
Tuneable reduction of CO2 – organocatalyzed selective formylation and methylation of amines†
Changyue
Ren
ab,
Constanza
Terazzi
a and
Thomas
Werner
 *ac
*ac
aLeibniz Institute for Catalysis e.V. at the University of Rostock, Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: Thomas.Werner@catalysis.de
bCollege of Pharmacy, Zunyi Medical University, Zunyi 563003, China
cDepartment of Chemistry, Paderborn University, Warburger Str. 100, D-33098 Paderborn, Germany. E-mail: th.werner@uni-paderborn.de
First published on 23rd November 2023
Abstract
The N-formylation and N-methylation of amines with carbon dioxide (CO2) are important types of transformations which give access to a wide range of key intermediates and compounds. Efficient catalytic methods for both reactions are rare because of the challenge of controlling selectivity. Herein, we report the environmentally benign organocatalyzed N-formylation and N-methylation of primary and secondary amines using CO2 as the C1 source in the presence of hydrosilanes as the reductant. Readily available methyltriphenylphosphonium methylcarbonate has been proven to be an efficient catalyst for the N-methylation of amines and CO2 in the presence of polymethylhydrosiloxane (PMHS) under mild conditions. In contrast, the selective N-formylation is achieved in the presence of trimethoxysilane as the reductant. In both transformations, a wide range of substrates were selectively converted. 15 primary and secondary amines were N-methylated and N-formylated and the corresponding products were obtained in yields of up to 98% and 94%, respectively. Moreover, benzimidazoles are accessible from diamines and CO2 in a one-pot reaction in yields of up to 83%. Mechanistic investigations revealed different reaction pathways for N-methylation and N-formylation depending on the reductant as well as the activation of CO2 by the catalyst.
Introduction
N-Methylamines and N-formamides are attractive chemicals that have found extensive applications in the pharmaceutical industry, biological sciences, and agriculture.1–8 Formamide derivatives, such as N-methylformamide and N,N-dimethylformamide (DMF), are among the most used solvents and important intermediates for the synthesis of other valuable materials,9 such as isocyanides10,11 and benzoheterocycles.12,13N-Methylamines are structural subunits that can be found in many significant bioactive compounds and drugs.14 The common C1 sources for N-methylation and N-formylation reactions include MeI,15 CH2N2,16 dimethyl carbonate,17 methanol,18 formic acid19 and CO2.20 In particular, the use of CO2 in combination with reducing agents is an attractive alternative to toxic and hazardous reagents (Scheme 1a). CO2 is a waste product of fuel combustion currently deemed as a promising surrogate of carbon feedstocks. Its utilization has been an active field of research in the past few decades. However, the kinetic inertness and thermodynamic stability of the CO2 molecule significantly hinder its efficient valorization under mild conditions.21–25 | ||
| Scheme 1 (a) N-Methylation and N-formylation of amines with CO2 and silane. (b) The formation of benzoheterocycles from diamines with CO2 and silane. | ||
Although a few catalysts have been reported to be able to perform the N-functionalization of amines to form both formamide and methylamine (Scheme 1a),26–29 there are several challenges to overcome in terms of these reactions. One of them is employing cost-efficient reductants.30 Phenylsilanes have been widely used in the fixation of CO2 into amines,26,28,29,31,32 but their use presents some disadvantages since they are expensive, and the byproducts formed are difficult to remove. Polymethylhydrosiloxane (PMHS) is an abundant, low-cost, non-toxic, and highly moisture-stable chemical waste product generated by the silicone industry.33–35 Even though it would be advantageous to use PMHS as the reductant in the reduction of CO2, very few organic catalysts are reported to be active for both PMHS and CO2 for the methylation reaction. Dyson et al. reported a thiazolium carbene-based catalyst for the N-formylation and N-methylation of amines using PMHS as a reducing agent, which allows the reaction to proceed under relatively mild conditions.36,37
The Xia group reported diazabicycloundecene (DBU) as an organocatalyst for the selective N-methylation and N-formylation of secondary amines, converting secondary amines, CO2 and PMHS into methylamines and formamides at 100 °C and 30 °C, respectively.38 Furthermore, benzoheterocycles can be prepared from o-phenylenediamine, CO2 and silane (Scheme 1b). The formation of the benzimidazole ring is plausible by monoformylation and subsequent imine formation with a suitable catalyst. However, due to the mismatched catalysis, the cyclization reaction normally requires two steps or harsher conditions than the N-formylation reaction.13,31,32,39,40 In the previous reports, the N-formylation and N-methylation reactions are assumed to proceed by a general base-catalysed reaction mechanism, in which hydrosilanes are activated by the basic catalyst or the carbamate salt formed.13 Most recently, the Song group reported the N-methylation and N-formylation reaction between CO2 and secondary amines in the presence of phenylsilane catalyzed by an ammonium carboxylate ionic liquid.41 In addition, Lin and coworkers reported Cs2CO3 to be an efficient inorganic catalyst for these reactions.42 These reports and our interest in using phosphonium salts as organocatalysts for the utilization of CO2 motivated us to explore a phosphorus-based organocatalyst in combination with PMHS as a reductant for the methylation of amines.43–46 We focused on methylcarbonate salts which were first reported as a CO2 surrogate in the preparation of propylene oxide in 2006,47 and subsequently have been employed in the transesterification48 and Henry reactions,49 and as vinylation reagents in the Wittig reaction.50
Results and discussion
Methyltriphenylphosphonium methylcarbonate is commercially available and can also be prepared from phosphines and dimethylcarbonate (DMC) according to the literature.50 PMHS was selected as the reducing agent and N-methylaniline (1a) as the model substrate for evaluating the activity of the catalyst towards methylation, utilizing CO2 as the carbon source at 1 bar and 70 °C (Table 1). In the absence of the catalyst, no conversion was observed (entry 1). A moderate yield of 53% and good selectivity for N-methylated product 2a were obtained in the presence of the catalyst (entry 2). In CH3CN, N-methylaniline (1a) gave the N-methylated product 2a and N-formylated product 3a with a yield of 20% and 6%, respectively (entry 3). In EtOAc and CH2Cl2, 1a failed to convert to either 2a or 3a (entries 4 and 5). Due to its polymeric nature, PMHS is less active than non-polymeric silanes. Thus, an excess of reductant is normally required.36–38,51 Full conversion was achieved by increasing the amount of PMHS and 2a was obtained in a very good yield of 87% (entry 6). Even under 1 atm of CO2 (balloon), 2a was accessible in 29% yield (entry 7). Replacing THF with the biomass-derived 2-methyltetrahydrofuran (Me-THF) gave 3a in 83% yield (entry 8). At a lower catalyst loading, an increase in the formation of the N-formylated product 3a was observed (entry 9). Finally, the impact of the reaction temperature was studied (entries 10–12). Neither an increase nor a decrease of the reaction temperature led to an improved yield of 2a. Notably, at lower temperatures (50 °C), the yield of 2a decreased to 55% but 3a was obtained in 38% yield (entry 12).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. (mol %) | Silane (equiv.) | Solvent | T (°C) | Yield 2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (%) a (%) |
Yield 3aa (%) |
| Reaction conditions: N-methylaniline (1a, 0.233 mmol, 1 equiv.), PMHS (4–10 equiv.), catalyst (0–10 mol%), p(CO2) = 1 bar, 50–80 °C, 16 h, solvent.a Yield was determined by 1H NMR using mesitylene as the internal standard.b CO2 balloon. | ||||||
| 1 | 0 | 4 | THF | 70 | 0 | 0 |
| 2 | 10 | 4 | THF | 70 | 53 | 0 |
| 3 | 10 | 4 | MeCN | 70 | 20 | 6 |
| 4 | 10 | 4 | EtOAc | 70 | 0 | 0 |
| 5 | 10 | 4 | CH2Cl2 | 70 | 0 | 0 |
| 6 | 10 | 10 | THF | 70 | 87 | 0 |
| 7b | 10 | 10 | THF | 70 | 29 | 0 |
| 8 | 10 | 10 | Me-THF | 70 | 83 | 10 |
| 9 | 5 | 10 | THF | 70 | 62 | 30 |
| 10 | 10 | 10 | THF | 80 | 72 | 0 |
| 11 | 10 | 10 | THF | 60 | 82 | 17 |
| 12 | 10 | 10 | THF | 50 | 55 | 38 |

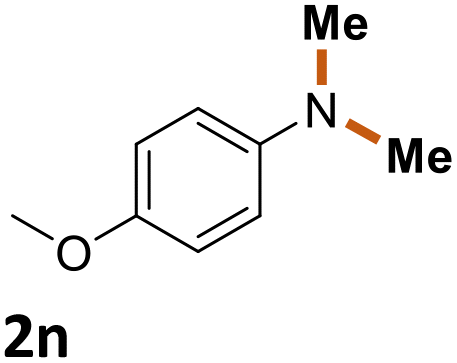
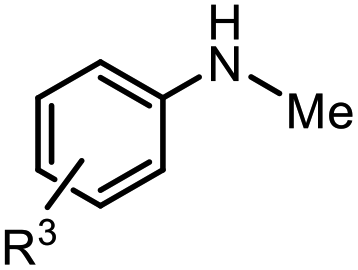
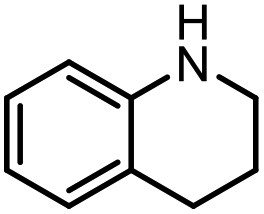
The scope of the reaction was next investigated by converting a variety of substituted secondary and primary amines 1 under the optimized conditions. While a reaction time of 16 h was required to achieve an appropriate yield for most substrates, full conversion was achieved within 4 h for a few substrates (Table 2). p-Methoxy-N-methylaniline (1b) gave the corresponding dimethylaniline 2b in 72% yield after 16 h, while 71% yield of o-methoxy-N,N-dimethylaniline (2c) was isolated after only 4 h (entries 1 and 2). Fluoro- and bromo-substituted N-methylanilines gave the products 2d and 2e in good yields of 86% and 77%, respectively (entries 3 and 4). Substrates with strong electron-withdrawing groups were challenging for the reaction: the methylated products of p-nitro- (2f) and p-acetyl-N-methylaniline (2g) were isolated in 19% and 42% yields, respectively (entries 5 and 6). Further functionalized substrates such as N-benzyl-, N-cyclohexyl-, N-butyl-, and N-allyl-anilines were converted to the corresponding methylated products 2h–2k in excellent yields (82–89%) (entries 7–10). Tetrahydroquinoline afforded the methylated product 2l in a yield of 76% in 4 h (entry 11). In comparison with secondary anilines, primary anilines normally exhibit lower nucleophilicity and form multiple reduction products42,52 However, with this catalytic system, aniline (1m) and p-methoxyaniline (1n) afforded dimethylanilines in yields of 90% and 42%, respectively (entries 12 and 13). Besides anilines, benzyl-N-methylamine (1o) was also tested. However, the formylated product 3o was obtained in 68% yield as the main product while the methylated product 2o was obtained in 30% yield (entry 14). Surprisingly, the sterically hindered substrate 2,2,6,6-tetramethylpiperidine was successfully converted to the methylated product pempidine 2p, which is a ganglion-blocking drug (entry 15).53
|
|
|||
|---|---|---|---|
| Entry | Substrate 1 | Product 2 | Yield |
| Reaction conditions: amine 1 (0.600 mmol), PMHS (10 equiv.), catalyst (10 mol%), p(CO2) = 1 bar, 70 °C, 16 h, THF. Isolated yields are given.a 4 h.b Yield was determined by 1H NMR using mesitylene as the internal standard.c The N-formylated product was obtained in 68% yield. | |||
| 1 |
|
2b, R3 = p-OCH3 | 72% |
| 2 | 2c, R3 = o-OCH3 | 71%a | |
| 3 | 2d, R3 = p-F | 86% | |
| 4 | 2e, R3 = p-Br | 77% | |
| 5 | 2f, R3 = p-NO2 | 19% | |
| 6 | 2g, R3 = p-Ac | 42% | |
| 7 |

|
2h, R2 = Bn | 89% |
| 8 | 2i, R2 = cyclohexyl | 83% | |
| 9 | 2j, R2 = n-butyl | 87% | |
| 10 | 2k, R2 = allyl | 82% | |
| 11 |
|
2l | 76%a,b |
| 12 |

|
|
90%b |
| 13 |
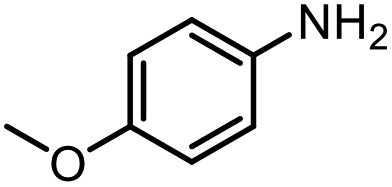
|
|
42% |
| 14 |
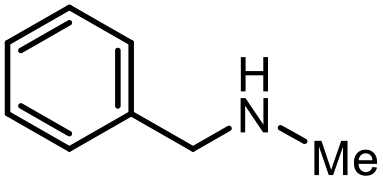
|
2o | 30%b,c |
| 15 |
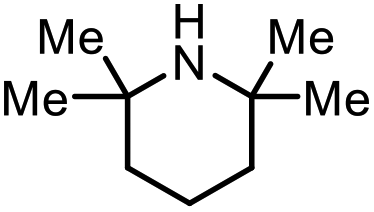
|
2p | 98%b |
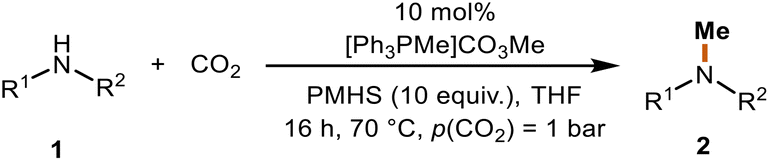
Since the formation of the N-formylated product 3a was also observed in significant amounts during the screening (Table 1, entry 12), we further explored the N-formylation of N-methylaniline (1a) catalyzed by [Ph3PMe]CO3Me (Table 3). Initially, we turned our focus to other silanes. PhSiH3 and Ph2SiH2 gave moderate yields of N,N-dimethylaniline (2a) and poor yields of N-methylformanilide (3a) (entries 1–5). PhSiH3 and Ph2SiH2 mainly formed methylated product 2a, and only 8% desired product 3a was observed (entries 1 and 2). Ph3SiH converted 1a to 3a in a yield of 39% and no 2a or 3a was observed with Et3SiH (entries 3 and 4). (MeO)3SiH afforded the N-formylated product in an excellent yield of 98% with outstanding selectivity (entry 5). Further screening of the reaction conditions was performed in the presence of (MeO)3SiH (entries 6–10). Reducing the reaction time to 4 h led to a comparable yield and selectivity (entry 6). 1a was quantitatively converted to the desired product 3a even with a much lower loading of the catalyst (entry 7). An excellent yield of the formylated product 3a (95%) was also obtained at 70 °C in 4 hours in the presence of 3 equiv. of (MeO)3SiH (entry 8). No product was obtained without the catalyst (entry 9). A lower amount of (MeO)3SiH was not sufficient to convert all the reactant (entry 10).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. (mol%) | Si–H (equiv.) | Silane | t (h) | Yield 2a (%) | Yield 3a (%) |
| Reaction conditions: N-methylaniline (1a, 0.233 mmol), silane (2–4 equiv.), catalyst (0–10 mol%), p(CO2) = 1 bar, 70 °C for 4–16 h, THF. The yield was determined by 1H NMR using mesitylene as the internal standard. | ||||||
| 1 | 10 | 4 | PhSiH3 | 16 | 31 | 8 |
| 2 | 10 | 4 | Ph2SiH2 | 16 | 55 | 8 |
| 3 | 10 | 4 | Ph3SiH | 16 | 0 | 39 |
| 4 | 10 | 4 | Et3SiH | 16 | 0 | 0 |
| 5 | 10 | 4 | (MeO)3SiH | 16 | 0 | 98 |
| 6 | 10 | 4 | (MeO)3SiH | 4 | 0 | 94 |
| 7 | 2 | 4 | (MeO)3SiH | 4 | 0 | 96 |
| 8 | 2 | 3 | (MeO) 3 SiH | 4 | 0 | 95 |
| 9 | 0 | 3 | (MeO)3SiH | 4 | 0 | 0 |
| 10 | 2 | 2 | (MeO)3SiH | 4 | 0 | 44 |
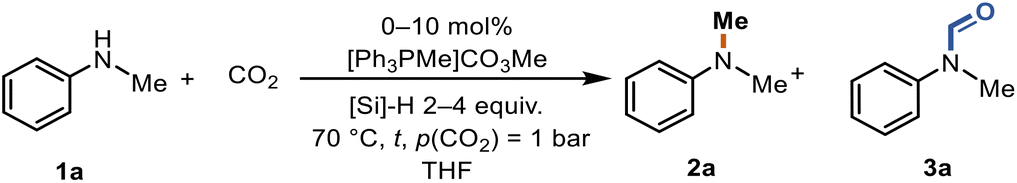
With the optimal conditions in hand, the scope and limitations of [Ph3PMe]CO3Me for the catalytic conversion of amines to their formylated products were studied at 70 °C in the presence of trimethoxysilane as the reductant (Table 4). Under these conditions, p-methoxyaniline (1b) was converted to 3b in a high yield of 86% (entry 1). In some cases, extension of the reaction time from 4 to 24 h was necessary. In contrast to the methylation reaction, o-methoxy-N-methylaniline (1c) needed 24 h to be converted to the corresponding formylated product in a good yield of 79% (entry 2). Electron-withdrawing groups were also evaluated: fluoro- and bromo-substituted anilines 1d and 1e afforded the corresponding N-methylformanilide 3d and 3e in good yields in 4 h; while nitro- and acetyl-substituted anilines 1f and 1g led to lower yields of 13% and 53%, respectively (entries 3–6). N-Substituted anilines were also tolerated, N-butyl- and N-allyl-substituted anilines 1j and 1k were successfully converted to their formylated products 3j and 3k in 84% and 