
A simple and convenient strategy for the oxidation of C(sp3)–H bonds based on γ-valerolactone†
Anwei
Wang
,
Jiayin
Huang
,
Chunsheng
Zhao
,
Yu
Fan
,
Junfeng
Qian
 ,
Qun
Chen
,
Mingyang
He
* and
Weiyou
Zhou
*
,
Qun
Chen
,
Mingyang
He
* and
Weiyou
Zhou
*
Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology, Changzhou University, 213164 Changzhou, China. E-mail: hemy_cczu@126.com; zhouwy426@126.com
First published on 21st November 2023
Abstract
The aerobic oxidation of C(sp3)–H bonds is an important issue in organic synthesis, and hydrogen atom transfer (HAT) is an effective process to initiate the oxidation reaction. Here, we demonstrate an innovative strategy for the oxidation of C(sp3)–H bonds based on γ-valerolactone, which can be produced from biomass. This reaction system promotes the aerobic oxidation of various benzylic and allylic C(sp3)–H bonds and alcohols. Excellent chemoselectivity for 1-isochromanone was achieved with the assistance of Ni2Al-layered double hydroxide by increasing the difference in bond dissociation energy between the 1- and 4-C–H bonds of isochroman. The excellent application potential of the reaction system has been demonstrated via the synthesis of pharmaceutical molecules, modification of steroidal substrates, scaled-up oxidation experiments, and the ready availability of γ-valerolactone from biomass. This work also reports on a new carbon-centered radical for the activation of C(sp3)–H bonds.
Introduction
The aerobic oxidation of C(sp3)–H bonds is an important topic in organic synthesis because it allows the production of high-value products and important intermediates directly from readily available precursors. Mechanistically, hydrogen atom transfer (HAT) is an important pathway for activating C(sp3)–H bonds1–3 and has thus attracted considerable research attention. Various reaction systems for C(sp3)–H bond activation have been developed based on N-oxyl compounds,4–7 peroxides,8,9 disulfides10,11 and halogen species,12,13 amides,14,15 and CBrCl3 analogues,16,17 among others, providing reactive O, S, X (X = Cl, Br, I), N-centered, and C-centered radicals as hydrogen atom abstractors. However, most of these compounds are consumed in the reaction, and more than one equiv. is required. Moreover, in practical applications, peroxides pose security risks, and halogen-containing compounds can corrode equipment. N-Hydroxyphthalimide (NHPI), a hydrogen abstractor bearing a N-hydroxy group, can be easily prepared and modified and it exhibits superior catalytic ability in many reactions.18–20 However, most reaction systems involving NHPI require the assistance of various catalysts or additives to improve the efficiency along with solvents such as acetonitrile and benzonitrile due to the high melting point of NHPI (233–235 °C). Moreover, it is difficult to separate NHPI from the reaction system for recycling. Owing to these reasons, existing reaction systems for the aerobic oxidation of C(sp3)–H bonds are complicated and costly, limiting their practical application. Convenient reaction systems for the aerobic oxidation of C(sp3)–H bonds with high economy thus need to be developed.In our ongoing research on the oxidative activation of various C–H bonds based on HAT or single-electron transfer processes,21,22 we identified a simple and convenient reaction system based on γ-valerolactone (GVL) that can promote the aerobic oxidation of various C(sp3)–H bonds via HAT. Notably, GVL can be produced from levulinic acid, and has been identified as one of the 12 important high-value bio-based building blocks23 by the United States Department of Energy and can be readily obtained through acid-catalyzed biomass degradation.24,25 GVL has received considerable attention because it is widely used as a food ingredient, bio-fuel additive, and renewable solvent (Fig. S2†).26,27 Here, for the first time, we demonstrate that GVL can be used as a catalyst of the reaction system for the aerobic oxidation of C(sp3)–H bonds (Scheme 1). Notably, this reaction system features a broad substrate scope and allows both the synthesis of pharmaceutical molecules and the modification of steroidal substrates. This work represents a major breakthrough and highlights the powerful synthetic potential of GVL as a reaction system for C–H activation.
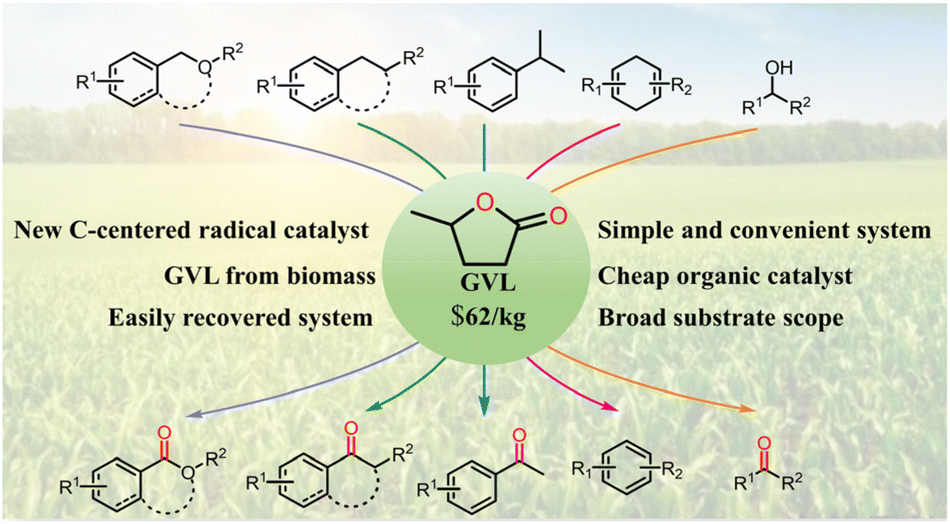 | ||
| Scheme 1 Aerobic oxidation of C(sp3)–H bonds over GVL. | ||
Results and discussion
Aerobic oxidation of isochroman over GVL
We first investigated the aerobic oxidation of isochroman, which is used to generate important structural units of bioactive natural products and pharmaceutical molecules. Several protocols relying on organocatalysts have been reported for the oxidation of isochroman using molecular oxygen as a green oxidant; however, most of the reaction systems are complex and costly (Scheme S1†). Furthermore, the chemoselectivity of the oxidation is frequently unsatisfactory because the oxidation can take place at the 4-site of isochroman in addition to the 1-site (the desired location). Thus, for practical applications, it is highly desirable to simplify these sophisticated multi-component catalyst systems and improve the reaction selectivity.In preliminary experiments, although GVL was found to be able to accelerate the aerobic oxidation of isochroman, the selectivity for the desired 1-isochromanone (P1) was only 61%, and for 4-isochromanone (P′1) it was 39% (Table 1, entry 1). Inspired by the effect of Lewis acids on the bond dissociation energy (BDE) of the O–H bond in NHPI,28,29 various acids and bases were tested as additives to improve the yield of 1-isochromanone (Table S1†). To our delight, these additives significantly affected the chemoselectivity and reactivity. Among the additives, Ni2Al-layered double hydroxide (LDH) offers the best selectivity for 1-isochromanone. Subsequently, the reaction conditions for the aerobic oxidation of isochroman were optimized (Fig. S1 and S3–S5†), and nearly quantitative yield of 1-isochromanone could be obtained under the optimized conditions (Table 1, entry 2). To our knowledge, this is the first example of using GVL as a catalyst to promote the aerobic oxidation of isochroman with Ni2Al-LDH as an additive to improve the chemoselectivity.
