
Selective synthesis of functionalized linear aliphatic primary amines via decarboxylative radical-polar crossover†
Robin
Cauwenbergh‡
 a,
Prakash Kumar
Sahoo‡
a,
Rakesh
Maiti‡
a,
Abra
Mathew
b,
Rositha
Kuniyil
b and
Shoubhik
Das
*a
a,
Prakash Kumar
Sahoo‡
a,
Rakesh
Maiti‡
a,
Abra
Mathew
b,
Rositha
Kuniyil
b and
Shoubhik
Das
*a
aDepartment of Chemistry, Organic Synthesis Division, University of Antwerp, 2020 Antwerp, Belgium. E-mail: shoubhik.das@uantwerpen.be
bDepartment of Chemistry, CompCat group, Indian Institute of Technology Palakkad, 678557, Kerala, India
First published on 24th November 2023
Abstract
Primary aliphatic amines are crucial, and at the very least, millions of tons are required every year. Therefore, significant interest has grown in finding new chemical routes for their synthesis. Additionally, to comply with sustainability requirements, synthetic routes using inexpensive feedstocks are highly prioritized. In this regard, the hydroaminoalkylation (HAA) reaction is highly desirable due to the wide availability and lower price of olefins. Although photoredox catalysis is already known for synthesizing amines, the synthesis of linear aliphatic amines is mostly limited to hydroaminomethylation (HAM) reactions and has never been applied to the synthesis of longer-chain linear aliphatic primary amines. Considering this, the synthesis of functionalized linear aliphatic primary amines is demonstrated by using the decarboxylative radical-polar crossover process, which yielded a wide range of functionalized linear aliphatic amines with excellent yields. The strength of this method is demonstrated by the synthesis of existing drugs and late-stage transformations of complex pharmaceuticals, and elaborated with detailed mechanistic studies.
Introduction
Amines are key compounds that have been used as bulk chemicals, materials, applied in biology, and have found many more applications.1–5 Expediently, amine functionalities are present in a large number of pharmaceuticals and play crucial roles such as bypassing the blood–brain barrier to achieve biological activities, solubility, and pharmacokinetics (Fig. 1A).6 In fact, >75% of the top selling drugs of the year 2021–22 contain amine/nitrogen moieties.7–9 Among different amines, primary amines are highly important due to their wide applications as precursors and valuable intermediates for the synthesis of advanced chemicals, pharmaceuticals, agrochemicals and materials.3,6 Particularly, linear aliphatic primary amines are vital and every year, at least a million tons are required to meet this demand.10–15 Therefore, there is significant interest in finding new chemical routes for the synthesis of aliphatic primary amines. Although a plethora of synthetic methods such as amination reactions,16–23 reductive amination reactions,24–27 Gabriel synthesis,28 Curtius rearrangement,29 the Deléphine reaction30 and others10,31 exist, these reactions suffer from poor selectivity in attaining primary amines over secondary and tertiary amines, require harsh reaction conditions, or involve toxic precursors. On the other hand, to comply with sustainability requirements, synthetic routes using inexpensive feedstock and non-toxic aminating reagents are highly prioritized. In this regard, metal-catalyzed alkene hydroamination32–35 and hydroaminoalkylation (HAA)36–46 have provided significant opportunities due to the wide availability of olefins (Fig. 1B).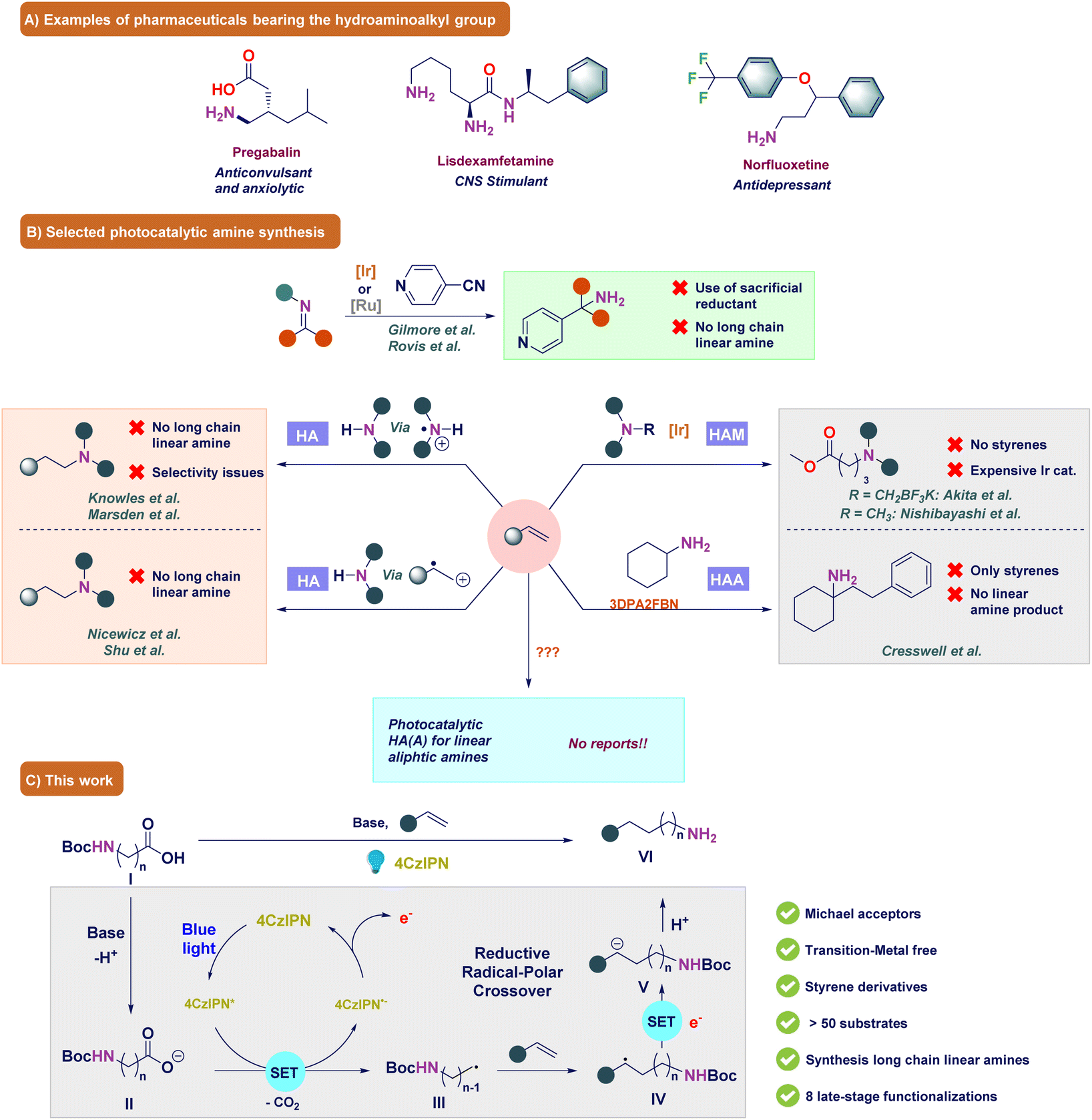 | ||
| Fig. 1 (A) Pharmaceutical compounds bearing the (amino)methyl group. (B) Selected state-of-the art methods. (C) Our strategy. | ||
Over the past decade, photoredox catalysis has become quite popular to attain sustainability under mild reaction conditions.47–51 One way of making primary amines is through the photochemical reduction of imines and in 2018, Gilmore et al. reported an excellent chemoselective approach using benzaldehydes and ammonia to form primary amines by reducing the in situ formed iminium ion to an α-amino radical.52 Later, in 2020, Rovis et al. expanded this method by using preformed imines or iminium salts.53 They developed excellent strategies but these procedures required a sacrificial and (over)stoichiometric amount of reagents and could not generate long chain aliphatic primary amines. Another way to synthesize primary amines is via the hydroamination reaction, i.e. direct coupling of amines with alkenes.54–56 Here, primary or secondary amines undergo a single electron oxidation of the amine to form an aminium radical cation or amine centered radical intermediate, which is not possible from the corresponding primary amines.57–63 However, by using ammonia as the aminating source, this problem has been circumvented. It should be noted that photocatalytic hydroamination towards primary amines still remains challenging as the final primary amine product is more electron-rich, and thus more prone to oxidation.64 Moreover, primary amines are more nucleophilic, which provide mixtures of secondary and tertiary amines through alkylation reactions. In this respect, Nicewicz et al. successfully bypassed these problems by oxidizing alkenes to the corresponding radical cation intermediate, which was reacted with a nucleophile such as ammonia.65,66 Later, Nicewicz's hydroamination procedure became the basis for the photocatalytic synthesis of amines.64,67,68 Although photocatalytic hydroamination has been studied extensively, still the synthesis of longer chain aliphatic primary amines remains underdeveloped.62,63
This problem could be resolved by functionalizing alkenes with an aminoalkyl group instead of an amino group using the HAA reaction. However, in the synthesis of linear aliphatic amines within the photocatalyzed HAA reactions, only hydroaminomethylation (HAM), where the olefin is functionalized with an aminomethyl group, has been reported to date. In fact, photocatalysis has shown highly regioselective HAM reactions under mild reaction conditions via the formation of α-aminoalkyl radicals from secondary or tertiary amines.69–75 These radicals underwent a Giese-type reaction with Michael acceptors such as malonates and malononitriles,72 α,β-unsaturated carbonyl compounds,76,77 esters,72,78 and alkenylpyridines79 (Fig. 1B). In addition, decarboxylation,71,80–85 desilylation70,86,87 and other pathways for α-aminomethyl radical generations88–92 have also been reported. Although the developed HAM reactions typically exhibit high product yields, certain limitations persist. Notably, the general importance of relying on a costly iridium photocatalyst remains paramount, while addressing the synthesis of longer chain aliphatic amines remains a persistent challenge in the realm of photoredox catalysis.71,92 This problem was partly solved by König et al., Cresswell et al., MacMillan et al. and Yuan et al. who reported the use of alkylamines for the HAA of olefins.71,85,91,93–95 Although a wide variety of alkylamines were functionalized, the König, Cresswell and MacMillan strategies generally required the generation of a secondary and tertiary α-amino radical center, whereas the Yuan strategy required an overstoichiometric amount of 1,3-bis-(2,6-diisopropylphenyl)-imidazolinium (iPrBr) as the hydride source, tertiary α-silylamines and expensive ligands, which limited the general synthesis of linear aliphatic amines. Moreover, the König strategy made use of highly specific starting materials and the MacMillan strategy was only successful for the highly activated diethyl malonates. These deficiencies were resolved by Jiang and coworkers, although for their strategy α-substituted vinylpyridines were necessary for obtaining moderate to high yields.92 Another approach for the synthesis of longer chain amines was introduced by Waser et al., where dipeptides reacted with ethynylbenziodoxolone (EBX) hypervalent iodine reagents to yield the alkenylated peptide.96 Although peptides generating primary α-amino radicals were selectively alkenylated at the C-terminus in moderate to excellent yields, the Ar-EBX compounds needed to be synthesized using three steps and resulted in a considerable amount of waste in the photoredox reaction.97 Given the literature above, the HAA reaction has garnered significant attention in the literature; however, existing reports predominantly rely on the generation of α-amino radicals as essential intermediates. In 2016, König and colleagues partially addressed this issue by employing non-stabilized primary radicals in alkylation reactions.85 While the authors did not conduct experimental trials to validate the suitability of their protocol for the synthesis of linear aliphatic amines, there is a reasonable conjecture that this methodology could be applicable for such purposes. Nevertheless, the protocol requires a high amount of the photocatalyst, it demands highly specialized starting materials, and results in a notable generation of waste products.85 These drawbacks can limit its applicability in large-scale synthesis. Therefore, a selective strategy for the generation of distant primary radicals and the subsequent synthesis of linear aliphatic primary amines is a big challenge to solve in photocatalysis.
In light of the gathered knowledge, the development of an organic photocatalyst-based HAM strategy, which is robust for both styrenes and Michael acceptors, assumes significant importance within the realm of organic synthesis. Furthermore, the expansion of this strategy to encompass the generation of distant primary radicals, i.e. radical species where the radical is formed on carbon centers positioned beyond the alpha position, for the synthesis of long chain aliphatic amines through the HAA method would add substantial value to the system. This extension would broaden the scope of applications, offering new avenues for the synthesis of long chain aliphatic amines. It is well known that 4CzIPN (Eox = +1.35 V vs. SCE) in its excited state form can readily oxidize organic molecules.98 We envisioned that a base-promoted decarboxylation of N-protected amino acids (I) could produce a primary (amino)alkyl radical (III) (Fig. 1C).99 We anticipated that the highly nucleophilic character of this radical will enable it to readily react with the olefin to form a new radical species (IV). Due to the highly reductive nature of the photocatalyst (Ered = −1.21 V), the radical is converted to the carbanion (V), which should form the linear aliphatic amine product (VI) upon protonation.98 Based on this strategy, we report a 4CzIPN-mediated reaction to afford linear aliphatic primary amines which tolerated diverse functional groups and generated the desired aliphatic amines in excellent yields.
Results and discussion
Optimization studies
At the outset of our reaction, 1,1-diphenylethylene (1) and boc-glycine (2) were chosen as model substrates to optimize the HAM reaction conditions (Table 1 and Tables S1–S11†). To our delight, when a solution of 1 and 2 with 1.0 eq. of cesium carbonate (Cs2CO3) in the presence of 5 mol% of 4CzIPN was stirred for 24 hours under a 40 W blue Kessil lamp at ambient temperature, the desired product was obtained in 94% yield (Table 1, entry 2). After a thorough base screening, a similar yield was obtained (95%) when much cheaper sodium hydroxide (NaOH) was used instead of Cs2CO3 (Table 1, entry 1). Next, different solvents were applied (Table 1, entries 4 and 5), where DMF also provided quantitative yield; however, owing to the greener aspect, DMSO was chosen as the solvent.100 Unfortunately, reducing the catalyst loading to 1 mol% resulted in a significant decrease in the product yield, down to 65% (Table 1, entry 7). Encouraged by these findings, control experiments were conducted to validate the significance of each reagent under the optimized reaction conditions. These experiments clearly demonstrated that the light source, photocatalyst (PC), and base were all indispensable for this transformation (Table 1, entry 8). Interestingly, under aerobic conditions, the yield decreased to 76%, indicating that anaerobic conditions were essential for obtaining excellent yields (Table 1, entry 9). Finally, the reaction was performed on a gram-scale, and excitingly, 91% yield of the product was obtained (Table 1, entry 10).|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yieldb (%) |
| a Reaction conditions: 1 (0.2 mmol, 1 equiv.), 2 (0.2 mmol, 1 equiv.). b Yield was determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as the internal standard. c No reaction was observed. d Reaction conditions: 1 (5.66 mmol), 2 (5.66 mmol, 1 equiv.), 72 hours. For a more extensive optimization study, see the ESI.† | ||
| 1 | None | 95 |
| 2 | Cs2CO3 (instead of NaOH) | 94 |
| 3 | DIPEA (instead of NaOH) | 0 |
| 4 | MeCN (1 mL) | 61 |
| 5 | DMF (1 mL) | 95 |
| 6 | 5 mol% 4CzIPN | 97 |
| 7 | 1 mol% 4CzIPN | 65 |
| 8 | No light, no base or no catalyst | 0c |
| 9 | Aerobic conditions | 76 |
| 10d | Gram scale | 91 |
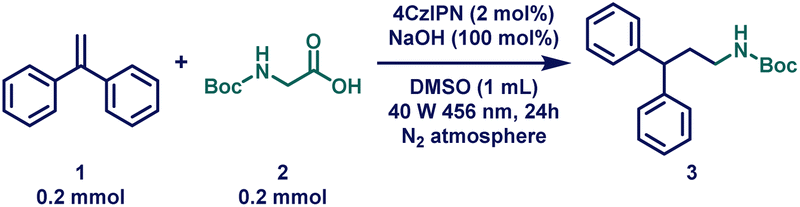
Reaction scope
With the optimized reaction conditions in hand, we sought to determine the substrate scope of the HAA reaction for the olefin component (Scheme 1A). At the beginning, non-hindered external vinylarenes were studied (Scheme 1A, 3–18) and our focus started with the activated styrenes (3 and 4), which provided excellent yields even when an electron-withdrawing group was present (94 and 95%, respectively). To show the applicability of this transformation, the formed product was deprotected in situ to obtain almost complete conversion (88%) to the linear aliphatic free amine product. After the successful transformations of activated external vinylarenes, our focus shifted towards different substituted styrenes (5–18). Delightfully, our mild reaction conditions were compatible with a wide range of substituted styrenes, involving both electron-donating and electron-withdrawing groups, which provided a versatile platform for further synthetic manipulations. This versatility in styrene substrates is a significant advancement from the previously reported HAM protocols, which, in most cases, only worked on Michael acceptors, highly activated 1,1-diphenylethylenes or vinylpyridines. In this regard, non-substituted styrene (5) afforded 81% yield. Further electron-withdrawing substituents such as p-bromo- (6), p-trifluoromethyl- (7), and p-cyano (8) substituents on styrene provided moderate to good yield (45–79% yield). After successfully exploring the electron-neutral and electron-withdrawing vinylarenes, curiosity drove us to explore electron-donating substrates. These substrates usually are difficult for this reaction because the formed carbanion in the benzylic position gets destabilized when electron-donating groups are present in the p-position of styrene.101 Delightfully, substituents such as tert-butyl (9), trimethylsilyl (10), and phenyl (11) were compatible with our system, yielding the corresponding products in moderate to good yields (42–75%). After achieving this, ortho- (12), tri- (13), and penta-substituted (14) styrenes worked in moderate to excellent yields (32–90% yield). Furthermore, external vinylarenes bearing α-substituents (15–18) were well tolerated and provided good to excellent yields (54–91%).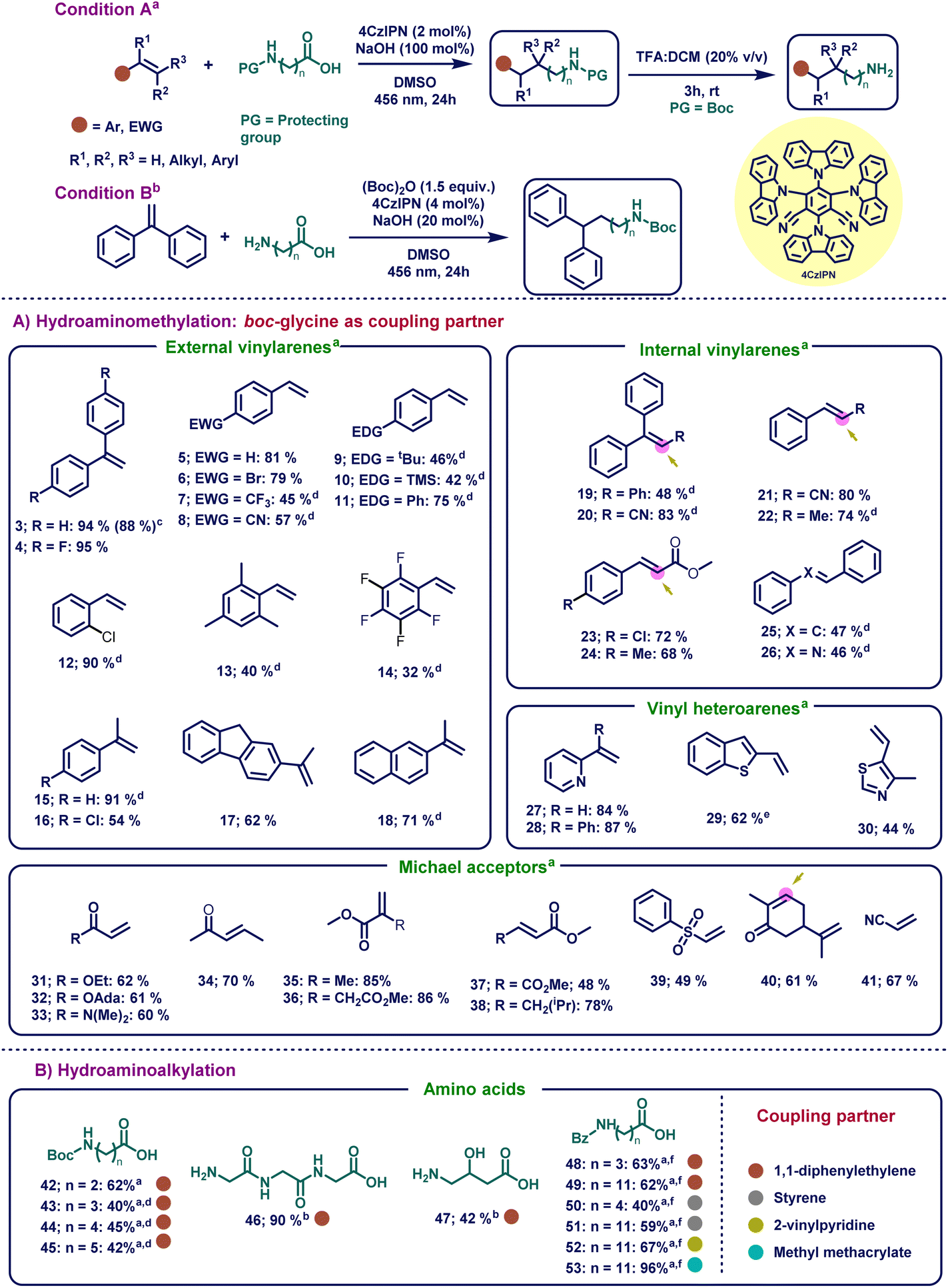 | ||
Scheme 1
a