85% yield, respectively, after 24 h (entries 7 and 8). Tetrahydroquinoline 1l gave the formylated product 3l in 94% yield (entry 9). The conversion of the benzylamines 1o and 1q was also successful and 92% and 75% yield, respectively, of the formylated methylbenzeneamine 3o and dibenzylamine 3q were isolated (entries 10 and 11). The piperazine derivative 1r afforded the formylated product 3r in a yield of 89% (entry 12). A moderate yield of 69% of the formylated product 3s was obtained from the pyridine-substituted amine 1s (entry 13). Aniline (1t) and pyridine-2-amine (1u) were selected as representative substrates of primary amines (entries 14 and 15). An excellent yield of 90% of N-formanilide 3t was obtained from aniline, while 53% of the N-formylated-pyridine-2-amine 3u was isolated.
|
|
|||
|---|---|---|---|
| Entry | Substrate 1 | Product 3 | Yield |
| Reaction conditions: amine 1 (0.600 mmol), trimethoxysilane (220 mg, 3 equiv.), catalyst (2 mol%), p(CO2) = 1 bar, 70 °C, 24 h, THF. Isolated yields are given.a 4 h. | |||
| 1 |
|
3b, R3 = p-OCH3 | 86%a |
| 2 | 3c, R3 = o-OCH3 | 79% | |
| 3 | 3d, R3 = p-F | 86%a | |
| 4 | 3e, R3 = p-Br | 76%a | |
| 5 | 3f, R3 = p-NO2 | 13% | |
| 6 | 3g, R3 = p-Ac | 53% | |
| 7 |

|
3j, R2 = n-butyl | 84% |
| 8 | 3k, R2 = allyl | 85% | |
| 9 |
|
3l | 94% |
| 10 |

|
3o | 92% |
| 11 |
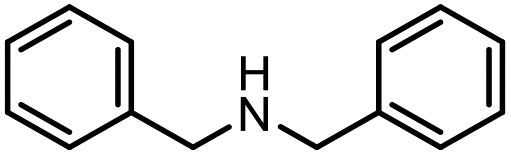
|
3q | 75% |
| 12 |
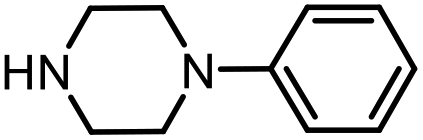
|
3r | 89% |
| 13 |
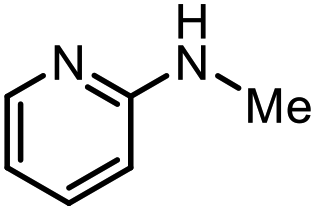
|
3s | 69% |
| 14 |

|
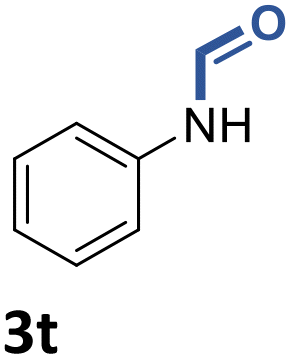
|
90% |
| 15 |
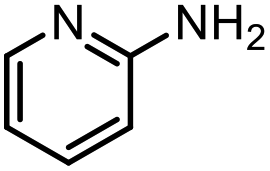
|

|
53% |

The excellent conversion and selectivity of aniline (1t) to N-phenylformamide (3t) encouraged us to study the possibility of the formation of benzimidazole by monoformylation of the o-phenylenediamine (4a) followed by subsequent intramolecular condensation with the amino group. Therefore, the reaction was performed under the same reaction conditions as for the formylation of aniline without other additives. To our delight, 83% of benzimidazole 5a was isolated from 1,2-diaminobenzene and CO2 in one step (Table 5, entry 1). This prompted us to investigate the synthesis of other imidazole derivatives 5. p-Methoxy-1,2-diaminobenzene (4b) afforded the desired product 5b in a yield of 47% (entry 2). N-Disubstituted substrates 1-N-methylbenzene-1,2-diamine (4c) and N-phenyl-o-phenylenediamine (4d) were evaluated in this protocol: 61% and 70% of the desired products 5c and 5d were obtained, respectively (entries 3 and 4). The lower yields were due to the formation of the monoformylation byproduct from the primary amino group that is less nucleophilic and reactive than secondary amino groups. There are only a few examples of the synthesis of quinazoline derivatives utilizing CO2.13,54,55 Under our conditions, the six-membered ring quinazoline derivative 5e was also isolated in a yield of 64% from o-(aminomethyl)aniline 4e (entry 5).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | Yield | ||
| Reaction conditions: diamine 4 (0.600 mmol), trimethoxysilane (3 equiv.), catalyst (2 mol%), p(CO2) = 1 bar, 70 °C for 24 h, CH3CN. Isolated yields are given. | |||||
| 1 |
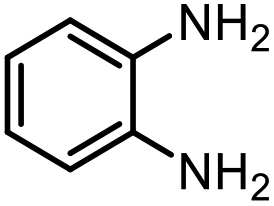
|
4a |
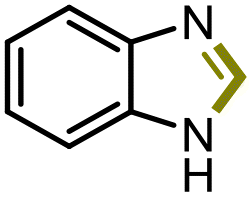
|
5a | 83% |
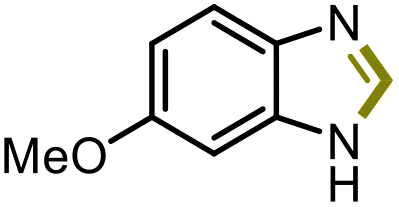
| 2 |
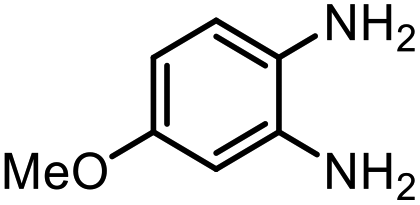
|
4b |
|
5b | 47% |
| 3 |

|
4c |
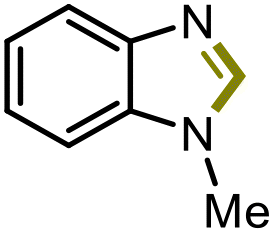
|
5c | 61% |
| 4 |

|
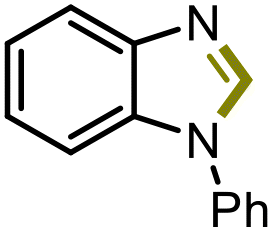
4d |
|
5d | 70% |
| 5 |
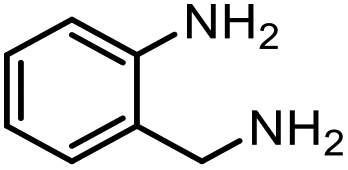
|
4e |
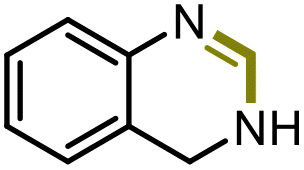
|
5e | 64% |

To gain insights into the reaction mechanism, control experiments were performed to identify the possible intermediates in the N-formylation and N-methylation reactions (Scheme 2). Initially, trimethoxysilane was reacted with CO2 in the presence of the catalyst [Ph3PMe]CO3Me, affording the silyl formate in 29% yield. Characteristic signals for silylformate were observed in the 1H and 13C NMR spectra at δ(SiOCHO) = 7.96 ppm and δ(SiOCHO) = 158.2 ppm, respectively (Scheme 2a and Fig. S1†).56 Notably, in the absence of the catalyst, this was not observed. Apart from silyl formate, tetramethoxysilane was also observed in the 1H NMR and 29Si NMR spectra at δ(SiOCH3) = 3.40 ppm and δ(SiOCH3) = −78.6 ppm, respectively (Fig. S1–S3†). The formation of tetramethoxysilane can be addressed by the over-reduction of silyl formate in the absence of amines.57 The resulting reaction mixture was purged with argon to replace the remaining CO2. N-Methylaniline (1a) was added and reacted at 70 °C for 6 h, affording N-methylformanilide (2a) in 69% yield (Scheme 2b). Without the catalyst, the silyl formate was not observed. Moreover, the silyl formate did not react with the amine 1a in the absence of the catalyst.
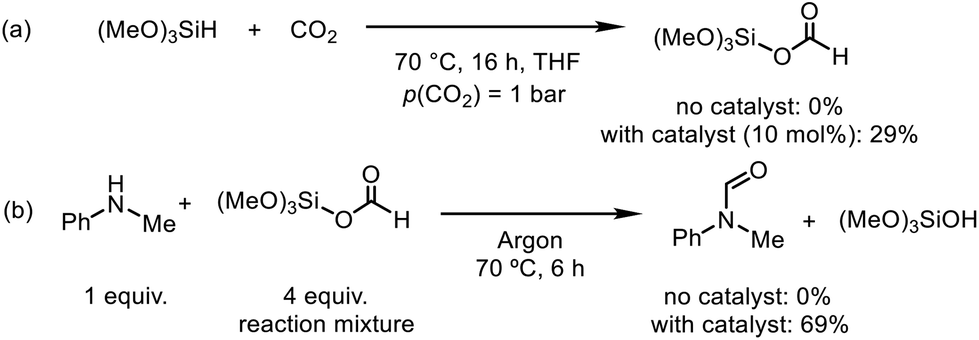 | ||
| Scheme 2 Control experiments for the N-formylation reaction. The yields were determined by 1H NMR using mesitylene as the internal standard. | ||
Thus we conclude that N-formylation proceeds via the silyl formate intermediate which has been reported before.58 However, for the N-methylation of amines by CO2 in the presence of organocatalysts, two possible pathways are discussed in the literature. Both pathways proceed via the initial formation of the silyl formate (Scheme 3). Path I: the N-formamide is formed and further reduced to N-methylamine by the silane. The majority of catalytic systems were proved to follow this pathway by experimental and computational evidence.26,29,41,59,60 Path II: the silane is reduced to silyl acetal, which reacts with the amine to afford the aminal. Subsequently, the aminal is reduced to the methylated product.28,38
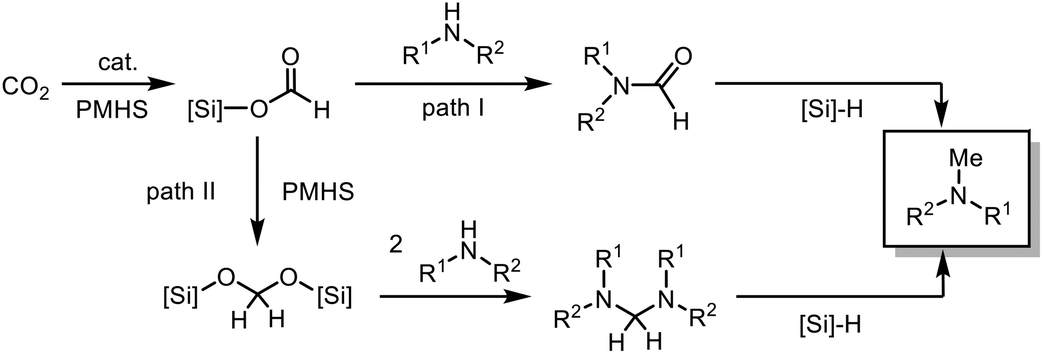 | ||
| Scheme 3 Possible pathways for the N-methylation reaction with PMHS. | ||
The two routes were evaluated by performing a series of test reactions. First, the conversion of N-formamide to N-methylamine was tested using PMHS as the reductant in the presence and absence of the catalyst under the methylation reaction conditions (Scheme 4a). In both cases, the product could not be obtained. Thus, path I was ruled out. Subsequently, we considered path II. We hypothesized that the intermediate formation of the aminal in the reaction mixture should be detectable by NMR. Indeed, it was possible to detect the formation of the aminal by 1H NMR spectroscopy. The spectra showed a characteristic signal for the aminal resonance at δ(N–CH3) = 2.86 ppm which agrees with previous reports.61 Under the optimized reaction conditions, the aminal was initially formed in 9% yield after 2 h (Table S1 and Fig. S5†). This supports the assumed formation of the aminal as an intermediate. Furthermore, the conversion of the aminal to the N-methylamine was studied. In this regard, N,N,N′,N′-tetramethylethanediamine (TMEDA), a commercially available aminal, was chosen to react with PMHS in the presence of CO2 since it is reported that CO2 contributed to the cleavage of the N–C bond.58 Under the reaction conditions, the aminal (TMEDA) and PMHS were converted and the corresponding N-methylated product was obtained in 50% yield in the presence of the catalyst, which demonstrated that path II is reasonable for this catalytic system (Scheme 4b and Fig. S6†). Only traces of the product were observed in the absence of CO2 or catalyst.
 | ||
| Scheme 4 Control experiments for the N-methylation reaction. The yield was determined by 1H NMR using mesitylene as the internal standard. | ||
Interestingly, despite being a milder reducing agent than trimethoxysilane, PMHS was efficient for the 6-electron reduction of CO2 in the presence of the catalyst62 This might be explained when path II is considered. In this route, due to the polymeric nature of PMHS, the formation of a bis silyl acetal can proceed via an intramolecular reduction of the silyl formate (Scheme S1†).
Once the different routes to the formation of products were established, we became interested in understanding the role of the catalyst in this reaction and the different selectivity observed for the reducing agents. Initially, a stoichiometric mixture of PMHS and catalyst [Ph3PMe]CO3Me was heated to 70 °C and analyzed by NMR spectroscopy (Fig. 1c). The characteristic signal at δ(Si–H) = 4.72 ppm in PMHS (see 1a) and the signal at δ(CO3CH3) = 3.37 ppm (see 1b) for the catalyst were not observed in the mixture. Instead, a new signal at δ = 8.58 ppm was observed in the mixture which can be attributed to the formation of the formate anion.63 A signal with the same chemical shift (δ(HCO2−) = 8.58 ppm) was observed in the mixture of trimethoxysilane and the catalyst (Fig. 2c). Also, in this case, the signals at δ(Si–H) = 4.07 ppm for the silane and for the catalyst δ(CO3CH3) = 3.37 ppm were not detectable in the mixture. The signals at 3.37 ppm and −78.60 ppm in the 29Si NMR spectra (Fig. S7†), respectively, indicate the formation of Si(OMe)4.64 The common signal in both mixtures at around 3.30 ppm (marked as *) can be addressed to a methoxy anion which is probably formed due to the decomposition of [Ph3PMe]CO3Me to CO2 and [Ph3PMe]OMe.49 These results indicate the conversion of the methylcarbonate anion and silane to formate. This led us to the assumption that the reaction is initiated in this step. Based on these results, we propose the mechanism shown in Scheme 5.
 | ||
| Fig. 1 (a) Section of the 1H NMR spectra of PMHS. (b) Section of the 1H NMR spectra of catalyst [Ph3PCH3]CO3Me. (c) Section of the 1H NMR spectra of a 1:1 mixture of PMHS and [Ph3PCH3]CO3Me after 16 h at 70 °C. * denotes the methoxy anion. | ||
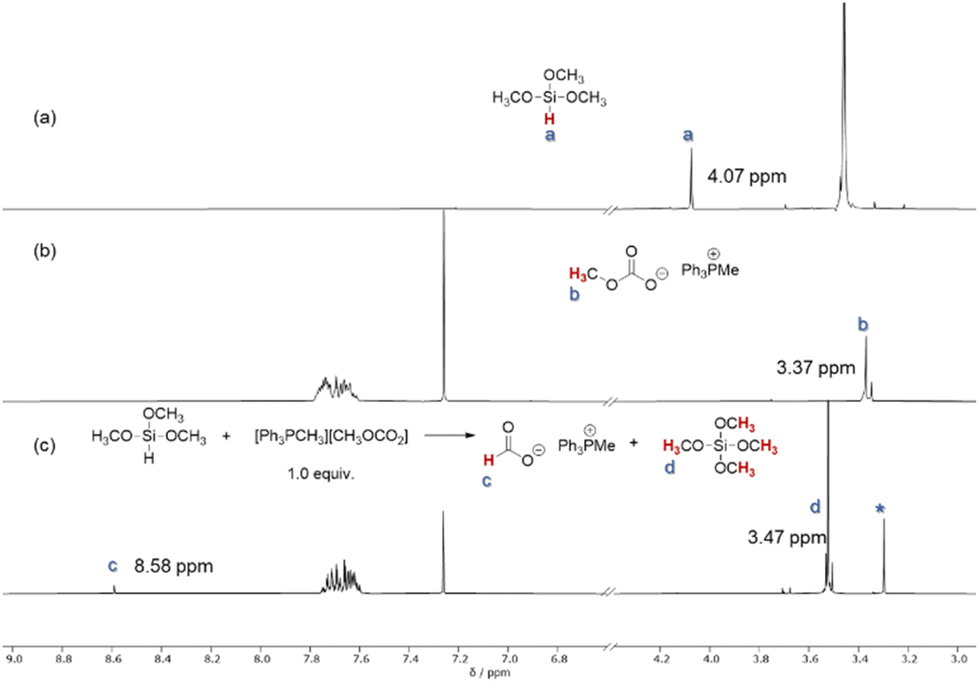 | ||
| Fig. 2 (a) Section of the 1H NMR spectra of PMHS. (b) Section of the 1H NMR spectra of the catalyst [Ph3PMe]CO3Me. (c) Section of the 1H NMR spectra of a 1:1 mixture of PMHS and [Ph3PMe]CO3Me after 16 h at 70 °C. * denotes the methoxy anion. | ||
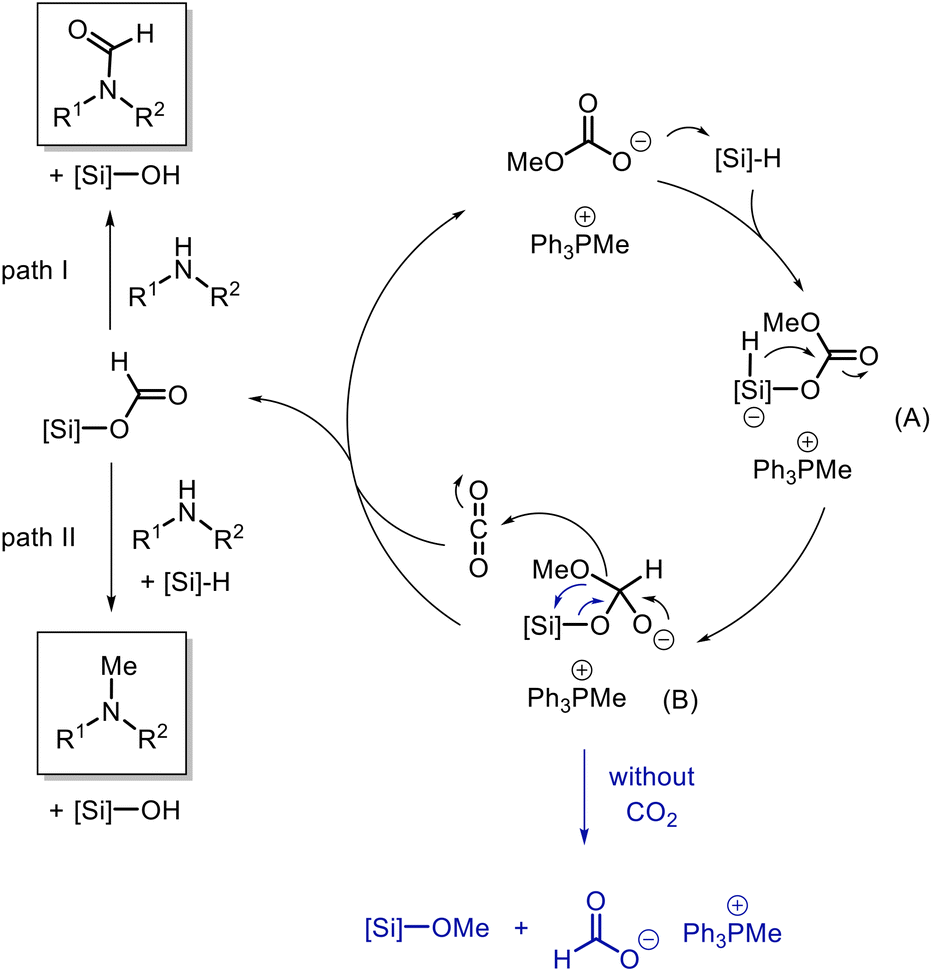 | ||
| Scheme 5 Mechanistic proposal for the selective conversion of amines with CO2 to formamides or methylamines with phosphonium methylcarbonate salts. | ||
The methylcarbonate anion attacks the silane forming A which facilitates the attack of the hydride to the carbonyl center and the generation of B. The decomposition of B leads to silyl formate and a methoxy anion which reacts with CO2 to regenerate methylcarbonate (catalyst) and it also leads to the regeneration of the catalyst in the following step, which has no negative impact on the catalytic efficiency. As indicated above, in the absence of CO2, this B can undergo a rearrangement resulting in the formate anion, e.g. tetramethoxysilane. In the presence of trimethoxysilane as the reducing agent, the formate further reacts with the amine through path I to the N-formylated product which is not further reduced. In contrast, if PMHS is used as the reducing agent, the N-formylated product further reacts to form bis silyl acetal which subsequently reacts with the amine and PMHS to give the N-methylated product.
Conclusions
Phosphonium methylcarbonate has been proven to be an efficient organocatalyst for the conversion of CO2 with primary and secondary amines and diamines. Notably, the selectivity of the reaction can be tuned by the choice of the reductant. In the presence of readily available PMHS, selective N-methylation was achieved under mild conditions. In contrast, the use of trimethoxysilane as the reducing agent leads to the selective N-formylation of the amines. Moreover, the one-pot synthesis of imidazoles was possible when diamines were used as the starting material. Mechanistic investigations revealed different reaction pathways for N-methylation and N-formylation depending on the reductant. N-Methylation proceeds most probably via silyl formate while N-formylation proceeds via a bissilyl acetal and aminal intermediate. The mechanistic studies also suggest that the catalyst directly activates CO2. This tuneable process was applied to a broad range of substrates. Under the optimized conditions, 15 primary and secondary amines and anilines were converted to N-methylated products in yields of up to 98%. The N-formylation of 15 amines led to the desired products in yields of up to 94%. Additionally, this procedure was evaluated for a one-pot N-formylation/condensation reaction of diamines leading to the corresponding heterocycles in yields of up to 83%. Thus, the presented mechanistic studies give insights into the N-methylation and N-formylation of amines with CO2. In addition, the protocols presented provide straightforward access to valuable intermediates that use CO2 as a C1 building block.Experimental
Synthesis of methyltriphenylphosphonium methylcarbonate
In a 10 mL Schlenk tube, triphenylphosphine (0.235 g, 0.896 mmol), dimethylcarbonate (DMC) (0.52 mL, 6.15 mmol), and methanol (0.52 mL) as solvent were introduced in the shortest time possible to limit the exposure of phosphine to air. The Schlenk tube was heated for 24 h at 140 °C. The reaction mixture was allowed to cool to an ambient temperature and volatiles were removed in a vacuum. The off-white solid was stirred under an inert atmosphere with cyclohexane (8.3 mL) at 50 °C for 2 hours. After filtration on a Gooch crucible, the product was obtained in 64% yield as a colorless solid (0.215 mg, 0.580 mmol). 1H NMR (300 MHz, CDCl3): δ = 7.82–7.58 (m, 15H), 3.37 (s, 3H) ppm. The methyl protons (P-CH3) were not detectable due to H/D exchange.501H NMR (300 MHz, CD3CN): δ = 7.98–7.81 (m, 3H), 7.79–7.62 (m, 12H), 3.29 (s, 3H), 2.89 (d, J = 15.7 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ = 134.96 (d, J = 2.9 Hz), 133.26 (d, J = 10.7 Hz), 130.39 (d, J = 12.9 Hz), 119.41 (d, J = 88.5 Hz), 52.20. 13C NMR (75 MHz, CD3CN): δ = 162.20, 140.32 (d, J = 3.1 Hz), 138.68 (d, J = 10.8 Hz), 135.47 (d, J = 12.9 Hz), 124.98 (d, J = 88.9 Hz), 56.17, 13.50 (d, J = 57.8 Hz) ppm. 31P NMR (122 MHz, CDCl3): δ = 21.92 ppm. HR-MS (MePh3P+): m/z calcd 277.1151, found 277.1153.Representative procedure for N-methylation – the synthesis of N-benzyl-N-methylaniline (2h)
In a stainless-steel autoclave, PMHS (414 μL) was added to a solution of the catalyst (21.4 mg, 60.0 μmol) and N-benzylaniline (1h, 110 mg, 0.600 mmol) in THF (5 mL). The autoclave was purged with CO2 and the pressure was kept constant at 1 bar. The reaction mixture was stirred at 70 °C for 16 h. Subsequently, CO2 was released slowly. The solvent was removed in a vacuum and the residue was purified by chromatography (SiO2, cyclohexane/ethyl acetate = 20/1) to yield N-benzyl-N-methylaniline (105 mg, 0.533 mmol, 89%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ = 7.37–7.28 (m, 2H), 7.28–7.19 (m, 5H), 6.81–6.69 (m, 3H), 4.55 (s, 2H), 3.03 (s, 3H).Representative procedure for N-formylation – the synthesis of N-benzylformamide (3o)
In a stainless-steel autoclave, trimethoxysilane (220 mg, 1.80 mmol) was added to a solution of the catalyst (4.23 mg, 12.0 μmol) and N-methyl-1-phenylmethanamine (72.7 mg, 0.600 mmol) in THF (0.6 mL). The autoclave was purged with CO2 and the pressure was kept constant at 1 bar. The reaction mixture was stirred at 70 °C for 24 h. Subsequently, CO2 was released slowly. The solvent was removed in a vacuum and the residue was purified by silica gel chromatography (cyclohexane/ethyl acetate = 10/1) to afford N-benzylformamide (82.0 mg, 0.552 mmol, 92%) as a colourless oil. 1H NMR (300 MHz, CDCl3) δ = 8.21 (s, 0.57H, maj), 8.08 (s, 0.43H, min), 7.38–7.04 (m, 5H), 4.44 (s, 0.89H, min), 4.31 (s, 1.19H, maj), 2.77 (s, 1.33H, min), 2.70 (s, 1.71H, maj).Representative procedure for the preparation of benzimidazoles – the synthesis of 1H-1,3-benzimidazole (4a)
In a stainless-steel autoclave, trimethoxysilane (220 mg, 1.80 mmol) was added to a solution of the catalyst (4.23 mg, 12.0 μmol) and o-phenylenediamine (64.9 mg, 0.600 mmol) in THF (6 mL). The autoclave was purged with CO2 and the pressure was kept constant at 1 bar. The reaction mixture was stirred at 70 °C for 24 h. Subsequently, CO2 was released slowly. Then the solvent was removed in a vacuum and the residue was purified by silica gel chromatography (cyclohexane/ethyl acetate = 1/1) to yield 1H-benzo[d]imidazole (58.8 mg, 0.498 mmol, 83%) as a colorless solid. 1H NMR (300 MHz, CDCl3) δ 8.11 (s, 1H), 7.79–7.59 (m, 2H), 7.35–7.28 (m, 2H), 6.28 (s, 1H).Author contributions
C. Ren performed the initial reaction screening. C. Ren and C. Terazzi performed the optimization of the reaction conditions and mechanistic studies. C. Ren performed the substrate scope. C. Ren, C. Terazzi and T. Werner wrote the manuscript.Conflicts of interest
There are no conflicts to declare.References
- J. Chatterjee, F. Rechenmacher and H. Kessler, Angew. Chem., Int. Ed., 2013, 52, 254–269 CrossRef CAS PubMed.
- M. Schou and C. Halldin, J. Labelled Compd. Radiopharm., 2012, 55, 460–462 CrossRef CAS.
- G. Antoni and B. Långström, in Positron Emission Tomography: Basic Sciences, ed. D. L. Bailey, D. W. Townsend, P. E. Valk and M. N. Maisey, Springer London, London, 2005, pp. 223–236, DOI:10.1007/1-84628-007-9_10.
- D. J. McCarthy, C. Halldin, J. D. Andersson and M. E. Pierson, in Annual Reports in Medicinal Chemistry, ed. J. E. Macor, Academic Press, 2009, vol. 44, pp. 501–513 Search PubMed.
- E. M. Isin, C. S. Elmore, G. N. Nilsson, R. A. Thompson and L. Weidolf, Chem. Res. Toxicol., 2012, 25, 532–542 Search PubMed.
- A. Del Vecchio, G. Destro, F. Taran and D. Audisio, J. Labelled Compd. Radiopharm., 2018, 61, 988–1007 CrossRef CAS PubMed.
- A. Del Vecchio, F. Caille, A. Chevalier, O. Loreau, K. Horkka, C. Halldin, M. Schou, N. Camus, P. Kessler, B. Kuhnast, F. Taran and D. Audisio, Angew. Chem., Int. Ed., 2018, 57, 9744–9748 CrossRef CAS PubMed.
- H. Bipp and H. Kieczka, in Ullmann's Encyclopedia of Industrial Chemistry, 2000, DOI:10.1002/14356007.a12_001.
- S. Kobayashi and K. Nishio, J. Org. Chem., 1994, 59, 6620–6628 CrossRef CAS.
- F. Brunelli, S. Aprile, C. Russo, M. Giustiniano and G. C. Tron, Green Chem., 2022, 24, 7022–7028 RSC.
- S. A. Salami, X. Siwe-Noundou and R. W. Krause, Molecules, 2022, 27, 6850 CrossRef CAS PubMed.
- M. L. Bennasar, T. Roca, M. Monerris and D. García-Díaz, J. Org. Chem., 2006, 71, 7028–7034 CrossRef CAS PubMed.
- M. Hulla, S. Nussbaum, A. R. Bonnin and P. J. Dyson, Chem. Commun., 2019, 55, 13089–13092 RSC.
- A. Sharma, A. Kumar, S. A. H. Abdel Monaim, Y. E. Jad, A. El-Faham, B. G. de la Torre and F. Albericio, Biopolymers, 2018, 109, e23110 CrossRef PubMed.
- M. J. Calverley, Synth. Commun., 1983, 13, 601–609 CrossRef CAS.
- E. Müller, H. Huber-Emden and W. Rundel, Justus Liebigs Ann. Chem., 1959, 623, 34–46 CrossRef.
- H. Seo, A.-C. Bédard, W. P. Chen, R. W. Hicklin, A. Alabugin and T. F. Jamison, Tetrahedron, 2018, 74, 3124–3128 CrossRef CAS.
- V. Goyal, N. Sarki, A. Narani, G. Naik, K. Natte and R. V. Jagadeesh, Coord. Chem. Rev., 2023, 474, 214827 CrossRef CAS.
- B. N. Atkinson and J. M. J. Williams, ChemCatChem, 2014, 6, 1860–1862 CrossRef CAS.
- G. Naik, N. Sarki, V. Goyal, A. Narani and K. Natte, Asian J. Org. Chem., 2022, 11, e202200270 CrossRef CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- J.-H. Ye, T. Ju, H. Huang, L.-L. Liao and D.-G. Yu, Acc. Chem. Res., 2021, 54, 2518–2531 CrossRef CAS PubMed.
- I. Sullivan, A. Goryachev, I. A. Digdaya, X. Li, H. A. Atwater, D. A. Vermaas and C. Xiang, Nat. Catal., 2021, 4, 952–958 CrossRef CAS.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- X.-F. Liu, R. Ma, C. Qiao, H. Cao and L.-N. He, Chem. – Eur. J., 2016, 22, 16489–16493 CrossRef CAS PubMed.
- C. Xie, J. Song, H. Wu, B. Zhou, C. Wu and B. Han, ACS Sustainable Chem. Eng., 2017, 5, 7086–7092 CrossRef CAS.
- X.-F. Liu, X.-Y. Li, C. Qiao, H.-C. Fu and L.-N. He, Angew. Chem., Int. Ed., 2017, 56, 7425–7429 CrossRef CAS PubMed.
- Y. Hu, J. Song, C. Xie, H. Wu, Z. Wang, T. Jiang, L. Wu, Y. Wang and B. Han, ACS Sustainable Chem. Eng., 2018, 6, 11228–11234 CrossRef CAS.
- X.-F. Liu, X.-Y. Li, C. Qiao and L.-N. He, Synlett, 2018, 29, 548–555 CrossRef CAS.
- R. Yao, Y. Li, J. Wang, J. Chen and Y. Xu, J. Catal., 2023, 418, 78–89 CrossRef CAS.
- H. Wu, W. Dai, S. Saravanamurugan, H. Li and S. Yang, Green Chem., 2020, 22, 5822–5832 RSC.
- N. M. Hein, Y. Seo, S. J. Lee and M. R. Gagné, Green Chem., 2019, 21, 2662–2669 RSC.
- R. O. Sauer, W. Scheiber and S. D. Brewer, J. Am. Chem. Soc., 1946, 68, 962–963 CrossRef CAS.
- N. J. Lawrence, M. D. Drew and S. M. Bushell, J. Chem. Soc., Perkin Trans., 1999, 3381–3391 RSC.
- F. D. Bobbink, S. Das and P. J. Dyson, Nat. Protoc., 2017, 12, 417–428 CrossRef CAS PubMed.
- S. Das, F. D. Bobbink, S. Bulut, M. Soudani and P. J. Dyson, Chem. Commun., 2016, 52, 2497–2500 RSC.
- G. Li, J. Chen, D.-Y. Zhu, Y. Chen and J.-B. Xia, Adv. Synth. Catal., 2018, 360, 2364–2369 CrossRef CAS.
- Z. Zhang, Q. Sun, C. Xia and W. Sun, Org. Lett., 2016, 18, 6316–6319 CrossRef CAS PubMed.
- X. Li, J. Zhang, Y. Yang, H. Hong, L. Han and N. Zhu, J. Organomet. Chem., 2021, 954–955, 122079 CrossRef CAS.
- W. Zhao, X. Chi, H. Li, J. He, J. Long, Y. Xu and S. Yang, Green Chem., 2019, 21, 567–577 RSC.
- C. Fang, C. Lu, M. Liu, Y. Zhu, Y. Fu and B.-L. Lin, ACS Catal., 2016, 6, 7876–7881 CrossRef CAS.
- T. Werner and H. Büttner, ChemSusChem, 2014, 7, 3268–3271 CrossRef CAS PubMed.
- H. Büttner, J. Steinbauer and T. Werner, ChemSusChem, 2015, 8, 2655–2669 CrossRef PubMed.
- J. Steinbauer, L. Longwitz, M. Frank, J. Epping, U. Kragl and T. Werner, Green Chem., 2017, 19, 4435–4445 RSC.
- C. Ren, A. Spannenberg and T. Werner, Asian J. Org. Chem., 2022, 11, e202200156 CrossRef CAS.
- A. Berkessel and M. Brandenburg, Org. Lett., 2006, 8, 4401–4404 CrossRef CAS PubMed.
- M. Hatano, Y. Tabata, Y. Yoshida, K. Toh, K. Yamashita, Y. Ogura and K. Ishihara, Green Chem., 2018, 20, 1193–1198 RSC.
- M. Fabris, M. Noè, A. Perosa, M. Selva and R. Ballini, J. Org. Chem., 2012, 77, 1805–1811 CrossRef CAS PubMed.
- L. Cattelan, M. Noè, M. Selva, N. Demitri and A. Perosa, ChemSusChem, 2015, 8, 3963–3966 CrossRef CAS PubMed.
- C. Lu, Z. Qiu, Y. Zhu and B.-L. Lin, Sci. Bull., 2019, 64, 723–729 CrossRef CAS PubMed.
- H. Zhang, Y. Zhang and K. Gao, Org. Chem. Front., 2023, 10, 2491–2497 RSC.
- A. Spinks and E. H. P. Young, Nature, 1958, 181, 1397–1398 CrossRef CAS.
- O. Jacquet, C. Das Neves Gomes, M. Ephritikhine and T. Cantat, ChemCatChem, 2013, 5, 117–120 CrossRef CAS.
- V. V. Phatake and B. M. Bhanage, Catal. Lett., 2019, 149, 347–359 CrossRef CAS.
- M. Tüchler, L. Gärtner, S. Fischer, A. D. Boese, F. Belaj and N. C. Mösch-Zanetti, Angew. Chem., Int. Ed., 2018, 57, 6906–6909 CrossRef PubMed.
- G. Feng, C. Du, L. Xiang, I. del Rosal, G. Li, X. Leng, E. Y. X. Chen, L. Maron and Y. Chen, ACS Catal., 2018, 8, 4710–4718 CrossRef CAS.
- M. Hulla and P. J. Dyson, Angew. Chem., Int. Ed., 2020, 59, 1002–1017 CrossRef CAS PubMed.
- S. Maji, A. Das and S. K. Mandal, Chem. Sci., 2021, 12, 12174–12180 RSC.
- Z. Huang, X. Jiang, S. Zhou, P. Yang, C.-X. Du and Y. Li, ChemSusChem, 2019, 12, 3054–3059 CrossRef CAS PubMed.
- M.-Y. Wang, N. Wang, X.-F. Liu, C. Qiao and L.-N. He, Green Chem., 2018, 20, 1564–1570 RSC.
- D. Addis, S. Das, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6004–6011 CrossRef CAS PubMed.
- H. Gao, J. Jia, C.-H. Tung and W. Wang, Organometallics, 2023, 42, 944–951 CrossRef CAS.
- D. C. Braddock, P. D. Lickiss, B. C. Rowley, D. Pugh, T. Purnomo, G. Santhakumar and S. J. Fussell, Org. Lett., 2018, 20, 950–953 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03993e |
| This journal is © The Royal Society of Chemistry 2024 |