|
|
|||
|---|---|---|---|
| Entry | Additive | Conv./% | Sel. of P1/% |
| a Reaction conditions: S1 (1 mmol), GVL (20 mmol), Ni2Al-LDH (100 mg), O2 (1 atm), other additives (2 eq.), 5 h, 110 °C. b Based on GC analysis using biphenyl as the internal standard substance. c Air (1 atm). d Ar (1 atm). e 18O2 (1 atm), 3 h. | |||
| 1 | — | >99 | 61 |
| 2 | Ni2Al-LDH | >99 | >99 |
| 3 | Ni2Al-LDH/DMAP | 74 | 47 |
| 4 | Ni2Al-LDH/pyridine | 94 | 88 |
| 5c | Ni2Al-LDH | 33 | 92 |
| 6d | Ni2Al-LDH | Trace | — |
| 7e | Ni2Al-LDH | 65 | >99 (93% 18O) |
| 8 | Ni2Al-LDH/BHT | Trace | — |
| 9 | Ni2Al-LDH/TEMPO | 10 | 89 |
| 10 | Ni2Al-LDH/CBrCl3 | 25 | 39 |
| 11 | Ni2Al-LDH/BQ | Trace | >99 |
To elucidate the function of Ni2Al-LDH, density functional theory (DFT) was used to calculate the BDE values of the 1- and 4-C–H bonds of isochroman based on the interactions between oxygen atoms in isochroman with the Lewis acid sites of Ni2Al-LDH, as reported previously.30 The results (Fig. S6† and Fig. 1a) show that both BDE values increased after introducing Ni2Al-LDH, and the difference in BDE between the 1- and 4-C–H bonds expanded from 6.5 to 8.5 kcal mol−1. Resultantly, the reaction rate ratio of vC(4)-H to vC(1)-H reduced from 0.4 to nearly zero, which can well explain the excellent selectivity of 1-isochromanone with the Ni2Al-LDH additive. The introduction of 4-dimethylaminopyridine (DMAP) or pyridine as a typical base into the reaction system caused the selectivity to markedly decrease (Table 1, entries 3 and 4), further verifying the importance of surface acidic sites. The results also suggest that C–H bond cleavage is a key step in the oxidation reaction.
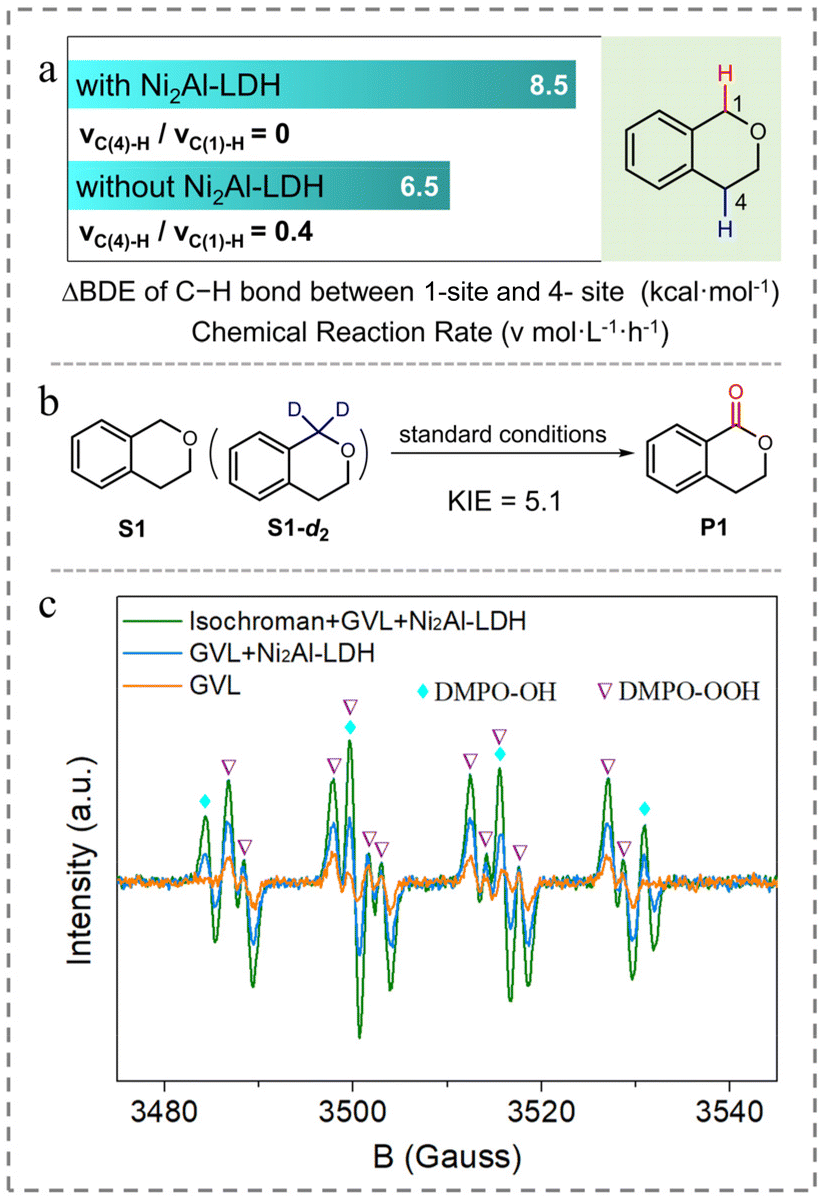 | ||
| Fig. 1 (a) Effect of Ni2Al-LDH on the difference in BDE between the 1- and 4-C–H bonds of isochroman. (b) Schematic of the KIE experimental reaction. (c) EPR analysis of the reaction system. | ||
To obtain further insights into the aerobic oxidation of C(sp3)–H bonds, a series of control experiments were conducted. When oxygen was replaced by air or argon, the conversion was significantly decreased (Table 1, entries 5 and 6), indicating the importance of molecular oxygen. Then, the source of oxygen was determined by an oxygen isotope labelling experiment. The GC-MS analysis indicated that product P1 (m/z = 148) was shifted two mass units higher to m/z = 150 (93% 18O labeled oxygen atom) when 16O2 was replaced by 18O2, strongly suggesting that the oxygen in product P1 is derived from molecular oxygen (entry 7). Introducing 3,5-di-tert-butyl-4-hydroxytoluene (BHT), 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), or CBrCl3 as a radical scavenger significantly suppresses the transformation of isochroman (Table 1, entries 8–10), suggestive of a radical-related oxidation process. When the reaction proceeded with benzoquinone (BQ), only trace of substrate converted, implying the formation of a hyperoxide intermediate (Table 1, entry 11).31 A kinetic isotope effect (KIE) experiment was conducted with a mixture of S1 and 1-deuterated S1-d2 as substrates under the standard conditions. After the full conversion of the substrate, an intramolecular KIE of 5.1 was calculated (Fig. 1b), implying that C–H bond cleavage is a greater contributor to the rate-determining step than electron transfer,32 and that HAT should occur predominantly for C–H bond cleavage. The results suggest that the reaction proceeds via a HAT process involving the formation of isochroman radicals.
The HAT reaction process was confirmed by performing the aerobic oxidation of 4-ethylanisole and 4-isopropylanisole under the standard conditions. As shown in Fig. S7,† the initial rate for the oxidation of 4-ethylanisole was apparently higher than that for 4-isopropylanisole, suggesting that the oxidation of isochroman with GVL was likely initiated by an HAT process,33–35 consistent with the above conclusion.
Subsequently, the possible catalytically active species were analyzed by electron paramagnetic resonance (EPR) spectroscopy (Fig. 1c). The EPR spectrum of the reaction solution indicates the presence of ˙OOH species (hydroperoxyl free radical, AN = 14.4 G, AHβ = 11 G, AHγ = 1.6 G)36 and ˙OH species (AHβ = AN = 15.6 G).37 In contrast, only ˙OOH species was observed in the pure GVL system, suggesting that ˙OOH species formed during the reaction. Combined with the ability of GVL to accelerate the oxidation reaction, we speculated that the reaction was initiated via a process in which triplet-state molecular O2 directly abstracts the H atom from GVL (Fig. S8†), generating a GVL radical and a hydroperoxyl free radical.38 1,1-Diphenylethylene was introduced into the GVL reaction system to capture the potential GVL radical.39 Gas chromatography-mass spectrometry analysis indicated the probable formation of the coupling product between 1,1-diphenylethylene and GVL (Fig. S9†) via free-radical addition, suggesting that the GVL radical might be generated during the reaction; however, it was difficult to determine the exact structure. Therefore, to identify the structure of the GVL radical, DFT calculations at the M06-2x/6-31G(2df,2p) level were performed with three possible radical structures, located at the α-, β- or γ-site of the GVL molecule.40,41 The transition states of the radicals were named TS1-α, TS1-β, and TS1-γ, respectively (Fig. S10a†). Compared with TS1-β, the free energies of TS1-α and TS1-γ were lower by 1.8 and 6.4 kcal mol−1, respectively (Fig. S10a†). Hence, INT-α, INT-γ, and ˙OOH species (i.e., the reactive intermediates that activate the substrate via an HAT process) were chosen for the calculation in the following step.
The free energies of TS3-1 and TS3-4 are significantly higher than those of the other transient states generated from INT1-γ and INT1-α, indicating that ˙OOH species can be excluded as the main active intermediate (Fig. S11†). Fig. S10b† shows the free energy landscape of isochroman activation. Notably, the difference in free energy between TS2-1 and TS2-4 generated from INT1-γ is 2.6 kcal mol−1, whereas the difference between TS4-1 and TS4-4 generated from INT1-α is 6.2 kcal mol−1. The difference in free energy will lead to differences in selectivity for 1-isochromanone and 4-isochromanone, and the larger the difference, the higher the selectivity. Considering the calculation results of TS1, we speculated that a high reaction temperature (>110 °C) benefits the formation of TS1-α and the activation of the 1-site of isochroman, whereas a low temperature (<110 °C) favors TS1-γ and leads to reduced selectivity for 1-isochromanone. The reaction results without Ni2Al-LDH verified this conclusion; the selectivity for 1-isochromanone increased as the reaction temperature increased (Fig. 2a). Therefore, it is reasonable to conclude that TS1-α is the predominant reaction intermediate under the selected conditions. To rule out the possibility of trace transition-metal involvement in the reaction, ICP-MS total element analysis of GVL and the reaction mixture was performed, and the results showed that the residual metals in GVL or the reaction mixture were all less than 1 ppm (Table S2†), excluding the possible effect of transition metals. Based on the above results, a possible reaction pathway for the aerobic oxidation of isochroman over GVL is proposed in Fig. 2b. In this reaction, molecular O2 abstracts the α-C–H atom from GVL under high temperatures and the γ-C–H atom under low temperatures to give the corresponding GVL radical (INT-α and INT-γ) and hydroperoxyl free radical (˙OOH). The two radicals generated from GVL then activate the isochroman molecule via HAT selectively for the 1-site with the assistance of Ni2Al-LDH. The hydroperoxyl free radical can further react with GVL to give the reactive intermediates INT1-α and INT1-γ along with hydrogen peroxide. The isochroman radicals thus formed are rapidly trapped by the reaction with oxygen to give peroxyl radical intermediates, which evolve into the oxidation products.42,43
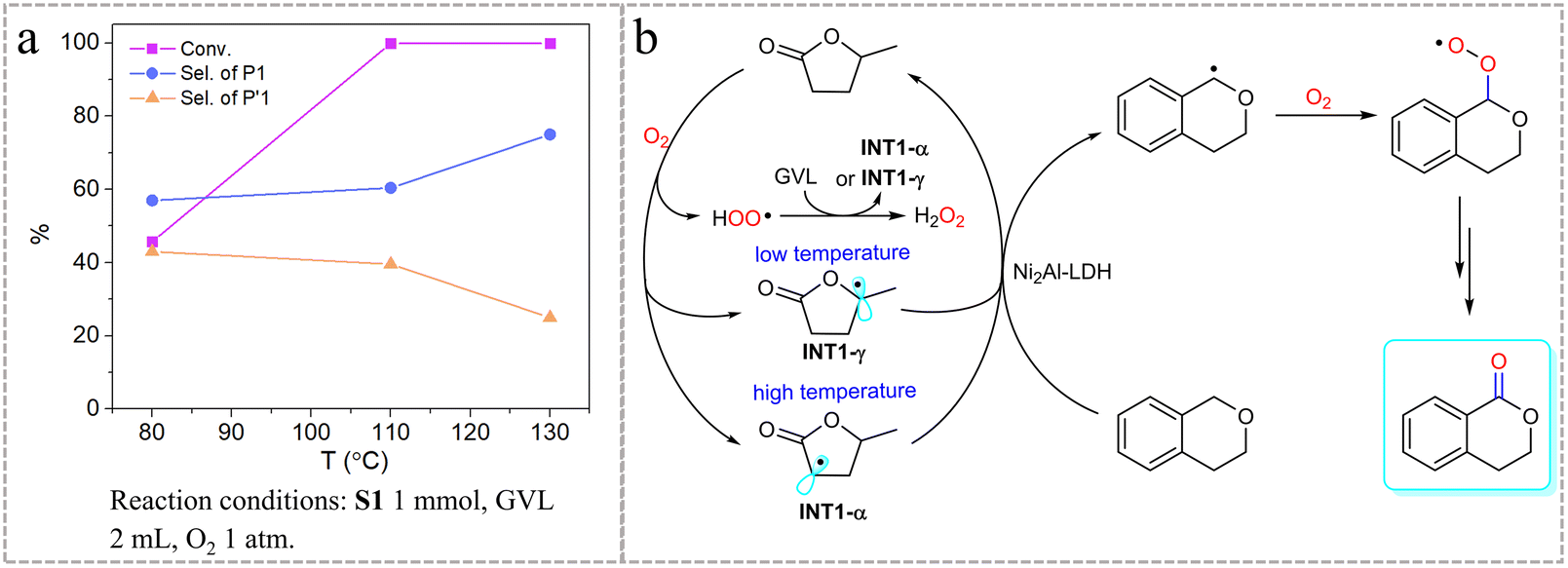 | ||
| Fig. 2 (a) Effect of reaction temperature on the oxidation of isochroman. (b) Proposed reaction mechanism. | ||
Scope of substrates
To demonstrate the substrate scope, various isochroman derivatives were first investigated (Scheme 2). The substrates with electron-donating or electron-withdrawing substituents were well tolerated by the GVL system with excellent yields in the presence of Ni2Al-LDH (P1–P9 and P11 and P12). However, isochroman with a trifluoromethyl group located at the 8-site did not give the corresponding product (P10), suggesting the presence of steric hindrance. Substrates with heterocyclic structures were also tolerated (P13 and P14). Notably, phthalide was obtained in excellent yield (P15). Subsequently, various benzyl ethers were tested in the absence of Ni2Al-LDH because there was only one benzylic site in the structure. Most of the substrates with various substituents gave high yields of the carbonation products in reasonable reaction times (P16–P28); substrate S22, which has a chloro substituent at the ortho-site, was an exception, likely due to the steric hindrance effect. Interestingly, 1,4-bis(methoxymethyl)-benzene gave the di-carbonized product in excellent yield (P29). With the exception of the substrate with a para-methoxyl substituent (S31), benzyl ethers with long carbon chains were also well tolerated in the present reaction system with good to excellent yields (P30, P32 and P33), higher than previously reported yields.44 Nevertheless, the GVL catalytic oxidation of 1-benzylpiperidine was not applicable and failed to afford the product 1-benzoylpiperidine, which was probably due to the presence of the nitrogenous heteroatom (see the ESI† for details).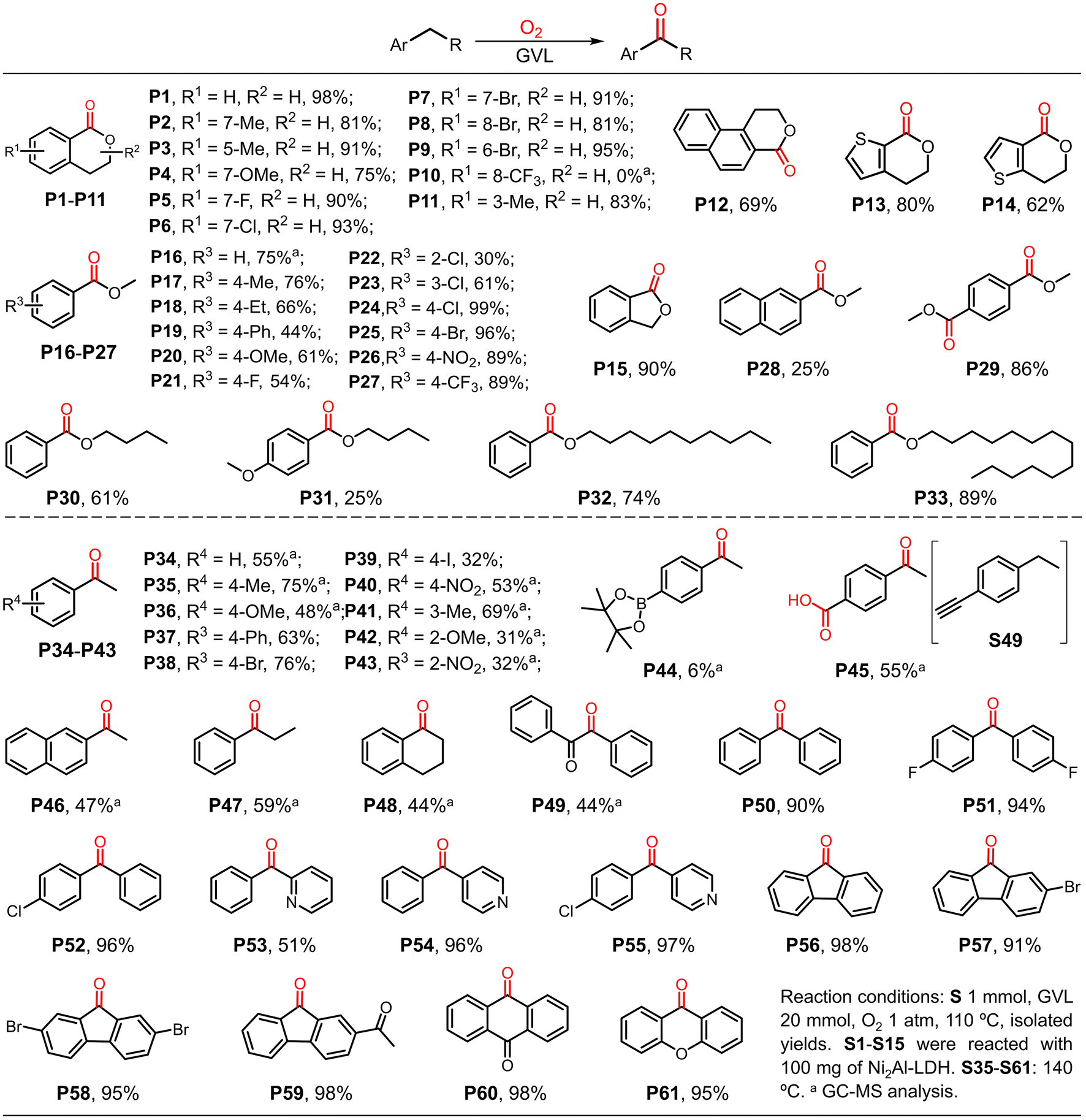 | ||
| Scheme 2 Scope of benzyl ether C–H bonds. | ||
We then turned our attention to the oxidation of other benzylic substrates without ether fragments, which always requires harsh reaction conditions or additives to proceed smoothly. Generally, strong oxidants such as H2O2 and tert-butyl hydroperoxide (TBHP) are employed in the presence of homo- or heterogeneous transition metal catalysts.45–47 The present simple reaction system gave moderate to good yields of the corresponding products (P34–P43 and P46–P49). Ethylbenzene derivatives substituted by different groups were also well tolerated. Of note, derivatives with groups located at the ortho-site resulted in lower yields of the oxygenation products than the substrates with groups located at the para-site (P42 and P43), and the boric acid ester-substituted substrate did not react well (P44). When 1-ethyl-4-ethynylbenzene was introduced into the reaction, 4-acetylbenzoic acid was obtained in good yield (P45), indicating that both sp3 C–H and sp C–H bonds can be oxidized in this reaction system. Diphenylmethane and its analogs were also well tolerated in the catalytic system, resulting in excellent yields of the corresponding oxidative products (P50–P61).
Having demonstrated the suitability of our convenient reaction system for the efficient oxidization of benzyl C–H bonds, additional substrates were investigated (Scheme 3). Tertiary benzylic C–H bonds were also smoothly oxidized to the corresponding carbonyl products. Isopropyl benzene and its analogues were transformed into ketones in good to excellent yields (P62, P63 and P66–P70) with the exception of substrates with n-butoxyl (P64) or phenoxyl (P65) substituents. Consistent with the above observation for ethylbenzene derivatives, substituents at the ortho-site (P71 and P72) suppressed the reaction due to steric hindrance. Interestingly, when 1,3,5-triisopropylbenzene was used as the substrate, the corresponding product 1,3,5-triacetylbenzene was obtained in excellent yield (81%), higher than that obtained using the previously reported method.6 In contrast, when 2,6-diisopropylnaphthalene was used as the substrate, only one tertiary C–H bond was oxidized in the main product (P74). Some substrates containing allylic C–H bonds were also investigated because the unsaturated enones resulting from oxidation can act as synthetic starting materials or drug precursors.48–50
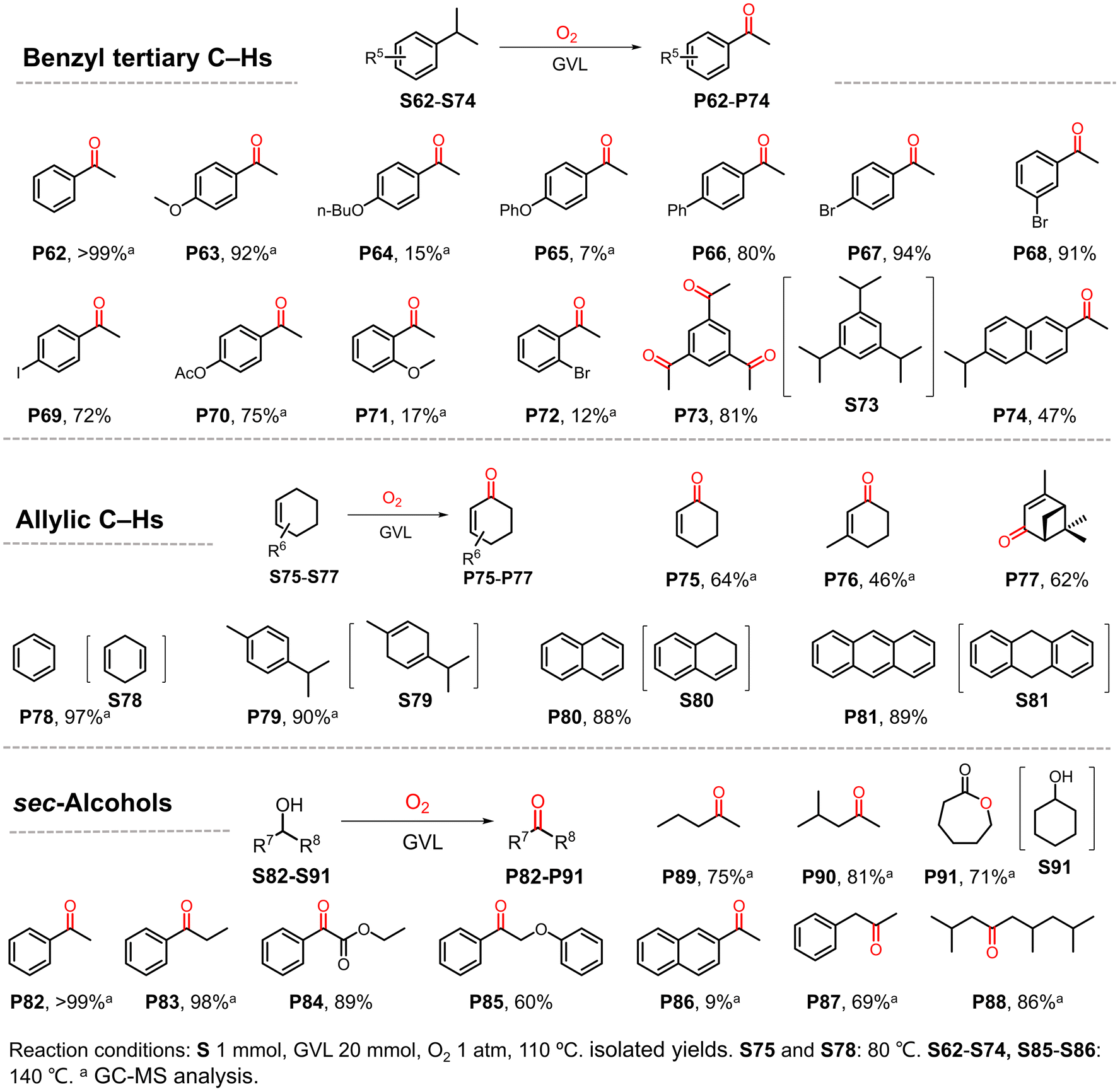 | ||
| Scheme 3 Scope of tertiary benzylic C–H bonds, allylic C–H bonds, and sec-alcohols. | ||
Cyclohexene and its derivatives gave the products of oxygenation at the α-site in moderate yields (P75–P77). In contrast, for substrates containing two unsaturated bonds in one six-membered ring, the dehydrogenative aromatization products were obtained in excellent yields (P78–P81). Some sec-alcohols were also tested. To our delight, various aromatic alcohols except for naphthalene ethanol were smoothly oxidized to ketones in good to excellent yields (P82–P86). Aliphatic alcohols were also well tolerated by the present GVL system (P87–P90), which produced better results than other reported reaction systems.51 Interestingly, oenantholacton was obtained when cyclohexanol was used, implying that a tandem reaction took place (P91). In this case, alcohol was first oxidized to ketone, which then underwent Baeyer–Villiger oxidation. The results also suggest the formation of peroxide species in this reaction.
Potential application of the reaction system
The power of the proposed reaction protocol was also demonstrated in the synthesis and modification of some valuable molecules (Scheme 4a). 7H-Dibenzo[c,e]oxepin-5-one (P92), an important intermediate for the preparation of curable polymeric materials,52 can be synthesized by oxidizing the corresponding ether precursor. However, this reaction generally requires a stoichiometric amount of highly toxic bromine, and the yield is only approximately 45%.53 In contrast, using the present reaction system, P92 was obtained in an excellent yield (80%). Menthone is an important fragrance that is usually produced via the oxidation of menthol.54–56 This transformation proceeded smoothly in a good yield (P93) in the GVL reaction system. The GVL system was also suitable for the oxidation of S94, a natural product-derived benzyl ether;57 the desired product was obtained in a good isolated yield. It is important to note that molecular oxygen was used as a green oxidant in the above transformations.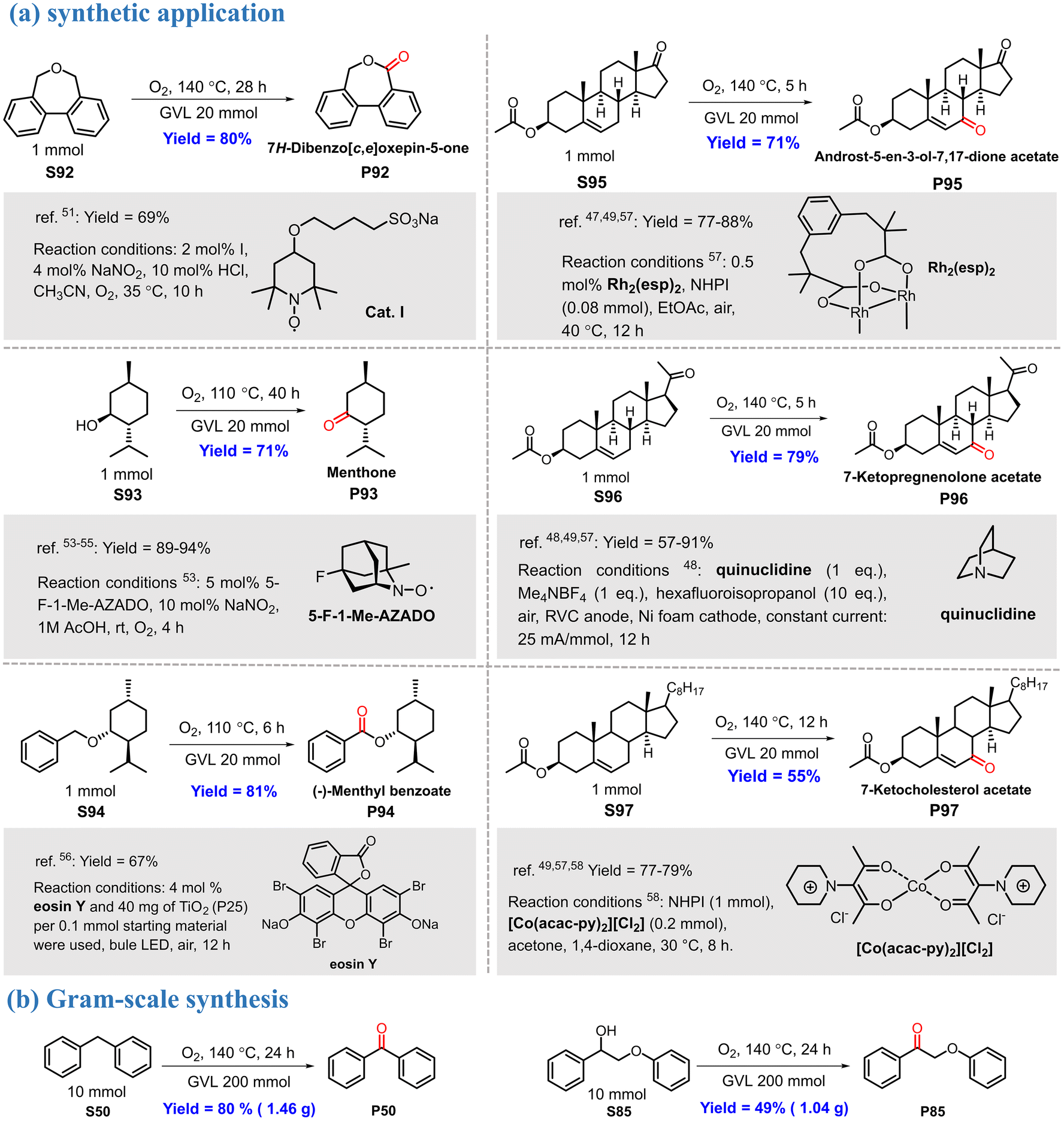 | ||
| Scheme 4 Synthetic application and gram-scale synthesis of the GVL reaction system. | ||
We were also pleased to find that our reaction conditions were amenable for the direct modification of some steroidal substrates via the oxidation of allylic C–H bonds. The oxidation was highly efficient for substrates bearing different functional groups, leading to the corresponding unsaturated enones in good yields (P95–P97). Compared with previously reported protocols,48–50,58 the GVL reaction system has the advantages of simplicity, environmental friendliness, and good yield. The reproducibility of the reaction was examined on a gram scale under similar reaction conditions; the products P50 (1.46 g, 80%) and P85 (1.04 g, 49%) were isolated with only a slight loss of yield (Scheme 4b).
To investigate the potential applications of the GVL reaction system, the reaction with isochroman as the substrate was scaled up 100 times, and the separation of the product along with the recyclability of GVL and Ni2Al-LDH were investigated (Fig. 3). When the oxidation reaction was finished, the mixture was filtered, and the residue was washed with ethyl acetate and dried, giving Ni2Al-LDH with an excellent recovery rate of 99%. The product and GVL were distilled in sequence from the filtrate under vacuum. The yield of the oxygenated product reached 92% with >99% selectivity, and the recovery rate of GVL was 92%. This model reaction suggests that the GVL reaction system has good application potential.
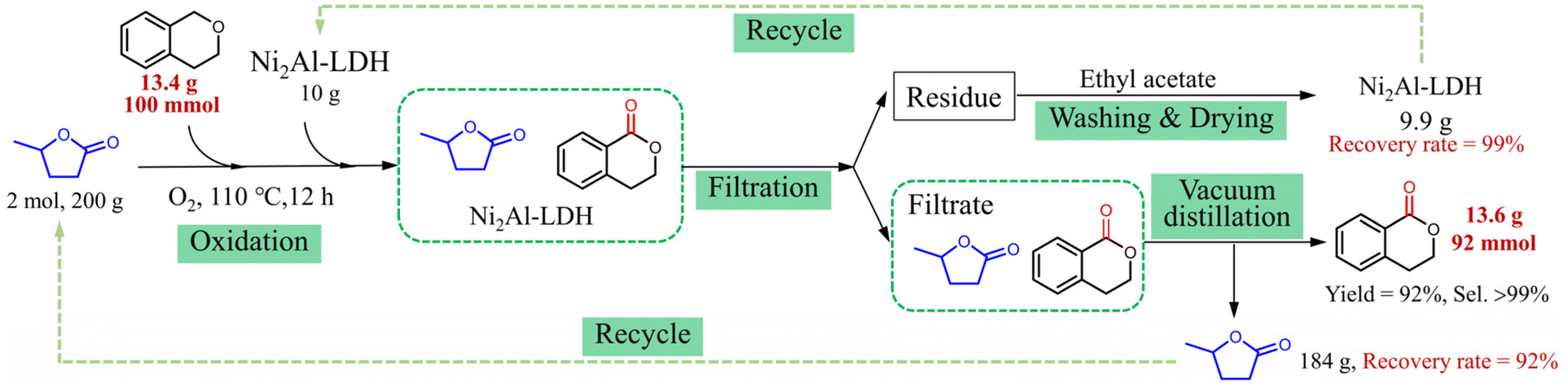 | ||
| Fig. 3 Scaled-up experiment for the oxidation of isochroman. | ||
Conclusions
In summary, we developed a simple and convenient reaction system for the aerobic oxidation of C(sp3)–H bonds based on GVL, which can accelerate oxidation via an HAT process. Under the optimized conditions, isochromans could be oxidized into the corresponding isochromanones with good chemoselectivity for the 1-site of the substrate with the assistance of Ni2Al-LDH. DFT calculations indicated that Ni2Al-LDH increased the selectivity by increasing the difference in BDE between the C–H bonds of the 1- and 4-sites of isochroman. A wide variety of C(sp3)–H bonds were compatible with the reaction system, which also provides good results in the synthesis and modification of some useful molecules. The successful scaling-up of the reaction system and the ready availability of GVL from biomass demonstrate the good application potential of the present reaction system. Moreover, a new carbon-centered radical based on GVL has been reported, which might have applications in various radical-mediated reactions.Author contributions
M. H., W. Z., Q. C. and J. Q. directed and secured the funding for this project. A. W. discovered the reaction, performed the experiments and analysed the data. J. H., C. Z. and Y. F. performed the synthesis experiments of substrates (S2–S14 and S17–S33). M. H. and W. Z. conceived and directed the project. W. Z. prepared the manuscript, while A. W. prepared the ESI.† All authors discussed the experimental results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Jiangsu Key Laboratory of Biomass Energy and Material (JSBEM202018), the Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology (BM2012110), a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the Natural Science Foundation for Colleges and Universities in Jiangsu Province (20KJA530003), the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX22_3024), and the Qinglan Project of Jiangsu Province and CNPC Innovation Foundation (2021DQ02-0707).Notes and references
- S. K. Kariofillis and A. G. Doyle, Acc. Chem. Res., 2021, 54, 988–1000 CrossRef CAS PubMed
.
- H. Huang, X. Jing, J. Deng, C. Meng and C. Duan, J. Am. Chem. Soc., 2023, 145, 2170–2182 CrossRef CAS PubMed
- F. Meyer, R. Frey, M. Ligibell, E. Sager, K. Schroer, R. Snajdrova and R. Buller, ACS Catal., 2021, 11, 6261–6269 CrossRef CAS
- F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561–574 CrossRef CAS
- H. Sterckx, B. Morel and B. U. W. Maes, Angew. Chem., Int. Ed., 2019, 58, 7946–7970 CrossRef CAS PubMed
- Z. Li, Y. Zhang, K. Li, Z. Zhou, Z. Zha and Z. Wang, Sci. China: Chem., 2021, 64, 2134–2141 CrossRef CAS
- Q. Zhang, J. Zhang, H. Qian and S. Ma, Org. Chem. Front., 2023, 10, 505–1511 Search PubMed
- J. Tan, T. Zheng, Y. Yu and K. Xu, RSC Adv., 2017, 7, 15176–15180 RSC
- S. Zhang, J. Zhang and H. Zou, Chem. Sci., 2022, 13, 1298–1306 RSC
- S. Geng, B. Xiong, Y. Zhang, J. Zhang, Y. He and Z. Feng, Chem. Commun., 2019, 55, 12699–12702 RSC
- X. Zhu, Y. Liu, C. Liu, H. Yang and H. Fu, Green Chem., 2020, 22, 4357–4363 RSC
- T. Dohi, S. Ueda, K. Iwasaki, Y. Tsunoda, K. Morimoto and Y. Kita, Beilstein J. Org. Chem., 2018, 14, 1087–1094 CrossRef CAS PubMed
- S. Guha and G. Sekar, Chem. – Eur. J., 2018, 24, 14171–14182 CrossRef CAS
- R. K. Quinn, Z. A. Konst, S. E. Michalak, Y. Schmidt, A. R. Szklarski, A. R. Flores, S. Nam, D. A. Horne, C. D. Vanderwal and E. J. Alexanian, J. Am. Chem. Soc., 2016, 138, 696–702 CrossRef CAS
- W. L. Czaplyski, C. G. Na and E. J. Alexanian, J. Am. Chem. Soc., 2016, 138, 13854–13857 CrossRef CAS
- J. W. Tucker, J. M. Narayanam, P. S. Shah and C. R. Stephenson, Chem. Commun., 2011, 47, 5040–5042 RSC
- P. Lu, T. Hou, X. Gu and P. Li, Org. Lett., 2015, 17, 1954–1957 CrossRef CAS
- Z.-X. Wu, G.-W. Hu and Y.-X. Luan, ACS Catal., 2022, 12, 11716–11733 CrossRef CAS
- M. A. Cribari, M. J. Unger and J. D. Martell, ACS Catal., 2022, 12, 12246–12252 CrossRef CAS PubMed
- M. M. Al-Taher, C. Kalamaras, M. A. Alqahtani and F. S. Alomar, Fuel, 2022, 324, 124563 CrossRef CAS
- W. Zhou, W. Lu, Z. Sun, J. Qian, M. He, Q. Chen and S. Sun, Appl. Catal., A, 2021, 624, 118322 CrossRef CAS
- W. Zhou, A. Wang, Z. Kong, X. Tian, Z. Xia, Z. Zhang, M. He, Q. Chen and S. Sun, Org.
Lett., 2021, 23, 6321–6325 CrossRef CAS
- T. Werpy and G. Petersen, Office of Scientific and Technical Information, 2004, vol. 69.
- Y. Lu, Y. Wang, Y. Wang, Q. Cao, X. Xie and W. Fang, Mol. Catal., 2020, 493, 111097 CrossRef CAS
- H. Liu, X. Cao, X. Tang, X. Zeng, Y. Sun, X. Ke and L. Lin, Green Chem., 2021, 23, 1983–1988 RSC
- Z. Zhang, ChemSusChem, 2016, 9, 156–171 CrossRef CAS PubMed
- R. Xu, K. Liu, H. Du, H. Liu, X. Cao, X. Zhao, G. Qu, X. Li, B. Li and C. Si, ChemSusChem, 2020, 13, 6461–6476 CrossRef CAS PubMed
- Y. Sun, W. Zhang, X. Hu and H. Li, J. Phys. Chem. B, 2010, 114, 4862–4869 CrossRef CAS
- H. Gao, L. Hu, Y. Hu, X. Lv, Y.-B. Wu and G. Lu, Catal. Sci. Technol., 2021, 11, 4417–4428 RSC
- L. Zhao, J. Duan, S. Yang, X. Li, Q. Liu and C. J. Martyniuk, Sep. Purif. Technol., 2018, 207, 231–239 CrossRef CAS
- W. Huang, B. C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 5438–5442 CrossRef CAS
- B. J. Foley, N. Bhuvanesh, J. Zhou and O. V. Ozerov, ACS Catal., 2020, 10, 9824–9836 CrossRef CAS
- A. M. Khenkin, L. Weiner, Y. Wang and R. Neumann, J. Am. Chem. Soc., 2001, 123, 8531–8542 CrossRef CAS PubMed
- S. Nakai, T. Uematsu, Y. Ogasawara, K. Suzuki, K. Yamaguchi and N. Mizuno, ChemCatChem, 2018, 10, 1096–1106 CrossRef CAS
- A. Wang, W. Zhou, Z. Sun, Z. Zhang, Z. Zhang, M. He and Q. Chen, Mol. Catal., 2021, 499, 111276 CrossRef CAS
- M. Danilczuk, S. Schlick and F. D. Coms, Macromolecules, 2013, 46, 6110–6117 CrossRef CAS
- W. P. Fagan, F. A. Villamena, J. L. Zweier and L. K. Weavers, Environ. Sci. Technol., 2022, 56, 3729–3738 CrossRef CAS PubMed
- J. Wu, J. Chen, L. Wang, H. Zhu, R. Liu, G. Song, C. Feng and Y. Li, Green Chem., 2023, 25, 940–945 RSC
- C. Hou, S. Sun, Z. Liu, H. Zhang, Y. Liu, Q. An, J. Zhao, J. Ma, Z. Sun and W. Chu, Adv. Synth. Catal., 2021, 363, 2806–2812 CrossRef CAS
- L. Xu, Y. Yi, S. Hu, J. Ye and A. Hu, Electrochim. Acta, 2022, 403, 139533 CrossRef CAS
- L. Zhang, L. Ma, J. Yuan, X.-M. Zhang and Z. Tang, Appl. Catal., B, 2023, 321, 122049 CrossRef CAS
- L. C. Finney, L. J. Mitchell and C. J. Moody, Green Chem., 2018, 20, 2242–2249 RSC
- A. Sagadevan, K. C. Hwang and M. D. Su, Nat. Commun., 2017, 8, 1812 CrossRef PubMed
- Z. Ma, H. Zhang, Z. Yang, G. Ji, B. Yu, X. Liu and Z. Liu, Green Chem., 2016, 18, 1976–1982 RSC
- S. K. Gupta and J. A. Choudhury, ChemCatChem, 2017, 9, 1979–1984 CrossRef CAS
- J. Zhou, M. Jia, M. Song, Z. Huang, A. Steiner, Q. An, J. Ma, Z. Guo, Q. Zhang, H. Sun, C. Robertson, J. Bacsa, J. Xiao and C. Li, Angew. Chem., Int. Ed., 2022, 61, 202205983 CrossRef
- C. Liu, H. Liu, X. Zheng, S. Chen, Q. Lai, C. Zheng, M. Huang, K. Cai, Z. Cai and S. Cai, ACS Catal., 2022, 12, 1375–1381 CrossRef
- Y. Kawamata, M. Yan, Z. Liu, D.-H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448–7451 CrossRef CAS PubMed
- S.-S. Zhu, Y. Liu, X.-L. Chen, L.-B. Qu and B. Yu, ACS Catal., 2021, 12, 126–134 CrossRef
- S. Zavahir, Q. Xiao, S. Sarina, J. Zhao, S. Bottle, M. Wellard, J. Jia, L. Jing, Y. Huang, J. P. Blinco, H. Wu and H.-Y. Zhu, ACS Catal., 2016, 6, 3580–3588 CrossRef CAS
- Z. Zhang, Y. Gao, Y. Liu, J. Li, H. Xie, H. Li and W. Wang, Org. Lett., 2015, 17, 5492–5495 CrossRef CAS
- S. Kobayashi, M. Kihara, T. Hashimoto and K. Kitamura, Yakugaku Zasshi, 1975, 95, 1449–1455 CrossRef CAS
- M. Shibuya, Y. Osada, Y. Sasano, M. Tomizawa and Y. Iwabuchi, J. Am. Chem. Soc., 2011, 133, 6497–6500 CrossRef CAS
- J. E. Steves and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 15742–15745 CrossRef CAS
- R. Zhang, R. Zhang, R. Jian, L. Zhang, M.-T. Zhang, Y. Xia and S. Luo, Nat. Commun., 2022, 13, 428 CrossRef CAS
- L. Ren, M.-M. Yang, C.-H. Tung, L.-Z. Wu and H. Cong, ACS Catal., 2017, 7, 8134–8138 CrossRef CAS
- Y. Wang, X. Chen, H. Jin and Y. Wang, Chem. – Eur. J., 2019, 25, 14273–14277 CrossRef CAS PubMed
- P. Zhang, C. Wang, Z. Chen and H. Li, Catal. Sci. Technol., 2011, 1, 1133–1137 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03639a |
| This journal is © The Royal Society of Chemistry 2024 |