
Unexpected stereoselective CuBr2-catalyzed cascade reaction of 2-ethynylanilines with silylynamides: facile and atom-economical access to N-vinylsilylindoles†
Zengzeng
Li‡
,
Fei
Lu‡
,
Qingchun
Xu
,
Gang
Liu
,
Ximei
Zhao
 * and
Guanghui
Wang
*
* and
Guanghui
Wang
*
School of Chemistry and Materials Science, Ludong University, Yantai 264025, China. E-mail: ximeizhao@ldu.edu.cn; wangguanghui@ldu.edu.cn
First published on 4th December 2023
Abstract
A stereoselective copper-catalyzed cascade reaction of 2-ethynylanilines with silylynamides has been developed. In this protocol, inexpensive CuBr2 is utilized as the catalyst. This simple, ligand-free, and cost-effective catalytic system furnishes valuable N-vinylsilylindoles in good to excellent yields, with perfect stereoselectivity in open air. Its potential synthetic applicability is illustrated by a large-scale experiment and down-stream synthetic transformation of the products.
Indoles are prevalent structural motifs in numerous natural products, pharmaceuticals, agrochemicals, and functional materials.1 Among them, N-vinylindoles are of particular importance, serving not only as core structures of various natural products and bioactive compounds,2 but also as valuable monomers for photoactive and polymeric materials.3 Therefore, the development of synthetic strategies for their preparation has attracted immense attention. Transition-metal-catalyzed cross-coupling reactions between indoles and coupling partners, such as vinyl halides,4 vinyl triflates,5 alkenes,6 and N-tosylhydrazones,7 represent reliable methods. The hydroamination of indoles with alkynes has been described by Verma and co-workers, furnishing N-vinylindoles in moderate to good yields.8 Ynamides are special alkynes with unique reactivity.9 Over the past few decades, they have become prominent building blocks in synthetic chemistry, enabling rapid assembly of a diverse array of N-containing molecules.10 For the vinylation of indoles with ynamides, in most cases, the reaction takes place exclusively at the C3 position due to its innate nucleophilic nature (Scheme 1a, left).11 In 2014, an unprecedented base-promoted N-vinylation of indoles with terminal ynamides was reported by Dodd and co-workers, providing a convenient access to N-vinylindoles (Scheme 1a, right).12 Despite these advances, current methodologies toward N-vinylindoles mainly focus on the direct N–H bond functionalization of indoles, whereas the more challenging one-step construction of indole skeletons together with installation of vinyl moieties at the N1 position is scarcely explored. Although a AuCl3/AgOTf-catalyzed cascade reaction of 2-alkynylanilines with terminal alkynes for the synthesis of N-vinylindoles was reported in 2007 by Li and co-workers, the products were limited to N-terminalvinylindoles (Scheme 1b).13 The introduction of a biologically important silyl group was impossible. Besides, their yields of most products were not so good (only one example over 80%).
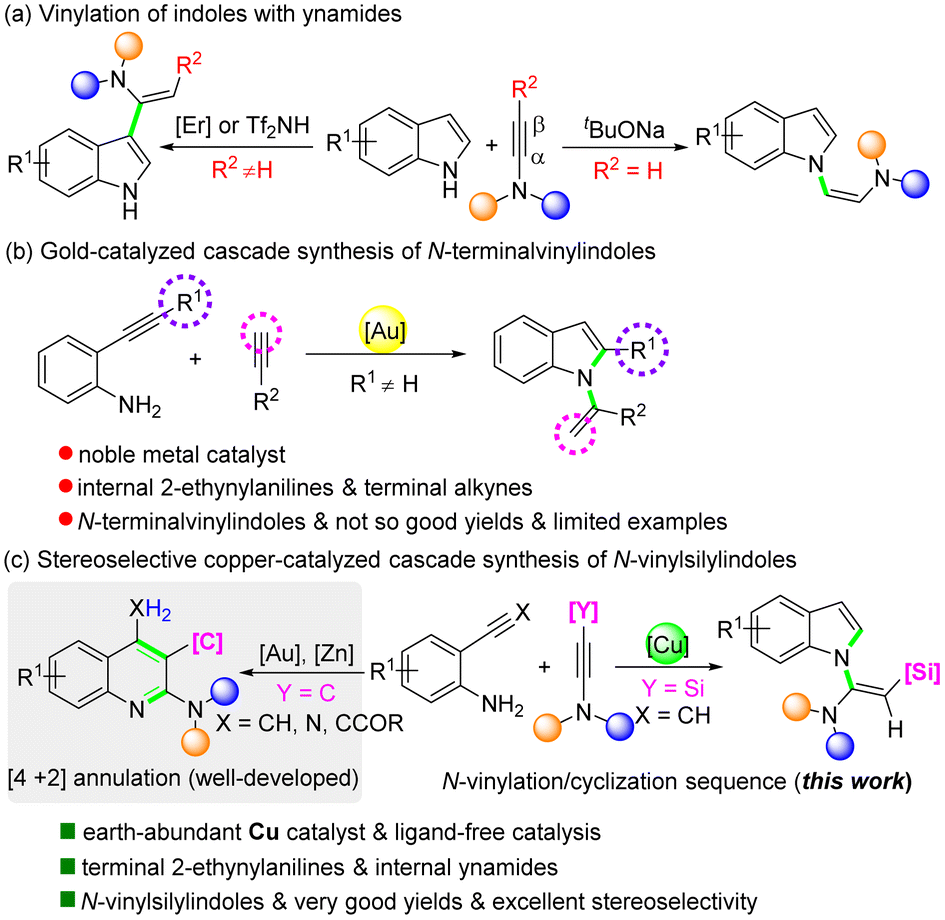 | ||
| Scheme 1 Synthesis of vinylindoles. | ||
Copper is an earth-abundant metal. Thus, its use as catalysts is more sustainable and more cost-effective than precious transition metal catalysts. In many organic transformations, copper has been considered as a first-line catalyst.14 In view of green and sustainable chemistry, and in continuation of our interest in aza-heterocycles and organosilanes,15 herein, we are pleased to report an unexpected stereoselective CuBr2-catalyzed cascade reaction of 2-ethynylanilines with silylynamides, allowing facile and atom-economical synthesis of N-vinylsilylindoles (Scheme 1c, right). It is interesting that this CuBr2-catalyzed reaction of 2-ethynylanilines with silylynamides did not occur in a common [4 + 2] mode for the reactions between aminoalkynes or aminonitriles and ynamides to give the quinoline products (Scheme 1c, left).16 Instead, unexpected but valuable N-vinylsilylindoles were obtained in good to excellent yields with perfect stereoselectivity (Scheme 1b, right). To our knowledge, this is the first stereoselective tandem synthesis of N-vinylindole scaffolds. In view of the important role of silicon atoms in bioactive compounds,17 this combination of indole frameworks with silyl groups might also make great sense for medicinal chemistry.
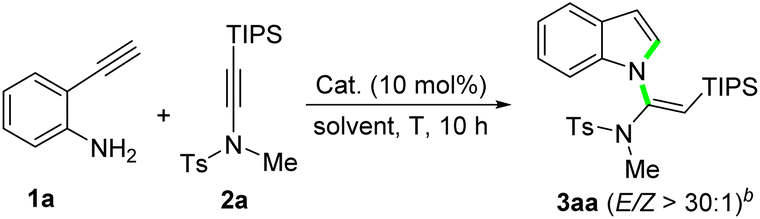
We initiated the study by using 2-ethynylaniline 1a and ynamide 2a as model substrates to optimize the reaction conditions (Table 1). Treatment of 2-ethynylaniline 1a and silylynamide 2a with 10 mol% ZnCl2 in 1,2-dichloroethane (DCE) at 100 °C for 10 h under a N2 atmosphere led to the desired product 3aa in 44% yield with perfect (E)-selectivity (entry 1). Simply changing ZnCl2 to ZnBr2 increased the yield to 64% (entry 2). ZnI2 and Zn(OTf)2 also afforded 3aa, but in lower yields (entries 3 and 4). Then we tried to perform the reaction by using CuBr2 as the catalyst. To our surprise, the yield rose to 96% (entry 5). But other screened copper salts, including Cu(OTf)2, CuCl and CuBr were much less efficient (entries 6–8). When PPh3AuCl/AgNTf2 was used as the catalyst, only a trace amount of the product 3aa was formed, and a large amount of the starting materials were recovered (entry 9). Changing the solvent to toluene, 1,4-dioxane, and dimethyl sulfoxide (DMSO) did not generate better results (entries 10–12). The yield remained virtually unchanged at a higher reaction temperature of 120 °C (entry 13), while decreasing the reaction temperature to 80 °C reduced the yield to 80% (entry 14). A lower dose of CuBr2 was also evaluated, but the yield dropped to 84% (entry 15). Finally, we tried the reaction in open vials, and found that there was no decrease in the yield (entry 16). Thus, the N2 atmosphere was unnecessary for this protocol.
|
|
||||
|---|---|---|---|---|
| Entry | Cat. (%) | Solv. | T (°C) | Yieldc (%) |
| a Reaction conditions: 1a (0.3 mmol), 2a (0.2 mmol), and catalyst (0.02 mmol) in solvent (1.0 mL) for 10 h under N2 atmosphere. Tf = trifluoromethylsulfonyl. b The E/Z ratio was determined by 1H NMR of the unpurified reaction mixture. c The NMR yield was measured by 1H NMR using 1,3,5-trimethoxybenzene as the internal reference. d Not detected. e With 0.01 mmol of catalyst. f The reaction was operated in open vials, and after operation the vials were closed. g Isolated yield. | ||||
| 1 | ZnCl2 | DCE | 100 | 44 |
| 2 | ZnBr2 | DCE | 100 | 64 |
| 3 | ZnI2 | DCE | 100 | 51 |
| 4 | Zn(OTf)2 | DCE | 100 | 12 |
| 5 | CuBr2 | DCE | 100 | 96 |
| 6 | Cu(OTf)2 | DCE | 100 | 25 |
| 7 | CuCl | DCE | 100 | 71 |
| 8 | CuBr | DCE | 100 | 78 |
| 9 | Ph3PAuCl/AgNTf2 | DCE | 100 | Trace |
| 10 | CuBr2 | Toluene | 100 | 25 |
| 11 | CuBr2 | 1,4-Dioxane | 100 | 65 |
| 12 | CuBr2 | DMSO | 100 | NDd |
| 13 | CuBr2 | DCE | 120 | 96 |
| 14 | CuBr2 | DCE | 80 | 80 |
| 15e | CuBr2 | DCE | 100 | 84 |
| 16 | CuBr 2 | DCE | 100 | 96 (95%) |
With the optimized conditions established (Table 1, entry 16), the scope of 2-ethynylanilines was then investigated. The reaction of differently substituted 2-ethynylanilines with silylynamide 2a proceeded smoothly, leading to various N-vinylsilylindoles in good to excellent yields with perfect stereoselectivity (Scheme 2). A broad range of electron-donating and electron-withdrawing groups on the phenyl ring, such as methoxyl (3ba), methyl (3ca, 3qa), bromide (3da, 3ka), chloride (3ea, 3la, 3ra), fluoride (3fa, 3ma), trifluoromethyl (3ga), ester (3ha), and nitro (3ia), were well compatible in this transformation. The position of the substituents had a slight impact on the yields of the products. An X-ray single-crystal structure analysis of 3ha fully confirmed the (E)-geometry of the N-vinylsilylindole products.18 To our delight, 2-ethynylanilines bearing two halogens, regardless of their relative positions, afforded the products 3na–3pa in excellent yields (91–94%). Interestingly, when 2,5-diethynylbenzene-1,4-diamine was employed as the substrate, a bidirectional cascade reaction occurred to give the N,N′-bis(vinylsilyl)pyrroloindole product 3sa in 70% yield. A phenyl-substituted internal alkyne was also subjected to the reaction. But to our disappointment, the reaction stopped after the N-vinylation step. Thus, the amidine intermediate (see 8 in Scheme 5), instead of the desired product 3ta, was obtained.
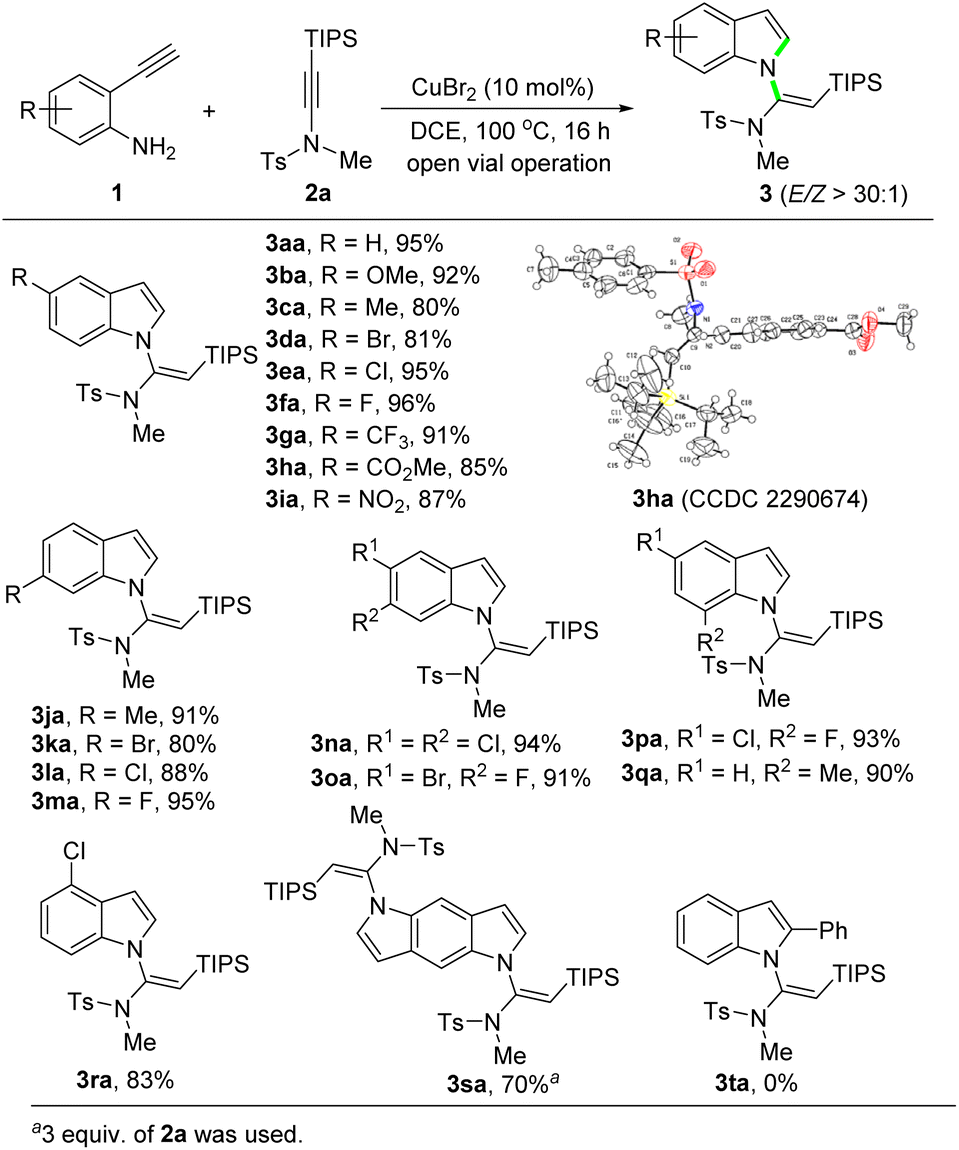 | ||
| Scheme 2 Substrate scope of 2-ethynylanilines. | ||
Subsequently, the scope with respect to the silylynamides was evaluated by using 2-ethynylaniline 1a as the reaction partner (Scheme 3). With regard to the functional group attached to the silylynamide nitrogen, in addition to methyl, n-propyl (3ab), and cyclopropyl (3ac) were well compatible, resulting in 65% and 71% yields, respectively, with excellent (E)-selectivity. The influence of the protecting group on the reactivity of silylynamides was also explored. Besides the tosyl group, substrates bearing benzenesulfonyl, 4-chlorobenzenesulfonyl, and mesyl protecting groups all reacted smoothly to afford the corresponding products in moderate to good yields with perfect stereoselectivity (3ad–3af). Interestingly, a 1,4-butanesultam derived silylynamide successfully underwent the cascade reaction to give the product 3ag in 85% as a single (E)-isomer. When an oxazolidinone derived silylynamide was employed as the substrate, the product 3ah was obtained in 45% yield with a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z ratio. Next, the reactivity of silylynamides decorated with different silyl groups were investigated. It was found that the steric bulk of the silyl group had a positive impact on the yield and stereoselectivity of the products. For instance, a substrate with a tert-butyldimethylsilyl gave N-vinylindole 3ai in 91% yield with a 11:1 E/Z ratio, whereas a less bulky triethylsilyl group (3aj) decreased the yield to 48% with a reduced E/Z ratio of 1.7:1.
:1 E/Z ratio. Next, the reactivity of silylynamides decorated with different silyl groups were investigated. It was found that the steric bulk of the silyl group had a positive impact on the yield and stereoselectivity of the products. For instance, a substrate with a tert-butyldimethylsilyl gave N-vinylindole 3ai in 91% yield with a 11:1 E/Z ratio, whereas a less bulky triethylsilyl group (3aj) decreased the yield to 48% with a reduced E/Z ratio of 1.7:1.
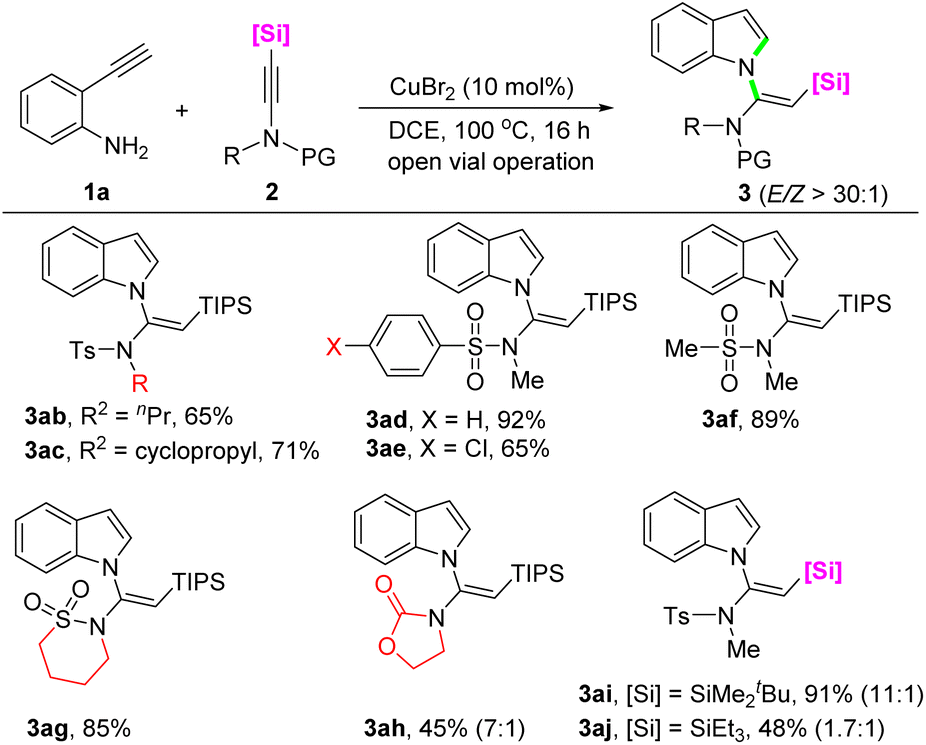 | ||
| Scheme 3 Substrate scope of ynamides. | ||
To demonstrate the potential synthetic applicability of this protocol, a gram-scale experiment, and synthetic transformation of the obtained product 3aa were conducted (Scheme 4). As shown in Scheme 4a, the reaction of 1a (3 mmol) with 2a (2 mmol) under standard reaction conditions delivered the product 3aa in 90% yield (0.87 g). Of note, the product 3aa can readily undergo a Er(OTf)3-catalyzed cis-hydroarylation reaction with ynamides, leading to the formation of 1,3-dialkenylindole 4 in 50% yield with a 14:1 Z/E ratio (Scheme 4b, left). Treatment of 3aa with oxalyl chloride in Et2O at 50 °C, followed by hydrolysis with aqueous NaHCO3, afforded acid 5 in 95% yield (Scheme 4b, right). Besides, the silyl group of 3aa can be easily removed in the presence of tetrabutylammonium fluoride (TBAF) to give terminal vinylindole 6 in 97% yield (Scheme 4b, lower part).
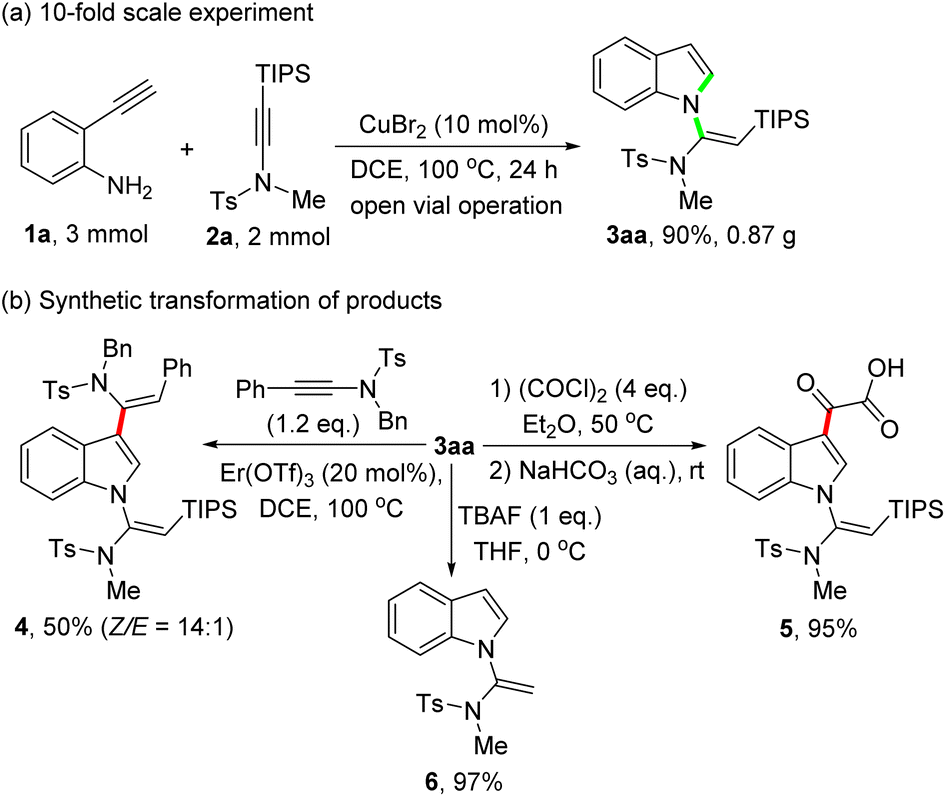 | ||
| Scheme 4 Gram-scale synthesis and further transformations. | ||
Finally, to shed light on the reaction mechanism, control experiments were carried out (Scheme 5). An intramolecular cyclization of 2-ethynylaniline 1a under standard conditions afforded indole 7 in 26% yield (Scheme 5a). But the reaction of indole 7 with silylynamide 2a under standard conditions did not give the desired product 3aa, only messy reaction mixtures being observed (Scheme 5b). This result excludes a cyclization/N-vinylation sequence as the reaction steps. We then tried to get the key reaction intermediate by conducting the reaction of 1a with 2a at a lower reaction temperature. To our delight, performing the reaction at room temperature afforded an amidine product 8 in 93% yield (Scheme 5c). Further treatment of 8 under standard conditions delivered the desired product 3aa in quantitative yield, whereas no reaction was observed when CuBr2 was not used (Scheme 5d). These results confirm a N-vinylation/cyclization sequence for this CuBr2-catalyzed tandem reaction of 2-ethynylanilines with silylynamides.
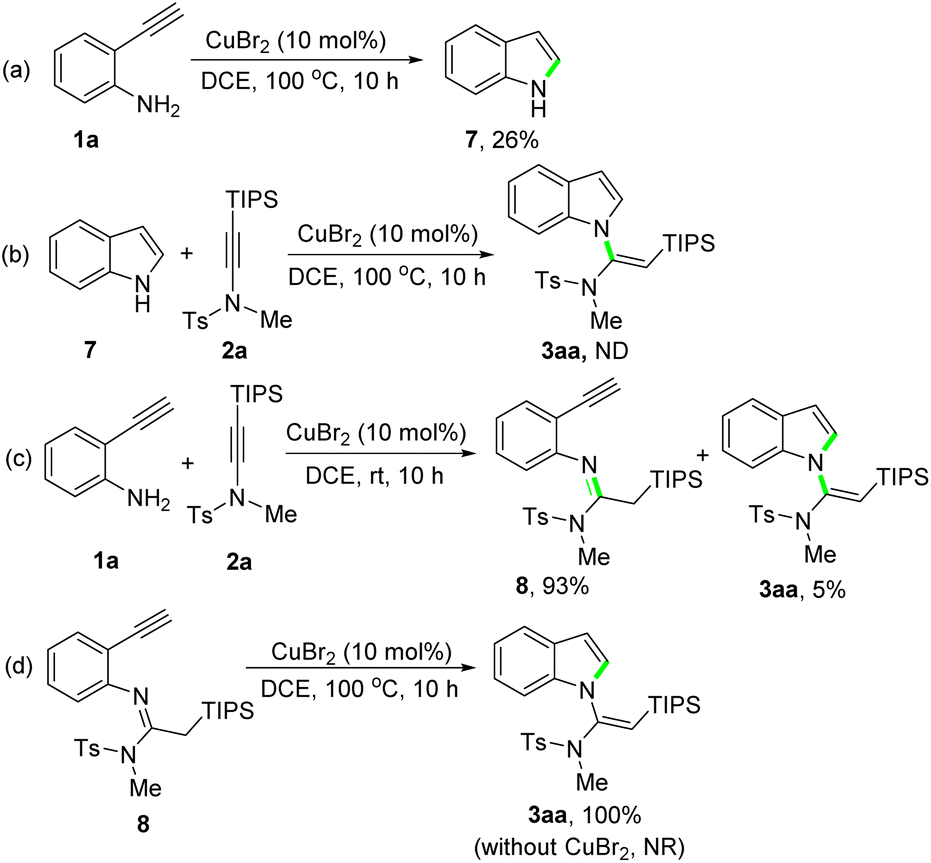 | ||
| Scheme 5 Control experiments. | ||
Based on the results of control experiments and previous reports,13 a plausible reaction mechanism for this new protocol is outlined in Scheme 6. Initially, the nucleophilic attack of NH2 group on 2-ethynylaniline 1a to the copper-activated silylynamide A/A′ gives vinyl-copper intermediate B, which after protodemetallation affords enamine intermediate C. A tautomerization of C forms imine intermediate 8, which can be obtained at a lower reaction temperature. Then, the copper catalyst coordinates to the alkyne unit of 8, leading to intermediate D, which undergoes a subsequent intramolecular cyclization via imine nitrogen attack to the copper-activated alkyne to generate intermediate E. To minimize the steric repulsion, the bulky TIPS group orients away from the protecting group and substituent attached to the ynamide nitrogen (E and E′). Finally, olefinization of E followed by protodemetallation delivers the desired N-vinylsilylindole 3aa as a single (E)-isomer. When less bulkily substituted ynamide nitrogen (3ah) or less bulky silyl groups are applied (3ai and 3aj), the steric repulsion decreases accordingly. Thus, the corresponding intermediates E and E′ can coexist in the reaction system, which finally evolves to the (E)- and (Z)-isomers of the products, respectively.
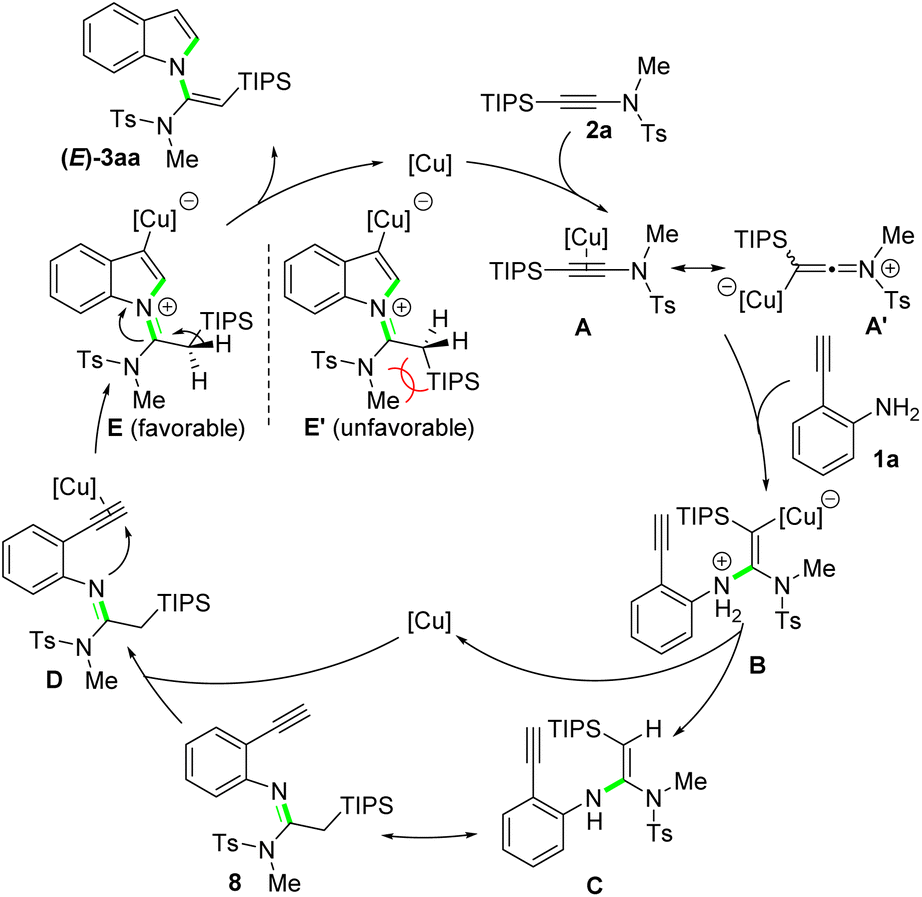 | ||
| Scheme 6 Proposed mechanism. | ||
Conclusions
In summary, we have developed a stereoselective and facile one-step synthesis of N-vinylsilylindoles via the CuBr2-catalyzed cascade reaction of 2-ethynylanilines with silylynamides. This strategy represents the first stereoselective tandem access to N-vinylindole scaffolds. This easy combination of biologically important indole frameworks and silyl groups would be highly attractive for medicinal chemists. The use of earth-abundant copper as the catalyst, the convenient open vial operation, the high yields of the products (up to 96%), and the excellent stereoselectivity (E/Z up to >30:1) are all highlights of this new protocol. Moreover, the broad substrate scope, good functional group compatibility, easy operation of gram-scale experiments, as well as the interesting down-stream synthetic conversion of the products make this methodology a practical tool for synthetic chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (22101107), the Natural Science Foundation of Shandong Province (ZR2023MB133), and Ludong University for financial support.References
- (a) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef CAS PubMed; (b) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608–9644 CrossRef CAS PubMed; (c) M. Shiri, Chem. Rev., 2012, 112, 3508–3549 CrossRef CAS PubMed.
- (a) M. Chbani, M. Païs, J.-M. Delauneux and C. Debitus, J. Nat. Prod., 1993, 56, 99–104 CrossRef CAS PubMed; (b) D. D. Belle, A. Tolvanen and M. J. T. Lounasmaa, Tetrahedron, 1996, 52, 11361–11378 CrossRef; (c) A. G. Schultz, W. P. Malachowski and Y. Pan, J. Org. Chem., 1997, 62, 1223–1229 CrossRef CAS; (d) G. Tasic, M. Simic, S. Popovic, S. Husinec, V. Maslak and V. Savic, Tetrahedron Lett., 2013, 54, 4536–4539 CrossRef CAS.
- (a) Y. Oshiro, Y. Shirota and H. J. P. J. Mikawa, Polym. J., 1974, 6, 364–369 CrossRef CAS; (b) M. Angiuli, F. Ciardelli, A. Colligiani, F. Greco, A. Romano, G. Ruggeri and E. Tombari, Appl. Opt., 2006, 45, 7928–7937 CrossRef CAS PubMed.
- (a) A. Y. Lebedev, V. V. Izmer, D. N. Kazyul'kin, I. P. Beletskaya and A. Z. Voskoboynikov, Org. Lett., 2002, 4, 623–626 CrossRef CAS PubMed; (b) Q. Liao, Y. Wang, L. Zhang and C. Xi, J. Org. Chem., 2009, 74, 6371–6373 CrossRef CAS PubMed.
- M. Movassaghi and A. E. Ondrus, J. Org. Chem., 2005, 70, 8638–8641 CrossRef CAS PubMed.
- G. Wu and W. Su, Org. Lett., 2013, 15, 5278–5281 CrossRef CAS PubMed.
- X. Zeng, G. Cheng, J. Shen and X. Cui, Org. Lett., 2013, 15, 3022–3025 CrossRef CAS PubMed.
- (a) A. K. Verma, M. Joshi and V. P. Singh, Org. Lett., 2011, 13, 1630–1633 CrossRef CAS PubMed; (b) M. Joshi, M. Patel, R. Tiwari and A. K. Verma, J. Org. Chem., 2012, 77, 5633–5645 CrossRef CAS PubMed; (c) M. Joshi, R. Tiwari and A. K. Verma, Org. Lett., 2012, 14, 1106–1109 CrossRef CAS PubMed.
- B. Zhou, T.-D. Tan, X.-Q. Zhu, M. Shang and L.-W. Ye, ACS Catal., 2019, 9, 6393–6406 CrossRef CAS.
- (a) F. L. Hong and L. W. Ye, Acc. Chem. Res., 2020, 53, 2003–2019 CrossRef CAS PubMed; (b) L.-W. Ye, X.-Q. Zhu, R. L. Sahani, Y. Xu, P.-C. Qian and R.-S. Liu, Chem. Rev., 2021, 121, 9039–9112 CrossRef CAS PubMed; (c) X. Zhao, M. Rudolph, A. M. Asiri and A. S. K. Hashmi, Front. Chem. Sci. Eng., 2019, 14, 317–349 CrossRef; (d) X.-N. Wang, H.-S. Yeom, L.-C. Fang, S. He, Z.-X. Ma, B. L. Kedrowski and R. P. Hsung, Acc. Chem. Res., 2014, 47, 560–578 CrossRef CAS PubMed.
- (a) Y. Zhang, Tetrahedron Lett., 2005, 46, 6483–6486 CrossRef CAS; (b) Y. Zhang, Tetrahedron, 2006, 62, 3917–3927 CrossRef CAS; (c) Y.-W. Liu, R.-J. Ma, Q.-E. Wang, C.-M. Si and B.-G. Wei, Tetrahedron, 2020, 76, 131649 CrossRef CAS.
- A. Hentz, P. Retailleau, V. Gandon, K. Cariou and R. H. Dodd, Angew. Chem., Int. Ed., 2014, 53, 8333–8337 CrossRef CAS PubMed.
- Y. Zhang, J. P. Donahue and C.-J. Li, Org. Lett., 2007, 9, 627–630 CrossRef CAS PubMed.
- (a) G. Anilkumar and S. Saranya, Copper Catalysis in Organic Synthesis, Wiley-VCH, 2020 CrossRef; (b) Q. Li, H. Zhu, Y. Liu, L. Yang, Q. Fan, Z. Xie and Z.-G. Le, RSC Adv., 2022, 12, 2736–2740 RSC; (c) X. Fang, C. Qi, X. Cao, Z.-G. Ren, D. J. Young and H.-X. Li, Green Chem., 2023, 25, 8068–8073 RSC; (d) A. Shen, Z. Wu, Y. Fang, J. Yang, H. Zhu and T. Tu, Asian J. Org. Chem., 2018, 7, 1113–1117 CrossRef CAS.
- (a) X. Zhao, X. Song, H. Jin, Z. Zeng, Q. Wang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2018, 360, 2720–2726 CrossRef CAS; (b) X. Zhao, B. Tian, Y. Yang, X. Si, F. F. Mulks, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2019, 361, 3155–3162 CrossRef CAS; (c) G. Wang, J. Jia, G. Liu, M. Yu, X. Chu, X. Liu and X. Zhao, Chem. Commun., 2021, 57, 11811–11814 RSC; (d) X. Zhao, Z. Li, J. Jia, M. Yu, G. Wang and G. Liu, Asian J. Org. Chem., 2022, 11, e202200458 CrossRef CAS; (e) G. Wang, H. Li, Y. Wang, Z. Li, G. Liu and X. Zhao, Org. Chem. Front., 2023, 10, 5705–5709 RSC.
- (a) N. D. Rode, A. Arcadi, A. Di Nicola, F. Marinelli and V. Michelet, Org. Lett., 2018, 20, 5103–5106 CrossRef CAS PubMed; (b) R. Vanjari, S. Dutta, M. P. Gogoi, V. Gandon and A. K. Sahoo, Org. Lett., 2018, 20, 8077–8081 CrossRef CAS PubMed.
- (a) G. A. Bikzhanova, I. S. Toulokhonova, S. Gately and R. West, Silicon Chem., 2006, 3, 209–217 CrossRef; (b) S. J. Barraza and S. E. Denmark, J. Am. Chem. Soc., 2018, 140, 6668–6684 CrossRef CAS PubMed; (c) R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779–3798 CrossRef CAS PubMed.
- CCDC 2290674† contains the supplementary crystallographic data (3ha) for this paper.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2290674. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc04244h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |