
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExplicating the effect of extraction methods on the techno-functional, structural, and in vitro prebiotic potential of soluble dietary fibers from mango and pomegranate peel
Shriya
Bhatt
ab and
Mahesh
Gupta
 *ab
*ab
aCSIR-Institute of Himalayan Bioresource Technology, Palampur-176061, Himachal Pradesh, India. E-mail: mgupta@ihbt.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
First published on 8th July 2024
Abstract
Peel is a major bio-waste and a potential source of numerous bioactive molecules, creating immense environmental issues but no commercial significance. Thus, different extraction conditions, including chemical, enzymatic, ultrasonication, microwave, and homogenization, with varied sample concentrations at 2%, 5%, and 10% (w/v) were employed for maximum soluble dietary fiber (SDF) extraction from both mango (Totapuri and Safeda) and pomegranate (Bhagwa and Daru) peel. The maximum SDF yield of 29.26 ± 0.25% was observed at 5% w/v for homogenization-assisted enzymatic extraction (HEE) from mango peel (Safeda). The proximate and techno-functional properties of SDF exhibited efficient activity with enhanced thermal stability and structural characteristics. Scanning electron microscopy revealed a loosened and porous structure. In addition, the samples demonstrated significant prebiotic activity with the synthesis of three major short-chain fatty acids (SCFAs) in the order of propionic (3.60 ± 0.08 mg mL−1) > acetic (2.64 ± 0.01 mg mL−1) > butyric acid (1.27 ± 0.01 mg mL−1), as quantified via ultra-performance liquid chromatography (UPLC). Thereby, this study highlights the role of waste fruit peel as a potent source of SDF, exhibiting profound prebiotic activity with imminent industrial application.
Sustainability spotlightThe present study provides substantial evidence to highlight the role of waste fruit peel as a potential source of SDF, exhibiting prebiotic activity with imminent industrial application. The study intended to decipher the knowledge for viable and reproducible methods having maximum soluble dietary fiber extraction yield with enhanced chemical and structural characteristics. The results highlight the potential role of extraction methods (homogenization and ultrasonication) as an effectual technique in augmenting the overall structural, thermal, and prebiotic properties of SDF with possible application in food product development. Consequently, the current research provides substantial evidence to highlight the role of waste fruit peel as a potential source of SDF, exhibiting prebiotic activity with imminent industrial application. |
Introduction
Fruits play a crucial role in sustaining a balanced lifestyle by delivering diverse essential nutrients to the body. In this case, the demand and production of fruits have increased considerably in the recent years.1 However, the production and processing of fruits generate waste, creating serious environmental concerns. Mango and pomegranate are two major fruits, generating about 15–20% and 40–50% of their waste in the form of a peel with no commercial significance.2,3 This waste is a potential source of numerous bioactive molecules, importantly dietary fiber (DF).3DF, a 7th fundamental nutrient, is generally classified as soluble dietary fiber (SDF) and insoluble dietary fiber (IDF) based on its solubility in water.4 The former has been earlier reported for its ability to bind toxic molecules, reduce blood glucose levels, and proliferate intestinal microbes with the prevention of diabetes and cardiovascular diseases.5–7 However, for a high-quality DF, the SDF content should not be less than 10%, providing effective processing characteristics along with positive health benefit.8 Consequently, there has been a continuous effort toward identifying optimum extraction techniques with a higher SDF yield.
The extraction of SDF is generally carried out using various conventional and non-conventional techniques,9 including thermal, enzymatic, chemical, chemo-enzymatic, ultrasonication, homogenization, and microwave-assisted extractions. According to literature reports, the dietary fiber from mango peel ranges from 28% to 78% with 13–28% of SDF and 14–50% of IDF. Similarly, the dietary fiber content in pomegranate peel ranges from 33% to 62% with an SDF content of 24.78%.10,11 However, DF from varied sources exhibits diverse yields, physicochemical, and functional properties, which entirely depend on the chemical composition and extraction method. Nowadays, the research focus mainly relies on the utilization of newer technologies, namely ultrasonication, homogenization, and microwave-assisted extraction. To the best of our knowledge, there have been no reports on comparing the effect of different extraction techniques on the yield, techno-functional, nutritional, structural, and prebiotic potential of soluble dietary fiber (SDF) extracted from mango and pomegranate fruit peels with molecular weight determination using MALDI-TOF/TOF-MS/MS.
Therefore, the aim of this study was to estimate the effect of chemical (acid, alkali), enzymatic (cellulase, α-amylase, protease, and amyloglucosidase), chemo-enzymatic, along with ultrasonication, homogenization, and microwave-assisted extractions on the structural and techno-functional properties of SDF. In addition, the in vitro proliferation of various probiotics was evaluated, along with the determination of major SCFAs. Overall, the study intended to interpret the knowledge for viable and reproducible methods having maximum extraction yield, along with effective prebiotic potential, generating the idea for the valorisation of waste to a potential bioactive ingredient.
Materials and methods
Material
Two mango varieties, i.e., Totapuri (A), Safeda (B), and two pomegranate varieties, i.e., Bhagwa (C), and Daru (D), were purchased from Himachal Pradesh Palampur (1472 meters), India (32.1109° N, 76.5363° E). The total dietary fiber kit (K-TDFR, 200A) was obtained from Megazyme (Wicklow, Ireland). The standards were from Sigma-Aldrich, India. The other chemicals and reagents were of analytic grade.The four bacterial strains, namely Wisella ciberia, Lactobacillus plantarum, Lactobacillus brevis, and Lactobacillus fermentum, were isolated previously in the laboratory, and WOW probiotic (a mixed consortium of 19 different Lactobacilli and Bifidobacteria) was procured from Palampur market, Himachal Pradesh, India.
Soluble dietary fiber extraction from peel
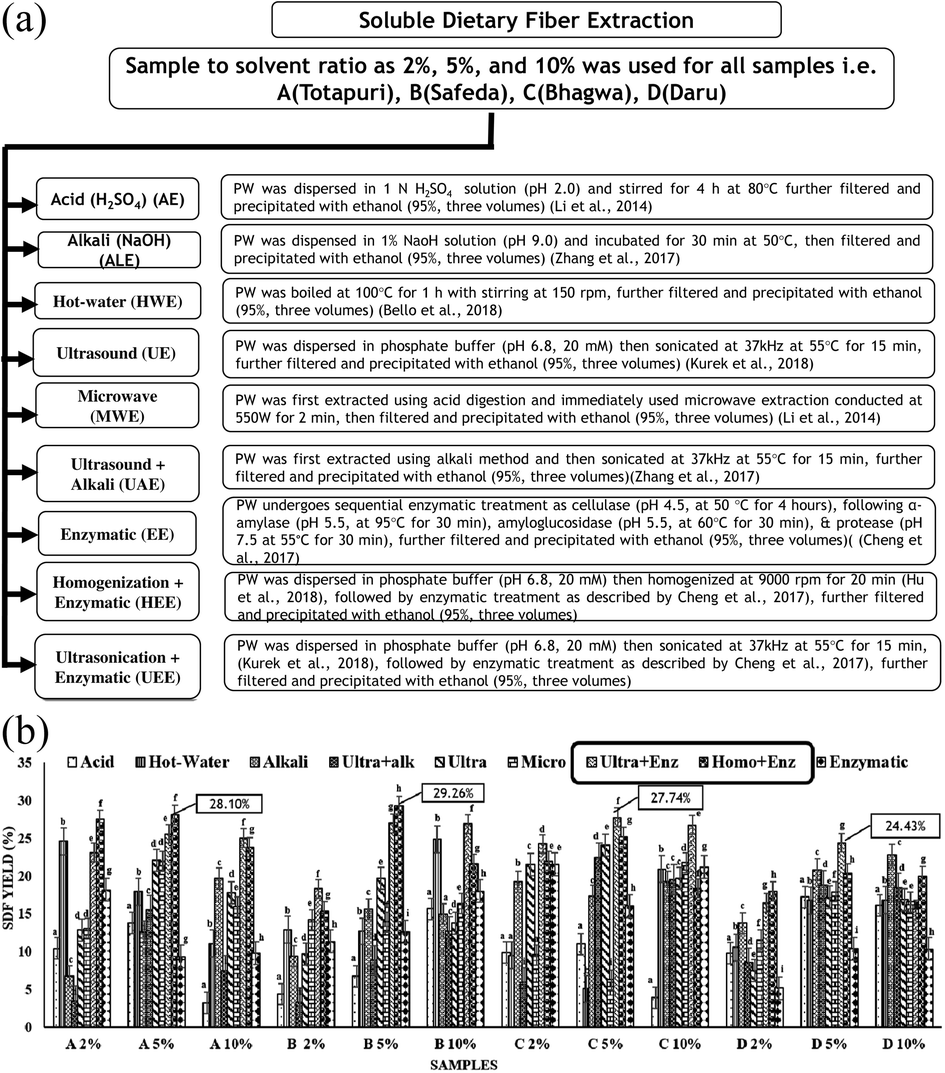 | ||
| Fig. 1 (A) Extraction of soluble dietary fiber using varied sample concentration and extraction conditions. PW: pre-treated peel powder. (B) Extraction yield of soluble dietary fiber. (A) Totapuri, (B) Safeda, (C) Bhagwa, and (D) Daru, 2%, 5%, and 10% indicate the sample concentration, acid: sulphuric acid (AE), alkali: sodium hydroxide (ALE), Ultra+Alk: ultrasound-assisted alkali extraction (UAE), ultra: ultrasound (UE), micro: microwave (MWE), Ult+Enz: ultrasound-assisted enzymatic extraction (UEE), Homo+Enz: homogenization-assisted enzymatic extraction (HEE). Values are the average of three replicates ± SD with significant difference (p < 0.05) indicated with different letters. | ||
The yield was calculated as described in the equation below:
| SDF yield (%) = (X − Z − T)/Y × 100 |
Proximate composition
The method described by Bhatt et al., 2021,18 was performed to estimate the moisture, fat, ash, and protein content. SDF was estimated using K-TDFR, 200A (Megazyme kit), following 991.43 AOAC methodology.Techno-functional and other characteristic properties
The methodology described by Dong et al., 2020,8 was followed for the oil absorption index (OAI), water solubility index (WSI), water absorption index (WAI), emulsion stability (ES), and emulsifying capacity (EC). The previously reported method by Kumari et al., 2021,20 was followed for water activity.The other characteristic properties, including the cholesterol-binding capacity (CBC), glucose adsorption capacity (GAC), sodium cholate binding capacity (SCBC), and cation exchange capacity (CEC), were determined as performed by Bhatt et al., 2022.12
Sugar profile
The monosaccharide composition of samples was quantified using high-performance anion-exchange chromatography-pulsed amperometric detection (HPAEC-PAD). Initially, the hydrolysis of samples was done following the method explained by Dong et al., 2020.8 The samples were further extracted using solid–liquid extraction without derivatization. Briefly, 10 mg sample was mixed with 1.5 mL distilled water (dH2O) and vortexed. Upon the addition of Carrez II (0.2 mL), the sample was mixed and vortexed (5 min), and then centrifuged for 20 min at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. The supernatant was transferred promptly to another tube. Afterwards, the second extraction was carried out using similar steps. The supernatant of both extractions was mixed to get the final aliquot. The HPAEC coupled to 945 Professional Detector Vario, an amperometric detector (Metrohm AG, Herisau, Suiza) in pulsed amperometric mode was used to analyse the monosaccharide composition of each sample. The separation of the standards, namely inositol, glucose, fructose, xylose, sucrose, and raffinose, and the samples was carried out using a Metrosep Carb 2 – 250/4.0 column programmed at 30 ± 0.1 °C. The mobile phase (isocratic) of 10 mM L−1 NaAc and 100 mM L−1 NaOH was followed at a flow rate of 0.5 mL min−1, with an injection volume of 20 μL and pressure of 15 MPa. The software A. MagIC Net (version 3.2) was used to analyse the samples.
000 rpm. The supernatant was transferred promptly to another tube. Afterwards, the second extraction was carried out using similar steps. The supernatant of both extractions was mixed to get the final aliquot. The HPAEC coupled to 945 Professional Detector Vario, an amperometric detector (Metrohm AG, Herisau, Suiza) in pulsed amperometric mode was used to analyse the monosaccharide composition of each sample. The separation of the standards, namely inositol, glucose, fructose, xylose, sucrose, and raffinose, and the samples was carried out using a Metrosep Carb 2 – 250/4.0 column programmed at 30 ± 0.1 °C. The mobile phase (isocratic) of 10 mM L−1 NaAc and 100 mM L−1 NaOH was followed at a flow rate of 0.5 mL min−1, with an injection volume of 20 μL and pressure of 15 MPa. The software A. MagIC Net (version 3.2) was used to analyse the samples.
Determination of the molecular weight using MALDI-TOF/TOF-MS/MS
The determination of the molecular weight was carried out using MALDI-TOF/TOF-MS/MS (Bruker, Ultraflextrame, USA). The sample preparation was done as defined by Lopez Garcia et al., 2016,19 with minor amendments. In brief, the 10 mg sample was dispersed in 1 mL dH2O and sonicated for 30 min, followed by centrifugation for 20 min at 8000 rpm. The supernatant was withdrawn and stored at 4 °C in glass vials until further analysis.Two matrixes, namely 2,5-dihydroxybenzoic acid (DHB) and 2′,4′,6′-trihydroxyacetophenone monohydrate (THAP), were used for analysis of samples. Each matrix of 10 mg mL−1 was dissolved in acetonitrile:water (80:20) prepared in 0.1% trifluoroacetic acid. 0.1 μL of sample and matrix (1:1) solution was loaded on the ATP Anchor Chip plate. Thereafter, the plate was allowed to dry at 37 °C for 10 min. Polytool software (Bruker Daltonics Inc., Germany) was used for the determination of the repeat unit, residue, the number of the average molecular weight (Mn), weight average molecular weight (Mw), and polydispersity index (PI). The TOF/TOF spectra were obtained at 2500 shots per spot with reflector positive ion mode within the mass range from 700 to 2000 Da.
Scanning electron microscopy (SEM) analysis
The sample mounting was carried out on the stub (aluminium stub) covered with carbon tape to carry out SEM analysis. The stubs thereafter were placed in the ion sputter (E1010 Hitachi, Japan). For conductivity, the samples were coated for 20 s with gold at a 10 Pa vacuum level. Finally, images were then analysed on SEM (S3400N; Hitachi) at 15 kV.Fourier transformation infrared spectroscopy (FT-IR) analysis
The structural characterization of the SDF samples was carried out via FT-IR spectroscopy (Shimadzu). The powdered sample (10 mg) was assessed in the 400–4000 cm−1 spectral range, and analysed with a maximum of 16 individual scans for each sample.Thermogravimetric (TGA) analysis
The thermal stability of all SDF samples was carried out using a TGA analyzer (NETZSCH Geratebau GmbH STA 449 F1 Jupiter) machine. In brief, 20 mg of sample was investigated in the nitrogen atmosphere via TGA with temperature ranging from 20 to 400 °C at 10 °C min−1 heating rate.Determination of flavonoid, phenolic content and antioxidant assays
The quantification of the total flavonoid and phenolic contents, along with antioxidant assays including 2,2-diphenyl-1-picrylhydrazyl (DPPH), 2,2′-azino-bis (3-ethylbenzothiazoline-6-sulphonic acid) (ABTS), were carried out as explained by Bhatt et al., 2021.21Colour assessment
A Hunter colorimeter (Chroma-Meter CR-400, KONICA MINOLTA) was used for the colorimetric assessment of samples. All of the experiments were performed in triplicate.Prebiotic potential
In vitro prebiotic activity
Probiotic strains, namely Wisella ciberia, Lactobacillus plantarum, Lactobacillus brevis, and Lactobacillus fermentum, along with WOW probiotic, were used to assess the prebiotic potential of various SDF samples at different time intervals (24 h and 48 h). The method described by Chen et al., 2020,23 was followed to evaluate the prebiotic potential of SDF samples on the proliferation of various probiotics. The bacterial growth calculated as log 10 CFU mL−1 was assessed both at 24 h and 48 h. Similarly, the medium was also evaluated for pH measurement at 24 h and 48 h using a pH meter (Eutech). Subsequently, the samples were kept at −20 °C until further use.SCFA quantification using ultra-performance liquid chromatography (UPLC)
The SCFA quantification was performed using UPLC. The methodology explained by Dobrowolska-Iwanek et al., 2020,24 was followed for SCFA quantification with minor modifications. The cultures were first centrifuged at 8000 rpm for 10 min. Furthermore, the supernatant was withdrawn, acidified to pH 2.5, and then filtered using PVDF 0.45 μm filter (Millipore, USA). Then, the samples were injected to a Waters Acquity UPLC-H system equipped with a STAR RP-18 end-capped column (100 × 2.1 mm, 2 μm particle size) at 25 °C monitored at 210 nm wavelength using an eλ PDA detector. The instrument functioned at a flow rate of 0.2 mL min−1 with (A) acetonitrile and (B) 0.1% of ortho-phosphoric acid prepared in deionized water, and degassed using an ultrasonicator. The gradient system included: 0 min: A 10%, 3 min: A 20%, 6 min: A 30%, 8 min: A 30%, 12.5 min: A 40%, 14 min: A 80%, 16 min: A 80%, and 18 min: A 10%. After each run, the column was equilibrated for 2 min. The acids were calculated through a comparison of the retention time with the standard. Different dilutions were prepared to determine the calibration curve for the quantification of each acid.Statistical analysis
Every experiment was done in triplicate, and signified as the mean ± standard deviation. To confirm the accuracy, a two-way Analysis of Variance (ANOVA) with a significance level of p < 0.05 was carried out by Tukey's multiple comparison test.Results and discussion
Effect of the extraction condition on soluble dietary fiber yield
Considering the yield as the assessment index, experiments were performed at varying sample concentrations and extraction methods (Fig. 1A). The extraction was carried out at three concentrations of 2%, 5%, and 10% w/v based on the previous reports5,25,26 with the maximum SDF yield in 5% samples as A5%, B5%, C5% and D5%, as shown in Fig. 1B. Different extraction conditions have been reported to affect the composition and structure of SDF, positively influencing the concomitant physical and chemical properties.5 Interestingly, the maximum extraction yield among both mango varieties was observed for HEE at 28.10 ± 0.04% in the A5% sample and 29.26 ± 0.25% in the B5% sample. However, UEE exhibited higher SDF yields for the two pomegranate varieties at 27.74 ± 0.04% (C5%) and 24.43 ± 0.01% (D5%), as shown in Fig. 1B. Fascinatingly, the maximum SDF yield observed in samples A5% and B5% highlights the potential effect of HEE in maximizing the yield. Compared to conventional extraction techniques, homogenization is thought to be a moderate extraction condition that exhibits high yield, continuous production, and an easy industrialization strategy.5 Furthermore, it has been demonstrated to disrupt the hydrogen bonds that form hemicellulose chains, transforming complex molecules into small-unit polysaccharides.26 The cellulose hydrolysis produced by the enzymatic activity also reduces the complex polysaccharides to small molecular polysaccharides.27 In addition, the density and rate of mass transfer probably could have altered the yield. Thereby, the combined effect of treatments and sample concentration enhanced the overall SDF yield. Conversely, the C5% and D5% samples exhibited the maximum SDF yield for UEE (Fig. 1B). The improvement in the SDF yield could possibly be due to the aforementioned effect of EE in conjunction with the breakdown of hydrogen bonds by ultrasonication, which breaks the intricate structures into considerably simpler components. Interestingly, our results are similar to findings reported by Hu et al., 2018,5 and Dong et al., 2020,8 where HEE & UEE unveiled maximum SDF yields. It is worth mentioning here that our study summarizes the effect of nine different extraction treatments on the SDF yield from the waste peel of mango and pomegranate. The samples with the maximum yield (i.e., A5% (HEE), was coded further as A5, B5% (HEE) as B5, C5% (UEE) as C5, and D5% (UEE) as D5) were analysed for other structural, techno-functional and prebiotic properties.Proximate composition
The samples exhibiting the maximum SDF yield (5% w/v) were evaluated for their proximate composition and purity (Table 1). The maximum amount of moisture content was observed as 9.34 ± 0.39% in sample C5. The ash content of all SDF ranged from 9.87% to 10.51%. However, a reduced protein content was observed between 1.05% and 1.28%, which could be ascribed to the enzymatic treatment by proteases, leading to the degradation of proteins. The fat content of all SDF samples was below 1% with a minimum content (0.51%) in sample D5, which is probably due to the sample pre-treatment with ether enhancing purity among the samples.| Sample | Moisture (%) | Fat (%) | Ash (%) | Protein (%) | Purity (%) | SDF (%) |
|---|---|---|---|---|---|---|
| a Values are the average of three replicates ± SD with significant difference (p < 0.05) indicated with different letters (a–d); A5: homogenization-assisted enzymatic extraction (HEE) of SDF from Totapuri, B5: homogenization-assisted enzymatic extraction (HEE) of SDF from Safeda, C5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Bhagwa, D5: ultrasonication-assisted enzymatic (UEE) extraction of SDF from Daru, SDF: soluble dietary fiber. | ||||||
| A5 | 7.58 ± 0.36a | 0.93 ± 0.11a | 5.32 ± 0.87a | 1.05 ± 0.05a | 85.12 ± 0.32a | 28.00 ± 0.04a |
| B5 | 8.51 ± 0.23a,b | 0.87 ± 0.08b | 5.33 ± 0.47b | 1.25 ± 0.02b | 84.04 ± 0.19b | 29.26 ± 0.25b |
| C5 | 9.34 ± 0.39b | 0.92 ± 0.13a | 4.68 ± 0.88c | 1.28 ± 0.05b | 83.78 ± 0.33b | 27.74 ± 0.03a |
| D5 | 9.16 ± 0.48b | 0.51 ± 0.21d | 5.12 ± 0.69c | 1.24 ± 0.05b | 83.97 ± 0.30b | 24.43 ± 0.01d |
Functional and other characteristic properties
The functional properties include WAI, WSI, and OAI, representing the hydration properties of the SDF samples. WAI indicates the binding of water that primarily depends on stability, density, chemical structure, amount, nature, and attachment site of SDF. In this study, the WAI of all SDF samples ranged from 4.07 to 7.11 g g−1 (Table 2), with significant difference (p < 0.05). The reason might be the disintegration of bonds (hydrogen bonds) among hemicellulose, liberating various hydrophilic groups for enhanced binding, along with improved absorption.26| Sample | WSI (%) | WAI (g g−1) | OAI (g g−1) | Water activity (aw) | EC (%) | ES (%) |
|---|---|---|---|---|---|---|
| a Values are the average of three replicates ± SD with significant difference (p < 0.05) indicated with different letters (a–d); A5: homogenization-assisted enzymatic extraction (HEE) of SDF from Totapuri, B5: homogenization-assisted enzymatic extraction (HEE) of SDF from Safeda, C5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Bhagwa, D5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Daru, WSI: water solubility index, WAI: water absorption index, OAI: oil absorption index, EC: emulsion capacity, ES: emulsion stability. | ||||||
| A5 | 61.50 ± 0.43a | 7.11 ± 0.04a | 4.27 ± 0.08a | 0.16 ± 0.00a | 55.00 ± 0.50a | 53.86 ± 0.66a |
| B5 | 64.20 ± 0.79b | 5.07 ± 0.02b | 3.51 ± 0.08b | 0.19 ± 0.00b | 47.66 ± 0.76b | 67.70 ± 0.70b |
| C5 | 63.70 ± 0.20b | 4.07 ± 0.04c | 3.07 ± 0.03c | 0.54 ± 0.00c | 74.83 ± 0.76c | 59.43 ± 0.56c |
| D5 | 69.03 ± 0.73c | 4.79 ± 0.04d | 3.12 ± 0.01c | 0.50 ± 0.01d | 85.50 ± 0.50d | 47.21 ± 0.64d |
Interestingly, in the present study maximum, WAI was observed in sample A5 as 7.11 ± 0.04 g g−1 higher than that of apple pomace and papaya peel reported previously.13,28 The maximum WSI was observed in sample D5 as 69.03 ± 0.73% (Table 2). The enhanced solubility may be attributed to the SDF sample's altered three-dimensional structure and simultaneous increase in the short-chain fiber. The OAI is dependent upon various factors, such as the hydrophobicity of the sample, its charge density and surface property. Thus, the maximum OAI was observed in sample A5 as 4.27 ± 0.08 g g−1 (Table 2). The difference in OAI may be attributed to the multifaceted effect of the chemo-mechanical treatment, which breaks down the insoluble fractions, resulting in open functional groups that maximize oil entrapment.4 The water activity of a sample relies on its moisture content. As presented in Table 2, the water activity of all samples ranged from 0.16 to 0.54, with minimum activity in sample A5. The low water activity exhibits greater stability besides its low susceptibility to degradation by microbes. The EC and ES exhibited significant differences (p < 0.05) among all samples, with the maximum EC observed in sample D5 and ES in sample B5, respectively. It is worth mentioning here that the samples exhibited higher EC compared to previously reported literature for citrus dietary fiber and potato pectin, making it a better alternative to commercial emulsifiers with potential health benefits.26,32
The other characteristic properties of dietary fiber include GAC, SCBC, CBC, and CEC. The maximum GAC, SCBC, and CBC were observed in samples B5 (8.98 ± 0.01 mg g−1), C5 (5.29 ± 0.08 mg g−1), and B5 (20.91 ± 0.72 mmol g−1), respectively (Table 3). The enhanced activity could be attributed to the combined effect of enzymatic and mechanical treatment, making the structure more loosened and porous, positively correlated to the results of SEM micrographs. This results in all of the non-polar and polar groups being uncovered, making them accessible for interaction and revealing potential health benefits. However, the maximum amount of CEC was observed in sample D5 as 3.05 ± 0.20 mg g−1. Fiber with free functional groups, such as hydroxyl and carboxyl phenols, influences CEC by enhancing the overall chelation characteristic by substituting cations for H+ ions.27 Thus, the present study highlights the imminent role of extracted SDF as a potential health ingredient.
| Sample | GAC (mg g−1) | SCBC (mg g−1) | CBC (mmol g−1) | CEC (mg g−1) |
|---|---|---|---|---|
| a Values are the average of three replicates ± SD with significant difference (p < 0.05) indicated with different letters (a–d); A5: homogenization-assisted enzymatic extraction (HEE) of SDF from Totapuri, B5: homogenization-assisted enzymatic extraction of SDF (HEE) from Safeda, C5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Bhagwa, D5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Daru. | ||||
| A5 | 8.00 ± 0.01a | 4.31 ± 0.09a | 18.47 ± 0.53a | 2.05 ± 0.15a |
| B5 | 8.98 ± 0.01b | 3.95 ± 0.12a | 20.91 ± 0.72b | 1.95 ± 0.25a |
| C5 | 5.33 ± 0.10c | 5.29 ± 0.08b | 18.42 ± 0.14a,b | 2.95 ± 0.23b |
| D5 | 6.79 ± 0.03d | 5.01 ± 0.06c | 18.67 ± 0.09a,b | 3.05 ± 0.20b |
Sugar profile and determination of molecular weight
The monosaccharide composition of all samples was quantified using HPAEC-PAD, as shown in Table 4. Analysis of the SDF samples revealed the presence of various monosaccharides with a maximum content of glucose (35.22 ± 0.01 mg g−1) and minimum of xylose (0.05 ± 0.01 mg g−1) observed in samples B and D, respectively. The most substantial quantity of glucose found in all SDF samples illustrates the effect of the extraction conditions influencing hemicellulose and cellulose, two non-pectic polysaccharides, which results in higher SDF yields and increased glucose content.8 Similarly, xylose was also observed in all SDF samples. As earlier reported, hemicellulose transformation to oligosaccharides yields xylose as the monomer sugar.29 Additionally, the occurrence of glucose and xylose have been reported in the side chain of the main pectin backbone.30 Therefore, this suggests that pectin—the main soluble dietary fiber found in fruit peel—is plausibly present in all SDF samples. The molecular weight determination was carried out to gain structural insights of all SDF samples (Table 5).| Sample code | Glucose (mg g−1) | Fructose (mg g−1) | Sucrose (mg g−1) | Xylose (mg g−1) | Raffinose (mg g−1) | Inositol (mg g−1) |
|---|---|---|---|---|---|---|
| a Values are the average of three replicates ± SD with significant difference (p < 0.05) indicated with different letters (a–d), ND: not detected. | ||||||
| A5 | 15.43 ± 0.26a | 8.76 ± 0.84a | ND | 0.24 ± 0.01a | 1.18 ± 0.01a | 0.26 ± 0.01a |
| B5 | 35.22 ± 0.93b | ND | 0.43 ± 0.02a | 0.26 ± 0.01a | 0.07 ± 0.01b | 0.25 ± 0.02a |
| C5 | 14.14 ± 0.25c | 6.05 ± 0.38b | 1.17 ± 0.09a | 0.05 ± 0.01a | ND | 0.18 ± 0.01a |
| D5 | 9.78 ± 0.29d | 3.54 ± 0.14c | 0.48 ± 0.01a | 0.57 ± 0.05a | 0.26 ± 0.02b | 0.26 ± 0.01a |
| Sample | Matrix | M n | M w | PI |
|---|---|---|---|---|
| a M n: number average molecular weight, Mw: weight average molecular weight, PI: polydispersity index; A5: homogenization-assisted enzymatic extraction (HEE) of SDF from Totapuri, B5: homogenization-assisted enzymatic extraction (HEE) of SDF from Safeda, C5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Bhagwa, D5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Daru. | ||||
| A5 | DHB | 195.130 | 198.906 | 1.019351 |
| B5 | 174.252 | 178.081 | 1.021974 | |
| C5 | 215.292 | 216.868 | 1.00732 | |
| 218.497 | 219.911 | 1.006471 | ||
| 199.256 | 202.235 | 1.014951 | ||
| D5 | 194.295 | 199.779 | 1.028225 | |
| A5 | THAP | 209.546 | 211.598 | 1.009793 |
| B5 | 188.125 | 190.717 | 1.013778 | |
| 378.506 | 381.219 | 1.007168 | ||
| 1563.89 | 1574.49 | 1.006778 | ||
| 1736.04 | 1740.14 | 1.002362 | ||
| C5 | 223.016 | 230.845 | 1.035105 | |
| D5 | 200.450 | 208.855 | 1.041931 | |
| 153.492 | 159.972 | 1.042217 | ||
Interestingly, there are no reports on the molecular weight determination of extracted soluble dietary fiber using MALDI-TOF/TOF-MS/MS. As depicted in Table 5, all of the SDF samples exhibited varied molecular weight range with different matrices. The two matrices, DHB and THAP, were employed in accordance with previously published studies due to their effective outcomes in the identification of polysaccharides with a wide mass range.19 The reflectron mode was used for all samples to enhance the resolution of the spectral data of low-mass oligomers (<5000 Da). It was observed that both matrices exhibited similar results in terms of the weight average molecular weight and maximum molecular weight of the SDF samples. The molecular weight of the samples ranged from 103.551 to 1923.382 Da.
The samples exhibited lower molecular weight, which might be due to the ultrasonication treatment disrupting the complex structure and rendering low molecular weight compounds.
Microstructure analysis
SEM is the most valued technique for investigating the microstructure of any material. In the present study, the effect of mechanical and enzymatic treatments was observed on the surface morphology of SDF samples with a maximum SDF yield, i.e., A5, B5, C5, and D5. As seen in Fig. 2, the samples exhibited loosened and porous structures along with voids. The size of the voids was measured using ImageJ software (NIH) with an average void size (μm) for samples A5, B5, C5, and D5 as 1.91 ± 0.64, 1.50 ± 0.54, 1.65 ± 0.57, 1.67 ± 0.74, respectively. In addition, the aspect ratio for the voids was calculated and observed in the range of 1–3.5 μm. This structural distortion may have been caused by the administration of mechanical treatments (ultrasonication and homogenization). These treatments are known to cause turbulence and shear stress, which might lead to the breaking of complex structures. In addition, enzymatic treatment has been reported to make structures porous and loosened, increasing their hydration capacity and making the inner structure more available for binding with water.23,26 Thereby, in this study, the microstructure analysis unveiled the role of various extraction conditions on morphology, positively correlating to increased techno-functional properties. | ||
| Fig. 2 Microstructure analysis of soluble dietary fiber using scanning electron microscopy. A5: homogenization-assisted enzymatic extraction (HEE) of SDF from Totapuri, B5: homogenization-assisted enzymatic extraction (HEE) of SDF from Safeda, C5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Bhagwa, D5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Daru; values are the average of three replicates ± SD with significant difference (p < 0.05) indicated with different letters (a–d). | ||
FT-IR analysis
The FT-IR spectra help to elucidate the occurrence of various functional groups and the bonding organization, illustrating the structural characteristic of fiber. Thus, the effect of varied extraction conditions was evaluated on the structural characteristics of the SDF samples (Fig. 3). The characteristic band, which spans 3200–3500 cm−1, shows that the –O–H bond is primarily stretched between hemicellulose and cellulose. Out of all of the SDF samples, the B5 and C5 samples showed a red shift of 21.82 cm−1 and 12.41 cm−1, respectively.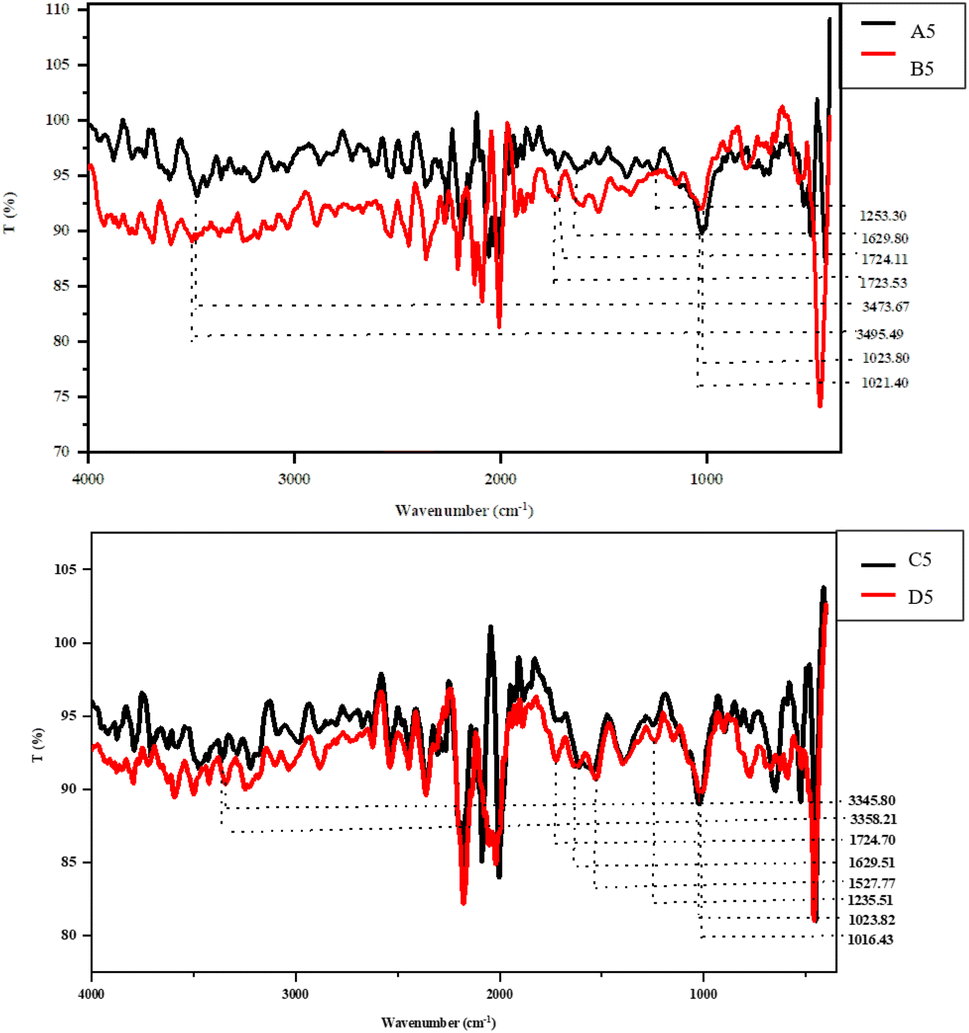 | ||
| Fig. 3 FT-IR spectra of soluble dietary fiber. A5: homogenization-assisted enzymatic extraction (HEE) of SDF from Totapuri, B5: homogenization-assisted enzymatic extraction (HEE) of SDF from Safeda, C5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Bhagwa, D5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Daru. | ||
This peak shift suggested a decrease in intra- and intermolecular hydrogen bonding and an increase in the stretching frequency of the –O–H bond, which had a beneficial impact on the total SDF yield (Fig. 3), demonstrating the plausible influence of extraction conditions on the degradation characteristics of complex polysaccharides.31 The peaks ranging from 1700 to 1500 cm−1 indicate the stretching vibrations amid the aromatic carbon skeleton C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, ester, and acetyl groups. It is worth mentioning here that a redshift of 0.58 cm−1 in the B5 sample, along with the complete disappearance of three peaks in this range, was observed among the B5 and C5 samples, depicting disintegration in the structure of the polysaccharides mainly between hemicellulose and lignin, increasing the overall SDF yields. The distinctive peaks between 1645 and 1612 cm−1, which are mainly found in lignin, explain the C–O stretching between the aromatic or conjugated ketones and flavones.31 The disappearance of the peak in samples B5 and C5 indicates the effect of extraction conditions in the disintegration of the complex fiber structure, increasing its overall yield. The range from 1400 to 1200 cm−1 designates the C–O and C–H stretching, CH2 bending, and stretching of methyl ester, primarily between pectin, cellulose, and hemicellulose.
C, ester, and acetyl groups. It is worth mentioning here that a redshift of 0.58 cm−1 in the B5 sample, along with the complete disappearance of three peaks in this range, was observed among the B5 and C5 samples, depicting disintegration in the structure of the polysaccharides mainly between hemicellulose and lignin, increasing the overall SDF yields. The distinctive peaks between 1645 and 1612 cm−1, which are mainly found in lignin, explain the C–O stretching between the aromatic or conjugated ketones and flavones.31 The disappearance of the peak in samples B5 and C5 indicates the effect of extraction conditions in the disintegration of the complex fiber structure, increasing its overall yield. The range from 1400 to 1200 cm−1 designates the C–O and C–H stretching, CH2 bending, and stretching of methyl ester, primarily between pectin, cellulose, and hemicellulose.
Following the previous observation, the complete removal of peaks was observed in this region for samples B5 and C5. Finally, the peaks from 1050 to 1000 cm−1 denote the stretching vibrations among –C–O bonds primarily due to collective vibrations among the C–O–C and C–O–H groups.31 Interestingly, a red shift of 2.4 cm−1 and 7.39 cm−1 was observed in samples B5 and C5, respectively, clearly depicting the effect of extraction conditions on the increased SDF yield. Therefore, the IR spectra validate the affirmative impact of the extraction conditions on the SDF yield, along with the structural and functional characteristics.
TGA analysis
The thermogravimetric analysis of all SDF samples (A5, B5, C5, and D5) was performed using a TGA analyzer (Fig. 4). The analysis was performed in the 50–400 °C temperature range, unveiling the characteristic degradation and weight loss in the sample. This degeneration could be divided into three main segments at 50–150 °C, 150–250 °C, and 250–400 °C. | ||
| Fig. 4 TGA analysis of the soluble dietary fiber. A5: homogenization-assisted enzymatic extraction (HEE) of SDF from Totapuri, B5: homogenization-assisted enzymatic extraction (HEE) of SDF from Safeda, C5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Bhagwa, D5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Daru. | ||
The primary stage, ranging from 50 to 150 °C, represents the significant weight loss among all SDF samples ascribed to the evaporation of all of the absorbed water from the samples, degradation of different low molecular weight polysaccharides, along with devolatilization, which mainly occurs at 121 °C.26 The next stage reveals the loss in sample weight in the range of 150–250 °C. The inception point from where a sudden loss in weight was observed in the degradation characteristics ranged from 152.80 °C to 178.68 °C. However, 20% weight loss among all samples was observed at this stage, with degradation temperatures ranging from 187.07 °C, 199.03 °C, 188.03 °C, and 223.89 °C for samples A5, B5, C5, and D5, respectively. This mass reduction among all samples could be accredited to fiber disintegration via reduction, dihydroxylation, or decarboxylation reactions.12 Comparable results were observed in previous studies, where soluble dietary fiber was extracted from coffee peels, showing a 20% mass reduction in the 130–250 °C temperature range.8 The final stage, ranging from 250 to 400 °C, resulted in an abrupt weight loss of up to 50% among all SDF samples. The major weight loss among samples A5, B5, C5, and D5 was observed within a temperature range of 293.07 °C, 294.03 °C, 301.03 °C, and 396.89 °C, respectively. The mass degradation might possibly be attributed to the thermal disintegration of hemicellulose, pectin, and lignin. The primary contributor of a mass loss is the pyrolytic disintegration of polysaccharides, especially hemicellulose and pectin.26 However, the residual masses of all SDF samples A5, B5, C5, and D5 at 398 °C were 39.27%, 36.50%, 43.79%, and 49.97%, respectively. Interestingly, in the present study, a significant residual mass of up to 50% was observed, which indicates the potential thermal stability of all samples. Thus, these results signify the thermal stability of all SDF samples, enhancing its potential application in the food industry.
Flavonoid, phenolic content and antioxidant assays
Table 6 describes the TFC and TPC of all SDF samples ranging from 1.71 to 8.57 μg RU per mg, and 5.40 to 10.11 μg GAE per mg, respectively. The results exhibit a significant difference (p < 0.05) with lower quantification. The reason for the reduced estimation could be the pretreatment of a peel with 70% ethanol that might have resulted in the leaching of all phytochemicals.12 Furthermore, prolonged ethanol-induced precipitation and filtration of SDF have been perceived for lower polyphenol concentration.33 For the estimation of the antioxidant activity, DPPH and ABTS assays were performed with the maximum scavenging activity in sample D (Table 6), which were positively correlated with the results of TPC and TFC.| Sample | Total phenolic content (μg GAE per mg) | Total flavonoid content (μg RU per mg) | IC50 of free radical scavenging activities (μg mL−1) | |
|---|---|---|---|---|
| DPPH | ABTS | |||
| a Values are the average of three replicates ± SD with significant difference (p < 0.05) indicated with different letters (a–d); A5: homogenization-assisted enzymatic extraction (HEE) of SDF from Totapuri, B5: homogenization-assisted enzymatic extraction (HEE) of SDF from Safeda, C5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Bhagwa, D5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Daru. | ||||
| A5 | 5.40 ± 0.38a | 1.71 ± 0.05a | 889.59 | 581.75 |
| B5 | 2.27 ± 0.09b | 1.81 ± 0.06a | 584.87 | 491.71 |
| C5 | 6.41 ± 0.44c | 4.85 ± 0.02c | 294.48 | 258.81 |
| D5 | 10.11 ± 0.06d | 8.57 ± 0.07d | 156.76 | 207.63 |
Color analysis
The color of all samples was quantified in terms of ‘L*’, ‘a*’, and ‘b*’ values with the color difference measured as ΔE. As depicted in Table 7, the ‘L*’ values exhibited significant differences (p < 0.05) among them, with maximum lightness in sample A5 (Table 7). Pretreatment of the peel that had possibly eliminated pigments and color could be the most likely cause of the lightness in the samples. The ‘a*’ values were observed to be significantly different (p < 0.05) with positive values among samples C5 and D5. Although no difference was observed for the samples, A5 and B5 represented negative values. The negative value among samples A5 and B5 described the greenness of the sample. The positive values among samples C5 and D5 exhibited the redness in the sample responsible for imparting the overall darkness to the sample and reduced L* value. However, ‘b*’ values exhibited a significant difference (p < 0.05), with positive values indicating the yellowness among samples. The color value exhibits a crucial role for application in different food products. Thereby, SDF samples could easily be incorporated into food products exhibiting possible health benefits.| Sample | L* | a* | b* | ΔE* |
|---|---|---|---|---|
| a Values are the average of three replicates ± SD with significant difference (p < 0.05) indicated with different letters (a–d); A5: homogenization-assisted enzymatic extraction (HEE) of SDF from Totapuri, B5: homogenization-assisted enzymatic extraction (HEE) of SDF from Safeda, C5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Bhagwa, D5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Daru. | ||||
| A5 | 85.43 ± 1.11a | −3.35 ± 0.01a | 8.58 ± 0.14a | 69.90 ± 1.07a |
| B5 | 81.94 ± 1.11b | −3.31 ± 0.25a | 12.46 ± 0.16b | 67.31 ± 0.13b |
| C5 | 51.33 ± 0.06c | 0.75 ± 0.02b | 7.48 ± 0.05c | 36.48 ± 0.03c |
| D5 | 48.55 ± 0.02d | 0.14 ± 0.13b | 7.35 ± 0.11c | 33.80 ± 0.03d |
Prebiotic potential
| Time interval | A5 | B5 | C5 | D5 | FOS | Inulin |
|---|---|---|---|---|---|---|
| a Values are average of replicates ± SD with significant difference (p < 0.05) indicated with different letters (a–c); A5: homogenization assisted enzymatic extraction (HEE) of SDF from Totapuri, B5: homogenization assisted enzymatic extraction (HEE) of SDF from Safeda, C5: ultrasonication assisted enzymatic extraction (UEE) of SDF from Bhagwa, D5: ultrasonication assisted enzymatic extraction (UEE) of SDF from Daru, FOS: fructo-oligosaccharide. | ||||||
| Alpha amylase activity (%) | ||||||
| 0 h | 7.23 ± 0.12a | 6.60 ± 0.01a | 7.15 ± 0.03a | 7.05 ± 0.06a | 7.60 ± 0.03a | 7.80 ± 0.03a |
| 3 h | 8.20 ± 0.03b | 6.73 ± 0.27a | 8.89 ± 0.06b | 7.01 ± 0.03a | 9.52 ± 0.09b | 8.73 ± 0.04b |
| 6 h | 8.32 ± 0.04b | 7.68 ± 0.08b | 8.95 ± 0.06b | 7.92 ± 0.06b | 12.29 ± 0.06c | 12.13 ± 0.12c |
|
||||||
| Gastric juice hydrolysis activity (%) | ||||||
| 0 h | 7.46 ± 0.13a | 6.64 ± 0.06a | 7.14 ± 0.03a | 7.00 ± 0.03a | 8.76 ± 0.10a | 8.49 ± 0.17a |
| 3 h | 7.48 ± 0.09a | 7.31 ± 0.02b | 7.40 ± 0.10a | 7.12 ± 0.12a | 17.82 ± 0.60b | 20.28 ± 0.65b |
| 6 h | 12.77 ± 0.13b | 7.27 ± 0.09b | 7.73 ± 0.06a | 7.73 ± 0.04b | 55.95 ± 0.06c | 66.72 ± 0.15c |
pH and cell growth analysis
The prebiotic effect of all SDF samples was further evaluated by fermenting the SDF samples with different probiotics monitored via cell growth measurements and pH. The heat map was drawn to elucidate the effect of probiotic proliferation on both cell growth and pH values. The heat map for cell growth was divided into a six-point scale, starting from less than 6 log10 CFU mL−1 as a bluish color and greater than 10 as reddish-orange color. The blue color indicates the minimum cell count range below 6 but not below 5.79 log10 CFU mL−1, and the reddish-orange color indicates the maximum cell count above 10 but not above 10.27 log10 CFU mL−1. In Fig. 5, it is apparent that the SDF samples, along with FOS, supported the growth and proliferation of all probiotic strains. Moreover, glucose and FOS were rapidly metabolized, supporting the proliferation of different probiotic strains at 24 h with a reduction at 48 h. Surprisingly, a substantial increase in the growth of Lactobacillus plantarum at 24 h was observed for sample A5, higher than that for glucose and FOS. This signifies the potential role of SDF in the proliferation of different probiotic strains. Interestingly, all SDF samples exhibited effective cell growth ranging between 5.79 log10 CFU mL−1 and 10.27 log10 CFU mL−1, illustrating the potential role of extracted SDF samples in the stimulation and proliferation of probiotics. | ||
| Fig. 5 pH, cell growth and SCFA quantification of SDF samples at 24 and 48 h. A5: homogenization-assisted enzymatic extraction (HEE) of SDF from Totapuri, B5: homogenization-assisted enzymatic extraction (HEE) of SDF from Safeda, C5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Bhagwa, D5: ultrasonication-assisted enzymatic extraction (UEE) of SDF from Daru, I: Lactobacillus fermentum, II: Lactobacillus plantarum, III: Wisella ciberia, IV: Lactobacillus brevis, V: mixed consortium, FOS: fructoligosaccharide. | ||
The pH of the samples exhibited a similar decreasing trend for all SDF samples, along with glucose and FOS (Fig. 5). The heat map was divided into a six-point scale for pH ranges starting from 4.11 to 6.83. The purple color indicates the minimum pH range below 4.5 but not below 4.11, and the bluish color indicates the maximum color above pH 6.5 but not above 7. The decrease in pH for MRS media supplemented with glucose demonstrated a steady decline compared to MRS supplemented with SDF and FOS samples, which is certainly correlated to cell growth measurement. This decrease in pH could be associated with the diverse metabolic pathways of microbes for utilizing the substrate in the media. A substantial decrease in pH could also be caused by catabolic suppression through the use of simple carbon sources like glucose.34 In addition, the production of various short-chain fatty acids (SCFAs), i.e., propionic, acetic, and butyric acid, during fermentation is mainly responsible for acidification, i.e., pH decrease. Thereby, the results indicate an effective decrease in pH with time that is positively correlated to the cell growth measurement.
SCFA quantification
Fig. 5 exemplifies the effect of fermentation on the production of three major SCFAs, viz., acetic, propionic, and butyric acid. The heat map for SCFA quantification was divided into a six-point scale starting from less than 0.128 mg mL−1 as purple color and greater than 3.660 mg mL−1 as purple-bluish color, where the purple color indicates the minimum SCFA range below 0.128 mg mL−1 but not below 0.123 mg mL−1, and the purple-bluish color indicates the maximum SCFA above 3.660 mg mL−1 but not above 3.784 mg mL−1. The SDF samples exhibited a significant increase (p < 0.05) at 24 h with a slight reduction at 48 h compared to the control samples, demonstrating a positive correlation between the fermentation conditions and SCFA production. The amount of SCFAs produced mainly depends upon the type of carbon source in the testing medium. Interestingly, in the present study, the highest amount of SCFAs produced was propionic acid, followed by acetic and butyric acid. Surprisingly, sample D5, which included the probiotic Lactobacillus brevis, showed the highest level of propionic acid (3.57 ± 0.03 mg mL−1) at 24 hours when compared to glucose and FOS, suggesting that it could be a potential source of prebiotics. Similarly, in the case of butyric acid, the highest concentration was observed in the MRS media supplemented with FOS. Interestingly, MRS supplemented with sample C5 containing probiotic Lactobacillus brevis unveiled an adequate butyric acid (1.16 ± 0.01 mg mL−1) content acting as an energy source for colonocytes, exhibiting health-associated benefits. However, in the present study, the highest amount of acetic acid (2.64 ± 0.01 mg mL−1) was observed at 24 h for sample A5 containing probiotic Wisella ciberia, directing it towards a potential prebiotic candidate. It is worth mentioning here that MRS supplemented with all SDF samples resulted in the maximum progression of almost all probiotic strains (Fig. 5). Therefore, the results emphasize the role of SDF extracted from fruit peels in the proliferation and production of various metabolites. This could be due to the enzymatic and mechanical treatment altering the structural characteristics of SDF samples, affecting its utilization and SCFAs production.34 Thus, SDF extracted from the waste peel may play a potential role in the amelioration of various inflammatory diseases via possible utilization as a prebiotic agent or in innovative synbiotic formulations.Conclusions
These results highlight the potential role of extraction methods (homogenization and ultrasonication) as an effectual technique in augmenting the overall structural, thermal, and prebiotic properties of SDF. Thus, the results emphasize the waste valorisation to a value-added ingredient, exhibiting adequate prebiotic activity with potential industrial application. Future research work will explore the opportunity for in vivo potential of the SDF candidates in the maintenance of intestinal inflammatory disorders.Author contributions
Shriya Bhatt: conceptualization, investigation, data curation, roles/writing: original draft. Mahesh Gupta: supervision, roles/writing: original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their gratitude to the Director, CSIR-Institute of Himalayan Bioresource Technology, for their exquisite support. S. B. expresses gratitude to ICMR for awarding a senior research fellowship, ICMR-SRF. The authors also acknowledge Dr Avnesh Kumari (Senior Technical Officer, Biotechnology Division, CSIR-IHBT) and Dr Robin Joshi (Senior Technical Officer, Biotechnology Division, CSIR-IHBT) for SEM and UPLC analysis, respectively, in manuscript number 5168.Notes and references
- M. V. Vilariño, C. Franco and C. Quarrington, Frontiers in Environmental Science, 2017, 5, 21 CrossRef.
- C. M. Ajila, S. G. Bhat and U. P. Rao, Food Chem., 2007, 102(4), 1006–1011 CrossRef.
- P. D. Pathak, S. A. Mandavgane and B. D. Kulkarni, Waste Biomass Valorization, 2017, 8, 1127–1137 CrossRef.
- M. Jia, J. Chen, X. Liu, M. Xie, S. Nie, Y. Chen, J. Xie and Q. Yu, Food Hydrocolloids, 2019, 99, 105349 CrossRef.
- H. Hu and Q. Zhao, RSC Adv., 2018, 8(72), 41117–41130 RSC.
- K. Lu, T. Yu, X. Cao, H. Xia, S. Wang, G. Sun, L. Chen and W. Liao, Front. Nutr., 2023, 10, 1253312 CrossRef PubMed.
- R. Khorasaniha, H. Olof, A. Voisin, K. Armstrong, E. Wine, T. Vasanthan and H. Armstrong, Food Hydrocolloids, 2023, 139, 108495 CrossRef CAS.
- W. Dong, D. Wang, R. Hu, Y. Long and L. Lv, Food Res. Int., 2020, 136, 10949 CrossRef PubMed.
- I. Buljeta, D. Šubarić, J. Babić, A. Pichler, J. Šimunović and M. Kopjar, Appl. Sci., 2023, 13(16), 9309 CrossRef.
- U. P. Mall and V. H. Patel, Food Chemistry Advances, 2023, 2, 100320 CrossRef.
- L. Serna-Cock, E. García-Gonzales and C. Torres-León, Food Rev. Int., 2016, 32(4), 364–376 CrossRef.
- S. Bhatt and M. Gupta, Biomass Convers. Biorefin., 2022, 1–16 Search PubMed.
- X. Li, X. He, Y. Lv and Q. He, J. Food Process Eng., 2014, 37(3), 293–298 CrossRef.
- B. Bello, S. Mustafa, J. S. Tan, T. A. T. Ibrahim, Y. J. Tam, A. B. Ariff, M. Y. Manap and S. Abbasiliasi, 3 Biotech, 2018, 8, 1–14 CrossRef PubMed.
- W. Zhang, G. Zeng, Y. Pan, W. Chen, W. Huang, H. Chen and Y. Li, Carbohydr. Polym., 2017, 172, 102–112 CrossRef PubMed.
- M. A. Kurek, S. Karp, J. Wyrwisz and Y. Niu, Food Hydrocolloids, 2018, 85, 321–330 CrossRef CAS.
- L. Cheng, X. Zhang, Y. Hong, Z. Li, C. Li and Z. Gu, Int. J. Biol. Macromol., 2017, 101, 1004–1011 CrossRef CAS PubMed.
- S. Bhatt, N. Kumari, V. Abhishek and M. Gupta, Journal of Food Measurement and Characterization, 2021, 15, 675–685 CrossRef.
- M. López-García, M. S. D. García, J. M. L. Vilariño and M. V. G. Rodríguez, Food Chem., 2016, 199, 597–604 CrossRef PubMed.
- R. Kumari, V. Abhishek and M. Gupta, Vegetos, 2021, 34, 205–211 CrossRef.
- S. Bhatt, V. Dadwal, Y. Padwad and M. Gupta, J. Food Process. Preserv., 2022, 46(1), e16137 Search PubMed.
- S. Bhatt, B. Singh and M. Gupta, J. Agric. Food Res., 2020, 2, 100069 Search PubMed.
- G. J. Chen, Q. Y. Hong, N. Ji, W. N. Wu and L. Z. Ma, Int. J. Biol. Macromol., 2020, 155, 674–684 CrossRef PubMed.
- J. Dobrowolska-Iwanek, R. Lauterbach, H. Huras, P. Paśko, E. Prochownik, M. Woźniakiewicz, S. Chrząszcz and P. Zagrodzki, Microchem. J., 2020, 155, 104671 CrossRef.
- J. Gan, Z. Huang, Q. Yu, G. Peng, Y. Chen, J. Xie, S. Nie and M. Xie, Food Hydrocolloids, 2020, 101, 105549 CrossRef CAS.
- Y. Zhang, J. Liao and J. Qi, Lwt, 2020, 128, 109397 CrossRef CAS.
- Y. Jiang, H. Yin, Y. Zheng, D. Wang, Z. Liu, Y. Deng and Y. Zhao, Food Res. Int., 2020, 136, 109348 CrossRef CAS PubMed.
- K. Wang, M. Li, Y. Wang, Z. Liu and Y. Ni, Food Hydrocolloids, 2021, 110, 106162 CrossRef CAS.
- L. C. Rojas-Pérez, P. C. Narváez-Rincón, M. A. M. Rocha, E. Coelho and M. A. Coimbra, Bioresources and Bioprocessing, 2022, 9(1), 105 CrossRef PubMed.
- J. Müller-Maatsch, M. Bencivenni, A. Caligiani, T. Tedeschi, G. Bruggeman, M. Bosch, J. Petrusan, B. Van Droogenbroeck, K. Elst and S. Sforza, Food Chem., 2016, 201, 37–45 CrossRef PubMed.
- N. George, A. A. Andersson, R. Andersson and A. Kamal-Eldin, NFS J., 2020, 21, 16–21 CrossRef.
- J. S. Yang, T. H. Mu and M. M. Ma, Food Chem., 2018, 244, 197–205 CrossRef CAS PubMed.
- G. Yu, J. Bei, J. Zhao, Q. Li and C. Cheng, Food Chem., 2018, 257, 333–340 CrossRef CAS PubMed.
- G. J. Chen, Q. Y. Hong, N. Ji, W. N. Wu and L. Z. Ma, Int. J. Biol. Macromol., 2020, 155, 674–684 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |