DOI:
10.1039/D3GC03592A
(Paper)
Green Chem., 2024,
26, 513-519
Visible light-driven highly atom-economical divergent synthesis of substituted fluorenols and cyclopropylcarbaldehydes†
Received
22nd September 2023
, Accepted 16th November 2023
First published on 24th November 2023
Abstract
Herein, we report a visible light-promoted intramolecular regiodivergent tandem radical reaction featuring cyclization/ring expansion followed by contraction to construct functionalized 9-fluorenol and naphthalene-fused cyclopropylcarbaldehyde derivatives. This method involves mild reaction conditions, a wide substrate scope (more than 34 examples), outstanding step efficiency with 100% atom economy, and excellent scalability. Moreover, it requires no external chemical oxidant. In the reaction, the alkyne moiety may act as a radical acceptor, reacting with the carbonyl group that has been activated through a combination of iridium photocatalysis and Lewis acid conditions under blue LED irradiation at room temperature. The generation of fluorenols and cyclopropyl-fused carbaldehydes directly from more stable and simpler conjugated enyne compounds has remained a challenge despite the highly desirable benefits of minimal prefunctionalization and increased operational safety. In the present study, the novelty of the designed protocol was demonstrated via the synthesis of chrysene analogs as well as other late-stage functionalizations.
Introduction
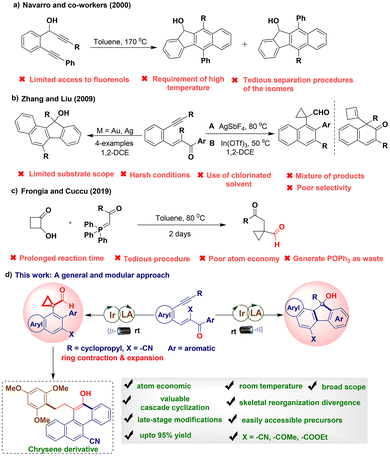
Molecular diversity has served as a fruitful platform in science and technology and is often achieved by assembling chemical building blocks through cascade cyclization using common precursors.1 Moreover, it can facilitate the formation of pharmaceutical and bioactive scaffolds2 and versatile intermediates in organic synthesis3 and provide access to natural products4 under sustainable reaction conditions. The construction of these motifs has drawn considerable attention for use in different reactions, including intermolecular–intramolecular cascade cyclization,5 ring expansion/contraction,6 transition-metal/metal-free catalysis,7 and radical chemistry.8 However, most of these preparative methods involve the multistep synthesis of starting precursors, generation of toxic waste, low atom economy, and harsh reaction conditions, limiting their applicability in synthetic organic chemistry. Hence, developing highly atom-economical reactions without using toxic/hazardous reagents and employing scalable protocols is highly desirable. Visible light-induced photocatalysis has gained attention as an energy-saving platform for developing diverse chemical transformations toward atom-economical synthesis; moreover, it offers an alternative to conventional synthetic methods owing to its low cost, safety, easy availability, and environmental friendliness.9 Additionally, iridium catalysts synergistic with Lewis acid under visible-light catalysis are emerging activation tools for generating highly-active open-shell radical species under mild conditions.10 Furthermore, polycyclic compounds are key building blocks for preparing biologically active natural products and versatile synthetic building blocks in materials science.11 In particular, polyaromatic hydrocarbons, such as fluorene derivatives, play a pivotal role as structural skeletons in optoelectronic material chemistry.12 In this regard, in 2000, Navarro and coworkers reported the thermal cycloaromatization of 1,6-diynes to construct fluorene derivatives (Scheme 1a).13a In 2009, Zhang and coworkers demonstrated a gold-catalyzed cyclization reaction for preparing benzo[a]fluorenes (only four examples). Moreover, they reported the Lewis acid-catalyzed regiodivergent synthesis of naphthalen-2(1H)-ones and cyclopropyl-fused carbaldehyde derivatives (Scheme 1b).13b Recently, Frongia and Cuccu reported a tandem Wittig reaction–ring contraction process for synthesizing cyclopropyl-fused carbaldehydes under conventional conditions (Scheme 1c).13c However, the abovementioned methods often suffer from harsh reaction conditions, environmentally unfriendly protocols, a mixture of regioisomers or a limited substrate scope, very low atom economy, and the generation of by-products. To address these challenges, our and other research groups rationally designed several protocols for enyne precursors bearing different groups,14,15 which are more readily accessible than the acyclic substrates required in conventional methods. Notably, achieving sequential introduction of two most valuable functionalities in a single cascade reaction in one step while enhancing molecular complexity using simple and easily accessible feedstocks, ideally with 100% atom economy, under mild conditions that ensure user-friendly operation represents a highly desirable yet challenging task in synthetic chemistry. Despite recent advances, no radical-based, atom-economical, and sustainable protocol is available for converting internal α,β unsaturated conjugated olefins into medium- to large-sized and related bridged ring libraries via ring contraction and expansion. In particular, access to fluorenols and cyclopropyl-fused carbaldehydes under visible-light photoredox catalysis remains challenging. Hence, in the present study, we focus on the development of visible light-mediated photocatalysis for synthesizing useful heterocyclic and cyclic compounds.13 Herein, we report the realization of a chemodivergent strategy involving 1,5-enyne precursors (1 or 3) for synthesizing fluorenols and cyclopropyl-fused carbaldehydes via carbon-skeleton reorganization in the presence of an Ir(III)-photocatalyst and BF3·OEt2 at room temperature (Scheme 1d). Our results include the following important features: (a) the intramolecular cascade transformations could be conducted at room temperature without the need for special precautions in a single synthetic operation; (b) the obtained products can be used for further synthesis of chrysene analogs in one step; (c) the reaction involves low loading of photocatalysts and Lewis acids under mild reaction conditions without the need for oxidants/additives; (d) the reaction is conducted under mild reaction conditions with a simple reaction setup, easy scale-up, and no by-product formation; and (e) the green metrics evaluation confirms that the developed photochemical process is simple, 100% atom economical, and environmentally benign.
 |
| | Scheme 1 Overview and comparison of previous studies and the present study. | |
Results and discussion
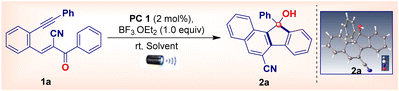
In our initial studies, we chose iridium(III) (PC1) (2 mol%) as the catalytic species. A screening of the reaction conditions for the intramolecular radical cascade protocol involving the model substrate (E)-2-benzoyl-3-(2-(phenylethynyl)phenyl)acrylonitrile 1a demonstrated that the use of 2 mol% of photocatalysts along with BF3·OEt2 (1.0 equiv.) in acetonitrile furnishes the corresponding fluorenol 2a in 81% yield under blue light-emitting diode (LED) (Kessil 40 W; λmax = 462 nm) irradiation for 24 h at room temperature (Table 1, entry 1). Additionally, the structure of 2a was unambiguously characterized using X-ray crystallography.16 A preliminary study was conducted using boron trifluoride etherate as an additive; notably, other additives did not produce higher yields of the desired product 2a (Table 1, entries 2–4). The yield of the product decreased when the additive loading was decreased (Table 1, entries 5 and 6). However, the protocol proved relatively robust regarding the choice of solvents. Substituting acetonitrile with PEG-400, methanol (MeOH), or dimethyl sulfoxide does not provide 2a in similar yields (Table 1, entries 7–9). Moreover, testing other photosensitizers did not reveal a suitable catalyst as the product yields obtained using these catalysts were lower than those obtained using PC1 (Table 1, entries 10 and 11).
Table 1 Optimization studiesa
|
|
| Entry |
Catalyst (X mol%) |
Additives (Y equiv.) |
Solvent |
Yieldb (%) |
|
Reaction conditions: 1a (0.2 mmol), PC 1 = [Ir{dFCF3ppy}2(bpy)]PF6 = Ir(III) (2.0 mol%), BF3·OEt2 (1.0 equiv.), acetonitrile (2.0 mL), 24 h stirring under blue LED irradiation at room temperature.
Isolated yields obtained through the isolation of products via a silica gel column. PC 2 = [Ru(bpy)3](PF6)2; PC 3 = TBADT.
|
| 1a |
None |
None |
Acetonitrile |
81 |
| 2 |
PC 1 (2) |
AlCl3 (1.0) |
Acetonitrile |
19 |
| 3 |
PC 1 (2) |
Zn(OTf)2 (1.0) |
Acetonitrile |
12 |
| 4 |
PC 1 (2) |
TfOH (1.0) |
Acetonitrile |
0 |
| 5 |
PC 1 (2) |
BF3·OEt2 (0.7) |
Acetonitrile |
62 |
| 6 |
PC 1 (2) |
BF3·OEt2 (0.5) |
Acetonitrile |
43 |
| 7 |
PC 1 (2) |
BF3·OEt2 (1.0) |
PEG-400 |
44 |
| 8 |
PC 1 (2) |
BF3·OEt2 (1.0) |
Methanol |
49 |
| 9 |
PC 1 (2) |
BF3·OEt2 (1.0) |
Dimethyl sulfoxide |
13 |
| 10 |
PC 2 instead of PC 1 |
BF3·OEt2 (1.0) |
Acetonitrile |
35 |
| 11 |
PC 3 instead of PC 1 |
BF3·OEt2 (1.0) |
Acetonitrile |
48 |
| 12 |
PC 1 (2) |
— |
Acetonitrile |
0 |
| 13 |
— |
BF3·OEt2 (1.0) |
Acetonitrile |
0 |
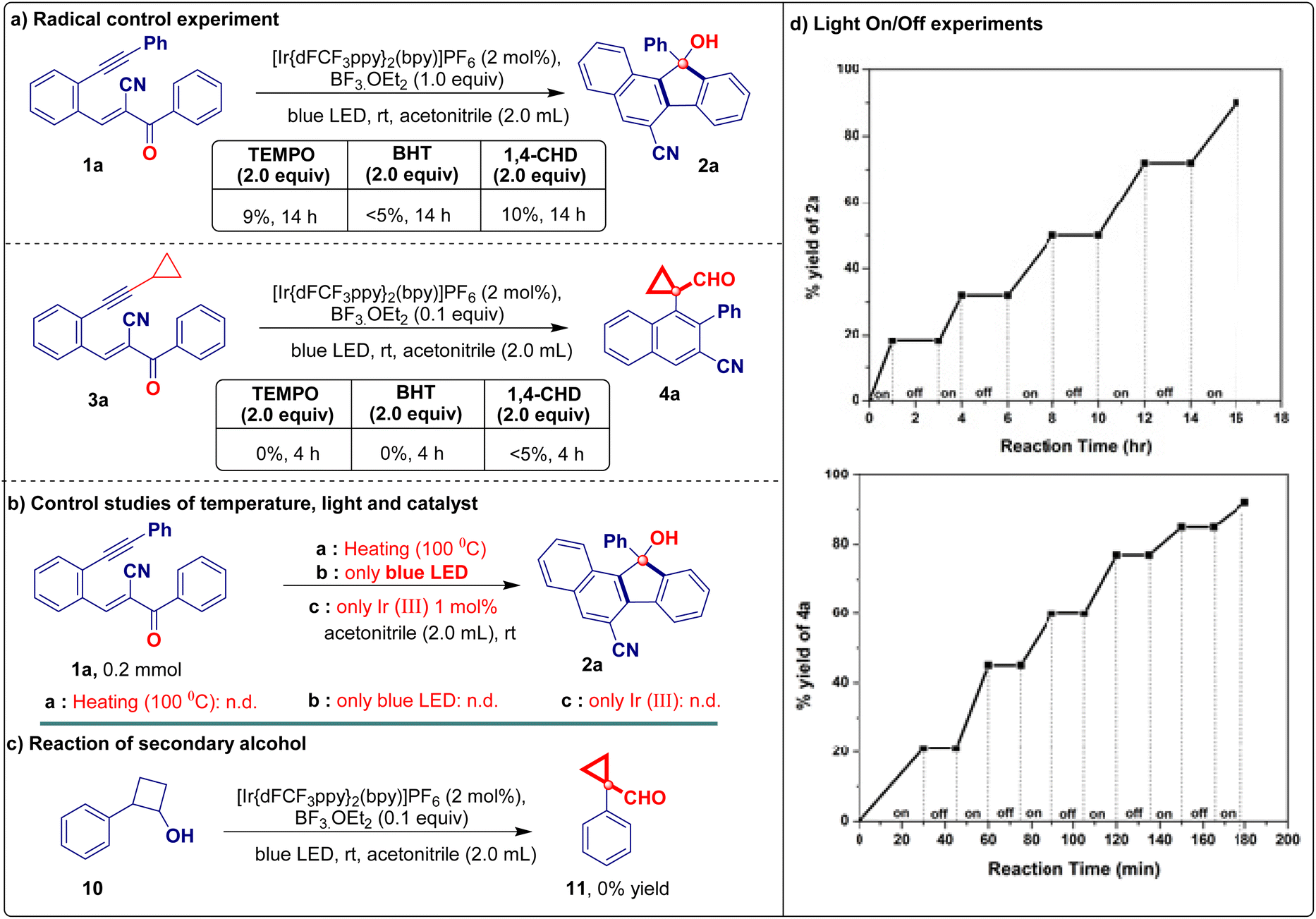
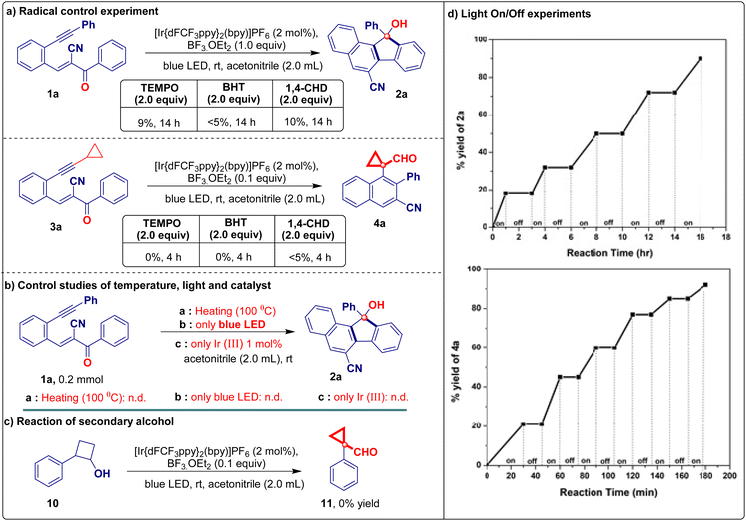
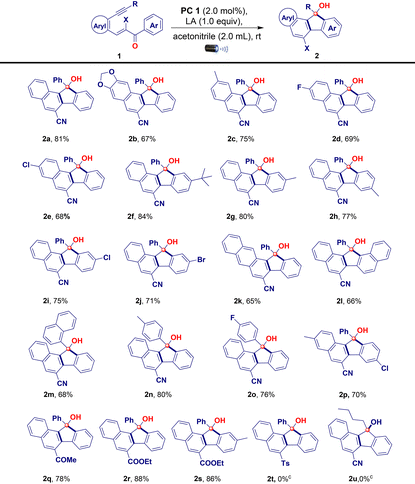
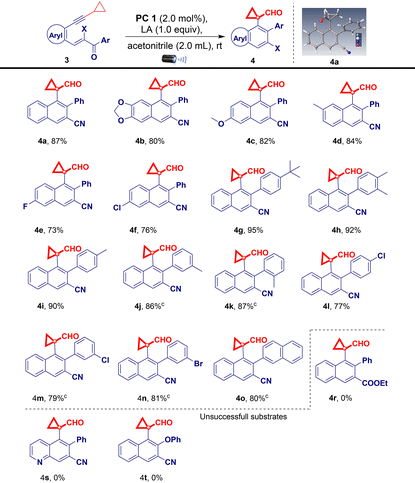
Control experiments demonstrated that both iridium(III) and the additive were indispensable for this reaction (Table 1, entries 12 and 13). To demonstrate the applicability of the developed visible light-promoted protocol, we prepared a library of fluorenols containing diverse functional groups attached to the aromatic/heteroaromatic ring of the acrylonitrile/ester/tosyl species (Table 2). The model reaction afforded product 2a in an isolated yield of 81%. As expected, electron-donating groups located at every position on the phenyl substituents were compatible with the photochemical cascade transformation protocol, providing compounds 2b, 2c, 2f–2h, and 2n in good yields (67%–84%) (Table 2). The tolerance of electron-withdrawing substituents was shown using halogen atoms such as fluorine (2d and 2o), chlorine (2e and 2i), and bromine (2j) (Table 2). The corresponding products were isolated in yields ranging from 68% to 76%. Remarkably, this catalytic system proved compatible with biphenyl (1k), naphthalene (1l and 1m), and disubstituted compounds (1p), all of which afforded the corresponding products in good yields (Table 2). A further evaluation was conducted by replacing the nitrile group with other functionalities in this transformation, aiming to assess the scope of its application. Interestingly, under standard reaction conditions, acryloketone/ester are compatible (1q–1s), producing the corresponding products in excellent yields (Table 2). However, the reaction of the tosyl and long chain alkyne derivatives (1t and 1u) produces a complex mixture of products. To further probe the synthetic utility of the protocol and explore the scope of Lewis acid and iridium photoredox-catalyzed intramolecular ring expansion/contraction cascade cyclizations, experiments performed under slightly modified reaction conditions were evaluated (Table 3, see the ESI† for full details). When we substituted aryl acetylenes with cyclopropyl acetylene (substrate 3), the reaction proceeded via ring expansion and cyclization followed by ring contraction to produce product 4 after the 6-exo-dig pathway (rather than 5-endo-trig vinyl radical trapping). This unique phenomenon enabled us to study the generality of the method under the developed reaction conditions using a series of electron-donating/withdrawing aromatic rings carrying different functionalities (Table 3). A wide variety of electron-donating (3b–3d and 3g–3k) and electron-withdrawing (3e, 3f, and 3l–3n) derivatives were well tolerated under optimized reaction conditions, including a simple derivative (3a), yielding the desired derivatives 4a–4n in 73%–95% yields (Table 3). Notably, the naphthalene compound also provided the corresponding cyclopropyl-fused carbaldehyde derivative 4o in a good yield (80%; Table 3). However, when the acryloester (3r), pyridine (3s), and phenylacrylate (3t) derivatives were subjected to intramolecular ring contraction/expansion, no desired products 4r (or a complex mixture), 4s, and 4t were obtained (Table 3). The structure of product 4a was unambiguously confirmed via X-ray crystallography.16 The 1,5-enyne products of the iridium- and BF3·OEt2-catalyzed cascade reactions serve as versatile building blocks for the synthesis of important decorated functionalities, as demonstrated by the modification of 2a and 4a (Scheme 2). Furthermore, hydroxy-fused benzo[a]fluorenes underwent reactions to form heteroatom-substituted fluorene products. For example, chlorination of fluorene 2a occurred when using 5 N HCl, affording product 5 in a quantitative reaction yield (Scheme 2a). Furthermore, 2a was smoothly methylated using sodium hydride and methyl iodide, furnishing product 6 (Scheme 2b). To demonstrate the synthetic utility of the developed protocol, we performed a large-scale and gram-scale reaction under standard reaction conditions, affording products 2a and 4a in 67% and 73% yields (Scheme 2c and d). The reduction of the carbonyl group of 4a afforded 4-(1-(hydroxymethyl)cyclopropyl)-3-phenyl-2-naphthonitrile (7) in 96% yield (Scheme 2e). Compound 4a was treated with an acid in the presence of an electron-donating arene (1,3,5-trimethoxybenzene) to facilitate the regioselective ring opening of cyclopropane followed by cyclization to afford the chrysene derivative 8 in 83% yield (Scheme 2f). Finally, selective oxidation of the formyl group by oxone in MeOH provided ester 9 in a good yield (Scheme 2g). The structure of compound 5 was confirmed via X-ray crystallography.16 To better understand the reaction mechanism, we performed several control studies. Initially, the effect of different radical scavengers was examined. Under standard reaction conditions (Scheme 3), only trace amounts of products 2a and 4a were produced when substrates 1a and 3a were treated independently with TEMPO, BHT, and 1,4-CHD, respectively. The obtained results clearly reveal that the reaction proceeds via a radical pathway (Scheme 3a). Additionally, we investigated the effect of temperature on the reaction by heating the reaction mixture at 100 °C under standard reaction conditions, revealing that no products were formed (2a) and the starting material was decomposed. Notably, when the reaction was conducted using only iridium catalysts or a blue LED, the starting material was recovered without the formation of 2a (Scheme 3b). We further investigated the reaction using alcohol 2-phenylcyclobutan-1-ol (10) under standard reaction conditions but did not obtain the desired product (11). This is likely because the starting material was not susceptible to the tested reaction conditions (Scheme 3c).
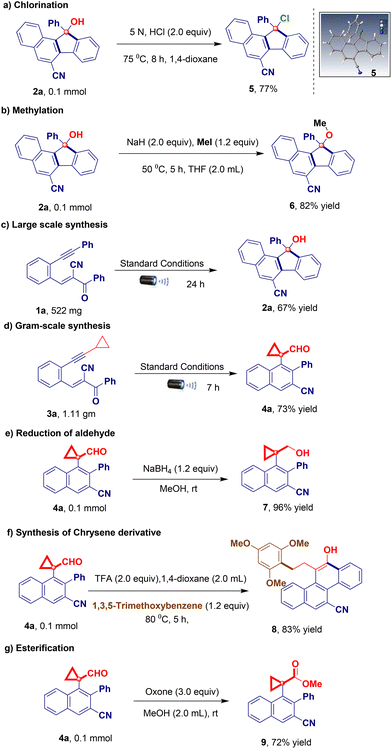 |
| | Scheme 2 Applications of the iridium-catalyzed cascade products 2a and 4a. | |
 |
| | Scheme 3 Mechanistic investigation. (a) Study of the effect of radical scavengers TEMPO, BHT, and 1,4-CHD on the reaction. (b) Control studies based on temperature, light, and the catalyst. (c) Reaction of secondary alcohol. (d) Light on/off experiments involving compounds 2a and 4a. | |
Table 2 Reaction scope for fluorenolsa,b
|
All reactions were conducted at a 0.2 mmol scale. 1a (0.2 mmol), PC 1 = [Ir{dFCF3ppy}2(bpy)]PF6 = Ir(III) (2.0 mol%), LA = BF3·OEt2 (1.0 equiv.), acetonitrile (2.0 mL) under blue LED exposure at room temperature for 24 h.
Isolated yield.
Observed complex mixture.
|
|
Table 3 Reaction scope for cyclopropyl-fused carbaldehydesa,b
|
All reactions were performed at a 0.2 mmol scale. 3 (0.2 mmol), PC 1 = [Ir{dFCF3ppy}2(bpy)]PF6 = Ir(III) (2.0 mol%), LA = BF3·OEt2 (0.1 equiv.), acetonitrile (2.0 mL) under blue LED irradiation at room temperature for 3–6 h.
Isolated yield.
A mixture of isomers was isolated.
|
|
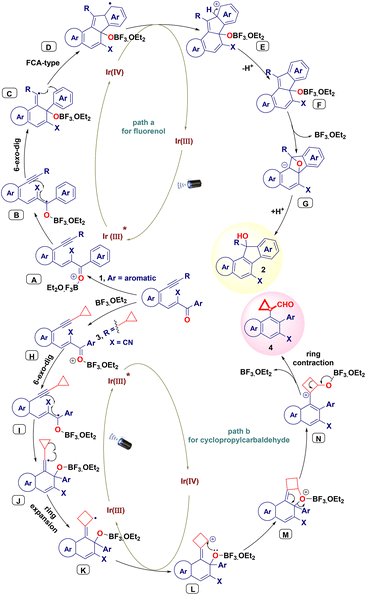
Subsequently, we conducted light on/off experiments using the standard substrate (1a or 3a) to examine the effect of light irradiation. The intramolecular cascade reaction came to a complete halt when the light was turned off and reactivated upon further irradiation (Scheme 3d). This allowed us to control the reaction duration. The experiment revealed that continuous exposure to light irradiation is essential to activating the iridium catalyst system to facilitate the transformation. To verify the greenness and sustainability of the present protocol, a quantitative green chemistry matrix17 for the synthesis of 4a was evaluated (Table 4), exhibiting a 0.42305 E-factor, 100% atom economy, 73% atom efficiency, 100% carbon efficiency, and 73% RME (see the ESI for more details in P. S10†). Based on the abovementioned mechanistic investigation and relevant literature reports,13b,c,18 a plausible working mechanism for the intramolecular reaction was hypothesized (Scheme 4). Initially, the photoredox catalyst Ir(III) is excited by photons under blue LED irradiation to generate an excited state Ir(III)*. Simultaneously, BF3·OEt2 is coordinated to the carbonyl group of substrates 1 or 3 and forms intermediate A or H, respectively. Then, the excited state Ir(III)* transfers electrons to intermediate A to produce ketyl radical intermediate B and Ir(IV). Intermediate B then undergoes intramolecular cyclization with the alkyne via a 6-exo-dig pathway, producing the intermediate vinyl radical species C. Subsequently, the Friedel–Crafts-type C–H functionalization of the benzene ring occurs through 5-endo-trig cyclization to generate benzylic radical intermediate D from six-membered vinyl radical species C. Ir(IV) undergoes single electron reduction to afford cationic intermediate E and releases Ir(III) for the next catalytic cycle. Intermediate F is generated via deprotonative aromatization, leading to the formation of epoxy intermediate G. Finally, the strain release from intermediate G followed by protonation affords product 2 (Scheme 4, path a). In path b, BF3·OEt2 coordinates with the ketyl radical intermediate, generating species I by gaining a single electron from the excited state Ir(III)*. Following this, 6-exo-dig cyclization produces vinyl cyclopropyl radical species J and Ir(IV). Active intermediate J subsequently undergoes ring expansion (K), and the loss of one electron affords cationic intermediate L and iridium(III) species for the next catalytic cycle. Intermediate L leads to the intramolecular cyclization of 6-5-4-membered unstable polycyclic species M, which is subsequently transformed into the cationic intermediate N. Finally, cationic intermediate N undergoes ring contraction to form the naphthalene-fused cyclopropyl carbaldehyde product 4 (Scheme 4, path b).
 |
| | Scheme 4 Plausible mechanism for fluorenols and cyclopropylcarbaldehydes. | |
Table 4 Evaluation of green chemistry metrics for the synthesis of 4a

|
| Reactant 1 |
(E)-2-Benzoyl-3-(2-(cyclopropylethynyl)phenyl)acrylonitrile (3a) |
1.11 g |
0.003733 mol |
FW 297.35 |
| Auxiliary |
Ir(III) |
0.0431 g |
0.00003736 mol |
FW 1155 |
| Product |
4-(1-Formylcyclopropyl)-3-phenyl-2-naphthonitrile (4a) |
0.8103 g |
0.002725 mol |
FW 297.35 |
| Product yield = 73% |
|
E-Factor = 1.11 g + 0.0431 g − 0.8103 g/0.8103 = 0.42305 kg waste per kg product |
| Atom economy = 297.35/297.35 × 100 = 100% |
| Atom efficiency = 73 × 100/100 = 73% |
| Carbon efficiency = 21/21 = 100% |
| Reaction mass efficiency = 0.8103 g/1.11 g × 100 = 73% |
Conclusions
We have disclosed a visible light-induced unprecedented radical cascade cycloaddition protocol for conjugated enynes, facilitating the robust, chemoselective, and divergent synthesis of highly functionalized fluorenols and naphthalene-fused cyclopropylcarbaldehydes in moderate to excellent yields with a wide substrate scope at room temperature under iridium and Lewis acid catalysis. A series of control experiments revealed that the reaction proceeded via radical pathways. Therefore, this novel visible light-induced intramolecular radical reaction supplements the ring expansion/contraction and rearrangement reactions of 1,5-enynes for the synthesis of polycyclic compounds. Furthermore, the method facilitates the synthesis of chrysene derivatives using readily available precursors in overall two steps, thus making it superior to previous literature-reported methods. Hence, the present work facilitates the future development of an efficient method for the synthesis of fluorenols and cyclopropylcarbaldehydes, which remains highly challenging. Moreover, the synthetic utility of the developed protocol was demonstrated through a direct transformation, yielding valuable core structures within complex molecules.
Author contributions
B.S.G. conceived and designed the project, performed and analysed the experimental data, and wrote the manuscript, ESI† and collected experimental data. L.-W.P., J.-H.L., and Y.-C.L. conducted experiments and synthesized starting materials. J.J.W. supervised and contributed to the relevant discussions while writing the manuscript. All authors discussed the results and approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge funding from the National Science and Technology Council (NSTC), Taiwan, and the Centre for Research and Development of Kaohsiung Medical University for 400 MHz NMR and X-ray crystallography analyses.
References
-
(a) H. Kim, T. T. Tung and S. B. Park, Org. Lett., 2013, 15, 5814 CrossRef CAS PubMed;
(b) K. T. Mortensen, T. J. Osberger, T. A. King, H. F. Sore and D. R. Spring, Chem. Rev., 2019, 119(17), 10288 CrossRef CAS PubMed;
(c) B. V. Varun, K. Vaithegi, S. Yi and S. B. Park, Nat. Commun., 2020, 11, 6308 CrossRef CAS PubMed;
(d) J. Sheng, H.-Q. Ni, S.-X. Ni, Y. He, R. Cui, G.-X. Liao, K.-J. Bian, B.-B. Wu and X.-S. Wang, Angew. Chem., Int. Ed., 2021, 60, 15020 CrossRef CAS PubMed;
(e) Y. Xia and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 9836 CrossRef CAS PubMed;
(f) S. L. Schreiber, Science, 2000, 287, 1964 CrossRef CAS PubMed;
(g) P. Prasanna, S. Perumal and J. C. Menéndez, Green Chem., 2013, 15, 1292 RSC;
(h) E. Jiménez-Núñeza and A. M. Echavarren, Green Chem., 2007, 333 Search PubMed.
-
(a) X. Luo, L. Qian, Y. Xiao, Y. Tang, Y. Zhao, X. Wang, L. Gu, Z. Lei, J. Bao, J. Wu, T. He, F. Hu, J. Z. H. Li, W. Zhu, L. Shao, X. Dong, Da. Chen, X. Qian and Y. Yang, Nat. Commun., 2019, 10, 258 CrossRef PubMed;
(b) H. Kim, T. T. Tung and S. B. Park, Org. Lett., 2013, 15, 5814 CrossRef CAS PubMed;
(c) S. Wetzel, R. S. Bon, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 10800 CrossRef CAS PubMed;
(d) M. C. García-González, E. Hernández-Vázquez, R. E. Gordillo-Cruza and L. D. Miranda, Chem. Commun., 2015, 51, 11669 RSC;
(e) Y. Choi, H. Kim and S. B. Park, Chem. Sci., 2019, 10, 569 RSC.
- W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 CrossRef PubMed.
-
(a) C. Cordier, D. Morton, S. Murrison, A. Nelson and C. O'Leary-Steele, Nat. Prod. Rep., 2008, 25, 719 RSC;
(b) S. Shang and D. S. Tan, Curr. Opin. Chem. Biol., 2005, 9, 248 CrossRef CAS PubMed.
-
(a) S. J. Gharpure, Padmaja, V. Prasath and Y. G. Shelke, Org. Lett., 2019, 21, 223 CrossRef CAS PubMed;
(b) S. Sahu, A. Banerjee, S. Kundu, A. Bhattacharyya and M. S. Maji, Org. Biomol. Chem., 2022, 20, 3029 RSC;
(c) Y.-Q. Zhang, X.-Q. Zhu, Y. Xu, H.-Z. Bu, J.-L. Wang, T.-Y. Zhai, J.-M. Zhou and L.-W. Ye, Green Chem., 2019, 21, 3023 RSC;
(d) P. Huang, Z. Yan, J. Ling, P. Li, J. Wang, J. Li, B. Sun and C. Jin, Green Chem., 2023, 25, 3989 RSC;
(e) A. Kumar, P. K. Mishra and A. K. Verma, Chem. Commun., 2023, 59, 7263 RSC.
- J. E. Hill, J. V. Matlock, Q. Lefebvre, K. G. Cooper and J. Clayden, Angew. Chem., Int. Ed., 2018, 57, 5788 CrossRef CAS PubMed and references cited therein.
-
(a) C.-M. Wang, L.-J. Qi, Q. Sun, B. Zhou, Z.-X. Zhang, Z.-F. Shi, S.-C. Lin, X. Lu, L. Gong and L.-W. Ye, Green Chem., 2018, 20, 3271 RSC;
(b) A. Thakur, Manisha, I. Kumar and U. Sharma, Asian J. Org. Chem., 2022, 11, e202100804 CrossRef CAS;
(c) G.-T. Song, N. McConnell, Z.-Z. Chen, X.-F. Yao, J.-H. Huang, J. Lei, H.-K. Lin, B. Frett, H.-y. Li and Z.-G. Xu, Adv. Synth. Catal., 2018, 360, 3655 CrossRef CAS.
-
(a) L. Li, Z.-L. Li, F.-L. Wang, Z. Guo, Y.-F. Cheng, N. Wang, X.-W. Dong, C. Fang, J. Liu, C. Hou, B. Tan and X.-Y. Liu, Nat. Commun., 2016, 7, 13852 CrossRef PubMed;
(b) S. Yuan, J. Zhang, D. Zhang, D. Wei, J. Zuo, J. Song, B. Yu and H.-M. Liu, Org. Lett., 2021, 23, 1445 CrossRef CAS PubMed;
(c) M. K. Lakshman and P. K. Vuram, Chem. Sci., 2017, 8, 5845 RSC.
-
(a) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748 CrossRef CAS PubMed;
(b) V. M. Lechner, M. Nappi, P. J. Deneny, S. Folliet, J. C. K. Chu and M. J. Gaunt, Chem. Rev., 2022, 122, 1752 CrossRef CAS PubMed;
(c) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586 CrossRef CAS PubMed.
-
(a) S.-Y. Hsieh and J. W. Bode, ACS Cent. Sci., 2017, 3, 66 CrossRef CAS PubMed;
(b) M. E. Daub, H. Jung, B. J. Lee, J. Won, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 9543 CrossRef CAS PubMed.
-
(a) S. Gould, N. Tamayo, C. R. Melville and M. C. Cone, J. Am. Chem. Soc., 1994, 116, 2207 CrossRef CAS;
(b) S. Gould and C. Melville, Bioorg. Med. Chem. Lett., 1995, 6, 51 CrossRef;
(c) G. Qabaja and G. B. Jones, J. Org. Chem., 2000, 65, 7187 CrossRef CAS PubMed;
(d) Y.-C. Wu, C.-Y. Duh, S.-K. Wang, K.-S. Chen and T.-H. Yang, J. Nat. Prod., 1990, 53, 1327 CrossRef CAS PubMed.
-
(a) K. D. Belfield, A. R. Morales, B.-S. Kang, J. M. Hales, D. J. Hagan, E. W. Van Stryland, V. M. Chapela and J. Percino, Chem. Mater., 2004, 16, 4634 CrossRef CAS;
(b) P. Laohapaisan, P. Chuangsoongnern, J. Tummatorn, C. Thongsornkleeb and S. Ruchirawat, J. Org. Chem., 2019, 84, 14451 CrossRef CAS PubMed.
-
(a) D. Rodríguez, A. Navarro, L. Castedo, D. Domínguez and C. Saá, Org. Lett., 2000, 2, 1497 CrossRef PubMed;
(b) L. Liu and J. Zhang, Angew. Chem., Int. Ed., 2009, 48, 6093 CrossRef CAS PubMed;
(c) F. Cuccu, L. Serusi, A. Luridiana, F. Secci, P. Caboni, D. J. Aitken and A. Frongia, Org. Lett., 2019, 21, 7755 CrossRef CAS PubMed.
-
(a) B. S. Gore, J.-H. Lin and J.-J. Wang, Green Chem., 2021, 23, 4144 RSC;
(b) B. S. Gore, C.-Y. Kuo and J.-J. Wang, Chem. Commun., 2022, 58, 4087 RSC;
(c) M. R. Mutra, V. S. Kudale, J. Li, W.-H. Tsai and J.-J. Wang, Green Chem., 2020, 22, 2288 RSC;
(d) M. R. Mutra and J.-J. Wang, Nat. Commun., 2022, 13, 2345 CrossRef CAS PubMed;
(e) B. S. Gore, J.-H. Lin and J.-J. Wang, Chem. Commun., 2023, 59, 5878 RSC.
-
(a) Z. Zhuang, C.-L. Li, Y. Xiang, Y.-H. Wang and Z.-X. Yu, Chem. Commun., 2017, 53, 2158 RSC;
(b) R. I. Patel, J. Singh and A. Sharma, ChemCatChem, 2022, 14, e202200260 CrossRef CAS and references cited therein.
- CCDC numbers for compounds 2a, 4a and 5 are 2258232, 2258234, and 2258237, respectively.†.
- See the ESI for details on green chemistry metrics.†.
-
(a) T. P. Yoon, Acc. Chem. Res., 2016, 49, 2307 CrossRef CAS PubMed;
(b) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886 CrossRef CAS PubMed;
(c) Y. Zhou, P. Zhao, X.-X. Yu, X. Geng, C. Wang, C. Huang, H. Yang, Z.-Y. Zhao and A.-X. Wu, Org. Lett., 2020, 22, 8359 CrossRef CAS PubMed;
(d) K. Livingstone, K. Siebold, S. Meyer, V. Martín-Heras, C. G. Daniliuc and R. Gilmour, ACS Catal., 2022, 12, 14507 CrossRef CAS PubMed;
(e) Y. Sun, X. Huang, X. Li, F. Luo, L. Zhang, M. Chen, S. Zheng and B. Peng, Adv. Synth. Catal., 2018, 360, 1082 CrossRef CAS;
(f) D. Moi, M. C. Cabua, V. Velichko, A. Cocco, A. Chiappone, R. Mocci, S. Porcu, M. Piras, S. Bianco, F. Pesciaioli and F. Secci, Molecules, 2022, 27, 7943 CrossRef CAS PubMed and references cited therein;
(g) A. N. Baumann, F. Schüppel, M. Eisold, A. Kreppel, R. d. Vivie-Riedle and D. Didier, J. Org. Chem., 2018, 83, 4905 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Lin-Wei
Pan‡
a,
Jun-Hao
Lin‡
a,
Yi-Chi
Luo
a,
Lin-Wei
Pan‡
a,
Jun-Hao
Lin‡
a,
Yi-Chi
Luo