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions A: alkene (0.2 mmol, 1 equiv.), protected amino acid (0.2 mmol, 1 equiv.), 4CzIPN (4 μmol, 0.02 equiv.), NaOH (0.2 mmol, 1 equiv.), DMSO (0.2 M) at rt for 24 hours under irradiation with 456 nm KESSIL lamp. bReaction conditions B: 1 (0.2 mmol, 1 equiv.), free amino acid (0.3 mmol, 1.5 equiv.), Boc-anhydride (0.3 mmol 1.5 equiv.), NaOH (0.04 mmol, 0.2 equiv.), 4CzIPN (8 μmol, 0.04 equiv.), DMSO (0.1 M) at rt for 4 h and subsequently for 24 hours under irradiation with 456 nm KESSIL lamp. Optimization studies for conditions in B are given in Tables S4–S7.†cYield given for free amine using TFA as the deprotecting agent. d48 hours. eCs2CO3 (0.2 mmol, 1 equiv.) was used as the base. fReaction conditions C: performed using conditions in A at 70 °C for 72 hours with 1 equiv. of Cs2CO3. Optimization studies for conditions C are given in Tables S8–S11.†1H NMR yields are given and was determined by using 1,3,5-trimethoxybenzene as the internal standard. See the ESI† for full experimental details. Reaction conditions A: alkene (0.2 mmol, 1 equiv.), protected amino acid (0.2 mmol, 1 equiv.), 4CzIPN (4 μmol, 0.02 equiv.), NaOH (0.2 mmol, 1 equiv.), DMSO (0.2 M) at rt for 24 hours under irradiation with 456 nm KESSIL lamp. bReaction conditions B: 1 (0.2 mmol, 1 equiv.), free amino acid (0.3 mmol, 1.5 equiv.), Boc-anhydride (0.3 mmol 1.5 equiv.), NaOH (0.04 mmol, 0.2 equiv.), 4CzIPN (8 μmol, 0.04 equiv.), DMSO (0.1 M) at rt for 4 h and subsequently for 24 hours under irradiation with 456 nm KESSIL lamp. Optimization studies for conditions in B are given in Tables S4–S7.†cYield given for free amine using TFA as the deprotecting agent. d48 hours. eCs2CO3 (0.2 mmol, 1 equiv.) was used as the base. fReaction conditions C: performed using conditions in A at 70 °C for 72 hours with 1 equiv. of Cs2CO3. Optimization studies for conditions C are given in Tables S8–S11.†1H NMR yields are given and was determined by using 1,3,5-trimethoxybenzene as the internal standard. See the ESI† for full experimental details. | ||
Next, we focused on exploring our catalytic system's versatility on sterically challenging internal vinylarenes. To our delight, a plethora of bulky internal vinylarenes underwent these reactions to provide good to excellent yields. Bulky trisubstituted compounds such as triphenylethylene (19) and 3,3-diphenylacrylonitrile (20) provided moderate to good yields (48–83%). On the other hand, styrenes having α-substitution of both electron-withdrawing groups such as –CN (21), –CO2Me (23 and 24), and donating groups such as –CH3 (22) moiety provided excellent yields (68–80%). It is worth noting that in the case of substrates where both Michael acceptor and styrene type moieties were present in the same molecule, the α-amino radical attacked selectively the styrene moiety and yielded regioselective products.102 In addition, stilbene (25) and internal imine 26 were tolerated in this photocatalytic reaction (47 and 46% yield, respectively). Expediently, heteroaromatic vinylarenes such as 2-vinylpyridine (27), 2-(1-phenylvinyl)pyridine (28), 2-vinylbenzo[b]thiophene (29), and 4-methyl-5-vinylthiazole (30) also underwent the HAM reaction, and provided the product in good to excellent yields (44–87%). Compared to previous reports, the scope could be extended to heteroaromatic olefins other than 2-vinylpyridine with good to excellent yields.
The robust catalytic system not only worked with styrene derivatives, but it was also able to functionalize various Michael acceptors. Simple Michael acceptors (31, 33–38, 40 and 41) along with bulky adamentyl acrylate (32) and (vinylsulfonyl)benzene (39) provided reasonable to excellent yields (48–86% yield). Remarkably, pregabalin (from 38), an anticonvulsant and anxiolytic drug, was synthesized by our method in 78% yield.103 Our newly developed method decreased the amount of steps for the initial industrial preparation of pregabalin tremendously from more than 8 steps to only one step using the cheap and commercially available (E)-methyl 5-methylhex-2-enoate.104 Furthermore, the natural product carvone (40), which is used as an air freshening product, can also undergo HAM leading to the desired product in good yield (61%).105 It is noteworthy that the newly developed method regioselectively underwent HAM with the conjugated double bond without interfering with the free double bond.
After investigating the HAM reaction, our interest shifted towards the synthesis of longer chain linear aliphatic amines. Here, long-chain amino acids were investigated under the same reaction conditions (Scheme 1B) for the generation of distant primary radical species. Interestingly, boc-protected 3-aminopropanoic (42), 4-aminobutanoic (43), 5-aminopentanoic (44) and 6-aminohexanoic (45) acids successfully yielded the corresponding distant primary radical, which attacked the olefin to yield the desired long-chain amine products in moderate to good yields (45–62%). Surprisingly, the tripeptide glycyl-glycyl-glycine (46) and γ-amino-β-hydroxybutyric acid (47) with free amine and hydroxy functional groups, respectively, were well-tolerated in our system by altering the conditions slightly (90 and 42%, respectively). Ultimately, robustness was proved when long-chain amino acids such as 4-benzamidobutanoic acid and 12-benzamidododecanoic acid were reacted with 1, styrene (5) and 2-vinylpyridine (27) to yield the desired products (48–52) in moderate to good yields (40–67%). Moreover, when methyl methacrylate (35) reacted with 12-benzoyldodecanoic acid, 96% product (53) was formed proving that our method also worked with Michael acceptors and long chain amino acids.
To further demonstrate the utility of our method, we showed effective conversion of our products into two drug molecules (prenylamine and pheniramine, Scheme 2A). Firstly, products 54 and 55 were synthesized using our catalytic system and were subsequently hydrolyzed in the presence of trifluoroacetic acid (TFA) to afford the free amines 56 and 57, respectively. After that, the amines were used in the next step without further purification. A reductive amination of 56 with benzyl acetone provided the drug prenylamine (58) in 68% overall yield. To obtain the drug pheniramine (59), formic acid mediated dimethylation of 57 was set up, providing 59 in 74% yield.
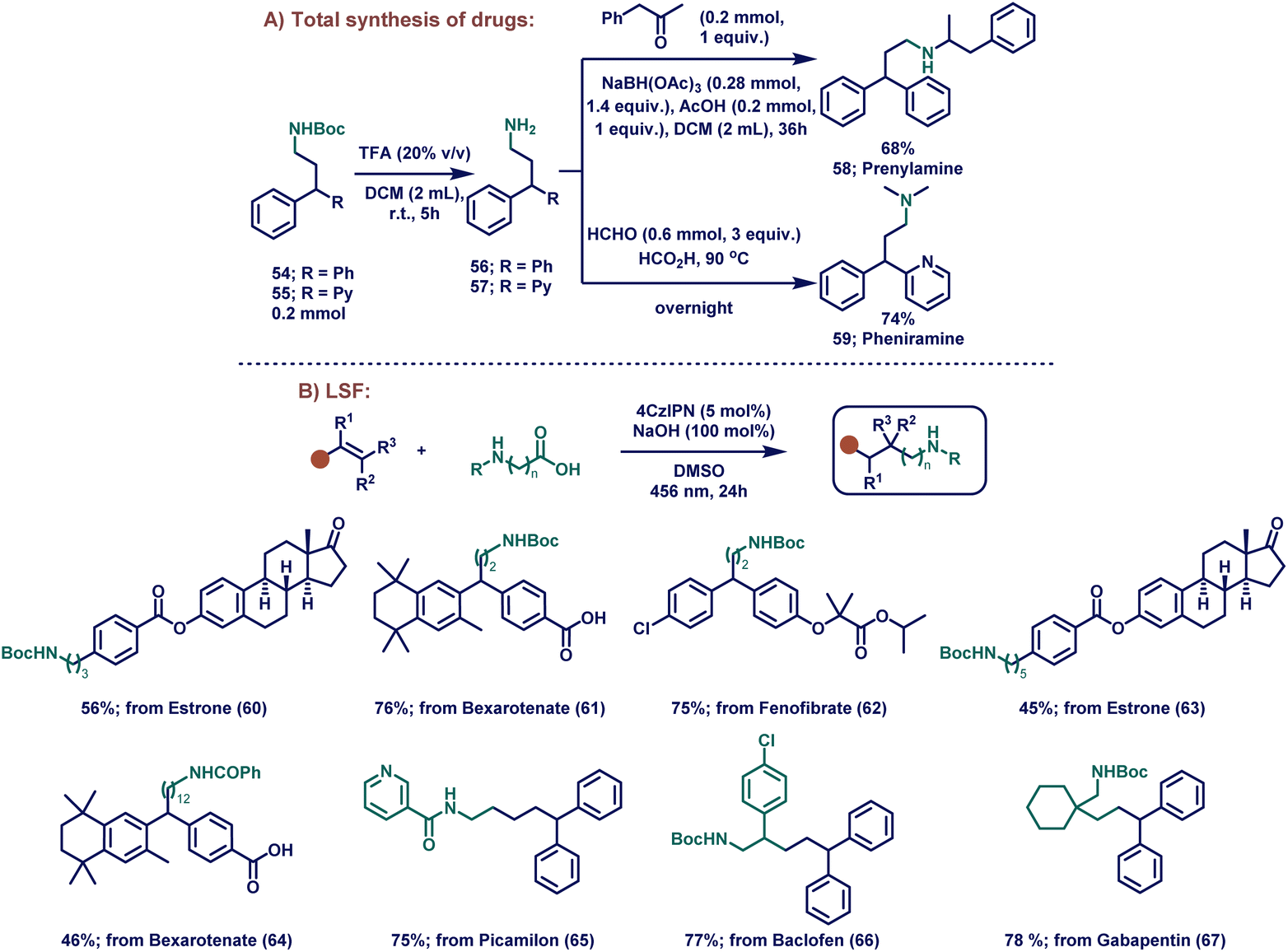 | ||
| Scheme 2 (A) Total synthesis of pharmaceuticals in a one-pot reaction and (B) late-stage modification of existing drugs. Reaction conditions: alkene (0.2 mmol, 1 equiv.), protected amino acid (0.2 mmol, 1 equiv.), 4CzIPN (4 μmol, 0.02 equiv.), NaOH (0.2 mmol, 1 equiv.), DMSO (0.2 M) at rt for 24 hours under irradiation with 456 nm KESSIL lamp. | ||
Next, we moved to assess the applicability of our method in late-stage functionalizations (LSFs). LSF is the chemoselective process of installing a particular functional group into a complex molecule. This modification facilitates the development of structure–activity relationships (SAR) and improves the physical properties such as solubility, stability etc.106 Since vinylarene is a common motif in various natural products and drug molecules, we selected large biomolecular derivatives for the LSF under our reaction conditions (Scheme 2B). Estrone, bexarotene, and fenofibrate derivatives were functionalized with boc-protected glycine and afforded the products (60–62) in moderate to excellent yields (Scheme 2B, 56–76%). Interestingly, boc-protected γ-aminobutyric acid with an estrone derivative also worked well (63; 45% yield). To our delight, when 12-benzamidododecanoic acid was employed with bexarotene under our catalytic conditions, the product 64 was obtained in 46% yield, proving the generality of the reaction for the synthesis of longer aliphatic amines. In addition, bioactive picamilon, drug baclofen, and gabapentin were also functionally modified with our catalytic conditions, providing the desired products in high yields (65 – 75%, 66 – 77%; 67 – 78%).
Mechanistic studies
After demonstrating the scope of our catalytic system, we became interested in proving the reaction mechanism (Fig. 1C). For this purpose, a series of control experiments were conducted (Scheme 3). The radical clock experiment with boc-protected glycine bearing a cyclopropane group at the α-position provided the mixture of the stereoisomeric ring-opened product 68, suggesting that the radical cascade reaction of cyclopropane ring opened to yield the α-aminoalkyl radical (III, Fig. 1C). In fact, the presence of radicals can be further confirmed by trapping the formed radical (III, Fig. 1C) with (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO). In this regard, when 1 equiv. of TEMPO was added to the catalytic reaction, no desired product was formed; instead, alkyl-TEMPO adduct 69 was detected in the HR-MS spectrum. Additional radical suppression experiments with the use of 2,6-di-tert-butyl-4-methylphenol (BHT, 1 equiv.) demonstrated the radical involvement in the reaction. Another interesting control experiment with D-glycine was performed to investigate the source of the H-atom at the α-position of the product VI (Fig. 1C), which provided the evidence of the reductive radical-polar crossover process. When the in situ formed boc-protected D-glycine molecule underwent photocatalytic HAM reaction with 1,1-diphenylethylene as a coupling partner, 75% deuteration at the α-position of the final product (70) was observed.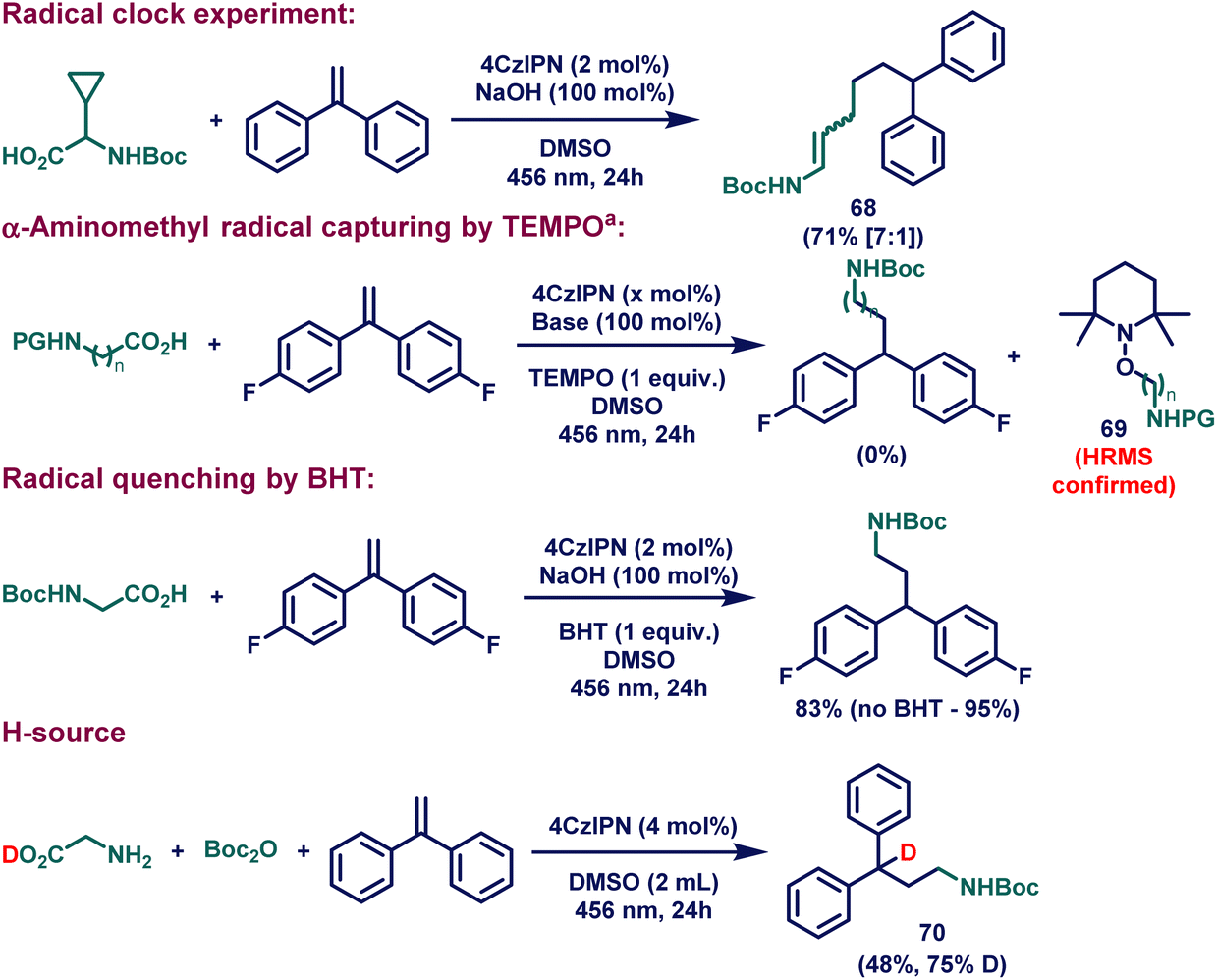 | ||
| Scheme 3 Mechanistic studies. See the ESI† for full experimental details. aReaction conditions A: boc-glycine (0.2 mmol, 1 equiv.), 4,4′-(ethene-1,1-diyl)bis(fluorobenzene) (0.2 mmol, 1 equiv.), 4CzIPN (4 μmol, 0.02 equiv.), NaOH (0.2 mmol, 1 equiv.), TEMPO (0.2 mmol, 1 equiv.) and DMSO (1 mL) at rt for 24 hours under irradiation with 456 nm KESSIL lamp. Reactions conditions B: 12-benzoyldodecanoic acid (0.2 mmol, 1 equiv.), 4,4′-(ethene-1,1-diyl)bis(fluorobenzene) (0.2 mmol, 1 equiv.), 4CzIPN (10 μmol, 0.05 equiv.), Cs2CO3 (0.2 mmol, 1 equiv.), TEMPO (0.2 mmol, 1 equiv.) and DMSO (2 mL) at 70 °C for 24 hours under irradiation with 456 nm KESSIL lamp. | ||
To further prove the mechanism, DFT calculations were performed for the photocatalytic decarboxylative radical polar crossover reaction of 1,1-diphenylethylene with boc-glycine (Fig. 2) at the PBE0-D3(BJ)/def2-TZVPP+SMD(DMSO)//PBE0-D3(BJ)/6-31G(d) level of theory.107–111,118 A long-lived triplet 4CzIPN species (B) with an energy of 51.3 kcal mol−1 was generated from the excitation and intersystem crossing (ISC) of the singlet 4CzIPN species (A). Afterwards, a single electron transfer (SET) process occurred from boc-glycine (C) to B leading to the formation of E along with the 4CzIPN radical anion (D) with a barrier of 6.8 kcal mol−1 (with respect to B). A barrierless decarboxylation from E generated an α-amino radical species (F). The spin natural orbitals of F clearly exhibited the stabilization of the radical by the α-amino group. This radical further underwent the C–C bond formation with 1,1-diphenylethylene (G) via the transition state TS1 with a barrier of 6 kcal mol−1 (from im1) to form a stable benzyl radical intermediate im2. The regeneration of the photocatalyst 4CzIPN (A) occurred through a feasible single electron transfer step from D to im2. The anionic intermediate im3 which formed during this step underwent protonation to form the stable product im4. A similar mechanistic pathway has been depicted for the HAA reaction involving boc-3-aminopropanoic acid and 1,1-diphenylethylene. In this case, the spin natural orbitals of (F′) showed the stabilization of the radical by the adjacent C–H bond (Fig. S6†).112
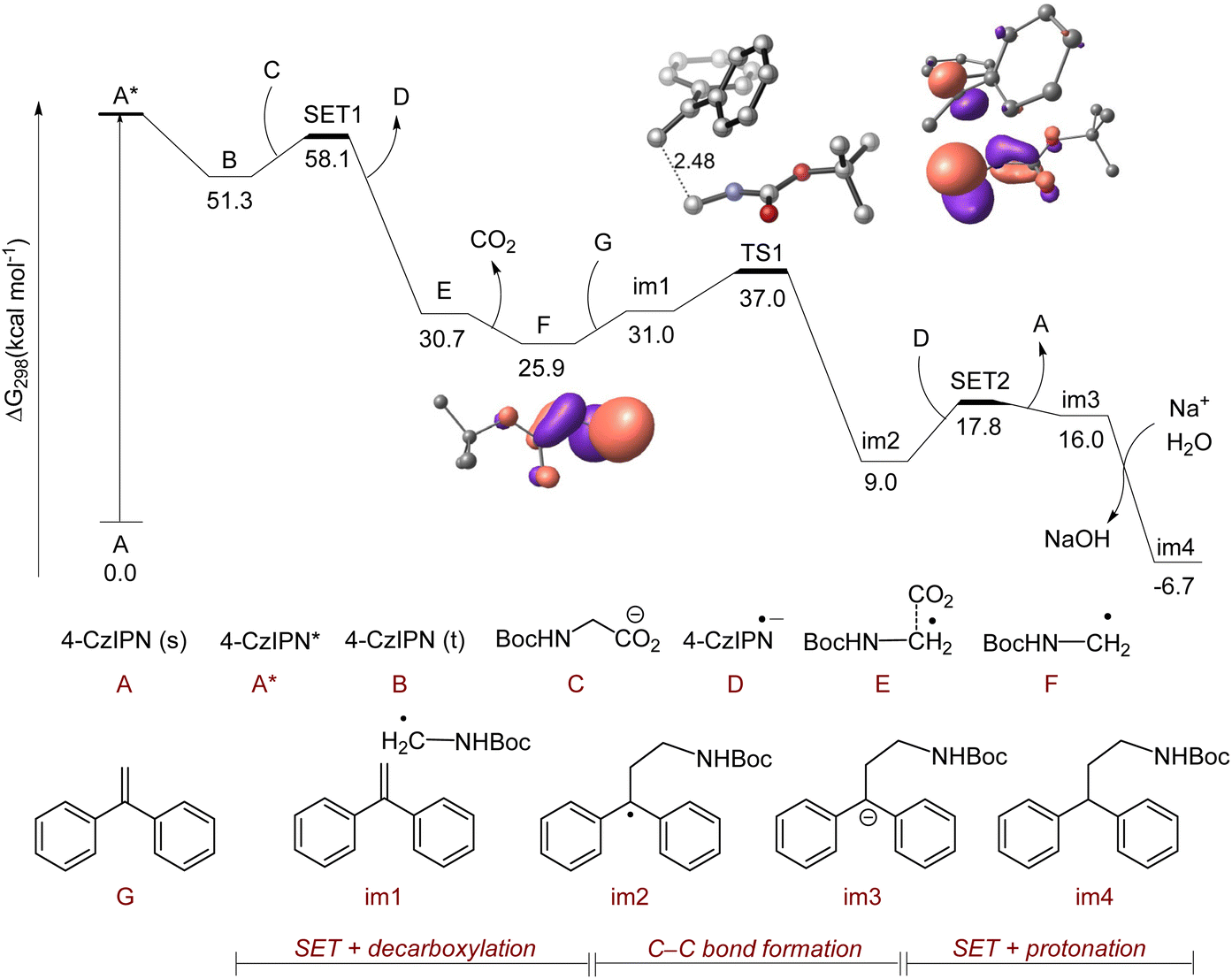 | ||
| Fig. 2 Computed Gibbs free energy profile diagram for the 4CzIPN catalyzed hydroaminoalkylation reaction of boc-glycine and 1,1-diphenylethylene at the PBE0-D3(BJ)/Def2-TZVPP+SMD(DMSO)//PBE0-D3(BJ)/6-31G(d) level of theory. The spin natural orbitals of species F and the transition state TS1 are visualized using an isovalue of 0.05. The bond lengths are given in Å. | ||
Greenness advancements
In organic chemistry, the transition towards more sustainable and environmentally conscious methodologies has gained considerable traction.113 In this section, the greenness advancements of our study will be discussed solely on the photocatalytic HAM reaction. The disadvantages of the noteworthy HAA reactions are described in the introduction.The photocatalytic HAM reaction, which has been studied extensively over the past decade, generally relies on the use of iridium photocatalysts. A notable advancement towards greener methods involves the replacement of conventional iridium-based photocatalysts with organic counterparts, such as 4CzIPN in our work.114
Atom economy is a central principle in green chemistry, assessing the efficiency of chemical reactions by measuring atom utilization and reducing the ecological impact (waste). Previous photocatalytic HAM methodologies generally made use of potassium alkyltrifluoroborates, phthalimide redox handles, Hantzsch esters and alkyltrimethylsilanes.70,86–92 These radical precursors generate a tremendous amount of waste, which decreases the greenness of the procedure. By using carboxylic acids, only carbon dioxide is released, which can be feasibly captured on bigger industrial scales.115 Moreover, the use of widely available and cheap amino acids as the radical source eliminates the (extensive) synthesis routes for the generation of the abovementioned radical precursors.
While the developed reaction represents a stride toward a more environmentally friendly synthetic process, it is important to acknowledge one drawback associated with this innovation – the production of CO2 as a byproduct. Although the generation of CO2 is typically viewed as a less desirable outcome due to its contribution to greenhouse gas emissions and global climate change, it is crucial to recognize that even in the context of “green” chemistry, some degree of waste production may be inevitable. Although CO2 formation as a byproduct is never seen as a positive aspect, it can be captured and utilized in various applications such as carbonation of beverages, enhanced growth in greenhouses, or in carbon capture and utilization (CCU) processes to mitigate its environmental impact.116,117
Conclusions
In conclusion, our research endeavors have significantly contributed to the existing knowledge on the HAM reaction. In this paper, we highlighted our novel visible-light mediated redox-neutral decarboxylative radical polar crossover process that addresses a significant gap in the existing literature on the HAM reaction. Notably, previous publications have predominantly focused on either the functionalization of styrene derivatives or the functionalization of Michael acceptors. However, our research has led to the discovery of a robust system capable of effectively catalyzing both substrate types in moderate to excellent yields. In addition, we eliminated the need for α-amino radicals and successfully created the positioning of the radical center at a greater distance from the nitrogen center, enabling the synthesis of long chain aliphatic amines from longer chain unnatural amino acids. To the best of our knowledge, this is the first reported example of a photoredox-mediated HAA reaction using distant primary carbon centered radicals to access linear aliphatic amines. This finding opens up new possibilities in the synthesis of diverse amine compounds and further underscores the broad potential of our developed system. Moreover, the robustness was proved by the late-stage functionalization of pharmaceuticals as well as the synthesis of drug compounds. Additionally, mechanistic studies clearly revealed the formation of radicals by the elimination of carbon dioxide from the amino acid. Lastly, DFT studies revealed the stabilization of the primary radical by the spin natural orbitals of F′ with the adjacent C–H bond orbitals.Author contributions
R.C. and S.D. conceived and designed the experiments. R.C., P.K.S. and R.M. performed the experiments, analysed the data and prepared the manuscript. R.C. performed the Stern–Volmer experiments. A.M and R.K did the DFT calculations. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the FWO research grant, FWO PhD fellowship, BOF-DOCPRO, Odysseus grant, and DST-SERB SRG grant (SRG/2022/000307). We thank Chandra High Performance Computing (HPC) facilities at IIT Palakkad.References
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Hydroamination: Direct Addition of Amines to Alkenes and Alkynes, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed.
- P. Vineis and R. Pirastu, Aromatic amines and cancer, Cancer Causes & Control, 1997, 8, 346–355 CrossRef CAS PubMed.
- H. Gröger, Biocatalytic concepts for synthesizing amine bulk chemicals: recent approaches towards linear and cyclic aliphatic primary amines and ω-substituted derivatives thereof, Appl. Microbiol. Biotechnol., 2019, 103, 83–95 CrossRef PubMed.
- S. L. Rice, R. R. Eitenmiller and P. E. Koehler, Biologically Active Amines in Food: A Review, J. Milk Food Technol., 1976, 39, 353–358 CrossRef CAS.
- T. Wang, P. M. Stein, H. Shi, C. Hu, M. Rudolph and A. S. K. Hashmi, Hydroxylamine-mediated C–C amination via an aza-hock rearrangement, Nat. Commun., 2021, 12, 7029 CrossRef CAS PubMed.
- T. Ubuka, in Handbook of Hormones (Second Edition), ed. H. Ando, K. Ukena and S. Nagata, Academic Press, San Diego, 2021, pp. 1035–1036, DOI:10.1016/B978-0-12-820649-2.00288-6.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- B. Buntz, 50 of 2021s best-selling pharmaceuticals. drugdiscoverytrends (2022). Available at: https://www.drugdiscoverytrends.com/50-of-2021s-best-selling-pharmaceuticals/. (Accessed: 16th August 2022).
- E. Vitaku, D. T. Smith and J. T. Njardarson, Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS.
- A. Trowbridge, S. M. Walton and M. J. Gaunt, New Strategies for the Transition-Metal Catalyzed Synthesis of Aliphatic Amines, Chem. Rev., 2020, 120, 2613–2692 CrossRef CAS.
- M. Ahmed, A. M. Seayad, R. Jackstell and M. Beller, Amines Made Easily: A Highly Selective Hydroaminomethylation of Olefins, J. Am. Chem. Soc., 2003, 125, 10311–10318 CrossRef CAS PubMed.
- S. Das, D. Addis, K. Junge and M. Beller, Zinc-Catalyzed Chemoselective Reduction of Tertiary and Secondary Amides to Amines, Eur. J. Chem., 2011, 17, 12186–12192 CrossRef CAS.
- S. Das, S. Zhou, D. Addis, S. Enthaler, K. Junge and M. Beller, Selective Catalytic Reductions of Amides and Nitriles to Amines, Top. Catal., 2010, 53, 979–984 CrossRef CAS.
- S. Das, D. Addis, S. Zhou, K. Junge and M. Beller, Zinc-Catalyzed Reduction of Amides: Unprecedented Selectivity and Functional Group Tolerance, J. Am. Chem. Soc., 2010, 132, 1770–1771 CrossRef CAS.
- S. Das, B. Wendt, K. Möller, K. Junge and M. Beller, Two Iron Catalysts are Better than One: A General and Convenient Reduction of Aromatic and Aliphatic Primary Amides, Angew. Chem., Int. Ed., 2012, 51, 1662–1666 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Selective Synthesis of Primary Amines Directly from Alcohols and Ammonia, Angew. Chem., Int. Ed., 2008, 47, 8661–8664 CrossRef CAS PubMed.
- A. Borzenko, N. L. Rotta-Loria, P. M. MacQueen, C. M. Lavoie, R. McDonald and M. Stradiotto, Nickel-catalyzed monoarylation of ammonia, Angew. Chem., Int. Ed., 2015, 54, 3773–3777 CrossRef CAS.
- R. A. Green and J. F. Hartwig, Nickel-Catalyzed Amination of Aryl Chlorides with Ammonia or Ammonium Salts, Angew. Chem., Int. Ed., 2015, 54, 3768–3772 CrossRef CAS.
- C. Zhu, G. Li, D. H. Ess, J. R. Falck and L. Kürti, Elusive Metal-Free Primary Amination of Arylboronic Acids: Synthetic Studies and Mechanism by Density Functional Theory, J. Am. Chem. Soc., 2012, 134, 18253–18256 CrossRef CAS.
- J. Peng, M. Chen, Z. Xie, S. Luo and Q. Zhu, Copper-mediated C(sp2)–H amination using TMSN3 as a nitrogen source: redox-neutral access to primary anilines, Org. Chem. Front., 2014, 1, 777–781 RSC.
- X. Tian, L. Song and A. S. K. Hashmi, Synthesis of Carbazoles and Related Heterocycles from Sulfilimines by Intramolecular C−H Aminations, Angew. Chem., Int. Ed., 2020, 59, 12342–12346 CrossRef CAS.
- J. Xie, S. Shi, T. Zhang, N. Mehrkens, M. Rudolph and A. S. K. Hashmi, A Highly Efficient Gold-Catalyzed Photoredox α-C(sp3)□H Alkynylation of Tertiary Aliphatic Amines with Sunlight, Angew. Chem., Int. Ed., 2015, 54, 6046–6050 CrossRef CAS PubMed.
- B. Górski, A.-L. Barthelemy, J. J. Douglas, F. Juliá and D. Leonori, Copper-catalysed amination of alkyl iodides enabled by halogen-atom transfer, Nat. Catal., 2021, 4, 623–630 CrossRef.
- T. Gross, A. M. Seayad, M. Ahmad and M. Beller, Synthesis of Primary Amines: First Homogeneously Catalyzed Reductive Amination with Ammonia, Org. Lett., 2002, 4, 2055–2058 CrossRef CAS PubMed.
- J. Gallardo-Donaire, M. Ernst, O. Trapp and T. Schaub, Direct Synthesis of Primary Amines via Ruthenium-Catalysed Amination of Ketones with Ammonia and Hydrogen, Adv. Synth. Catal., 2016, 358, 358–363 CrossRef CAS.
- J. Gallardo-Donaire, M. Hermsen, J. Wysocki, M. Ernst, F. Rominger, O. Trapp, A. S. K. Hashmi, A. Schäfer, P. Comba and T. Schaub, Direct Asymmetric Ruthenium-Catalyzed Reductive Amination of Alkyl–Aryl Ketones with Ammonia and Hydrogen, J. Am. Chem. Soc., 2018, 140, 355–361 CrossRef CAS.
- Z. Wu, W. Wang, H. Guo, G. Gao, H. Huang and M. Chang, Iridium-catalyzed direct asymmetric reductive amination utilizing primary alkyl amines as the N-sources, Nat. Commun., 2022, 13, 3344 CrossRef CAS PubMed.
- M. S. Gibson and R. W. Bradshaw, The Gabriel Synthesis of Primary Amines, Angew. Chem., Int. Ed. Engl., 1968, 7, 919–930 CrossRef CAS.
- A. K. Ghosh, M. Brindisi and A. Sarkar, The Curtius Rearrangement: Applications in Modern Drug Discovery and Medicinal Chemistry, ChemMedChem, 2018, 13, 2351–2373 CrossRef CAS.
- I. M. Lapina, L. M. Pevzner and A. A. Potekhin, Reactions of alkyl (halomethyl)furancarboxylates with hexamethylenetetramine, Russ. J. Gen. Chem., 2006, 76, 1304–1309 CrossRef CAS.
- P. L. Anelli, L. Lattuada and F. Uggeri, One-Pot Mitsunobu-Staudinger Preparation of 3-Aminocholan-24-oic Acid Esters from 3-Hydroxycholan-24-oic Acid Esters, Synth. Commun., 1998, 28, 109–117 CrossRef CAS.
- T. E. Müller and M. Beller, Metal-Initiated Amination of Alkenes and Alkynes, Chem. Rev., 1998, 98, 675–704 CrossRef.
- A. E. Strom and J. F. Hartwig, One-Pot Anti-Markovnikov Hydroamination of Unactivated Alkenes by Hydrozirconation and Amination, J. Org. Chem., 2013, 78, 8909–8914 CrossRef CAS PubMed.
- S. Guo, J. C. Yang and S. L. Buchwald, A Practical Electrophilic Nitrogen Source for the Synthesis of Chiral Primary Amines by Copper-Catalyzed Hydroamination, J. Am. Chem. Soc., 2018, 140, 15976–15984 CrossRef CAS PubMed.
- Y. Zhou, X. Xu, H. Sun, G. Tao, X.-Y. Chang, X. Xing, B. Chen and C. Xu, Development of highly efficient platinum catalysts for hydroalkoxylation and hydroamination of unactivated alkenes, Nat. Commun., 2021, 12, 1953 CrossRef CAS PubMed.
- W. Reppe and H. Vetter, Carbonylierung VI. Synthesen mit Metallcarbonylwasserstoffen, Justus Liebigs Ann. Chem., 1953, 582, 133–161 CrossRef CAS.
- C. Chen, X.-Q. Dong and X. Zhang, Recent progress in rhodium-catalyzed hydroaminomethylation, Org. Chem. Front., 2016, 3, 1359–1370 RSC.
- D. Crozet, M. Urrutigoïty and P. Kalck, Recent Advances in Amine Synthesis by Catalytic Hydroaminomethylation of Alkenes, ChemCatChem, 2011, 3, 1102–1118 CrossRef CAS.
- P. Kalck and M. Urrutigoïty, Tandem Hydroaminomethylation Reaction to Synthesize Amines from Alkenes, Chem. Rev., 2018, 118, 3833–3861 CrossRef CAS PubMed.
- S. Hanna, J. C. Holder and J. F. Hartwig, A Multicatalytic Approach to the Hydroaminomethylation of α-Olefins, Angew. Chem., Int. Ed., 2019, 58, 3368–3372 CrossRef CAS.
- R. Kubiak, I. Prochnow and S. Doye, [Ind2TiMe2]: A Catalyst for the Hydroaminomethylation of Alkenes and Styrenes, Angew. Chem., Int. Ed., 2010, 49, 2626–2629 CrossRef CAS.
- P. W. Roesky, Catalytic Hydroaminoalkylation, Angew. Chem., Int. Ed., 2009, 48, 4892–4894 CrossRef CAS PubMed.
- S. B. Herzon and J. F. Hartwig, Hydroaminoalkylation of Unactivated Olefins with Dialkylamines, J. Am. Chem. Soc., 2008, 130, 14940–14941 CrossRef CAS PubMed.
- M. Y. S. Ibrahim and M. Abolhasani, Recyclable cooperative catalyst for accelerated hydroaminomethylation of hindered amines in a continuous segmented flow reactor, Nat. Commun., 2022, 13, 2441 CrossRef CAS.
- R. C. DiPucchio, S.-C. Rosca and L. L. Schafer, Hydroaminoalkylation for the Catalytic Addition of Amines to Alkenes or Alkynes: Diverse Mechanisms Enable Diverse Substrate Scope, J. Am. Chem. Soc., 2022, 144, 11459–11481 CrossRef CAS.
- P. Eisenberger, R. O. Ayinla, J. M. P. Lauzon and L. L. Schafer, Tantalum–Amidate Complexes for the Hydroaminoalkylation of Secondary Amines: Enhanced Substrate Scope and Enantioselective Chiral Amine Synthesis, Angew. Chem., Int. Ed., 2009, 48, 8361–8365 CrossRef CAS PubMed.
- R. Cauwenbergh and S. Das, Photocatalysis: A Green Tool for Redox Reactions, Synlett, 2021, 129–149, DOI:10.1055/s-0040-1706042.
- W. Schilling, D. Riemer, Y. Zhang, N. Hatami and S. Das, Metal-Free Catalyst for Visible-Light-Induced Oxidation of Unactivated Alcohols Using Air/Oxygen as an Oxidant, ACS Catal., 2018, 8, 5425–5430 CrossRef CAS.
- Y. Zhang, N. Hatami, N. S. Lange, E. Ronge, W. Schilling, C. Jooss and S. Das, A metal-free heterogeneous photocatalyst for the selective oxidative cleavage of C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in aryl olefins via harvesting direct solar energy, Green Chem., 2020, 22, 4516–4522 RSC.
C bonds in aryl olefins via harvesting direct solar energy, Green Chem., 2020, 22, 4516–4522 RSC. - R. Cauwenbergh and S. Das, Photochemical reduction of carbon dioxide to formic acid, Green Chem., 2021, 23, 2553–2574 RSC.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis?, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS.
- J. Rong, P. H. Seeberger and K. Gilmore, Chemoselective Photoredox Synthesis of Unprotected Primary Amines Using Ammonia, Org. Lett., 2018, 20, 4081–4085 CrossRef CAS PubMed.
- M. C. Nicastri, D. Lehnherr, Y.-h. Lam, D. A. DiRocco and T. Rovis, Synthesis of Sterically Hindered Primary Amines by Concurrent Tandem Photoredox Catalysis, J. Am. Chem. Soc., 2020, 142, 987–998 CrossRef CAS.
- M. C. Wood, D. C. Leitch, C. S. Yeung, J. A. Kozak and L. L. Schafer, Chiral Neutral Zirconium Amidate Complexes for the Asymmetric Hydroamination of Alkenes, Angew. Chem., Int. Ed., 2007, 46, 354–358 CrossRef CAS PubMed.
- A. Tillack, I. Garcia Castro, C. G. Hartung and M. Beller, Anti-Markovnikov Hydroamination of Terminal Alkynes, Angew. Chem., Int. Ed., 2002, 41, 2541–2543 CrossRef CAS.
- D. C. Leitch, P. R. Payne, C. R. Dunbar and L. L. Schafer, Broadening the Scope of Group 4 Hydroamination Catalysis Using a Tethered Ureate Ligand, J. Am. Chem. Soc., 2009, 131, 18246–18247 CrossRef CAS PubMed.
- D. C. Miller, J. M. Ganley, A. J. Musacchio, T. C. Sherwood, W. R. Ewing and R. R. Knowles, Anti-Markovnikov Hydroamination of Unactivated Alkenes with Primary Alkyl Amines, J. Am. Chem. Soc., 2019, 141, 16590–16594 CrossRef CAS PubMed.
- J. M. Ganley, P. R. D. Murray and R. R. Knowles, Photocatalytic Generation of Aminium Radical Cations for C–N Bond Formation, ACS Catal., 2020, 10, 11712–11738 CrossRef CAS PubMed.
- D. Francis, A. Nelson and S. P. Marsden, Synthesis of β-Diamine Building Blocks by Photocatalytic Hydroamination of Enecarbamates with Amines, Ammonia and N−H Heterocycles, Eur. J. Chem., 2020, 26, 14861–14865 CrossRef CAS.
- A. J. Musacchio, B. C. Lainhart, X. Zhang, S. G. Naguib, T. C. Sherwood and R. R. Knowles, Catalytic intermolecular hydroaminations of unactivated olefins with secondary alkyl amines, Science, 2017, 355, 727–730 CrossRef CAS PubMed.
- A. J. Musacchio, L. Q. Nguyen, G. H. Beard and R. R. Knowles, Catalytic Olefin Hydroamination with Aminium Radical Cations: A Photoredox Method for Direct C–N Bond Formation, J. Am. Chem. Soc., 2014, 136, 12217–12220 CrossRef CAS PubMed.
- A. J. Chinn, K. Sedillo and A. G. Doyle, Phosphine/Photoredox Catalyzed Anti-Markovnikov Hydroamination of Olefins with Primary Sulfonamides via α-Scission from Phosphoranyl Radicals, J. Am. Chem. Soc., 2021, 143, 18331–18338 CrossRef CAS PubMed.
- Z.-P. Ye, Y.-Z. Hu, P.-J. Xia, H.-Y. Xiang, K. Chen and H. Yang, Photocatalytic intermolecular anti-Markovnikov hydroamination of unactivated alkenes with N-hydroxyphthalimide, Org. Chem. Front., 2021, 8, 273–277 RSC.
- Y.-D. Du, B.-H. Chen and W. Shu, Direct Access to Primary Amines from Alkenes by Selective Metal-Free Hydroamination, Angew. Chem., Int. Ed., 2021, 60, 9875–9880 CrossRef CAS PubMed.
- T. M. Nguyen and D. A. Nicewicz, Anti-Markovnikov Hydroamination of Alkenes Catalyzed by an Organic Photoredox System, J. Am. Chem. Soc., 2013, 135, 9588–9591 CrossRef CAS PubMed.
- T. M. Nguyen, N. Manohar and D. A. Nicewicz, Anti-Markovnikov Hydroamination of Alkenes Catalyzed by a Two-Component Organic Photoredox System: Direct Access to Phenethylamine Derivatives, Angew. Chem., Int. Ed., 2014, 53, 6198–6201 CrossRef CAS.
- H. Ishizuka, K. Adachi, M. Odagi and K. Nagasawa, Synthesis of Isoxazolidines by Intramolecular Hydroamination of N-Alkoxyamides in the Presence of a Visible-Light Photoredox Catalyst, Bull. Chem. Soc. Jpn., 2019, 92, 1447–1449 CrossRef CAS.
- T. Taeufer, R. Hauptmann, F. El-Hage, T. S. Mayer, H. Jiao, J. Rabeah and J. Pospech, Pyrimidopteridine-Catalyzed Hydroamination of Stilbenes with Primary Amines: A Dual Photoredox and Hydrogen Atom Transfer Catalyst, ACS Catal., 2021, 11, 4862–4869 CrossRef CAS.
- X. Chai, X. Hu, X. Zhao, Y. Yin, S. Cao and Z. Jiang, Asymmetric Hydroaminoalkylation of Alkenylazaarenes via Cooperative Photoredox and Chiral Hydrogen-Bonding Catalysis, Angew. Chem., Int. Ed., 2022, 61, e202115110 CrossRef CAS.
- Z. Wu, S. N. Gockel and K. L. Hull, Anti-Markovnikov hydro(amino)alkylation of vinylarenes via photoredox catalysis, Nat. Commun., 2021, 12, 5956 CrossRef CAS PubMed.
- L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, Carboxylic Acids as A Traceless Activation Group for Conjugate Additions: A Three-Step Synthesis of (±)-Pregabalin, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS.
- Y. Miyake, K. Nakajima and Y. Nishibayashi, Visible-Light-Mediated Utilization of α-Aminoalkyl Radicals: Addition to Electron-Deficient Alkenes Using Photoredox Catalysts, J. Am. Chem. Soc., 2012, 134, 3338–3341 CrossRef CAS PubMed.
- J. Ye, I. Kalvet, F. Schoenebeck and T. Rovis, Direct α-alkylation of primary aliphatic amines enabled by CO2 and electrostatics, Nat. Chem., 2018, 10, 1037–1041 CrossRef CAS PubMed.
- S. M. Thullen and T. Rovis, A Mild Hydroaminoalkylation of Conjugated Dienes Using a Unified Cobalt and Photoredox Catalytic System, J. Am. Chem. Soc., 2017, 139, 15504–15508 CrossRef CAS PubMed.
- J. Zheng and B. Breit, Regiodivergent Hydroaminoalkylation of Alkynes and Allenes by a Combined Rhodium and Photoredox Catalytic System, Angew. Chem., Int. Ed., 2019, 58, 3392–3397 CrossRef CAS PubMed.
- J. B. McManus, N. P. R. Onuska and D. A. Nicewicz, Generation and Alkylation of α-Carbamyl Radicals via Organic Photoredox Catalysis, J. Am. Chem. Soc., 2018, 140, 9056–9060 CrossRef CAS PubMed.
- J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Asymmetric catalytic formation of quaternary carbons by iminium ion trapping of radicals, Nature, 2016, 532, 218–222 CrossRef CAS PubMed.
- A. Trowbridge, D. Reich and M. J. Gaunt, Multicomponent synthesis of tertiary alkylamines by photocatalytic olefin-hydroaminoalkylation, Nature, 2018, 561, 522–527 CrossRef CAS PubMed.
- H. B. Hepburn and P. Melchiorre, Brønsted acid-catalysed conjugate addition of photochemically generated α-amino radicals to alkenylpyridines, Chem. Commun., 2016, 52, 3520–3523 RSC.
- P. Fernandez-Rodriguez, F. Legros, T. Maier, A. Weber, M. Méndez, V. Derdau, G. Hessler, M. Kurz, A. Villar-Garea and S. Ruf, Photoinduced Decarboxylative Radical Addition Reactions for Late Stage Functionalization of Peptide Substrates, Eur. J. Org. Chem., 2021, 782–787 CrossRef CAS.
- A. Millet, Q. Lefebvre and M. Rueping, Visible-Light Photoredox-Catalyzed Giese Reaction: Decarboxylative Addition of Amino Acid Derived α-Amino Radicals to Electron-Deficient Olefins, Eur. J. Chem., 2016, 22, 13464–13468 CrossRef CAS.
- Y.-F. Si, X.-L. Chen, X.-Y. Fu, K. Sun, X. Song, L.-B. Qu and B. Yu, Divergent g-C3N4-catalyzed Reactions of Quinoxalin-2(1H)-ones with N-Aryl Glycines under Visible Light: Solvent-Controlled Hydroaminomethylation and Annulation, ACS Sustainable Chem. Eng., 2020, 8, 10740–10746 CAS.
- J. Zheng, A. Nikbakht and B. Breit, Dual Palladium/Photoredox-Catalyzed Enantioselective and Regioselective Decarboxylative Hydroaminoalkylation of Allenes, ACS Catal., 2021, 11, 3343–3350 CrossRef CAS.
- J. Zheng, N. Tang, H. Xie and B. Breit, Regio-, Diastereo-, and Enantioselective Decarboxylative Hydroaminoalkylation of Dienol Ethers Enabled by Dual Palladium/Photoredox Catalysis, Angew. Chem., Int. Ed., 2022, 61, e202200105 CrossRef CAS PubMed.
- J. Schwarz and B. König, Metal-free, visible-light-mediated, decarboxylative alkylation of biomass-derived compounds, Green Chem., 2016, 18, 4743–4749 RSC.
- Z.-Y. Cao, T. Ghosh and P. Melchiorre, Enantioselective radical conjugate additions driven by a photoactive intramolecular iminium-ion-based EDA complex, Nat. Commun., 2018, 9, 3274 CrossRef PubMed.
- E. Le Saux, D. Ma, P. Bonilla, C. M. Holden, D. Lustosa and P. Melchiorre, A General Organocatalytic System for Enantioselective Radical Conjugate Additions to Enals, Angew. Chem., Int. Ed., 2021, 60, 5357–5362 CrossRef CAS PubMed.
- K. Miyazawa, T. Koike and M. Akita, Hydroaminomethylation of Olefins with Aminomethyltrifluoroborate by Photoredox Catalysis, Adv. Synth. Catal., 2014, 356, 2749–2755 CrossRef CAS.
- J. Ma, J. Lin, L. Zhao, K. Harms, M. Marsch, X. Xie and E. Meggers, Synthesis of β-Substituted γ-Aminobutyric Acid Derivatives through Enantioselective Photoredox Catalysis, Angew. Chem., Int. Ed., 2018, 57, 11193–11197 CrossRef CAS PubMed.
- F. Gu, W. Huang, X. Liu, W. Chen and X. Cheng, Substituted Hantzsch Esters as Versatile Radical Reservoirs in Photoredox Reactions, Adv. Synth. Catal., 2018, 360, 925–931 CrossRef CAS.
- S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. MacMillan, Decarboxylative alkylation for site-selective bioconjugation of native proteins via oxidation potentials, Nat. Chem., 2018, 10, 205–211 CrossRef CAS.
- Y. Yin, Y. Dai, H. Jia, J. Li, L. Bu, B. Qiao, X. Zhao and Z. Jiang, Conjugate Addition–Enantioselective Protonation of N-Aryl Glycines to α-Branched 2-Vinylazaarenes via Cooperative Photoredox and Asymmetric Catalysis, J. Am. Chem. Soc., 2018, 140, 6083–6087 CrossRef CAS.
- H. E. Askey, J. D. Grayson, J. D. Tibbetts, J. C. Turner-Dore, J. M. Holmes, G. Kociok-Kohn, G. L. Wrigley and A. J. Cresswell, Photocatalytic Hydroaminoalkylation of Styrenes with Unprotected Primary Alkylamines, J. Am. Chem. Soc., 2021, 143, 15936–15945 CrossRef CAS.
- S. Zheng, W. Wang and W. Yuan, Remote and Proximal Hydroaminoalkylation of Alkenes Enabled by Photoredox/Nickel Dual Catalysis, J. Am. Chem. Soc., 2022, 144, 17776–17782 CrossRef CAS PubMed.
- A. S. H. Ryder, W. B. Cunningham, G. Ballantyne, T. Mules, A. G. Kinsella, J. Turner-Dore, C. M. Alder, L. J. Edwards, B. S. J. McKay, M. N. Grayson and A. J. Cresswell, Photocatalytic α-Tertiary Amine Synthesis via C−H Alkylation of Unmasked Primary Amines, Angew. Chem., Int. Ed., 2020, 59, 14986–14991 CrossRef CAS PubMed.
- M. Garreau, F. Le Vaillant and J. Waser, C-Terminal Bioconjugation of Peptides through Photoredox Catalyzed Decarboxylative Alkynylation, Angew. Chem., Int. Ed., 2019, 58, 8182–8186 CrossRef CAS PubMed.
- C. Chen, X. Wang and T. Yang, Recent Synthetic Applications of the Hypervalent Iodine(III) Reagents in Visible-Light-Induced Photoredox Catalysis, Front. Chem., 2020, 8, 551159 CrossRef CAS PubMed.
- T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Recent advances of 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) in photocatalytic transformations, Chem. Commun., 2019, 55, 5408–5419 RSC.
- Y. Jin and H. Fu, Visible-Light Photoredox Decarboxylative Couplings, Asian J. Org. Chem., 2017, 6, 368–385 CrossRef CAS.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Tools and techniques for solvent selection: green solvent selection guides, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- R. E. Gawley, Overview of Carbanion Dynamics and Electrophilic Substitutions in Chiral Organolithium Compounds, Top. Stereochem., 2010, 26, 93–133 CAS.
- N. A. Larionova, J. M. Ondozabal, E. G. Smith and X. C. Cambeiro, A Photocatalytic Regioselective Direct Hydroaminoalkylation of Aryl-Substituted Alkenes with Amines, Org. Lett., 2021, 23, 5383–5388 CrossRef CAS PubMed.
- J. E. Frampton and R. H. Foster, Pregabalin, Drugs, 2005, 65, 111–118 CrossRef PubMed.
- M. S. Hoekstra, D. M. Sobieray, M. A. Schwindt, T. A. Mulhern, T. M. Grote, B. K. Huckabee, V. S. Hendrickson, L. C. Franklin, E. J. Granger and G. L. Karrick, Chemical Development of CI-1008, an Enantiomerically Pure Anticonvulsant, Org. Process Res. Dev., 1997, 1, 26–38 CrossRef CAS.
- C. C. C. R. de Carvalho and M. M. R. da Fonseca, Carvone: Why and how should one bother to produce this terpene, Food Chem., 2006, 95, 413–422 CrossRef CAS.
- J. Börgel and T. Ritter, Late-Stage Functionalization, Chem, 2020, 6, 1877–1887 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- I. V. Alabugin, G. dos Passos Gomes and M. A. Abdo, Hyperconjugation, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1389 Search PubMed.
- P. Anastas and N. Eghbali, Green Chemistry: Principles and Practice, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- D. C. J. Constable, Green chemistry metrics, in Handbook of green chemistry, ed. D. J. C. Constable and C. Jimenéz-González, Wiley BCH, Weinheim, 2018, vol. 11, pp. 1–27 Search PubMed.
- W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A.-C. Roger, R. Amal, H. He and S.-E. Park, Industrial carbon dioxide capture and utilization: state of the art and future challenges, Chem. Soc. Rev., 2020, 49, 8584–8686 RSC.
- J. Vansant, in Carbon Dioxide Recovery and Utilization, ed. M. Aresta, Springer Netherlands, Dordrecht, 2003, pp. 3–50 Search PubMed.
- R. J. Allam, R. Bredesen and E. Drioli, in Carbon Dioxide Recovery and Utilization, ed. M. Aresta, Springer Netherlands, Dordrecht, 2003, pp. 53–120, DOI:10.1007/978-94-017-0245-4_2.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03187j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |