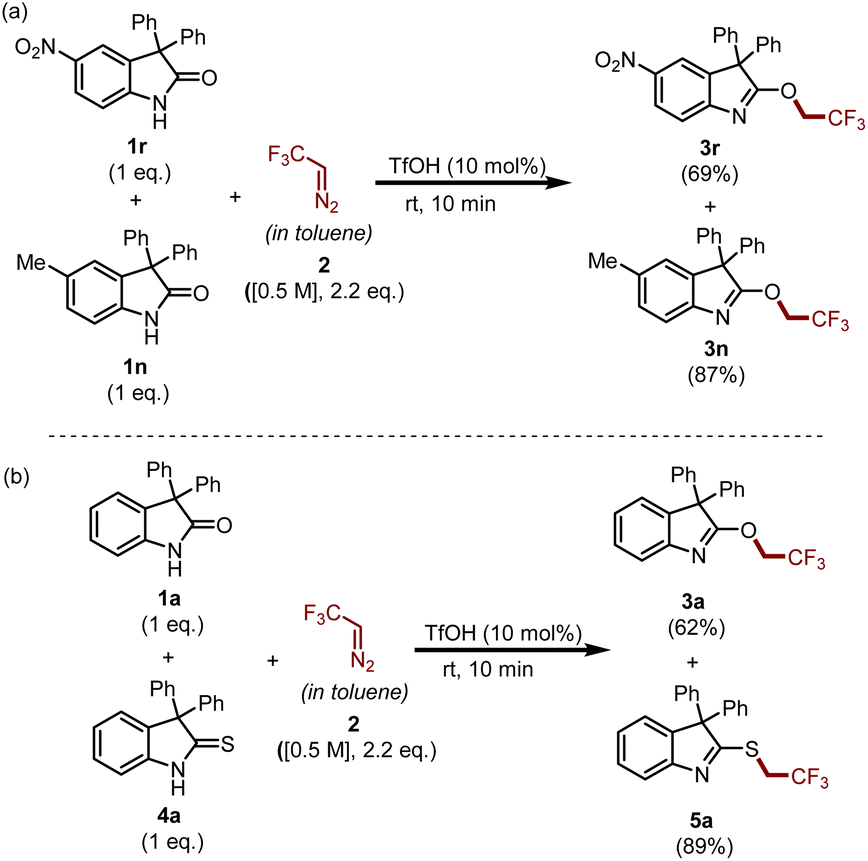 | ||
| Scheme 6 Competitive experiments: (a) electron-deficient vs. electron-rich oxindoles; (b) oxindole vs. thio-oxindoles. | ||
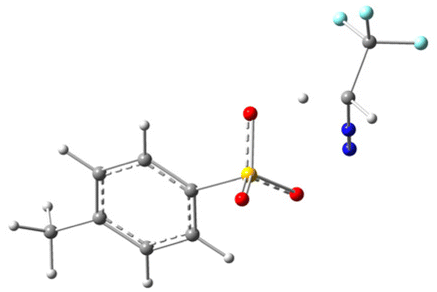
To gain deeper mechanistic insights, we conducted theoretical studies of different acids at the B3LYP/6-31G(d) level to examine our hypothesis. All structures have been optimized at the B3LYP/6-31G(d) level and the details are presented in Table 3 of the ESI.† During the preliminary examination, it was found that the process of abstracting a proton from a Brønsted acid is thermodynamically endergonic, which demands an input of energy. As a result, this step is a rate-determining step for the reaction. In this context, we specifically examined the proton abstraction capability of three Brønsted acids, i.e. TfOH, TFA, and p-TSA, using N2CHCF32, and the results are summarized in Table 2 (or Table 1 of ESI†). The data presented in Table 2 indicate that the H-abstraction from TfOH results in the lowest energy barrier, whereas TFA has the highest. It is noteworthy that all the reactions are thermodynamically feasible, as the transition state is attainable at room temperature. Nevertheless, the kinetics of the reaction varies depending on the reaction pathway. In this process, the transition states (TSs) of the complex (2 + TfOH) have also been verified by the imaginary frequency and intrinsic reaction coordinate (IRC) calculations as shown in Table 2 of the ESI.† The ground state (GS) and transition state (TS) of the complex (2 + TfOH) are located at 4 and 0 respectively; however, protonated 2 (N2CH2CF3) is located at −4. The uphill trajectory observable in the reaction plays a crucial role in governing its kinetics. This trajectory is consistent with those observed for the other two acids as well. As a result, the reaction involving TfOH delivers the corresponding product fastest compared to the other two Brønsted acids.
| 2 + Acid | Ground state (GS) | Transition state (TS) | ΔG‡ |
|---|---|---|---|
| 2 + TfOH |
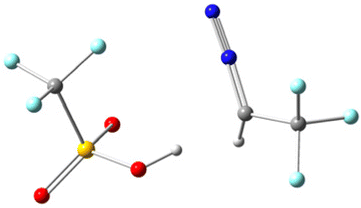
|
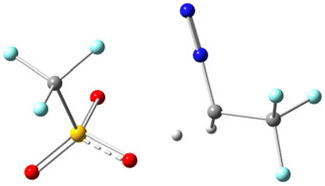
|
7.13 kcal mol−1 |
| 2 + p-TSA |
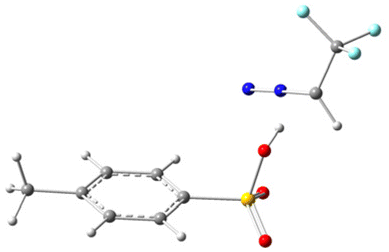
|
|
14.83 kcal mol−1 |
| 2 + TFA |
|
|
17.51 kcal mol−1 |
Next, a plausible reaction mechanism for the chemoselective O- and S-trifluoroethylation of oxindoles 1 and thio-oxindoles 4, respectively, is outlined in Scheme 7. Initially, the catalyst TfOH protonated 2,2,2-trifluorodiazomethane 2 and formed an activated intermediate I-a. A nucleophilic attack on the intermediate I-a through the O or S-centre (chemoselectively) of the compounds 1 or 4 resulted in a new intermediate I-b. Finally, proton abstraction from 1-b by the triflate ion leads to the formation of the final products 3 or 5.
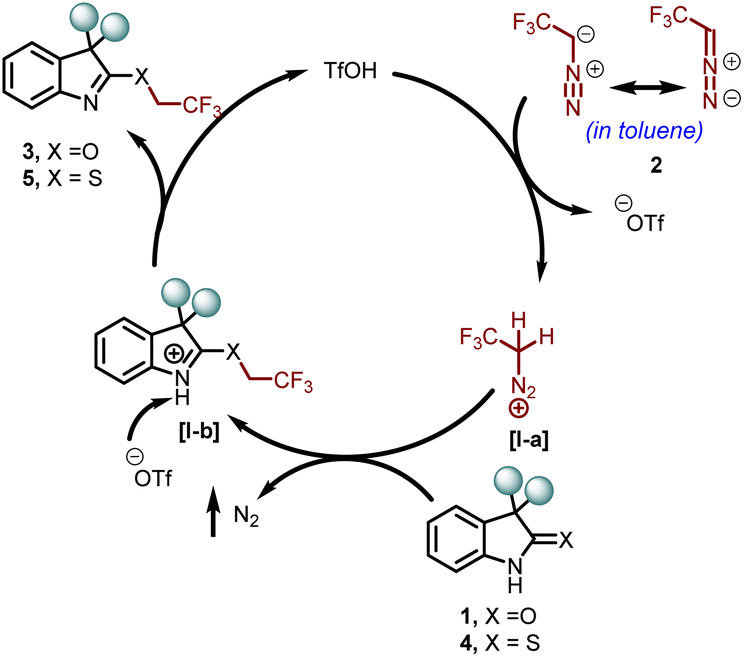 | ||
| Scheme 7 Plausible mechanistic pathway. | ||
To investigate the applicability of the proposed methodology, a preparative-scale synthesis of compound 3a was conducted. To our delight, we obtained 3.0 g of the desired product with magnificent chemoselectivity (Scheme 8).
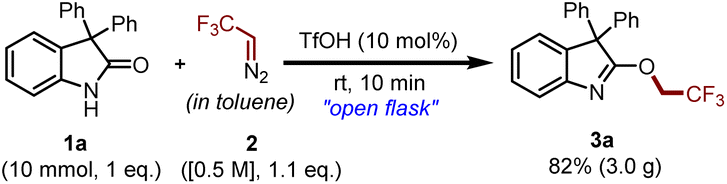 | ||
| Scheme 8 Gram scale synthesis of 3a. | ||
Thereafter, we determined the various green chemistry metrics (E factor, atom economy, carbon efficiency, and reaction mass efficiency) for the reaction protocol taking compound 1a as a model substrate (Table 3). Gratifyingly, the developed approach has high atom economy (93%), high carbon efficiency (100%), high reaction mass efficiency (74%), and low E-factor (0.40 kg waste per kg product), indicating the greenness of the methodology. In addition, we conducted an EcoScale evaluation based on six parameters, namely yield, temperature/time, price of reaction components, technical setup, safety, workup, and purification.18 The result of this evaluation, as presented in Table 4, indicates a remarkable EcoScale value of 86, signifying that an excellent green synthesis protocol is implemented that is safe, technically sound, and environmentally friendly. This C–O bond-forming chemoselective transformation does not generate any hazardous by-products, rendering it a viable option for green synthesis protocols. Furthermore, the reaction requires none of the expensive metals or harsh reaction conditions commonly employed in many synthetic methods, making it a cost-effective and safe approach.19 The reaction setup is straightforward, and the reaction can be executed efficiently at room temperature within a few minutes. These key features make this process an exemplary and eco-friendly green process.
|
|
|||
|---|---|---|---|
| Chemicals | Molecular weight (g mol−1) | Weight (mg) | |
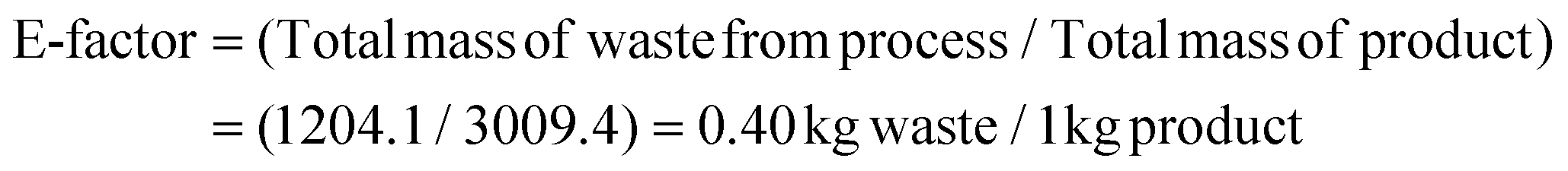
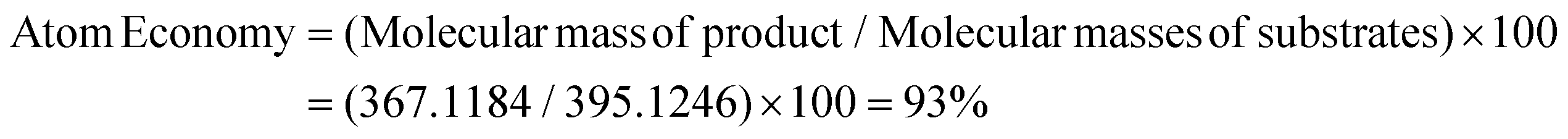
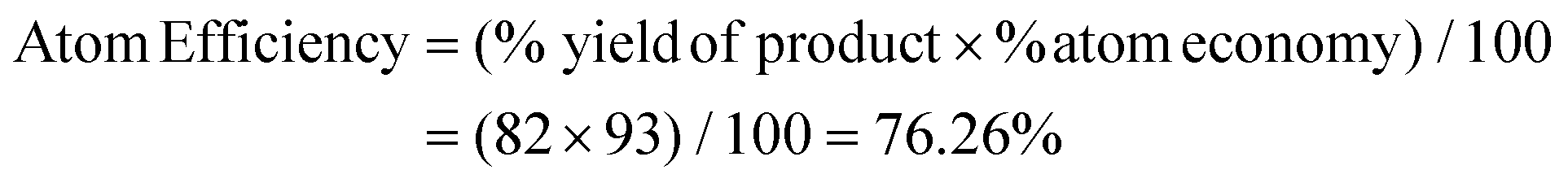
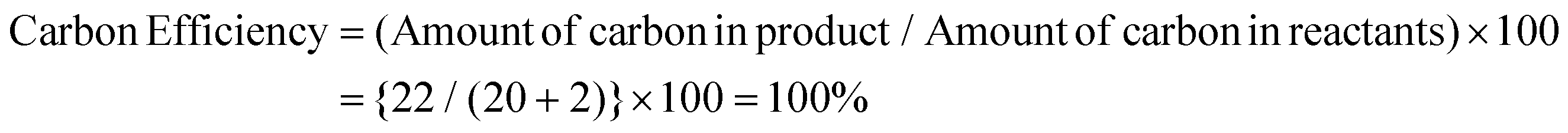
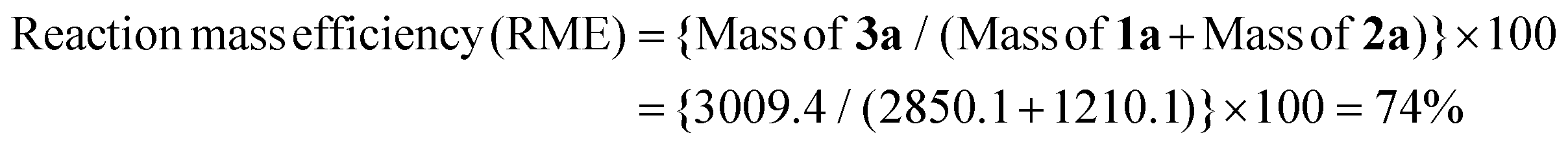
|
|||
| Substrate 1a | 3,3-Diphenylindolin-2-one | 285.1154 | 2850.1 mg |
| Substrate 2a | 2,2,2,-Trifluorodiazoethane | 110.0092 | 1210.1 mg |
| Catalyst | Trifluoromethanesulfonic acid | 150.08 | 150 mg |
| Solvent used | Toluene | 92.14 | 1724 mg |
| Solvent recovered | Toluene | 92.14 | 1720.69 |
| Product 3a | 3,3-Diphenyl-2-(2,2,2-trifluoroethoxy)-3H-indole | 367.1184 | 3009.4 mg (82%) |
| Waste (byproducts) | — | — | 1204.1 mg (18%) |
| Parameters | Penalty points | |
|---|---|---|
| The scoring criteria are as follows: >75, excellent; >50, acceptable; and <50, inadequate. | ||
| 1 | Yield (100–82% yield)/2 = 9 | =9 |
| 2 | Total cost of synthesis of 10 mmol of 3a <$50 | =0 |
| 3 | Safety, 2,2,2-trifluorodiazoethane | =5 |
| 4 | Technical setup (simple setup) | =0 |
| 5 | Temperature/time (room temperature, <1 h) | =0 |
| 6 | Workup and purification (classical chromatography) | =0 |
| Total of individual penalties in EcoScale calculation = 100 − sum of individual penalties = 100 − 14 = 86 | =14 | |
Conclusions
In this study, we have successfully introduced a straightforward and effective procedure for the chemoselective O-trifluoroethylation of 3,3-disubstituted, monosubstituted, unsubstituted, and chalcone-based oxindoles and isoindolines and S-trifluoroethylation of thio-oxindoles under mild reaction conditions, without the need for hazardous fluorinating reagents. The products are obtained in excellent yields, and in the case of chalcone-based oxindoles, E-selective products were obtained. Remarkable functional group compatibility and exclusive chemoselectivity clearly indicate the potentiality of the protocol. This is the first time that such a method has been developed as we used an environmentally benign catalyst, such as TfOH. Unlike previous synthetic trifluoroethylation protocols, our method involves the use of renewable and inexpensive feedstock chemicals without solvent, making it more sustainable. Furthermore, we calculated various green chemistry metrics, such as atom economy (93%), carbon efficiency (100%), reaction mass efficiency (74%), and E-factor (0.40 kg waste per kg product), all of which indicate the protocol's “greenness.” The EcoScale evaluation demonstrates the simplicity, efficacy, and environmental friendliness of this methodology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the financial support from SERB, Department of Science and Technology (Award No. CRG/2022/008154), New Delhi, India, and IIT Ropar for infrastructural facilities. The authors wish to thank Dr T. J. Dhilip Kumar and Apoorv Kushwaha, IIT Ropar, for the theoretical calculations. We would like to thank the CRF facilities of IIT Ropar for the characterization of compounds. ML and PRS would like to thank IIT Ropar for their fellowship.References
- (a) K. Müller, F. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed; (c) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- (a) R. Bahleda, J. E. Grilley-Olson, R. Govindan, F. Barlesi, L. Greillier, M. Perol, I. Ray-Coquard, D. Strumberg, B. Schultheis, G. K. Dy, G. Zalcman, G. J. Weiss, A. O. Walter, M. Kornacker, P. Rajagopalan, D. Henderson, H. Nogai, M. Ocker and J. C. Soria, Br. J. Cancer, 2017, 116, 1505–1512 CrossRef CAS; (b) S. Jain, R. Song and J. Xie, OncoTargets Ther., 2017, 10, 1645–1653 CrossRef CAS; (c) N. N. M. Anuar, N. S. N. Hisam, S. L. Liew and A. Ugusman, Front. Pharmacol., 2020, 11, 564108 CrossRef CAS.
- A. S. Nair, A. K. Singh, A. Kumar, S. Kumar, S. Sukumaran, V. P. Koyiparambath, L. K. Pappachen, T. M. Rangarajan, H. Kim and M. Bijo, Processes, 2022, 10, 2054 CrossRef CAS.
- F. Francis and F. Wuest, Molecules, 2021, 26, 6478 CrossRef CAS PubMed.
- (a) R. E. Thompson, X. Liu, N. Alonso-García, P. J. Pereira, K. A. Jolliffe and R. J. Payne, J. Am. Chem. Soc., 2014, 136, 8161–8164 CrossRef CAS; (b) H. F. Motiwala, A. M. Armaly, J. G. Cacioppo, T. C. Coombs, K. R. Koehn, V. M. Norwood IV and J. Aubé, Chem. Rev., 2022, 122, 12544–12747 CrossRef CAS PubMed.
- (a) S. Barata-Vallejo, B. Lantaño and A. Postigo, Chem. – Eur. J., 2014, 20, 16806–16829 CrossRef CAS PubMed; (b) C. Yang, A. Hassanpour, K. Ghorbanpour, S. Abdolmohammadi and E. Vessally, RSC Adv., 2019, 9, 27625–27639 RSC; (c) H. Xiao, Z. Zhang, Y. Fang, L. Zhu and C. Li, Chem. Soc. Rev., 2021, 50, 6308–6319 RSC; (d) J. Charpentier, N. Fruh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed.
- (a) G. L. Tolnai, A. Székely, Z. Makó, T. Gáti, J. Daru, T. Bihari, A. Stirling and Z. Novák, Chem. Commun., 2015, 51, 4488–4491 RSC; (b) K. G. Andrews, R. Faizova and R. M. Denton, Nat. Commun., 2017, 8, 15913 CrossRef PubMed; (c) C. L. Zhao, Q. Y. Han and C. P. Zhang, Org. Lett., 2018, 20, 6480–6484 CrossRef CAS PubMed; (d) C. Lu, Z. Qiu, M. Xuan, Y. Huang, Y. Lou, Y. Zhu, H. Shen and B. L. Lin, Adv. Synth. Catal., 2020, 362, 4151–4158 CrossRef CAS; (e) X. Li, X. Gao, C. Y. He and X. Zhang, Org. Lett., 2021, 23, 1400–1405 CrossRef CAS PubMed; (f) X. Yang, W. D. Meng, X. H. Xu and Y. Huang, Org. Chem. Front., 2021, 8, 6597–6602 RSC; (g) Z. Deng, L. Y. Qiu, J. Chen, H. Zhang, W. Cao and X. J. Tang, Adv. Synth. Catal., 2023, 365, 693–698 CrossRef CAS.
- Y. M. Khetmalis, M. Shivani, S. Murugesan and K. V. Sekhar, Biomed. Pharmacother., 2021, 141, 111842 CrossRef CAS PubMed.
- (a) E. J. Han, Y. Sun, Q. Shen, Q. Y. Chen, Y. Guo and Y. G. Huang, Org. Chem. Front., 2015, 2, 1379–1387 RSC; (b) S. Y. Yan, Z. Z. Zhang and B. F. Shi, Chem. Commun., 2017, 53, 10287–10290 RSC; (c) H. Zhang, H. Y. Wang, Y. Luo, C. Chen, Y. Cao, P. Chen, Y. L. Guo, Y. Lan and G. Liu, ACS Catal., 2018, 8, 2173–2178 CrossRef CAS; (d) H. Li, J. Sheng, G. X. Liao, B. B. Wu, H. Q. Ni, Y. Li and X. s. Wang, Adv. Synth. Catal., 2020, 362, 5363–5367 CrossRef CAS; (e) K. Zhao, Z. Y. Zhang, X. L. Cui, Y. X. Wang, X. D. Wu, W. M. Li, J. X. Wu, L. L. Zhao, J. Y. Guo and T. P. Loh, Org. Lett., 2020, 22, 9029–9035 CrossRef CAS PubMed; (f) B. He, Q. Pan, Y. Guo, Q. Y. Chen and C. Liu, Org. Lett., 2020, 22, 6552–6556 CrossRef CAS PubMed; (g) L. Hou, X. Jing, H. Huang and C. Duan, Chem. Commun., 2023, 59, 3407–3410 RSC; (h) Y. N. Li, M. X. Zhou, J. B. Wu, Z. Wang and Y. F. Zeng, Org. Biomol. Chem., 2022, 20, 9613–9617 RSC; (i) Y. F. Zeng, M. X. Zhou, Y. N. Li, X. Wu, Y. Guo and Z. Wang, Org. Lett., 2022, 24, 7440–7445 CrossRef CAS PubMed; (j) L. Ruyet, M. I. Lapuh, V. S. Koshti, T. Földesi, P. Jubault, T. Poisson, Z. Novák and T. Besset, Chem. Commun., 2021, 57, 6241 RSC; (k) L. Dong, T. Feng, D. Xiong, Z. Xu, J. Cheng, X. Xu, X. Shao and Z. Li, Org. Lett., 2022, 24, 1913–1917 CrossRef CAS PubMed.
- (a) Y. Zhao and J. Hu, Angew. Chem., Int. Ed., 2012, 51, 1033–1036 CrossRef CAS PubMed; (b) D. A. Culkin and J. F. Hartwig, Organometallics, 2004, 23, 3398–3416 CrossRef CAS.
- B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 938–941 CrossRef CAS PubMed.
- P. K. Mykhailiuk, Chem. Rev., 2020, 120, 12718–12755 CrossRef CAS PubMed.
- H. Luo, G. Wu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 14503–14507 CrossRef CAS PubMed.
- (a) B. Gopal, P. R. Singh, M. Kumar and A. Goswami, J. Org. Chem., 2023, 88, 132–142 CrossRef CAS PubMed; (b) P. R. Singh, M. Lamba, D. Singh and A. Goswami, Adv. Synth. Catal., 2023, 365, 1685–1692 CrossRef.
- P. Thanigaimalai, K. C. Lee, V. K. Sharma, N. Sharma, E. Roh, Y. Kim and S. H. Jung, Chem. Pharm. Bull., 2011, 59, 1285–1288 CrossRef CAS.
- (a) Y. Hirata, Y. Ito, M. Takashima, K. Yagyu, K. Oh-Hashi, H. Suzuki, K. Ono, K. Furuta and M. Sawada, ACS Chem. Neurosci., 2020, 11, 76–85 CrossRef CAS; (b) S. K. Suthar, S. Bansal, N. Narkhede, M. Guleria, A. T. Alex and A. Joseph, Chem. Pharm. Bull., 2017, 65, 833–839 CrossRef CAS.
- R. A. Altman, A. M. Hyde, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 9613–9620 CrossRef CAS PubMed.
- (a) K. J. Hock, L. Mertens, F. K. Metze, C. Schmittmanna and R. M. Koenigs, Green Chem., 2017, 19, 905–909 RSC; (b) A. D. Sharapov, R. F. Fatykhov, I. A. Khalymbadzha, V. V. Sharutin, S. Santra, G. V. Zyryanov, O. N. Chupakhin and B. C. Ranu, Green Chem., 2022, 24, 2429–2437 RSC; (c) A. N. V. Satyanarayana, N. Mukherjee and T. Chatterjee, Green Chem., 2023, 25, 779–788 RSC.
- (a) D. M. Gale, W. J. Middleton and C. G. Krespan, J. Am. Chem. Soc., 1965, 87, 657 CrossRef CAS; (b) W. J. Middleton, D. M. Gale and C. G. Krespan, J. Am. Chem. Soc., 1968, 90, 6813 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2243514, 2257366, 2257371, 2269435, 2288102 and 2285990. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc03702a |
| This journal is © The Royal Society of Chemistry 2024 |