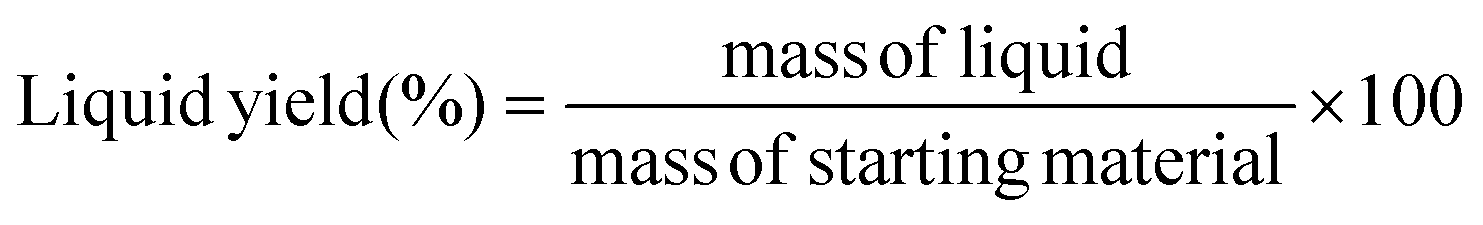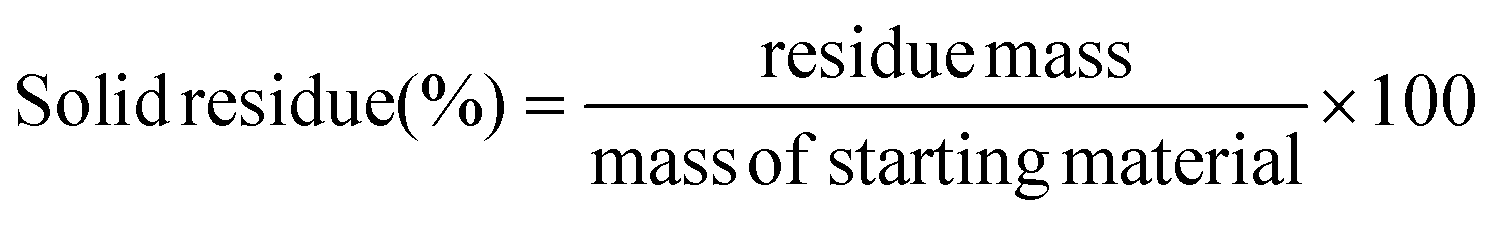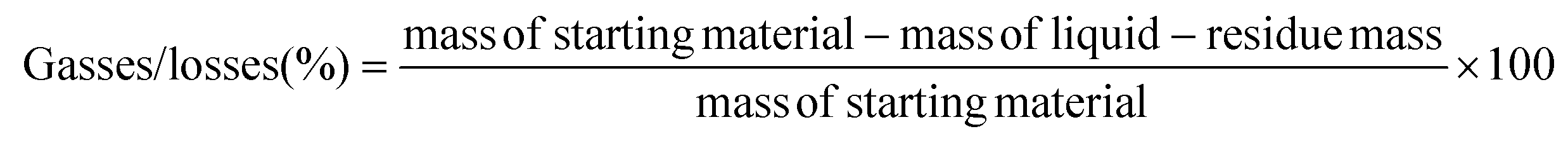DOI:
10.1039/D3GC02407E
(Paper)
Green Chem., 2024,
26, 340-352
Total revalorization of high impact polystyrene (HIPS): enhancing styrene recovery and upcycling of the rubber phase†
Received
4th July 2023
, Accepted 23rd November 2023
First published on 29th November 2023
Abstract
High impact polystyrene (HIPS) is a two-phase polymeric material that consists of a free polystyrene (PS) matrix and rubber particles. HIPS occupies a substantial portion of plastic waste. Even though HIPS waste could potentially be employed as an efficient feedstock for the recovery of the styrene monomer via pyrolysis, several challenges must be overcome first, like the low styrene yield (<50%) and the generation of char due to the presence of rubber. To tackle these challenges, a green fractionation process using ethyl acetate (EtOAc) as an efficient solvent to separate rubber from the free PS matrix in HIPS is envisioned, which is carried out under mild conditions. The subsequent pyrolysis of the fractionated sample at 300 °C led to a 20% increase in styrene selectivity compared to the pyrolysis of untreated HIPS. Moreover, the revalorization of the rubber particles was accomplished by ethenolysis metathesis, in which after 4 h at 100 °C, polybutadiene was split to produce 1,5-hexadiene as a major product (60% yield) and isolated PS, which was further thermally degraded, achieving a styrene selectivity of 70%.
1. Introduction
Plastics are among the most commonly used materials in modern society, with their production rapidly increasing during the past decades from 2.3 million tons in 1950 to 367 million tons in 2020.1–3 As a consequence, the amount of plastic waste keeps rising: in Europe alone, 29.5 million tons of plastic post-consumer waste are collected yearly.4 Since recycling rates remain low, a sizeable fraction of all plastic waste ends up in landfills, or is simply discarded in nature,4 with about 8 million tons of plastic waste ending up in the oceans each year.5
It is easy to understand why plastics are a major environmental concern, since their accelerated growth has such a severe negative impact. Among the variety of plastic waste generated, a large fraction originates from polystyrene-based materials,6 as they represent around 7% of the global plastic market share.7 Within the types of polystyrene (PS), high impact polystyrene (HIPS) corresponds to 30–40% of the worldwide PS production, finding applications in packaging, containers, appliance parts, household items and electronic equipment.8–12 Actually, the waste produced by electronic equipment, known as “waste electrical and electronic equipment” (WEEE), reached 59 million tons by 2022, with HIPS representing 42% of all WEEE.13,14 Current valorization techniques for polyolefins (including PS) involve pyrolysis reactions, perhaps the most common approach,15 metathesis reactions, like ethenolysis,16 hydrogenolysis over metal catalysts,17 hydrocracking over bifunctional catalysts,18 among others. However, in all of these systems, there is still room for growth and further upgrading before industrial implementation.15
HIPS is a two-phase material consisting of a free polystyrene matrix and rubber particles, which contain micron-sized polybutadiene (PB) particles (3–10 wt% of the total material), surrounded by covalently grafted PS.19–23 The role of the PB particles is to enhance the mechanical properties of the polymer (a method known as rubber toughening), which provides the HIPS with higher impact strength, elongation at break, and fracture toughness. Moreover, the improved polymeric material also shows a different behavior under heat, light, and irradiation.24 Due to the high PS content in HIPS, this waste stream could be considered as an obvious feedstock to recover the styrene monomer in a closed-loop process. However, in almost all efforts to thermally recycle HIPS, the styrene selectivity and yield are lower than in the thermal degradation of pure PS samples.25–35 This can be attributed to the presence of the PB particles, which contribute to the formation of char nuclei during the thermal degradation process and affect the thermal decomposition mechanism, decreasing in turn the styrene yield.25–35 A possible solution to this problem could be the separation of the rubber particles from the free PS matrix before the pyrolysis process. In a lab context, such fractionation can be carried out employing the Ruffing technique. In this method, HIPS is dissolved in toluene, mixed at 60 °C for two hours and centrifuged at the same temperature for one hour.36 It is important to note that the Ruffing technique is in fact an analytical technique and it would, most likely, not be suitable for potential large scale applications. Besides the Ruffing technique, additional methods have been developed,36–40 some of them employing high temperatures (210 °C),37 others, large amounts of solvents (mixtures of toluene/methyl ethyl ketone),38 high centrifugation speeds (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–27000 rpm, 70 min) and/or low temperatures (−17 °C).39 However, the harsh conditions of these processes, in addition to the use of non-green solvents, also render them inapplicable in potential large scale applications.
000–27000 rpm, 70 min) and/or low temperatures (−17 °C).39 However, the harsh conditions of these processes, in addition to the use of non-green solvents, also render them inapplicable in potential large scale applications.
Moreover, even after the fractionation, a crucial consideration is how to valorize the rubber particle fraction. In this sense, ethenolysis, defined as cross metathesis with ethylene, is a 100% atom efficient, green reaction, which can be performed at relatively low pressure and temperature. It can be used to split long unsaturated carbon chains or to produce α,ω-dienes from cyclic olefins.41 Ethylene can be produced from ethanol, which can be obtained from biomass.42,43 Ethenolysis of PB has been effectively performed using homogeneous Grubbs catalysts, as demonstrated by Watson and Wagener.44 Hence, we envisage that this reaction could be used to depolymerize the PB in the rubber phase while leaving the PS intact for a subsequent degradation process.
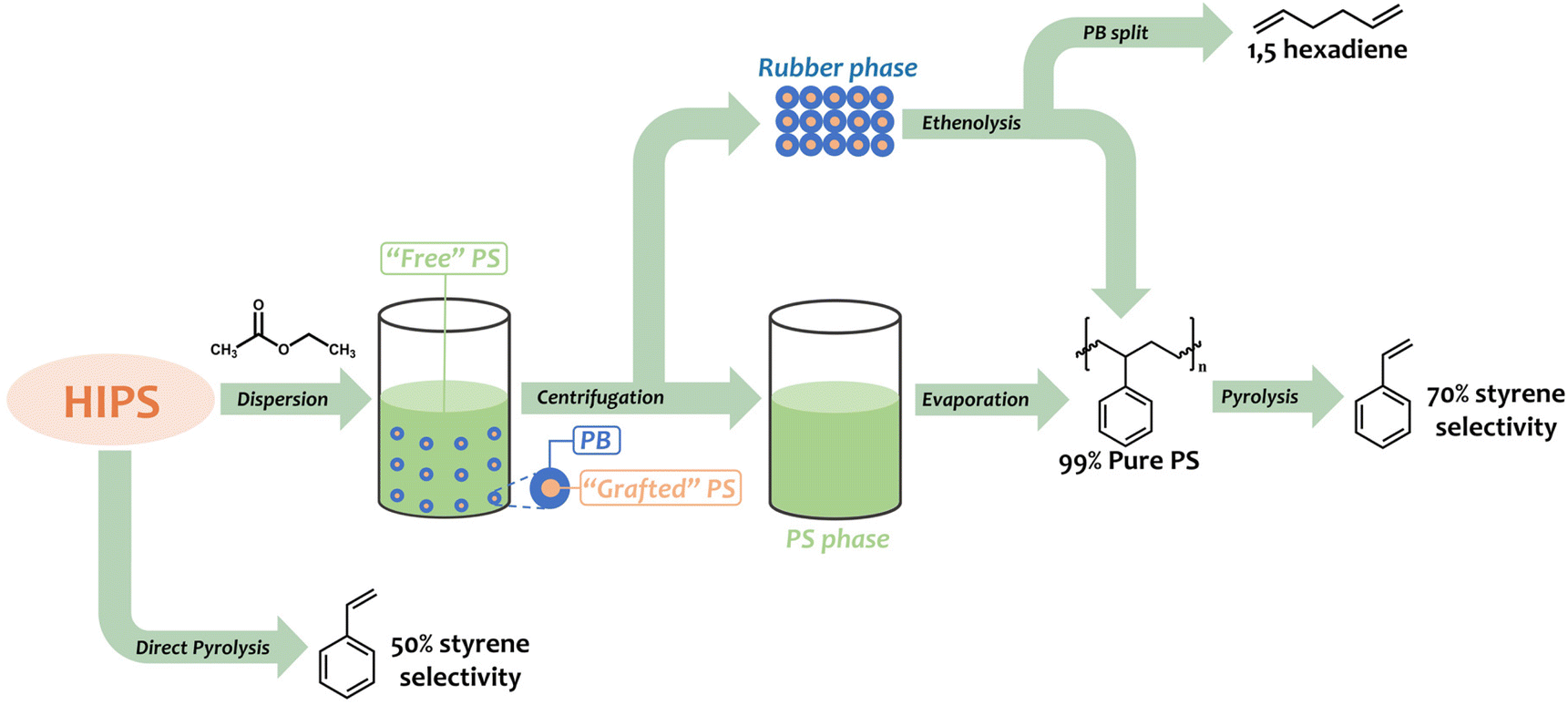
In this work, we aim at the total revalorization of HIPS, enhancing styrene recovery by solvent pre-fractionation and upcycling of the PB phase. To this end, ethyl acetate is used as an efficient green solvent for the isolation of the free PS matrix from the rubber phase in HIPS, which was complemented by computational studies modeling the interaction of the PS, PB, and the rubber phase (PB with grafted PS) with ethyl acetate and other possible solvents. After isolating large amounts of free PS from HIPS, the effect of the pre-fractionation process on the selectivity to styrene in the thermal degradation of PS is evaluated. The valorization of the rubber phase is achieved using ethenolysis metathesis, where the grafted PS is separated from the rubber phase and further thermally degraded, while the PB is split to produce α,ω-dienes in the presence of ethylene gas and a Grubbs catalyst. A summary of the proposed approach is presented in Scheme 1.
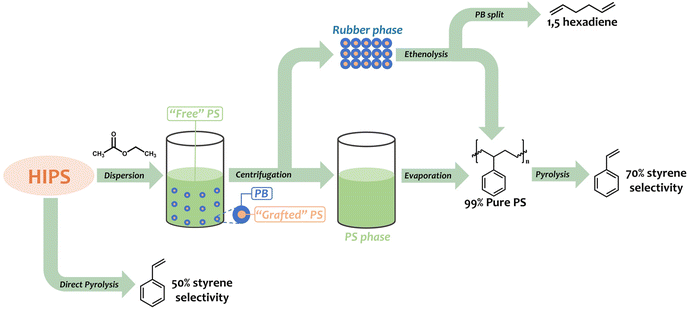 |
| | Scheme 1 Summarized description of the HIPS revalorization process presented in this work. | |
2. Experimental
2.1. Materials
Commercial pellet-type HIPS was provided by a styrenics manufacturer (8.5 wt% PB, 3.3 wt% of mineral oil, 500 ppm zinc stearate and 500 ppm stabilizer Irganox 1076). The commercial PS sample, denoted PS-192 (where 192 stands for Mw = 192 kDa), was purchased from Sigma Aldrich. Ethyl acetate (EtOAc, Fisher chemicals) was used as the pre-fractionation solvent, while solvents such as benzene (Carl Roth), toluene (Fischer Chemicals), xylene (Fischer Chemicals), tetrahydrofuran (THF, Sigma Aldrich), methyl ethyl ketone (MEK, Acros Organics) and chloroform (CHCl3, Fischer Chemicals) were also evaluated. For the ethenolysis metathesis, ethylene and nitrogen gases were supplied by Air Liquide, the Grubbs M202 catalyst and CDCl3 were purchased from Sigma Aldrich, and methanol (MeOH, Fischer chemicals) was used as anti-solvent to precipitate the PS. Toluene (Fischer Chemicals) was used as an internal standard to quantify the ethenolysis products. All the proton nuclear magnetic resonance (1H-NMR) experiments were conducted in CDCl3. Finally, the liquid composition percentages, after the thermal degradation of the PS samples, were determined by gas chromatography (GC), using mesitylene (Alfa Aesar) as internal standard.
2.2. Pre-fractionation process: isolation of the free PS from HIPS samples
In a Nalgene polypropylene (PPCO) 250 ml bottle, 10 g of HIPS and 200 ml of EtOAc were added and stirred for two hours. The dispersion was then centrifuged at 10000 rpm for 15 min at room temperature, and subsequently, a first large portion of the supernatant solution (160 ml) was poured into a 1000 ml glass beaker, leaving 40 ml of the supernatant solution in the Nalgene bottle. An additional amount of EtOAc (150 ml) was used to re-disperse the leftover supernatant solution (containing the rubber phase and remaining PS), and after 1 h of stirring the sample was centrifuged again at 10000 rpm for 15 minutes at room temperature. The two supernatant solutions were then combined in the glass beaker for evaporation of the solvent, while the (solid) rubber phase was kept in the Nalgene bottle. The two phases were placed in an oven at 70 °C overnight before 1H-NMR and Fourier-transform infrared spectroscopy (FTIR) analyses. After every fractionation cycle, 8 g of the free PS (denoted as PS-1) and 2 g of the rubber phase (PS plus grafted PB) were isolated.
2.3. Ethenolysis: PS recovery from the rubber phase and revalorization of PB
The rubber phase (120 mg) was stirred for 1 h at 60 °C, in 6 ml CDCl3, to ensure the fine dispersion of the polymer before being transferred to a stainless steel reactor (total volume 15 ml), together with 7 mg of Grubbs catalyst M202 and a stirring bar. The headspace of the reactor was first flushed with N2, then with ethylene three times, before being pressurized to 8 bars of ethylene and placed in a pre-heated heating block at 100 °C. Reaction starts at the moment the reactor reaches the desired temperature (15 minutes) and is conducted for 4 h. After reaction, the reactor was cooled down in a water bath and an aliquot of the product mixture was analyzed by 1H-NMR (using toluene as internal standard), gas chromatography-mass spectrometry (GC-MS) on an Agilent 6890N GC instrument, equipped with a HP-5 MS column, coupled to a 5973N mass spectrometer and GC on a Shimadzu GC-2010 instrument equipped with a CP-SIL 5 CB column and FID detector. The remainder of the crude liquid product was transferred to a glass vial to recover the PS employing 8 ml of methanol (MeOH) as anti-solvent. The precipitated PS was recovered by centrifugation (3500 rpm for 5 minutes) and washed two times with MeOH before drying overnight at 70 °C. The obtained PS powder (denoted PS-2) was weighed and characterized by 1H-NMR and FTIR. On average, 65 mg of dried PS-2 were obtained per reaction cycle.
2.4. Thermal degradation: styrene recovery from HIPS and PS samples
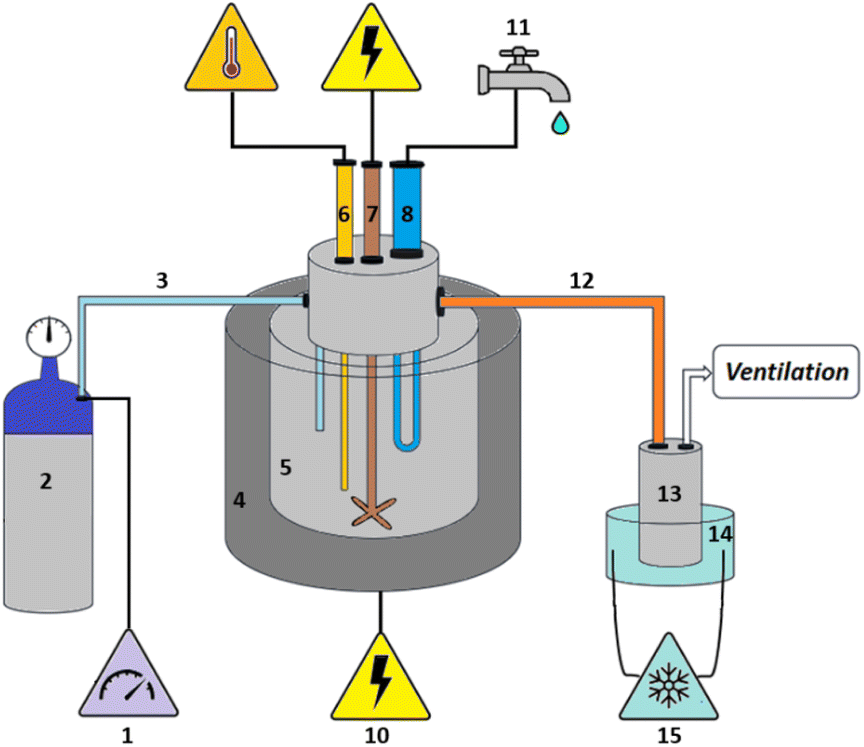
The plastic samples (10 g) were thermally degraded in a pyrolysis reactor equipped with a water cooling system (Fig. 1 and Fig. S1 ESI†). The reaction was carried out at 300 °C (heating rate 2.5 °C min−1) under continuous N2 flow (1 ml min−1) for 1 h, under mechanical stirring at 300 rpm. The gaseous products were removed from the reactor and collected in the condenser (1.6 °C). After reaction, the energy supply was stopped and the reactor was cooled down until 70 °C. The liquid products were recovered from the condenser, weighed and analyzed by GC-MS on an Agilent 6890N GC instrument, equipped with a HP-5 MS column, coupled to a 5973N mass spectrometer and GC (using mesitylene as internal standard) on a Shimadzu GC-2010 instrument equipped with a CP-SIL 5 CB column and FID detector. The solid residue remaining in the reactor was collected with EtOAc, dried until constant weight and analyzed by 1H-NMR and GC-MS. The liquid yield, solid residue, and gasses/losses percentages were calculated according to eqn (1)–(3). To determine the composition of the gasses/losses, a 1 L Tedlar bag was connected to the ventilation line throughout the reaction (Fig. 1), and the content of each bag was analyzed on a Shimadzu GC-2014 instrument equipped with a SH-l-1MS column and an FID detector.| | 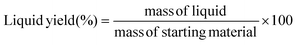 | (1) |
| | 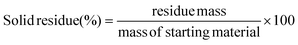 | (2) |
| | 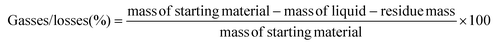 | (3) |
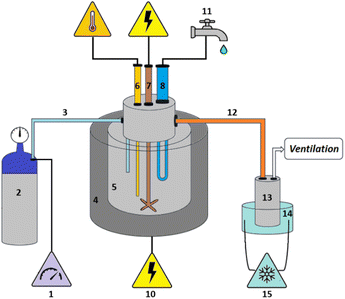 |
| | Fig. 1 Reactor for the pyrolysis of polystyrene: (1) nitrogen flow regulator, (2) nitrogen gas cylinder, (3) nitrogen flow tube, (4) heat jacket, (5) reaction vessel, (6) Pt-100 thermocouple, (7) stirring mechanism, (8) water-cooling system, (9) two-loop eurotherm temperature control system, (10) power supply, (11) water supply, (12) heated tube (T = 80 °C), (13) stainless steel condenser, (14) condenser cooler using ethylene glycol/water mixture (T = 1.6 °C), (15) cooling pump and ethylene glycol/water mixture reservoir. | |
2.5. Characterization of the polymeric materials
PS-192, the phases obtained from the pre-fractionation (PS-1, rubber phase), and the ethenolysis products, were analyzed by 1H-NMR spectroscopy in a Bruker 400 MHz or 600 MHz instrument. Analysis of the 1H-NMR spectra allowed the determination of the wt% of PS and PB in the HIPS sample (ESI†). FTIR spectra of PS-192, PS-1, PS-2 and the rubber phase were collected in an Agilent Cary 630 ATR-FTIR spectrometer in attenuated total reflectance (ATR) mode. Finally, the molecular weights of PS-1, PS-2 were determined by employing an Agilent gel permeation chromatography (GPC) instrument equipped with a PL 1110–6500 column, calibrated with commercial PS standards ranging from 162 Da to 364 kDa.
2.6. Interaction of the solvents with PS, PB and PS-grafted PB polymeric chains: computational studies
Molecular dynamics simulations were performed using GROMACS v2021.3 and the OPLS-AA force field,45–47 whereby extra dihedral parameters were generated by LigParGen.48–50 The simulations for solubility prediction were performed according to a procedure outlined in a previously published benchmark study; for clarity the setup is briefly outlined here as well.51 The PS and PB polymer chains were composed of 200 backbone atoms, i.e. 100 and 50 monomers, respectively. This polymer length was confirmed to provide a good compromise between computational efficiency and reliable polymer behavior. A PS-grafted PB chain was made by attaching a PS chain of 100 monomers onto a PB chain of 50 monomers (200 backbone atoms each).52 The solvate module implemented in GROMACS was used to solvate the polymer chains in a pre-equilibrated solvent box with dimensions 24.50 Å × 24.50 Å × 24.50 Å, while periodic boundary conditions were applied in all directions. The simulation boxes were then relaxed through a steepest descent energy minimization for 10000 steps, after which they were equilibrated during a 2-step procedure. First, a 100 ps NVT simulation was performed at 298.15 K, followed by another 100 ps NPT simulation at 298.15 K and 1.0 bar. A simulation step of 1 fs was used throughout and the Berendsen thermostat and barostat were applied with a time constant of 0.2 ps for the former and a coupling constant of 1.0 ps for the latter.53 Finally, a production run was performed in the NPT ensemble for 20 ns for the PS and PB and 30 ns for the grafted PS with a timestep of 2 fs, while applying constraints on all bonds using the LINCS algorithm.54 The temperature was fixed at 298.15 K using the extended-ensemble Nose–Hoover scheme with a 2.0 ps time constant, while the pressure was controlled at 1 bar using the Parrinello–Rahman coupling method with a time constant of 5.0 ps.55–57 Non-bonded interactions were calculated using van der Waals and coulombic (through the particle-mesh Ewald summation) terms with a cutoff of 1.3 nm. Snapshots were collected every 100 ps and the last 10 ns of the simulation were used for post processing.
3. Results and discussion
3.1. Pre-fractionation process
Solvent screening.
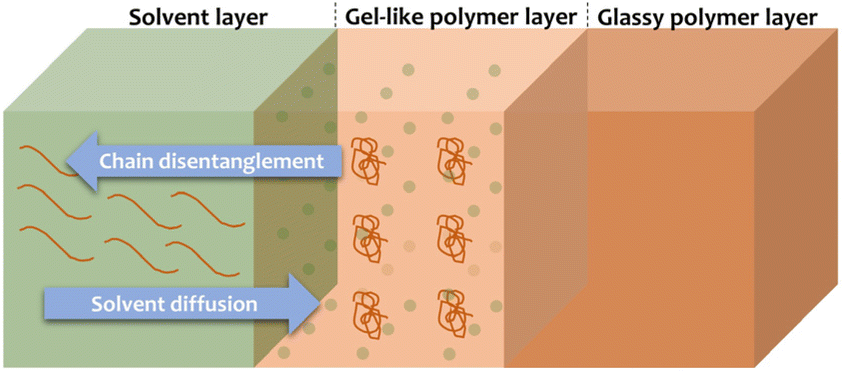
Since the usual procedures for HIPS fractionation serve analytical purposes, and are most likely not suitable for upscaling,36–40 our initial target was to isolate the free PS from the rubber phase by applying milder conditions than those usually employed and a green solvent for potential large scale applications. The choice of a solvent that is able to selectively dissolve a polymer constitutes an extremely important topic since polymers are among the most ubiquitous materials.58,59 Membrane science, microlithography, plastic recycling, drug delivery and tissue regeneration are exemplary fields that are strongly dependent on the polymer dissolution.59 Rationalizing the behavior of polymers in specific solvents helps improving and optimizing the desired physical properties of the material according to the application.59 However, understanding the dissolution of polymers is not a simple task as it is governed by multiple factors. In general, two types of transport processes occur during the dissolution of a polymer: solvent diffusion and chain disentanglement.58,59 When a polymer interacts with a thermodynamically favored solvent, diffusion of the solvent into the polymer takes place.59 This results in the plasticization of the polymer and consequently, the swollen part of the polymer forms a gel-like layer between the glassy polymer layer and the solvent (as presented in Fig. 2).59 After some time, the polymer dissolves as the chain disentanglement progresses.59
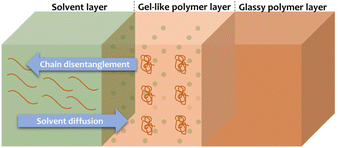 |
| | Fig. 2 Representation of the polymer dissolution process.59 | |
Therefore, in a HIPS fractionation process, it is essential to use a solvent that is able to selectively dissolve PS (but not PB or the rubber phase). Overall, PS is soluble in aromatic solvents, terpenes (limonene, terpinene, phellandrene, etc.) and several oxygenated solvents like ethers, esters or ketones.60 On the other hand, the rubber phase consists of PS-grafted PB. On its own, PB is soluble in ethylbenzene or toluene but poorly compatible with oxygenated solvents,61 while solvents such as cyclohexane, methylcyclohexane, benzene and toluene are suitable for styrene butadiene rubbers (SBR).62 Considering this, the usage of a green oxygenated solvent would possible lead to HIPS fractionation. It is also important that a low boiling solvent is selected because during evaporation, thermal degradation of the polymer could take place at high temperatures.58 Hence, EtOAc emerged as a green solvent choice due to its low boiling point (77 °C), good affinity for PS,60 relatively low toxicity and low cost.63 Its pre-fractionation performance was compared to that of other organic solvents such as benzene, toluene, xylene, tetrahydrofuran (THF) and methyl ethyl ketone (MEK). Table 1 shows that with EtOAc, the highest purity of the free PS phase can be obtained. 1H-NMR was employed to determine the wt% of PB remaining in each specific phase. The calculation of the polymer composition was achieved using the integrals of the aromatic PS protons and the vinylic PB protons since they are the most distinctive signals, and do not overlap with other peaks (Fig. S2–S9 ESI†). The nominal value for the total PB content of the initial HIPS sample equals to 8.5 wt% and it is used as a comparison point to determine the fractionation efficiency.
Table 1 Composition of the supernatant isolated from HIPS after the fractionation using different organic solventsa
| |
PS (wt%) |
PB (wt%) |
|
1 g of HIPS dispersed in 20 ml of solvent, stirred overnight, and centrifuged at room temperature. The analysis of the supernatant and the subsequent determination of the wt% of each phase were done by 1H-NMR. Composition of HIPS is 91.5% (PS) and 8.5% (PB).
|
| CHCl3 |
92.0 |
8.0 |
| THF |
92.2 |
7.8 |
| Xylene |
93.7 |
6.3 |
| Toluene |
94.3 |
5.7 |
| Benzene |
94.3 |
5.7 |
| MEK |
98.5 |
1.5 |
| EtOAc |
99.3 |
0.7 |
Only with EtOAc and MEK, two distinct phases were obtained after centrifugation (Fig. S10 ESI†), while with the other solvents, almost full dissolution of the HIPS is observed (Fig. S11 ESI†), and only a negligible amount of the rubber phase is separated. This could be linked to the affinity of both polymers (PS, PB) for the aforementioned solvents. In view of this, we decided to first study such affinity by the Hansen solubility sphere model (HSP), one of the well-known models employed in polymer chemistry,64 to at least partially explain the higher fractionation efficiency obtained when the process is carried out employing EtOAc (and MEK) as solvents.
Polymer–solvent interactions and molecular dynamics simulations.
Considering HIPS as a mixture of PS and SBR, the Ra/Ro values for each of polymer were estimated and are presented in Table 2.59,65 In general, the lower the Ra/Ro value, the higher the affinity of the solvent with the polymer, and an Ra/Ro value close to or higher than 1.0, indicates low affinity. Analyzing the values from Table 2, it is understandable that solvents like CHCl3, xylene, toluene and benzene will dissolve both the PS and the SBR, since all of the Ra/Ro obtained are well below 1.0, which consequently leads to the full dissolution of the HIPS. Conversely, based on the Ra/Ro values, EtOAc and MEK effectively dissolve PS, but poorly dissolve SBR, which corresponds to the two distinct phases obtained after centrifugation during the fractionation experiment. An unexpected result is obtained when employing THF as fractionation solvent. Based on the Ra/Ro values, THF is expected to preferentially dissolve the PS phase. However, under the studied fractionation conditions, THF fails to effectively separate the two polymer phases. While this contradiction is expected considering the model's several assumptions,66 it motivated us to pursue computational studies to obtain a deeper understanding of the behavior of the polymer phases in the different studied solvents.
Table 2
R
a/Ro calculated using the Hansen solubility parameters according to the HSP modela
| Solvent |
R
a/Ro (PS) |
R
a/Ro (SBR) |
R
a/Ro (PB) |
|
The values are calculated using the Hansen solubility parameters from Table S1, ESI.†
|
| CHCl3 |
0.30 |
0.46 |
0.38 |
| THF |
0.44 |
0.91 |
0.91 |
| Xylene |
0.40 |
0.37 |
0.22 |
| Benzene |
0.47 |
0.58 |
0.49 |
| Toluene |
0.38 |
0.34 |
0.30 |
| MEK |
0.48 |
1.05 |
1.16 |
| EtOAc |
0.52 |
0.92 |
0.91 |
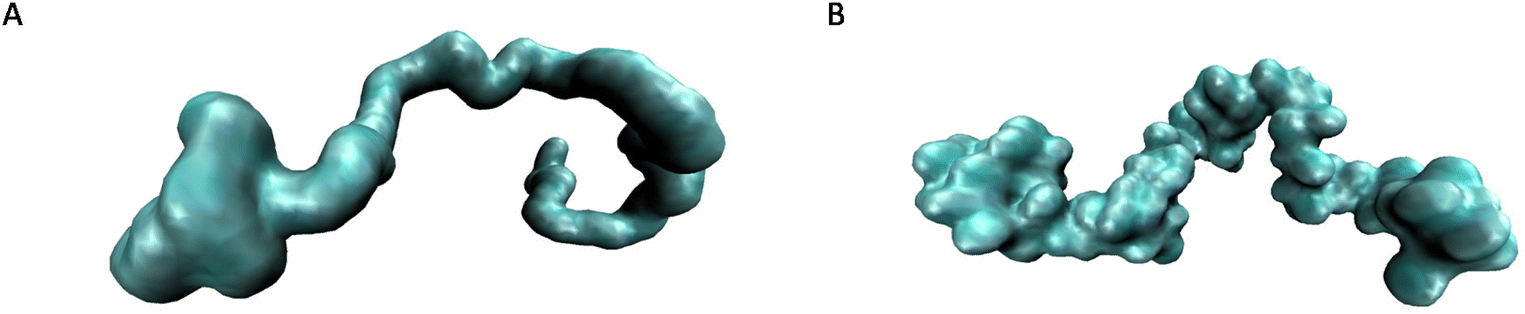
Molecular dynamics simulations were performed to assess the solvation behavior of PS, grafted PS (SBR) and PB in THF, xylene and EtOAc. Only these three solvents were considered to keep the computational cost reasonable. For each of these systems (9 systems in total), the relative solvent accessible surface area (rSASA) was calculated, which was shown to be a reliable proxy for solvation free energies in a previous work.51 The solvent accessible surface area is the surface area of the polymer that is in contact with the solvent; it is routinely calculated from molecular dynamics trajectories by rolling a sphere over the van der Waals surface of the target molecule. As such, solvent accessible surface area gives an idea about the conformation of the polymer chain, as a smaller value indicates a more collapsed structure with less exposure to the solvent. The rSASA is calculated as the ratio between the average solvent accessible surface area throughout the simulation and the maximum solvent accessible surface area observed for the polymer chain of interest across the different solvents. By definition, a value close to 1 represents a polymer chain that prefers to be in full contact with the solvent, while a lower value indicates that the polymer is trying to reduce its contact with the solvent by collapsing on itself. The results for the investigated systems are shown in Table 3. It can be seen that generally all three polymers are well dissolved in the three considered solvents with only slight differences between them. Overall, THF shows a slight preference to dissolve PB and SBR over PS, while xylene shows similar dissolution behavior for the three considered polymers. EtOAc, on the other hand, shows a preference to dissolve PS over PB and SBR. In Fig. 3, the solvent accessible surface area is visualized for PB and PS in EtOAc. It can be seen that PB clumps together at the left outer end of the chain, while PS remains in full contact with the solvent. Note that the surface of PS displays lumps along its backbone as a result of its phenyl side chains, while PB is linear with a smoother surface as a result. Importantly, the harsh conditions needed to separate these polymers suggest that solubility and separation are indeed governed by very subtle changes in polymer–polymer and polymer–solvent interactions. Evidently, PS is slightly more stable in full contact with EtOAc than PB and SBR, and those small differences in solubility, as shown by the experimental results, can be exploited by applying a subsequent method for particle isolation (e.g. in the form of centrifugation), resulting in the efficient separation of PS from PB.
 |
| | Fig. 3 Visualization of the polymer surface in contact with the solvent for PB (A) and PS (B) in EtOAc, as calculated from molecular dynamics trajectories. | |
Table 3 Relative solvent accessible surface area (rSASA) calculated using the Hansen solubility parameters according to the HSP modela
| Solvent |
rSASA (PS) |
rSASA (SBR) |
rSASA (PB) |
|
A value close to 1 indicates good solubility behavior.
|
| THF |
0.87 |
0.91 |
0.93 |
| Xylene |
0.93 |
0.92 |
0.92 |
| EtOAc |
0.92 |
0.88 |
0.89 |
Scale-up and 1H-NMR analysis of the phases.
Guided by the results obtained during the solvent screening (Table 1), EtOAc was chosen for a scale-up of the fractionation procedure using up to 10 g of HIPS sample per batch. We started by investigating the optimal amounts of fractionation solvent necessary for an efficient PS recovery at this scale (Table 4). When employing 200 ml of EtOAc, free PS could be collected with >99% purity, and after a second extraction cycle (additional wash with 150 ml EtOAc), 8 g of free PS could be isolated (denoted as PS-1), while 2 g of the rubber phase remained (Fig. S12 ESI†).
Table 4 Effect of the EtOAc/HIPS mass ratio on the purity of the obtained dissolved PS-rich fractiona
| EtOAc (ml) |
wt% styrene monomersb |
wt% butadiene monomersb |
|
10 g of HIPS dispersed in the specified amount of EtOAc.
The wt% in the dried supernatant phase were determined by 1H-NMR.
5 g of HIPS.
|
| 80 |
94.5 |
5.5 |
| 100 |
96.8 |
3.2 |
| 120 |
98.5 |
1.5 |
| 150 |
98.9 |
1.1 |
| 200 |
99.1 |
0.9 |
| 200c |
99.1 |
0.9 |
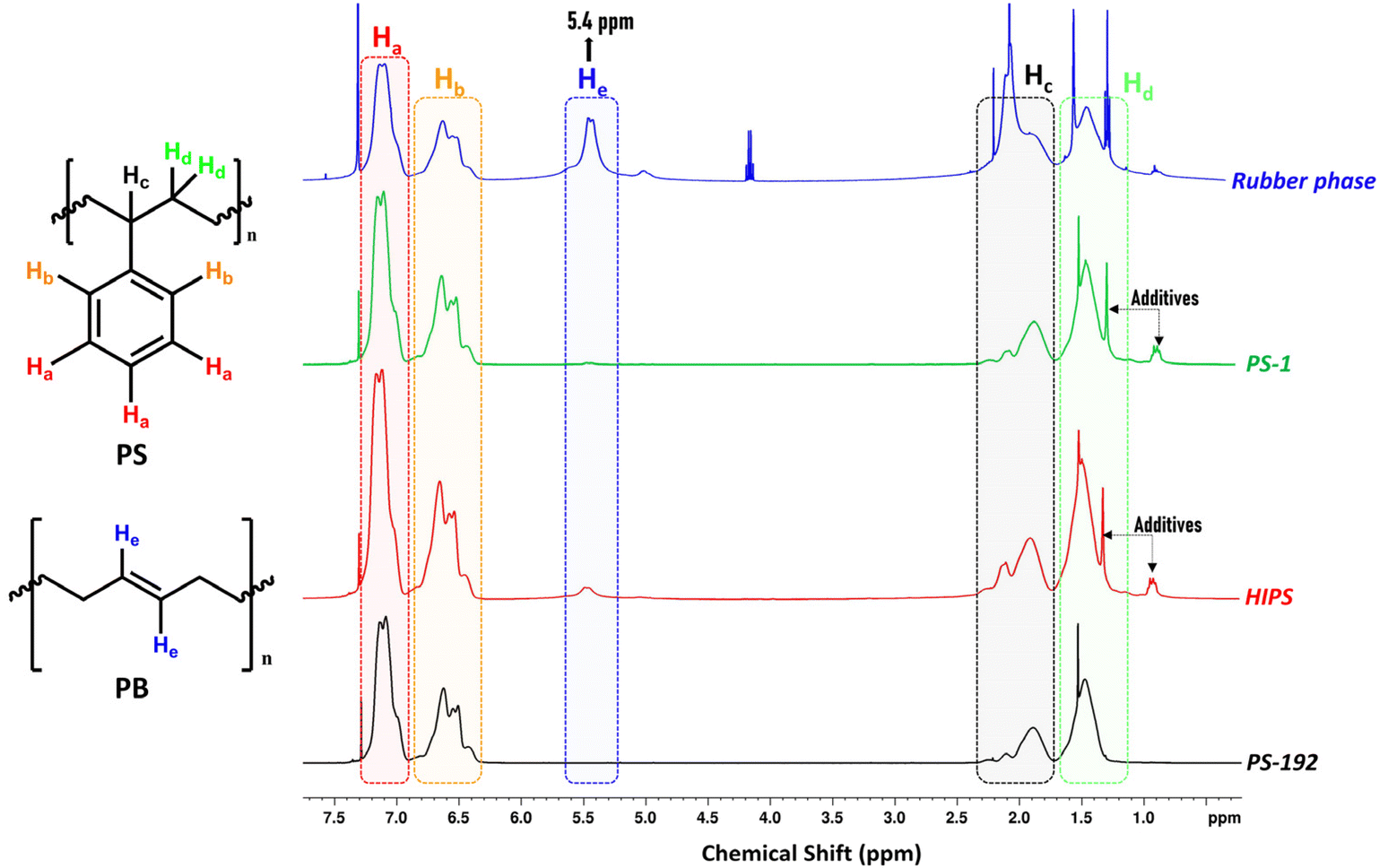
Like in small scale experiments, 1H-NMR was used to determine the composition and purity of the isolated phases. Fig. 4 presents the 1H-NMR spectra of HIPS, the commercial (and pure) PS (PS-192), and the fractions isolated from HIPS, viz. PS-1 and the rubber phase. The spectra reveal a prominent difference in the vinylic proton region, around 5.4 ppm. Compared to the original HIPS sample, only a negligible signal is observed for PS-1 in this region (Fig. S13 ESI†), while in the case of the rubber phase (Fig. S14 ESI†), the relative intensity of the signal corresponding to the vinylic He is significantly increased, indicating a high PB content in the sample. In both the HIPS and the PS-1 sample, the signals at 0.9 ppm, 1.3 ppm (the multiplet and singlet, respectively) can be attributed to the additives present in HIPS (zinc stearate, Irganox 1076),67,68 whereas for the rubber phase, the signal at 0.9 ppm can be assigned to –CH3 terminal groups in PB.
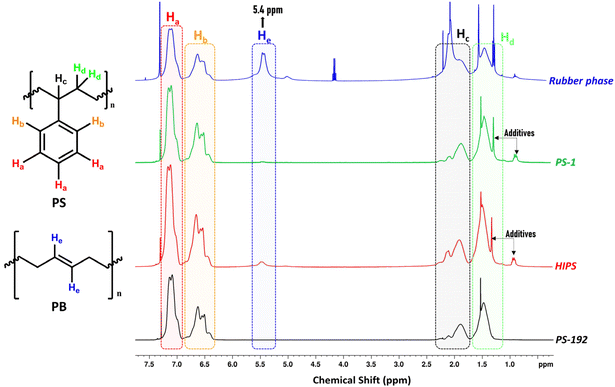 |
| | Fig. 4
1H-NMR comparison of commercial PS-192, HIPS, PS-1 (isolated PS), and the rubber phase. The spectra illustrate the most distinctive proton signals of both polymers contained in HIPS (400 MHz, solvent CDCl3). | |
3.2. Rubber phase upcycling: ethenolysis metathesis and isolation of the grafted PS
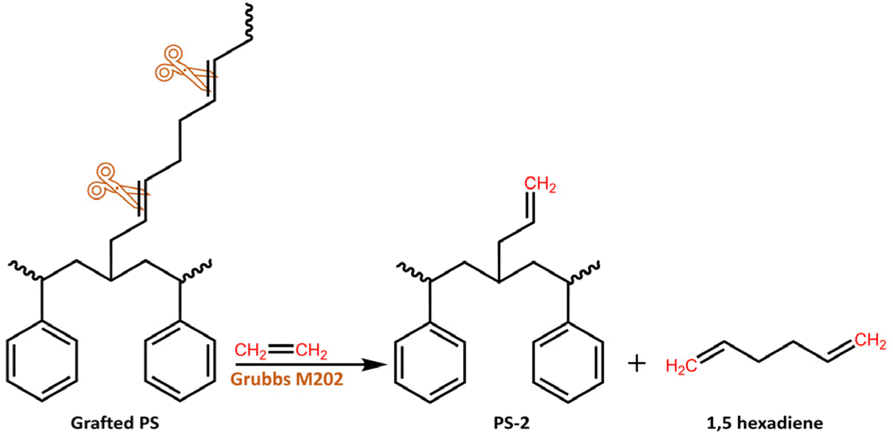
The next objective was to revalorize the rubber phase by recovering the grafted PS and using the PB for obtaining α,ω-dienes through ethenolysis-metathesis, as presented in Scheme 2.
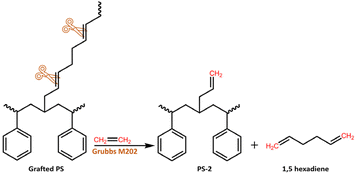 |
| | Scheme 2 Schematic representation of the ethenolysis-metathesis using the rubber phase as a starting material. | |
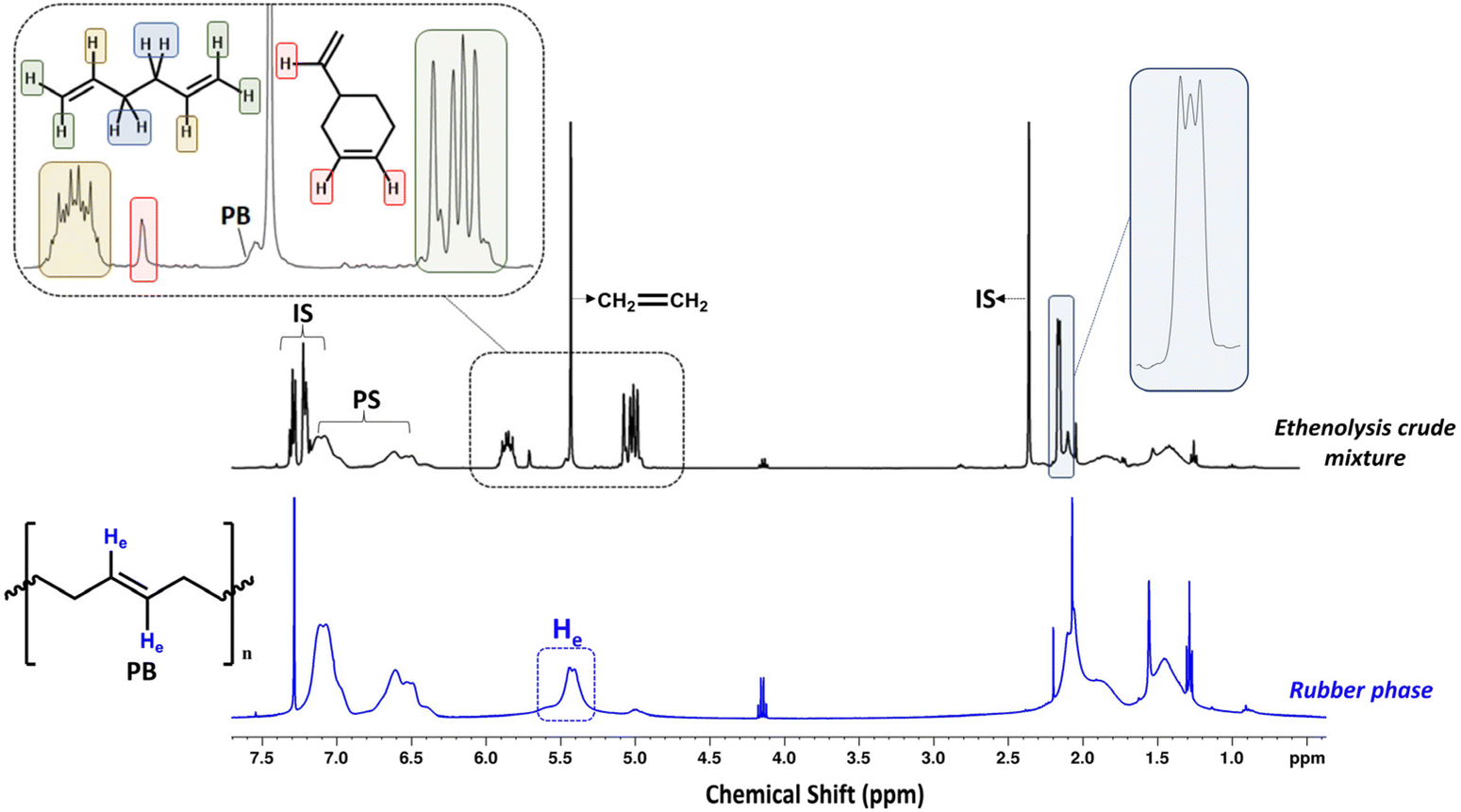
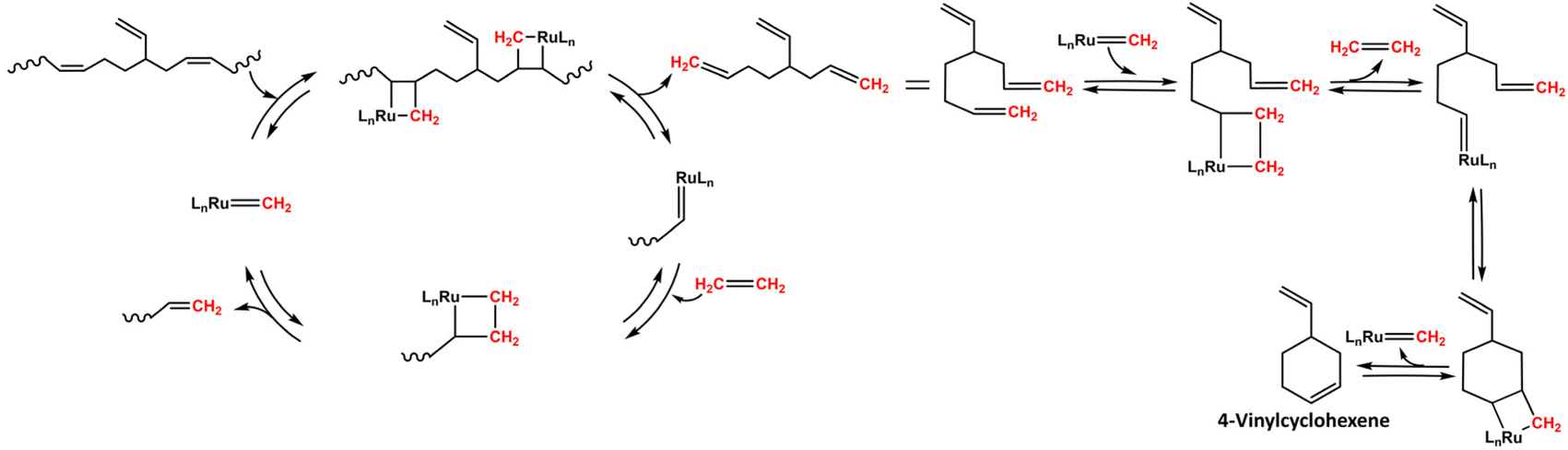
After 4 h of reaction time, the crude ethenolysis mixture was analyzed by 1H-NMR (Fig. 5), which showed a sharp decrease in the main peak of the PB vinylic protons (5.4 ppm) and the appearance of new multiplets at 2.2, 5.0, 5.7 and 5.8 ppm. These multiplets can be ascribed to the protons of 1,5-hexadiene, the major product expected from the ethenolysis of PB.44 The presence of 4-vinylcyclohexene, a potential side-product of the reaction, formed according to Scheme 3,41,69 was evidenced by the appearance of a signal at 5.7 ppm and confirmed by GC-MS. The yields of the ethenolysis products were calculated by 1H-NMR and found to be 60% (of 1,5-hexadiene) and 5% (of 4-vinylcyclohexene) with respect to the initial 37.5 wt% PB content included in the rubber phase (Fig. S15 ESI†). In comparison, using a commercial PB sample with molecular weight of 200 kDa, the yield of 1,5-hexadiene reaches 72%, while the yield of 4-vinylcyclohexene is below 1% (Fig. S16 ESI†). These discrepancies can be due to structural differences between commercial PB and the one present in the rubber, e.g. a different degree of 1,2- versus 1,4-incorporation of butadiene. This would explain the increased relative formation of 4-vinylcyclohexene from the rubber.
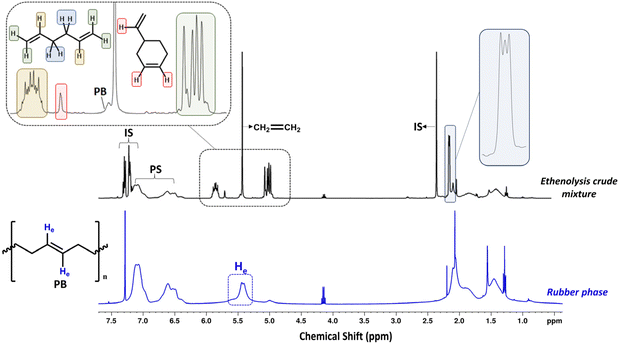 |
| | Fig. 5
1H-NMR comparison of the rubber phase and the crude ethenolysis reaction mixture. Signals at 7.3, 7.2, 2.4 ppm in the ethenolysis crude reaction mixture spectrum correspond to toluene (internal standard), while the signal at 5.4 ppm corresponds to leftover ethylene gas (400 MHz, CDCl3). | |
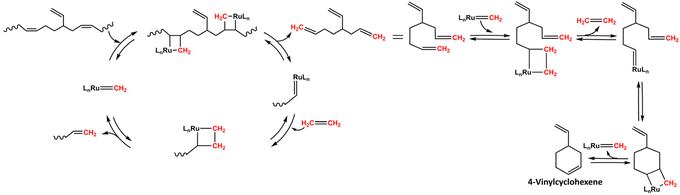 |
| | Scheme 3 Proposed mechanism for the formation of 4-vinylcyclohexene.41,69 | |
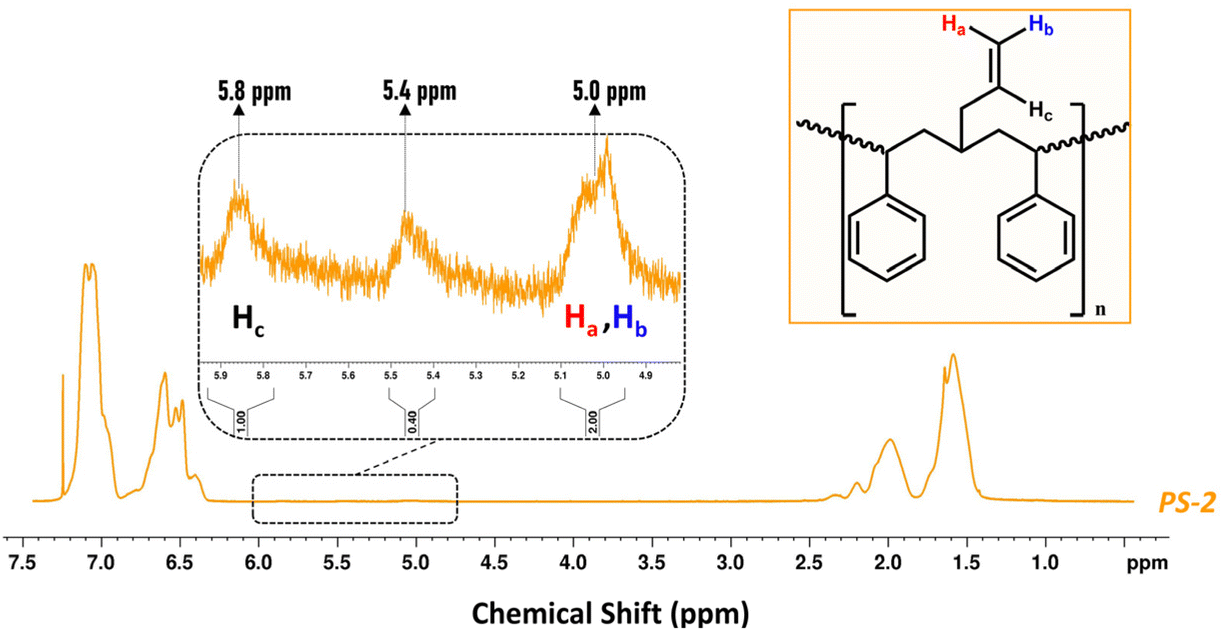
On the other hand, the isolated PS resulting from the ethenolysis metathesis process (denoted PS-2) was recovered by addition of MeOH (as anti-solvent) with an overall efficiency of almost 90% (Fig. S17 ESI†). It was first characterized by 1H-NMR (Fig. 6 and Fig. S17 ESI†). Close inspection of the spectrum reveals the appearance of two weak signals in the region between 5.0 and 6.0 ppm (vinylic region), which are due to the 2-propenyl side chains on the backbone of PS-2, in accordance with Scheme 2. The diastereotopic protons Ha, Hb are detected at 5.0 ppm, while Hc is detected at 5.8 ppm.70 In addition, the signal at 5.4 ppm probably originates from the –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) of lower molecular weight PB that is either attached to the PS-2 backbone or co-precipitated upon the addition of MeOH (Fig. 6 and Fig. S17 ESI†).
of lower molecular weight PB that is either attached to the PS-2 backbone or co-precipitated upon the addition of MeOH (Fig. 6 and Fig. S17 ESI†).
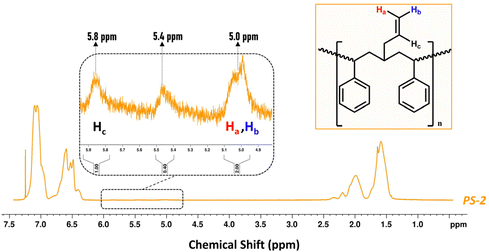 |
| | Fig. 6
1H-NMR spectrum of PS-2 isolated from the ethenolysis reaction mixture (600 MHz, solvent CDCl3). | |
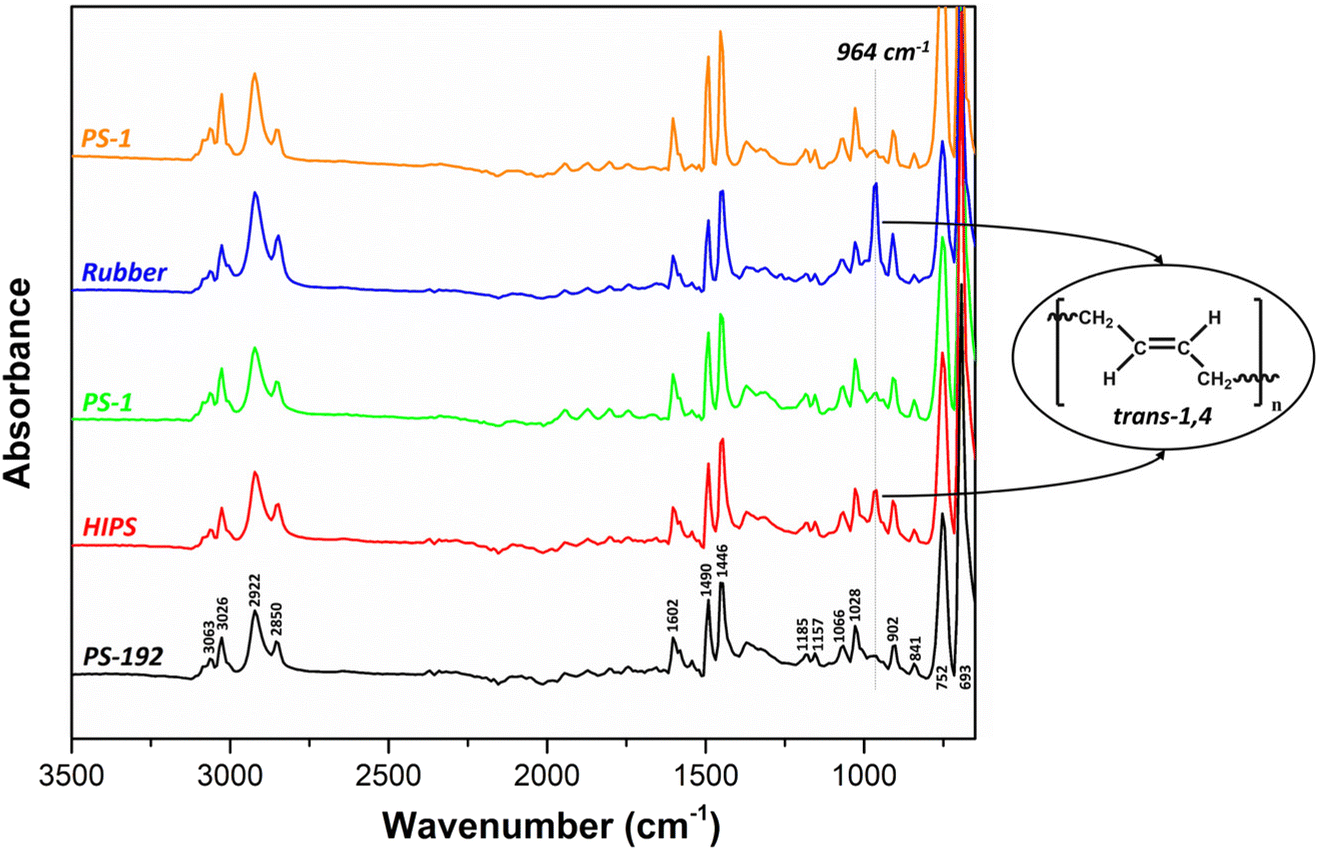
The characterization of the isolated fractions PS-1 and PS-2, was further expanded to ATR-FTIR measurements. The FTIR spectra of HIPS, PS-192, PS-1, PS-2 and the rubber phase revealed the presence of distinctive bands, assigned to the PS backbone of the samples (Fig. 7): C–H stretching between 3200–2800 cm−1, with aromatic C–H stretching vibrations at 3086, 3063 and 3026 cm−1, asymmetrical and symmetrical stretching vibrations of the methylene –CH2 groups at 2922 and 2850 cm−1, respectively; C–C stretching vibrations in the benzene ring at 1602, 1490, 1446 cm−1; C–C stretching and C–H bending vibrations at 1185, 1157 cm−1; in-plane C–H bending of the aromatic ring at 1066 and 1028 cm−1; and out-of-plane C–H bending vibrations between 900–675 cm−1.71 The two most intense bands at 752 and 693 cm−1 reveal that the samples contain monosubstituted benzene rings.71 Although the FTIR spectra of all samples look very similar, the intensity of the band around 964 cm−1 differs. This band is related to the PB fraction and it is attributed to the CC vibration of trans-1,4 double bonds.72 Accordingly, the band is present with the highest intensity in the spectrum of the rubber phase, and it is also observed in the spectrum of the HIPS sample. However, it is not observed in the spectra of the samples PS-1 and PS-2, which can be attributed to the low PB content (0.9 wt% in PS-1 and 0.05 wt% in PS-2) attached to the PS backbone (or lower molecular weight PB) in these samples.
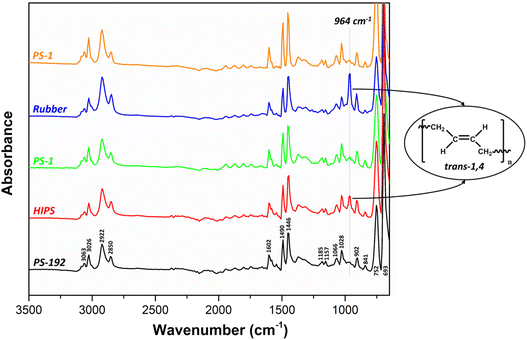 |
| | Fig. 7 ATR-FTIR spectra of PS-192, HIPS, PS-1, PS-2 and the rubber phase. Major differences are observed in the intensity of the 964 cm−1 band, attributed to the CC vibration of trans-1,4 double bonds. | |
3.3. Thermal degradation of the PS phase
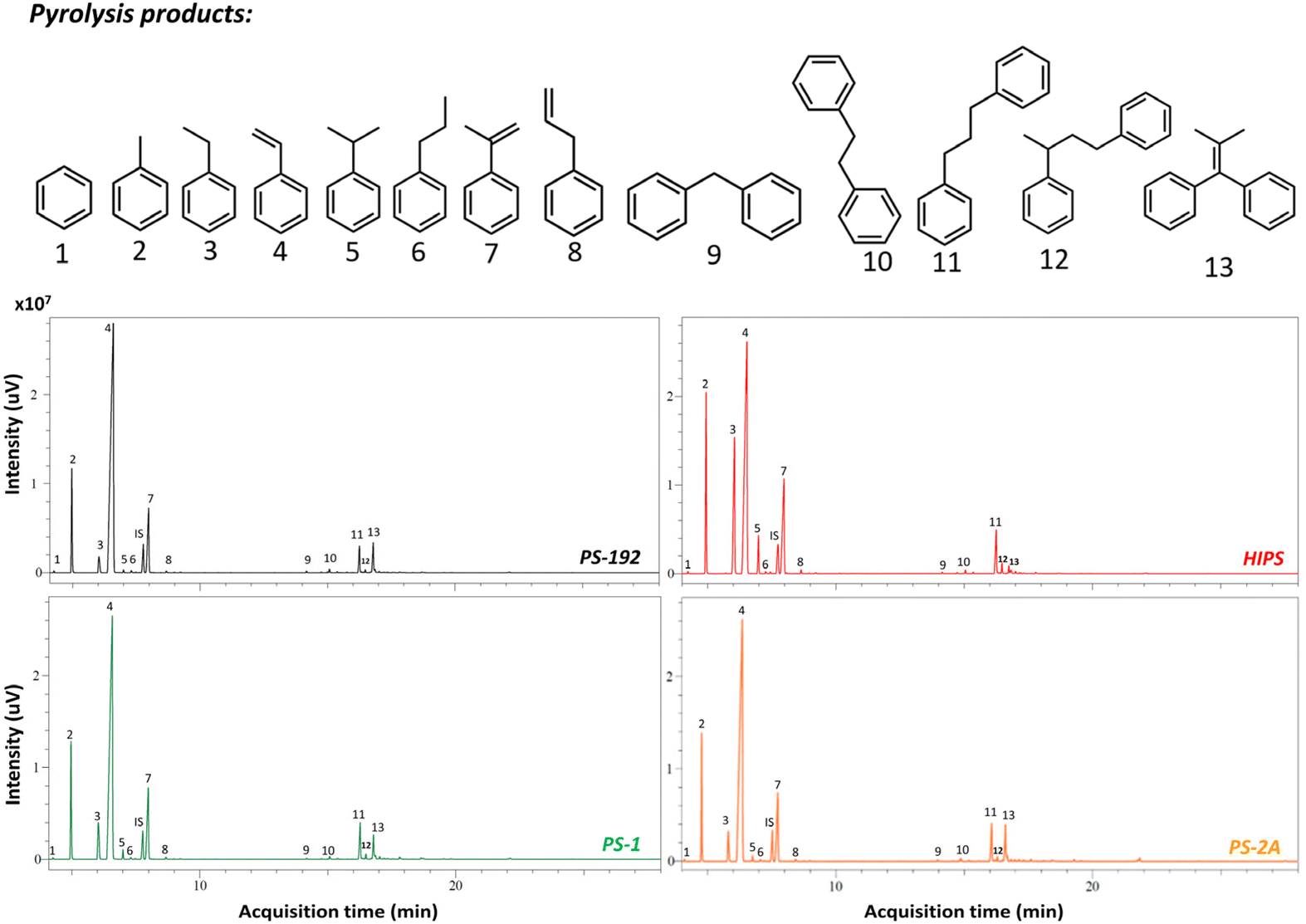
After isolation of the two PS fractions, PS-1 and PS-2, and with a circular economy and chemical recycling to monomer process in mind,73 we envisioned the pyrolysis of such phases and evaluated the impact of the pre-fractionation process on the thermal degradation of HIPS. In order to determine the average molecular weight of PS-1 and PS-2, GPC measurements were carried out, showing that PS-1 has an average molecular weight of 180 kDa, while PS-2 has an average molecular weight of 217 kDa (Fig. S18 and S19 ESI†), and therefore, the aforementioned samples are expected to give similar thermal degradation results as PS-192. To this end, the pyrolysis of the sample PS-192 was studied as a reference. Fig. 8 presents the GC chromatogram obtained after the pyrolysis of 10 g PS-192 at 300 °C for 1 h, where 13 major products are detected (Fig. S20 ESI†), with styrene being the most abundant. Conversely, in the case of the thermal degradation of HIPS, the generated amounts of ethylbenzene, toluene and α-methyl-styrene increased, while the selectivity to styrene decreased. Specifically, the ethylbenzene, toluene and α-methylstyrene selectivity rose from 2% to 16%, 10% to 14%, and 9% to 13%, respectively; the styrene selectivity on the other hand dropped from 74% to 50%, as shown in Table 5. These changes in the product selectivity are a clear indication of the effect of the presence of the PB particles on the thermal degradation mechanism; they contribute to the formation of stable tertiary radicals in the backbone of the grafted PS, which eventually end up preferentially producing toluene, ethylbenzene and α-methylstyrene.74 Remarkably, the composition of the product mixture obtained from the thermal degradation of the recovered PS from HIPS (PS-1) was very similar to that of the reference PS-192, with selectivities to toluene, ethylbenzene, styrene, α-methylstyrene of 11%, 5%, 69%, and 10%, respectively. The small difference in styrene selectivity (74% for PS-192 v. 69% for PS-1) can be attributed to the remaining 0.9 wt% of PB, which, as previously discussed, has a negative effect on the styrene recovery.
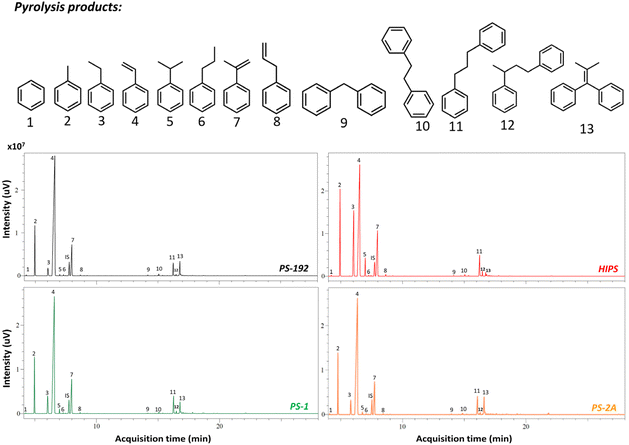 |
| | Fig. 8 Comparison of the GC chromatograms obtained after the thermal degradation of PS-192, HIPS, PS-1, PS-2A. | |
Table 5 Thermal decomposition of PS and HIPS samples
| Liquid composition (%) |
PS-192 |
HIPSa |
PS-1b |
PS-2Ac |
|
12.5 g HIPS (corresponding to 10 g of pure PS).
10 g PS-1 (PS isolated from HIPS).
PS-2A consists of 9 g of PS-1 and 1 g of PS-2 (PS isolated after ethenolysis).
Styrene yield calculated from the liquid yield and the styrene selectivity from GC and GC-MS analyses.
|
| Benzene (1) |
0.2 |
0.2 |
0.2 |
0.2 |
| Toluene (2) |
10.2 |
14.4 |
11.1 |
10.8 |
| Ethylbenzene (3) |
2.4 |
16.1 |
4.7 |
3.5 |
|
Styrene (4)
|
73.9
|
49.7
|
69.1
|
69.6
|
| Cumene (5) |
0.2 |
2.6 |
0.6 |
0.4 |
| Propylbenzene (6) |
0.2 |
0.2 |
0.2 |
0.2 |
| α-Methylstyrene (7) |
8.9 |
13.0 |
9.6 |
9.6 |
| Allylbenzene (8) |
0.2 |
0.3 |
0.2 |
0.2 |
| Diphenylmethane (9) |
0.1 |
0.1 |
0.1 |
0.1 |
| 1,2-Diphenylethane (10) |
0.2 |
0.2 |
0.2 |
0.2 |
| 1,3-Diphenylpropane (11) |
1.5 |
2.5 |
2.3 |
2.4 |
| Butane-1,3-diyldibenzene (12) |
0.1 |
0.4 |
0.2 |
0.2 |
| 2-Methyl-1,1-diphenyl propene (13) |
1.9 |
0.3 |
1.5 |
2.6 |
| |
| Liquid yield (%) |
75.0 |
60.1 |
75.4 |
75.2 |
| Gasses/losses (%) |
7.8 |
8.2 |
9.4 |
9.2 |
| Residue (%) |
17.2 |
31.8 |
15.2 |
15.6 |
|
Styrene yield (%)
|
55.4
|
29.9
|
52.1
|
52.3
|
Additional experiments were performed mixing the two recovered PS phases, PS-1 and PS-2 (sample PS-2A). In this case, the selectivity to styrene in the pyrolysis experiment was slightly increased to 70%, compared to 69% with pure PS-1. This increment, while rather low, could be explained by the lower PB content of the PS-2 fraction, as shown in Table S2 of the ESI.† Overall, the results presented in Table 5 highlight the positive effect of the pre-fractionation of PS from HIPS, which leads to an enhanced styrene yield (almost 20% higher compared to the non-fractionated, HIPS sample). Besides the composition of the pyrolysis liquid, the pre-fractionation procedure also affects the composition of the gas phase. Analysis of the Tedlar gas collection bags for PS-192, HIPS and PS-1 showed the presence of C1–C4 gases along with some toluene, ethylbenzene and styrene. In the case of HIPS, the percentages of C1–C4 are significantly higher than those obtained for the commercial sample PS-192, with a lower amount of styrene. After the fractionation, the amount of C1–C4 decreases from 77% to 56% and the amount of styrene increases from 7% to 18% (Fig. S21 and ESI, Table S3†), indicating once again a more efficient styrene recovery due to the pre-fractionation process. It should be pointed out that the mass balance cannot be fully closed since there are around 5% losses after each thermal degradation experiment (Table 5 and Table S3†).
Moreover, analysis of the solid residue by GC-MS and 1H-NMR (Fig. S22 ESI†) reveals the presence of several aromatic molecules, including styrene and α-methyl-styrene. Remarkably, the 1H-NMR analysis provides important evidence of the positive effect of the pre-fractionation, regarding the decrease of char formation. In particular, after the thermal degradation of HIPS large amounts of char are formed (32%, Table 5), remaining in the reactor along with the degraded PB particles (Fig. S23 ESI†), which would complicate the cleaning of the reactor with a potential industrial application in sight.75–77 Conversely, minimal amounts of char remained after the thermal degradations of PS-192, PS-1, PS-2A. Considering these facts, we can infer that the pre-fractionation process is not only important for the styrene recovery, but would also contribute to ease and simplify a potential industrial HIPS recycling process.
3.4. A total revalorization approach and future prospects
Overall, we have presented an alternative approach for the revalorization of HIPS to styrene, aiming at a circular economy, by maximizing PS recovery and recycling, not only via the proposed green fractionation of HIPS with EtOAc, but also through two well-known and established processes: pyrolysis of PS and ethenolysis of PB (Fig. S24 ESI†). However, it is worth noting that there are still several opportunities for further improving these processes, which are important to consider before implementing the proposed upcycling route, besides the ones currently being explored by various chemical companies.78 Regarding the pyrolysis of PS, which currently has a high energy demand and a complex reaction mechanism, the process could be upgraded by the inclusion of a catalyst. The presence of an effective catalyst would allow to obtain a narrower product distribution under less stringent conditions.6 Regarding the ethenolysis of PB, already some advancements have been achieved by designing novel immobilized catalysts.79–81 However, there is certainly room for improvement, especially in the preparation of more active and selective catalysts, with higher chemical stability, recyclability and robustness.41 In this sense, more studies on catalytic routes towards polystyrene recycling and green catalytic routes for CC cleavage would uncover their full potential.
4. Conclusions
We have developed a new strategy for the total revalorization of HIPS, by isolating the free PS matrix from the rubber phase by addition of EtOAc as a pre-fractionation solvent. The thermal degradation of the isolated free PS matrix led to an enhanced styrene yield compared to the non-fractionated HIPS sample, with a concomitant decrease in the amount of generated char. Ethenolysis metathesis was applied for the valorization of the rubber phase, producing 1,5-hexadiene in high yields and resulting in the isolation of the grafted PS, which can be further thermally degraded without affecting the styrene yield.
All in all, the insights obtained from the pre-fractionation procedure and the ethenolysis metathesis, could provide a route towards a more efficient way to recycle HIPS at an industrial level, aiming at a circular economy, especially considering that the described process allows for an increase of the styrene selectivity and yield of up to 20%, compared to the non-fractionated HIPS. Besides the positive effects on the styrene recovery, the fractionation procedure minimizes the formation of char, which simplifies the cleaning process in industrial reactors and would help circumvent the use of toxic solvents, thus decreasing the negative environmental impact of the depolymerization.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was performed in the framework of the Moonshot cluster SBO project PREFER (HBC.2020.2609 “The plastics refinery: no more waste”), with the financial support of VLAIO (Flemish Agency for Innovation and Entrepreneurship) via the Flemish spearhead cluster Catalisti. F. D. P. and M. D. acknowledge the Research Foundation – Flanders (FWO) for financial support through SBO grant S001819N. The computational resources and services used in this work were provided by the VSC (Flemish Supercomputing Center), funded by the Research Foundation – Flanders (FWO) and the Flemish Government. J. V. acknowledges the Research Foundation – Flanders (FWO) for a junior postdoctoral mandate (project no. 12E6423N). The authors thank Margot Houbrechts for assistance during gel permeation chromatography measurements.
References
- N. Simon, K. Raubenheimer, N. Urho, S. Unger, D. Azoulay, T. Farrelly, J. Sousa, H. Van Asselt, G. Carlini, C. Sekomo, M. L. Schulte, P. O. Busch, N. Wienrich and L. Weian, A binding global agreement to address the life cycle of plastics, Science, 2021, 373(6550), 43–47 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3(7), e1700782 CrossRef PubMed.
- L. Lebreton and A. Andrady, Future scenarios of global plastic waste generation and disposal, Palgrave Commun., 2019, 5(6), 1–11 Search PubMed.
- EuropePlastics. Plastics – the Facts 2021. An analysis of European plastics production, demand andwaste data, retrieved from: https://plasticseurope.org/knowledge-hub/plastics-the-facts-2021/.
- Y. Qin, T. Zhang, H. Y. V. Ching, G. S. Raman and S. Das, Integrated strategy for the synthesis of aromatic building blocks via upcycling of real-life plastic wastes, Chem, 2022, 8(9), 2472–2484 CAS.
- C. Marquez, C. Martin, N. Linares and D. E. De Vos, Catalytic routes towards polystyrene recycling, Mater. Horiz., 2023, 10, 1625–1640 RSC.
-
T. A. Farrelly and I. C. Shaw, Polystyrene as Hazardous Household Waste, in Household Hazardous Waste Management, ed. D. Mmereki, InctechOpen, Rijeka, 2017, pp. 45–60 Search PubMed.
- F. Parres, L. Sánchez, R. Balart and J. López, Determination of the photo-degradation level of high impact polystyrene (HIPS) using pyrolysis–gas chromatography–mass spectrometry, J. Anal. Appl. Pyrolysis, 2007, 78(2), 250–256 CrossRef CAS.
- A. Marcilla, A. Gómez-Siurana, J. C. García Quesada and D. Berenguer, Characterization of styrene–butadiene copolymers by catalytic pyrolysis over Al-MCM-41, J. Anal. Appl. Pyrolysis, 2009, 85(1–2), 327–333 CrossRef CAS.
- S. Thakur, A. Verma, B. Sharma, J. Chaudhary, S. Tamulevicius and V. K. Thakur, Recent developments in recycling of polystyrene based plastics, Curr. Opin. Green Sustain. Chem., 2018, 13, 32–38 CrossRef.
- F. Lin, M. Zhang, X. Li, S. Mao and Y. Wei, Synergistic Effects of Diatoms on Intumescent Flame Retardant High Impact Polystyrene System, Polymers, 2022, 14(20), 4453 CrossRef CAS PubMed.
- Y. V. Vazquez and S. E. Barbosa, Compatibilization Strategies for Recycling Applications of High Impact Polystyrene/Acrylonitrile Butadiene Blends, J. Polym. Environ., 2017, 25(3), 903–912 CrossRef CAS.
- World Economic Forum. (2020). Projected electronic waste generation worldwide from 2019 to 2030 (in million metric tons)*. Statista. Statista Inc. Accessed: June 22, 2023. Retrieved from: https://www.statista.com/statistics/1067081/generation-electronic-waste-globally-forecast/.
- J. A. Salbidegoitia, E. G. Fuentes, M. P. González-Marcoset and J. R. González-Velasco, Recycle of plastic residues in cellular phones through catalytic hydrocracking to liquid fuels, J. Mater. Cycles Waste Manage., 2017, 19(2), 782–793 CrossRef CAS.
- J. Wei, J. Liu, W. Zeng, Z. Dong, J. Song, S. Liu and G. Liu, Catalytic hydroconversion processes for upcycling plastic waste to fuels and chemicals, Catal. Sci. Technol., 2023, 13, 1258–1280 RSC.
- N. M. Wang, G. Strong, V. DaSilva, L. Gao, R. Huacuja, I. A. Konstantinov, M. S. Rosen, A. J. Nett, S. Ewart, R. Geyer, S. L. Scott and D. Guironnet, Chemical Recycling of Polyethylene by Tandem Catalytic Conversion to Propylene, J. Am. Chem. Soc., 2022, 144(40), 18526–18531 CrossRef CAS PubMed.
- G. Zichittella, A. M. Ebrahim, J. Zhu, A. E. Brenner, G. Drake, G. T. Beckham, S. R. Bare, J. E. Rorrer and Y. Román-Leshkov, Hydrogenolysis of Polyethylene and Polypropylene into Propane over Cobalt-Based Catalysts, JACS Au, 2022, 2, 2259–2268 CrossRef CAS PubMed.
- J. E. Rorrer, A. M. Ebrahim, Y. Questell-Santiago, J. Zhu, C. Troyano-Valls, A. S. Asundi, A. E. Brenner, S. R. Bare, C. J. Tassone, G. T. Beckham and Y. Román-Leshkov, Role of Bifunctional Ru/Acid Catalysts in the Selective Hydrocracking of Polyethylene and Polypropylene Waste to Liquid Hydrocarbons, ACS Catal., 2022, 12, 13969–13979 CrossRef CAS.
- F. Vilaplana, S. Karlsson, A. Ribes-Greus, C. Schade and N. Nestle, NMR relaxation reveals modifications in rubber phase dynamics during long-term degradation of high-impact polystyrene (HIPS), Polymer, 2011, 52(6), 1410–1416 CrossRef CAS.
- L. Song, Y. Zhang, J. Ren, Y. Li, B. Yang, E. Xing, Y. Wang and Y. Shi, Effect of Entanglement Density on Mechanical Properties and the Deformation Mechanism of Rubber-Modified PPO/PS Blends, Macromol. Mater. Eng., 2022, 307(10), 2200325 CrossRef CAS.
- Y. Israeli, J. Lacoste, J. Lemaire, R. P. Singh and S. Sivaram, Photo- and thermoinitiated oxidation of high-impact polystyrene. I. Characterization by FT-IR spectroscopy, J. Polym. Sci., Part A: Polym. Chem., 1994, 32(3), 485–493 CrossRef CAS.
- F. Soriano, G. Morales and R. Díaz de León, Recycling of high impact polystyrene in coextruded sheet: Influence of the number of processing cycles on the microstructure and macroscopic properties, Polym. Eng. Sci., 2006, 46(12), 1698–1705 CrossRef CAS.
- E. P. Chang and A. Takahashi, Factors influencing the impact strength of high impact polystyrene, Polym. Eng. Sci., 1978, 18(5), 350–354 CrossRef CAS.
- A. Vishwa Prasad and R. P. Singh, Recent Developments in the Degradation and Stabilization of High-Impact Polystyrene, J. Macromol. Sci., Part C, 1997, 37(4), 581–598 CrossRef.
- S. H. Jung, S. J. Kim and J. S. Kim, The influence of reaction parameters on characteristics of pyrolysis oils from waste high impact polystyrene and acrylonitrile–butadiene–styrene using a fluidized bed reactor, Fuel Process. Technol., 2013, 116, 123–129 CrossRef CAS.
- C. Ma, J. Yu, B. Wang, Z. Song, J. Xiang, S. Hu, S. Su and L. Sun, Catalytic pyrolysis of flame retarded high impact polystyrene over various solid acid catalysts, Fuel Process. Technol., 2017, 155, 32–41 CrossRef CAS.
- C. Ma, J. Yu, Q. Yan, Z. Song, K. Wang, B. Wang and L. Sun, Pyrolysis-catalytic upgrading of brominated high impact polystyrene over Fe and Ni modified catalysts: Influence of HZSM-5 and MCM-41 catalysts, Polym. Degrad. Stab., 2017, 146, 1–12 CrossRef CAS.
- E. V. Antonakou, K. G. Kalogiannis, S. D. Stephanidis, K. S. Triantafyllidis, A. A. Lappas and D. S. Achilias, Pyrolysis and catalytic pyrolysis as a recycling method of waste CDs originating from polycarbonate and HIPS, Waste Manage., 2014, 34(12), 2487–2493 CrossRef CAS.
- C. Khaobang and C. Areeprasert, Investigation on thermal decomposition and kinetics study of recovered oil from electronic waste by thermogravimetric analysis, Energy Procedia, 2017, 138, 506–511 CrossRef CAS.
- N. Cai, X. Li, S. Xia, L. Sun, J. Hu, P. Bartocci, F. Fantozzi, P. T. Williams, H. Yang and H. Chen, Pyrolysis-catalysis of different waste plastics over Fe/Al2O3 catalyst: High-value hydrogen, liquid fuels, carbon nanotubes and possible reaction mechanisms, Energy Convers. Manage., 2021, 229, 113794 CrossRef CAS.
- C. Muhammad, J. A. Onwudili and P. T. Williams, Catalytic pyrolysis of waste plastic from electrical and electronic equipment, J. Anal. Appl. Pyrolysis, 2015, 113, 332–339 CrossRef CAS.
- S. Thakur, A. Verma, B. Sharma, J. Chaudhary, S. Tamulevicius and V. K. Thakur, Recent developments in recycling of polystyrene based plastics, Curr. Opin. Green Sustain. Chem., 2018, 13, 32–38 CrossRef.
- A. da R. D. Monteiro, D. M. V. De Miranda, J. C. C. Da Silva Pinto and J. J. Soto, Life Cycle Assessment of the Catalytic Pyrolysis of High-Density Polyethylene (HDPE) and High-Impact Polystyrene (HIPS), Macromol. React. Eng., 2022, 16(6), 2200037 CrossRef CAS.
- Q. Kong, R. Lv and S. Zhang, Flame retardant and the degradation mechanism of high impact polystyrene/Fe-montmorillonite nanocomposites, J. Polym. Res., 2008, 15(6), 453–458 CrossRef CAS.
- E. Jakab, Md. A. Uddin, T. Bhaskar and Y. Sakata, Thermal decomposition of flame-retarded high-impact polystyrene, J. Anal. Appl. Pyrolysis, 2003, 68–69, 83–99 CrossRef CAS.
-
N. R. Ruffing, US Pat., 3243481, 1966 Search PubMed.
- R. A. Hall, Measurement of rubber particle fraction in high-impact polystyrene, J. Mater. Sci., 1990, 25(1), 183–186 CrossRef CAS.
- J. Rovere, C. A. Correa, V. G. Grassi and M. F. Dal Pizzol, Role of the rubber particle and polybutadiene cis content on the toughness of high impact polystyrene, J. Mater. Sci., 2008, 43(3), 952–959 CrossRef CAS.
- Y. Subbaiah, V. B. Raghavendra, S. Puttamadappa, S. Bandyopadhyay, P. Mukhopadhyay and A. Kumar, Influence of submicron tiny gel rubber particles on the estimation of grafting and rubber efficiency in high-impact polystyrene, J. Elastomers Plastics, 2017, 49(8), 684–695 CrossRef CAS.
- V. G. Grassi, M. F. Dal Pizzol, M. M. C. Forte and S. C. Amico, Influence of small rubber particles on the environmental stress cracking of high impact polystyrene, J. Appl. Polym. Sci., 2011, 121(3), 1697–1706 CrossRef CAS.
- J. Bidange, C. Fischmeister and C. Bruneau, Ethenolysis: A Green Catalytic Tool to Cleave Carbon–Carbon Double Bonds, Chem. – Eur. J., 2016, 22(35), 12226–12244 CrossRef CAS PubMed.
-
Z. Wu, W. Xie, C. Xie, Q. Liu, X. Mu, J. Zhang, Y. Luo, X. Shu and C. Yan, US Pat, 0281363, 2009 Search PubMed.
- J. Spekreijse, J. Le Nôtre, J. van Haveren, E. L. Scott and J. P. M. Sanders, Simultaneous production of biobased styrene and acrylates using ethenolysis, Green Chem., 2012, 14, 2747–2751 RSC.
- M. D. Watson and K. B. Wagener, Acyclic diene metathesis (ADMET) depolymerization: ethenolysis of 1,4-polybutadiene using a ruthenium complex, J. Polym. Sci., Part A: Polym. Chem., 1999, 37(12), 1857–1861 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- W. L. Jorgensen and J. Tirado-Rives, The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin, J. Am. Chem. Soc., 1988, 110(6), 1657–1666 CrossRef CAS PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, GROMACS: Fast, flexible, and free, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS.
- W. L. Jorgensen and J. Tirado-Rives, Potential energy functions for atomic-level simulations of water and organic and biomolecular systems, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(19), 6665–6670 CrossRef CAS.
- L. S. Dodda, J. Z. Vilseck, J. Tirado-Rives and W. L. Jorgensen, 1.14*CM1A-LBCC: Localized Bond-Charge Corrected CM1A Charges for Condensed-Phase Simulations, J. Phys. Chem. B, 2017, 121(15), 3864–3870 CrossRef CAS PubMed.
- L. S. Dodda, I. Cabeza de Vaca, J. Tirado-Rives and W. L. Jorgensen, LigParGen web server: an automatic OPLS-AA parameter generator for organic ligands, Nucleic Acids Res., 2017, 45(W1), W331–W336 CrossRef CAS.
- M. Denayer, J. Vekeman, F. Tielens and F. De Proft, Towards a predictive model for polymer solubility using the noncovalent interaction index: polyethylene as a case study, Phys. Chem. Chem. Phys., 2021, 23(44), 25374–25387 RSC.
- K. Hu, Z. K. Cui, Y. Yuan, Q. Zhuang, T. Wang, X. Liu and Z. Han, Synthesis, structure, and properties of high-impact polystyrene/octavinyl polyhedral oligomeric silsesquioxane nanocomposites, Polym. Compos., 2016, 37(4), 1049–1055 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, Molecular dynamics with coupling to an external bath, J. Chem. Phys., 1984, 81(4), 3684–3690 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, LINCS: A linear constraint solver for molecular simulations, J. Comput. Chem., 1997, 18(12), 1463–1472 CrossRef CAS.
- D. J. Evans and B. L. Holian, The Nose–Hoover thermostat, J. Chem. Phys., 1985, 83(8), 4069–4074 CrossRef CAS.
- M. Parrinello and A. Rahman, Polymorphic transitions in single crystals: A new molecular dynamics method, J. Appl. Phys., 1981, 52(12), 7182–7190 CrossRef CAS.
- S. Nosé and M. L. Klein, Constant pressure molecular dynamics for molecular systems, Mol. Phys., 1983, 50(5), 1055–1076 CrossRef.
- Y. B. Zhao, X. D. Lv and H. G. Ni, Solvent-based separation and recycling of waste plastics: A review, Chemosphere, 2018, 209, 707–720 CrossRef CAS PubMed.
- B. A. Miller-Chou and J. L. Koenig, A review of polymer dissolution, Prog. Polym. Sci., 2003, 28(8), 1223–1270 CrossRef CAS.
- I. Tsampanakis and A. O. White, The Mechanics of Forming Ideal Polymer–Solvent Combinations for Open-Loop Chemical Recycling of Solvents and Plastics, Polymers, 2022, 14, 112 CrossRef CAS PubMed.
- I. M. Balashova, R. G. Buduen and R. P. Danner, Solubility of organic solvents in 1,4-cis-polybutadiene, Fluid Phase Equilib., 2012, 334, 10–14 CrossRef CAS.
- E. Díez, G. Ovejero, M. D. Romero and I. Díaz, Polymer–solvent interaction parameters of SBS rubbers by inverse gas chromatography measurements, Fluid Phase Equilib., 2011, 308(1–2), 107–113 CrossRef.
- R. F. Cella, G. D. Mumbach, K. L. Andrade, P. Oliveira, C. Marangoni, A. Bolzan, S. Bernard and R. A. F. Machado, Polystyrene recycling processes by dissolution in ethyl acetate, J. Appl. Polym. Sci., 2018, 135(18), 46208 CrossRef.
- Y. Ihara, H. Yamagishi, M. Naito and Y. Yamamoto, Machine learning of organic solvents reveals an extraordinary axis in Hansen space as indicator of spherical precipitation of polymers, Aggregate, 2023, e365 CrossRef.
- Values available at: https://www.hansen-solubility.com/HSP-science/sphere.php.
-
C. M. Hansen, Hansen Solubility Parameters: A User's Handbook, CRC Press, Boca Raton, 2nd edn, 2007, p. 39 Search PubMed.
- K. S. Park, Y. J. Kim and E. K. Cho, Composition Characterization of Fatty Acid Zinc Salts by Chromatographic and NMR Spectroscopic Analyses on Their Fatty Acid Methyl Esters, J. Anal. Methods Chem., 2019, 7594767, 1–11 Search PubMed.
- Spectrum available at: https://sdbs.db.aist.go.jp/sdbs/cgi-bin/landingpage?sdbsno=16053.
- M. Yu, S. Lou and F. Gonzalez-Bobes, Ring-Closing Metathesis in Pharmaceutical Development: Fundamentals, Applications, and Future Directions, Org. Process Res. Dev., 2018, 22, 918–946 CrossRef CAS.
- D. Michel, W. Böhlmann, J. Roland and S. Mulla-Osman, Study of conformation and dynamics of molecules adsorbed in zeolites by 1H-NMR, Lect. Notes Phys., 2004, 634, 232–236 Search PubMed.
- D. Olmos, E. V. Martín and J. González-Benito, New molecular-scale information on polystyrene dynamics in PS and PS–BaTiO3 composites from FTIR spectroscopy, Phys. Chem. Chem. Phys., 2014, 16, 24339 RSC.
- F. J. Arráez, M. L. Arnal and A. J. Müller, Thermal degradation of high-impact polystyrene with pro-oxidant additives, Polym. Bull., 2019, 76(3), 1489–1515 CrossRef.
- G. W. Coates and Y. D. Y. L. Getzler, Chemical recycling to monomer for an ideal, circular polymer economy, Nat. Rev. Mater., 2020, 5(7), 501–516 CrossRef CAS.
-
M. Blazso, Composition of Liquid Fuels Derived from the Pyrolysis of Plastics, in Feedstock Recycling and Pyrolysis of Waste Plastics: Converting Waste Plastics into Diesel and Other Fuels, 2006, p. 330 Search PubMed.
- A. Matuszewska, M. Owczuk and K. Biernat, Current Trends in Waste Plastics’ Liquefaction into Fuel Fraction: A Review, Energies, 2022, 15(8), 2719 CrossRef CAS.
- R. V. Nanditta, K. Goyal, B. N. Rahul and G. Balaji, Review on Process Development and Challenges in Biomass Pyrolysis, J. Phys.: Conf. Ser., 2021, 2054(1), 012043 CrossRef.
- J. Y. M. Chewa, S. J. Tonneijk, W. R. Paterson and D. I. Wilson, Solvent-based cleaning of emulsion polymerization reactors, Chem. Eng. J., 2006, 117(1), 61–69 CrossRef.
- A. H. Tullo, Plastic has a problem; is chemistry the solution?, Chem. Eng. News, 2019, 97, 29–34 Search PubMed.
- J. Tomasek and J. Schatz, Olefin metathesis in aqueous media, Green Chem., 2013, 15, 2317–2338 RSC.
- M. Nagyházi, G. Turczel, A. Balla, G. Szálas, I. Tóth, G. T. Gál, B. Petra, P. T. Anastas and R. Tuba, Towards Sustainable Catalysis – Highly Efficient Olefin Metathesis in Protic Media Using Phase Labelled Cyclic Alkyl Amino Carbene (CAAC) Ruthenium Catalysts, ChemCatChem, 2020, 12, 1953–1957 CrossRef.
- H. Clavier, K. Grela, A. Kirschning, M. Mauduit and S. P. Nolan, Sustainable Concepts in Olefin Metathesis, Angew. Chem., Int. Ed., 2007, 46, 6786–6801 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Kwinten
Janssens
a,
Kwinten
Janssens