
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the potential of a covalent triazine framework based on [1]benzothieno[3,2-b][1]benzothiophene (DPhBTBT-CTF) as a metal-free heterogeneous photocatalyst†
M. Carmen
Borrallo-Aniceto
a,
Mercedes
Pintado-Sierra
b,
Antonio
Valverde-González‡
 a,
Urbano
Díaz
c,
Félix
Sánchez
b,
Eva M.
Maya
*a and
Marta
Iglesias
*a
a,
Urbano
Díaz
c,
Félix
Sánchez
b,
Eva M.
Maya
*a and
Marta
Iglesias
*a
aInstituto de Ciencia de Materiales de Madrid, CSIC, C/ Sor Juana Inés de la Cruz, 3, 28049, Madrid, Spain
bInstituto de Química Orgánica General, CSIC, C/ Juan de la Cierva, 3, 28006, Madrid, Spain
cInstituto de Tecnología Química, Universitat Politècnica de València, Consejo Superior de Investigaciones Científicas, Avenida de los Naranjos s/n, E-46022 Valencia, Spain
First published on 21st December 2023
Abstract
The development of photocatalysts that are effective in different organic processes is a topic of great interest to researchers in materials chemistry. Herein, we report the design and synthesis of a new covalent triazine framework (CTF) built by acid-catalyzed trimerization of 4,4′-(benzo[b]benzo[4,5]thieno[2,3-d]thiophene-2,7-diyl)dibenzonitrile activated by microwaves that generates a new organic polymer, DPhBTBT-CTF. This CTF exhibits visible-light absorption due to the electron-donating BTBT units and the extended π-conjugated framework with the electron-acceptor triazine node and proves to be an efficient and versatile metal-free heterogeneous photocatalyst for different organic processes such as selective oxidations of both sulfides and benzyl alcohols to sulfoxides and benzaldehydes, respectively or the oxidative bromination of electron rich aromatic compounds. Moreover, two different proof of concept reactions were tested: the Diels–Alder cycloaddition and the hydroxylation of boronic acids. Heterogeneity studies confirmed that this photocatalyst can be recycled without significant loss of selectivity, and also some mechanistic tests suggested the preferred oxidation pathway.
Introduction
Following the emergence of the principles of green chemistry, catalytic processes have been widely applied in different industries. In this sense, photocatalyzed reactions have received increasing attention in recent years due to the mild reaction conditions and their fit within the principles of sustainable chemistry. Thus, different types of photocatalysts have been reported from molecular organic dyes or transition-metal complexes to solid heterogeneous catalysts.1–7 The design and preparation of highly stable photocatalysts with high performance have aroused great interest. Besides, photoredox catalysts based on semiconductors have made it possible to carry out processes that are difficult to do with other catalysts, such as multistep reactions.8 In the last decade, different organic porous polymers (POPs) have been applied in different photocatalytic processes such as oxidations, reductions, couplings, cyclizations, polymerizations and asymmetric reactions among others.9–11 From a structural point of view, POPs can be classified as crystalline covalent organic frameworks (COFs), covalent triazine frameworks (CTFs) or amorphous porous aromatic frameworks (PAFs), conjugated microporous polymers (CMPs), polymers of intrinsic microporosity (PIMs) and hyper-crosslinked polymers (HCPs). CTFs are a promising type of material which have a 1,3,5-triazine ring covalently bonded to organic units with remarkable photophysical properties12,13 that make them useful to be applied in different photocatalytic reactions.14 CTFs have been employed as photocatalysts in aerobic oxidation reactions (sulfides,15–17 benzyl alcohols18) and oxidative coupling of amines or dehalogenation reactions,19 and also are effective for oxidative degradation (methylene blue, safranin T or rhodamine B).20 However, in order to increase the versatility of these materials, it is necessary to introduce other photoactive groups into the framework.Fused π-conjugated [1]benzothieno[3,2-b][1]benzothiophene (BTBT) derivatives are an important class of organic semiconductors due to their robustness and physicochemical characteristics.21–24 Also, it has been reported that organic semiconductors based on 2,7-diphenyl[1]-benzothieno[3,2-b][1]-benzothiophene derivatives (DPhBTBT)25 show significantly improved field effect transistor (FET) properties either because of its functionalization with methoxy groups (BOP–BTBT)26 or because of the π-extension27 structure. Therefore, the incorporation of BTBT units into organic frameworks is an interesting approach for developing stable organic semiconductor polymers since the solid-state properties can be improved (conjugation, charge transport, etc.). Some polymers based on thiophene,28 benzodithiophene,29 or thieno–thiophene units have been reported and have been applied as electroactive polymers,30 photoluminiscent devices, or film transistors,31,32 or proved as photocatalysts for hydrogen33 and H2O2 production.34,35 However, there are scarce examples in which these materials have been applied as heterogeneous photocatalysts for organic synthesis.36,37
In this work, we have synthesized a new material based on a triazine core with linkages of diphenylBTBT (Fig. 1) (DPhBTBT-CTF) by an acid-catalyzed cyclotrimerization reaction activated by microwaves. Although DPhBTBT-CTF is amorphous, the coexistence of BTBT (electron donor) and triazine (electron acceptor) moieties has a synergistic effect producing an improved photocatalytic performance that does not occur with its molecular counterparts. To the best of our knowledge, the application of BTBT-based polymers as photocatalysts for organic transformations has been little studied; thus, we have demonstrated that DPhBTBT-CTF activates a great diversity of substrates being an effective and versatile catalyst for selective aerobic oxidations, bromination of aromatic compounds, Diels–Alder cycloadditions or even hydroxylation of boronic acids.
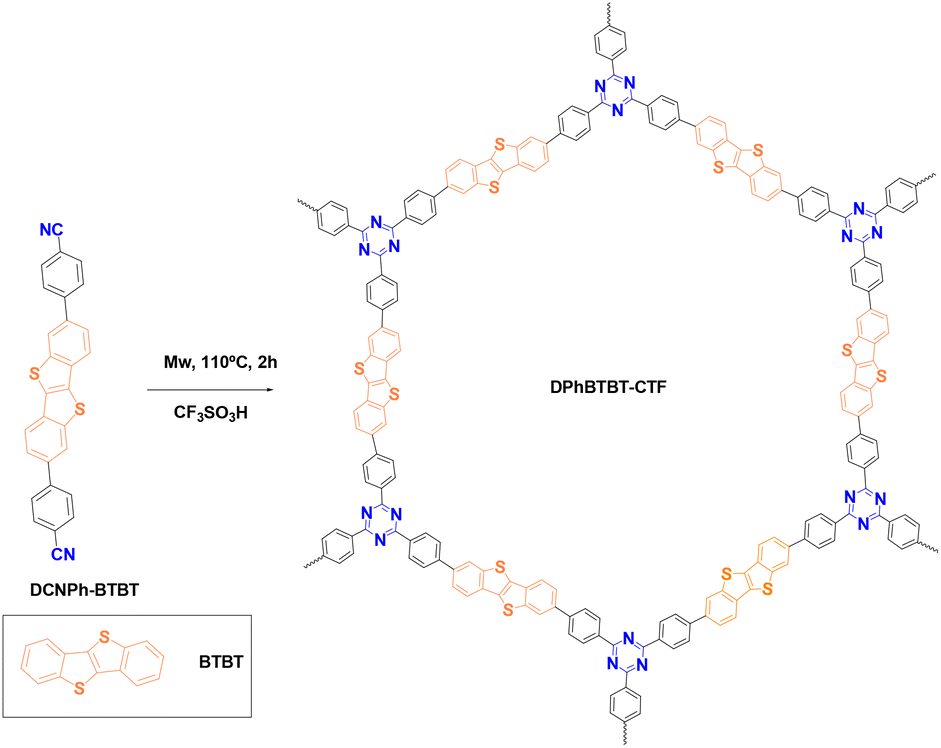 | ||
| Fig. 1 Synthesis and idealized structure of the DPhBTBT-conjugated covalent triazine framework. | ||
Results and discussion
Synthesis and characterization
A diphenylthienothiophene based CTF (DPhBTBT-CTF) was synthesized by cyclotrimerization of 4,4′-(benzo[b]benzo[4,5]thieno[2,3-d]thiophene-2,7-diyl)dibenzonitrile (DCNPh–BTBT). The synthetic schemes to prepare the monomer and the corresponding polymer are shown in Scheme S1† and Fig. 1, respectively. DCNPh–BTBT was obtained through the Suzuki–Miyaura coupling reaction using (4-cyanophenyl)boronic acid and 2,7-dibromobenzo[b]benzo[4,5]thieno[2,3-d]thiophene (Br2-BTBT)38 catalyzed by Pd(dppf)2Cl2. The resulting product was purified by recrystallization from dimethylacetamide (DMA) to get the pure compound as white powder. The chemical structure was confirmed by NMR, FT-IR, mass spectrometry (MS) and elemental analysis (see ESI, Fig. S2†).It has been demonstrated that superacid-catalyzed polymerization under mild reaction conditions conserves the semiconductive properties of an aromatic dinitrile monomer.39 Therefore, DPhBTBT-CTF was obtained by cyclotrimerization of dinitrile DCNPh–BTBT at a > 70% yield in two hours assisted by trifluoromethanesulfonic acid (TFMS) under microwave activation (200 W, 110 °C, 2 h),15,40 and it was insoluble in water and in usual organic solvents. The chemical structure was determined by Fourier transform infrared spectroscopy (FT-IR), solid 13C-NMR spectroscopy and analytical techniques of the solid state. The resulting material showed a N/S ratio (calculated from experimental elemental analysis) that agrees well with the theoretical value obtained from the ideal formula (see the ESI†). Powder X-ray diffraction (PXRD) shows a pattern with a broad diffraction peak centered at ∼23° that indicates an amorphous structure for the CTF (Fig. S3† right). As it has been reported for other CTFs, XRD shows a main peak at 2θ 23.2° ascribed to the π–π stacking in the conjugated aromatic framework.41,42
The FT-IR spectrum (Fig. 2a) shows the disappearance of the νC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N vibration at 2225 cm−1 which confirms the high degree of cyclization of the dinitrile monomer and the presence of the characteristic C
N vibration at 2225 cm−1 which confirms the high degree of cyclization of the dinitrile monomer and the presence of the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching bands of the triazine ring at 1358 and 1504 cm−1. The signal around 1600 cm−1 is assigned to the aromatic rings and the bands at 1031, 765 (ν(C–H)) and 804 cm−1 (ν(C–S)) correspond to the thiophene skeleton. Solid-state 13C-NMR obtained by cross-polarization magical angle spinning (CP-MAS) confirms that both moieties (triazine and DPhBTBT) are present in the framework (Fig. 2b). The signal at δ 168.9 ppm, which corresponds to sp2 C atoms in the triazine ring (CN), is wider and high-field shifted compared to the previously reported naphthalene-based triazines (170.4, 169.9 ppm, Fig. S4†). This suggests a higher electron density, possibly associated with the AA stacking type, as recently reported.43 The signal corresponding to the carbons of the thiophene ring (C–S) appears at 143 ppm and the intense signals between 120 and 140 ppm are ascribed to the aromatic carbons on the phenyl ring. The disappearance of the signal at 110 ppm, corresponding to the nitrile group of the monomer, confirmed the effectiveness of the polymerization. The thermal stability was measured by TGA (thermogravimetrical analysis) under air (Fig. 2c). The thermogram shows the high thermal stability of DPhBTBT-CTF with a degradation temperature above 300 °C.
N stretching bands of the triazine ring at 1358 and 1504 cm−1. The signal around 1600 cm−1 is assigned to the aromatic rings and the bands at 1031, 765 (ν(C–H)) and 804 cm−1 (ν(C–S)) correspond to the thiophene skeleton. Solid-state 13C-NMR obtained by cross-polarization magical angle spinning (CP-MAS) confirms that both moieties (triazine and DPhBTBT) are present in the framework (Fig. 2b). The signal at δ 168.9 ppm, which corresponds to sp2 C atoms in the triazine ring (CN), is wider and high-field shifted compared to the previously reported naphthalene-based triazines (170.4, 169.9 ppm, Fig. S4†). This suggests a higher electron density, possibly associated with the AA stacking type, as recently reported.43 The signal corresponding to the carbons of the thiophene ring (C–S) appears at 143 ppm and the intense signals between 120 and 140 ppm are ascribed to the aromatic carbons on the phenyl ring. The disappearance of the signal at 110 ppm, corresponding to the nitrile group of the monomer, confirmed the effectiveness of the polymerization. The thermal stability was measured by TGA (thermogravimetrical analysis) under air (Fig. 2c). The thermogram shows the high thermal stability of DPhBTBT-CTF with a degradation temperature above 300 °C.
 | ||
| Fig. 2 Characterization data: (a) FTIR spectra and (b) solid-state 13C NMR spectra of the monomer (red line) and DPhBTBT-CTF (black line), (c) TGA, (d) XPS traces, (e) EPR, (f) SEM and (g) TEM images of DPhBTBT-CTF. | ||
Structural composition was also analyzed by X-ray photoelectron spectroscopy (XPS) (Fig. S3† left and Fig. 2d). The XPS spectrum indicates that nitrogen, carbon and sulfur are present in DPhBTBT-CTF. The N 1s spectrum exhibits a single peak centered at 400.9 eV assigned to the effective formation of pyridinic N from the triazine moiety (CN–C). The C 1s spectrum shows a peak at 285.9 corresponding to CC, C–N, and C–S carbons. Moreover, the S 2p spectrum appears divided into two peaks at 165.0 and 166.3 eV corresponding to the S 2p3/2 and S 2p1/2 binding energies, respectively. An additional peak at 169.6 eV could be due to some oxidation of sulfur on the BTBT unit.44
Electron paramagnetic resonance (EPR) is used to detect radicals in CTFs. EPR of DPhBTBT-CTF (Fig. 2e) displays a single Lorentzian line corresponding to an unpaired electron (g = 2.02) on the aromatic rings typical of organic polymers with extended π-conjugation.45 This spectrum is similar to that obtained previously from naphthalene-based covalent triazine frameworks.15
Regarding porosity, we have found that the N2 adsorption isotherm at 77 K of DPhBTBT-CTF (Fig. S5† left) shows low N2 adsorption which results in a low Brunauer–Emmett–Teller (BET) surface area of 57.8 m2 g−1. This has also been observed previously in other CTFs prepared by acid-mediated synthesis probably due to the rapid acid-catalyzed polymerization that favored a reduced internal porosity.16,46 Therefore, we have additionally measured the adsorption of CO2 at 273 K. Fig. S5† indicates that DPhBTBT-CTF exhibits microporosity with a Langmuir surface area of 280 m2 g−1. Scanning electron microscopy (SEM) images show that DPhBTBT-CTF exhibits a compact sponge-like structure, along with a distinct layered morphology similar to other CTFs synthesized using microwave activation (Fig. 2f).15 TEM images confirm the probable laminar morphology of the covalent organic materials, showcasing distinct layers intricately arranged as stacked sheets. The observed disorder among these superposed layers underscores the complex nature of the material's structure, contributing to its porous characteristics. Noteworthy is the existence of internal microporosity within these layers, suggesting the presence of sinuous channels and void spaces, characteristic of disordered covalent triazine frameworks (Fig. 2g).
The optical properties of DPhBTBT-CTF have been analyzed by ultraviolet-visible absorption and diffuse reflectance (UV-Vis, DRS) spectroscopy at room temperature showing a continuous absorption between 220 and 600 nm with maxima at 329, 465 nm (Fig. 3) and a red-shift behavior when compared to that of the DCNPh–BTBT monomer. The wide absorption and band gap showed by DPhBTBT-CTF are due to the extended π-conjugated donor–acceptor structure, which produces an electron push–pull effect. The material exhibits fluorescence with an emission maximum at 520 nm (Fig. S6†). These spectroscopic results evidenced the possibilities of this material to be used as an efficient photocatalyst for organic reactions.
 | ||
| Fig. 3 Top: (a) UV–Vis and diffuse reflectance spectra and (b) Tauc plots obtained with the Kubelka–Munk function and the linear fit for optical band gaps. Down: (c) HOMO and LUMO energy levels and (d) Nyquist plot (applied voltage E = 1.8 V). | ||
The Tauc plot obtained from the Kubelka–Munk function gives the estimated band gap energy that has a value of 2.20 eV for the CTF and 2.92 eV for the dinitrile precursor (Fig. 3b). Additionally, the corresponding highest occupied molecular orbital (HOMO) potentials (vs. Ag/AgCl) (Fig. 3c) were obtained by electrochemical cyclic voltammetry measurements (Fig. S7 and Table S1†). The HOMO is estimated from the onset of the oxidation potentials as 1.66 V for the dinitrile precursor and 1.38 V for DPhBTBT-CTF. Thus, the corresponding lowest occupied molecular orbital (LUMO) potentials were estimated to be −1.25 V and −0.81 V, respectively. The values obtained for the DPhCN–BTBT precursor agree well with those previously reported for other BTBT-compounds (diphenyl–BTBT and dimethoxyphenyl–BTBT).26,47 Electrochemical impedance spectroscopy (EIS) measurements were performed and the corresponding Nyquist plot was obtained (Fig. 3d). A broad semicircular pattern showing the charge transfer along the material is observed. It should be notice that as the photocatalytic process takes place at the surface of the particles the distance crossed by the charges are long and the obtained charge transfer resistance is high. We have applied different potentials and as shown in Fig. S7,† the semicircle is only observed when a potential of 1.8 V is applied (which corresponds to the oxidation peak in the cyclic voltammogram).48
