
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational element-doping of FeOOH-based electrocatalysts for efficient ammonia electrosynthesis†
Haifan
Wang
abc,
Menglei
Yuan
*ad,
Jingxian
Zhang
ab,
Yiling
Bai
ef,
Ke
Zhang
g,
Bin
Li
*g and
Guangjin
Zhang
 *ab
*ab
aCAS Key Laboratory of Green Process and Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: zhanggj@ipe.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cSchool of Chemical Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
dState Key Laboratory of Solidification Processing and School of Materials Science and Engineering, Northwestern Polytechnical University, Xi’an 710072, China. E-mail: mlyuan@nwpu.edu.cn
eCAS Key Laboratory of Carbon Materials, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China
fSynCat@Beijing, Synfuels China Technology Co. Ltd, Beijing 101407, China
gZhengzhou Tobacco Research Institute of CNTC, Zhengzhou 450001, China. E-mail: lib@ztri.com.cn
First published on 1st November 2023
Abstract
Electrocatalysis has been intensively studied in nitrogen (N2) reduction for its sustainable power and stable catalytic performance, but it is still limited by weak activation of N2 at the catalytic sites, and the competition from the hydrogen evolution reaction (HER). The special d-orbital electron arrangement of transition metals and the tuning of the microenvironment provide possible strategies to enhance the activation of N2, while improving the selectivity of the eNRR. Herein, FeO(OH, S) with high spin state and Mo–FeOOH with low spin state were designed around the FeOOH-based catalysts through elemental doping, which could achieve excellent ammonia yield performance of 80.1 ± 4.0 μg h−1 mgcat−1 (FE 36.9 ± 0.5%) and 86.8 ± 4.1 μg h−1 mgcat−1 (FE 29.1 ± 0.8%) in 0.1 M LiClO4 at −0.6 V vs. RHE, respectively, coupled with polyethylene glycol (PEG) to inhibit the HER. Based on theoretical calculations to investigate the adsorption of N2 on Fe sites, the FeO(OH, S) catalyst has stronger adsorption ability, which may originate from the high spin effect, which means that the more isolated and highly active eg orbital electrons are more beneficial to realize the electronic feedback mechanism, promoting the d–π* orbital interaction with N2.
Broader contextTo investigate different d-orbital electron structures of Fe active sites, FeO(OH, S) with high spin state (t2g3eg2) and Mo–FeOOH with low spin state (t2g5eg0) catalysts were successfully synthesized by elemental doping strategies. With PEG inhibiting the HER, FeO(OH, S) and Mo–FeOOH catalysts achieved outstanding electrocatalytic nitrogen reduction reaction (eNRR) performance with NH3 yield rates of 80.1 ± 4.0 μg h−1 mgcat−1 (FE 36.9 ± 0.5%) and 86.8 ± 4.1 μg h−1 mgcat−1 (FE 29.1 ± 0.8%) in 0.1 M LiClO4 + 20% PEG, respectively. DFT calculations reveal that FeO(OH, S) exhibits the strongest N2 adsorption capability, which is mainly because more isolated eg orbital electrons could promote d–π* orbital interaction to activate N2. This possible high spin effect may provide positive guidance for the electronic structure design of other TM-based electrocatalysts for the eNRR. |
1. Introduction
As the main component of the natural nitrogen element cycle, nitrogen (N2) makes up 78% of the composition of air. Unfortunately, N2 cannot be utilized directly, but has to be converted into nitrate (NO3−, NO2−) and ammonia (NH3, NH4+) by lightning and biological fixation reactions. The establishment of the Haber–Bosch ammonia synthesis process (N2 + 3H2 → 2NH3) provides a successful human intervention for nitrogen fixation and an efficient chemical synthesis route to form ammonia. However, the Haber–Bosch technique consumes a lot of energy from raw hydrogen (H2) production and requires harsh reaction conditions (350–550 °C, 150–350 atm),1 while generating carbon dioxide (CO2), which is a serious burden to the carbon cycle in nature. With the development of green catalytic research, photocatalytic,2–4 electrocatalytic, and enzyme catalytic5 systems have emerged as new green alternatives to the Haber–Bosch route. Among them, electrocatalytic nitrogen reduction (eNRR) has received more attention due to its sustainable power force, electricity, and relatively stable catalytic performance. However, there are still major challenges that currently limit the industrial application of the eNRR, such as the low diffusion rate of N2 in aqueous electrolytes, the hindrance of slow N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond cleavage, and the competition from the side hydrogen evolution reaction (HER).6
N triple bond cleavage, and the competition from the side hydrogen evolution reaction (HER).6
With the aim of reducing HER kinetics and increasing the efficiency of the eNRR process, the construction of a suitable catalytic microenvironment is considered an efficient strategy to reduce the proton concentration on the catalyst surface by controlling the diffusion of protons or proton carriers,7–9 such as the introduction of special electrolyte additives, including polyethylene glycol (PEG), methanol (CH3OH), etc. Electrostatic potential analysis of PEG shows that a large number of negative oxygen sites (C–O–C) can form hydrogen bonds with the H atom in water molecules (H2O, H–O–H) or [H3O]+, thus forming a local hydrophobic layer and hindering the diffusion of H2O to the electrode surface.10,11 Furthermore, the unique d-orbital electronic configuration of transition metals (TMs) located in IIIB to VIII groups provides a possible mechanism for enhancing the activation of N2. For example, Fe,12,13 Co,14,15 Ni,16,17 Mo,18,19 V,20,21 Ti,22,23 and other elements24,25 are widely used to develop high performance ammonia electrocatalysts. Specifically, when N2 chemisorbs to the active sites of TM-based catalysts (TMs-N), the electrons from the σ orbitals of the N2 molecule can transfer to the unoccupied d orbitals of the TMs (d–σ). Meanwhile, feedback electrons from the occupied d orbitals of TMs are also donated to the empty anti-bond π* orbitals of N2 (d–π*). This electronic “donation–backdonation” mechanism will reduce the triple bond level and activate N2.26–28 Therefore, as with TM-based electrocatalysts, rational design of the d-orbital electron arrangement would facilitate the adsorption and activation of N2. And the change in the orbital electron arrangement is often reflected in the spin state and effective paramagnetic moment (μeff) of the corresponding metal. In addition, much literature indicates that atomic doping can introduce new active sites, and optimize the local charge distribution and electronic structure of the catalytic centers, which is a valid strategy to modulate the spin state of the materials.29–34
FeOOH is a common iron oxide and has excellent electrocatalytic activities, including in water splitting (HER,35 oxygen evolution reaction (OER)36,37),38 the eNRR, and batteries.39,40 Zhu et al. reported that FeOOH and β-FeO (OH, F) electrocatalysts could achieve NH3 yields of 23.32 μg h−1 mgcat−1 (FE 6.7%) at −0.75 V vs. RHE and 42.38 μg h−1 mgcat−1 (FE 9.02%) at −0.6 V vs. RHE in 0.5 M LiClO4.41,42 Also using the atomic doping method, Tan et al. developed a Zr–FeOOH electrocatalyst to achieve superior eNRR performance (1.39 × 10−10 mol s−1 cm−2, FE 35.63%) at −0.5 V vs. RHE in 0.1 M Na2SO4.43 In non-aqueous systems, Ren et al. constructed CH3OH + 0.16% H2O electrolyte to improve the eNRR efficiency of FeOOH-CNT to a higher level (262.5 μg h−1 mgcat−1, FE 75.9%).44 In this article, based on the elemental doping strategy, FeO(OH, S) with a high spin state and Mo–FeOOH with a low spin state were successfully synthesized, which could achieve excellent NH3 yield performances of 80.1 ± 4.0 μg h−1 mgcat−1 (FE 36.9 ± 0.5%) and 86.8 ± 4.1 μg h−1 mgcat−1 (FE 29.1 ± 0.8%) at -0.6 V vs. RHE in 0.1 M LiClO4 + 20% PEG, respectively, coupling with PEG to inhibit the HER. Compared with the aqueous system, this reactive system we reported can further improve the ammonia yield rates and efficiencies of the electrocatalysts, which indicates that the FeO(OH, S) and Mo–FeOOH catalysts exhibited better eNRR performance than most recent electrocatalysts listed in Table S3 (ESI†). But for non-aqueous systems, such as the methanol system, there is still a certain gap in the performance of ammonia synthesis, which is due to the fact that the appropriate introduction of PEG only partially inhibits the occurrence of water splitting to hydrogen, rather than decreasing the activity of protons totally. Furthermore, when Fe sites were chosen as the main active center, theoretical calculations revealed that FeO(OH, S) has a stronger N2 adsorption energy, which is mainly due to more isolated and highly active eg orbital electrons. As for FeOOH-based electrocatalysts, this possible high spin effect could enhance d–π* orbital interaction to activate N2.
2. Experimental
2.1. Materials
Iron chloride [FeCl3, AR, 99.9%], molybdenum pentachloride [MoCl5, AR], polyethylene glycol [PEG, AR], sodium hypochlorite [NaClO, AR], sodium citrate [C6H5Na3O7, AR], salicylic acid [C7H6O3, AR], sodium nitroferricyanide(III)dihydrate [C5H4FeN6Na2O3, AR], p-dimethylaminobenzaldehyde [C9H11NO, AR], ammonium chloride [NH4Cl, 14N(AR), 15N(98%, AR)], fumaric acid [C4H4O4, AR, 99.5%] and lithium perchlorate [LiClO4, AR, ≥99.99%] were purchased from Aladdin Chemical Reagent Co., Ltd. Urea [CO(NH2)2, AR], hydrochloric acid [HCl, AR], hydrazine hydrate [N2H4·H2O, AR, 85%], sodium hydroxide [NaOH, AR] and anhydrous ethanol [C2H6O, AR] were purchased from Sinopharm Chemical Reagent Co., Ltd. Thiourea [CH4N2S, AR] was purchased from Xilong Scientific Co., Ltd. Sodium chloride [NaCl, AR] was purchased from Beijing Chemical Industry Group Co., Ltd. Water was purified by the Millipore system and ethanol was utilized without further purification.2.2. Preparation of FeOOH nanorods
10 g FeCl3 was first dissolved in 80 mL DI and was stirred for 1 h. Subsequently, this obtained solution was transferred into a 100 mL hydrothermal autoclave at 120 °C for 12 h. The resulting product was washed several times with DI and ethanol. The final sample FeOOH was dried in an oven at 60 °C for 12 h.2.3. Preparation of FeO(OH)x and FeO(OH, S)
2.6 g FeCl3 was first dissolved in 35 mL DI and 35 mL ethanol, and then this mixture was stirred for 1 h. Subsequently, this obtained solution was transferred into a 100 mL hydrothermal autoclave at 120 °C for 5 h. The resulting product was washed several times with DI and ethanol. The final sample FeO(OH)x was dried in an oven at 60 °C for 12 h.100 mg FeO(OH)x sample was dispersed into 40 mL DI and sonicated for 1 h to make the suspension homogeneous. After that, 1 g thiourea was dissolved into the above suspension and stirred for 30 min, and then the mixture was transferred to a 50 mL hydrothermal autoclave at 120 °C for 3 h. When the reaction finished, the resulting product was washed several times with DI and ethanol. The final yellow sample FeO(OH, S) was dried in an oven at 60 °C for 12 h.
2.4. Preparation of FeOOH–oil bath (FeOOH-o) and Mo–FeOOH
1.35 g FeCl3·6H2O, 0.75 g urea, and 2.9 g NaCl were dissolved in 25 mL DI and stirred for 1 h. The solution was then transferred to a 50 mL round-bottom flask with a reflux condenser and placed in a thermostat oil bath cauldron at 100 °C for 4 h. When the reaction finished, the resulting product continued to age at 100 °C for 1 h. After aging, the yellow sample was washed several times with DI and ethanol. Then the FeOOH-o catalyst was placed in a vacuum oven and dried at 60 °C for 12 h.Based on similar synthesis methods, 1.23 g FeCl3·6H2O, 109.3 mg MoCl5, 0.75 g urea, and 2.9 g NaCl were the precursors of the Mo–FeOOH catalyst.
2.5. Electrochemical experiments
The H-type electrochemical reaction cell and three electrode system were used to measure the electrochemical ammonia synthesis. The catalyst ink for the working electrode was prepared by dispersing about 3 mg of catalyst in a mixed solution of 30 μL Nafion (0.5 wt%), 500 μL ethanol, and 470 μL water followed by sonication for 1 h. The prepared catalyst loaded on a piece of pretreated carbon cloth (1 × 1.5 cm2) was used as the working electrode with a mass loading of 0.3 mg cm−2. To avoid contamination with nitrogen-containing species in the air, electrodes were used either immediately after preparation or kept in a vacuum before being used in electrochemical experiments. Meanwhile, a graphite rod and Ag/AgCl electrode (saturated KCl electrolyte) were employed as counter electrodes and reference electrodes, respectively.3. Results and discussion
3.1. Characterization of the catalysts
Pristine FeOOH, FeO(OH)x, and FeO(OH, S) samples were synthesized by the typical solvent-thermal method. In order to dope more sulfur atoms, FeO(OH)x with partial hydroxyl deletion was first synthesized by the ethanol solvent-thermal method. And then FeO(OH)x as the precursor was further treated with thiourea to obtain the final sample FeO(OH, S). As shown in the X-ray diffraction (XRD) diagram, all the samples exhibit similar diffraction patterns, which is well consistent with the tetragonal phase of FeOOH (JCPDS: 75-1594)41 (Fig. 1a1). Specifically, the characteristic diffraction peaks of FeO(OH, S) located at 11.85°, 16.91°, 26.84°, 34.17°, 35.32°, 39.30°, 46.55° and 56.09° correspond to the (110), (200), (130), (400), (211), (301), (411) and (251) crystal planes. The crystallinity of FeO(OH, S) is lower than that of pristine FeOOH obtained by the direct hydrothermal route, which may be due to the insufficient hydrolysis of FeCl3 to Fe(OH)3 in the ethanol solution, which affected the formation of the subsequent FeOOH crystal during the synthesis of the precursor FeO(OH)x by the ethanol–water solvent thermal method.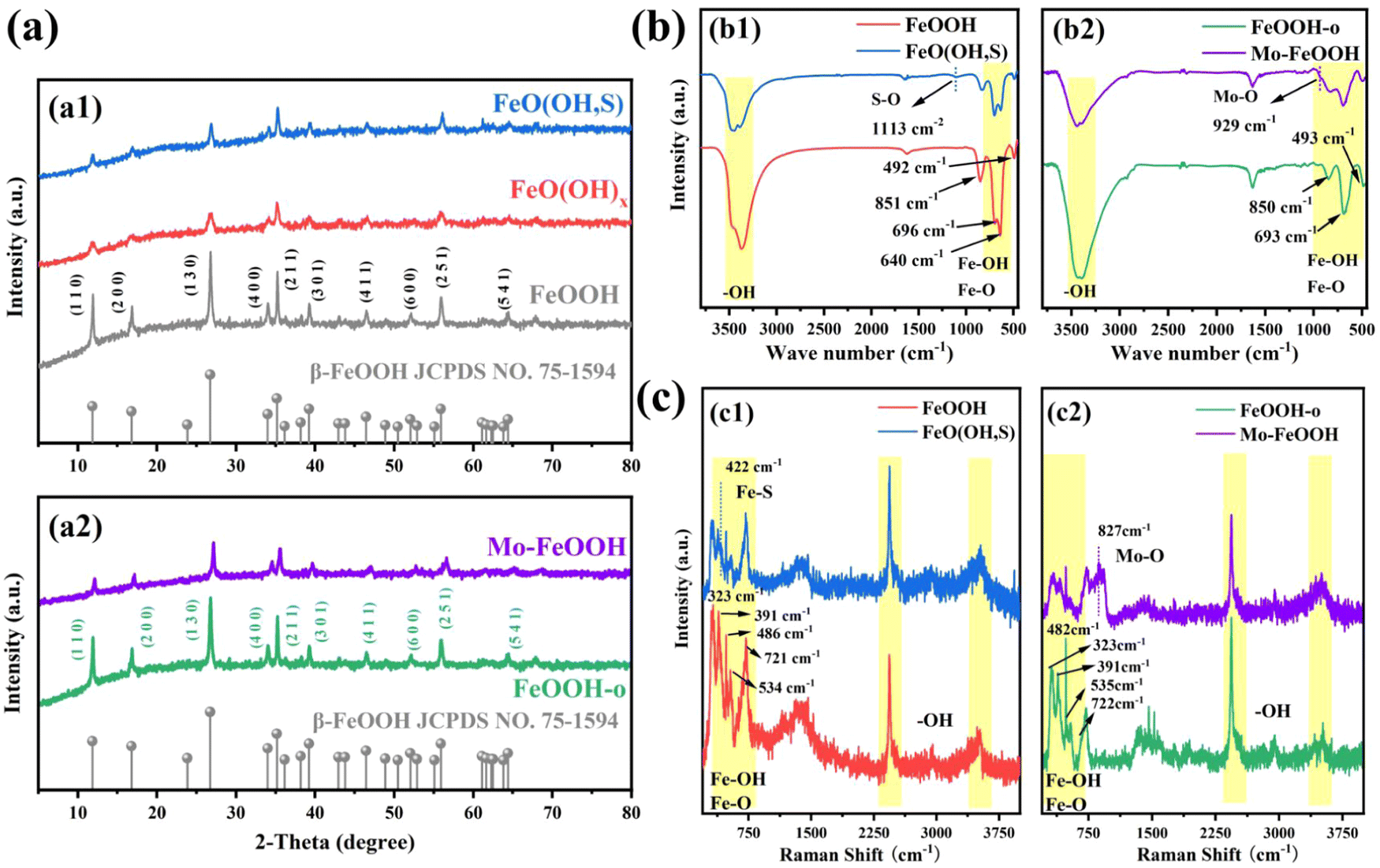 | ||
| Fig. 1 (a) XRD patterns of different FeOOH samples: (a1) pristine FeOOH and FeO(OH,S) catalysts; (a2) FeOOH-o and Mo–FeOOH(5%) catalysts; (b) FTIR spectra of different FeOOH samples: (b1) pristine FeOOH and FeO(OH,S) catalysts; (b2) FeOOH-o and Mo–FeOOH(5%) catalysts; (c) Raman spectra of different FeOOH samples: (c1) pristine FeOOH and FeO(OH,S) catalysts; (c2) FeOOH-o and Mo–FeOOH(5%) catalysts. | ||
In Fig. 1b1, the FTIR spectra show the stretching vibrations of –OH groups (3379 cm−1) and Fe–O bonds (640 cm−1, 696 cm−1). Meanwhile, the Fe–OH bending vibrations45 also appear at 851 cm−1 and 492 cm−1. As can be seen from the Raman spectrum in Fig. 1c1, the peaks at 323 cm−1, 391 cm−1, 486 cm−1, 534 cm−1, and 721 cm−1 can be assigned to the vibrations of Fe–OH and Fe–O bonds. In addition, the peaks of –OH groups46 are located at 2,436 cm−1 and 3,520 cm−1. Compared to pristine FeOOH, FeO(OH, S) presents extra characteristic peaks at 1113 cm−1 in the FTIR spectrum and at 422 cm−1 in the Raman spectrum, which correspond to S–O vibration47 and the Fe–S Tg mode,48 respectively. Similar to the XRD results, the intensity of the Fe–O and Fe–OH peaks observed in the FTIR and Raman spectra also decreases. And the reasons can be ascribed to two aspects: (i) the ethanol solvent thermal method limited the complete formation of FeOOH, resulting in the deletion of partial groups; (ii) the introduction of sulfur successfully replaced partial –OH groups in FeOOH after thiourea treatment.
As for the FeOOH-o and Mo–FeOOH samples synthesized by the oil-bath route, the XRD diagram (Fig. 1a2) reveals that the characteristic diffraction peaks of the FeOOH-o and Mo–FeOOH(5%) samples also match well with the tetragonal phase of FeOOH. Meanwhile, the peak intensity of the Mo–FeOOH sample decreases compared with the FeOOH-o sample because the introduction of Mo reduces the amount of Fe source supply and replaces partial Fe sites. The composition of the groups on the catalyst surface was also verified by FTIR and Raman spectra. It is found that the group peak positions of FeOOH-o and Mo–FeOOH(5%) are similar to the results of pristine FeOOH and FeO(OH, S) samples, including –OH groups, Fe–O and Fe–OH bonds. However, the peaks at 927 cm−1 in the FTIR spectrum (Fig. 1b2) and 827 cm−1 in the Raman spectrum (Fig. 1c2) are attributed to the formation of Mo–O bonds.49,50 Due to the partial replacement of Fe sites by Mo atoms, the intensity of the main group peaks of Mo–FeOOH(5%) in the FTIR and Raman spectra decreased, compared to the FeOOH-o sample.
To realize the microscopic morphology of the synthesized catalyst series, the relevant samples were characterized by scanning electron microscopy (SEM). In Fig. 2a and Fig. S2 (ESI†), the FeO(OH, S) sample exhibits a short nanorod structure with a length of about 100 nm, and EDS analysis shows that elemental species of Fe, O and S are uniformly distributed in the prepared samples with the mass fraction of sulfur being about 1.6 wt% (Fig. S3 and Table S1, ESI†). In addition, the lattice of FeO(OH, S) is further observed by high-resolution TEM (HRTEM). As shown in Fig. 2b, an interplanar distance of the FeO(OH, S) sample is measured to be 0.532 nm, corresponding to the (200) crystal plane. Using the same testing instruments, the FeOOH-o catalyst shows a nano-flower structure, while the morphology of the Mo–FeOOH catalyst changes from nano-flower to nano-plate with the increase of the relative Mo content, as shown in Fig. 2c and Fig. S4 (ESI†). In this study, the Mo–FeOOH(5%) catalyst is chosen as the main research catalyst and is directly named Mo–FeOOH. Then, the results of the Mo mass fraction in the Mo–FeOOH catalyst tested by EDS analysis and ICP are 16.60 wt% and 13.86 wt%, respectively (Fig. S5 and Table S2, ESI†). In Fig. 2d, the HRTEM image of Mo–FeOOH exhibits that the interplanar distance is about 0.7554 nm, which agrees well with the (110) plane of the crystal.
 | ||
| Fig. 2 (a) SEM image of the FeO(OH, S) catalyst and corresponding EDS mapping results; (b) HRTEM image of the FeO(OH, S) catalyst; (c) SEM image of the Mo–FeOOH(5%) catalyst and corresponding EDS mapping results; (d) HRTEM images of the Mo–FeOOH(5%) catalyst. | ||
X-Ray photoelectron spectroscopy (XPS) is a common technique to characterize the surface chemical state of samples. The XPS survey spectrum shown in Fig. S6(a) (ESI†) indicates that the FeO(OH, S) contains Fe and O elements, but the S 2p peaks are not observed due to the tiny amount of S content. In the Fe 2p spectrum of pristine FeOOH (Fig. 3a), two different peak signals centered at 710.6 and 724.3 eV are attributed to Fe 2p3/2 and Fe 2p1/2, respectively.41,51 In addition, there are two satellite peaks located at 719.1 and 733.1 eV. Compared with the pristine FeOOH, the signal peak positions of Fe 2p3/2 and Fe 2p1/2 in FeO(OH, S) exhibit a slight shift toward higher binding energy position, which stems from the Fe–S interaction. And the O 1s spectra (Fig. S6(b), ESI†) show three peak signals at 530.2, 531.6, and 533.6 eV, which can be ascribed as the lattice O and Fe–OH bonds,52,53 as well as the –OH groups attached to the catalyst surface due to the aqueous phase synthesis process. As for FeO(OH, S), the introduction of S atoms leads to a negative shift of the overall O 1s signal peaks, which results from the formation of the S–O bond. Fe–S and S–O bonds were further demonstrated by S 2p spectra, and as revealed in Fig. 3b, the intense signal peak at 168.1 eV is attributed to the S–O bonds, while the signal peaks at 163.8 eV and 162.5 eV can be assigned to S 2p1/2 and S 2p3/2, representing S2− and the formation of Fe–S bonds.54
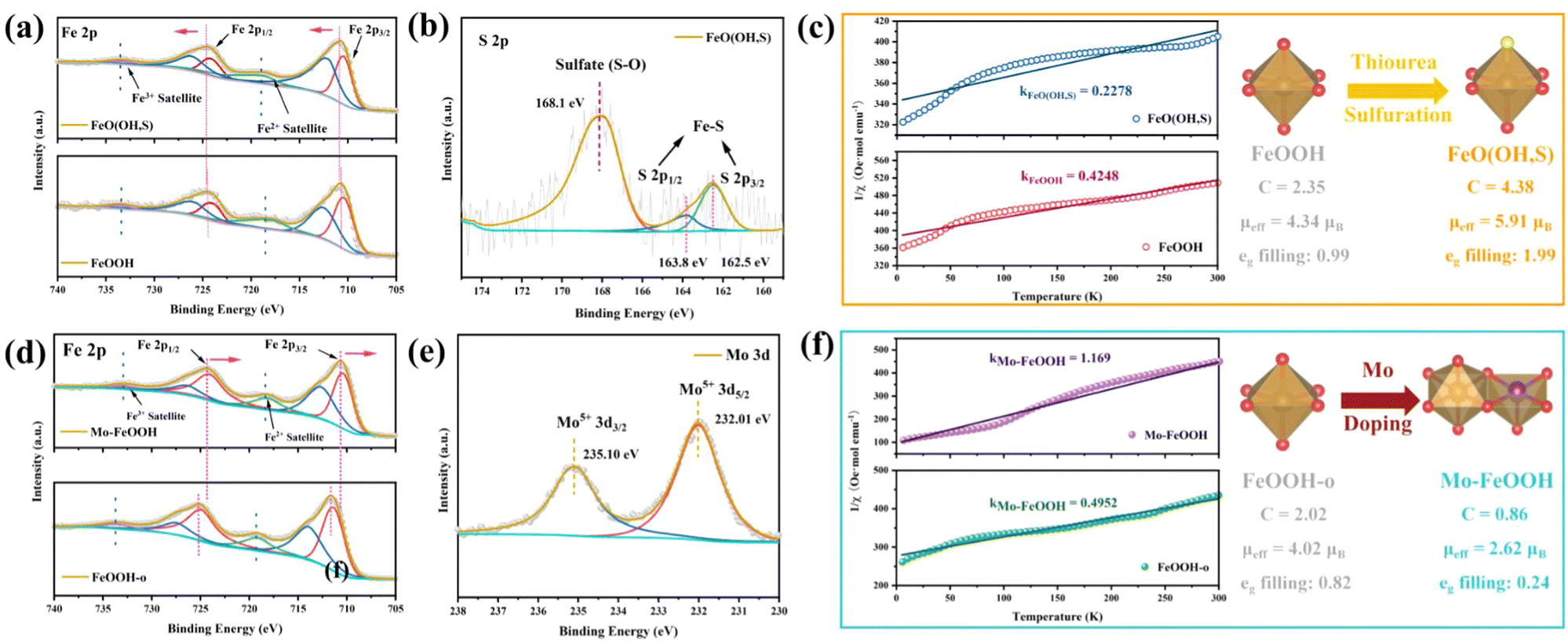 | ||
| Fig. 3 (a) High-resolution Fe 2p spectra of pristine FeOOH and FeO(OH, S) catalysts; (b) high-resolution S 2p spectra of the FeO(OH, S) catalyst; (c) temperature-dependent inverse susceptibility (1/χ) curves of pristine FeOOH and FeO(OH, S) catalysts; (d) high-resolution Fe 2p spectra of the FeOOH-o and Mo–FeOOH catalysts; (e) high-resolution Mo 3d spectra of the Mo–FeOOH catalyst; (f) temperature-dependent inverse susceptibility (1/χ) curves of the FeOOH-o and Mo–FeOOH catalysts. | ||
Focusing on the Mo–FeOOH sample, the presence of Fe, O, and Mo elements can be observed in the XPS survey spectrum in Fig. S7(a) (ESI†), which is in good agreement with the EDS results and proves the successful preparation of Mo–FeOOH. In the Fe 2p spectrum (Fig. 3d), two different peak signals centered at 711.6 and 725.2 eV are attributed to Fe 2p3/2 and Fe 2p1/2, respectively.55,56 Furthermore, the shift of the Fe 2p signal peaks towards negative positions in Mo–FeOOH is attributed to the electronic tuning effect of the Mo sites and the charge redistribution around Fe sites. In Fig. 3e, the 3d spectra of Mo show that the characteristic peaks at 232.01 eV and 235.1 eV belong to the 3d5/2 and 3d3/2 of Mo5+, further confirming the successful introduction of Mo atoms.57 The O 1s spectrum (Fig. S7(b), ESI†) shows two peaks at 529.8 eV and 531.5 eV for FeOOH-o, which can be attributed to the lattice O and Fe–OH bonds.58 Similarly, the O 1s signal peaks in the Mo–FeOOH are negatively shifted, which may be due to the formation of Mo–O bonds.
Based on the crystal field theory and the transition metal compound model, the electrostatic interaction between the central metal ion and the ligands causes the metal d orbitals to undergo energy level splitting. As for the octahedral field constructed by six ligands, five degenerate d orbitals are split into two high-energy eg orbitals and three low-energy t2g orbitals. The effective paramagnetic moment (μeff) is visual data to analyze the spin state of transition metal compounds and further understand the arrangement of orbital electrons. Therefore, to verify that the elemental doping strategy can modulate the spin state of the Fe sites for FeOOH-based materials, the ZFC-FC magnetic measurement technique was used to quantitatively evaluate the spin state of the relative catalysts: the magnetization (χ) of pristine FeOOH, FeO(OH, S), FeOOH-o and Mo–FeOOH samples was measured at a given external magnetic field (H = 500 Oe for pristine FeOOH and FeO(OH, S); H = 300 Oe for FeOOH-o and Mo–FeOOH), and then the inverse magnetization (1/χ) curves with temperature were plotted to fit the Curie–Weiss law (χ = C/(T − θ)) to obtain the Curie constant C, and further calculate the μeff to calculate the electron orbital arrangement.59
As shown in Fig. 3c, the μeff values for pristine FeOOH and FeO(OH, S) are 4.34μB and 5.91μB, respectively, indicating that the S doping successfully improves the spin state of Fe sites and the eg orbitals show an electron half-filled state (t2g3eg2). From a crystal field theory perspective, this phenomenon is attributed to the fact that weak field ligands, such as F−, S2− and so on, can reduce the orbital splitting energy (EΔ) of the central metal ion, thus contributing electrons to fill higher energy orbitals. Using similar curve fitting and calculation methods, the μeff values of FeOOH-o and Mo–FeOOH are obtained as 4.02μB and 2.62μB in Fig. 3f, respectively. The decreasing spin state of the Mo–FeOOH can be attributed to the electronic tuning of the Mo sites,32,60 indicating that the eg orbitals of the Fe sites in Mo–FeOOH are basically empty (t2g5eg0). Additionally, theoretical calculation can also determine changes of the spin state by the net spin-up state density. As shown in Fig. S30 (ESI†), compared with Fe sites (in FeO(OH, S)), the decrease of net spin up (Δspin-up) in Fe sites (in FeOOH or Mo–FeOOH) confirms the successful spin state transition from high spin state to intermediate and low spin state of the FeO6 octahedron. In conclusion, we have successfully synthesized FeO(OH, S) and Mo–FeOOH catalysts by elemental doping, and demonstrated the validity of this approach in regulating the spin state of the active center.
3.2. The electrocatalytic activity of the catalysts
The ammonia electrosynthesis activity of the samples in 0.1 M LiClO4 + 20% PEG electrolyte was evaluated in an H-shaped cell, which is separated by a Nafion 211 membrane and equipped with a three-electrode system. The feed gases (N2 and Ar) and the reactor must be rigorously pretreated prior to the experiments in order to reduce the influence of relevant impurities (NH3, NOx) on the measurement results. The main product ammonia was detected by the indophenol blue method, the standard curves of which are shown in Fig. S8(a and b) (ESI†). Initially, the linear sweep voltammetry (LSV) tests were used to analyze the potential of eNRR activities for FeO(OH, S) and Mo–FeOOH catalysts. As shown in Fig. 4(a), both FeO(OH, S) and Mo–FeOOH catalysts show an increase in current density under N2-saturated electrolyte compared to the LSV profile measured in Ar-saturated electrolyte, which indicates the occurrence of the eNRR process on the corresponding catalysts. Chronoamperometry measurements were then performed in the range of −0.4 to −0.8 V vs. RHE to quantify the ammonia yield rates and Faraday efficiencies at each reaction potential for FeO(OH, S) and Mo–FeOOH catalysts. According to Fig. 4(b and c), FeO(OH, S) and Mo–FeOOH catalysts exhibited the highest NH3 yield rate of 80.1 ± 4.0 and 86.8 ± 4.1 μg h−1 mgcat−1 at −0.6 V vs. RHE with FE of 36.9 ± 0.5% and 29.1 ± 0.8%, respectively. In addition, FeO(OH, S) and Mo–FeOOH catalysts also show better performance than that of pristine FeOOH and FeOOH-o catalysts shown in Fig. 4(d and e), which can be attributed to the larger electrochemical active surface (ECAS) and higher charge transfer efficiency shown in Fig. 4(h and i).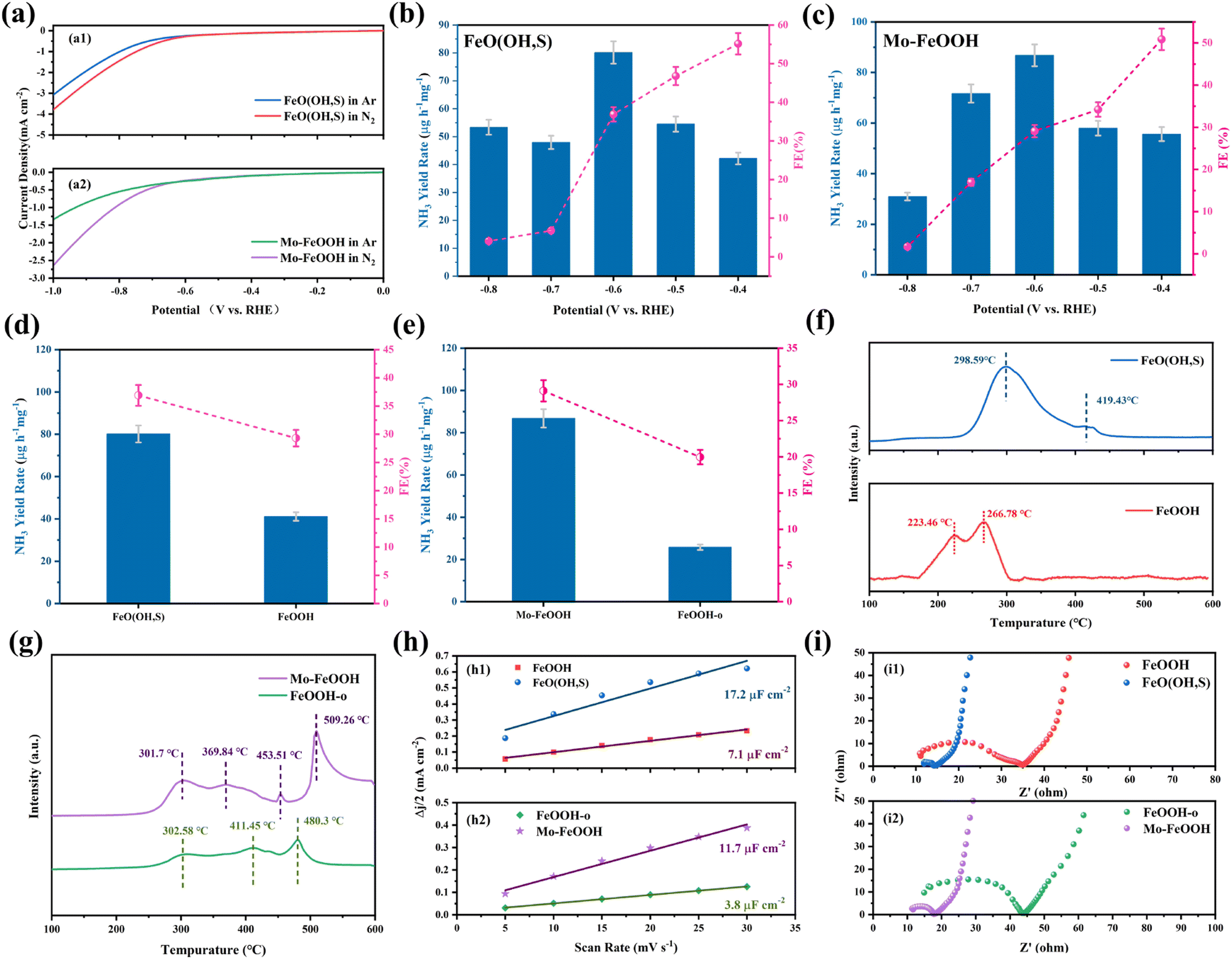 | ||
| Fig. 4 (a) The LSV of different FeOOH samples in N2 and Ar saturated electrolyte: (a1) FeO(OH, S) catalyst; (a2) Mo–FeOOH catalyst; (b) the ammonia yield rates and Faradaic efficiencies of the FeO(OH, S) catalyst at various potentials; (c) the ammonia yield rates and Faradaic efficiencies of the Mo–FeOOH catalyst at various potentials; (d) the ammonia yield rate and efficiency of the pristine FeOOH and FeO(OH, S) catalysts; (e) the ammonia yield rate and efficiency of the FeOOH-o and Mo–FeOOH catalysts; (f) N2-TPD of the pristine FeOOH and FeO(OH, S) catalysts; (g) N2-TPD of the FeOOH-o and Mo–FeOOH catalysts; (h) Δj/2 of different FeOOH samples plotted against various scan rates: (h1) pristine FeOOH and FeO(OH, S) catalysts; (h2) FeOOH-o and Mo–FeOOH catalysts; (i) Nyquist plots of electrochemical impedance spectra (EIS) of different FeOOH samples: (i1) pristine FeOOH and FeO(OH, S) catalysts; (i2) FeOOH-o and Mo–FeOOH catalysts. | ||
Based on the same methods, we again measured the ammonia electrosynthesis activities of FeO(OH, S) and Mo–FeOOH catalysts in 0.1 M LiClO4 electrolyte. Compared with the LSV curves and chronoamperometric curves of FeO(OH, S) in N2-saturated electrolytes in Fig. S14(a and b) (ESI†), the decreased current density with the introduction of PEG reflects that the HER is inhibited during the whole reaction. As shown in Fig. S14(c) (ESI†), it was observed that the FeO(OH, S) sample exhibited better eNRR activity under 20% PEG at −0.6 V vs. RHE than that without PEG, and the corresponding ammonia yield and FE were improved by 4.1 and 5.3 times. In Fig. S15 (ESI†), the Mo–FeOOH catalyst also showed similar performance differences in N2-saturated electrolyte with different content of PEG.
From another aspect, the temperature-programmed N2 desorption (N2-TPD) measurement can also be performed to demonstrate the nitrogen chemisorption capacities of the catalysts. Fig. 4f shows an obvious hierarchy with the desorption temperature following a sequence of FeO(OH, S) > pristine FeOOH, indicating that S doping would enhance N2 adsorption at Fe sites. However, Mo–FeOOH shows opposite properties in Fig. 4g. The temperature of the desorption peaks corresponding to FeOOH-o decreases, while a sharp peak appears at 509.3 °C, resulting from the introduction of Mo sites. Therefore, Mo doping would weaken the N2 adsorption on Fe sites, but provide new active centers to promote the eNRR process.
As for the catalytic stability over 10 h chronoamperometry shown in Fig. S16 and S17 (ESI†), the current densities of the FeO(OH, S) and Mo–FeOOH catalysts show significant fluctuations only at the initial stage but maintain good current stability for the rest of the time. In addition, after long-term electrolysis, the results of XRD, XPS, TEM or SEM show that the FeO(OH, S) and Mo–FeOOH catalysts still maintained a similar crystal structure, microscopic morphology and chemical state. However for the Mo–FeOOH catalyst, compared with the O 1s pattern before electrolysis, a new signal peak of –OH groups appears at 532.8 eV after the reaction, which can be attributed to the absorption of H2O on the catalyst surface after the aqueous phase reaction. In terms of experimental repeatability, four independent electrolysis experiments were carried out on FeO(OH, S) and Mo–FeOOH catalysts under the optimum reaction potential of −0.6 V vs. RHE and 0.1 M LiClO4 + 20% PEG electrolyte. The results of these repeated measurements show little variation of NH3 yield rate and selectivity in Fig. S18 and S19 (ESI†), proving that the related catalysts have stable and reliable performance in ammonia electrosynthesis under ambient conditions.
Meanwhile, the by-product N2H4 was detected by the Watt–Chrisp method at −0.6 V vs. RHE for FeO(OH, S) and Mo–FeOOH catalysts and it is found that almost no obvious peaks appear in the UV-Vis absorption spectrum in Fig. S23(a) and S24(a) (ESI†), which means little amounts of N2H4 were produced during the eNRR process. Furthermore, in order to avoid environmental contaminants from leading to false positive results, including NOx and impurity ammonia in the atmospheric environment, a series of comparison experiments were set up to confirm that the ammonia detected in the above experiments originated from the eNRR process of the FeO(OH, S) and Mo–FeOOH catalysts. As shown in Fig. S23(b) and S24(b) (ESI†), when the bare carbon cloth or the electrode loaded with catalysts were inserted into the N2-saturated electrolyte at open circuit potential (OCP), the electrolytes were detected by UV-Vis spectrophotometer after 2 h reaction and the peaks were found to be lower than the 0 ppm standard curve, which means that no impurity ammonia contaminated the catalyst, carbon cloth and reactor.
Moreover, when the catalyst was used as the working electrode, the signal peaks were significant only in the N2-saturated electrolytes and not in the Ar-saturated electrolytes at −0.6 V vs. RHE. These above phenomena suggest that the ammonia detected in this work was mainly produced by the catalysts in the N2-saturated electrolyte under a reasonable operating potential, and prove that the corresponding experimental results are accurate. In fact, the 1H NMR spectrum is more intuitive to verify the nitrogen source of the ammonia electrosynthesis. As expected in Fig. S27(a) and S28(a) (ESI†), no other 14NH3 peak signals were observed when 15N2 was used as a nitrogen source, suggesting that the N element in NH3 electrosynthesized in the eNRR process comes only from the feed gas. In addition, the 1H NMR spectrum is an auxiliary technique to confirm the accuracy of the UV-Vis spectra for ammonia detection. Also as shown in Fig. S27(b, c) and S28(b, c) (ESI†), the 14NH3 concentration detected by 1H NMR spectroscopy was basically in agreement with the results of UV-Vis absorption spectroscopy, illustrating the reliability of the above experimental methods.
3.3. DFT calculation
Density functional theory (DFT) calculations were utilized to directly observe the ammonia synthesis process for FeO(OH, S) and Mo–FeOOH. Herein, the XRD results indicate that all the synthesized FeOOH-based species belong to the tetragonal phase with the space group of I4/m, so this computational model (mp-1237867, Materials Project) was chosen as the original cell. Moreover, based on the results of EDS and ICP measurement, reasonable amounts of Mo and S atoms selectively replace the original Fe sites and hydroxyl groups in the FeOOH cell.Firstly, as for the FeO(OH, S) model, two possible sulfur-substituted sites are mainly considered in the FeOOH (400) crystal plane expansion cell, as shown in Fig. 5a. Based on the above two models, the adsorption energies of N2 on the Fe sites under the end-on and side-on configurations were calculated and compared in Fig. 5b, which shows that N2 prefers to be adsorbed on the Fe site under the end-on configuration (−0.271 eV) and generate obvious charge interaction (Fig. 5c), when S atoms replaced the internal –OH groups of FeOOH. Also compared with pristine FeOOH (−0.142 eV) in Fig. 5(d), FeO(OH, S-i) has stronger N2 adsorption capacity, which is consistent with the results of the N2-TPD tests. Finally, the Gibbs free energies were calculated for each basic reaction of the N2 reduction as shown in Fig. 5h. The rate-determining steps (RDS) for both hydrogenation pathways are the formation of *NNH with an energy barrier of 1.96 eV. In addition, the conversion of *NNH to *HNNH (0.12 eV) is easier than the conversion of *NNH to *NNH2 (0.8 eV), so the alternating hydrogenation mechanism is more advantageous for the whole hydrogenation process for FeO(OH, S). Similarly, in order to investigate the difference of N2 adsorption between Fe and Mo sites in Mo–FeOOH, the adsorption energy calculations show that N2 is more readily chemisorbed on the Mo sites with the end-on configuration (−0.656 eV) and on the Fe sites of Mo–FeOOH (−0.139 eV) and pristine FeOOH (−0.142 eV), as shown in Fig. 5(e–g). Based on the calculation of the Gibbs free energies in Fig. 5i, the RDS for the distal- and alter-hydrogenation pathways is still the formation of *NNH (1.11 eV). However, the conversion of *NNH to *NNH2 (−0.16 eV) has more negative free energy changes than the process from *NNH to *HNNH (0.35 eV), so the distal route is more likely to proceed.
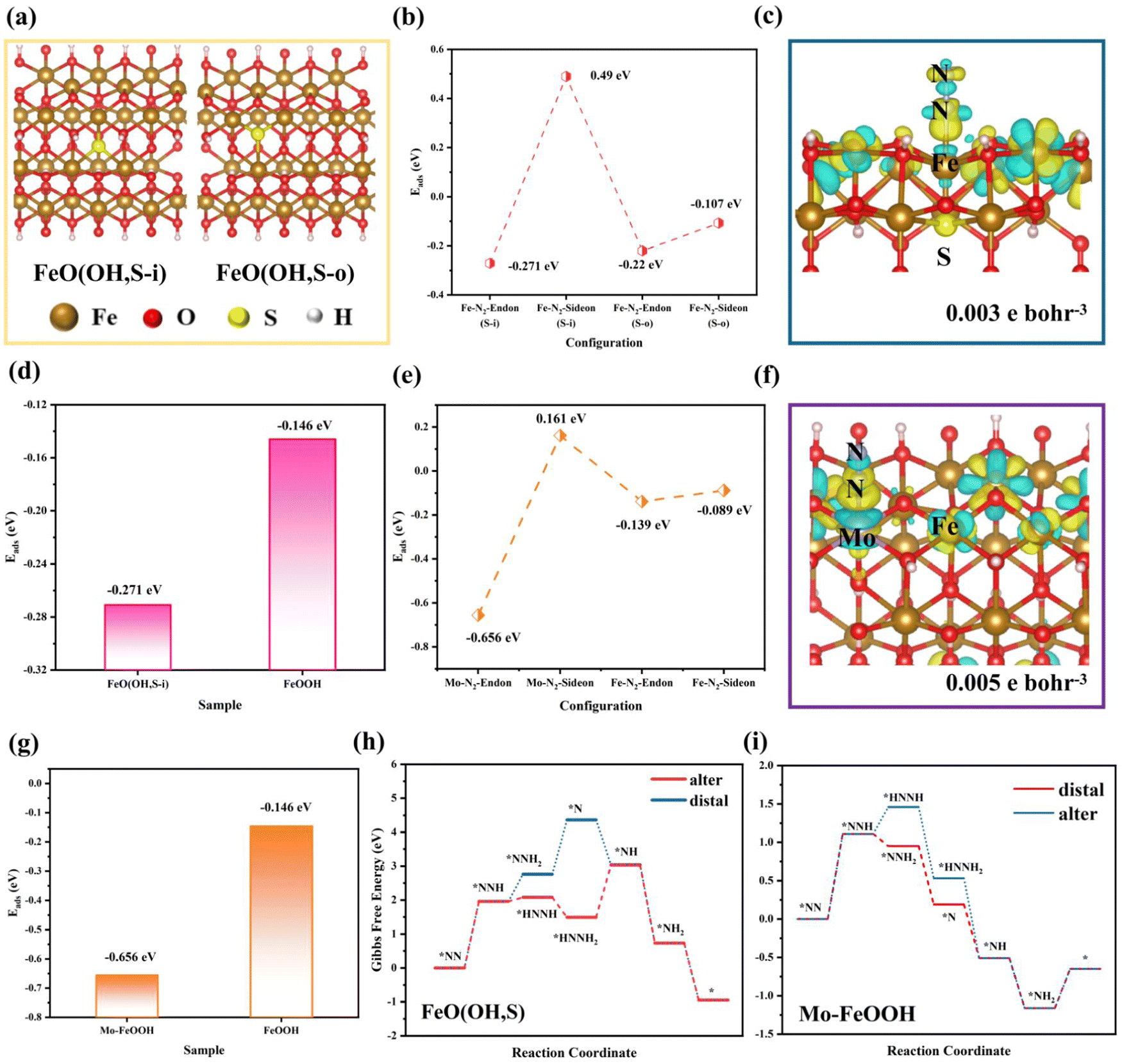 | ||
| Fig. 5 (a) Two sulfur coordination of FeO(OH, S) models; (b) the adsorption energy diagram for N2 chemisorbed on Fe sites of FeO(OH, S) with different sulfur coordination and adsorption configurations; (c) the charge density difference of N2 chemisorbed on the Fe site of FeO(OH, S-i) (the yellow and cyan color indicate electron accumulation and depletion, respectively); (d) the adsorption energy diagram for N2 chemisorbed on Fe sites of pristine FeOOH and FeO(OH, S-i); (e) the adsorption energy diagram for N2 chemisorbed on Fe and Mo sites of Mo–FeOOH with different adsorption configurations; (f) the charge density difference of N2 chemisorbed on the Mo site of Mo–FeOOH; (g) the adsorption energy diagram for N2 chemisorbed on the active sites of FeOOH-o and Mo–FeOOH; (h) the Gibbs energies of electrolytic ammonia production over FeO(OH, S-i); (i) the Gibbs energies of electrolytic ammonia production over Mo–FeOOH. | ||
Moreover, we continued to compare the Gibbs free energies for each basic reaction in pristine FeOOH and Mo–FeOOH (Fe as the main reactive center). As shown in Fig. S31(a) (ESI†), pristine FeOOH follows the alternating hydrogenation route for the whole hydrogenation process. Fortunately, Mo–FeOOH(Fe) in Fig. S31(b) (ESI†) also exhibits a similar reaction route, which offers a basis for comparison among FeOOH, FeO(OH, S) and Mo–FeOOH(Fe). In Fig. S32 (ESI†), the RDS is still the formation of *NNH for FeOOH, FeO(OH, S) and Mo–FeOOH(Fe) species. However, FeOOH presents the largest energy barrier (3.3 eV), which significantly inhibits its hydrogenation reaction. While for FeO(OH, S) and Mo–FeOOH(Fe), the formation energy barriers of *NNH are similar, and Mo–FeOOH(Fe) is more beneficial for the second hydrogenation to form *HNNH. In general, when Fe sites are chosen as the main reactive centers, Mo–FeOOH(Fe) is more conducive to the hydrogenation in the eNRR process.
Impressively, when N2 was fixed on the Fe sites with the end-on configuration, FeO(OH, S) exhibits the strongest adsorption capacity (−0.271 eV), which exceeds that of pristine FeOOH (−0.142 eV) and Mo–FeOOH (−0.139 eV), and is consistent with the changes of Fe spin states in Fig. 6a. In particular, based on the magnetic measurements of FeOOH, FeO(OH, S) and Mo–FeOOH, Fig. 6c illustrates that the Fe sites in FeOOH exhibit the medium spin state (t2g4eg1), while the Fe sites in FeO(OH, S) and Mo–FeOOH exhibit the high spin state (t2g3eg2) and low spin state (t2g5eg0), respectively. Based on the electronic “donation–backdonation” mechanism of transition metals activating N2, empty eg orbitals are more favorable to accept the σ orbital electrons from N2 during the first step of activation due to low electron repulsion,61,62 but half-occupied eg orbitals also have a weaker ability to accommodate these electrons. As shown in Fig. 6b, although the peak intensity of the d–σ orbital interaction of FeO(OH, S) with half-occupied eg orbitals is lower than that of Mo–FeOOH with empty eg orbitals, this electronic donation process for FeO(OH, S) still exists. However, the high filling state of the electrons in the eg orbitals is conducive to the second step of the d–π* feedback mechanism, which can promote the transfer of electrons to the anti-bond π* orbitals of N2 and enhance the activation of N2 in general. Therefore, there are almost no pronounced peaks at the d–π* orbital interaction below the Fermi level in the -pCOHP diagram for Mo–FeOOH due to the lack of isolated and active electrons in the eg orbitals.
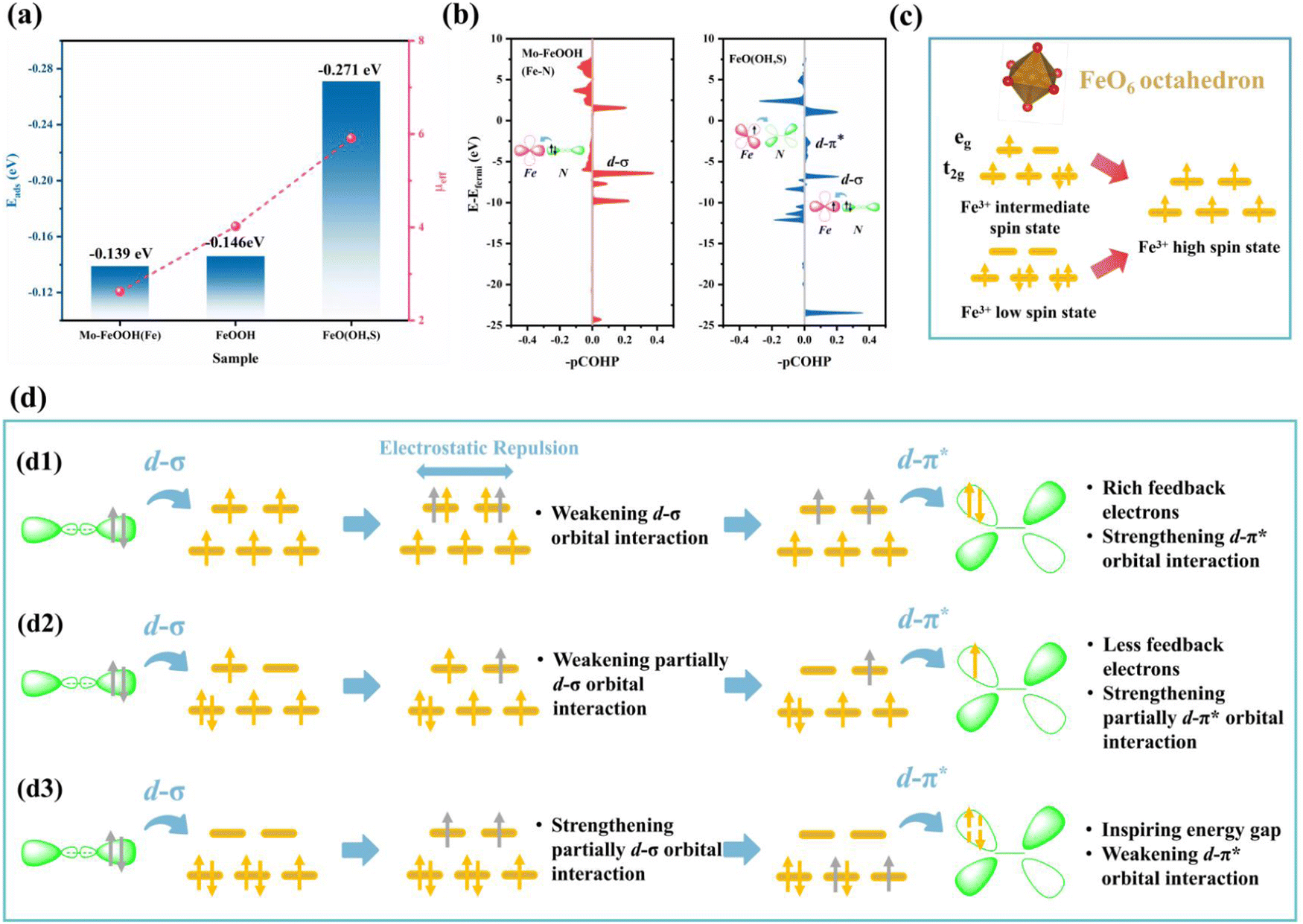 | ||
| Fig. 6 (a) Spin states and N2 adsorption energies of Fe sites for different FeOOH-based catalysts; (b) the –pCOHP of Fe–N interactions and the electron donation-acceptance process between Fe sites and N2 for FeO(OH, S) and Mo–FeOOH; (c) spin states and corresponding d-orbital electronic structures of Fe sites; (d) the possible mechanisms of the spin state effect for the N2 activation process. | ||
In order to reveal the electronic interaction mechanism in-depth, in Fig. 6d, we speculate that a high spin effect exists in FeOOH-based electrocatalysts, which can enhance the N2 adsorption and activation ability of Fe sites, and the driving force may be the internal electrostatic repulsion. Based on the above experimental and calculation results, for FeO(OH, S)(d1) when the electrons from the σ orbitals of N2 transfer to the high-energy eg orbitals, although the electron repulsion would weaken the d–σ orbital interaction, this driving force also causes the electrons originally occupying the eg orbitals to transfer to the anti-bonding π* orbitals of N2. The remaining σ electrons would then restore the electronic structure of the Fe sites to the initial state. In contrast, FeOOH(d2) with the medium spin state (t2g4eg1) can only contribute one electron to promote d-π* orbital interaction, while Mo–FeOOH(d3) needs to excite electrons in lower-energy t2g orbitals to realize d–π* interaction, which may be more difficult and explains the differences in the N2 adsorption energy among the three catalysts.
4. Conclusion
Herein, FeO(OH, S) and Mo–FeOOH catalysts with different spin states were designed by the elemental doping method. With PEG inhibiting the HER kinetics, both FeO(OH, S) and Mo–FeOOH catalysts could achieve superior NH3 yields at −0.6 V vs. RHE of 80.1 ± 4.0 μg h−1 mgcat−1 (FE 36.9 ± 0.5%) and 86.8 ± 4.1 μg h−1 mgcat−1 (FE 29.1 ± 0.8%), respectively, in 20% PEG + 0.1 M LiClO4. Furthermore, the theoretical calculations showed that the high spin state FeO(OH, S) catalyst exhibited the most negative N2 adsorption energy when Fe sites served as the main active center for N2 fixation, which actually originated from the fact that high-spin Fe3+ has a half-occupied eg orbital filling state to facilitate the electronic “donor-backdonation” activation mechanism with molecular orbitals of N2. In addition, low-spin Fe can accelerate the hydrogenation reaction in general. In other words, this article comprehensively discussed the potential role of the spin state or electronic orbital modulation on the N2 activation process around FeOOH-based electrocatalysts by element doping, which would be beneficial to improve the eNRR performance. In addition, relative strategies and conclusions may provide positive guidance for the electronic structure design of other TM-based electrocatalysts in the eNRR research field.Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.Author contributions
Haifan Wang: writing original script, experiment design, characterization analysis, and theoretical calculation; Menglei Yuan, Jingxian Zhang, Yiling Bai and Ke Zhang: data collection and characterization analysis; Bin Li: draft review, editing and funding acquisition; Guangjin Zhang: supervision, experiment design, draft review, and editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (no. 22178361; 22378402; 52302310), the International Partnership Project of CAS (039GJHZ2022029GC), the National Key R&D Program of China (no. 2020YFA0710200), the foundation of the Innovation Academy for Green Manufacture Institute, Chinese Academy of Sciences, under grand no. IAGM2022D07, Qin Chuangyuan Cites High-level Innovation and Entrepreneurship Talent Programs (QCYRCXM-2022-335), the Fundamental Research Funds for the Central Universities (G2022KY05111) and the Open Project Program of Anhui Province International Research Center on Advanced Building Materials (grant no. JZCL2303KF).References
- B. Sun, S. Lu, Y. Qian, X. Zhang and J. Tian, Carbon Energy, 2023, 5, e305 CrossRef CAS.
- S. Shang, W. Xiong, C. Yang, B. Johannessen, R. Liu, H.-Y. Hsu, Q. Gu, M. K. H. Leung and J. Shang, ACS Nano, 2021, 15, 9670–9678 CrossRef CAS PubMed.
- R. Guan, X. Cheng, Y. Chen, Z. Wu, Z. Zhao, Q. Shang, Y. Sun and Z. Sun, Nano Res., 2023, 16, 10770–10778 CrossRef CAS.
- Y. Zhang, L. Guo, Y. Wang, T. Wang, T. Ma, Z. Zhang, D. Wang, B. Xu and F. Fu, J. Mater. Sci. Technol., 2022, 110, 152–160 CrossRef CAS.
- Y. Zhang, J. Zhao, D. Yang, B. Wang, Y. Zhou, J. Wang, H. Chen, T. Mei, S. Ye and J. Qu, Nat. Chem., 2022, 14, 46–52 CrossRef CAS PubMed.
- Q. Liu, T. Xu, Y. Luo, Q. Kong, T. Li, S. Lu, A. A. Alshehri, K. A. Alzahrani and X. Sun, Curr. Opin. Electrochem., 2021, 29, 100766 CrossRef CAS.
- C. Du, C. Qiu, Z. Fang, P. Li, Y. Gao, J. Wang and W. Chen, Nano Energy, 2022, 92, 106784 CrossRef CAS.
- P. Shen, X. Li, Y. Luo, Y. Guo, X. Zhao and K. Chu, ACS Nano, 2022, 16, 7915–7925 CrossRef CAS PubMed.
- L. Wen, K. Sun, X. Liu, W. Yang, L. Li and H.-L. Jiang, Adv. Mater., 2023, 35, 2210669 CrossRef CAS.
- Y. Guo, J. Gu, R. Zhang, S. Zhang, Z. Li, Y. Zhao, Z. Huang, J. Fan, Z. Chen and C. Zhi, Adv. Energy Mater., 2021, 11, 2101699 CrossRef CAS.
- J. Xie, Z. Liang and Y.-C. Lu, Nat. Mater., 2020, 19, 1006–1011 CrossRef CAS.
- Y. Zhao, S. Zhang, C. Han, Q. Lu, Q. Fu, H. Jiang, L. Yang, Y. Xing, Q. Zheng, J. Shen, L. Yan and X. Zhao, Chem. Eng. J., 2023, 468, 143517 CrossRef CAS.
- Y. Kong, L. Wu, X. Yang, Y. Li, S. Zheng, B. Yang, Z. Li, Q. Zhang, S. Zhou, L. Lei, G. Wu and Y. Hou, Adv. Funct. Mater., 2022, 32, 2205409 CrossRef CAS.
- N. Q. Tran, X. Liu, Y. Cho, L. T. Duy, L. Zheng, J. Yu, S. Ajmal, X. Shao, J. Lee and H. Lee, J. Mater. Chem. A, 2022, 10, 8432–8439 RSC.
- J. Yu, X. Ren, J. Lu, H. Bai, X. Wang, J. Hu and H. Huang, J. Alloys Compd., 2022, 902, 163862 CrossRef CAS.
- F. Bai, X. Qu, C. Li, S. Liu, J. Sun, X. Chen and W. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 28033–28043 CrossRef CAS.
- M. Yuan, Q. Li, J. Zhang, J. Wu, T. Zhao, Z. Liu, L. Zhou, H. He, B. Li and G. Zhang, Adv. Funct. Mater., 2020, 30, 2004208 CrossRef CAS.
- H. Fei, R. Liu, J. Wang, T. Guo, Z. Wu, D. Wang and F. Liu, Adv. Funct. Mater., 2023, 2302501 CrossRef CAS.
- L. Li, W. Yu, W. Gong, H. Wang, C.-L. Chiang, Y. Lin, J. Zhao, L. Zhang, J.-M. Lee and G. Zou, Appl. Catal., B, 2023, 321, 122038 CrossRef CAS.
- L. Zhao, Y. Xiong, X. Wang, R. Zhao, X. Chi, Y. Zhou, H. Wang, Z. Yang and Y.-M. Yan, Small, 2022, 18, 2106939 CrossRef CAS PubMed.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Wang, S. Li, H. He and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10910–10918 CrossRef CAS PubMed.
- H.-j Chen, Z.-q Xu, S. Sun, Y. Luo, Q. Liu, M. S. Hamdy, Z.-s Feng, X. Sun and Y. Wang, Inorg. Chem. Front., 2022, 9, 4608–4613 RSC.
- Y. Sun, Y. Han, X. Zhang, W. Cai, Y. Zhang, Y. Zhang, Z. Li, B. Li, J. Lai and L. Wang, Appl. Catal., B, 2022, 319, 121933 CrossRef CAS.
- B. Jiang, Y. Guo, F. Sun, S. Wang, Y. Kang, X. Xu, J. Zhao, J. You, M. Eguchi, Y. Yamauchi and H. Li, ACS Nano, 2023, 17, 13017–13043 CrossRef CAS PubMed.
- B. Jiang, H. Xue, P. Wang, H. Du, Y. Kang, J. Zhao, S. Wang, W. Zhou, Z. Bian, H. Li, J. Henzie and Y. Yamauchi, J. Am. Chem. Soc., 2023, 145, 6079–6086 CrossRef CAS.
- H. Li, M. Xia, B. Chong, H. Xiao, B. Zhang, B. Lin, B. Yang and G. Yang, ACS Catal., 2022, 12, 10361–10372 CrossRef CAS.
- X. Lv, W. Wei, H. Wang, F. Li, B. Huang, Y. Dai and T. Jacob, J. Mater. Chem. A, 2020, 8, 20047–20053 RSC.
- Z. Wen, H. Lv and X. Wu, ACS Appl. Mater. Interfaces, 2022, 14, 52079–52086 CrossRef CAS.
- H. Wang, J. Qi, N. Yang, W. Cui, J. Wang, Q. Li, Q. Zhang, X. Yu, L. Gu, J. Li, R. Yu, K. Huang, S. Song, S. Feng and D. Wang, Angew. Chem., Int. Ed., 2020, 59, 19691–19695 CrossRef CAS PubMed.
- J. Zhang, S. Geng, R. Li, X. Zhang, Y. Zhou, T. Yu, Y. Wang, S. Song and Z. Shao, Chem. Eng. J., 2021, 420, 130492 CrossRef CAS.
- G. Song, R. Gao, Z. Zhao, Y. Zhang, H. Tan, H. Li, D. Wang, Z. Sun and M. Feng, Appl. Catal., B, 2022, 301, 120809 CrossRef CAS.
- Y. Wang, W. Cheng, P. Yuan, G. Yang, S. Mu, J. Liang, H. Xia, K. Guo, M. Liu, S. Zhao, G. Qu, B.-A. Lu, Y. Hu, J. Hu and J.-N. Zhang, Adv. Sci., 2021, 8, 2102915 CrossRef CAS PubMed.
- J. Ran, L. Wang, M. Si, X. Liang and D. Gao, Small, 2023, 19, 2206367 CrossRef CAS.
- Y. Li, Y. Ji, Y. Zhao, J. Chen, S. Zheng, X. Sang, B. Yang, Z. Li, L. Lei, Z. Wen, X. Feng and Y. Hou, Adv. Mater., 2022, 34, 2202240 CrossRef CAS.
- C. Yang, W. Zhong, K. Shen, Q. Zhang, R. Zhao, H. Xiang, J. Wu, X. Li and N. Yang, Adv. Energy Mater., 2022, 12, 2200077 CrossRef CAS.
- X. Chen, Q. Wang, Y. Cheng, H. Xing, J. Li, X. Zhu, L. Ma, Y. Li and D. Liu, Adv. Funct. Mater., 2022, 32, 2112674 CrossRef CAS.
- X. Yin, R. Cai, X. Dai, F. Nie, Y. Gan, Y. Ye, Z. Ren, Y. Liu, B. Wu, Y. Cao and X. Zhang, J. Mater. Chem. A, 2022, 10, 11386–11393 RSC.
- J.-Q. Lv, X. Chen, Y. Chang, Y.-G. Li and H.-Y. Zang, ACS Appl. Mater. Interfaces, 2022, 14, 52877–52885 CrossRef CAS PubMed.
- J. Lu, Z. Wang, Y. Guo, Z. Jin, G. Cao, J. Qiu, F. Lian, A. Wang and W. Wang, Energy Storage Mater., 2022, 47, 561–568 CrossRef.
- B. C. Park, J. Cho, J. Zhang, M. Amedzo-Adore, D. B. Lee, S.-C. Kim, J. S. Bae, Y. R. Uhm, S.-O. Kim, J. Koo, Y.-M. Kang and Y. K. Kim, J. Mater. Chem. A, 2022, 10, 17740–17751 RSC.
- X. Zhu, Z. Liu, Q. Liu, Y. Luo, X. Shi, A. M. Asiri, Y. Wu and X. Sun, Chem. Commun., 2018, 54, 11332–11335 RSC.
- X. Zhu, Z. Liu, H. Wang, R. Zhao, H. Chen, T. Wang, F. Wang, Y. Luo, Y. Wu and X. Sun, Chem. Commun., 2019, 55, 3987–3990 RSC.
- J. Tan, X. He, F. Yin, X. Liang, G. Li and Z. Li, Appl. Surf. Sci., 2021, 567, 150801 CrossRef CAS.
- Y. Ren, C. Yu, X. Han, X. Tan, Q. Wei, W. Li, Y. Han, L. Yang and J. Qiu, ACS Energy Lett., 2021, 6, 3844–3850 CrossRef CAS.
- X. Wei, Q. Liu, H. Zhang, Z. Lu, J. Liu, R. Chen, R. Li, Z. Li, P. Liu and J. Wang, Dalton Trans., 2017, 46, 15746–15756 RSC.
- M. R. Chowdhury, V. Fester, G. Kale and O. Cespedes, J. Nanopart. Res., 2014, 16, 2412 CrossRef.
- Y. Sun, M. Danish, M. Ali, A. Shan, M. Li, Y. Lyu, Z. Qiu, Q. Sui, X. Zang and S. Lyu, Chem. Eng. J., 2020, 394, 124830 CrossRef CAS.
- J. Sun, C. Liu, W. Kong, J. Liu, L. Ma, S. Li and Y. Xu, J. Mater. Sci. Technol., 2022, 110, 161–166 CrossRef CAS.
- A. D. Hadigavabar, K. Tabatabaeian, M. A. Zanjanchi and M. Mamaghani, React. Kinet., Mech. Catal., 2018, 124, 857–871 CrossRef CAS.
- M. Isaac, N. Santha and V. U. Nayar, J. Raman Spectrosc., 1991, 22, 237–239 CrossRef CAS.
- X. Chen, X. Shi, M. Cheng, M. Zhang, J. Ma, Z. Liang, X. Zhai and Y. Du, CrystEngComm, 2023, 25, 1599–1607 RSC.
- L. Zhang, H. Li, B. Yang, N. Han, Y. Wang, Z. Zhang, Y. Zhou, D. Chen and Y. Gao, J. Solid State Electrochem., 2020, 24, 905–914 CrossRef CAS.
- X. Zhang, Y. Liu, S. Dong, Z. Ye and Y. Wei, J. Alloys Compd., 2018, 744, 507–515 CrossRef CAS.
- R. Guo, Y. He, T. Yu, P. Cheng, J. You, H. Lin, C.-T. Chen, T. Chan, X. Liu and Z. Hu, Chem. Eng. J., 2021, 420, 127587 CrossRef CAS.
- F. N. I. Sari, H.-S. Chen, A. K. Anbalagan, Y.-J. Huang, S.-C. Haw, J.-M. Chen, C.-H. Lee, Y.-H. Su and J.-M. Ting, Chem. Eng. J., 2022, 438, 135515 CrossRef CAS.
- H. Gwon, S. Park, Q. Lu, H. J. Choi and S. Lee, J. Ind. Eng. Chem., 2023, 124, 279–286 CrossRef CAS.
- M. Kawase, K. Akaike, K. Aoyama, Y. Ito, M. Tamura and K. Kanai, Appl. Catal., B, 2020, 273, 119068 CrossRef CAS.
- J. Hu, S. Li, J. Chu, S. Niu, J. Wang, Y. Du, Z. Li, X. Han and P. Xu, ACS Catal., 2019, 9, 10705–10711 CrossRef CAS.
- G. Shen, R. Zhang, L. Pan, F. Hou, Y. Zhao, Z. Shen, W. Mi, C. Shi, Q. Wang, X. Zhang and J.-J. Zou, Angew. Chem., Int. Ed., 2020, 59, 2313–2317 CrossRef CAS PubMed.
- Y. Zhang, X. Wang, T. Liu, Q. Dang, L. Zhu, Y. Luo, J. Jiang and S. Tang, J. Mater. Chem. A, 2022, 10, 23704–23711 RSC.
- M. Yuan, J. Chen, Y. Xu, R. Liu, T. Zhao, J. Zhang, Z. Ren, Z. Liu, C. Streb, H. He, C. Yang, S. Zhang and G. Zhang, Energy Environ. Sci., 2021, 14, 6605–6615 RSC.
- M. Yuan, J. Chen, H. Zhang, Q. Li, L. Zhou, C. Yang, R. Liu, Z. Liu, S. Zhang and G. Zhang, Energy Environ. Sci., 2022, 15, 2084–2095 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00208j |
| This journal is © The Royal Society of Chemistry 2024 |