
The synergy of in situ-generated Ni0 and Ni2P to enhance CO adsorption and protonation for selective CH4 production from photocatalytic CO2 reduction†
Xuemei
Liu‡
a,
Chaonan
Cui‡
b,
Shuoshuo
Wei
a,
Jinyu
Han
a,
Xinli
Zhu
 a,
Qingfeng
Ge
*c and
Hua
Wang
*a
a,
Qingfeng
Ge
*c and
Hua
Wang
*a
aCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Key Laboratory for Green Chemical Technology of the Ministry of Education, School of Chemical Engineering and Technology, Tianjin University, Tianjin, China. E-mail: tjuwanghua@tju.edu.cn
bState Key Laboratory for Structural Chemistry of Unstable and Stable Species, Institute of Chemistry, Chinese Academy of Sciences, Beijing, China
cDepartment of Chemistry and Biochemistry, Southern Illinois University, Carbondale, IL, 62901, USA. E-mail: qge@chem.siu.edu
First published on 8th December 2023
Abstract
The selective photocatalytic reduction of CO2 to CH4 remains a challenge because there is a need for not only strong adsorption sites for the intermediates, but also optimal proton-feeding sites on the photocatalyst surface. Herein, a synergistic dual-site function between in situ-generated Ni0 and Ni2P on carbon nitride nanosheets (CN) for photocatalytic reduction of CO2 to CH4 is presented. The highest CH4 production rate of 69.03 μmol g−1 h−1 is achieved on Ni2P/CN-0.5 in an aqueous suspension. Detailed analyses show that the promotion of CH4 is closely correlated with the formation of Ni0 sites due to light irradiation, which is confirmed by tracking the compositions of Ni2P/CN-0.5 through XPS and HRTEM characterization. Density functional theory calculations have been combined with CO-TPD and in situ FTIR spectra to reveal the synergy between in situ-generated Ni0 sites and Ni2P. It shows that the Ni0 sites can stabilize the key intermediate *CO, while the Ni0–Ni2P interface can promote *H transfer from Ni2P to Ni0. Therefore, the CO intermediates are rapidly protonated to form CHO* instead of being desorbed from the surface to produce CO, and subsequently CHO* will be converted into CH4. This work demonstrates a new strategy of designing highly efficient photocatalysts with synergistic catalytic sites for CO2 conversion to hydrocarbons.
1. Introduction
Converting CO2 into carbon-neutral fuels such as CH4 is an extremely effective solution to the energy crisis and environmental problems. Among various approaches, photocatalysis could convert CO2 into value-added chemicals using renewable solar energy without resorting to a secondary energy storage medium.1,2 However, converting CO2 to CH4 is a complex electrochemical process, requiring 8 proton-coupled electron transfer processes (e− + H+).3–5 Along the way, the two-electron reduction products CO and HCOOH may desorb from the catalyst instead of further hydrogenating to CH4, resulting in low methane selectivity.6 Therefore, an efficient photocatalyst for selective CH4 formation from CO2 reduction should possess excellent photochemical properties, enriched electrons on active sites with the optimal proton-feeding and adsorption of intermediates.7 So far, developing such a highly efficient photocatalyst to selectively reduce CO2 to CH4 remains a great challenge. Carbon nitride (CN) is an environmentally friendly semiconductor with excellent photochemical properties and stability.8 In addition, the introduction of a metal-based co-catalyst to CN has been approved as one of the effective strategies to improve the photocatalytic activity. The additional metal components on CN not only inhibit the recombination of photogenerated electrons and holes, but also provide the active sites for the adsorption and activation of CO2, as well as the subsequent hydrogenation steps.9Transition metal phosphides (TMPs), owing to their lower cost, unique electronic structure, and excellent electrical conductivity, have been widely regarded as promising alternatives to Pt for hydrogen production from photocatalytic water splitting.10,11 Recently, TMPs were also attempted for CO2 reduction. Wang et al. constructed NiCoOP NPs@MHCF catalysts; the optimized trimetallic oxyphosphide catalysts exhibited considerable activity in photosensitive CO2 reduction with a CO evolution rate.12 Calvinho et al. prepared NixPy for the electrocatalytic reduction of CO2 at low potentials using a solid-state method, and their results indicated that NixPy, especially Ni2P, has an excellent activity in the electrocatalytic reduction of CO2 to hydrocarbon products.13 Li et al. found that the co-modification with Ni2P and NiO of CN caused the increase of CH4 yield.14 These studies demonstrated that Ni2P could act as a proton-feeding site to promote the kinetics of the protonation step – the key step for CH4 production in the photocatalytic reduction of CO2 to CH4.15,16
In this work, we develop a dual active site photocatalyst Ni0–Ni2P/CN boosting CH4 formation from the photocatalytic reduction of CO2. Firstly, Ni2P/CN photocatalysts were prepared through a hydrothermal reaction using red phosphorus as the P source. Ni0 sites were in situ-generated under light irradiation, which exhibited a close link with the enhanced CH4 yield. The conditions of introducing Ni2P to CN have been optimized to maximize the CH4 production. It is demonstrated that these Ni0 sites work synergistically with Ni2P sites to promote the formation of CH4. In situ FTIR spectra, CO-TPD and theoretical calculations were used to gain insights into the reaction mechanism of CO2 reduction on the Ni0–Ni2P/CN photocatalyst.
2. Experimental and computational methods
2.1 Synthesis of CN and Ni2P/CN-x
Carbon nitride was synthesized by the reported method.17 Specifically, 10.0 g of urea was dissolved in 20 mL of H2O. After the temperature stabilized to room temperature, 1.0 g of ammonium oxalate, which generates NH3 and CO2 during thermal decomposition, was used as a pore builder in this system to obtain the porous g-C3N4, which was added to the above solution while stirring. The mixed solution was stirred at room temperature for 1 h and then raised to 80 °C for 16 h to obtain a white precursor. The precursors were ground and transferred to a 50 ml ceramic crucible and calcined in a muffle furnace at a heating rate of 5 °C min−1 for 2 h at 550 °C to obtain orange carbon nitride nanosheets, named CN.Ni2P/CN-x samples were synthesized by a simple hydrothermal method. Typically, 0.3 g of CN and 0.5 mmol of Ni(NO3)2·6H2O were dispersed in 65 mL of H2O by sonication for 1 h. 5.0 mmol of P was added to the above solution and stirred for 1 h at room temperature. Next, the resulting suspension was loaded into a 100 mL Teflon autoclave and maintained at 140 °C for 10 h. The precipitates were separated by centrifugation and washed several times with water and ethanol, and then vacuum dried at 60 °C for 12 h. The resulting gray solid was labeled as Ni2P/CN-0.5. By changing the amount of Ni(NO3)2·6H2O and P in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, other Ni2P/CN-x samples were obtained, where x represents the amount of Ni(NO3)2·6H2O (mmol) used. The preparation process is shown in Fig. 1a.
:10, other Ni2P/CN-x samples were obtained, where x represents the amount of Ni(NO3)2·6H2O (mmol) used. The preparation process is shown in Fig. 1a.
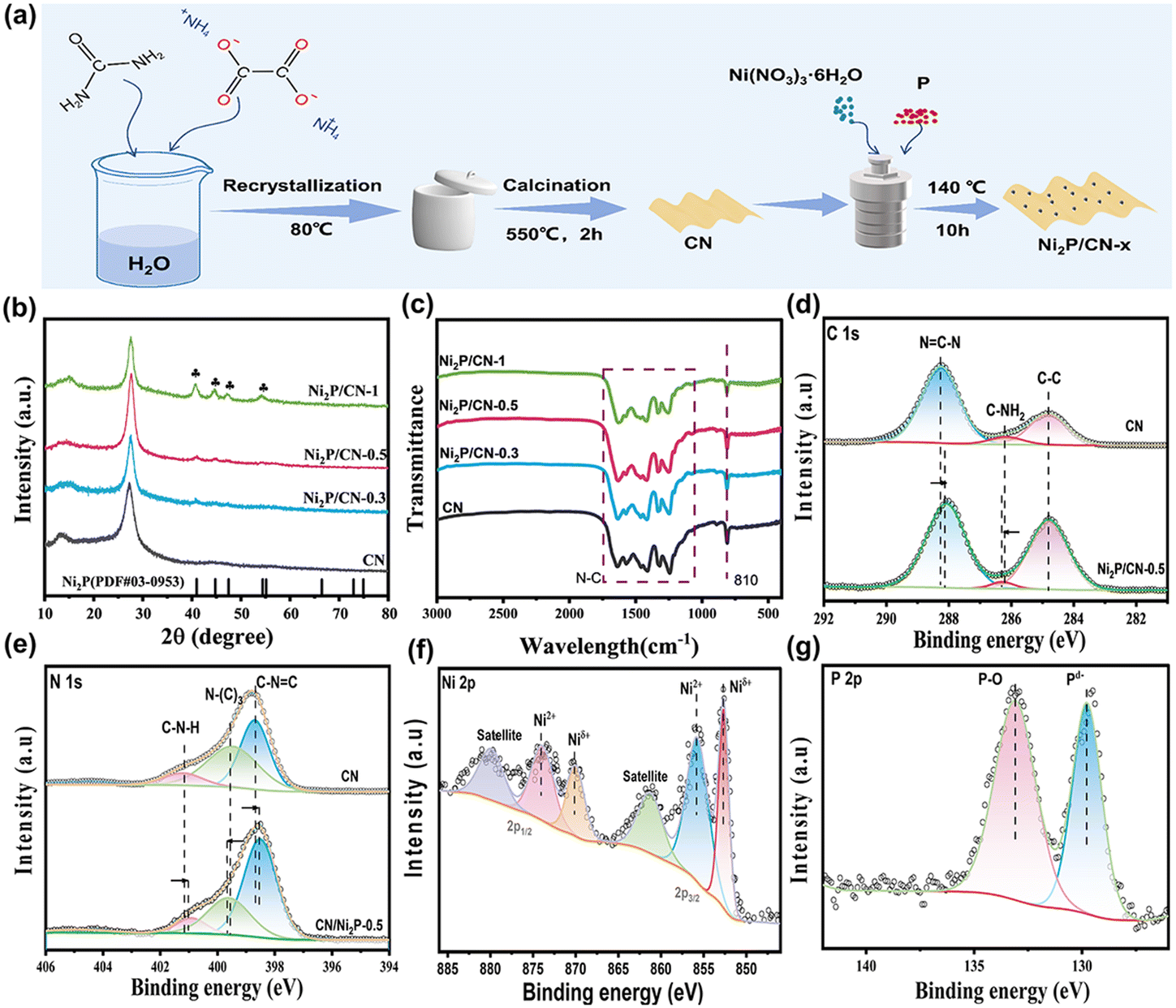 | ||
| Fig. 1 (a) Schematic diagram of the synthesis process of Ni2P/CN-x; (b) XRD patterns; and (c) FTIR spectra of CN and Ni2P/CN-x; and (d–g) XPS spectra of the elements in CN and Ni2P/CN-0.5. | ||
2.2 Material characterization
An X-ray diffractometer (XRD, D8-Focus, Bruker AXS) equipped with a Cu Kα (λ = 1.54056 Å) radiation source was used to record the crystalline structure of the samples with a scanning speed of 5° min−1 and a scanning range (2θ) of 10–80°. Fourier transform infrared spectroscopy (FTIR, Magna 670, Nicolet) was used to detect the functional group of the samples, using KBr as a reference. The sample morphology was examined using a field emission scanning electron microscope (SEM, S-4800, Hitachi). A 200 kV field emission transmission electron microscope (TEM, JEM-2100F, Japan Electronics) was used to obtain scanning transmission electron microscopy (STEM) and high-resolution transmission electron microscopy (HRTEM) images and selected area energy dispersive X-ray spectroscopy (EDS) spectra of the samples. An X-ray photoelectron spectrometer (XPS, ESCALAB Xi+, Thermo Fisher Scientific) equipped with Al Kα radiation was used to analyze the surface chemical composition of the samples, and the binding energy was calibrated by carbon C 1s (284.8 eV). An ultraviolet-visible diffuse reflectance spectrophotometer (UV-vis DRS, T2600, Youke) was used to acquire the absorption spectra of the samples. The photoluminescence (PL) spectra of the samples were obtained using a steady-state fluorescence spectrometer (Fluorolog 3-21, Jobin Yvon Inc.) with an excitation wavelength of 420 nm. Nitrogen adsorption–desorption isotherms were obtained with a nitrogen physisorption instrument (Tristar 3000 Micromeritics), and CO2 adsorption isotherms were obtained with a specific surface and pore size analyzer (BELSORP-max, MicrotracBEL, Japan) at 25 °C. All samples were vacuum pretreated at 200 °C for 3 hours prior to the measurement.2.3 Performance of photocatalytic CO2 reduction
The photocatalytic CO2 reduction reaction was carried out in a 250 mL Pyrex reactor with a UV-vis light source (300 W Xe lamp), and the reaction temperature was maintained at 15 °C using a cooling water circulator. Specifically, 25 mg of the as-prepared sample were dispersed into a mixed solution containing 20 mL of 0.1 M NaOH and 10 mL of TEOA. Prior to the illumination, the reaction system was purged with high purity CO2 for 30 min to expel the air, while allowing the reaction solution to be saturated with CO2. The reaction was carried out under UV-vis light (320 nm < λ < 780 nm, 200 mW cm−2) with continuous stirring. During the reaction, 1 mL of gas was extracted with a syringe at 40 min intervals and the products were analyzed by gas chromatography (GC 9790 II Plus, Fuli), and the results were averaged from three repeating tests (more details can be seen in the ESI†). A schematic diagram of the photocatalytic reaction system is shown in Fig. S1.†2.4 Density functional theory (DFT) calculations
DFT calculations were performed using the spin-polarized Vienna Ab initio Simulation Package (VASP) with projector-augmented wave pseudopotential (PAW).18 The generalized gradient approximation (GGA)19 in the Perdew–Burke–Ernzerhof (PBE)20 form was adopted, with a cutoff energy of 400 eV for the plane-wave basis set. The ionic convergence limit was set to 0.02 eV Å−1, while the electronic convergence limit was set to 10–5 eV. The optimization of gas molecules was performed in a cubic box of 20 Å in each direction and using a single k point (Γ point) for the Brillouin-zone integration. For periodic models, a vacuum of 20 Å in the Z direction was set to prevent the interaction between two periodic units. A k-point grid of 3 × 3 × 1 was set for Brillouin-zone integration of the catalysts.The Gibbs free energy change (ΔG) of each elementary step was calculated by using the following equation:
| ΔG = ΔE + ΔEZPE − TΔS, | (1) |
3 Results and discussion
3.1 Crystal structure and chemical composition
The phase structures of the as-prepared CN and Ni2P/CN-x were revealed from XRD spectra, and the results are shown in Fig. 1b.All samples presented the CN characteristic diffraction peaks at 13.7° and 27.2° attributed to the periodic in-plane tri-s-triazine motif stacking and the interlayer stacking of CN.27 In addition, Ni2P/CN-x samples display the peaks at 40.8°, 44.6°, 47.3° and 54.2°, ascribed to the (111), (201), (210) and (300) crystal planes of the Ni2P hexagonal crystal structure (PDF #03-0953), respectively. The intensity of the diffraction peaks attributed to Ni2P increases with the amount of Ni and P precursors, indicating the successful introduction of Ni2P into the substrate CN.28,29 From the FTIR spectra (Fig. 1c), the featured absorption peaks of CN can be observed. The peak at 810 cm−1 corresponds to the respiratory vibration peak of the triazine ring in CN, and the band at 1250–1650 cm−1 corresponds to the stretching mode of the CN heterocycle, indicating that the introduction of Ni2P does not alter the molecular structure of CN. The above results indicated that the composited photocatalysts Ni2P/CN were successfully prepared.