Catalytic activity
With all the characterization results mentioned above, we set out to investigate the possibilities and scope of DPhBTBT-CTF as a photocatalyst, starting with the photooxidation of sulfides.The initial screening experiments were performed with thioanisole as a model substrate, oxygen as an oxidant, room temperature, acetonitrile as the solvent and irradiation with blue LED light (2 × 30 W). As shown in Fig. 4, the photooxidation of thioanisole achieved a conversion >99% yielding selectively the corresponding sulfoxide, which is comparable to other reported catalysts (Table S4†).49
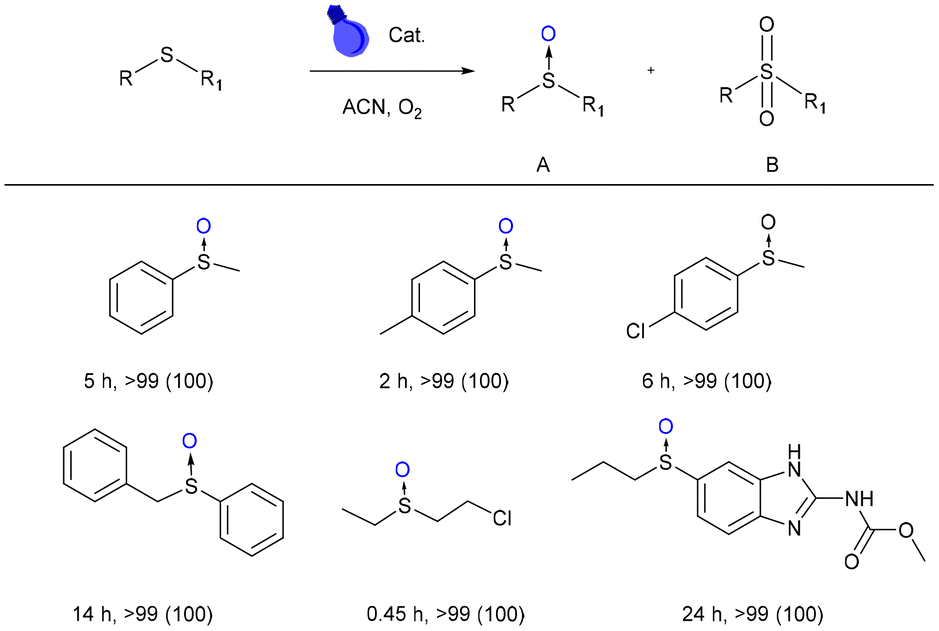 | ||
| Fig. 4 Photocatalytic performance in the sulfoxidation reaction (in parentheses, selectivity to sulfoxide). Conditions: catalyst (4.4 mg), sulfide (0.2 mmol), acetonitrile (0.5 mL), irradiated by blue LED light (2 × 30 W) at RT under an O2 balloon atmosphere. Conversion and selectivity determined by GC-MS and 1H-NMR. | ||
To compare the efficiency of the catalyst with respect to those based on naphthalene (NDP-CTFs) previously reported by us, we have oxidized methylphenylsulfide using the conditions of this work (5 mmol% catalyst and blue LED light (2 × 30 W)) against those previously reported (20 mg catalyst and blue LED (50 W))15 and we found that DPhBTBT-CTF is more efficient since the sulfoxide is obtained in only 2 h of reaction versus 5 h needed with NDP-CTF.
The substitution of oxygen by air also leads selectively to the sulfoxide although longer reaction times are necessary (9 h). DPhBTBT-CTF is also effective for the oxidation of 4-methythioanisole (with an electron-donating substituent) yielding selectively the sulfoxide in only 2 h. Oxidation of a 4-chlorothioanisole (with an electron withdrawing group) affords the corresponding sulfoxide after a longer reaction time (6 h). Oxidation of phenyl benzyl sulfide, a substrate with high steric hindrance, is also effectively affording the sulfoxide in 14 h. Interestingly, (2-chloroethyl)(ethyl)sulfane (mustard gas simulant) is the fastest to get oxidized to its sulfoxide. Besides, to expand the applicability of DPhBTBT-CTF, we have oxidized the albenzadole obtaining selectively the corresponding sulfoxide after 24 h. Some representative chromatograms and NMR spectra of these reactions are collected in Fig. S9 and S10.† Control experiments without light, a catalyst or an oxidant yield only traces of sulfoxide after 24 h (Table S2,† entries 1–3), which demonstrates that all components are necessary for the reaction to take place. The reaction using the dinitrile precursor as a catalyst only leads to traces of sulfoxide after 12 h of irradiation (Table S2,† entry 4) what confirms the synergic effect of both the triazine core and the BTBT.
In general, photocatalytic oxidation processes involving polymeric organic frameworks implies the participation of singlet oxygen and superoxide (1O2, O2˙−) reactive species.7 To determine the main oxygen species involved and thus elucidate the photocatalytic mechanism, we conducted several control experiments under standard conditions in the presence of various types of scavengers (Table S2, entries 5–9 and Fig. S8†). The oxidation in the presence of 2,2,6,6-tetramethylpiperidine, TEMPO, and p-benzoquinone (radical scavengers), 1,4-diazabicyclo[2.2.2]octane, DABCO (a singlet oxygen scavenger), and KI (holes, h+ scavenger) are largely inhibited and the presence of CuSO4 (e− scavenger) only yields 15.4% product which indicates that every species (1O2, O2˙− and h+) participated in the process.
Taking into account these results and other reported data,42 the mechanism proposed implies a charge separation after irradiation beside the formation of e−–h+ couples. Then, by an electron and energy transfer, oxygen was activated as radical O2˙− and singlet oxygen 1O2 species. Holes produce an intermediate cationic radical from sulfide and then O2˙− and 1O2 attack it to give an intermediate which reacts with the sulfide to produce the sulfoxide (Fig. S10†).
The photoredox ability of DPhBTBT-CTF was also tested in the oxidation of benzyl alcohols, yielding quantitatively and with high selectivity the corresponding benzaldehydes (Fig. 5). The oxidation of 4-methylbenzyl alcohol and 4-methoxybenzyl alcohol (with an electron-donating group) leads to the corresponding benzaldehyde with high selectivity. The 4-NO2–benzyl alcohol led to a lower conversion due to the strong electron-withdrawing effect. Chromatograms of some reactions are collected in Fig. S11.†
 | ||
| Fig. 5 Photooxidation of benzylic alcohols (in parentheses, selectivity to aldehydes). Conditions: substrate (0.2 mmol), catalyst (4.4 mg), acetonitrile (0.5 mL), O2 and blue LED light (2 × 30 W) (conversion and selectivity determined by GC-MS). | ||
Similarly, control experiments in the presence of scavengers (TEMPO, DABCO, KI or CuSO4) (Fig. S12†) indicated that the photogenerated electrons activate the oxygen generating the radical O2˙− and singlet oxygen 1O2 species. Therefore, in the oxidation of benzyl alcohols both species contribute. Considering these results, the proposed mechanism is similar to that of the sulfide oxidation. Upon irradiation, an electron was photogenerated that activates O2 to form radical and singlet oxygen species, which removes one proton from the alcohol giving a ˙OOH species. This anion is oxidized by the h+ to an anionic radical, and then the ˙OOH oxidizes this radical to the corresponding aldehyde (Fig. S13†).
To further determine the photocatalytic versatility of DPhBTBT-CTF, it was tested in the photocatalytic bromination of aromatic compounds. This reaction is an alternative strategy to obtain a class of important intermediates in synthetic organic chemistry as halogenated hydrocarbons.41,50 We have performed the halogenation of a series of aromatic compounds using HBr (47%) or HCl (37%) as the halide source under an oxygen atmosphere and a blue LED as the irradiation source (Fig. 6). Monobrominated compounds can be selectively obtained by controlling the reaction time. Again, control experiments without a catalyst or light do not lead to appreciable amounts of product. Chlorination with hydrochloric acid was also performed, yielding the corresponding chlorinated compounds although longer reaction times are necessary to achieve quantitative yields (Fig. 6), which demonstrate the versatility of DPhBTBT-CTF as a photocatalyst.
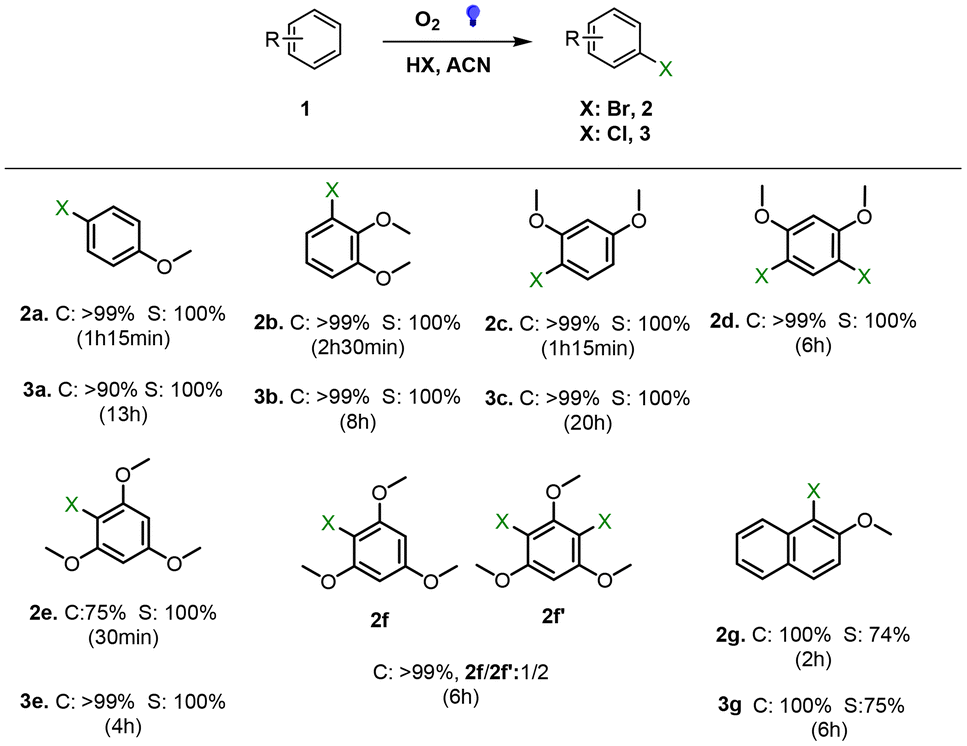 | ||
| Fig. 6 Photocatalyzed halogenation of aromatic compounds: substrate (0.15 mmol), HBr 47% (1.05 mmol) or HCl 37% (1.05 mmol), catalyst (4.4 mg), acetonitrile (0.5 mL), a LED blue lamp, r.t.; conversion was followed by GC and yields are determined by 1H-NMR (selected GC chromatograms and NMR spectra are shown in Fig. S15–S21 and S23–S27†). | ||
The proposed mechanism for this reaction involves a radical pathway on the basis of previously reported data for other CTFs.41,51 Under the light irradiation, the photogenerated electron could activate the oxygen to O2˙− and singlet oxygen 1O2 species, while the h+ oxidized the substrate to its cationic radical, which reacted with Br− to give an adduct intermediate. Then, the brominated product was obtained after the subsequent oxidation of the intermediates by oxygen species (Fig. S15†).
We have also performed two proof of concept reactions that confirm the versatility of this novel platform. Firstly, the Diels–Alder (D–A) photocycloaddition reaction because of the great importance in the organic synthesis of a metal-free heterogeneous photocatalyzed process involves the formation of cycles.52,53 As a model reaction we have chosen the D–A cycloaddition between trans-anethole and 2,3-dimethyl-1,3-butadiene. The D–A reaction was carried out in acetonitrile at room temperature, in air, using blue LED light irradiation and the cycloadduct was selectively obtained after 7 h (the corresponding kinetic profile is shown in Fig. 7b). GCs and 1H-NMR are collected in Fig. S17.† Control experiments without a catalyst, light or under an inert atmosphere indicate that all of them are necessary for both yield and selectivity. The trans-anethole![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2,3-dimethyl butadiene ratio to afford selectively the [4 + 2] adduct was found to be 1:3.
:2,3-dimethyl butadiene ratio to afford selectively the [4 + 2] adduct was found to be 1:3.
 | ||
| Fig. 7 Top: D–A reaction between trans-anethole (0.13 mmol – 1 eq.) and 2,3-dimethyl-1,3-butadiene (0.4 mmol – 3 eq.) in 0.5 mL of ACN with 4.4 mg catalyst loading and kinetic profile. Down: Fluorescence quenching experiments. | ||
Additionally, we have carried out fluorescence quenching experiments in the presence of trans-anethole and 2,3-dimethyl-1,3-butadiene (Fig. 7c and Table S3†). We have found that DPhBTBT-CTF interacts with trans-anethole and not with the diene, with a KSV of 2080.6 M−1versus 17.5 M−1. According to these data, the mechanism for the reaction should be similar to that previously reported.53
The second proof of concept reaction is the synthesis of phenols through the photocatalytic oxidative hydroxylation of arylboronic acids which is a very effective and sustainable method,54 resulting in selective phenol formation after 24 h of irradiation (Fig. 8 and S18†).
 | ||
| Fig. 8 Hydroxylation of phenylboronic acid. Reactions conditions: substrate (0.2 mmol), triethylamine (0.6 mmol), catalyst (4.4 mg) and tetrahydrofuran (0.5 mL), an O2 atmosphere, blue light. | ||
Recycling
The results demonstrate the versatility and broad applicability of DPhBTBT-CTF as an organic catalyst for the photoredox processes. An important characteristic of a heterogeneous photocatalyst is the recyclability. Thus, we have studied its recyclability in the oxidation of mustard gas simulant as the substrate under the optimized conditions. After the substrate was totally converted into nontoxic sulfoxide, we checked the reusability of the catalyst by addition of more sulfide to the reaction mixture. It was irradiated towards total oxidation to sulfoxide (Fig. 9a). The same amount of substrate was added in each cycle and the result shows that the selectivity of DPhBTBT-CTF was maintained for 6 cycles although a gradual loss of activity was observed and longer reaction times are necessary to obtain high conversions. On the other hand, the reusability and stability of the catalyst are further verified by separation of DPhBTBT-CTF from the reaction mixture via a centrifugation method. The recovered solid was washed with acetonitrile to remove the residual organic compounds and dried, and then used in a new reaction. It was observed that the sulfoxide was quantitatively obtained although the time needed for the reaction to be complete increased with each new run (Fig. 9b). The catalyst maintains its structure since the BTBT and triazine moieties were observed by FTIR, EPR and morphology (SEM and TEM images) analyses which are similar to those exhibited by the pristine material (Fig. S19†). | ||
| Fig. 9 Left. Oxidation of mustard gas simulant in the presence of DPhBTBT-CTF under a blue LED with six consecutive injections of substrate. Right: Recycling of DPhBTBT-CTF toward mustard gas simulant. | ||
Finally, a comparison of the photocatalytic performance of DPBTBT-CTF with that of other reported effective photocatalysts (Tables S4 and S5†)55,56 shows that both the catalytic activity and selectivity of DPhBTBT-CTF are superior or similar to those of state-of-the-art catalysts.
Conclusions
We report the design and preparation of a novel conjugated covalent triazine framework (DPhBTBT-CTF) by microwave activated acid-catalyzed cyclotrimerization of 4,4′-(benzo[b]benzo[4,5]thieno[2,3-d]thiophene-2,7-diyl) dibenzonitrile. The combination of the stronger electron-donating ability of the BTBT unit and the acceptor capability of triazine makes the material a very effective and versatile photocatalyst for different reactions such as the selective oxidation of sulfides to sulfoxides or benzylic alcohols to aldehydes, bromination of anisoles, and other reactions as the Diels–Alder cycloaddition or the hydroxylation of phenyl boronic acid under an oxygen atmosphere. In addition, DPhBTBT-CTF is robust and was recycled for at least six times. This work shows that an adequate design of polymers at the molecular level combining donor and acceptor structural building units leads to highly efficient metal-free catalytic systems that can be used in many processes of interest in organic chemistry under sustainable conditions. Moreover, the present work provides the possibility of combining the BTBT units with other acceptor moieties to build new porous organic polymers with great potential as photocatalysts.Experimental section
General
All chemicals and solvents were analytical reagents obtained from commercial sources and used without further purification unless otherwise indicated. The precursor 2,7-dibromobenzo[b]benzo[4,5]thieno[2,3-d]thiophene was synthesized according to a previous report.57 Experimental details, characterization methods and apparatus are given in the ESI.†Synthesis of the diphenylBTBT-covalent triazine framework DPhBTBT-CTF
To a 10 mL MW vial fitted with a magnetic stirrer, dinitrile, DCNPh–BTBT, (300 mg, 0.66 mmol, 1 eq.) and trifluoromethane sulfonic acid (2 mL, 33.9 mmol, 50 eq.) were added. The mixture was stirred at 200 W and 110 °C for 2 h (CEM Discover). After that, the reaction mixture was poured into an ice/water mixture (70 mL) and a water/ammonia solution (20:1) (35 mL) was added until pH 6–7. Then, the product was filtered, washed with water, ethanol and THF and dried at 100 °C under vacuum. The final brown solid was obtained in 72% yield (216 mg). The characterization data are collected throughout the study and given in the ESI.†
Catalytic activity
Details for the photocatalyzed Diels–Alder cycloaddition and the hydroxylation of arylboronic acids will be found in the ESI.†
Author contributions
M. Carmen Borrallo-Aniceto: synthesis, characterization and catalytic experiments; Mercedes Pintado-Sierra: synthesis and characterization; Félix Sánchez: design and review & editing; Antonio Valverde: formal analysis, investigation, and writing the original draft; Urbano Díaz: characterization and writing – review; Eva M. Maya: formal analysis, investigation, funding acquisition and writing – review & editing; Marta Iglesias: conceptualization, funding acquisition and writing – review. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the grants PID2020-112590GB-C22 and PID2020-112590GB-C21 funded by MCIN/AEI/ 10.13039/501100011033. The authors also acknowledge Dr M Luisa Ferrer and Laura González for the electrochemical analysis.References
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wang and J. Tang, Chem. Soc. Rev., 2020, 49, 4135–4165, 10.1039/d0cs00278j.
- C. C. Nguyen, N. N. Vu and T. O. Do, J. Mater. Chem. A, 2015, 3, 18345–18359, 10.1039/c5ta04326c.
- M. Xiao, Z. Wang, M. Lyu, B. Luo, S. Wang, G. Liu, H. M. Cheng and L. Wang, Adv. Mater., 2019, 31, 1–23, DOI:10.1002/adma.201801369.
- M. Debruyne, V. Van Speybroeck, P. Van Der Voort and C. V. Stevens, Green Chem., 2021, 23, 7361–7434, 10.1039/d1gc02319e.
- Z. Zhou, Y. Xiao, J. Tian, N. Nan, R. Song and J. Li, J. Mater. Chem. A, 2023, 11, 3245–3261, 10.1039/d2ta09582c.
- S. Liu, M. Wang, Y. He, Q. Cheng, T. Qian and C. Yan, Coord. Chem. Rev., 2023, 475, 214882, DOI:10.1016/j.ccr.2022.214882.
- G. Kumar, B. Cai, S. Ott and H. Tian, Chem. Phys. Rev., 2023, 4, 011307, DOI:10.1063/5.0123282.
- A. Savateev and M. Antonietti, ACS Catal., 2018, 8, 9790–9808, DOI:10.1021/acscatal.8b02595.
- Z. Zhang, J. Jia, Y. Zhi, S. Ma and X. Liu, Chem. Soc. Rev., 2022, 51, 2444–2490, 10.1039/d1cs00808k.
- B. Wang, R. B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416, DOI:10.1021/jacs.0c06473.
- Y. N. Gong, X. Guan and H. L. Jiang, Coord. Chem. Rev., 2023, 475, 214889, DOI:10.1016/j.ccr.2022.214889.
- L. Liao, M. Li, Y. Yin, J. Chen, Q. Zhong, R. Du, S. Liu, Y. He, W. Fu and F. Zeng, ACS Omega, 2023, 8, 4527–4542, DOI:10.1021/acsomega.2c06961.
- M. Liu, L. Guo, S. Jin and B. Tan, J. Mater. Chem. A, 2019, 7, 5153–5172, 10.1039/c8ta12442f.
- R. Sun and B. Tan, Chem. Res. Chin. Univ., 2022, 38, 310–324, DOI:10.1007/s40242-022-1468-4.
- B. Fuerte-Díez, A. Valverde-González, M. Pintado-Sierra, U. Díaz, F. Sánchez, E. M. Maya and M. Iglesias, Sol. RRL, 2022, 6, 1–10, DOI:10.1002/solr.202100848.
- B. Wu, Y. Liu, Y. Zhang, L. Fan, Q. Y. Li, Z. Yu, X. Zhao, Y. C. Zheng and X. J. Wang, J. Mater. Chem. A, 2022, 10, 12489–12496, 10.1039/d2ta01441f.
- Y. Wang, X. Li, X. Dong, F. Zhang, D. Jiang, Y. Sun, R. Wang and X. Lang, Adv. Energy Sustainability Res., 2023, 4, 2200129, DOI:10.1002/aesr.202200129.
- W. Huang, B. C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 5438–5442, DOI:10.1021/acscatal.7b01719.
- W. Huang, N. Huber, S. Jiang, K. Landfester and K. A. I. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18368–18373, DOI:10.1002/anie.202007358.
- J. Xie, S. A. Shevlin, Q. Ruan, S. J. A. Moniz, Y. Liu, X. Liu, Y. Li, C. C. Lau, Z. X. Guo and J. Tang, Energy Environ. Sci., 2018, 11, 1617–1624, 10.1039/c7ee02981k.
- P. Xie, T. Liu, J. Sun and J. Yang, Adv. Funct. Mater., 2022, 32, 2200843–2200870, DOI:10.1002/adfm.202200843.
- W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489–1502, 10.1039/b813123f.
- A. Sanzone, S. Mattiello, G. M. Garavaglia, A. M. Calascibetta, C. Ceriani, M. Sassi and L. Beverina, Green Chem., 2019, 21, 4400–4405, 10.1039/c9gc01071h.
- C. Niebel, Y. Kim, C. Ruzié, J. Karpinska, B. Chattopadhyay, G. Schweicher, A. Richard, V. Lemaur, Y. Olivier, J. Cornil, A. R. Kennedy, Y. Diao, W. Y. Lee, S. Mannsfeld, Z. Bao and Y. H. Geerts, J. Mater. Chem. C, 2015, 3, 674–685, 10.1039/c4tc02158d.
- K. Takimiya, H. Ebata, K. Sakamoto, T. Izawa, T. Otsubo and Y. Kunugi, J. Am. Chem. Soc., 2006, 128, 12604–12605, DOI:10.1021/ja064052l.
- C. Yao, X. Chen, Y. He, Y. Guo, I. Murtaza and H. Meng, RSC Adv., 2017, 7, 5514–5518, 10.1039/c6ra28074a.
- T. Yamamoto, T. Nishimura, T. Mori, E. Miyazaki, I. Osaka and K. Takimiya, Org. Lett., 2012, 14, 4914–4917, DOI:10.1021/ol302243t.
- S. Zhang, F. Zhao, G. Yasin, Y. Y. Dong, J. Zhao, Y. Guo, P. Tsiakaras and J. Zhao, J. Colloid Interface Sci., 2023, 637, 41–54, DOI:10.1016/j.jcis.2023.01.066.
- F. Yi, Q. Yang, X. Li, Y. Yuan, H. Cao, K. Liu and H. Yan, J. Solid State Chem., 2023, 318, 123769, DOI:10.1016/j.jssc.2022.123769.
- I. Fouad, Z. Mechbal, K. I. Chane-Ching, A. Adenier, F. Maurel, J. J. Aaron, P. Vodicka, K. Cernovska, V. Kozmik and J. Svoboda, J. Mater. Chem., 2004, 14, 1711–1721, 10.1039/b308779b.
- M. Sugiyama, S. Jancke, T. Uemura, M. Kondo, Y. Inoue, N. Namba, T. Araki, T. Fukushima and T. Sekitani, Org. Electron., 2021, 96, 106219, DOI:10.1016/j.orgel.2021.106219.
- C. Lô, A. Adenier, K. I. Chane-Ching, F. Maurel, J. J. Aaron, B. Kosata and J. Svoboda, Synth. Met., 2006, 156, 256–269, DOI:10.1016/j.synthmet.2005.12.003.
- X. Han, Y. Zhang, Y. Dong, J. Zhao, S. Ming and J. Zhang, RSC Adv., 2021, 12, 708–718, 10.1039/d1ra07916f.
- L. Wang, J. Sun, M. Deng, C. Liu, S. Ataberk Cayan, K. Molkens, P. Geiregat, R. Morent, N. De Geyter, J. Chakraborty and P. Van Der Voort, Catal. Sci. Technol., 2023, 13, 6463–6471, 10.1039/d3cy01175e.
- M. Deng, J. Sun, A. Laemont, C. Liu, L. Wang, L. Bourda, J. Chakraborty, K. Van Hecke, R. Morent, N. De Geyter, K. Leus, H. Chen and P. Van Der Voort, Green Chem., 2023, 25, 3069–3076, 10.1039/D2GC04459E.
- H. Zhang, Q. Huang, W. Zhang, C. Pan, J. Wang, C. Ai, J. Tang and G. Yu, ChemPhotoChem, 2019, 3, 645–651, DOI:10.1002/cptc.201900095.
- X. Li, X. Ma, F. Zhang, X. Dong and X. Lang, Appl. Catal., B, 2021, 298, 120514, DOI:10.1016/j.apcatb.2021.120514.
- H. Monobe, L. An, P. Hu, B. Q. Wang, K. Q. Zhao and Y. Shimizu, Mol. Cryst. Liq. Cryst., 2017, 647, 119–126, DOI:10.1080/15421406.2017.1289443.
- X. Zhu, C. Tian, S. M. Mahurin, S. H. Chai, C. Wang, S. Brown, G. M. Veith, H. Luo, H. Liu and S. Dai, J. Am. Chem. Soc., 2012, 134, 10478–10484, DOI:10.1021/ja304879c.
- S. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Adv. Mater., 2012, 24, 2357–2361, DOI:10.1002/adma.201200751.
- Y. Zou, S. Abednatanzi, P. Gohari Derakhshandeh, S. Mazzanti, C. M. Schüßlbauer, D. Cruz, P. Van Der Voort, J. W. Shi, M. Antonietti, D. M. Guldi and A. Savateev, Nat. Commun., 2022, 13, 1–13, DOI:10.1038/s41467-022-29781-9.
- H. Hao, F. Zhang, X. Dong and X. Lang, Appl. Catal., B, 2021, 299, 120691, DOI:10.1016/j.apcatb.2021.120691.
- C. Kang, Z. Zhang, A. K. Usadi, D. C. Calabro, L. S. Baugh, K. Yu, Y. Wang and D. Zhao, J. Am. Chem. Soc., 2022, 144, 3192–3199, DOI:10.1021/jacs.1c12708.
- B. Cai, L. Cao, R. Zhang, N. Xu, J. Tang, K. Wang, Q. Li, B. Xu, Y. Liu and Y. Fan, ACS Appl. Energy Mater., 2023, 6, 930–938, DOI:10.1021/acsaem.2c03322.
- S. Fischer, J. Schmidt, P. Strauch and A. Thomas, Angew. Chem., Int. Ed., 2013, 52, 12174–12178, DOI:10.1002/anie.201303045.
- Y. Hu, W. Huang, H. Wang, Q. He, Y. Zhou, P. Yang, Y. Li and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 14378–14382, DOI:10.1002/anie.202006618.
- J. Wang, Y. He, S. Guo, M. U. Ali, C. Zhao, Y. Zhu, T. Wang, Y. Wang, J. Miao, G. Wei and H. Meng, ACS Appl. Mater. Interfaces, 2021, 13, 12250–12258, DOI:10.1021/acsami.0c21286.
- M. Hao, Y. Xie, X. Liu, Z. Chen, H. Yang, G. I. N. Waterhouse, S. Ma and X. Wang, JACS Au, 2023, 3, 239–251, DOI:10.1021/jacsau.2c00614.
- H. Chen, H. S. Jena, X. Feng, K. Leus and P. Van Der Voort, Angew. Chem., Int. Ed., 2022, 61, e20220493, DOI:10.1002/anie.202204938.
- Y. Markushyna, C. Teutloff, B. Kurpil, D. Cruz, I. Lauermann, Y. Zhao, M. Antonietti and A. Savateev, Appl. Catal., B, 2019, 248, 211–217, DOI:10.1016/j.apcatb.2019.02.016.
- R. Li, J. Byun, W. Huang, C. Ayed, L. Wang and K. A. I. Zhang, ACS Catal., 2018, 8, 4735–4750, DOI:10.1021/acscatal.8b00407.
- N. Huber, R. Li, C. T. J. Ferguson, D. W. Gehrig, C. Ramanan, P. W. M. Blom, K. Landfester and K. A. I. Zhang, Catal. Sci. Technol., 2020, 10, 2092–2099, 10.1039/d0cy00016g.
- Y. Zhao and M. Antonietti, Angew. Chem., Int. Ed., 2017, 56, 9336–9340, DOI:10.1002/anie.201703438.
- G. B. Wang, Y. J. Wang, J. L. Kan, K. H. Xie, H. P. Xu, F. Zhao, M. C. Wang, Y. Geng and Y. Bin Dong, J. Am. Chem. Soc., 2023, 145, 4951–4956, DOI:10.1021/jacs.2c13541.
- H. Zhang, W. Wei and K. A. I. Zhang, Chem. Commun., 2023, 59, 9167–9181, 10.1039/d3cc02081a.
- A. López-Magano, S. Daliran, A. R. Oveisi, R. Mas-Ballesté, A. Dhakshinamoorthy, J. Alemán, H. Garcia and R. Luque, Adv. Mater., 2023, 35, 2209475, DOI:10.1002/adma.202209475.
- M. Saito, I. Osaka, E. Miyazaki, K. Takimiya, H. Kuwabara and M. Ikeda, Tetrahedron Lett., 2011, 52, 285–288, DOI:10.1016/j.tetlet.2010.11.021.
Footnotes |
| † Electronic supplementary information (ESI) available: Full synthesis and characterization details. See DOI: https://doi.org/10.1039/d3gc03529h |
| ‡ Present Address: Sorbonne Université, CNRS, Institut Parisien de Chimie Moléculaire, Equipe Chimie des Polymères, 4 Place Jussieu, 75005 Paris, France. |
| This journal is © The Royal Society of Chemistry 2024 |