
Acid catalyst screening for hydrolysis of post-consumer PET waste and exploration of acidolysis†
Patrícia
Pereira
 a,
Phillip E.
Savage
*a and
Christian W.
Pester
*ab
a,
Phillip E.
Savage
*a and
Christian W.
Pester
*ab
aDepartment of Chemical Engineering, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: psavage@psu.edu; pester@psu.edu
bDepartment of Chemistry and Department of Materials Science and Engineering, The Pennsylvania State University, University Park, Pennsylvania 16802, USA
First published on 8th January 2024
Abstract
Efficient recycling of polyethylene terephthalate (PET) plastics is a global concern due to the growing volume of plastic waste and its environmental impact. We studied PET hydrolysis and acidolysis processes to recover the PET monomer terephthalic acid (TPA) using various acid catalysts (zeolites, inorganic acids, ionic liquids, carboxylic acids, metal salts, and CO2) below the PET melting point and under identical conditions. TPA yield depended largely on the solution pH for some catalysts, especially aliphatic carboxylic acids, nitric acid, and CO2. However, TPA yields from hydrolysis with metal salts, ionic liquids, sulfuric acid, and aromatic carboxylic acids are also influenced by factors such as solubility limits, oxidation, and anion effects (for metal salts). Under mild hydrolysis conditions at 200 °C for 2 hours, carboxylic acids and metal salts achieved TPA yields > 80%, outperforming nitric acid, which required much more corrosive conditions at pH = 0.7. Zeolites have minimal impact on TPA yields in hydrolysis below the PET melting point. CO2 as a catalyst precursor to carbonic acid did not increase TPA yields significantly. We also explored using acetic acid as the sole reaction medium (acidolysis), which exhibited high TPA yields and a similar environmental energy impact to acid-catalyzed hydrolysis. Propanoic acid showed comparable efficiency, offering promising avenues for chemical recycling of PET.
1. Introduction
Municipal solid waste landfills in the United States contain approximately 26 wt% plastics and textiles.1,2 This proportion is anticipated to increase, in part due to the growing volume of discarded polyethylene terephthalate (PET) from items such as bottles and fast fashion clothing.3,4 Mechanical recycling handles only 28% of PET waste due to the complexity and cost involved in producing recycled products of sufficient quality.5,6 Chemical recycling offers an alternative pathway by producing value-added chemicals or recovering monomers from a polymer. Nevertheless, this technique has not gained significant traction at a commercial scale for post-consumer PET recycling due to concerns related to its economic viability, including aspects related to collection, sorting, transportation, and reprocessing.7Hydrolysis can decompose PET into terephthalic acid (TPA) and ethylene glycol, i.e., two monomers for the industrial production of PET.8–10 It is significantly faster when PET is in the molten state.9 Below the melting point of PET (≈250 °C) it often involves the use of acid or base catalysts. Alkaline PET hydrolysis requires a subsequent acidification process, which is an additional step for TPA recovery not necessary for acid hydrolysis.11 Although mineral acid catalysts have been extensively studied, their oxidative effect and tendency to cause carbonization decrease product yields.9,12–15 Hydrolysis can take place within 70–100 °C in the presence of highly concentrated sulfuric or nitric acids but the reaction times can extend to several days, presenting various engineering challenges.13–16 These challenges encompass managing highly corrosive solutions, the necessity to recycle substantial volumes of acid, and the production of salt waste.17–20
Beyond mineral acids, other acid catalysts have been explored but only to a very limited extent. Examples include solid acids (e.g., zeolites), acidic ionic liquids, carboxylic acids, and metal salts.21–24 There is one report on the hydrolysis of PET using zeolites.24 With microwave-heating the hydrogenated alumina silica zeolite HZSM-5 gave higher TPA yields than runs conducted without catalyst.24 Hydrolysis of PET involving Brønsted acidic ionic liquids (BAILs) functionalized with a sulfonic acid group (IL-SO3H), demonstrated higher monomer yields than with sulfuric acid under similar acid concentrations.21,22 There has also been some limited prior exploration of organic acids as catalysts.25 Additionally, TPA was shown to autocatalyze PET hydrolysis.7 It is worth noting that low concentrations of acetic acid did not facilitate this reaction,12 but acetic acid enhanced PET decomposition through acetolysis and aminolysis.26,27 Prior research has also explored the use of metal salts for PET depolymerization, but primarily via glycolysis and aminolysis. There has been much less work on hydrolysis. These materials are believed to ionize and form complexes with the carbonyl group of the ester, thus promoting bond scission.23,27–31 Among these, zinc acetate proved more effective than sodium acetate.12 Alternatives such as NaCl, CaCl2, NaHCO3 or KHCO3 were explored for their more environmentally benign characteristics compared to traditional heavy metal acetates (e.g., zinc, cobalt, copper, cadmium).23
While the acid catalysts mentioned above have demonstrated promise in PET hydrolytic depolymerization, comparing these catalysts across various studies has proven challenging due to the divergence in reaction conditions. These disparities encompass factors such as reaction temperatures, durations, heating methods, and catalyst loadings, all of which contribute to varying reaction pH levels. As a result, this scarcity of comprehensive and comparable data on the performance of each catalyst class impedes direct comparisons and hampers the development of novel catalytic depolymerization processes. This study presents a screening analysis to evaluate PET hydrolysis, primarily below the PET melting point, using different classes of potential acid catalysts under consistent reaction conditions, while also assessing their green chemistry metrics. Further, this investigation identifies the effectiveness of PET depolymerization into TPA through acidolysis employing acetic and propanoic acids without the presence of water.
2. Experimental section
2.1. PET samples, chemicals, and reagents
Green bottles that had contained Perrier® sparkling water (16.9 oz) served as a representative post-consumer PET source. Labels and caps were removed, and entire bottles were cut into small quadrilateral chips with average dimensions of 5.6 ± 2.1 mm × 8.4 ± 2.4 mm. The thickness of the body of the bottle was 0.5 mm, while the bottom was thicker (2 mm).Dimethyl terephthalate (DMT) with 99% purity was from Acros Organics as white crystals measuring about 4.0 mm. The zeolites ZSM-5 (CBV 5524G), Y (CBV 300), and β (CP814E*) were all purchased from Zeolyst International in the ammonium form. Their particle size is 125–500 μm. Zeolites were calcined in air at 550 °C for 4 h to convert the ammonium to the hydrogen form prior to use. The acidic ionic liquids 1-ethyl-3-methylimidazolium hydrogen sulfate (denominated herein as IL, 98%) and 1-propylsulfonic-3-methylimidazolium hydrogen sulfate (denominated herein as IL-SO3H, 99%, powder), were purchased from Alfa Aesar.
Other acid catalysts or catalyst precursors examined were glacial acetic acid (Fisher Scientific), benzoic acid (99%, Thermo Scientific Chemicals, powder), 4-formyl benzoic acid (4-FBA, 97%, Sigma Aldrich), TPA and isophthalic acid (both 99% purity, TCI), glycolic acid (98%, Alfa Aesar), propanoic acid (>99.5%, Sigma Aldrich), stearic acid (Sigma Aldrich), nitric acid (99.9%, Sigma Aldrich), sulfuric acid (75% v/v, Ricca Chemical), zinc sulfate 7-hydrate (Ward's Science, powder), zinc iodide (98%, Thermo Scientific Chemicals, powder), and CO2 (dry ice purchased from the Penn State Creamery). Dimethyl sulfoxide (DMSO) was purchased from Millipore Sigma. Matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-ToF-MS) used 2,5-dihydroxybenzoic acid (98%, Sigma Aldrich). Deionized water was from an in-house water purification system composed of ion exchange, reverse osmosis, high-capacity ion exchange, UV sterilization, and submicron filtration units.
2.2. Characterization of materials
Characterization of the plastic bottle chips is discussed in detail in our previous publication.32 The melting point of the post-consumer PET (Tm,PET) was measured as 250 °C. A Ross Ultra pH/ATC triode electrode was used to measure the pH of the aqueous medium at room temperature after calibration with pH 4 and pH 7 buffer solutions.X-ray diffraction (XRD) provided information about the zeolite crystal structure. Powders were front-loaded into a silicon, zero-background holder. Diffraction data were collected from 5 to 70° 2θ using a Malvern Panalytical Empyrean® instrument with a Cu K-alpha source. Data were collected with a nominal step size of 0.026° 2θ.
Temperature-programmed desorption of NH3 (NH3-TDP, Micromeritics Autochem II 2920 Chemisorption analyzer) was used to determine the total acidity of the zeolites and the relative acid strength. 0.2 g of catalyst was degassed at 300 °C (10 °C min−1) for 2 h in flowing helium and then returned to ambient temperature. The samples were then treated with 50 mL min−1 of 15 v% NH3-He for 1 h at room temperature to saturate the surface with NH3. The desorption profile was measured by a thermal conductivity detector as He flowed over the sample as it was heated at 10 °C min−1 to a final temperature of 500 °C or 700 °C, which was then maintained for 1 h. The strengths of the different acid sites were determined by peak deconvolution and subsequent integration. The temperature regions 70–110 °C, 130–230 °C, and 260–580 °C were taken to correspond to desorption from weak, medium, and strong acid sites, respectively (Fig. S1 and Table S1†).
The hydrothermal stability of 4-formylbenzoic acid was determined by loading a reactor with 4-FBA and 2.9 mL of water and then placing it in the sandbath at 200 °C for 2 h. The solids in the reactor were recovered by filtration and then dried. Dissolving the solids in DMSO allowed quantification by High-Performance Liquid Chromatography (HPLC) following the procedure outlined in prior literature29 but with phosphoric acid instead of sulfuric acid as a component in the mobile phase.
2.3. Experimental procedure for PET hydrolysis
The hydrolysis reaction was performed in stainless-steel Swagelok reactors which comprised a port connector and caps of 1/2 in. nominal size, resulting in 4 mL reactor volume. Experiments with added dry ice used reactors that also include a 15 ± 3 cm length of stainless-steel tubing and a valve for venting gas post-reaction. All hydrolysis experiments used a fixed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 mass ratio of PET (or DMT) to deionized water. Table S2† shows the water and catalyst loadings used in the experiments. An isothermal Techne fluidized sand bath held the sealed reactors for the desired batch holding time at the hydrolysis temperature. For PET depolymerization with acetic acid and propanoic acid, 4 mL reactors were loaded with 0.2 g of PET and the desired quantity of organic acid.
:10 mass ratio of PET (or DMT) to deionized water. Table S2† shows the water and catalyst loadings used in the experiments. An isothermal Techne fluidized sand bath held the sealed reactors for the desired batch holding time at the hydrolysis temperature. For PET depolymerization with acetic acid and propanoic acid, 4 mL reactors were loaded with 0.2 g of PET and the desired quantity of organic acid.
Performing PET hydrolysis at low pH creates safety concerns that must be managed. Low pH at these elevated temperatures can cause corrosion of stainless steel. Each reactor was inspected carefully after use and reactors were discarded if they had experienced noticeable corrosion. This issue is even more significant if continuous operation was targeted as a reactor wall may weaken over time and no longer withstand the high system pressures.
Immediately after removing the reactors from the sand bath, the reaction was quenched by submerging the reactors in room-temperature water. Due to the higher pressure in the reactors with added CO2, they were then placed in a freezer, so the liquid water became ice before opening. This step prevented loss of non-gaseous material that might otherwise exit the reactor with the vented CO2. The method for product extraction is described in detail elsewhere.32 The aqueous phase was separated from the solid phase by filtration. The aqueous-phase samples were then dried in an oven to recover any water-soluble solids.
The water-insoluble, solid phase contained catalyst (if used), unreacted PET, oligomers, TPA, and other byproducts. Dimethyl sulfoxide (DMSO) was added to dissolve these solids and recover TPA. The remaining water- and DMSO-insoluble components, apart from any spent solid catalyst, are referred to as undissolved solids. Eqn (1) gives the yield (Y) of this product fraction, where mi represents the mass of substance i loaded into, or recovered, from the reactor.
 | (1) |
The zeolite catalyst (e.g., HY) was in powder form and easily recovered from the undissolved solids, when desired, by manually removing the larger particles of unreacted PET and oligomers. This powder was used a second time for a recyclability test of HY where PET was reacted, and the products extracted as above.
2.4. Characterization of products
HPLC was used to determine TPA concentration (in DMSO). The TPA yield (YTPA, eqn (2)) is the ratio of the mass of TPA produced (mTPA) to the maximum TPA available stoichiometrically, presuming the post-consumer material is entirely PET. | (2) |
The stoichiometry of the hydrolysis reaction is such that complete hydrolysis of a given mass of pure PET (mPET) would give 86% of that mass in TPA, and the balance would be ethylene glycol (EG). EG would be formed in a 1:1 molar ratio with TPA. When TPA was loaded into the reactor as a potential catalyst, the loaded TPA mass was subtracted from the total mass recovered at the end of the experiment to calculate the mass of TPA produced by hydrolysis. The TPA yield from hydrolysis of DMT was also calculated with eqn (2) with the mass of DMT (mDMT) loaded into the reactor used in place of the mass of PET (mPET).
We define byproducts as the sum of DMSO-soluble solids that are not TPA plus the aqueous-phase products recovered by evaporating the water. The yield of byproducts was obtained using
 | (3) |
Matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-ToF MS, UltrafleXtreme Bruker) showed the molecular weight and identities of the repeat units and end groups for oligomers in the undissolved solids. A Bruker NMR DPX400 chemically characterized samples of about 6 mg of dried solids dissolved in 0.6 mL of deuterated DMSO at 400 MHz with a pulse length (90 °C) of 12.7 μs, 2 s delay, 32 scans, and 4800 Hz spectral width. A Shimadzu LCMS-8030 liquid chromatography–mass spectrometry instrument was used to analyze products in the aqueous phase.32
2.5. Green chemistry metrics
The environmental energy impact, ξ in eqn (4), is a metric that assesses the potential environmental impacts of PET depolymerization under different process conditions.9,33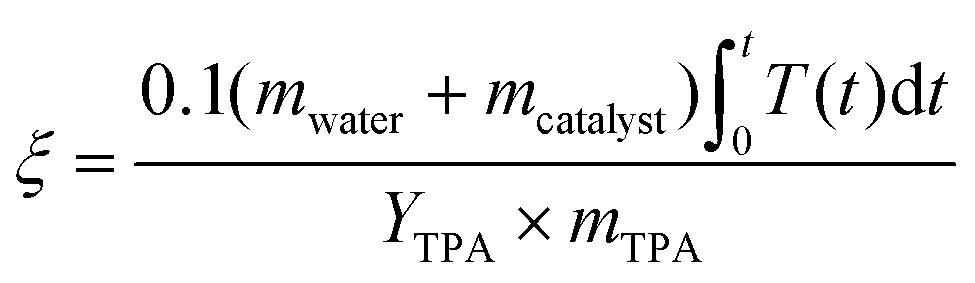 | (4) |
3. Results and discussion
We investigated the TPA yield produced through PET hydrolysis employing diverse catalyst classes under uniform reaction conditions, facilitating direct performance comparisons. We previously showed that the material recovery and analysis protocols outlined above recovered 95.7 ± 0.4 wt% of the TPA in a reactor in a control experiment.323.1. Hydrolysis
 | ||
| Fig. 1 TPA yield from hydrolysis of PET chips and DMT (1/5/50 mass ratio of zeolite/PET or DMT/water). (a) 200 °C, 2 h, (b) 270 °C, 30 min. | ||
The unchanged yields at 200 °C can likely be attributed to PET remaining in a separate solid phase during the reaction, thus limiting its effective interaction with the porous catalysts. To investigate this hypothesis, we conducted hydrolysis experiments using dimethyl terephthalate (DMT), a small-molecule mimic of PET. At 200 °C, the hydrolysis of DMT occurred in a molten state (melting point of DMT is 145 °C). Since DMT is a smaller molecule than PET, it might interact more effectively with the surfaces within the pores of the zeolites, given the size-dependent nature of zeolite catalysis. This enhanced interaction could explain the higher TPA yields compared to PET hydrolysis.
At 270 °C, HY exhibited YTPA of 85% and 96% for hydrolysis of PET and DMT, respectively, marking it as the top-performing zeolite among the tested group. HY also provided the highest yields from hydrolysis conducted at 200 °C. Several factors, including zeolite pore size, surface area, and stability in hot liquid water, likely contributed to these outcomes. The effectiveness of HY might be attributed to it having the largest average pore size (12 Å) and surface area (925 m2 g−1) among the tested zeolites (Table S3†), which can allow for more contact between the catalyst and the reactants. Additionally, HY has a low Si/Al ratio of 5.1 and features a faujasite framework, which is typically more stable than other frameworks in aqueous environments at elevated temperature and pressure.35,36
An examination of TPA yield with HZSM-5 reveals that the concentration of strong acid sites appears to strongly influence PET hydrolysis. At 270 °C, HZSM-5 demonstrated a TPA yield from PET and DMT hydrolysis that was comparable to that achieved with Hβ, despite its smaller pores, lower surface area, and higher Si/Al ratio. An explanation for the efficacy of HZSM-5 lies in its higher proportion of acidic sites (see Table S1 and Fig. S1†). Kang et al.24 proposed that PET hydrolysis primarily occurred on the external acid sites of ZSM-5 since PET molecules were too bulky to penetrate its pores. The TPA yield from DMT with HZSM-5 surpassed that from PET, which aligns with the notion that DMT, being a smaller molecule, can more readily access the internal surface area and highly acidic active sites of HZSM-5.
Since HY gave the highest TPA yields for both PET and DMT, HY was characterized by X-Ray Diffraction before and after hydrolysis to determine the effect of the hydrothermal conditions on the zeolite structure. Fig. S2† shows that HY had a change in the zeolite structure after being used for PET hydrolysis at 270 °C for 30 min. Some characteristic peaks decreased, and others appeared that cannot be assigned to the virgin HY structure. This change is increasingly pronounced in HY used twice (with no post-run catalyst treatment or regeneration in between). Changes in the zeolite structure can impact its ability to catalyze the reaction, potentially affecting the yield of TPA. Indeed, TPA yield after a second use of HY (YTPA = 17%), was much lower than the 85% yield obtained with the fresh catalyst (Fig. S3†). Beyond possible degradation, this decreased performance could also result from pore blockage, which could, however, possibly be ameliorated by calcining the used catalyst prior to re-use. This approach was shown by Mo et al.,36 who regenerated HZSM-5 after use in hydrothermal media with fatty acids. Zeolites showed little impact, however, on TPA yield from PET hydrolysis at 200 °C, which is below the PET melting point (250 °C). Therefore, we did not perform additional experiments to analyze changes in zeolite structure and lack of recyclability at the higher temperatures studied (270 °C).
 | ||
| Fig. 2 Influence of pH (measured at room temperature) on the TPA yield from PET hydrolysis with sulfuric acid, nitric acid, and ionic liquids (200 °C, 2 h, and 1/10 mass ratio PET/water). | ||
The TPA yield from hydrolysis with sulfuric acid increases as pH is reduced but it reached a plateau at approximately 40% around pH = 1.5. The yields of undissolved solids and byproducts (Fig. S4a†) exhibit a similar plateau-like trend. These results align with those observed by Tabekh et al.,37 who also reported a maximum yield in their experiments with H2SO4. In contrast, hydrolysis with HNO3, showed a different pattern, as YTPA continued to increase with decreasing pH, and the yield of undissolved solids continued to decrease (Fig. 2 and Fig. S4b†). On average, the use of HNO3 resulted in less formation of byproducts (Ybyproducts = 20%) compared to H2SO4 (32%), Fig. S4.† Such byproducts could be resultant from oxidation reactions, which can lead to coloration of the final product.37 Both sulfuric acid and nitric acid are potent oxidizing agents, which could explain the observed coloration during hydrolysis in H2SO4 (pH ≤ 1.6) and in HNO3 (pH < 1.4; Fig. S5†). HPLC analysis (Fig. S6†) indicated the presence of several peaks that could be potential color bodies. Additionally, carboxylic acids (like TPA) can be fully oxidized to produce CO2 and water, which would result in a higher gas percentage. This was evidenced experimentally by the need to carefully open the reactors from H2SO4 and HNO3 catalyzed reactions to avoid losing liquid with the vented gas due to pressurization from increased gas formation.
Against our expectations, the molecular weight of undissolved solids across the different pH values was statistically similar and independent of the catalyst used, resulting in a degree of polymerization of 7 to 9 PET repeating units (observed by matrix-assisted laser desorption/ionization-time of flight mass spectrometry, MALDI-ToF MS, Table S4†).
For pH > 1.6, the YTPA from PET hydrolysis with IL or with IL-SO3H was not statistically different. However, the yield of undissolved solids (Fig. S7†) remained ≈ 50% for IL-SO3H and decreased to 46% (from 90%) for IL between 0.7 < pH < 2.9. Hydrolysis with IL-SO3H resulted in a higher yield of byproducts (compared to IL) at pH > 1.55. This suggests that IL-SO3H tends to favor PET depolymerization into byproducts over TPA production. At 1.6 < pH < 1.8, the YTPA obtained with both ionic liquids was 10% higher than with sulfuric acid. This observation is consistent with the finding reported by Liu et al.22 where IL-SO3H yielded higher TPA yields than sulfuric acid for PET hydrolysis, likely due to its dual role as a solvent and catalyst. Experiments confirmed that PET did not dissolve or leach (no measurable mass loss) into the different acidic solutions at room temperature, even after two weeks. Such tests could not be performed at the reaction conditions due to the inability to separate PET mass loss by dissolution from PET mass loss by hydrolysis. Additionally, the reaction products from IL and IL-SO3H at pH < 1.6 were dark, and the reactors from these runs had to be carefully opened due to pressurization during the reaction, again indicating the production of gaseous byproducts. These phenomena were not observed with any of the other catalyst classes used in this study. They are consistent with IL and IL-SO3H inducing oxidation reactions, as we hypothesize was the case for H2SO4 and HNO3.
 | ||
| Fig. 3 Influence of carboxylic acid catalyst loading on the TPA yield from PET hydrolysis (200 °C, 2 h, 1/10 mass ratio PET/water). The dashed line represents the TPA yield average without catalyst, and the shaded area the standard deviation. | ||
Notably, TPA yield increased with the concentration of aromatic acids even after surpassing their solubility limits. For example, the solubility (mole fraction) of TPA in water at 200 °C is less than 0.002.38 This maximum solubility is exceeded at a TPA loading of 0.0012 molTPA gPET−1 because 2.48 mL of water would be required to dissolve the 0.041 g TPA added, but only 2.07 mL were actually added. Likewise, the solubility of 4-FBA at 200 °C (2.471 g per 100 g water)39 is exceeded at a catalyst loading of 0.003 mol gPET−1 where only 0.05 g of 4-FBA would be dissolved in 2.07 mL of water, but 0.1 g were introduced to the system. Additionally, the melting points of these aromatic carboxylic acids are all above the reaction temperature of 200 °C, implying that the aqueous reaction medium likely consists of solid PET and both dissolved and undissolved aromatic acids.
The TPA yield increasing beyond the solubility of the aromatic acids cannot be attributed to solid–solid interactions alone, as control experiments showed that PET did not react at 200 °C and 2 h solely in the presence of these aromatic acids. Furthermore, this behavior cannot be explained by side reactions occurring after catalyst decomposition, given their stability in hot, compressed water. Benzoic acid and TPA remained stable in water at temperatures as high as 350 °C and 300 °C.40 HPLC analysis showed 4-FBA decomposed slightly in water at 200 °C for 2 h. There were product peaks at 2 min 24 seconds and 10 min 18 seconds, representing approximately 12% and 0.2% of the 4-FBA peak area (at 3 min 48 seconds), respectively (Fig. S8†).
To achieve about 60% YTPA at the conditions studied, an aromatic carboxylic acid loading of about 8 × 10−3 molcat gPET−1 is required. Table S2† shows the organic acid loadings could exceed 1 g, which means the volume in the reactor headspace is lower and the pressure would be higher than for reactions with the inorganic acids. Though pressure has minimal influence on PET uncatalyzed hydrolysis,32 we examined whether pressure might play a role for the catalyzed hydrolysis. A set of experiments was done by loading more water in the reactor to achieve a higher pressure at reaction conditions of around 35 MPa, instead of 1.6 MPa. These runs were done at a molcatalyst gPET−1 ratio that gave TPA yields of less than 20% in Fig. 3. Table S5† shows the TPA yields from the high-pressure experiments were not statistically different from the lower-pressure runs for the tested catalysts (4-FBA, TPA, propanoic acid, acetic acid, and benzoic acid).
Fig. 3 shows YTPA increased from 17% to 64% when the TPA loading increased from 0.001 to 0.005 molTPA gPET−1. When the reaction time at the higher loading was extended from 2 h to 3 h, the TPA yield reached 98%, much higher than the yield of 25% from non-catalytic hydrolysis (Fig. S9†). This yield is comparable to that reported in a previous study with added TPA (YTPA (220 °C, 3 h, 0.005 molTPA gPET−1) = 95.5%),7 and it shows the potential for TPA-catalyzed hydrolysis of PET.
Below a 0.003 molcat gPET−1 loading, the aromatic carboxylic acids also led to lower yields of undissolved solids and higher yields of byproducts than did the aliphatic carboxylic acids (Fig. S10†). This indicates that aromatic carboxylic acids favor a PET depolymerization with less side products. Isophthalic acid and bis(2-hydroxyethyl) terephthalate were identified as byproducts (Fig. S11 and S12†). In contrast to the other carboxylic acids, adding more stearic acid led to increasing yields of byproducts (Fig. S13†) but low YTPA (5 ± 3% on average). This observation suggests that stearic acid promotes PET decomposition but not TPA production.
The present findings suggest that organic acids show potential to catalyze PET depolymerization below its melting point, though high loadings are needed to achieve high TPA yields. The color of the products obtained with carboxylic acids was consistently white (based solely on visual observation) and there was no pressurization effect. As such, discoloration, and oxidation side products (as present with e.g., H2SO4) are mitigated. The TPA product is not pure, however, as there are other peaks in the HPLC chromatograms (exemplified in Fig. S11† for PET hydrolysis in the presence of 4-FBA). TPA purity can be assessed through acid–base titrations.26 In industrial processes, TPA purification involves hydrogenation of crude TPA product, re-crystallization, filtration, and drying.41 Why continually increasing TPA yields are produced with increasing catalyst loading, even beyond the solubility limit in the reaction medium, remains subject to further studies.
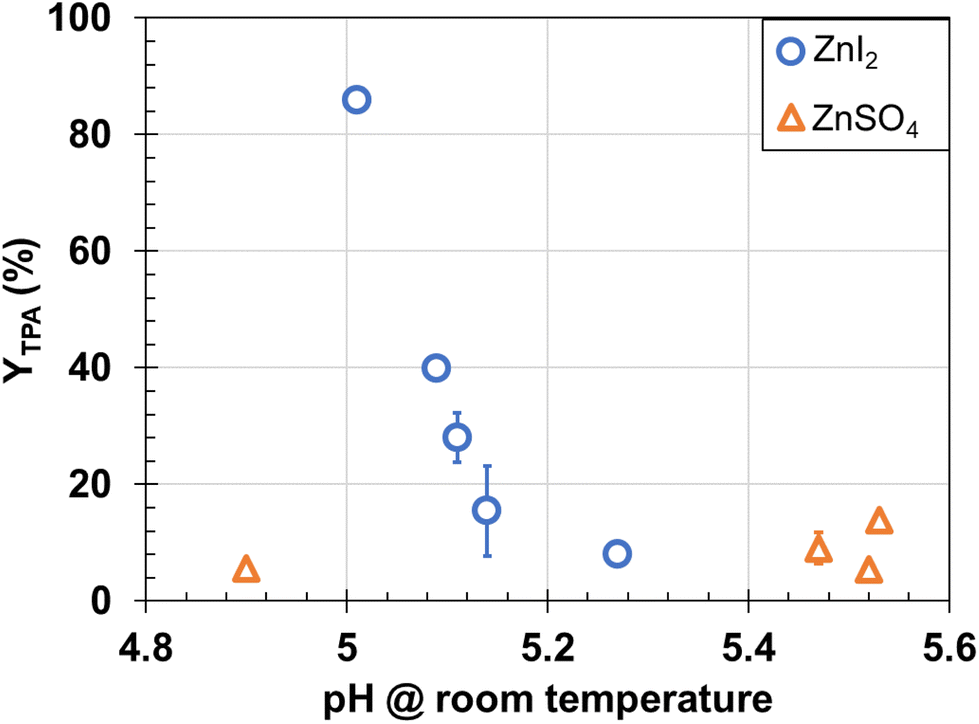 | ||
| Fig. 4 Effect of ZnI2 and ZnSO4 on TPA yield from PET hydrolysis (200 °C, 2 h, 1/10 mass ratio PET/water). | ||
The Lewis acid catalyst coordinates with the oxygen atoms in the ester groups of PET. This coordination activates the ester linkage and allows water molecules to break the activated PET ester groups into the monomers TPA and ethylene glycol (EG).47 Both the metal cation and anion can influence this reaction. Campanelli et al.12 observed an increase in the reaction rate in the presence of zinc salts for hydrolysis above the PET melting point. Stanica-Ezeanu and Matei23 observed a higher PET hydrolysis rate in marine water (that contained Na+, Mg2+, Ca2+, and K+) with higher salinity. Both previous studies hypothesized that the enhanced PET depolymerization with cations could be related to electronic destabilization of the polymer–water interface resulting in a greater interfacial area available for the hydrolysis reaction. They did not evaluate the effect of the anions. Although the metal cation serves as the Lewis acid, the anion (which does not seem to directly participate in the catalytic reaction itself) greatly affected the TPA yield. For these Lewis acids, pH is also not the sole contributing factor and anions might also provide favorable electronic destabilization to facilitate PET depolymerization.
We considered the Hofmeister series to provide insights into the effect of the metal salts.49 This series ranks ions based on their ability to influence the properties of water and its interactions with solutes. Ions are categorized as chaotropic (structure-breakers) or kosmotropic (structure-makers), based on their effects on solubility, protein stability, and other properties of aqueous solution.49 Chaotropic anions weaken the hydrogen bonding, potentially leading to changes in the solvation of reactants and products, impacting the effective concentration of reactants at the active sites of the catalyst and thereby contribute to solvation and destabilization of hydrophobic particles.50 Kosmotropic anions have a stronger hydrogen bonding with water molecules and may promote the formation of stable solvent structures.51,52 The mechanisms behind the Hofmeister series remain poorly understood, and theories such as the site binding model and the cavity model have been proposed to explain them.51,52
The anion that resulted in higher YTPA (I−) is chaotropic whereas SO42− is kosmotropic.49 We hypothesize that increasing the solvation of PET and oligomers though addition of a chaotropic salt, increases the likelihood of water molecules effectively attacking and breaking PET ester bonds. Nevertheless, to draw definitive conclusions regarding the impact of various cations and anions on PET hydrolysis, additional research involving different combinations of cations and anions is necessary.
3.2 Comparison between acid catalysts
Fig. 5 suggests that TPA yields from PET hydrolysis with glycolic, propanoic, acetic acid and nitric acid appear related to the pH of the solution. These results suggest that pH is the dominant factor for these catalysts. At pH = 3.3 YTPA is negligible, which correlates well with reactions with added CO2. For the other water-soluble Brönsted acid catalysts used in this study (ionic liquids and sulfuric acid) the TPA yields do not follow this trend, suggesting pH is not the sole contributing factor. The deviation seems to be connected to side reactions from oxidation. Additionally, the solubility of PET in the reaction medium affects PET hydrolysis rates,53 so if some of the acids facilitated PET dissolution, this differential dissolution could be a confounding factor. However, their direct comparison with water-soluble acid catalysts is difficult to make due to inability to measure solution pH at the reaction conditions and PET dissolution. | ||
| Fig. 5 Effect of pH on TPA yield from PET hydrolysis with different aliphatic carboxylic acids and nitric acid (200 °C, 2 h, 1/10 mass ratio PET/water). | ||
Another way to compare the catalysts is to examine the TPA yields achieved at comparable mass loadings. Fig. S15† displays the TPA yields from the catalysts (zeolites excepted) as a function of the catalyst mass loading. At loadings below 0.3 gcat gPET−1 nitric acid is the most effective catalyst. At higher loadings, ZnI2 and carboxylic acids also give TPA yields exceeding 80%.
3.3 Acidolysis
Considering the high YTPA from PET hydrolysis with acetic acid as a catalyst, we investigated PET depolymerization in glacial acetic acid (no water) at temperatures below the melting point of PET. This exploratory work on using acetic acid as a solvolytic reagent is a natural extension of the previous section. Acetic acid is inexpensive and can be produced from bio-renewable sources. PET acetolysis yields TPA and ethylene glycol diacetate as the primary products from depolymerization. We are aware of only one very recent prior study of PET acetolysis,26 which was published as this manuscript was being prepared. In addition, we conducted experiments using propanoic acid in the absence of water. It also catalyzed PET hydrolysis and we desired to determine whether it would also enable solvolytic depolymerization of PET.Fig. 6 shows the yield of TPA from PET depolymerization by acetic acid or propanoic acid. YTPA was above 80% at low PET/acetic acid ratios, but it decreased to almost zero as the ratio increased. All the experiments were done with excess acetic acid, as the stoichiometric ratio is 1.6 gPET gAcetic acid−1. PET acetolysis resulted in other byproducts (Fig. S16†) that contain aromatic structures (Fig. S17†) and the product increased coloration in correlation with the escalating mass ratio of acetic acid to PET (Fig. S18†). Peng et al.26 also reported high TPA yield (95.8%) and 100% PET conversion from the acetolysis of PET, but above its melting point (280 °C, 2 h, and 0.19 gPET gAcetic acid−1). Similar to acetic acid, the TPA yield with propanoic acid decreased from a high of 71 ± 13% at 0.1 gPET gacid−1 to 34 ± 4% at 0.48 gPET gacid−1.
 | ||
| Fig. 6 Effect of PET/HOAc or propanoic acid ratio on TPA yield from PET acidolysis (200 °C, 2 h, 0.2 gPET). | ||
3.4. Green chemistry metrics
Table 1 displays the ξ values for the catalysts that showed the higher YTPA values in this study. Generally, the addition of catalysts to the reaction medium decreased the environmental energy impact of PET hydrolysis at 200 °C by two orders of magnitude relative to uncatalyzed hydrolysis. None of the catalysts tested in the present study are greatly superior to any others based on this metric. Using the zeolite HY for PET hydrolysis at 270 °C (T > Tm,PET) led to the lowest environmental energy impact (ξ = 1.3 × 104 °C min) for the catalysts studied herein.| Ref. | Reaction | Catalyst | Temp (°C) | Time (min) | gPET gsolvent−1 | ξ 104 (°C min) |
|---|---|---|---|---|---|---|
| a Additional solvent [Bmim]Cl/water. | ||||||
| Below PET melting temperature | ||||||
| This study | Hydrolysis | None | 200 | 120 | 0.1 | 587 |
| Hydrolysis | Nitric Acid, pH = 1.4 | 200 | 120 | 0.1 | 8.5 | |
| Hydrolysis | TPA, 0.005 molTPA gPET−1 | 200 | 180 | 0.1 | 4.5 | |
| Hydrolysis | 4-FBA, 0.01 mol4-FBA gPET−1 | 200 | 120 | 0.1 | 10.2 | |
| Hydrolysis | Benzoic Acid, 0.07 molBA gPET−1 | 200 | 120 | 0.1 | 7.0 | |
| Hydrolysis | Acetic acid, 0.17 molAA gPET−1 | 200 | 120 | 0.1 | 4.4 | |
| Hydrolysis | ZnI2, pH = 5.0 | 200 | 120 | 0.1 | 4.0 | |
| Acetolysis | None | 200 | 120 | 0.2 | 3.1 | |
| Yang et al.25 | Hydrolysis | PTSA, 16 gcatalyst gPET−1 | 150 | 90 | 0.05 | 6.1 |
| Liu et al.22 | Hydrolysis | [HSO3-pmin][HSO4] a 1/5 gcatalyst gPET−1 | 170 | 270 | 0.75 | 2.4 |
| W. Yang et al.7 | Hydrolysis | TPA, 0.005 molTPA gPET−1 | 220 | 180 | 0.125 | 5.6 |
| Above PET melting temperature | ||||||
| This study | Hydrolysis | None | 270 | 30 | 0.1 | 5.7 |
| Hydrolysis | HY | 270 | 30 | 0.1 | 1.3 | |
| Peng et al.26 | Acetolysis | None | 280 | 120 | 0.2 | 2.4 |
The use of acetic acid as a solvent (with no catalyst) led to a value of ξ = 3.1 × 104 °C min from acetolysis at 200 °C. This ξ value is slightly higher than that from recently published results from optimized acetolysis of PET at a higher temperature (280 °C, i.e., above Tm,PET). These results indicate that acetic acid could be a viable alternative to water for PET depolymerization as it provided lower ξ than did acid-catalyzed hydrolysis. Further analysis is necessary to evaluate the technoeconomic feasibility and the TPA product quality, as the final product was brown, which suggests the need for additional product purification. According to Peng et al.26 using activated carbon to remove color bodies for the product can achieve an average of 99.7% TPA purity.
4. Conclusions
At a given set of reaction conditions, the choice of acid catalyst can significantly affect the yields of TPA and byproducts. The pH of the reaction mixture plays a crucial role in TPA production from PET hydrolysis. TPA yields from hydrolysis with nitric acid, several aliphatic acids, and CO2 shared a common correlation with pH, but yields with the other acid catalysts (e.g., ionic liquids and sulfuric acid) did not follow this correlation most likely due to oxidation reactions, as evidenced by the production of gaseous byproducts and discoloration of reaction products.Organic acids and zinc iodide show promise as catalysts for PET hydrolysis. The aromatic carboxylic acids examined gave higher yields of TPA and lower yields of PET oligomers than did aliphatic carboxylic acids at similar catalyst loadings. The mechanism for the increased TPA yields with increasing catalyst loading, even when the aromatic carboxylic acid is not soluble in the reaction medium, remains unclear. It does not seem to be dependent on the pressure or side reactions from decomposition of the acid catalyst, and there is no reaction between solid PET and solid carboxylic acid. More research is needed to elucidate the mechanism by which solid carboxylic acid catalysts are effective.
TPA is especially interesting as a potential catalyst. Its addition resulted in a 98% yield of TPA from PET hydrolysis at 200 °C and it is the main depolymerization product. One could envision a process wherein the reactor effluent, which would contain TPA, is recycled to provide the catalyst needed for the PET hydrolysis reaction. TPA possesses a distinct advantage over other carboxylic acids due to its stability at the reaction conditions and inherent ability to avoid complex product/catalyst separation processes.
For a given cation (Zn2+), iodide led to higher yields of TPA from PET hydrolysis than did SO42−. We hypothesize that iodide, being chaotropic increases the solvation of PET and oligomers leading to the likelihood of water molecules effectively attacking and breaking PET ester bonds. However, additional work with other metal salts is needed to more fully assess and understand the role of these additives in hydrolytic depolymerization of PET. Acid catalysts provided environmental energy impact metrics that were lower than those for uncatalyzed hydrolysis at the same conditions and were similar with values for that metric calculated from literature results.
The present preliminary examination of acidolysis of PET showed that TPA yields of over 80% can be achieved at 200 °C from solid PET. Acetolysis provided an environmental energy factor similar to acid-catalyzed hydrolysis. Acetic acid is abundant, inexpensive, and can come from bio-renewable sources. Additional research into acidolysis over a broader range of reaction conditions is needed to assess this approach further. It may provide a viable option for chemical recycling of PET. Acidolysis with propanoic acid yielded similar TPA yields as acetic acid, suggesting that catalysts responding similarly to the pH effect in PET hydrolysis exhibit similar behavior during acidolysis at the same pH.
Zeolites are active catalysts for ester hydrolysis at 200 °C, as evidenced by the yield of TPA from DMT increasing from less than 20% after 2 h with no catalyst to greater than 60% with the zeolites examined herein. These solid acid catalysts showed little impact, however, on TPA yield from PET hydrolysis at 200 °C, which is below the PET melting point. At 270 °C, where PET was in a molten state, the different zeolites provided higher TPA yields, with zeolite HY giving the highest (85%). CO2 increased PET depolymerization but did not affect the TPA yield due to pH limitations.
We posit that information about depolymerization alone is not sufficient to identify an optimal catalyst. One would also need to consider product purity, byproduct formation, and the downstream separation processes that would be needed to produce purified terephthalic acid.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant EFRI E3P number 2029397. We thank Dr Yanyu Mu and Dr Robert Rioux for helpful discussions on HPLC use and Michael Lemelin and Dr Ezra Clark for the use of and discussion about LC-MS. The authors acknowledge the Penn State Materials Characterization Lab, especially Dr Ekaterina Bazilevskaya, Dr Bob Hengstebeck, and Gino Tamborine, for using thermogravimetric differential scanning calorimetry, X-ray diffraction, and ammonia temperature-programed desorption. The authors are also thankful to the Proteomics and Mass Spectrometry Core Facility at the Huck Institute of Life Sciences, especially to Dr Tatiana Laremore for assisting in preparing samples and interpreting MALDI-TOF MS data. Dr Gina Noh provided helpful insights into the characterization of zeolites.References
- EPA, Advancing Sustainable Materials Management: 2018 Fact Sheet Assessing Trends in Materials Generation and Management in the United States, 2020 Search PubMed.
- EPA, U.S. State and Local Waste and Materials Characterization Reports, 2023 Search PubMed.
- J. M. Hawley, in Handbook of Recycling: State-of-the-art for Practitioners, Analysts, and Scientists, ed. E. Worrell and M. A. Reuter, Elsevier, 2014, pp. 211–217 Search PubMed.
- FAQs on Plastics - Our World in Data, https://ourworldindata.org/faq-on-plastics, (accessed 28 January 2021).
- F. Welle, Resour., Conserv. Recycl., 2011, 55, 865–875 CrossRef.
- S. Kumartasli and O. Avinc, in Sustainable Textiles: Production, Processing, Manufacturing & Chemistry, ed. S. S. Muthu, Springer, Cham, 2020, pp. 1–19 Search PubMed.
- W. Yang, R. Liu, C. Li, Y. Song and C. Hu, Waste Manage., 2021, 135, 267–274 CrossRef CAS PubMed.
- R. A. F. Tomás, J. C. M. Bordado and J. F. P. Gomes, Chem. Rev., 2013, 113, 7421–7469 CrossRef PubMed.
- E. Barnard, J. Jonathan, R. Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 RSC.
- K. Lee, Y. Jing, Y. Wang and N. Yan, Nat. Rev. Chem., 2022, 6, 635–652 CrossRef PubMed.
- H. Abedsoltan, Polym. Eng. Sci., 2023, 63, 2651–2674 CrossRef CAS.
- J. R. Campanelli, D. C. Cooper and M. R. Kamal, J. Appl. Polym. Sci., 1994, 53, 985–991 CrossRef CAS.
- T. Yoshiaka, N. Okayama and A. Okuwaki, Ind. Eng. Chem. Res., 1998, 37, 336–340 CrossRef.
- S. Donnini Mancini and M. Zanin, Polym.-Plast. Technol. Eng., 2007, 46, 135–144 CrossRef.
- S. Mishra, A. S. Goje and V. S. Zope, Polym. React. Eng., 2003, 11, 79–99 CrossRef CAS.
- P. Benyathiar, P. Kumar, G. Carpenter, J. Brace and D. K. Mishra, Polymers, 2022, 14, 2366 CrossRef CAS PubMed.
- H. Li, H. A. Aguirre-Villegas, R. D. Allen, X. Bai, C. H. Benson, G. T. Beckham, S. L. Bradshaw, J. L. Brown, R. C. Brown, V. S. Cecon, J. B. Curley, G. W. Curtzwiler, S. Dong, S. Gaddameedi, J. E. Estela-García, I. Hermans, M. S. Kim, J. Ma, L. O. Mark, M. Mavrikakis, O. O. Olafasakin, T. A. Osswald, K. G. Papanikolaou, H. Radhakrishnan, M. A. S. Castillo, K. L. Sánchez-Rivera, K. N. Tumu, R. C. Van Lehn, K. L. Vorst, M. M. Wright, J. Wu, V. M. Zavala, P. Zhou and G. W. Huber, Green Chem., 2022, 24, 8899–9002 RSC.
- Y.-M. Zhang, Y.-Q. Sun, Z.-J. Wang and J. Zhang, S. Afr. J. Sci., 2013, 109, 1–4 Search PubMed.
- G. P. Karayannidis, A. P. Chatziavgoustis and D. S. Achilias, Adv. Polym. Technol., 2002, 21, 250–259 CrossRef CAS.
- A. M. Al-Sabagh, F. Z. Yehia, G. Eshaq, A. M. Rabie and A. E. ElMetwally, Egypt. J. Pet., 2016, 25, 53–64 CrossRef.
- A. S. Amarasekara, J. A. Gonzalez and V. C. Nwankwo, J Ionic Liq., 2022, 2, 100021 CrossRef.
- F. Liu, X. Cui, S. Yu, Z. Li and X. Ge, J. Appl. Polym. Sci., 2009, 114, 3561–3565 CrossRef CAS.
- D. Stanica-Ezeanu and D. Matei, Sci. Rep., 2021, 11, 4431 CrossRef CAS PubMed.
- M. J. Kang, H. J. Yu, J. Jegal, H. S. Kim and H. G. Cha, Chem. Eng. J., 2020, 398, 125655 CrossRef CAS.
- W. Yang, J. Wang, L. Jiao, Y. Song, C. Li and C. Hu, Green Chem., 2022, 24, 1362–1372 RSC.
- Y. Peng, J. Yang, C. Deng, Y. Fu and J. Deng, Nat. Commun., 2023, 14, 3249 CrossRef CAS PubMed.
- S. R. Shukla and A. M. Harad, Polym. Degrad. Stab., 2006, 91, 1850–1854 CrossRef CAS.
- R. López-Fonseca, I. Duque-Ingunza, B. De Rivas, S. Arnaiz and J. I. Gutiérrez-Ortiz, Polym. Degrad. Stab., 2010, 95, 1022–1028 CrossRef.
- S. R. Shukla and A. M. Harad, J. Appl. Polym. Sci., 2005, 97, 513–517 CrossRef CAS.
- N. D. Pingale, V. S. Palekar and S. R. Shukla, J. Appl. Polym. Sci., 2010, 115, 249–254 CrossRef CAS.
- N. D. Pingale and S. R. Shukla, Eur. Polym. J., 2009, 45, 2695–2700 CrossRef CAS.
- P. Pereira, C. W. Pester and P. E. Savage, ACS Sustainable Chem. Eng., 2023, 11, 7203–7209 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- J. J. Rubio Arias and W. Thielemans, Green Chem., 2021, 23, 9945–9956 RSC.
- L. Zhang, K. Chen, B. Chen, J. L. White and D. E. Resasco, J. Am. Chem. Soc., 2015, 137, 11810–11819 CrossRef CAS PubMed.
- N. Mo and P. E. Savage, ACS Sustainable Chem. Eng., 2014, 2, 88–94 CrossRef CAS.
- H. Tabekh, Y. Koudsi and Z. Ajji, Rev. Roum. Chim., 2012, 57, 1029–1034 Search PubMed.
- Y. Takebayashi, K. Sue, S. Yoda, Y. Hakuta and T. Furuya, J. Chem. Eng. Data, 2012, 57, 1810–1816 CrossRef CAS.
- W. Sun, W. Qu and L. Zhao, J. Chem. Eng. Data, 2010, 55, 4476–4478 CrossRef CAS.
- J. B. Dunn, M. L. Burns, S. E. Hunter and P. E. Savage, J. Supercrit. Fluids, 2003, 27, 263–274 CrossRef CAS.
- H. L. Lee, C. W. Chiu and T. Lee, Chem. Eng. J. Adv., 2021, 5, 100079 CrossRef CAS.
- S. E. Hunter and P. E. Savage, Chem. Eng. Sci., 2004, 59, 4903–4909 CrossRef CAS.
- S. E. Hunter and P. E. Savage, Ind. Eng. Chem. Res., 2003, 42, 290–294 CrossRef CAS.
- C. M. Comisar, S. E. Hunter, A. Walton and P. E. Savage, Ind. Eng. Chem. Res., 2008, 47, 577–584 CrossRef CAS.
- PubChem, Zinc Sulfate, https://pubchem.ncbi.nlm.nih.gov/compound/ZINC-sulfate.
- Zinc iodide, 98+%, pure, Thermo Scientific Chemicals, https://www.fishersci.com/shop/products/zinc-iodide-98-pure-thermo-scientific/AC208060500.
- B. Liu, W. Fu, X. Lu, Q. Zhou and S. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 3292–3300 CrossRef CAS.
- J. March, Advanced Organic Chemistry Reactions, Mechanisms, and Structure, J. Wiley & Sons, New York, 4th edn, 1992 Search PubMed.
- K. P. Gregory, G. R. Elliott, H. Robertson, A. Kumar, E. J. Wanless, G. B. Webber, V. S. J. Craig, G. G. Andersson and A. J. Page, Phys. Chem. Chem. Phys., 2022, 24, 12682–12718 RSC.
- S. Moelbert, B. Normand and P. D. L. Rio, Biophys. Chem., 2004, 112, 45–57 CrossRef CAS PubMed.
- V. Mazzini and V. S. J. Craig, Chem. Sci., 2017, 8, 7052–7065 RSC.
- R. L. Baldwin, Biophys. J., 1996, 71, 2056–2063 CrossRef CAS PubMed.
- F. Liu, X. Cui, S. Yu, Z. Li and X. Ge, J. Appl. Polym. Sci., 2009, 114, 3561–3565 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NH3 TPD data; catalyst and water loadings; zeolite properties; XRD of HY zeolite; yields of undissolved solids and byproducts from PET hydrolysis with inorganic acids, ionic liquids, and carboxylic acids; color of dissolved solids from hydrolysis with nitric acid and sulfuric acid; MALDI-TOF MS analysis of oligomers for different PET hydrolysis experiments; effect of pressure on TPA yields for carboxylic acid catalyzed PET hydrolysis; HPLC effect of adding TPA on PET hydrolysis; chromatograms of products from PET hydrolysis and acetolysis; effect of CO2 on PET hydrolysis; 1H NMR spectra of DMSO-soluble compounds for PET acetolysis experiments. See DOI: https://doi.org/10.1039/d3gc03906d |
| This journal is © The Royal Society of Chemistry 2024 |