To investigate the surface composition and chemical states of the samples, XPS spectra of CN and Ni2P/CN-0.5 were obtained (Fig. 1d–g). For pure CN, the C 1s spectrum displays three distinct peaks at 284.80, 286.20, and 288.27 eV (Fig. 1d), which are assigned to the surface adventitious carbon, C–NH2 and sp2 hybridized carbon (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N), respectively.30 The N 1s spectrum (Fig. 1e) exhibits three peaks at 398.68, 399.54, and 401.17 eV, corresponding to C–NC, N–(C)3, and C–NH in CN.31,32 By comparison, the corresponding peaks for Ni2P/CN-0.5 slightly shifted, indicating a strong interaction between Ni2P and CN.33 The Ni 2p spectrum (Fig. 1f) was spin split into Ni 2p3/2 and Ni 2p1/2. In the Ni 2p3/2 region, three peaks appeared at 852.65, 855.89, and 861.40 eV, corresponding to the Niδ+ species in Ni2P, the Ni2+ species and the satellite peak, respectively. In the Ni 2p1/2 region, there were three peaks, at 870.10, 874.00, and 880.00 eV, which could be attributed to Niδ+, Ni2+, and satellite peaks, respectively.34 In the P 2p spectrum (Fig. 1g), the peak at 129.78 eV was attributed to the Pδ− species in Ni2P and the peak at 133.11 eV corresponded to the formation of P–O bonds.28,35 Note that the reason for the presence of Ni2+ and P–O bonds is twofold: the surface oxidation of Ni2P and the formation of partially hydrated surface phosphates (e.g. Ni3(PO4)2) using a hydrothermal phosphating method.13 Therefore, the XPS results confirm the successful loading of Ni2P on CN and the strong electronic interaction between them.36,37
C–N), respectively.30 The N 1s spectrum (Fig. 1e) exhibits three peaks at 398.68, 399.54, and 401.17 eV, corresponding to C–NC, N–(C)3, and C–NH in CN.31,32 By comparison, the corresponding peaks for Ni2P/CN-0.5 slightly shifted, indicating a strong interaction between Ni2P and CN.33 The Ni 2p spectrum (Fig. 1f) was spin split into Ni 2p3/2 and Ni 2p1/2. In the Ni 2p3/2 region, three peaks appeared at 852.65, 855.89, and 861.40 eV, corresponding to the Niδ+ species in Ni2P, the Ni2+ species and the satellite peak, respectively. In the Ni 2p1/2 region, there were three peaks, at 870.10, 874.00, and 880.00 eV, which could be attributed to Niδ+, Ni2+, and satellite peaks, respectively.34 In the P 2p spectrum (Fig. 1g), the peak at 129.78 eV was attributed to the Pδ− species in Ni2P and the peak at 133.11 eV corresponded to the formation of P–O bonds.28,35 Note that the reason for the presence of Ni2+ and P–O bonds is twofold: the surface oxidation of Ni2P and the formation of partially hydrated surface phosphates (e.g. Ni3(PO4)2) using a hydrothermal phosphating method.13 Therefore, the XPS results confirm the successful loading of Ni2P on CN and the strong electronic interaction between them.36,37
3.2 Morphology and microstructure
SEM and TEM images provide the morphology and the microstructure of the samples (Fig. 2). Fig. 2a and b indicate that CN has a thin sheet structure with curled edges. Ni2P/CN-0.5 maintains the nanosheet morphology (Fig. 2c) and the edges are further curled due to the hydrothermal treatment.38 The TEM and STEM images of Ni2P/CN-0.5 (Fig. 2d and e) demonstrate that Ni2P nanoparticles (the bright part) are distributed on the CN sheet.39,40 In the HRTEM image (Fig. 2f), the lattice spacing of 0.221 nm is indexed to the (111) crystal plane of Ni2P.41,42 The EDS maps of Ni2P/CN-0.5 are shown in Fig. 2g, indicating the presence of C, N, Ni, and P elements.27 The above results confirmed that Ni2P nanoparticles are successfully dispersed on the CN nanosheets. | ||
| Fig. 2 (a and b) SEM and TEM of CN, and (c–f) the SEM, TEM, STEM and HRTEM image of Ni2P/CN-0.5. (g) The STEM-EDS elemental mapping of Ni2P/CN-0.5. | ||
3.3 Optical and electrical properties
The UV-Vis diffuse reflectance spectra of the as-prepared samples are shown in Fig. 3a. Compared to CN, the increased visible light absorption of Ni2P/CN-x is attributed to the intrinsic absorption of Ni2P.43 There is a slight red shift in the absorption edge of Ni2P/CN-x, an indication of narrowing of the band gaps (Eg) of Ni2P/CN-x from that of CN.44 The band gaps of CN, Ni2P/CN-0.3, Ni2P/CN-0.5 and Ni2P/CN-1 were calculated to be 2.60 eV, 2.54 eV, 2.50 eV and 2.46 eV, according to the plots of (αhν)x (x = 2) vs. band energy (hν).45 As shown in Fig. S2,† the Mott–Schottky (MS) curves exhibit a positive slope, a characteristic feature of the n-type semiconductor.46 The flat-band potential (Vfb) was determined to be −1.15 and −1.07 V (vs. NHE, pH = 7) of CN and Ni2P/CN-0.5. According to the equation ECB = Vfb − 0.2 V, the conduction band potentials (ECB) of CN and Ni2P/CN-0.5 were −1.35 and −1.27 V (vs. NHE, pH = 7), respectively.47 The valence band potential (EVB) could then be determined based on the equation EVB = Eg − ECB.48 A summary of these results led to the alignment of the bands of CN and Ni2P/CN-0.5, as shown in Fig. 3b. Clearly, both CN and Ni2P/CN-0.5 could drive both CO2 reduction and H2O oxidation reactions.37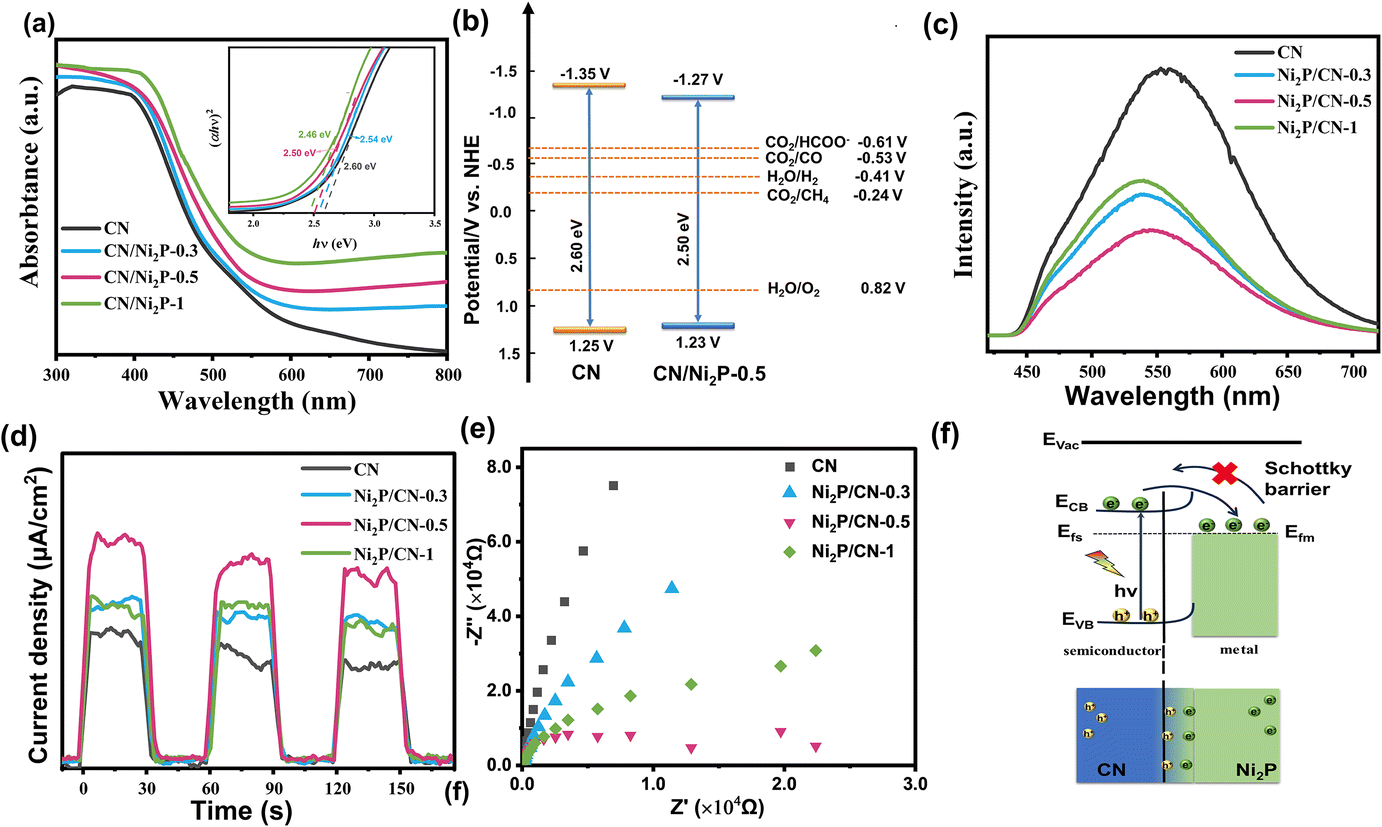 | ||
| Fig. 3 (a) UV–vis absorption spectra and band gaps of the as-prepared samples (α, h and ν represent the absorption coefficient, Planck constant, and light frequency, respectively). (b) Band positions of CN and Ni2P/CN-0.5. (c) PL spectra; (d) photocurrent response curves; and (e) EIS plots of CN and Ni2P/CN-x. (f) The diagram of the Schottky junction and electron transfer path between Ni2P and g-C3N4. | ||
The PL spectra, photocurrent density profile and EIS plots were used to evaluate the ability of the separation and transport of photocarriers.49 As shown in Fig. 3c, the Ni2P/CN-x composites all show a significant decrease in emission intensity compared to CN, and Ni2P/CN-0.5 exhibits the lowest emission intensity. The quenching of fluorescence indicates that the complexation of photogenerated electron–hole pairs is effectively suppressed following the successful loading of Ni2P. In addition, the change in the energy distribution of Ni2P/CN-x surface states caused a slight blue shift of the PL peaks.50,51
As shown in Fig. 3d, all Ni2P/CN composites exhibit a higher photocurrent than CN, demonstrating an effective transfer of photogenerated electrons to the surface. The impedance arc radius of the Ni2P/CN composites was smaller compared to that of pure CN, indicating less resistance to the charge transport in Ni2P/CN than in CN (Fig. 3f), with Ni2P/CN-0.5 showing the smallest radius.52 The above results demonstrate that the Ni2P/CN-0.5 photocatalyst exhibits the most efficient separation of the photogenerated electrons and holes, which is essential to facilitate the photocatalytic CO2 reduction.
Due to the metallic properties and excellent electrical conductivity of Ni2P, the enhanced separation and transfer of photogenerated electrons and holes in the composite catalyst can be attributed to the formation of a Schottky junction between CN and Ni2P (Fig. 3f).53 Previous studies showed that the Fermi level of CN was higher than that of Ni2P.54 When CN and Ni2P are in contact, the difference in Fermi levels drives the electron transfer from CN to Ni2P until the equilibrium is reached and a Schottky junction is formed. The existence of the Schottky barrier prevents the electrons to flow back to CN, effectively facilitating the separation and transfer of the photogenerated charge carriers, as illustrated in Fig. 3f. These findings are demonstrated by the density of states (DOS) analysis of C3N4 and Ni2P (Fig. S3 and S4†). As a result, Ni2P acted as an electron trap and enriched the active sites with a sufficient number of electrons, which drives the reduction of CO2 to CH4.
3.4 Photocatalytic performance for CO2 reduction
The photocatalytic performance of the as-prepared photocatalysts for CO2 reduction was evaluated in an aqueous solution of NaOH and TEOA under UV-vis irradiation for 200 min. During the reaction, the gas was sampled at an interval of 40 min and detected by the GC method, and the GC chromatograms of gas produced during the photocatalytic reduction are shown in Fig. S5.† The detected gas products included CH4 and CO from CO2 reduction as well as H2 from the competitive H2O reduction. As shown in Fig. 4a–c, CN exhibits the lowest yields of CH4, H2 and CO. All Ni2P/CN-x photocatalysts show sharply enhanced CH4 and H2 yields, but a very low CO yield, suggesting that loading Ni2P has a positive effect on the adsorption and further hydrogenation of CO. Notably, for the Ni2P/CN-x photocatalysts, the CH4 production displays a different trend from H2 with increasing Ni2P loading. As shown in Fig. 4d, the CH4 evolution showed a peak yield of 69.03 μmol g−1 h−1 on Ni2P/CN-0.5, wherever H2 yields increase monotonously to the highest on Ni2P/CN-1. Accordingly, Ni2P/CN-0.5 provides the highest selectivity of CH4. It can be explained that the number of reduced protons increases as the Ni2P loading increases, and when the formation of the reduced protons is beyond their consumption by the hydrogenation of CO to CH4, the excessive H will recombine and desorb as H2. Therefore, the loading of Ni2P has to be optimal to maximize the CH4 selectivity from CO2 reduction. Moreover, by a contrastive analysis of the yield-time profiles before and after the irradiation time of 40 min in Fig. 4a and c, it is observed that there is a sharp change of yield for either CH4 (from a slow yield increase to a fast increase) or H2 (from a fast yield increase to a slow increase). This interesting finding will be discussed in the next section with regard to the identification of active sites and the mechanism. | ||
| Fig. 4 The product yield profiles with the reaction time of (a) CH4, (b) CO and (c) H2; (d) the average evolution rates of CH4, CO and H2 on CN and Ni2P/CN-x within a reaction time of 4 hours; (e) the results of the condition controlled experiments; and (f) the gas evolution rates of the reference samples. | ||
Control experiments were conducted on the photocatalyst Ni2P/CN-0.5 to confirm the source of the resulting CH4 (Fig. 4e). Either under dark conditions or in the absence of Ni2P/CN-0.5, no gaseous product was detected, indicating that light irradiation and the photocatalyst Ni2P/CN-0.5 are essential for the reactions. Only hydrogen was detected if CO2 was not introduced into the reaction system. Therefore, CO2 was the source of CH4 and CO in the products. In addition, without NaOH, the yields of CH4 and CO decreased significantly, whereas the yield of H2 increased. The presence of NaOH increased the solubility of CO2 from the neutral aqueous condition (pH of the different reaction solution as shown in Table S1†).55 The role of the sacrificial agent (TEOA) was also tested. In the absence of TEOA, only a small quantity of products was detected due to the high recombination rate of the photogenerated charge carriers when the holes were not consumed efficiently. The test under the visible light irradiation (>420 nm) showed that the photocatalytic activity was substantially lower than that under the light in the wavelength range of UV-vis (320–780 nm). This demonstrated that the energy from the visible light was not enough to generate the sufficient photocharge carriers. The above results indicated that the CH4 and CO products originated from the photocatalytic reduction of CO2 on the photocatalyst Ni2P/CN-0.5 under UV-vis light irradiation.
The additional reference samples were used to eliminate or quantify the effect of the other possible contributions to the performance of the Ni2P/CN-0.5 catalyst.56 As shown in Fig. 4f, hydrothermal treatment of CN nanosheets (Hydro-CN) alone does not improve the catalytic performance for either CH4 or H2 production notably.57 When adding Ni(NO3)2 only for modifying CN, the obtained sample Ni/CN provided a modest increase in CH4 production. In contrast, H2 became the dominant product on P/CN, with P being the only promoter. The XRD spectra of both Ni/CN and P/CN in Fig. S6a† demonstrated that the diffraction peaks assigned to the CN nanosheet structure remained well. The very different product distribution with a single component promoter (Ni or P) clearly showed that the Ni species are key to the formation of CH4. These results also confirmed that the successful loading of Ni2P on the CN surface was the main reason for the high photocatalytic activity of the composite catalysts. And that P or Ni(NO)3 alone with CN could not generate high-performance composite catalysts under hydrothermal conditions. Further analysis focuses on understanding the nature as well as the role of the surface species of the Ni2P/CN-0.5 photocatalyst for catalytic CO2 reduction.
3.5 Identification of the active sites promoting CH4 formation: in situ-generated Ni0 induced by light irradiation
The average rate of CH4 and H2 in every interval and the selectivity of CH4 along with irradiation time on Ni2P/CN-0.5 are shown in Fig. 5a. Ni/CN showed a period of activation, but not as noticeably as Ni2P/CN (Fig. S6b†), suggesting that the new active sites should be relevant to Ni species. In order to probe the new active site due to light irradiation, the XPS spectra of the fresh Ni2P/CN-0.5 and the light induced generated one (Ni0–Ni2P/CN-0.5) were compared, as shown in Fig. S7† and Fig. 5c. As displayed in Fig. S7a and b,† in comparison with the fresh Ni2P/CN-0.5, Ni0–Ni2P/CN-0.5 showed positive shifts of the binding energy for both the peak of sp2 hybridized C (NC–N) in the C1s spectrum and the peak of CN in the N 1s spectrum. It is indicative that CN nanosheets act as the electron donor in the composite Ni0–Ni2P/CN-0.5 under light irradiation.58 In addition, note that the peaks corresponding to the P–O bond in the P 2p spectra (Fig. S7c†) decreased and the binding energy negatively shifted. Importantly, the notably different Ni 2p spectra of Ni0–Ni2P/CN-0.5 from Ni2P/CN-0.5 can be observed (Fig. 5c). For Ni0–Ni2P/CN-0.5, the peaks ascribed to Ni2+ decreased significantly, and a new pair of peaks at 850.2 and 867.6 eV corresponding to the metallic Ni (Ni0) appeared. Meanwhile, the formation of Ni0 in Ni0–Ni2P/CN-0.5 was again observed from the HRTEM image, which exhibited lattice fringes with a spacing of 0.176 nm and 0.221 nm assignable to the Ni (200) and Ni2P (111) facets, respectively.29 The combined XPS and HRTEM results clearly confirm the in situ formation of the Ni0 sites. Considering both the principle of the hydrothermal phosphating method and the decreased peak intensity of Ni2+, P–O bond in the XPS spectra of Ni0–Ni2P/CN-0.5, these Ni0 sites are attributed to the reduction of the hydrated surface nickel phosphorylate (e.g. Ni3(PO4)2) under light irradiation. The presence of Ni0 sites is believed to increase the CH4 yield as Ni0 plays an active role in the methanation of CO.59,60 Consequently, the Ni0–Ni2P/CN photocatalytic system is in situ generated by light irradiation. The coexisting active sites of Ni2P and Ni0 synergistically work to enhance the production of CH4.61,62
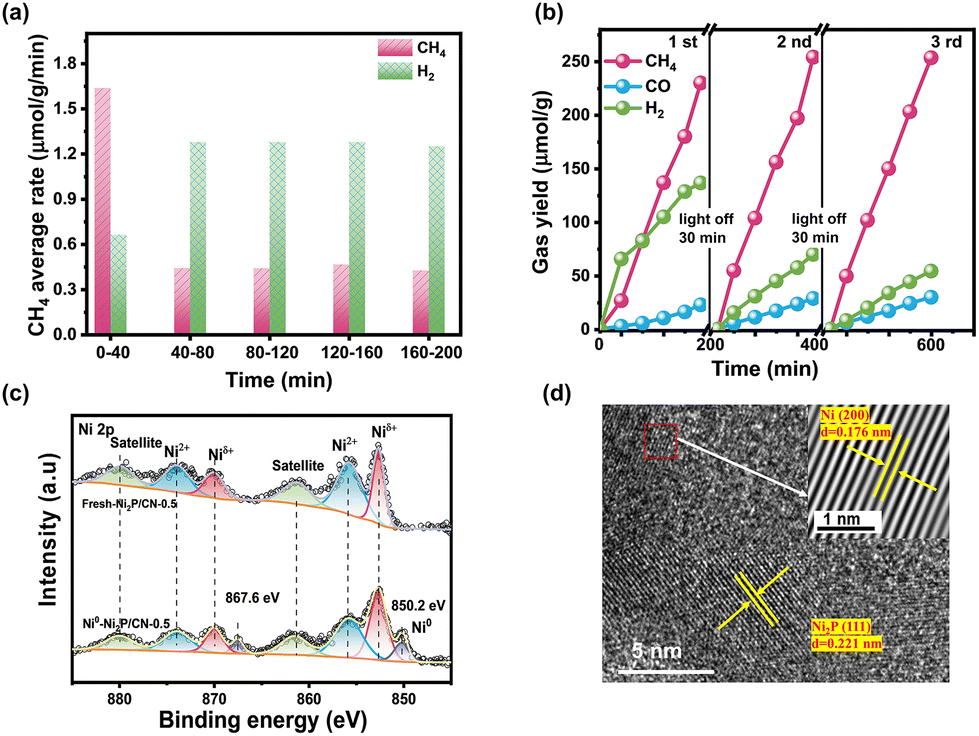 | ||
| Fig. 5 (a) The average rate of CH4 and H2 and the selectivity of CH4 in every interval on Ni2P/CN-0.5; (b) the product yield profiles in the 3-cycle experiments on Ni2P/CN-0.5; (c) the Ni 2p spectra of the fresh Ni2P/CN-0.5 and Ni0–Ni2P/CN-0.5 (b); and (d) HRTEM of Ni0–Ni2P/CN-0.5. | ||
3.6 Understanding the roles of light induced generated Ni0 in Ni0–Ni2P/CN-0.5
The CO2 adsorption capacity of the photocatalysts play an important role in determining their photocatalytic CO2 reduction performances. The CO2 adsorption isotherm of CN, Ni2P/CN-0.5 and Ni0–Ni2P/CN-0.5 at 25 °C is shown in Fig. 6a. Clearly, the CO2 adsorption capacity of Ni2P/CN-0.5 was higher than that of CN, indicating that the modification of CN by Ni2P is beneficial for the absorption of CO2. Clearly, even higher CO2 adsorption capacity was achieved over Ni0–Ni2P/CN-0.5, indicative of Ni sites also having the strong affinity of CO2.63 Moreover, the nitrogen isothermal adsorption–desorption isotherms were conducted, and the results are shown in Fig. S8 and Table S2.† For comparison, the results of CN, Ni2P/CN-0.5 and Ni0–Ni2P/CN-0.5 are displayed in Fig. 6b; it can be seen that their similar BET areas could exclude the effect of physical structures on the adsorption of CO2. Therefore, the generated Ni0 sites on Ni0–Ni2P/CN-0.5 enhance CO2 adsorption, thereby improving the ability of activating CO2 molecules. | ||
| Fig. 6 (a) CO2-adsorption isotherms; (b) N2 adsorption–desorption isotherms; (c) CO-TPD spectra on CN, Ni2P/CN-0.5 and Ni0–Ni2P/CN-0.5; and (d) in situ FTIR spectra of CO2 and H2O interacting under light irradiation. | ||
Furthermore, CO-TPD was used to evaluate the adsorption properties of CO, a key intermediate in photocatalytic CO2 reduction, on the CN, Ni2P/CN-0.5 and Ni0–Ni2P/CN-0.5 samples.64 As shown in Fig. 6c, CN displays insignificant CO desorption features compared to the other two samples. This is consistent with the fact that CO is the dominant product on CN (Fig. 4b). The Ni2P modified CN, i.e., both Ni2P/CN-0.5 and Ni0–Ni2P/CN-0.5, shows much higher CO desorption peaks, indicating the enhanced ability to adsorb CO. Furthermore, the CO desorption of Ni0–Ni2P/CN-0.5 is increased compared to Ni2P/CN-0.5, which could be attributed to the appearance of Ni0 sites induced by light irradiation. Therefore, the presence of Ni0 enhances the adsorption of CO and facilitates subsequent hydrogenation to CH4. Therefore, the CO-TPD result is indicative of the presence of the light-induced Ni0 leading to an enhancement of CO adsorption. Besides, as shown in Table S3,† we compared the results with previous studies. In other systems where transition metal phosphides are used as co-catalysts and the solvent is water or other sacrificial agents, the products are mostly CO with few multielectron hydrocarbons. In contrast, our catalyst has excellent catalytic activity for CH4 under mild reaction aqueous conditions. Combining the above results and product yields, and the excellent performance for CH4 is attributed to the generation of Ni0 for enhanced CO2 and CO adsorption.
In addition, in situ FTIR spectroscopy was performed to track the intermediates in the photocatalytic CO2 reduction process, and the results are shown in Fig. 6d. The peaks corresponding to HCO3− (1444 cm−1), m-CO32− (1373 cm−1) and b-CO32− (1309 cm−1, 1530 cm−1) increase with prolonged illumination, demonstrating enhanced CO2 adsorption and activation due to the appearance of Ni0.65,66 Moreover, the adsorbed *COOH is generally regarded as the initial intermediate in the CO2 reduction reaction. The bands for CO stretching and O–H deformation of *COOH can be observed at 1620 cm−1, and 1220 cm−1.67 It is noted that the band attributed to *CO (2100 cm−1) is very weak throughout the test, which suggests the enhanced kinetics of the conversion of *CO to the successive intermediates. Further hydrogenation of the adsorbed *CO leads to the formation of intermediates, such as *CHO and CH3O*, before forming CH4. As shown in the enlarged view in Fig. 6d, the H–CO bending mode of *CHO and the C–O stretching mode of the adsorbed CH3O* species can be observed at 1100 cm−1 and 1125 cm−1, respectively.68 These results provide further evidence that Ni0 plays an essential role in activating CO2 and facilitating the adsorption and further hydrogenation of CO. The identification of the CH3O* intermediate supports that the photocatalytic CO2 reduction on Ni2P/CN-0.5 follows the formaldehyde-mediated reaction pathway (CO2 (g) → *CO2 → *COOH → *CO → *CHO → *CH2O → CH3O* → CH4 (g)), as shown in Scheme S1.†69
DFT calculations were conducted to further evaluate the role of the Ni0 site in photocatalytic CO2 reduction to CH4 on Ni2P/CN-0.5. A Ni4 cluster supported on Ni2P (111) (namely Ni4/Ni2P) was constructed to represent the Ni0 active site (Fig. S9 and S10†). The first reduction step of CO2 was considered to be the formation of the *COOH intermediate. As shown in COHP of Ni2P(111) and Ni4/Ni2P(111) (Fig. 7a and b), the antibonding feature of Ni4/Ni2P(111) near the Fermi level was more pronounced than Ni2P(111). More charges have been transferred between *COOH and the surface on Ni4/Ni2P(111) than on Ni2P (111) based on the Bader charge analysis. The integrated COHP (namely ICOHP) of O–Ni and C–Ni on Ni4/Ni2P and Ni2P (111) surfaces can be used as a quantitative measure of bond strength where a more negative ICOHP value corresponds to a stronger bond.70,71 As shown in Fig. 7c, the ICOHPs of both O–Ni and C–Ni bonds on Ni4/Ni2P (111) are more negative than those on Ni2P (111), further demonstrating that the presence of Ni4 can facilitate the adsorption of *COOH.72,73
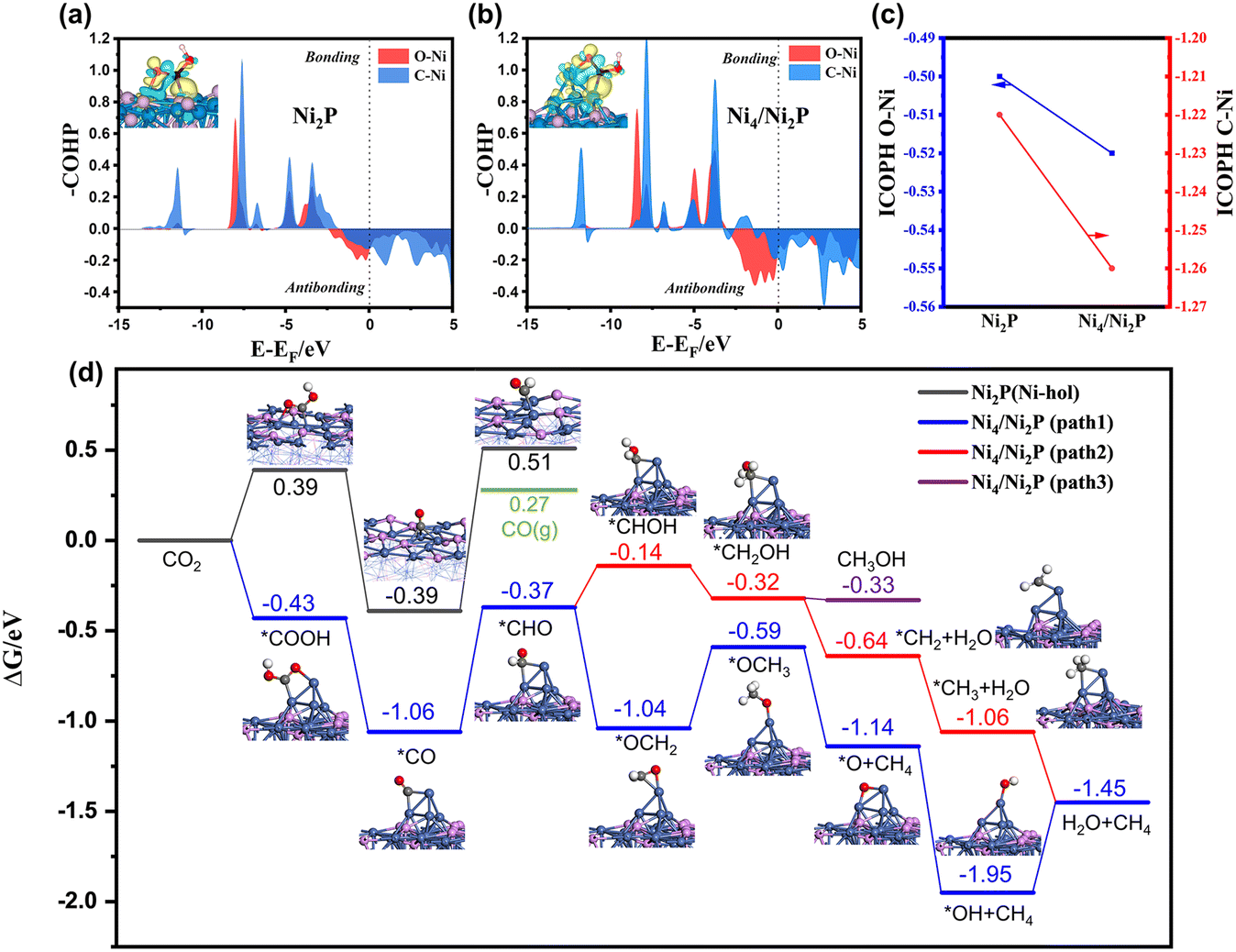 | ||
| Fig. 7 The crystal orbital Hamilton population (COHP) of adsorbed *COOH for the O–Ni and C–Ni bonds on the (a) Ni2P(111) surface and (b) Ni4/Ni2P(111). The insets show the charge density difference of *COOH on these two catalysts. (c) The integrated-COHP (ICOHP) of the O–Ni and C–Ni bonds on Ni2P (111) and Ni4/Ni2P (111), respectively. (d) Reaction pathways for CO2 reduction to CH4 on Ni2P (Ni-hol) and Ni4/Ni2P catalysts. | ||
The reaction free energy diagrams along the pathway for CO2 reduction to CH4 on Ni4/Ni2P and Ni2P are shown in Fig. 7d and Fig. S11.† On the Ni2P (111) surface, CO2 prefers to adsorb at the Ni-hollow site with a *COOH formation energy of 0.39 eV, which can be converted to *CO after releasing one water molecule. The formation of *CHO by the hydrogenation of *CO on Ni2P(111) is endergonic with a value of 0.90 eV and raises the free energy to a level higher than that of the desorbed CO molecule. In contrast, *COOH on Ni4/Ni2P (111) is highly stabilized with an adsorption energy of −0.43 eV by combining with two Ni atoms of Ni4 cluster. The enhanced stabilization of the adsorbed *CO on Ni4/Ni2P (111) is consistent with the CO-TPD results. Further hydrogenation will form *CHO with a formation energy of −0.37 eV, which is much lower than that for CO desorption (0.27 eV). Therefore, the CO (g) formation will be suppressed and the hydrogenation of *CO will be facilitated. Among three subsequent reaction pathways considered, two pathways (path 1 and 2) leading to CH4 are favorable. Bader charge analysis of the intermediates through paths 1 and 2 (Fig. S12†) shows that Ni4 acts like an electron donor, while larger charge variations are observed on path 1 than on path 2.
On the other hand, the enhancement of *CO adsorption on Ni4/Ni2P (111) results in decrease of the free energy barrier of the rate-determining step, which is related to the formation of *CHO on both Ni4/Ni2P and Ni2P catalysts, from 0.90 to 0.69 eV. The lower energy barrier promotes the CO2 reduction reaction more competitively than the hydrogen evolution reaction (HER) (Fig. S13†), elucidating the selectivity enhanced by the Ni0 site formation on Ni2P. Furthermore, the process for H transfer from the Ni2P surface to the Ni4 cluster for *CHO formation on Ni4/Ni2P (111) has been calculated (Fig. S14†). It indicates that the emergence of the Ni0 site can facilitate the process by overcoming a moderate barrier (∼1.05 eV) and thus enhance the subsequent CH4 synthesis. To sum up, the DFT results reveal that the synergy of Ni0 and Ni2P contributes to the stabilization of the *COOH and *CO intermediates, which enhance the hydrogenation process, resulting in the efficient formation of CH4.
Based on the above results and previous reports, the photocatalytic CO2 reduction reaction over Ni2P/CN-x catalysts followed a typical co-catalytic mechanism. As shown in Fig. 8, at the initial stage under irradiation, the photogenerated electrons were separated and transferred to the Ni2P particles, causing the competitive hydrogen evolution reaction (HER) to produce gaseous H2 as the dominant product, whereas very low yields of CO and CH4 were obtained from CO2 reduction. Meanwhile, some photoelectrons on Ni2P particles were consumed by the reduction of the hydrated surface nickel phosphorylate to generate Ni0 sites. Once the ensemble of Ni0 sites reaches a critical size, it will act synergistically with Ni2P to stabilize *COOH and *CO and facilitate subsequent hydrogenation reactions. Since CO2 consumes *H more rapidly in the reduction process, HER is greatly inhibited. The photocatalyst presents a sharp increase in CH4 evolution accompanied by the significant decrease of H2 yield in the initial irradiation period, until CH4 production becomes completely dominant over H2 evolution. Therefore, selective production of CH4 on Ni0–Ni2P/CN can be attributed to (a) favorable photochemical properties in separating and transferring the photogenerated electrons and holes and (b) enhanced CO adsorption and hydrogenation activity due to the in situ generated Ni0 sites. The synergy between Ni0 and Ni2P also contributes to the stabilization of the hydrogenation intermediates and facilitates sequential hydrogenation, leading to the efficient formation of CH4.
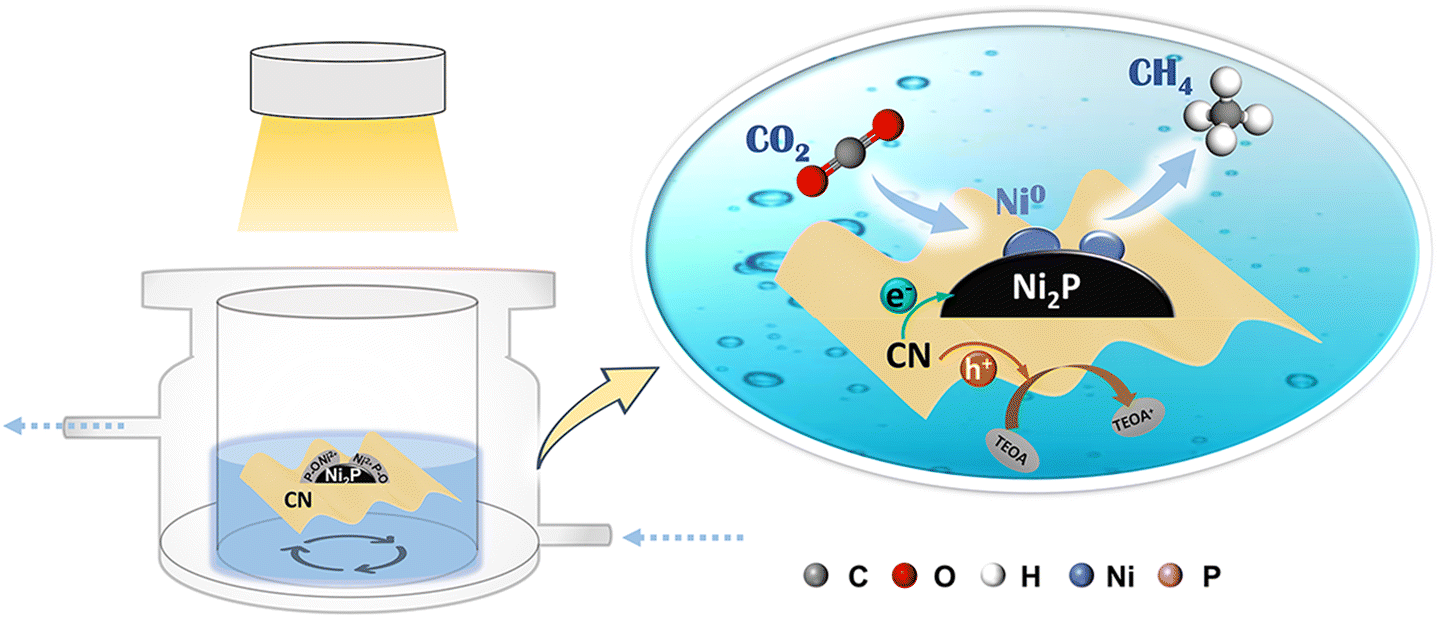 | ||
| Fig. 8 Schematic diagram of the mechanism of the reduction of CO2 to CH4 on the Ni0–Ni2P/CN photocatalyst. | ||
4. Conclusion
A dual active site photocatalyst Ni0–Ni2P/CN for selective CH4 formation has been successfully synthesized. It shows high efficiency in separating and transferring the photogenerated electrons and holes. The conditions of introducing Ni2P to CN have been optimized to maximize the CH4 production. Importantly, the in situ generated Ni0 sites under light irradiation have been confirmed, which is closely correlated with the promotion of CH4. In situ FTIR spectra, CO-TPD and theoretical calculations were used to gain insights into the synergy between the in situ-generated Ni0 sites and Ni2P sites. The results reveal that the Ni0–Ni2P interface can promote the H transfer from Ni2P to Ni0, while the Ni0 sites can stabilize the key intermediate *CO and reduce the energy barrier for *CO to *CHO. Therefore, the subsequent protonation steps can be facilitated, giving rise to the production of the multi-electron product CH4 and the suppressed side reaction of hydrogen evolution. This work demonstrated the success of designing a highly efficient photocatalyst with multiple active sites for CO2 conversion to hydrocarbons.Author contributions
Xuemei Liu: writing – original draft, methodology, investigation analysis, and data curation. Chaonan Cui: software, methodology, investigation analysis, data curation and formal analysis. Shuoshuo Wei: investigation analysis and data curation. Jinyu Han: writing – review and editing. Xinli Zhu: writing – review and editing. Qingfeng Ge: supervision and writing – review and editing. Hua Wang: conceptualization, supervision, writing – review and editing, and project administration. X. L. and C. C. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities, the National Natural Science Foundation of China (No. 22178266 and 22272180) and the Beijing Natural Science Foundation (Grant No. 2232035).References
- B. Zhao, Y. Zhao, P. Liu, Y. Men, X. Meng and Y. Pan, Jiégòu huàxué, 2022, vol. 41, pp. 12–21 Search PubMed.
- X. Meng, C. Peng, J. Jia, P. Liu, Y. Men and Y. Pan, J. CO2 Util., 2022, 55, 101844 CrossRef CAS.
- J. Nai, S. Wang and X. W. D. Lou, Sci. Adv., 2019, 5, x5095 CrossRef PubMed.
- P. Liu, Y. L. Men, X. Y. Meng, C. Peng, Y. Zhao and Y. X. Pan, Angew. Chem., Int. Ed., 2023, 62, e202309443 CrossRef CAS PubMed.
- W. Gao, S. Li, H. He, X. Li, Z. Cheng, Y. Yang, J. Wang, Q. Shen, X. Wang, Y. Xiong, Y. Zhou and Z. Zou, Nat. Commun., 2021, 12, 4747 CrossRef CAS PubMed.
- J. Li, H. Huang, W. Xue, K. Sun, X. Song, C. Wu, L. Nie, Y. Li, C. Liu, Y. Pan, H. Jiang, D. Mei and C. Zhong, Nat. Catal., 2021, 4, 719–729 CrossRef CAS.
- S. Bai, L. Tan, C. Ning, G. Liu, Z. Wu, T. Shen, L. Zheng and Y. Song, Small, 2023, 19, 2300581 CrossRef CAS PubMed.
- B. Dam, B. Das and B. K. Patel, Green Chem., 2023, 25, 3374–3397 RSC.
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- W. Wang, X. Zhao, Y. Cao, Z. Yan, R. Zhu, Y. Tao, X. Chen, D. Zhang, G. Li and D. L. Phillips, ACS Appl. Mater. Interfaces, 2019, 11, 16527–16537 CrossRef CAS PubMed.
- F. Zhang, J. Zhang, J. Li, X. Jin, Y. Li, M. Wu, X. Kang, T. Hu, X. Wang, W. Ren and G. Zhang, J. Mater. Chem. A, 2019, 7, 6939–6945 RSC.
- Y. Wang, S. Wang and X. W. D. Lou, Angew. Chem., Int. Ed., 2019, 58, 17236–17240 CrossRef CAS PubMed.
- K. U. D. Calvinho, A. B. Laursen, K. M. K. Yap, T. A. Goetjen, S. Hwang, N. Murali, B. Mejia-Sosa, A. Lubarski, K. M. Teeluck, E. S. Hall, E. Garfunkel, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2018, 11, 2550–2559 RSC.
- Q. Li, W. Feng, Y. Liu, D. Chen, Z. Wu and H. Wang, J. Mater. Chem. A, 2022, 10, 15752–15765 RSC.
- G. Huang, Q. Niu, J. Zhang, H. Huang, Q. Chen, J. Bi and L. Wu, Chem. Eng. J., 2022, 427, 131018 CrossRef CAS.
- D. Sun, J. Li, T. Shen, S. An, B. Qi and Y. Song, ACS Appl. Mater. Interfaces, 2022, 14, 16369–16378 CrossRef CAS PubMed.
- S. Zhang, Y. Liu, P. Gu, R. Ma, T. Wen, G. Zhao, L. Li, Y. Ai, C. Hu and X. Wang, Appl. Catal., B, 2019, 248, 1–10 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- C. Cui, H. Wang, X. Zhu, J. Han and Q. Ge, Sci. China: Chem., 2015, 58, 607–613 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- M. Yu and D. R. Trinkle, J. Chem. Phys., 2011, 134, 64111 CrossRef PubMed.
- V. Wang, N. Xu, J. Liu, G. Tang and W. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- M. Li, S. Zhang, X. Liu, J. Han, X. Zhu, Q. Ge and H. Wang, Eur. J. Inorg. Chem., 2019, 2019, 2058–2064 CrossRef CAS.
- P. Wen, K. Zhao, H. Li, J. Li, J. Li, Q. Ma, S. M. Geyer, L. Jiang and Y. Qiu, J. Mater. Chem. A, 2020, 8, 2995–3004 RSC.
- Z. Li, X. Zhang, J. Liu, R. Shi, G. I. N. Waterhouse, X. D. Wen and T. Zhang, Adv. Mater., 2021, 33, 2103248 CrossRef CAS PubMed.
- X. Jiang, W. Wang, H. Wang, H. Hong, Y. Yang, K. Wang, L. Zhao and B. Han, Green Chem., 2022, 24, 7652–7660 RSC.
- L. Liu, Y. Qi, J. Yang, W. Cui, X. Li and Z. Zhang, Appl. Surf. Sci., 2015, 358, 319–327 CrossRef CAS.
- W. Weng, S. Wang, W. Xiao and X. W. D. Lou, Adv. Mater., 2020, 32, 2001560 CrossRef CAS PubMed.
- N. Chen, X. Jia, H. He, H. Lin, M. Guo, J. Cao, J. Zhang and S. Chen, Chin. J. Catal., 2022, 43, 276–287 CrossRef CAS.
- Y. Yang, P. Zhang, Y. Lei, C. Zhou, J. Liu, S. Guo, S. Li and L. Chen, Int. J. Hydrogen Energy, 2021, 46, 10346–10355 CrossRef CAS.
- H. Sun, X. Xu, Z. Yan, X. Chen, F. Cheng, P. S. Weiss and J. Chen, Chem. Mater., 2017, 29, 8539–8547 CrossRef CAS.
- Z. Lin, Y. Zhao, J. Luo, S. Jiang, C. Sun and S. Song, Adv. Funct. Mater., 2019, 30, 1908797 CrossRef.
- S. Cui, X. Wang, L. Wang and X. Zheng, Dalton Trans., 2021, 50, 5978–5987 RSC.
- C. Chen and J. Wu, ACS Appl. Energy Mater., 2022, 5, 9733–9741 CrossRef CAS.
- C. Li, Y. Du, D. Wang, S. Yin, W. Tu, Z. Chen, M. Kraft, G. Chen and R. Xu, Adv. Funct. Mater., 2017, 27, 1604328 CrossRef.
- Y. Guo, Q. Wang, M. Wang, M. Shen, L. Zhang and J. Shi, Catal. Commun., 2021, 156, 106326 CrossRef CAS.
- A. Wang, C. Wang, L. Fu, W. Wong-Ng and Y. Lan, Nano-Micro Lett., 2017, 9, 47 CrossRef PubMed.
- L. Gao, X. Song, J. Ren and Z. Yuan, Inorg. Chem. Front., 2022, 9, 1964–1972 RSC.
- Y. Yang, P. Zhang, Y. Lei, C. Zhou, J. Liu, S. Guo, S. Li and L. Chen, Int. J. Hydrogen Energy, 2021, 46, 10346–10355 CrossRef CAS.
- Q. Gai, S. Ren, X. Zheng, W. Liu, Q. Dong and R. Gao, New J. Chem., 2020, 44, 4332–4339 RSC.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed.
- X. Jia, J. Cao, H. Lin, M. Zhang, X. Guo and S. Chen, Appl. Catal., B, 2017, 204, 505–514 CrossRef CAS.
- Y. Zou, J. Shi, L. Sun, D. Ma, S. Mao, Y. Lv and Y. Cheng, Chem. Eng. J., 2019, 378, 122192 CrossRef CAS.
- M. Li, S. Zhang, L. Li, J. Han, X. Zhu, Q. Ge and H. Wang, ACS Sustainable Chem. Eng., 2020, 8, 11465–11476 CrossRef CAS.
- Y. Bai, M. Li, X. Liu, J. Han, X. Zhu, Q. Ge and H. Wang, Ind. Eng. Chem. Res., 2022, 61, 8724–8737 CrossRef CAS.
- A. Gokarna, N. R. Pavaskar, S. D. Sathaye, V. Ganesan and S. V. Bhoraskar, J. Appl. Phys., 2002, 92, 2118–2124 CrossRef CAS.
- Y. Tian, W. Tang, H. Xiong, T. Chen, B. Li, X. Jing, J. Zhang and S. Xu, J. Lumin., 2020, 228, 117616 CrossRef CAS.
- M. Li, H. Wang, X. Li, S. Zhang, J. Han, A. F. Masters, T. Maschmeyer and X. Liu, ChemCatChem, 2018, 10, 581–589 CrossRef CAS.
- J. Zhou, J. Liang, Z. Fan, W. Tan, X. Sun, R. Ding, P. Gao and Y. Zhang, Int. J. Hydrogen Energy, 2023 DOI:10.1016/j.ijhydene.2023.10.209.
- Z. Chen, Y. Yu, X. She, K. Xia, Z. Mo, H. Chen, Y. Song, J. Huang, H. Li and H. Xu, Appl. Surf. Sci., 2019, 495, 143528 CrossRef CAS.
- M. R. Karimi Estahbanati, N. Mahinpey, M. Feilizadeh, F. Attar and M. C. Iliuta, Int. J. Hydrogen Energy, 2019, 44, 32030–32041 CrossRef CAS.
- H. Li, Q. Song, S. Wan, C. W. Tung, C. Liu, Y. Pan, G. Luo, H. M. Chen, S. Cao, J. Yu and L. Zhang, Small, 2023, 19, e2301711 CrossRef PubMed.
- Z. Gu, Z. Cui, Z. Wang, K. S. Qin, Y. Asakura, T. Hasegawa, S. Tsukuda, K. Hongo, R. Maezono and S. Yin, Appl. Catal., B, 2020, 279, 119376 CrossRef CAS.
- M. Lei, N. Wang, L. Zhu, Q. Zhou, G. Nie and H. Tang, Appl. Catal., B, 2016, 182, 414–423 CrossRef CAS.
- K. W. Feng and Y. Li, ChemSusChem, 2023, 16, e202202250 CrossRef CAS PubMed.
- X. Xiong, C. Mao, Z. Yang, Q. Zhang, G. I. N. Waterhouse, L. Gu and T. Zhang, Adv. Energy Mater., 2020, 10, 2002928 CrossRef CAS.
- C. Lv, L. Xu, M. Chen, Y. Cui, X. Wen, Y. Li, C. Wu, B. Yang, Z. Miao, X. Hu and Q. Shou, Front. Chem., 2020, 8, 32 CrossRef PubMed.
- A. I. Tsiotsias, N. D. Charisiou, I. V. Yentekakis and M. A. Goula, Nanomaterials, 2021, 11, 28 CrossRef CAS PubMed.
- M. Wang, D. Chen, N. Li, Q. Xu, H. Li, J. He and J. Lu, Adv. Mater., 2022, 34, 2202960 CrossRef CAS PubMed.
- M. Muir and M. Trenary, J. Phys. Chem. C, 2020, 124, 14722–14729 CrossRef CAS.
- J. Sheng, Y. He, J. Li, C. Yuan, H. Huang, S. Wang, Y. Sun, Z. Wang and F. Dong, ACS Nano, 2020, 14, 13103–13114 CrossRef CAS PubMed.
- L. Cheng, X. Yue, L. Wang, D. Zhang, P. Zhang, J. Fan and Q. Xiang, Adv. Mater., 2021, 33, 2105135 CrossRef CAS PubMed.
- Y. Zhang, D. Yao, B. Xia, H. Xu, Y. Tang, K. Davey, J. Ran and S. Qiao, Small Sci., 2021, 1, 2000052 CrossRef CAS.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- P. Liu, Z. Huang, X. Gao, X. Hong, J. Zhu, G. Wang, Y. Wu, J. Zeng and X. Zheng, Adv. Mater., 2022, 34, 2200057 CrossRef CAS PubMed.
- C. Cui, H. Zhang, R. Cheng, B. Huang and Z. Luo, ACS Catal., 2022, 12, 14964–14975 CrossRef CAS.
- R. Dronskowski and P. E. Blochl, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS.
- Y. Li, S. Wang, X. Wang, Y. He, Q. Wang, Y. Li, M. Li, G. Yang, J. Yi, H. Lin, D. Huang, L. Li, H. Chen and J. Ye, J. Am. Chem. Soc., 2020, 142, 19259–19267 CrossRef CAS PubMed.
- P. Liu, L. Niu, Y. Men, C. Peng, Z. Liu, X. Meng and Y. Pan, Appl. Catal., B, 2023, 320, 121985 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03549b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |