
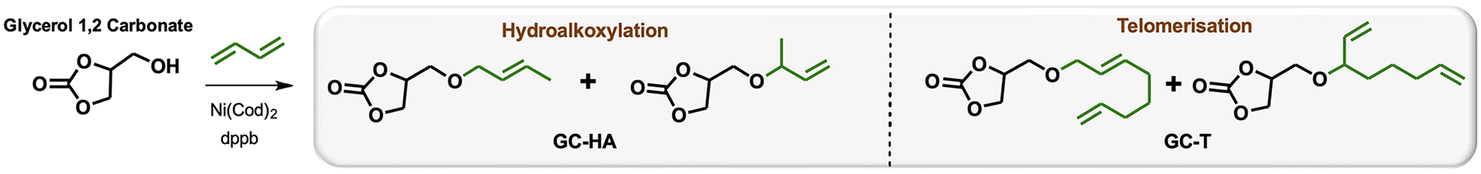
Efficient nickel-catalysed telomerisation on glycerol carbonate: a new linker route for lignin functionalisation†
Tiphaine
Richard
ab,
Walid
Abdallah
a,
Xavier
Trivelli
c,
Mathieu
Sauthier
 b and
Clément
Dumont
*ab
b and
Clément
Dumont
*ab
aUniversité de Lille, CNRS, Centrale Lille, ENSCL, Université Artois, UMR 8181-UCCS-Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: clement.dumont@icam.fr
bIcam School of Engineering, Lille campus, 6 rue Auber, B.P 10079, 59016 Lille Cedex, France
cCNRS, INRAE, Centrale Lille, Université Artois, FR 2638 – IMEC – Institut Michel-Eugène Chevreul, Université de Lille, 59000 Lille, France
First published on 29th November 2023
Abstract
An effective method of grafting functionalities onto lignin based on glycerol carbonate has been developed. This biobased grafting reagent reacts with lignin without generating co-products. Alkenyl groups were added to the glycerol carbonate moiety from 1,3-butadiene chemistry using the nickel-catalysed hydroalkoxylation reaction and the newly developed nickel-catalysed telomerisation reaction under solvent-free conditions. The high-atom economical reactions lead to three different sets of glycerol carbonate ethers with various chain lengths that can be separated from the catalyst to avoid lignin contamination with metals. Kraft, organosolv and soda lignins, without prior purification, were grafted via ether functions using these linkers in a clean and efficient procedure. The technical and modified lignins were characterised by NMR, IR and SEC, and their thermal properties were determined by DSC. The glass transition temperatures of the modified lignins are significantly reduced, opening up possibilities of shaping, for example, on hot presses. In addition, these modifications offer the possibility of inserting new reactive functions within the lignin, including unsaturations and alcohols, allowing a new range of lignin-based resins to be developed.
Introduction
Petroleum-based materials are industrially produced on a very large scale and widely used for the production of goods. For a more sustainable economy, the use of renewable carbon to produce materials is particularly attractive as it allows recycling and storage of CO2. Lignocellulosic biomass is one of the major alternative sources to oil, is not an edible resource and is available in large quantities. Lignin represents about 20% of lignocellulosic biomass and 100 million tons of lignin are already industrially produced as a side product of the paper industry and second-generation bioethanol production plants. Lignin is a bio-based polymer with high aromatic content and thus is clearly very attractive for the plastic industry. Still, this natural product is essentially used as low-value fuel and is currently 98% burned to generate electricity and heat.1 The valorisation of technical lignins for materials production is indeed largely impeded by weak mechanical properties and poor compatibility with synthetic polymers. In this context, the chemical functionalisation of lignin is required to access materials that can be processed, mixed with other polymeric materials,2,3 or used as macromonomers.4 The functionalisation of lignin's phenolic and aliphatic hydroxyl groups improves its properties and thus offers access to new valorisation routes.5 Several functionalisation approaches can be applied based on the available alcohols of lignin. The most commonly used routes involve esterification and, to a lesser extent, etherification reactions. These latter reactions offer interesting advantages, such as good robustness towards water as the ether group is more reluctant to undergo hydrolysis than the ester function, even in the presence of bases. However, lignin ethers are mainly obtained from the Williamson reaction with alkyl halides or epichlorohydrin,6,7 generating stoichiometric quantities of salts or co-products as waste. In an effort to develop more eco-friendly lignin functionalisation methods, our laboratory has employed innovative homogeneous catalytic reactions such as the telomerisation of 1,3-butadiene.8 This industrial reaction, catalysed by palladium, generates C8 allyl ethers with two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C unsaturations within the biopolymer9–12 that are interesting for further chemical transformations and have thus attracted more recent interest.13–15 This highly atom-economical reaction under mild conditions uses 1,3-butadiene, an industrial reagent available from alkane cracking or bioethanol.16 As the main advantages in comparison with the Williamson reaction, the telomerisation reaction does not release salts during the synthesis of the ether moieties and the gaseous 1,3-butadiene can be easily removed from the product by simply venting the reactor. However, two shortcomings may impede the use of this kind of process for lignin transformation at an industrial scale: the high price of the palladium catalyst and its persistence in the final material due to the difficulties of separating it from lignin at the end of the reaction. To solve these problems, this article proposes an innovative grafting route through a grafting agent, namely a “linker” that will support the catalytic reaction and can be easily removed from the catalyst at the end of the reaction (Scheme 1). To preserve the benefits of catalytic reactions, this linker should not generate salts or co-products either during its synthesis or when grafted onto lignin. This can be achieved with ring-opening reactions. The available rings known in the literature for lignin grafting are epoxides,17,18 cyclic carbonates19–21 and anhydrides.22,23 Linkers based on glycerol carbonate are particularly attractive as this derivative is biobased, made from glycerol, a byproduct of the biofuel industry, and its synthesis from glycerol is green and straightforward.24–29 Moreover, several studies have been performed on cyclic carbonate opening reactions with lignin hydroxyl groups. This transformation known as the oxyalkylation reaction is generally performed at high temperatures under basic conditions and generates only CO2 as a co-product.19,30,31 It is noteworthy that glycerol carbonate has an available hydroxyl that can be etherified thanks to catalytic reactions such as telomerisation, thus allowing the introduction of unsaturated aliphatic tails in the structure. To date, there are no effective alternative catalysts to the noble metal palladium for the telomerisation of 1,3-butadiene to give OC8 ethers. The rare examples with rhodium32 or nickel33 have shown very limited success by reaching yields that have never exceeded 30%. However, we turned our attention toward the use of nickel-based catalysts as a potential inexpensive and metal-abundant solution for the salt-free synthesis of ethers. Nickel catalysts are indeed known to selectively promote the hydroalkoxylation of 1,3-butadiene with an alcohol that allows the synthesis of butenyl ethers.33–36 This reaction was thus studied in this article to convert glycerol carbonate into the corresponding butenyl ether (OC4). By tuning the reaction conditions, this nickel-based catalysis also allowed efficient synthesis of octadienyl ethers (OC8), thus giving a first example of high-yield telomerisation of 1,3-butadiene with nickel. Both families of ethers were successfully used to graft lignins.
C unsaturations within the biopolymer9–12 that are interesting for further chemical transformations and have thus attracted more recent interest.13–15 This highly atom-economical reaction under mild conditions uses 1,3-butadiene, an industrial reagent available from alkane cracking or bioethanol.16 As the main advantages in comparison with the Williamson reaction, the telomerisation reaction does not release salts during the synthesis of the ether moieties and the gaseous 1,3-butadiene can be easily removed from the product by simply venting the reactor. However, two shortcomings may impede the use of this kind of process for lignin transformation at an industrial scale: the high price of the palladium catalyst and its persistence in the final material due to the difficulties of separating it from lignin at the end of the reaction. To solve these problems, this article proposes an innovative grafting route through a grafting agent, namely a “linker” that will support the catalytic reaction and can be easily removed from the catalyst at the end of the reaction (Scheme 1). To preserve the benefits of catalytic reactions, this linker should not generate salts or co-products either during its synthesis or when grafted onto lignin. This can be achieved with ring-opening reactions. The available rings known in the literature for lignin grafting are epoxides,17,18 cyclic carbonates19–21 and anhydrides.22,23 Linkers based on glycerol carbonate are particularly attractive as this derivative is biobased, made from glycerol, a byproduct of the biofuel industry, and its synthesis from glycerol is green and straightforward.24–29 Moreover, several studies have been performed on cyclic carbonate opening reactions with lignin hydroxyl groups. This transformation known as the oxyalkylation reaction is generally performed at high temperatures under basic conditions and generates only CO2 as a co-product.19,30,31 It is noteworthy that glycerol carbonate has an available hydroxyl that can be etherified thanks to catalytic reactions such as telomerisation, thus allowing the introduction of unsaturated aliphatic tails in the structure. To date, there are no effective alternative catalysts to the noble metal palladium for the telomerisation of 1,3-butadiene to give OC8 ethers. The rare examples with rhodium32 or nickel33 have shown very limited success by reaching yields that have never exceeded 30%. However, we turned our attention toward the use of nickel-based catalysts as a potential inexpensive and metal-abundant solution for the salt-free synthesis of ethers. Nickel catalysts are indeed known to selectively promote the hydroalkoxylation of 1,3-butadiene with an alcohol that allows the synthesis of butenyl ethers.33–36 This reaction was thus studied in this article to convert glycerol carbonate into the corresponding butenyl ether (OC4). By tuning the reaction conditions, this nickel-based catalysis also allowed efficient synthesis of octadienyl ethers (OC8), thus giving a first example of high-yield telomerisation of 1,3-butadiene with nickel. Both families of ethers were successfully used to graft lignins.
 | ||
| Scheme 1 Overall scheme of Ni-catalysed telomerisation of the glycerol-based “linker” for lignin functionalisation. | ||
Results and discussion
Glycerol is a versatile and commonly used organic compound derived from vegetal sources. Glycerol is also a co-product of the biodiesel industry, making it a readily available and inexpensive reactant. The glycerol carbonate derivative was synthesised through a low-temperature reaction using dimethylcarbonate (DMC) without requiring additional solvents by following a conventional procedure.24 This makes the production of glycerol carbonate environmentally friendly, as DMC is considered a recommended aprotic solvent.37 An optimisation study of the 1,3-butadiene hydroalkoxylation reaction with nickel was performed using glycerol carbonate as the starting material (Table 1). Standard conditions from previous laboratory investigations34 were used involving 1.5 mol% Ni(cod)2, 1.5 equivalents per nickel of dppb (1,4-bis(diphenylphosphino)butane) and 2 equivalents of 1,3-butadiene at 60 °C for 1 hour (entry 1). Under these conditions, 29% of the glycerol carbonate (GC) was converted into linear and branched C4 hydroalkoxylated ethers (GC-HA) with a 2/8 ratio. To improve the conversion of the substrate, the impact of the reaction time was first studied. The conversion of glycerol carbonate increases with the reaction duration and reaches a maximum of 93% after 24 hours of reaction (entries 1–5). Interestingly, the selectivity between GC-HA and GC-T is also reaction time-dependent. High proportions of GC-T were obtained with long reaction times; GC-HA was, for example, selectively obtained after 3 hours of reaction (entry 2), while GC-T was the major product after 16 hours (55% yield – entry 4) and 24 h of reaction (65% yield – entry 5). This shows a transformation of GC-HA into GC-T, which is consistent with a reversible hydroalkoxylation reaction, followed by the incorporation of a second equivalent of 1,3-butadiene for the formation of GC-T.35 A proposed mechanism is discussed in the ESI.† This reversibility of hydroalkoxylation also explains the variations observed in the linear to branched ratio (L/B) with reaction time (entries 1–5). A maximum between C4 and C8 products is reached with a ratio of around 70/30 (GC-T/GC-HA). The dominant telomerisation product was, in that case, obtained with 65% yield. The comparative study of the reaction at different temperatures showed that a decrease in temperature to 40 °C reduces the overall conversion of glycerol carbonate from 94% to 52% (entries 4 and 6) but favours the obtention of GC-HA with yields and L/B ratios similar to those obtained after 3 h at 60 °C (entries 2 and 6). On the other hand, increasing the temperature to 80 °C had a negative effect on the conversion, which is reduced to 74%. The product of telomerisation appears to be favoured at 60 °C with a yield of 54% in GC-T compared to 20% in GC-HA (compare entries 4 and 7). Concerning GC-HA, the increase in temperature increases the L/B ratio from 3/7 to 5/5. Increasing the number of equivalents of 1,3-butadiene equivalents from 2 to 5 at 80 °C boosted the GC conversion from 74% to 88% while maximising the GC-T yield to 70% (compare entries 7 and 8). Finally, the addition of a large excess of 1,3-butadiene (10 equivalents), although improving GC conversion, did not alter the final ratio of the products obtained, which was still similar to those obtained at 60 °C, i.e. a 70/30 ratio in favour of the telomer. It should be noted that the increase in the number of equivalents of 1,3-butadiene had no impact on the L/B selectivity of the isomers (entries 7–9). This balance of the GC-HA/GC-T ratio was confirmed by kinetic studies carried out at 60 °C and 80 °C (see Fig. SI1 and 2†). Interestingly, during tests on the effect of the catalyst loading, a maximum efficiency was observed with 1.5 mol% loading.|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | [Ni] (mol%) | Butadiene (eq) | T (°C) | Time (h) | GC conv.a % | Yield GC-HAa % | Yield GC-Ta % | Ratio GC-HA L/Ba | Ratio GC-T L/Ba |
| Conditions: 0.4 mL glycerol carbonate dried and freeze pumped, solvent-free, Ni(cod)2, dppb/Ni = 1.5 or PPh3/Ni = 3, N2 atmosphere, a Schlenk tube equipped with a Rotaflo stopcock.a Conversions, yields and ratios are determined by gas chromatography.b Reaction performed on 0.2 mL of glycerol carbonate. | |||||||||
| 1 | 1.5 | 2 | 60 | 1 | 29 | 29 | 0 | 2/8 | — |
| 2 | 1.5 | 2 | 60 | 3 | 62 | 62 | 0 | 1/9 | — |
| 3 | 1.5 | 2 | 60 | 6 | 84 | 71 | 13 | 2/8 | 6/4 |
| 4 | 1.5 | 2 | 60 | 16 | 94 | 39 | 55 | 3/7 | 6/4 |
| 5 | 1.5 | 2 | 60 | 24 | 93 | 28 | 65 | 4/6 | 5/5 |
| 6 | 1.5 | 2 | 40 | 16 | 52 | 50 | 2 | 1/9 | 6/4 |
| 7 | 1.5 | 2 | 80 | 16 | 74 | 20 | 54 | 5/5 | 6/4 |
| 8 | 1.5 | 5 | 80 | 16 | 88 | 18 | 70 | 5/5 | 6/4 |
| 9b | 1.5 | 10 | 80 | 16 | 95 | 28 | 67 | 5/5 | 6/4 |
| 10 | 0.8 | 2 | 80 | 16 | 29 | 29 | 0 | 2/8 | — |
| 11 | 2.3 | 2 | 80 | 16 | 55 | 26 | 29 | 5/5 | 6/4 |
| 12 | 3 | 2 | 80 | 16 | 23 | 20 | 3 | 5/5 | 1/9 |
Indeed, decreasing the catalytic loading drastically reduced the GC conversion to 29%, while a higher loading, such as 2.3 mol% and 3 mol%, also generated progressive decreases in conversion to 55% and 23%, respectively (compare entries 7 and 10–12). The additional study of different phosphines and the impact of solvent are detailed in the ESI† and do not provide significant results. The optimisation of the hydroalkoxylation reaction of glycerol carbonate with nickel thus demonstrated a novel efficiency in the telomerisation of 1,3-butadiene, allowing the formation of C8 ethers with the advantage of using an abundant metal and thus inexpensive catalyst. The choice of reaction parameters allows tuning the selectivity toward either the GC-HA or the GC-T product.
To explore the grafting of these new compounds onto lignin, three products were synthesised and isolated thanks to this nickel-catalysed protocol. Although nickel is abundant, purification by distillation or silica gel column chromatography allows catalyst separation from the linker, thus avoiding contamination of the final lignin-based material with a metal. GC-T with an L/B ratio of 20/1 and GC-HA product with an L/B ratio of 2/8 were isolated by chromatography on silica gel and used for lignin grafting (Table 2). In addition, a mixture of the two allyl ethers GC-T and GC-HA with the GC-T/GC-HA ratio of 70/30 has also been synthesised for reaction with lignin. Using the mixture advantageously avoids product separation steps. Notably, for this synthesis, the reaction upscaling on a scale of several grams slightly shifted the linear to branched and GC-T to GC-HA ratios to 7/4 and 3/7, respectively (compare with Table 1 entry 9).
|
|
|||||
|---|---|---|---|---|---|
| Name | Scheme (major) | GC-T content % | GC-HA content % | Ratio GC-T L/B | Ratio GC-HA L/B |
| GC-T |
|
100 | 0 | 20/1 | — |
| GC-HA |

|
0 | 100 | — | 2/8 |
| Mix | GC-T + GC-HA | 71 | 29 | 7/4 | 3/7 |

In this study, our attention was focused on the effect of the modification of the physicochemical properties of lignin by grafting these three different carbonates. The grafting was first performed and optimised with GC-T, the isolated telomerised compound. The reaction between cyclic carbonates and lignin showed different reaction pathways that depend on the nature of the reacting hydroxyl groups (Scheme 2).19,30,38 Phenols are mainly involved in ring-opening reactions that lead to two ethers whose structures depend on the carbon attacked by the nucleophile. The reaction is, in that case, accompanied by CO2 release. Aliphatic alcohols react with the carbonyl group and yield two possible acyclic carbonates. It should also be noted that carboxylic acids are also reactive toward cyclic carbonate ring-opening reactions and lead to esters. Undesirable secondary cross-linking reactions may also be present via transcarbonation reactions, which may impact the lignin's final properties and also reduce its solubility in common organic solvents.
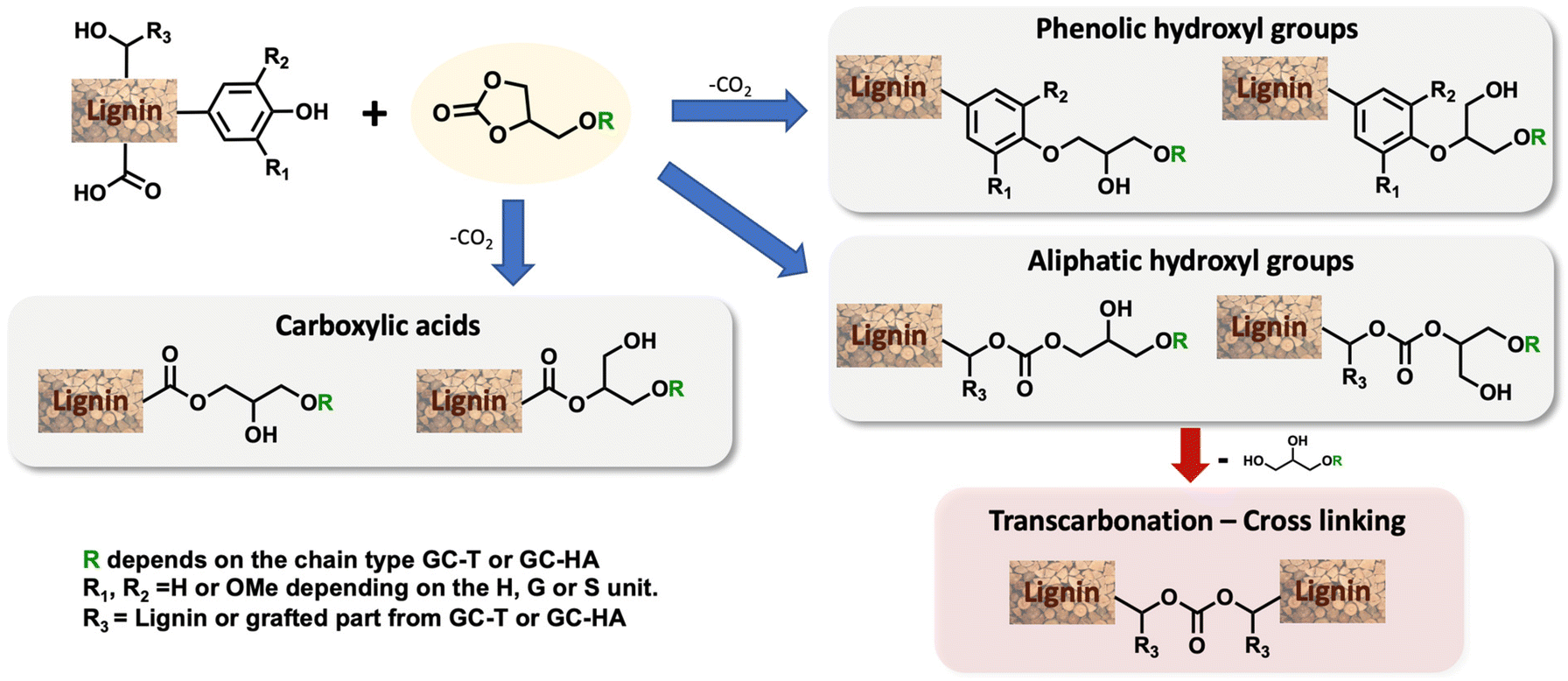
The optimisation study aimed to determine the most convenient reaction conditions for the grafting of GC-T onto kraft lignin (Table 3). The substitution degrees of the phenolic, aliphatic and carboxylic hydroxyl groups were determined by 31P NMR. The efficient grafting of lignin induced a significant mass increase of material. The lignin %weight in the final product was thus determined from the substitution degrees measured by 31P NMR correlated with the quantification of the grafted unsaturated chains, which is determined by 1H NMR. Finally, conversion and mass data allow the estimation of the number of substituted hydroxyl groups other than those from by GC-T, which was here attributed to transcarbonation reactions. All calculations are reported in the ESI.† It is noteworthy that the substitution degree observed in 31P NMR for all the following reactions was in accordance with the number of unsaturated chains grafted onto the lignin as determined by proton NMR. This suggests that the hydroxyl groups involved in the transcarbonation reactions were overwhelmingly from the glycerol part added to the lignin. The initial parameters for the grafting of GC-T onto lignin were chosen from previous studies that used cyclic carbonates without additional chains, such as glycerol carbonate, propylene carbonate or vinyl carbonate.19,39 Kraft lignin was first reacted with 6 equivalents of GC-T with respect to the alcohols (aliphatic and phenolic) of the lignin at a temperature of 150 °C and in the presence of 0.1 equivalents of K2CO3 (Table 3, entry 1). To improve the homogeneity of the mixture, up to 9 equivalents of DMSO were added relative to the lignin OH groups. With these parameters, 93% of the phenolic and none of the aliphatic alcohols were substituted, representing 64% of the total alcohol content of the lignin. All carboxylic acids were also converted and the final material contained 52% lignin. Initial estimations of transcarbonation indicate that 17% of additional hydroxyl groups were involved in carbonate cross-links. A longer reaction time of 15 hours was used without significant changes in the results (entry 2). The effect of temperature was then studied and no conversion was obtained when the reaction was carried out at 120 °C (entry 3). On the other hand, increasing the temperature to 170 °C generated total conversion of the phenolic groups of the lignin, combined with low conversion of aliphatic alcohols up to 6% (entry 4).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Base | T (°C) | T (h) | Substitution degreea [%] | Lignin wt% | Estimated transcarb.b [% OH] | |||
| OH tot. | OHAl. | OH Ph. | COOH | ||||||
Conditions: 0.5 g of kraft lignin, 6 eq. of modified carbonate relative to lignin total OH, DMSO (9 eq./OH), 0.1 eq. of the chosen base, N2 atmosphere.a Based on the 31P NMR results, the calculation method is presented in Fig. SI16.†b Determined due to the difference between the concentration of OH obtained and the theoretical concentration (in mmol g−1) considering the %weight of lignin in the final product. The calculation is presented in Fig. SI16.† *![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Simplified scheme not representing all possibilities of Scheme 2. Simplified scheme not representing all possibilities of Scheme 2. |
|||||||||
| 1 | K2CO3 | 150 | 3 | 64 | 0 | 93 | 100 | 52 | 17 |
| 2 | K2CO3 | 150 | 15 | 65 | 0 | 94 | 100 | 52 | 12 |
| 3 | K2CO3 | 120 | 3 | 0 | — | — | — | n.d | n.d |
| 4 | K2CO3 | 170 | 3 | 71 | 6 | 100 | 100 | 50 | 13 |
| 5 | Na2CO3 | 170 | 3 | 75 | 19 | 100 | 100 | 49 | 8 |
| 6 | DBU | 170 | 3 | 75 | 20 | 100 | 100 | 49 | 13 |
| 7 | TEA | 170 | 3 | n.d | — | — | — | n.d | n.d |
| 8 | K2CO3 | 170 | 8 | n.d | — | — | — | n.d | n.d |

These temperature conditions did not significantly affect the cross-linking reactions due to transcarbonation, which was limited to around 13% of the hydroxyl groups present in the modified lignin. A change of base from K2CO3 to Na2CO3 slightly affected the conversion of aliphatic alcohols, whose degree of substitution increased to 19%, followed by a decrease in transcarbonation to 8% (entry 5). In contrast, the use of DBU, known to limit the presence of transcarbonation in the case of propylene carbonate,30 did not significantly reduce this reaction in the case of GC-T (entry 6). The use of triethylamine resulted in major insolubility of the lignin in usual solvents, hindering comparative quantification (entry 7). Analysis of the very small soluble fraction indicated that chains are indeed grafted in similar quantities to other bases. This insolubility can therefore potentially be explained by the significant impact of triethylamine in generating cross-linking through carbonates. Similar solubility issues were observed when an extended reaction time of 8 hours was applied with potassium carbonate as a base (compare entries 4 and 8). The transcarbonation occurred at a lower rate than the other ring-opening reactions and likely becomes important when most of the phenolic hydroxyl groups are converted. The combination of reaction temperature and reaction times is critical to limit this side reaction.
After optimisation, the 31P NMR pattern following the opening reactions of the cyclic carbonates shows complete removal of the signals at 134–136 ppm and 137–145 ppm corresponding, respectively, to the carboxylic acids and the different phenols available in the lignin (Fig. 1). Ring opening of the cyclic carbonate generates a new aliphatic hydroxyl group derived from the glycerol part of the grafted chain. Consequently, while signals corresponding to the phenols and carboxylic acids (COOH in Fig. 1) disappear, a broad signal appears in the aliphatic alcohol region between 145 and 147 ppm. The aliphatic hydroxyl groups initially present in the lignin structure are mostly unreacted and appear as a broad signal at 147–149 ppm in both spectra.
 | ||
| Fig. 1 Comparison between 31P NMR spectra of kraft lignin (blue curve) and modified lignin (red curve) from Table 1 entry 4 – using the Argyropoulos et al. method.44 | ||
For the structural characterisation of the lignin modifications, a first 1H NMR investigation was conducted. Following the same protocol, the model lignin molecule guaiacol was modified and isolated to facilitate characterisation. The NMR spectrum obtained from the modified guaiacol was compared with the spectrum obtained for the modified lignin (Fig. 2). In both spectra, the signals in the aliphatic region of the spectrum indicate the presence of methyl and methylenic groups that are characteristic of an octadienyl chain (see peaks 6 and 7). Strong signals at 4.8–5.1 ppm and 5.5–6.0 ppm also clearly reveal the presence of the olefinic protons (peaks 5 and 8). The weak signal at 5.15 ppm on the 1H NMR spectrum is consistent with the presence of branched octadienyl structures. The broad signal between 3.3 ppm and 4.2 ppm corresponds to the methoxy groups found in the lignin structure stacked on the protons derived from the glycerol part and the OCH2 protons of the octadienyl chain (see 1, 2, 3 and 4). The very weak signals around 4.5 ppm correspond to the other ring-opening reactions of the cyclic carbonate (see Scheme 2). The low intensity of these signals indicates that this kind of cyclic carbonate opening is largely limited in the reaction.
 | ||
| Fig. 2 Comparison between 1H NMR spectra of guaiacol and modified lignin from Table 1 entry 4. | ||
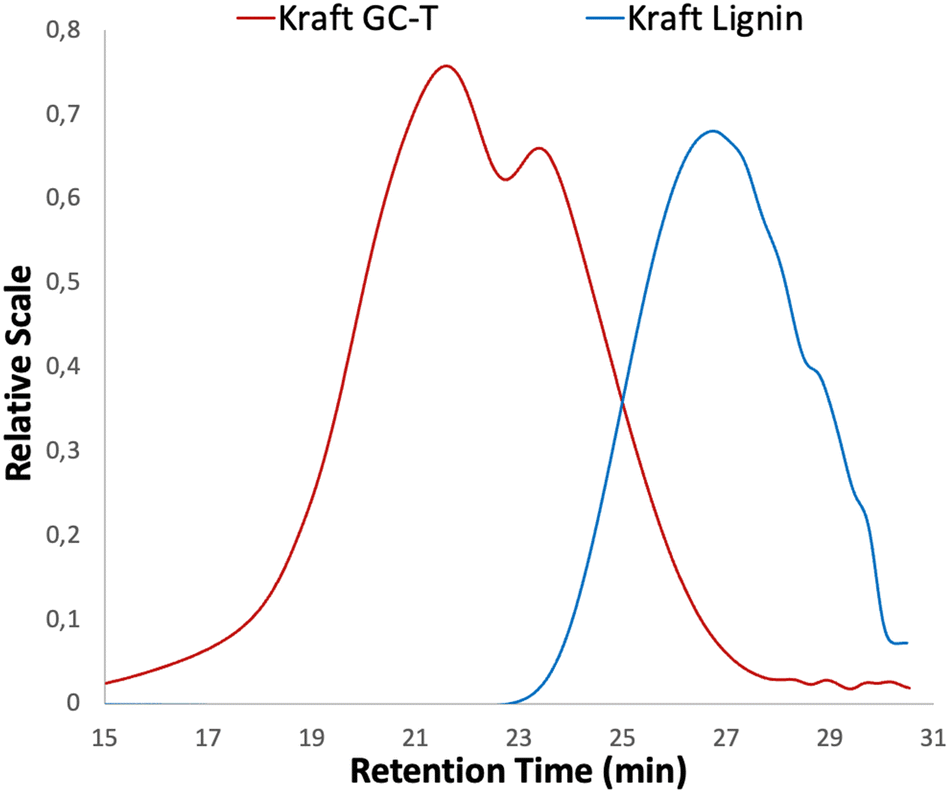
SEC measurements were performed on the materials to determine the number average molecular weight Mn and the dispersity Đ of lignins. SEC analysis of technical kraft lignin used as starting material revealed a number average molecular weight Mn of 1400 g mol−1 with a dispersity Đ of 1.6 (Fig. 3, see the blue curve). After the reaction with GC-T, a significant shift towards higher molecular weights (Mn = 6700 g mol−1) corresponding to the large number of grafted chains on the macromolecule is observed (Fig. 3, see the red curve). Although shifted, the profile of the SEC curves is not similar, indeed, the dispersity of the modified macromolecule increases drastically at Đ = 8.4 and is clearly bimodal, as already observed in the literature with cyclic carbonates.30 These profiles associated with high dispersion of SEC curves suggest the presence of cross-linking due to transcarbonation reactions. Although the dispersity is high, the lignin showed no solubility issues after the reaction. However, fractionation methods can be envisaged to reduce the dispersity depending on the intended applications.40,41
 | ||
| Fig. 3 Comparison between SEC of technical kraft lignin and modified kraft GC-T. | ||
To provide clearer evidence of the formation of cross-linking carbonate groups, a study was performed comparing kraft lignin and modified lignin using 13C NMR (Fig. 4). Investigation of the 140–160 ppm region reveals various signals associated with aromatics and possible carbonates. According to the literature,30 the signal observed in the kraft lignin at 146–148 ppm is attributed to carbons bearing methoxy groups in the G- and C5-substituted units of the lignin structure (G1 and C51, see the blue curve). After the reaction, the initial signal disappeared in favour of two signals at 148–150 ppm and 151–153 ppm, corresponding, respectively, to the G2 and C52 carbons of the modified lignin. A prominent signal is also present at around 155 ppm corresponding to carbonate carbon. This indicates a significant amount of carbonate in the modified lignin as estimated by 31P NMR. As the aliphatic alcohols were not converted in this sample, they may be attributed to cross-linking transcarbonation. The complete 13C NMR and 2D HSQC NMR results are available in the ESI (see Fig. SI34–37†), and these showed no unexpected transformations in the lignin structure.
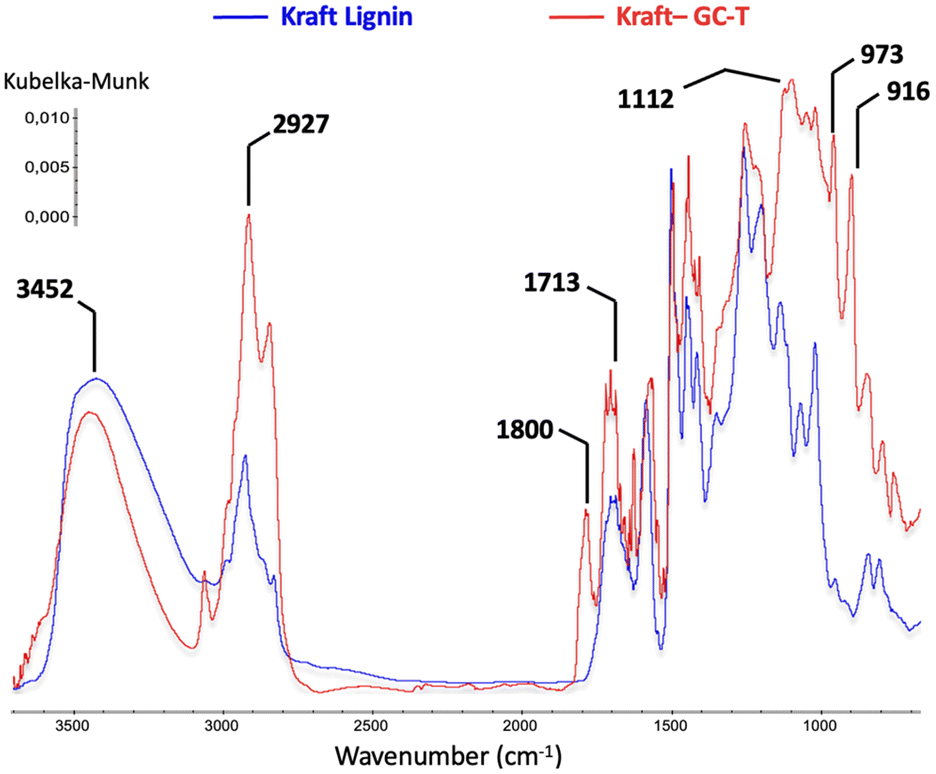 | ||
| Fig. 4 Comparison between 13C NMR spectra of kraft lignin (blue) and kraft GC-T (red). | ||
The comparison of the IR spectra of lignin before and after the reaction also clearly indicates the presence of new carbonyl groups with a characteristic band at 1800 cm−1 (Fig. 5). The increase in band intensities at 2900 cm−1 and 1100 cm−1 is consistent with the presence of new alkane chains coupled to ether functions. In addition, the signals at 973 and 916 cm−1 are assigned to alkene sp2 C–H bending patterns for monosubstituted and trans-disubstituted olefin functions, respectively.
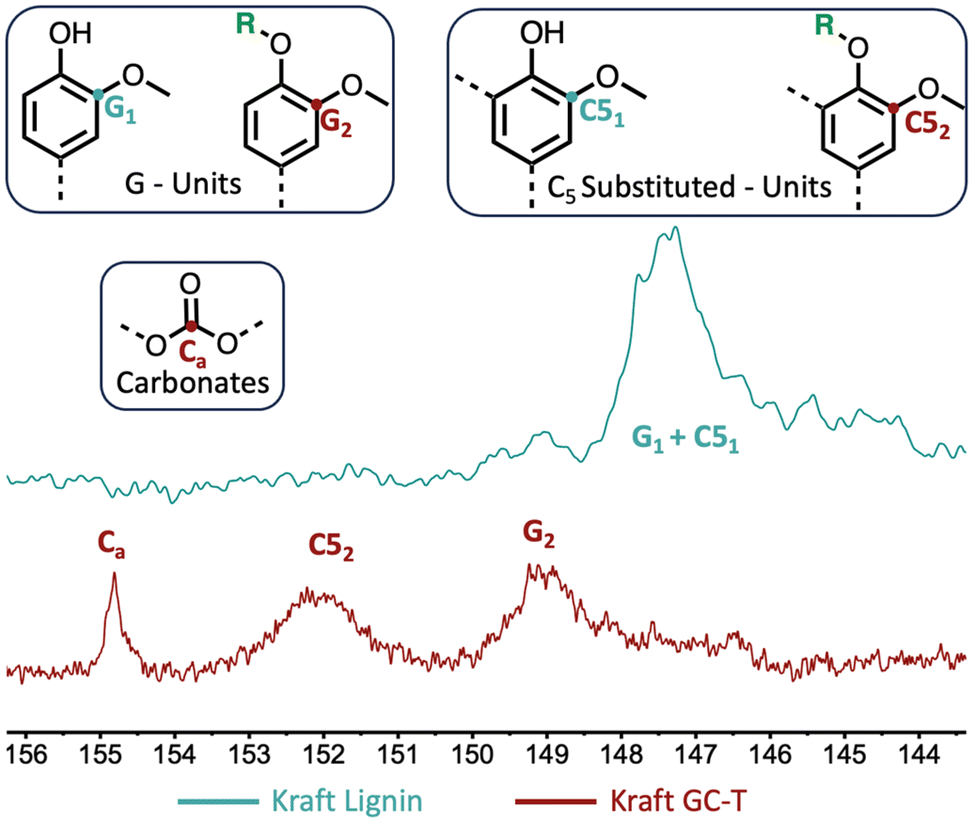 | ||
| Fig. 5 Comparison between FTIR spectra of technical kraft lignin and modified lignin (Table 1 entry 4). | ||
This new procedure of lignin transformation with linkers derived from glycerol carbonate ethers was then applied to different industrial lignins (Table 4).
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Lignin | Linker | Conversiona [%] | Carb. trans.a | DSC | SEC | ||||
| OHtot | OHal | OHph | COOH | [% OH] | T g |
M
n (app)b |
Đ | |||
| Conditions: 3 h, 170 °C, 0.1 eq. K2CO3, DMSO (2 mL), 0.5 g lignin (1 eq./OH), 6 eq. of modified carbonate relative to lignin total OH. The composition of the modified glycerol carbonate mixture is indicated in Table 2 (71% GC-T/29% GC-HA).a Conversion and proportion of transcarbonation are determined by 31P NMR.b Mn apparent values have been determined by size exclusion chromatography (SEC) analysis in THF using polystyrene standards. *Simplified scheme not representing all possibilities of Scheme 2. |
||||||||||
| 1 | Kraft | GC-T | 71 | 6 | 100 | 100 | 13 | 45 °C | 6700 | 8.4 |
| 2 | Soda | GC-T | 71 | 0 | 100 | 100 | 16 | 48 °C | 4700 | 2.9 |
| 3 | Organosolv | GC-T | 54 | 0 | 100 | 100 | 17 | 48 °C | 4500 | 3.7 |
| 4 | Kraft | GC-HA | 81 | 38 | 100 | 100 | 19 | 45 °C | 5300 | 5.1 |
| 5 | Soda | GC-HA | 84 | 44 | 100 | 100 | 24 | 41 °C | 3900 | 3.0 |
| 6 | Organosolv | GC-HA | 78 | 50 | 100 | 100 | 21 | 49 °C | 4200 | 5.2 |
| 7 | Kraft | Mix | 88 | 60 | 100 | 100 | 6 | 37 °C | 6500 | 5.0 |
| 8 | Soda | Mix | 81 | 31 | 100 | 100 | 12 | 35 °C | 5400 | 6.8 |
| 9 | Organosolv | Mix | 82 | 59 | 100 | 100 | 12 | 49 °C | 5300 | 3.8 |

The initial specifications of these different technical lignins are reported in the ESI (Table SI2†), and lignins are used without prior purification. Under the optimised reaction conditions obtained with kraft lignin, 71% of the total hydroxyl groups of soda lignin were substituted with GC-T as reagent (entry 2). Such as that in the case of the kraft lignin, 100% of the phenols and 100% of the COOH groups were converted while the aliphatic hydroxyl groups mainly remained untouched. The phenolic groups were also fully substituted when organosolv lignin was reacted (entry 3). With this lignin, the higher proportion of aliphatic hydroxyls in comparison with phenolic ones explains the lower total substitution degree (54% with organosolv lignin). Note that the percentage of substituted hydroxyl groups involved in cross-linking carbonate groups was similar to all the lignins used in the study (compare entries 1–3). A slight difference was observed between the different lignins in the steric exclusion chromatography results. Indeed, kraft lignin systematically had significantly higher Mn and polydispersity values than soda or organosolv lignins, from 6700 g mol−1 to 4700 and 4500 g mol−1, and Đ of 8.4 to 2.9 and 3.7, respectively (compare entries 1–3). Nevertheless, the profile of the SEC curves of these modified lignins remained bimodal as on the kraft GC-T lignin (see Fig. SI30–33†). GC-HA was then subsequently used as reagent in the grafting reactions to determine the impact of the grafted chain size on the reaction output and lignin properties. For kraft lignin, the phenolic hydroxyl groups and COOH were fully substituted (entries 4 and 7). Surprisingly, using a shorter chain (GC-HA) greatly enhanced the grafting on the aliphatic hydroxyl groups of the lignin reaching 38% substitution (compare entries 1 and 4). Soda lignin followed the same trend with 44% of substituted aliphatic hydroxyl groups and 84% of the total substitution degree (entry 5). This substitution rate was even more important with organosolv lignin and reached 50% of the aliphatic hydroxyl groups with 78% of the conversion of total alcohols. The aliphatic hydroxyl groups were also slightly more impacted regarding the transcarbonation reactions, increasing for example from 13% to 19% in the case of kraft lignin (compare entries 1 and 4) without impacting solubility. The molar masses were of the same magnitude as previously observed with GC-T, which, despite the shorter chains grafted, was balanced by a higher overall substitution rate. In addition, to overcome the separation of telomerisation and hydroalkoxylation products and to allow a more efficient scale-up, the mixture of GC-T and GC-HA linkers, referred to as the “Mix” in Table 2, was directly used to functionalise lignin.
For kraft lignin, grafting the mixture significantly improved the substitution degree of aliphatic hydroxyl groups, which increased up to 60%, while maintaining the total conversion of the other two reactive groups (entry 7). In the case of soda lignin, the substitution degree results were quite similar to those observed with GC-HA (compare entries 5 and 8) with a substitution of total hydroxyl groups up to 81%. The organosolv lignin followed the same trend with 59% of substitution degree of its aliphatic alcohols along with 82% of the total conversion of the hydroxyl groups. In contrast to the use of GC-HA, the use of the linker mixture (GC-HA and GC-T) showed a reduction in the proportion of hydroxyl groups modified via secondary transcarbonation reactions. Consequently, only a maximum of 12% of the hydroxyl groups were cross-linked in this type of secondary reaction. As expected, the combination of the grafting of longer chains, coupled with a higher substitution degree of the aliphatic hydroxyl groups increased the overall Mn on the different lignins studied. Thus, the kraft lignin reached an Mn = 6500 g mol−1 while the soda and organosolv lignins approached 5300 g mol−1 for Đ values between 3.8 and 5.
The 1H NMR analyses performed on the different linkers grafted onto the lignin showed no modification of the reactivity towards the carbonate cycle. Thus, the chain length grafted onto the glycerol carbonate does not influence the opening direction of the carbonate cycle. Indeed, the absence of characteristic peaks around 4.5 ppm in the 1H NMR spectrum (Fig. 6) is comparable to that observed with GC-T. In the GC-HA grafting case, the NMR spectrum profile is close to the one obtained with GC-T. The signals between 4.75 and 6 ppm still attest to the large amount of unsaturations added within the lignin structure (Fig. 6, blue curve).
 | ||
| Fig. 6 Comparative 1H NMR spectra of modified kraft lignins: kraft GC-T, kraft GC-HA and kraft mix. | ||
Two aliphatic signals at 0.9–1.4 ppm and 1.4–1.9 ppm indicate the simultaneous presence of branched and linear butenyl (C4) chains, respectively. The integration of these signals further indicates a 2/8 ratio in favour of branched chains corresponding to the ratio initially present in the GC-HA (see Table 2). Thus, there seems to be no particular opening selectivity of the cyclic carbonates depending on the chain length grafted onto the glycerol carbonate. Finally, by using the mixture of GC-T and GC-HA, the proton NMR spectrum shows the presence of both types of grafted chains on lignin by the distinction of the peaks around 1.8–2.3 ppm and 0.9–1.4 ppm corresponding to the chains from GC-T and GC-HA, respectively (Fig. 6, green curve).
The transformations performed with linkers of different chain lengths have a significant impact on the thermal properties of the lignin. Thus, the glass transition temperature (Tg) values of technical lignins are initially ranging from 80 to 150 °C (ref. 42 and 43) and drop substantially between 35 °C and 45 °C after the modifications (see Table 4). The degradation temperatures of technical lignins are much too close to the glass transition temperatures, which drastically limits the shaping possibilities. This reduction of the glass transition temperature after chemical modification allowed the modified lignins to be sufficiently viscous below the degradation temperature to be processed and thus used as biobased plastic materials. Therefore, shaping on a hot press became possible, for example, from GC-T grafted kraft lignin, of which an application is shown in Fig. 7. This material, composed of 100% lignin/GC-T without any additives has a glossy aspect that can even be engraved with a laser cutter. As lignin macromolecules are small, the material remains relatively brittle. Still, the experiments showed that shaping is possible and the presence of two different types of polymerisable functions (free hydroxyl groups and alkene functions, see Fig. 7) opens up interesting new opportunities in the field of resins for numerous applications, such as polyurethanes, polyepoxides and composite materials.
 | ||
| Fig. 7 Example of GC-T grafted kraft lignin material. | ||
Materials and methods
General considerations
Kraft lignin (UPM BioPiva 100 – Softwood) was provided by UPM Biochemicals, organosolv lignin by ChemicalPoint UG, Germany and soda lignin by GreenValue Enterprises LLC. Lignins were dried under reduced pressure at 60 °C for 48 h before use. Chemicals were purchased from Sigma-Aldrich, Alfa Aesar, Acros Organics and Strem Chemicals. 1,3-Butadiene was purchased from Linde Gas France. NMR spectra were recorded in CDCl3 (99.8% atom D), which was purchased from Sigma-Aldrich. 1H NMR spectra were obtained at either 300 or 400 MHz, and deeper characterisation studies were performed; NMR experiments were run at 303 K on Bruker AVANCEIIIHD 600 (13C = 151 MHz) and AVANCE NEO 900 (13C = 226 MHz) spectrometers equipped with 5 mm cryo-1H/2H/13C/15N/19F-QCI, broad-band-BBO and cryo 1H/2H/13C/15N-TCI probes. The samples were dissolved in DMSO-d6. Mono-dimensional 1H and 13C{1H}, and bi-dimensional 1H–13C-HSQC-DEPT and 1H–13C-HSQC-TOCSY (60 or 80 ms mixing time) spectra were recorded. Chemical shifts (1H and 13C) were referenced in parts per million relative to TMS (δ = 0.00 ppm) and were referenced to the residual solvent peak (1H, DMSO-d5 2.50 ppm; 13C, DMSO-d6 39.52 ppm). Diffuse reflectance infrared spectra were collected with a Harrick cell on a Nicolet Avatar spectrometer fitted with an MCT detector. Typically, 64 scans were accumulated for each spectrum (a resolution of 4 cm−1).The number-average (Mn) and weight-average (Mw) molecular weights and the dispersity (Đ) of the product were determined by size exclusion chromatography (SEC) in tetrahydrofuran at 40 °C at a flow rate of 1 mL min−1. The sample concentration was 0.50 wt%. Mn, Mw, and Đ were determined from the refractive index (RI) signal using a calibration curve based on polystyrene (PS) standards from Polymer Standards Service, on a Waters apparatus equipped with Waters Styragel columns HR2, HR3, HR5 and HR5E.
DSC measurements were carried out using a PerkinElmer DSC 4000 differential scanning calorimeter. The measurements were carried out with a heating rate of 10 K min−1 in the temperature range from 243 K to 453 K, under nitrogen flow (20 mL min−1).
The GC analyses were performed on a GC Shimadzu® 2010 equipped with an HT 5 column (30 m, 0.25 mm i.d., 0.25 μm thickness) and flame ionisation detector (FID). Conditions: injector and detector = 250 °C, gas = nitrogen at 0.64 mL min−1, temperature = 150 °C, 100 °C hold for 1 min then ramp of 10 °C min−1 up to 250 °C and hold for 10 min at 250 °C.
Titration of the hydroxyl groups in the technical lignins and modified lignins
31P NMR experiments were carried out following a previously reported method:44 lignin (30 mg) was dissolved in a CDCl3/pyridine mixture (1:1.6 v/v, 0.5 mL). The phosphitylation reagent, 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (150 μL), and the internal standard N-hydroxy-6-norbornene-2,3-dicarboximide (100 μL of 0.1 M solution in 1:1.6 v/v CDCl3/pyridine mixture) were added successively. Chromium(III) acetylacetonate (100 μL of 0.014 M solution) in the same CDCl3/pyridine mixture was added to the solution to homogenise and accelerate phosphorus relaxation. Spectra were recorded with a 15 s relaxation time and an average number of 512 scans. Chemical shifts are relative to the signal of the phospholane hydrolysis product at 132.2 ppm. The integrated value of the internal standard was used for the calculation of the absolute amount of each functional group. A comparison with the amounts of functional groups in technical lignins leads to the characterisation of modified lignins. Calculations of the substitution degree, mass percentages of lignin and percentages of transcarbonation are described in the ESI.†
General procedure of 1,3-butadiene hydroalkoxylation/telomerisation of glycerol carbonate (GC-T and GC-HA)
Glycerol carbonate (freeze pumped) (5 g, 42.3 mmol, 1 eq.), Ni(cod)2 (196 mg, 713 μmol, 1.5%) and dppb (460 mg, 1079 μmol, 2.25%) were gathered in an autoclave inside a glove box. Then, condensed 1,3-butadiene (20 mL, 229 mmol, 5 eq.) at −50 °C was inserted into the autoclave under nitrogen, which was then placed at 80 °C for 15 h. After the reaction, the reactor was cooled to room temperature and the excess gas was evacuated. The desired products were isolated by column chromatography on silica gel with petroleum ether/ethyl acetate 80:20 eluant affording 98% isolated yield, 29% GC-HA (68% branched–32% linear) and 71% GC-T (38% branched–62% linear).
General procedure of lignin grafting using glycerol carbonate derivatives
Lignin (0.5 g), modified carbonate (6 eq./lignin OH) and base (0.1 eq.) were gathered in a Schlenk tube which was then placed under an inert atmosphere. Then, DMSO (2 mL) was added. The reaction was heated at 170 °C (preheated disposal) during the time chosen. After the reaction, the Schlenk tube was cooled and modified lignin was precipitated from the crude product by diluting the mixture into 2 mL of THF and using a 1/1/1 mixture of diethyl ether/petroleum ether/distilled water. The precipitation was repeated 3 times to remove all the glycerol carbonate and the DMSO.Preliminary shaping study of kraft GC-T lignin
By using a PINETTE PEI manual press with heated plates, 15 g of kraft GC-T lignin powder was hot-pressed at 120 °C under a pressure of 5 bars for 30 minutes in a rectangular PTFE mould. The images in Fig. 7 were obtained directly after demoulding at room temperature.Conclusions
A new strategy for the clean and efficient transformation of lignin has been developed. This involves the use of linkers, i.e. grafting agents, which are obtained from glycerol carbonate and represent a promising method for grafting a wide range of functionalities onto lignin. Glycerol carbonate is efficiently etherified using nickel catalysis, with Ni being an abundant and inexpensive metal, initially through the hydroalkoxylation reaction with 1,3-butadiene. Surprisingly, this resulted in the first efficient example of telomerisation of 1,3-butadiene with nickel with 95% conversions and over 70% selectivity in telomer chains. This nickel-catalysed atom-economical reaction under solvent-free conditions provides two glycerol carbonate derivatives with C4 or C8 chains, which can be isolated as a mixture. These molecules are then grafted onto industrial kraft, organosolv and soda lignins with substitution degrees of up to 88% of the total hydroxyl groups. Using these linkers allows the introduction of unsaturated ether chains on lignin without any salt formation and without any traces of metal in the final material as nickel is removed from the linker before lignin grafting. In addition, the thermal properties of lignin are substantially modified with lower glass transition temperature values that range between 35 °C and 49 °C, making lignin malleable and shapable when heated. Besides the above properties, these new lignin macro-molecules also bear new reactive polymerisable functions. These new functionalities offer numerous possibilities for applications in polyester, polyurethane or polyepoxy materials due to the presence of the two unsaturations and the free aliphatic alcohol function. Thus, new promising perspectives are opening up in creating new high-performance biobased materials based on lignin, which could potentially bring lignin native properties such as flame retardancy or UV protection.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Ministère de l'Enseignement Supérieur, de la Recherche et de l'Innovation, the CNRS, the Hauts-de-France Region and Icam for their financial support. Financial support from the IR INFRANALYTICS FR2054 for conducting the NMR research is gratefully acknowledged. We are thankful for the support for the NMR facility from the European Union with the European Regional Development Fund (ERDF), the Hauts de France Regional Council (contract no. 17003781), Métropole Européenne de Lille (contract no. 2016_ESR_05), the French State (contract no. 2017-R3-CTRLPhase 1), the Pasteur Institute of Lille, the University of Lille and the CNRS. We acknowledge C. Delabre for HR-MS analysis. We would also like to acknowledge COST Action CA17128 (LignoCOST) for promoting the exchange and dissemination of knowledge and expertise in the field of lignin valorisation.References
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS.
- Chemical Modification, Properties, and Usage of Lignin, ed. T. Q. Hu, Springer US, Boston, MA, 2002 Search PubMed.
- D. Kun and B. Pukánszky, Eur. Polym. J., 2017, 93, 618–641 CrossRef CAS.
- C. Wang, S. S. Kelley and R. A. Venditti, ChemSusChem, 2016, 9, 770–783 CrossRef CAS PubMed.
- A. E. Kazzaz, Z. H. Feizi and P. Fatehi, Green Chem., 2019, 21, 5714–5752 RSC.
- W. Ren, X. Pan, G. Wang, W. Cheng and Y. Liu, Green Chem., 2016, 18, 5008–5014 RSC.
- C. Asada, S. Basnet, M. Otsuka, C. Sasaki and Y. Nakamura, Int. J. Biol. Macromol., 2015, 74, 413–419 CrossRef CAS PubMed.
- C. Dumont, R. M. Gauvin, F. Belva and M. Sauthier, ChemSusChem, 2018, 11, 1649–1655 CrossRef CAS PubMed.
- A. Behr, M. Becker, T. Beckmann, L. Johnen, J. Leschinski and S. Reyer, Angew. Chem., Int. Ed., 2009, 48, 3598–3614 CrossRef CAS PubMed.
- D. Peruzzo, M. Drelon, C. Dumont, A. Mortreux, I. Suisse and M. Sauthier, Mol. Catal., 2021, 502, 111369 CrossRef CAS.
- M. S. Ibn El Alami, I. Suisse, S. Fadlallah, M. Sauthier and M. Visseaux, C. R. Chim., 2016, 19, 299–305 CrossRef.
- N. D. Clement, L. Routaboul, A. Grotevendt, R. Jackstell and M. Beller, Chem. – Eur. J., 2008, 14, 7408–7420 CrossRef CAS PubMed.
- M. Jawerth, M. Lawoko, S. Lundmark, C. Perez-Berumen and M. Johansson, RSC Adv., 2016, 6, 96281–96288 RSC.
- L. C. Over and M. A. R. Meier, Green Chem., 2015, 18, 197–207 RSC.
- A. Duval and L. Avérous, ChemSusChem, 2017, 10, 1813–1822 CrossRef CAS PubMed.
- G. Pomalaza, M. Capron, V. Ordomsky and F. Dumeignil, Catalysts, 2016, 6, 203 CrossRef.
- P. Giannì, H. Lange and C. Crestini, ACS Sustainable Chem. Eng., 2020, 8, 7628–7638 CrossRef PubMed.
- M. A. Delgado, E. Cortés-Triviño, C. Valencia and J. M. Franco, Tribol. Int., 2020, 146, 106231 CrossRef CAS.
- A. Duval and L. Avérous, ACS Sustainable Chem. Eng., 2017, 5, 7334–7343 CrossRef CAS.
- L.-Y. Liu, M. Cho, N. Sathitsuksanoh, S. Chowdhury and S. Renneckar, ACS Sustainable Chem. Eng., 2018, 6, 12251–12260 CrossRef CAS.
- X. Zhang, Y. Kim, I. Elsayed, M. Taylor, T. L. Eberhardt, E. B. Hassan and R. Shmulsky, Ind. Crops Prod., 2019, 141, 111797 CrossRef CAS.
- C. Scarica, R. Suriano, M. Levi, S. Turri and G. Griffini, ACS Sustainable Chem. Eng., 2018, 6, 3392–3401 CrossRef CAS.
- F. Monteil-Rivera and L. Paquet, Ind. Crops Prod., 2015, 65, 446–453 CrossRef CAS.
- P. de Caro, M. Bandres, M. Urrutigoïty, C. Cecutti and S. Thiebaud-Roux, Front. Chem., 2019, 7, 308 CrossRef CAS PubMed.
- A. Kaur, R. Prakash and A. Ali, Talanta, 2018, 178, 1001–1005 CrossRef CAS PubMed.
- A. Dibenedetto, A. Angelini, M. Aresta, J. Ethiraj, C. Fragale and F. Nocito, Tetrahedron, 2011, 67, 1308–1313 CrossRef CAS.
- J. R. Ochoa-Gómez, O. Gómez-Jiménez-Aberasturi, B. Maestro-Madurga, A. Pesquera-Rodríguez, C. Ramírez-López, L. Lorenzo-Ibarreta, J. Torrecilla-Soria and M. C. Villarán-Velasco, Appl. Catal., A, 2009, 366, 315–324 CrossRef.
- J. R. Ochoa-Gómez, O. Gómez-Jiménez-Aberasturi, C. Ramírez-López and B. Maestro-Madurga, Green Chem., 2012, 14, 3368 RSC.
- A.-C. Simao, B. Lynikaite-Pukleviciene, C. Rousseau, A. Tatibouet, S. Cassel, A. Sackus, A. P. Rauter and P. Rollin, Lett. Org. Chem., 2006, 3, 744–748 CrossRef CAS.
- I. Kühnel, B. Saake and R. Lehnen, Ind. Crops Prod., 2017, 101, 75–83 CrossRef.
- I. Kühnel, B. Saake and R. Lehnen, Macromol. Chem. Phys., 2018, 219, 1700613 CrossRef.
- K. C. Dewhirst, J. Org. Chem., 1967, 32, 1297–1300 CrossRef CAS.
- S. Bigot, M. S. I. El Alami, A. Mifleur, Y. Castanet, I. Suisse, A. Mortreux and M. Sauthier, Chem. – Eur. J., 2013, 19, 9785–9788 CrossRef CAS PubMed.
- A. Mifleur, H. Ledru, A. Lopes, I. Suisse, A. Mortreux and M. Sauthier, Adv. Synth. Catal., 2016, 358, 110–121 CrossRef CAS.
- A. Mifleur, D. S. Mérel, A. Mortreux, I. Suisse, F. Capet, X. Trivelli, M. Sauthier and S. A. Macgregor, ACS Catal., 2017, 7, 6915–6923 CrossRef CAS.
- G. Tran and C. Mazet, Org. Lett., 2019, 21, 9124–9127 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS.
- A. Duval and L. Avérous, ACS Sustainable Chem. Eng., 2016, 4, 3103–3112 CrossRef CAS.
- Q. Hua, L.-Y. Liu, M. Cho, M. A. Karaaslan, H. Zhang, C. S. Kim and S. Renneckar, Biomacromolecules, 2023, 24, 592–603 CrossRef CAS PubMed.
- A. Duval, G. Layrac, A. van Zomeren, A. T. Smit, E. Pollet and L. Avérous, ChemSusChem, 2021, 14, 387–397 CrossRef CAS PubMed.
- S. Imman, P. Khongchamnan, W. Wanmolee, N. Laosiripojana, T. Kreetachat, C. Sakulthaew, C. Chokejaroenrat and N. Suriyachai, RSC Adv., 2021, 11, 26773–26784 RSC.
- B. Thomas, S. Geng, M. Sain and K. Oksman, Nanomaterials, 2021, 11, 653 CrossRef CAS PubMed.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03511e |
| This journal is © The Royal Society of Chemistry 2024 |