
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding charge carrier dynamics in organic photocatalysts for hydrogen evolution
Jinhyuk
Choi†
a,
Wooteak
Jung†
a,
Soranyel
Gonzalez-Carrero
 b,
James R.
Durrant
b,
Hyojung
Cha
*c and
Taiho
Park
*a
b,
James R.
Durrant
b,
Hyojung
Cha
*c and
Taiho
Park
*a
aDepartment of Chemical Engineering, Pohang University of Science and Technology (POSTECH), 77 Cheongam-Ro, Nam-Gu, Pohang, Gyeongbuk 37673, Republic of Korea. E-mail: taihopark@postech.ac.kr
bDepartment of Chemistry and Centre for Processable Electronics, Imperial College London, London, W12 0BZ, UK
cDepartment of Hydrogen and Renewable Energy, Kyungpook National University, Daegu 41566, Republic of Korea. E-mail: hcha@knu.ac.kr
First published on 20th July 2024
Abstract
Hydrogen generation using particulate photocatalysts is a promising approach to achieving sustainable energy solutions to energy shortages and environmental challenges. Among them, organic semiconductor-based photocatalysts offer significant advantages, including tunable structural and optoelectronic properties, easy of processibility, and cost-effectiveness over the inorganic photocatalysts. However, their photocatalytic efficiency is limited by inherent properties, such as the formation of Frenkel excitons, the presence of numerous energetic defects, and low charge separation efficiency. To enhance performance and expand the potential of organic photocatalysts, it is crucial to understand the relationship between molecular structure and charge carrier dynamics in photocatalytic processes. In this reason, Recent research about organic photocatalysis for hydrogen evolution has focused on uncovering limiting factors and comprehending the fundamental charge carrier behaviors that determine the performance of organic photocatalysts by utilizing the use of advanced time-resolved analysis tools. This review desicribes charge behaviors, including photocharge generation and transport within the bulk of the organic photocatalyst, as well as charge transfer and charge-induced redox reactions at the interface within photocatalytic system. Their characteristics, according to the molecular structures, are summarized based on time-resolved analysis methodologies, including transient spectroscopy with the aim to provide an understanding of the correlation between charge carrier dynamics, molecular structure, and photocatalytic performance.
Broader contextHydrogen production via water splitting using particulate photocatalysts has garnered significant interest as a renewable energy. Initially, TiO2 was implicated in hydrogen generation, leading to the development of various inorganic semiconductors. Despite notable increases in efficiency, their large bandgap characteristics and harsh synthetic process have posed practical obstacles. Consequently, recent research efforts have shifted towards the development of organic semiconductor-based photocatalysts with advantages of tunable electronic structure, favorable process. Furthermore, their potential as a tool for studying the relationship between structure, properties, and performance in photocatalytic processes is being increasingly recognized, accentuating the current necessity for a fundamental approach to organic photocatalysis. To give comprehensive insight about organic photocatalyst, this review focuses on the charge flows in conjugated organic photocatalytic system and clarify the relation to molecular structures. We believe that the systematic investigation of the structure – charge dynamic properties – performance correlation will help boost molecular design strategies to develop organic photocatalysts. |
1. Introduction
Increasing concerns regarding environmental pollution and the depletion of energy resources are predominantly attributed to the surge in carbon emissions and the growing demand for energy.1 Consequently, the development of renewable and sustainable energy sources has become imperative to mitigate dependence on fossil fuels. Solar energy, the primary source of natural energy, has emerged as a vital solution to address this global challenge.2,3 Intensive research efforts are focussed on harnessing distributed solar energy and converting it into usable energy forms, with solar cells serving as a prototypical example of a device that converts sunlight into electrical energy.4–7 Recent decades have witnessed significant progress in solar cell technology, leveraging insights from materials and device physics. Nonetheless, a fundamental challenge remains: solar cells do not directly store the electrical energy they produce, necessitating further investment in storage and distribution infrastructures. This limitation has garnered interest in solar-to-fuel conversion, particularly in the storage of solar energy as chemical energy in molecular fuels8,9 such as hydrogen. This advancement highlights the role of green hydrogen as a crucial energy vector and a chemical intermediate in the shift towards more sustainable energy systems.10Three main technologies facilitate hydrogen generation through solar water splitting: photovoltaic (PV) electrolysis,11,12 photoelectrocatalysis (PEC)13,14 and photocatalysis15,16 (Fig. 1). Among these technologies, PV electrolysis demonstrates the highest solar-to-hydrogen conversion efficiency. However, despite its efficiency, the complexity of its device structure, which combines PV cells17,18 with electrocatalysis, and the high costs associated with electrolysers, restrict its viability as an alternative to fossil fuels. In the PEC approach, the separation and transport of photogenerated holes and electrons to the photocathode and photoanode, respectively, can be enhanced by an external bias that promotes reduction and oxidation reactions. Both PEC and photocatalysis are considered economically viable methods for hydrogen production, largely due to their low projected costs. The Department of Energy (DOE) has set a target production cost of $1 per kilogram of hydrogen to be considered practical.19 Particulate photocatalysts, with their straightforward device architecture, namely dispersion in water, present a significant opportunity for cost reduction. Therefore, photocatalytic systems are strong candidates for fulfilling the criteria for commercial viability. The photocatalytic water-splitting reaction was first discovered in 1972 by Akira and Kenichi Honda, marking a significant milestone in demonstrating hydrogen generation from water using a TiO2 photoelectrode.20 This seminal research laid the foundation for subsequent advancements in the field. Since then, researchers have primarily investigated inorganic semiconductor-based materials as potential photocatalysts for water-splitting, with metal oxides and chalcogenides emerging as promising candidates. Among these, various forms of TiO2, including N-doped TiO221 and TiO2 hybrids,22 in conjunction with different organic semiconductors, such as covalent organic frameworks, have been developed to enhance the catalytic performance through nanoparticle dispersion and junction formation.
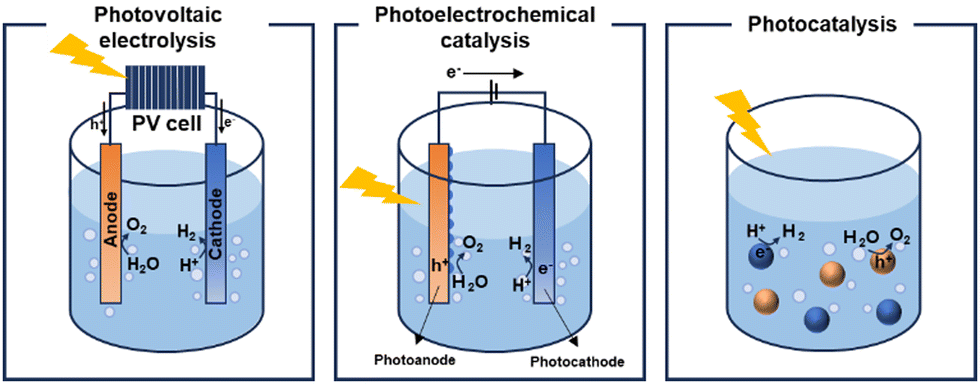 | ||
| Fig. 1 Schematic illustration of the solar-driven water-splitting system. PV electrolysis (left), photoelectrochemical catalysis (middle), and photocatalysis (right). | ||
Recent achievements include a notable breakthrough by a group led by Domen, who reported a CdTe quantum dot and vanadium-doped indium sulfide (CdTe/V-In2S3) photocatalyst with an internal quantum efficiency exceeding 100% and an apparent quantum yield (AQY) of 73%.23 Furthermore, an aluminium-doped strontium titanate (SrTiO3:Al) photocatalyst incorporating Rh/Cr2O3 and CoOOH cocatalysts, demonstrated nearly 100% AQY at a wavelength of 350 nm.24 The AQY metric reflects the intrinsic ability of a photocatalyst to convert incident photons into molecular fuel, calculated as: the number of electrons used for H2 generation/the number of incident photons. However, despite the availability of low-cost inorganic photocatalysts with high AQYs, their application is limited to the UV region, which constitutes only 4.4% of the solar spectrum due to their wide bandgaps. Consequently, research efforts have increasingly focused on reducing the band gaps of semiconductors, marking a crucial trend in the pursuit of high-performance photocatalysts. Although recent advancements have successfully tailored inorganic materials for improved efficiencies in visible-light activation, their practical implementation remains hindered by the use of expensive materials and complex fabrication processes.25
From a thermodynamic perspective, metal–oxide semiconductors, commonly used as photocatalysts, feature a valence band primarily composed of oxygen 2p orbitals. They offer a high ionisation potential (IP) beneficial for the oxygen evolution reaction (OER) by providing sufficient potential to overcome kinetic limitations.26 However, these inorganic semiconductors encounter challenges in the photocatalytic hydrogen evolution reaction (HER) at longer wavelengths. For example, anatase TiO2,27,28 a well-known inorganic photocatalyst for hydrogen evolution, possesses a conduction band edge that is sufficiently negative to facilitate proton reduction. However, it is limited to wavelengths below 400 nm due to its wide band gap (3.2 eV).
The exploration of organic photocatalysts has addressed the issue of band gap tuning and provided alternatives to high-temperature processing and structural rigidity. In contrast, organic semiconductors face a considerable challenge in increasing their IP beyond 1.23 eV to facilitate the OER, which often results in diminished OER activity.29 Nonetheless, carbon-based organic materials generally exhibit lower electron affinities (EAs) and higher lowest unoccupied molecular orbitals (LUMOs) than metal oxides. Moreover, adjusting the LUMO energy level above the proton reduction potential in organic semiconductors is more achievable than in metal oxides owing to their flexible compositional and structural modifications. Consequently, conjugated organic semiconductor-based photocatalysts have been developed by leveraging their tuneable optical properties to enhance the HER. Besides the diverse optoelectronic properties of organic materials, their tuneable morphologies (ranging from one- to three-dimensional porous structures), coupled with solvent processability, make them compelling candidates for photocatalysis. Based on these features, current research efforts aim to elucidate the mechanisms and explore the relationship among the structure, properties, and performance of conjugated organic photocatalysts using systematic molecular design strategies.
The integration of organic semiconductors into photocatalysis began with the synthesis of poly(p-phenylene) (PPP) in 1985,30 leading to the exploration of various organic photocatalysts, including carbon nitrides,31,32 covalent triazine frameworks,33,34 conjugated organic frameworks,35,36 and conjugated microporous polymers.37,38 However, challenges such as limited processability and less-defined structures have hindered precise adjustments of their structure and properties. Recently, there has been a shift in focus towards linear conjugated polymers, primarily due to their tuneable optoelectronic properties that enable absorption across a broad solar spectrum, including the visible region (approximately 44% of the entire solar spectrum).39 The inherent structural diversity and tunability of these polymers provide a systematic approach for designing photocatalysts, facilitating a better understanding of the correlation between the molecular structure and photocatalytic performance. Substantial progress has been made in the development of linearly conjugated polymer-based photocatalysts that exhibit remarkable hydrogen generation activities at wavelengths greater than 600 nm. However, despite these advancements, a comprehensive understanding of the hydrogen reduction process in organic photocatalysts remains lacking. This poses a crucial barrier to further enhancing their performance and widespread application in photocatalysis. For instance, the roles of cocatalysts, even in trace amounts, and the intimate interactions of the photocatalyst with water are recognised as critical factors in the photocatalytic proton reduction process.
To date, most studies have relied on sacrificial electron donors with the goal of eventually substituting these donors for economically-viable oxidation reactions or reversible couples for integration into tandem devices. However, few studies have explored these aspects in relation to the molecular structure of photocatalysts while considering charge dynamics in the photocatalytic process.40 Therefore, investigating the relationship between molecular structure and charge dynamics within the components of conjugated organic semiconductor-based photocatalytic systems is essential for designing optimised molecular structures that promote efficient photocatalysis.
In this review, we focus on elucidating the fundamental mechanisms of photocatalytic hydrogen generation and charge carrier dynamics at each stage of the process. Drawing from recent progress in photocatalytic material development, our objective was to enhance the understanding of how molecular structure influences photocatalytic efficiency. By investigating notable examples, we aimed to clarify the intricate interplay between these factors. Additionally, we discussed the inherent challenges in material development and the practical constraints that will guide future advancements in high-performance water-splitting photocatalysts.
2. Basic principle of photocatalytic water splitting
Water splitting consists of two half reactions: the OER (which oxidizes H2O to produce O2) and the HER (which reduces protons to H2).41| OER: 2H2O → O2 + 4H+ + 4e− |
| HER: 4H+ + 4e− → 2H2 |
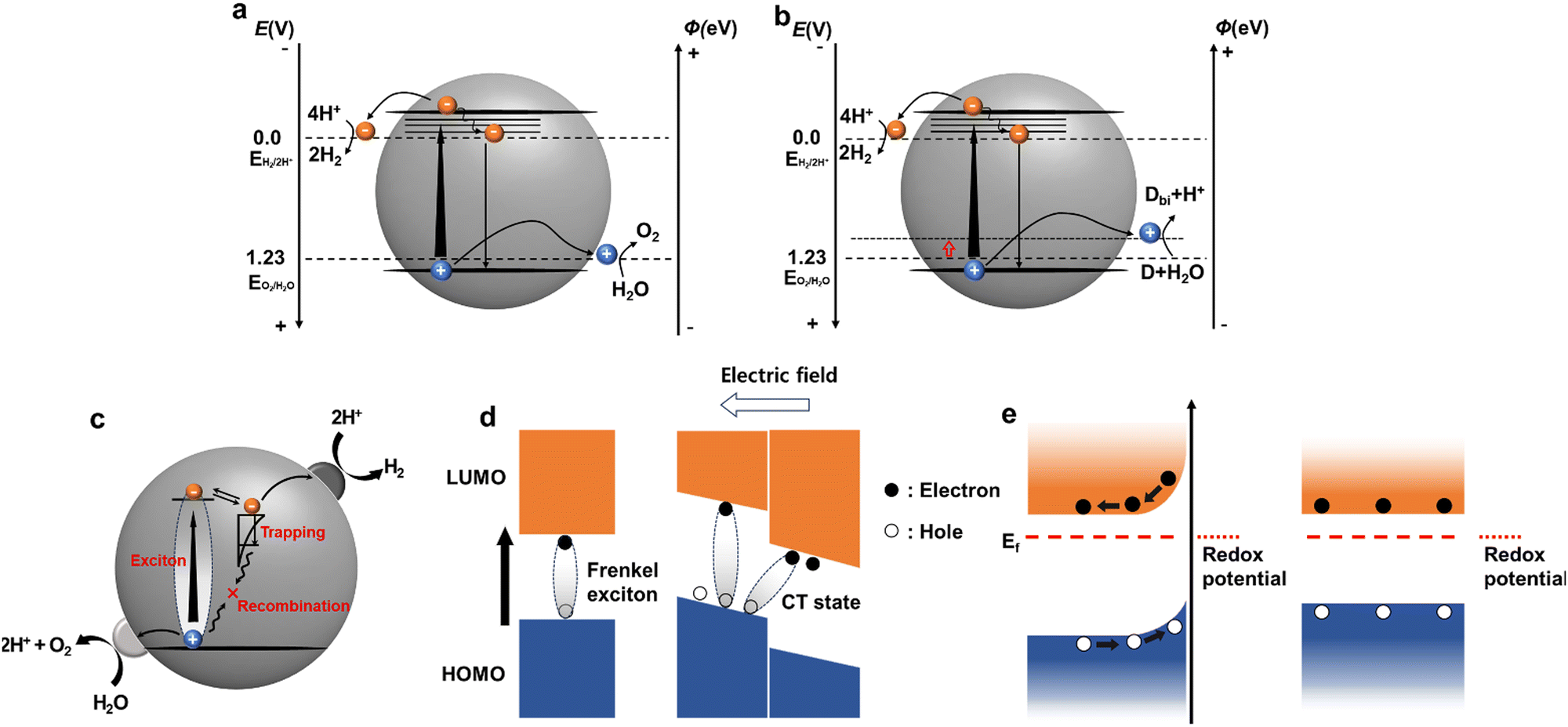 | ||
| Fig. 2 Energy and potential diagrams of (a) photocatalytic overall water splitting and (b) half hydrogen reduction with a sacrificial donor (denoted as D). (Φ = −eE (vs. NHE) −4.44 eV, e is an elementary charge). Dbi means the biproduct in oxidation reaction of compound D. (c) Schematic of the photocatalytic redox reaction processes. (d) Exciton photogeneration at a single domain and heterojunction. (e) Band structures at different interface conditions. | ||
However, the overall water splitting (OWS) process is thermodynamically uphill (requiring 237 kJ mol−1). In particular, the OER, which involves the transfer of four electrons, requires a significant overpotential, rendering the process both thermodynamically and kinetically demanding. Photocatalysis is a complex process that includes charge recombination and trapping (Fig. 2c).42 The photocatalytic proton-reduction process generally involves three critical steps: (1) light absorption and exciton generation, (2) exciton diffusion and charge transport to photocatalytically active sites, and (3) charge transfer to the reactant for photocatalytic redox reactions. During that process charge trapping and recombination are also involved (Fig. 2c).42 Hence, for catalytic reactions within a photocatalytic system to proceed efficiently, the photogenerated charges must reach the active sites at the photocatalyst/electrolyte interface within their decay lifetimes. Unlike inorganic photocatalysts, the exact mechanism by which organic photocatalysts produce hydrogen has not been conclusively identified. These mechanisms continue to be investigated using various approaches that consider the intrinsic properties of materials. In this section, we examine the step-by-step mechanism of hydrogen production using molecular and systemically-modified organic photocatalysts.
2.1. Light absorption and exciton generation
The effective conversion of solar energy into chemical energy requires effective light absorption across a broad spectral range. Given that a substantial portion of the solar spectrum lies within the visible range of 400–750 nm, materials with broad absorption in this region are essential for achieving efficient hydrogen generation. The absorption properties of materials are determined by their band gap, which represents the energy difference between the highest occupied molecular orbital and the LUMO levels, which are intrinsic properties of the materials. The optoelectronic properties of organic semiconductors are intimately linked to their conjugated systems and molecular packing structures, allowing for the facile manipulation of frontier orbital energy levels and band gaps through adjustments in the building blocks or molecular packing.The first linear conjugated polymer applied in the photocatalytic HER was PPP.30 PPP exhibited modest photocatalytic efficiency with an AQY of 0.04% under irradiation at λ > 290 nm and low H2-production efficiency at λ > 400 nm due to the band gap of PPP (2.76 eV). Subsequently, PPP derivatives, such as polypyridine with nitrogen heteroatoms, showed slightly enhanced photocatalytic activity in the visible-light region.43 Notably, significant process was achieved by copolymerising a dibenzo[b,d]thiophene sulfone unit with a benzene unit, narrowing the band gap to 2.70 eV, resulting in a remarkable HER of 3680 μmol h−1 g−1. However, sulfone-based polymers exhibit low solubilities in organic solvents such as chloroform, tetrahydrofuran and chlorobenzene owing to the high polarity of the backbone without sidechains. In addition to the sulfone unit, the benzothiadiazole (BT) unit has emerged as an effective building block for photocatalysts because of its high solubility. This highlights the ongoing exploration of materials with improved photocatalytic performances.
In organic semiconducting polymers, light absorption and subsequent excitation do not immediately yield free charge carriers.44 Instead, Frenkel excitons are formed, where electrons and holes are coulombically bound together, with binding energies typically ranging from 100 meV to 1 eV, exceeding the thermal energy at room temperature (Fig. 2d).45 This behaviour is attributed to the lower dielectric constant (2–4) of organic semiconductor materials compared to the higher dielectric constant of inorganic semiconductors (such as 11.7 for Si), which inhibits the formation of free charges in organic semiconductor-based photocatalysts. Moreover, the reorganisation energy resulting from structural relaxation (electron–phonon coupling) leads to the creation of localised polarons—electrons and holes associated with the polarisation of the surrounding molecular structure—further hindering charge transport. Consequently, the availability of active electrons for reduction reactions are limited, thereby reducing the photocatalytic performance.46 Furthermore the soft properties of organic semiconductors introduce several competitive pathways, including charge trapping and exciton/charge recombination, at the surface or within the bulk.47 This poses significant challenges in promoting exciton and charge dissociation while minimising competing recombination processes.
2.2. Exciton diffusion and charge transport to photocatalytic active sites
The exciton diffusion length represents the maximum distance that an exciton can travel before decaying to its ground state. In organic semiconductors, the efficient dissociation of photogenerated excitons into free charges depends on the interface within this diffusion length, despite potential recombination pathways. However, the inherent structural disorder in organic semiconductors typically limits the exciton diffusion length to 5–10 nm, significantly shorter than the light absorption depth (100–200 nm). This spatial discrepancy restricts the conversion of photogenerated excitons into free charges mostly to the near-surface or interface regions, forming what is termed an ‘exciton dead zone’.48 Strategies to enhance the efficiency of hydrogen generation from proton reduction in the reaction medium include the use of high-dipole sidechains, such as ethylene glycol sidechains, which stabilise electron polarons, as discussed later. Additionally, introducing a highly planar structure into a polymer can reduce the binding energy of excitons, thereby modulating the charge-transfer pathway and facilitating electron transfer within the polymeric backbone.49Effective charge separation is crucial for producing photocatalytically active free charges. The separation efficiency is fundamentally determined by charge dynamics, which play a pivotal role in the performance of organic semiconductor-based optoelectronics. When a photoinduced exciton reaches the surface of an organic photocatalyst without decay, a charge transfer process occurs between the photocatalyst and sacrificial donor or cocatalyst. These processes, known as reductive and oxidative quenching, are instrumental in separating the excitons and generating free charges. Reductive quenching involves the photocatalyst accepting an electron from a sacrificial donor, leading to the oxidation of the donor. Conversely, oxidative quenching occurs when electrons are transferred to the cocatalyst or reactant, resulting in the oxidation of the photocatalyst itself.50,51
Once free charges are formed, they migrate to the active site at the reaction centre based on their energy levels. Unlike inorganic materials, where atoms are rigidly bonded in lattice structures via covalent bonds, organic semiconductors feature molecular packing predominantly governed by weaker van der Waals interactions, including π–π stacking. In particular, conjugated polymers display flexible chains with varying conformations and lengths introducing dynamic disorder that affects charge transport through thermal fluctuations. Furthermore, static disorder arising from the polycrystalline and amorphous morphologies of organic semiconductors complicates the application of the Drude model, typically used for inorganic materials,52 to describe charge-carrier transport. This energetic disorder creates tail states within the density of states, trapping photogenerated charges in lower-energy states. Charges near the band edge may transition into transport states via thermal energy, undergoing successive trapping and detrapping processes. Organic semiconductors feature energetically distributed trap states throughout deep within band gaps. Filling these deep trap states can lead to trap-assisted recombination sites, further impeding charge transport.53,54 Due to this energetic discontinuity, charge transport in organic semiconductors occurs primarily through a thermally-assisted hopping mechanism rather than band-like transport.55,56
2.3. Charge transfer to the reactants for the photocatalytic redox reaction
Upon reaching active sites with adequate potentials, photogenerated charge carriers initiate photocatalytic reactions at the interface between the semiconductor and aqueous solution, leading to the production of the desired products. The process of charge transfer between the semiconductor and electrolyte plays a critical role in determining the rate and direction of photocatalytic reactions. In inorganic semiconductors, interaction with the electrolyte results in the formation of a space charge layer, driven by the energy difference between the Fermi level of the semiconductor and the redox potential of the electrolyte. This energy discrepancy induces band bending at the surface, generating a built-in electric field that facilitates charge-carrier separation and transport.57–60 Conversely, organic semiconductors exhibit reduced charging effects because of their low doping levels and charge density, resulting in a diminished driving force compared to their inorganic counterparts (Fig. 2e). Organic materials present specific challenges in accurately characterising the electronic states at their surfaces. Factors such as voids and the penetrable nature of organic semiconductors can obscure defining surface states, leading to ambiguities in characterisation. To mitigate these challenges, organic polymer catalysts often require metallic cocatalysts (such as Pd residues in the Suzuki–Miyaura coupling) to participate in reaction systems. For example, Pt can enhance the hydrogen evolution rate of polymer catalysts through in situ photodeposition by illuminating dispersed particles in an aqueous solution of metal precursors or during synthesis. The role and significance of cocatalysts in enhancing charge transfer during the photocatalytic process are further discussed in the following section. Ensuring close contact between the photocatalyst and cocatalyst is crucial for efficient charge transfer. For instance, Pt precursors, such as K2PtCl6, typically exist as PtCl62− with negative charges in aqueous phases. Therefore, photocatalysts with a net positive surface charge are preferred for effective interaction with Pt cocatalysts.58 Hu et al. demonstrated that a conjugated polyelectrolyte tethering an ammonium cation exhibited enhanced electron transfer to a Pt cocatalyst, which contrasted with the results achieved for the sulfonate-containing polyelectrolyte.61Despite these complexities, it is evident that the interface between the organic photocatalyst and aqueous solution plays a crucial role in photocatalytic reactions. Furthermore, the solution environment, particularly redox reactions involving sacrificial agents, significantly influences the process. In the following section, we discuss these factors and their implications for organic photocatalysis, with a focus on polymers. Specifically, we investigate the impact of these factors on charge dynamics during the photocatalytic process, as this directly affects the overall photocatalytic performance.
3. Experimental observation of charge-carrier dynamics in a photocatalytic system
Understanding the dynamics of charge carriers in photocatalytic systems is crucial for addressing theoretical challenges and improving the performance. For film-type photoelectrodes, photodiodes, and PVs, X-ray-based technologies, such as X-ray scattering and X-ray photoelectron spectroscopy are widely employed to analyse the fine structure of devices62 or to study the changes in the atomic electronic environment.63,64 However, analysing organic photocatalysts using X-ray techniques is challenging due to their non-uniformly dispersed particle nature. Moreover, the analysis conditions for organic photocatalysts considerably differ from their actual operating environments, as discussed later in this section. Consequently, analysing the internal and interface structures of organic photocatalysts is not straightforward, prompting researchers to explore operating mechanisms through charge dynamics analysis. This approach leverages the high light absorption properties of organic semiconductors, which has become a major research focus.Photocatalytic processes involve complex charge transfer, energy transfer, and transport mechanisms that are particularly pronounced in organic semiconductor-based photocatalysts. These materials may involve additional pathways, such as geminate and non-geminate recombination, intermediate charge transfer, and diverse trapping mechanisms.65 These processes occur over a broad range of timescales, ranging from femtoseconds to milliseconds, covering light absorption, charge generation, and subsequent photocatalytic reactions. To drive catalytic redox reactions with photogenerated charges effectively, the charge-carrier lifetime must exceed the timescale of the interfacial photocatalytic reaction. Charge recombination and annihilation at various sites act as barriers to charge-carrier movement within the photocatalyst, thereby limiting the photocatalytic efficiency. Therefore, understanding the mechanisms and dynamics of charge carriers is crucial for the rational design of photocatalyst materials.
Typically, the analysis of charge dynamics employs time-resolved techniques to observe the transient responses of samples as a function of the delay time following excitation with a pulsed laser. Time-resolved spectroscopic methods, such as time-resolved photoluminescence (TRPL), transient absorption spectroscopy (TAS), and photoinduced absorption spectroscopy (PIAS), play a pivotal role in investigating charge dynamic behaviour in photocatalytic processes and identifying the key factors influencing photocatalytic performance (Fig. 3).
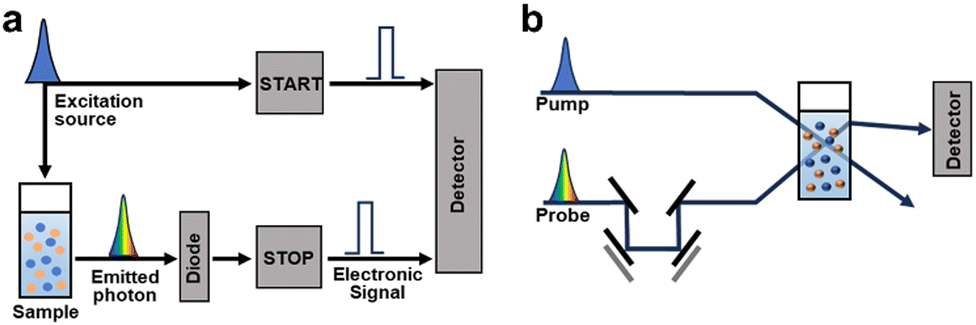 | ||
| Fig. 3 Schematic illustration of the instrument of (a) TCSPC and (b) TAS. A time delay (Δt) between the pump and probe pulses is made by moving mirrors. | ||
Given the extensive range of materials addressing an analysis using time-resolved spectroscopic techniques, this section provides a brief introduction to these methods.66 When semiconductors are exposed to electromagnetic radiation with energies surpassing their band gap, they absorb the radiation. This excites the electrons from their ground state to higher energy states, creating holes in the ground states. Then, photoexcited materials undergo various decay pathways that dissipate energy into their surroundings. If the photoexcited electrons and holes recombine radiatively, they emit photons in a process known as photoluminescence (PL). TRPL traces this PL decay by observing the charge-carrier behaviour in the emission state and measuring the radiative decay over time following excitation with a monochromatic pulsed laser. Time-correlated single-photon counting (TCSPC) is frequently employed in TRPL measurements (Fig. 3a). In this method, an MHz pulsed laser excites the sample, generating an electronic signal.67 The emitted photons from the PL of the sample halt the electronic signal, enabling the instrument to record each emitted single photon and its corresponding delay time repeatedly. By analysing these decay times, TRPL sheds light into charge and energy transfer processes, which are influenced by the chemical structure, solvent environment, and interfacial interactions of the chromophore.
Conversely, TAS examines the transient absorption of excited states, providing kinetic data on: exciton states, charge transfer states (which are partially bound exciton states at donor–acceptor (D–A) interfaces), and polaron states. Therefore, it offers valuable insights into the exciton diffusion lengths and dynamics of intermediate states during photocatalytic reactions that make it a useful tool for mechanistic studies. TAS experiments involve two successive laser pulses: a pump pulse to excite the sample and a probe pulse to induce the absorption of the excited state after a time delay ranging from femtoseconds to microseconds, depending on the measurement objective (Fig. 3b). TAS employs a pump pulse with a peak intensity significantly higher than that of standard solar irradiation (AM 1.5G, 100 mW cm−2). This is to ensure a sufficient concentration of excited states for detectable transient absorption signals. However, comparing the photogenerated charge dynamics under TAS measurement conditions with those under actual photocatalytic conditions is challenging. TAS data capture the absorption difference between the absorbance caused by the probe pulse following the pump pulse and steady-state absorbance in the absence of the pump pulse, measured across various times and wavelengths.
The wavelength-dependent transient absorption spectrum provides critical insights into the dynamics of charge carriers and excited states in photocatalytic systems. This spectrum is characterised by both negative and positive absorption signals. The negative signal is caused by ground-state bleaching (GSB), which results from the depletion of the ground-state population following photoexcitation, with stimulated emission contributing to this negative signal. Conversely, the positive absorption signal corresponds to excited state absorption (ESA), which is induced by various processes, including singlet and triplet exciton states, and absorption by charge-separate species.68 The absorption kinetics can be observed by plotting the variations in absorbance as a function of the delay time at specific wavelengths. These kinetics can be modelled using different functions for analytical purposes. Extensive discussions on the interpretation of TAS data are available in several reviews, providing a comprehensive understanding of this subject matter.69,70
Given that photo-redox reactions on the surface of photocatalysts unfold over longer periods than charge dynamics within the bulk, it is crucial to examine charge behaviours over millisecond to second timescales. PIAS is a pump–probe technique that employs a longer, lower-energy pump pulse compared to TAS (which typically spans a few seconds with a continuous probe). This method facilitates the detection of accumulated charges in a quasi-steady state and aids in identifying bottlenecks within the photocatalytic process.71,72 The subsequent discussion will delve into the development of linear polymer photocatalysts for sacrificial hydrogen evolution and strategies based on the charge dynamics at each step of the photocatalytic process (as elucidated by these sophisticated spectroscopic analysis tools) (Scheme 1).
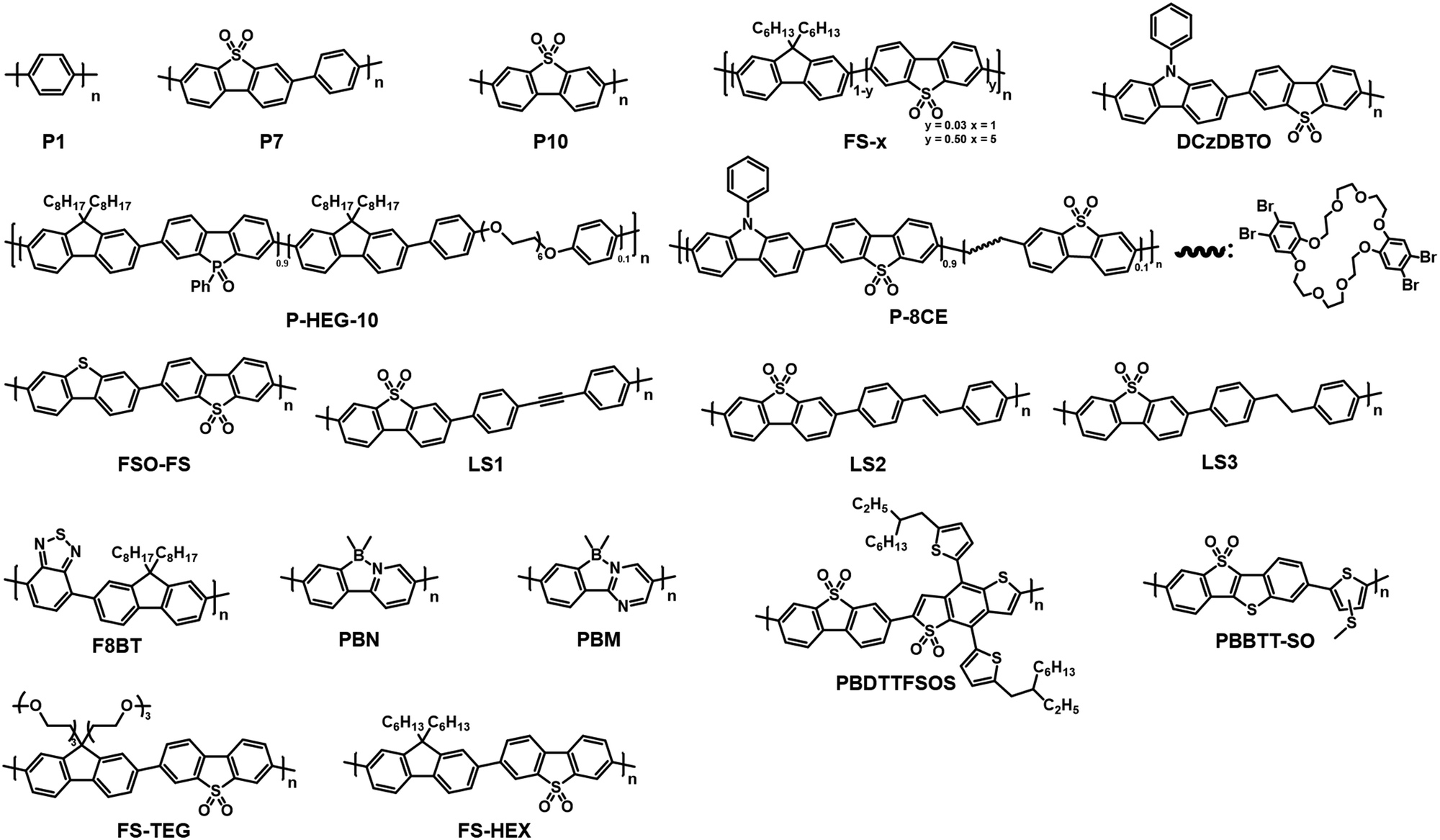 | ||
| Scheme 1 Molecular structures of linear polymeric photocatalysts. | ||
4. Case studies on charge-carrier dynamics in organic photocatalysis systems
4.1. Backbone design and its charge carrier dynamics
As mentioned previously, the structure of the polymers used for hydrogen production lags behind that of organic solar cells. Therefore, understanding the mechanisms of charge generation and hydrogen production is crucial for developing polymers with diverse backbone structures for efficient solar fuel production. One of the earliest studies focused on a sulfone-based polymer copolymerised with a benzene unit, which exhibited an enhanced HER performance. This enhancement was attributed to the efficient charge separation and transport facilitated by the extended conjugation and planarization of the polymer backbone.73 Moreover, the sulfone groups increase the local dipole moment in the polymer backbone, enabling facile charge transfer both kinetically and thermodynamically. In addition to charge generation and transport in the bulk of photocatalysts, the hydrophilicity of the polymer backbone significantly affects charge dynamics at the semiconductor/electrolyte interface by regulating interactions with the surrounding environment. Cooper et al. investigated these effects by comparing polymer photocatalysts with varying sulfone unit proportions (P1, P7, and P10).40 Their results indicated that the HER efficiency increased with higher sulfone contents. The corresponding femtosecond TAS (fs-TAS) data obtained under photocatalytic conditions (Fig. 4a) revealed that the negative polarons produced by reductive quenching with TEA play a significant role in the photocatalytic process. Notably, the intensity of the electron polaron peak at 630 nm increased in the order: P1, P7, and P10. The absorption kinetics at 630 nm showed rapid polaron formation within 100 ps, highlighting fast reductive quenching by TEA (Fig. 4b). Moreover, over a relatively long timescale from microseconds to milliseconds, the polaron absorption dynamics at 630 nm revealed a higher peak intensity for P10 than for P7 (Fig. 4c). This observation suggests that the polar sulfone group contributes to the generation of long-lived and active charges. Based on these analyses, combined with theoretical calculations, the authors demonstrated that the enhanced permeability of polar sulfone to water facilitated efficient polaron generation by providing a driving force for hole transfer to TEA. This study emphasises the significance of the surrounding environment in photocatalytic hydrogen generation, highlighting that the design of the polymer backbone and its interaction with the environment are critical factors for optimising photocatalytic performance.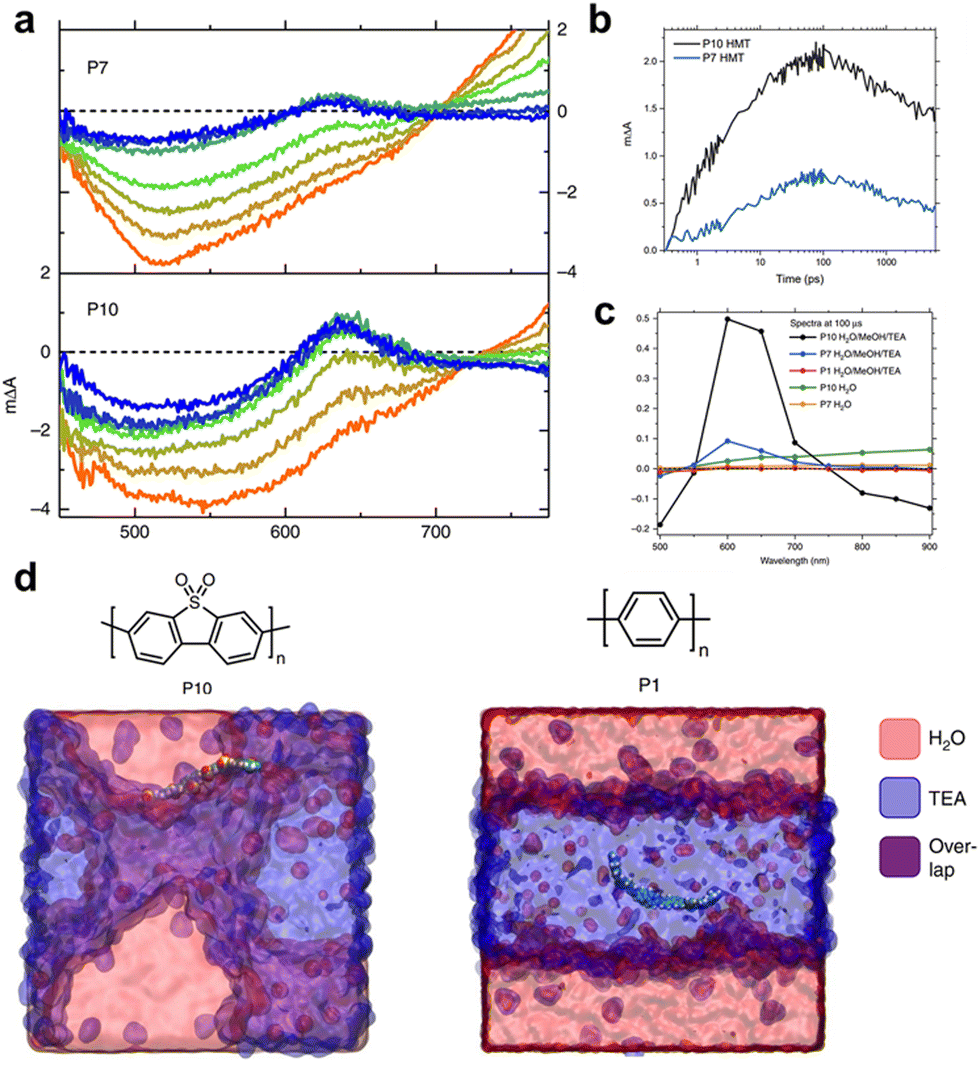 | ||
| Fig. 4 (a) Transient absorption spectra of P7 and P10 suspensions in a solvent mixture of equal volumes of H2O, MeOH, and TEA on a time scale of fs–ns. (b) Transient absorption kinetics at time scale of ps–ns. (c) Transient absorption spectra probed at 633 nm at 100 μs. All spectra data were obtained at an excitation wavelength of 355 nm and fluence of 0.08 mJ cm−2 for (a), (b) and 0.32 mJ cm−2 for (c). (d) Atomistic molecular dynamics snapshots of polar P10 and nonpolar P1. While P10 resides at the interface between water and TEA, P1 is embedded in the TEA phase. Reproduce with permission.40 Copyright 2018, Springer Nature. | ||
Recently, Hillman et al. investigated solution-processable polymers (FS1–FS5) with varying ratios of sulfone and 9,9-di-n-hexyl-fluorene monomers. They aimed to precisely analyse the effect of the sulfone content on the photocatalytic process (Fig. 5).74 Under identical photocatalytic conditions involving H2O/TEA/MeOH, increasing the sulfone content positively impacted the HER, which was consistent with the results reported in previous studies. Besides enhancing light absorption ability, the polar sulfone group also influences the solvent environment surrounding the photocatalysts. To quantify this effect on the local environment, molecular dynamic simulations and experiments were employed to analyse an influence of surrounding conditions and permittivities of the polymers (Fig. 5a). Remarkably, the polar FS5 exhibited a relatively large permittivity of 7.1, indicating a high water content in its vicinity. And the excited excitonic hole and electron (EA* and IP*) of polymers was stabilised as a permittivity of the surrounding solvent increases (conditions to be surrounded by more water molecules) (Fig. 5b). Using density functional theory (DFT) calculations, the authors estimated the feasibility of the photocatalyst and the oxidation potential as functions of the solvent permittivity of the polymer. Based on the DFT results, they identified the threshold permittivity constant at which the excited hole potential (EA*) of the polymer exceeded the oxidation potential of TEA, as depicted in Fig. 5c. Employing fs-TAS measurements to probe the formation of free electrons, the positive absorption peak intensities corresponding to singlet excitons above 620 nm for FS5 and 700 nm for FS1 were observed to increase within 1 ps after excitation (Fig. 5d). Deconvolution of the transient absorption kinetics at 600 nm revealed distinctive features of FS5, including a rapid rising signal within 1 ps, which corresponded to an intermediate excited state, and a peak after 5 ps, which was attributed to the formation of an electron polaron (Fig. 5e). This latter feature was not observed for FS1, indicating that only FS5 possessed the driving force for the TEA-mediated reductive quenching. Conversely, the photocatalyst with a higher sulfone content showcased an accelerated polaron decay in the presence of the Pd cocatalyst, indicating that sulfone facilitates charge extraction to Pd (Fig. 5f). Notably, a linear correlation was established between the intensity at 600 nm on a microsecond timescale and HER, suggesting that the ability of sulfone-containing photocatalysts to generate hydrogen is limited by free electron generation. These results indicate that the local solvent environment influences both the EA* of the photocatalyst and oxidation potential of TEA, which alters the thermodynamics of the system and is directly related to the photocatalytic performance.
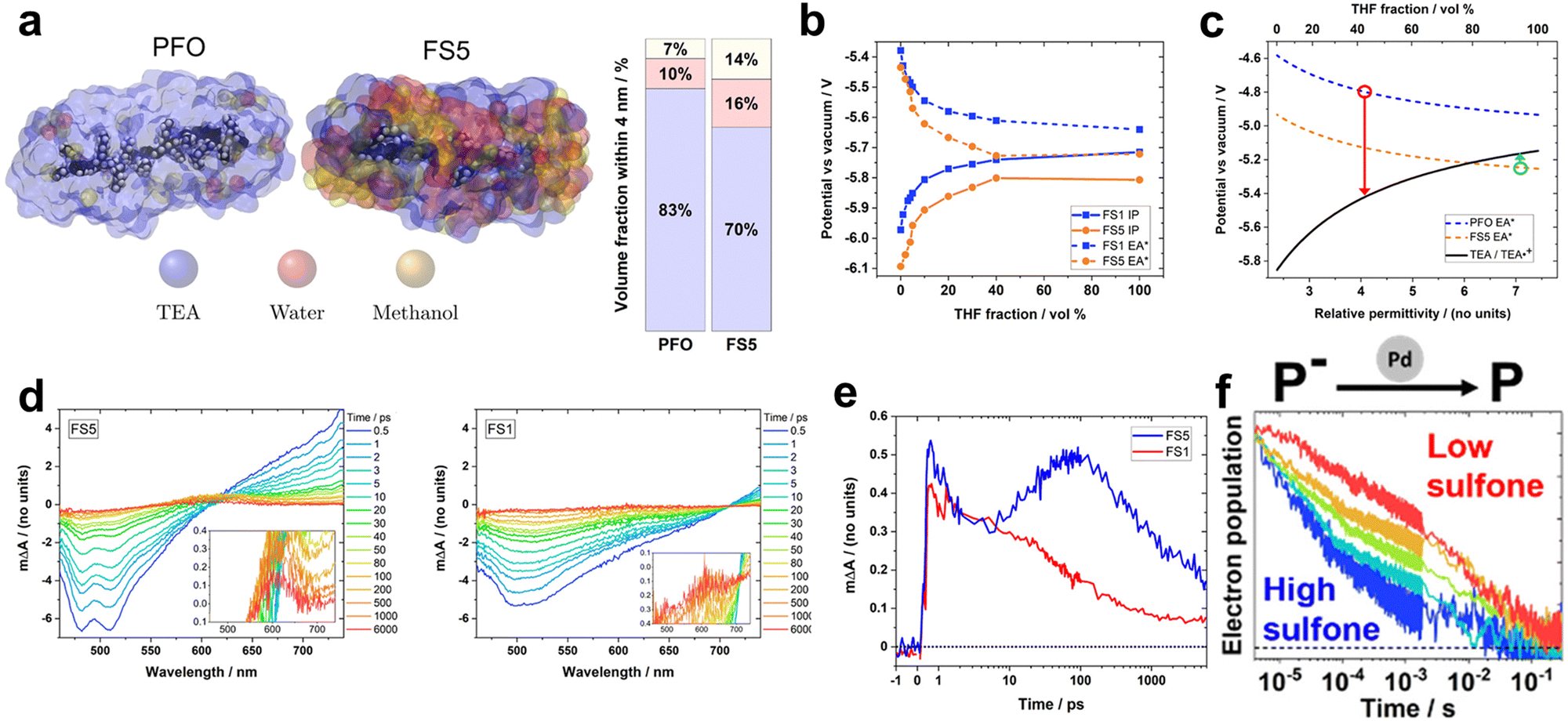 | ||
Fig. 5 (a) Molecular dynamics (MD) simulation of poly(9,9-di-n-octyl-9H-fluorene) (PFO) and FS5 oligomers in an H2O/TEA/methanol 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 vol% mixture. (b) Calculated ionization potential and excited state hole potential (EA*) for FS1 and FS5, as functions of the volume fraction of THF using differential pulse voltammetry. (c) Simulated potential of PFO and PS5 with TEA oxidation potential as a function of the relative permittivity. The red and green circles indicate the potentials of PFO and FS5 at relative permittivity at a radial distance of 4 nm from the mass center of the polymer, respectively. (d) Transient absorption spectroscopy under 420 nm excitation and laser fluence of 90 μJ cm−2 for FS5 (left) and FS1 (right) dispersion in an H2O/TEA/MeOH solution. (e) Deconvoluted electron/charge transfer-state absorption kinetics at 600 nm for PS1 and PS5 extracted from the spectra data (d). (f) Normalized 600 nm transient absorption kinetics of FSn films, excited to match the electron density at 4 μs in each film (1.3–1.9 μΔA/d). The “ΔA” and “d” letters indicate absorbance change at 4 μs and the film thickness, respectively. Reproduced with permission.74 Copyright 2022, ACS Publication. :1:1 vol% mixture. (b) Calculated ionization potential and excited state hole potential (EA*) for FS1 and FS5, as functions of the volume fraction of THF using differential pulse voltammetry. (c) Simulated potential of PFO and PS5 with TEA oxidation potential as a function of the relative permittivity. The red and green circles indicate the potentials of PFO and FS5 at relative permittivity at a radial distance of 4 nm from the mass center of the polymer, respectively. (d) Transient absorption spectroscopy under 420 nm excitation and laser fluence of 90 μJ cm−2 for FS5 (left) and FS1 (right) dispersion in an H2O/TEA/MeOH solution. (e) Deconvoluted electron/charge transfer-state absorption kinetics at 600 nm for PS1 and PS5 extracted from the spectra data (d). (f) Normalized 600 nm transient absorption kinetics of FSn films, excited to match the electron density at 4 μs in each film (1.3–1.9 μΔA/d). The “ΔA” and “d” letters indicate absorbance change at 4 μs and the film thickness, respectively. Reproduced with permission.74 Copyright 2022, ACS Publication. | ||
It was concluded that a high-dipole substituent in the polymer could stabilise the generated exciton lifetime. The introduction of hydrophilic glycol into the polymer structure, either in the sidechain or backbone, is a potent strategy because hydrophilic glycol can interact with water to enhance water contact. Chou's group implemented this idea by incorporating hydrophilic hexaethylene glycol into a hydrophobic conjugated polymer backbone of PFBPO, which was equipped with a phenylbenzo[b]phosphindole-5-oxide acceptor moiety. This backbone engineering resulted in high photocatalytic performance, with an HER of 10.82 mmol h−1 g−1 and an AQY of 18.19%.75 Similarly, Chou's group introduced a crown ether to produce main-chain-engineered discontinuously conjugated polymers (DCPs). These DCPs exhibited longer lifetimes than DCzDBTO, indicating that the exciton lifetime increased owing to the high dipole generated by the trapped Na+ in seawater. Consequently, the P-8CE, which was one of the DCPs, showed HERs of 41.8 and 38.9 mmol h−1 g−1 in natural seawater.76 This conclusion contrasts completely with mechanisms related to electron transfer pertaining to organic solar cells, where non-conjugated backbones can act as insulators, potentially interfering with charge transport along the polymer backbone. It suggests that a simple increase in conjugation length does not significantly impact hydrogen production. This finding indicates that the hydrogen generation mechanism is controlled by a wide variety of factors that require further elucidation.
In single-component photocatalysts, achieving the separation of electrons and holes is challenging due to the strong electron and hole binding energies of localised excitons, as well as significant charge–lattice coupling in the amorphous or semi-crystalline morphology. The generated Frenkel excitons, where electrons and holes are tightly bound, experience rapid radiative and non-radiative decay within picoseconds to nanoseconds. Consequently, achieving rapid charge separation within this timescale is crucial for effective photocatalytic processes. Designing D–A-alternating conjugated structures or promoting backbone planarization can facilitate both charge separation and migration by establishing built-in electric fields, through the push–pull effect between the donor and acceptor. In addition, electronic coupling between the donor and acceptor enables the broadening of the absorption range of the photocatalyst into the visible region.
Accordingly, researchers have attempted to develop sulfone-based D–A copolymers to enhance the photoinduced charge-carrier generation and photocatalytic activity in the visible region. For example, D–A-structured dibenzo[b,d]thiophene sulfone copolymerised with fluorene displayed red-shifted absorption with an optical band gap of 2.07 eV, resulting in a high HER of 5.04 mmol h−1 g−1.77 Xin Li's group investigated the effect of D–A-type structures on charge separation and migration by copolymerising pyrene with different comonomers, including fluorenone, carbazole, and dibenzothiophene, using the Suzuki–Miyaura coupling reaction.78 Among these, the electron-deficient fluorenone copolymer demonstrated an enhanced photocatalytic performance (43.22 mmol h−1 g−1), which was attributed to fast charge separation and migration facilitated by the built-in electric field in the D–A copolymer. In addition, they reported faster charge dynamics in a D–A copolymer with a fluorenone acceptor unit, as determined by fs-TAS measurements. The broad positive absorption signal in the visible region corresponding to the exciton-state absorption of the PyFO copolymer decayed rapidly within 146 ps, indicating rapid exciton dissociation. Further studies have investigated the impact of charge-transfer state dynamics and exciton binding energy on the photocatalytic activity of D–A copolymers. For example, Wang's group modulated the exciton binding (Eb) and charge transfer state energies (ECT) by controlling the local D–A structures of D–A-conjugated copolymers.49 They synthesised four different sulfone-based copolymers using various donor units. Then, they demonstrated that copolymers with planarized structures and extended conjugation facilitated exciton delocalisation and exhibited a reduced Eb, thereby enhancing charge transfer. Thus, the FSO-FS copolymer, which incorporates a sulphur bridge, exhibited an outstanding photocatalytic performance compared with that of a copolymer featuring a fluorene donor unit with a sp3 carbon bridge. These studies highlighted that D–A structures with planarized backbones and extended conjugation can significantly reduce the Eb and enhance exciton dissociation, thereby improving the overall photocatalytic efficiency.
In addition, introducing various linker units can be a powerful strategy for boosting photocatalytic effects. Sprick et al. explored the impact of different linkers by incorporating alkyl and alkyne linkers into sulfone- and fluorene-based polymers. They pointed out that all sulfone-based polymers exhibited relatively longer exciton lifetimes than their fluorene-based counterparts, which is consistent with previous literature. In addition, LS2 demonstrated a higher HER of 3.334 mmol h−1 g−1 compared with that of LS3 (1.980 mmol h−1 g−1), which was attributed to its superior visible light absorption and hydrophilicity.79 Similarly, Chen's group investigated the effect of introducing a series of π-bridges into conjugated microporous polymers. Among these, DBD-S showed the highest HER at 20.3 mmol h−1 g−1 with the longest exciton lifetime of 0.69 ns. This study highlighted the significance of regulating the push–pull interaction between the donor and acceptor units by using heteroatom π-bridges, demonstrating their critical role in optimising photocatalytic performance.80 These findings underscore the potential of linker unit modifications to enhance the photocatalytic efficiency by influencing exciton lifetimes, light absorption properties, and the hydrophilicity of the materials.
Although the hydrogen generation mechanism of the BT unit shares many similarities with that of sulfone-based polymers, the focus of BT-based polymers is primarily on hydrogen transport within the polymer backbone. Tian's group first reported a BT-based linear polymer photocatalyst with an impressive initial rate constant of 8.3 mmol h−1 g−1.81 In addition, they systematically investigated the role of BT as an active site for photocatalysis. Using DFT calculations, they proposed that the nitrogen component of BT serves as a proton-binding site for photocatalytic reactions, demonstrating its potential as an electrocatalyst for hydrogen generation without the need for additional cocatalysts. Furthermore, by incorporating a thiophene bridge into PFBT, the hydrogen evolution rate was increased to 50 mmol h−1 g−1.82 Tian et al. elucidated the mechanism of hydrogen generation in BT moieties using real-time cyclic voltammetry (CV). They introduced the BT derivative 2,1,3-BT-4,7-dicarbonitrile to avoid potential side reactions from occurring during the analysis. Two distinct redox events appeared in the CV waves at −1.06 and −1.88 V (versus ferrocene/ferrocenium) in acetonitrile, implying that two electrons participated in the HER. They also emphasised that hydrogen atoms must attach to both nitrogen atoms in the BT structure.83 This hypothesis was further supported by analysing the molecular dynamics simulations of excited BT structures by Chung's group.84 Charge dynamics regarding BT-based polymers is discussed in more detail in the next section, as numerous studies have focused on this aspect.
In contrast to the initial developments in semiconducting polymer-based photocatalysts that started in 2016, which primarily focused on a limited number of representative active cores, recent research has expanded to explore hydrogen production using various new structures. For example, Pan's group reported boron (B)-substituted conjugated polymers designed to enhance the separation and transport of photo-induced electron/hole pairs. Owing to the high polarity of the B–N bond in the polymer backbone, PBN showed the highest HER of 50.8 mmol h−1 g−1 among their series.85 Similarly, study on another type of B–N-based polymer, PBM, achieved an improved HER of 68.8 mmol h−1 g−1.86 Pan et al. emphasised that the enhanced electron transfer to reactants and improved interface contact ability were facilitated by the high polarity of the B–N bond, which was supported by both experimental results and calculations. Various other structures have also been investigated. Chou's group synthesised all-acceptor copolymers, with PBDTTTSOS achieving the highest efficiency in the thin film polymer (PBDTTTSOS, 150.7 mmol h−1 m−2).87 In addition, they developed BBTT by incorporating a thiophene unit into the sulfone structure. This asymmetric acceptor core in PBBTT-1SO achieved an HER of up to 222.03 mmol h−1 g−1 and AQY of 27.5% at 500 nm.88 These examples highlight the active progress in exploring new active structures, moving beyond the initial research focus on structures similar to those present in various organic semiconductor materials. Nevertheless, to fully uncover the universal mechanism of hydrogen production, it is essential not only to continue developing diverse structures but also to conduct concurrent research on the hydrogen production mechanisms specific to each structure. This dual approach enhances our understanding and paves the way for the optimisation of hydrogen generation using organic photocatalysts.
4.2. Substituent engineering and its charge carrier dynamics
In water-splitting semiconducting polymers, substituent modifications can be classified as the substitution of heterogeneous elements or alteration of ethylene glycol sidechains. To substitute heterogeneous elements, Xiang et al. utilised a theoretical approach to enhance the performance of fluorinated BT-based photocatalysts. Using DFT calculations, they demonstrated that the photocatalytic hydrogen evolution process proceeded via proton-coupled electron transfer facilitated by coupling between the p orbitals of the meta-positioned carbon of the BT unit and nitrogen p orbital (regarded as the hydrogen-binding site). Introducing para-substituted fluorine enlarges the meta-positioned carbon p orbital, thereby enhancing the coupling and reducing the activation barrier for hydrogen evolution. In addition, meta-substituted methoxy, which aligns with para-substituted fluorine, further reduced the barrier, resulting in a 3.1-fold enhancement compared with that of the non-substituted material.89 Fluorination in the thiophene bridge of BT presents an alternative strategy to increase the hydrogen evolution rate. For example, fluorine-substituted dithiophene, P3 (26 mmol h−1 g−1), was introduced to promote highly ordered lamellar stacking. Moreover, the high polarity of fluorine facilitates the observation of long-lived polarons via TAS.90The sidechains of organic photocatalysts significantly influence the local solvent environment and overall performance of the photocatalytic materials. Nelson et al. investigated the impact of different sidechains, including hexyl (FS-Hex) and triethylene glycol (FS-TEG), in sulfone-based polymers.91 The analysis of the fs-TA spectra and comparison of the charge dynamics in these conjugated polymers, which were dispersed in an H2O/MeOH/TEA solution, revealed similar shapes that were attributed to their shared backbone structure (Fig. 6a). However, a significant difference in the transient absorbance amplitudes at 600 nm, corresponding to the polaron peak, was observed under identical conditions. Specifically, the peak intensity of FS-TEG was four times greater than that of FS-Hex after 100 ps (Fig. 6b) because of the higher extinction absorption coefficient of FS-TEG. To deconvolute the effect of the absorbance difference on the charge behaviour, simulations equating the maximum exciton densities of both samples demonstrated similar polaron lifetimes for FS-TEG and FS-Hex within the picosecond timescale (Fig. 6c).
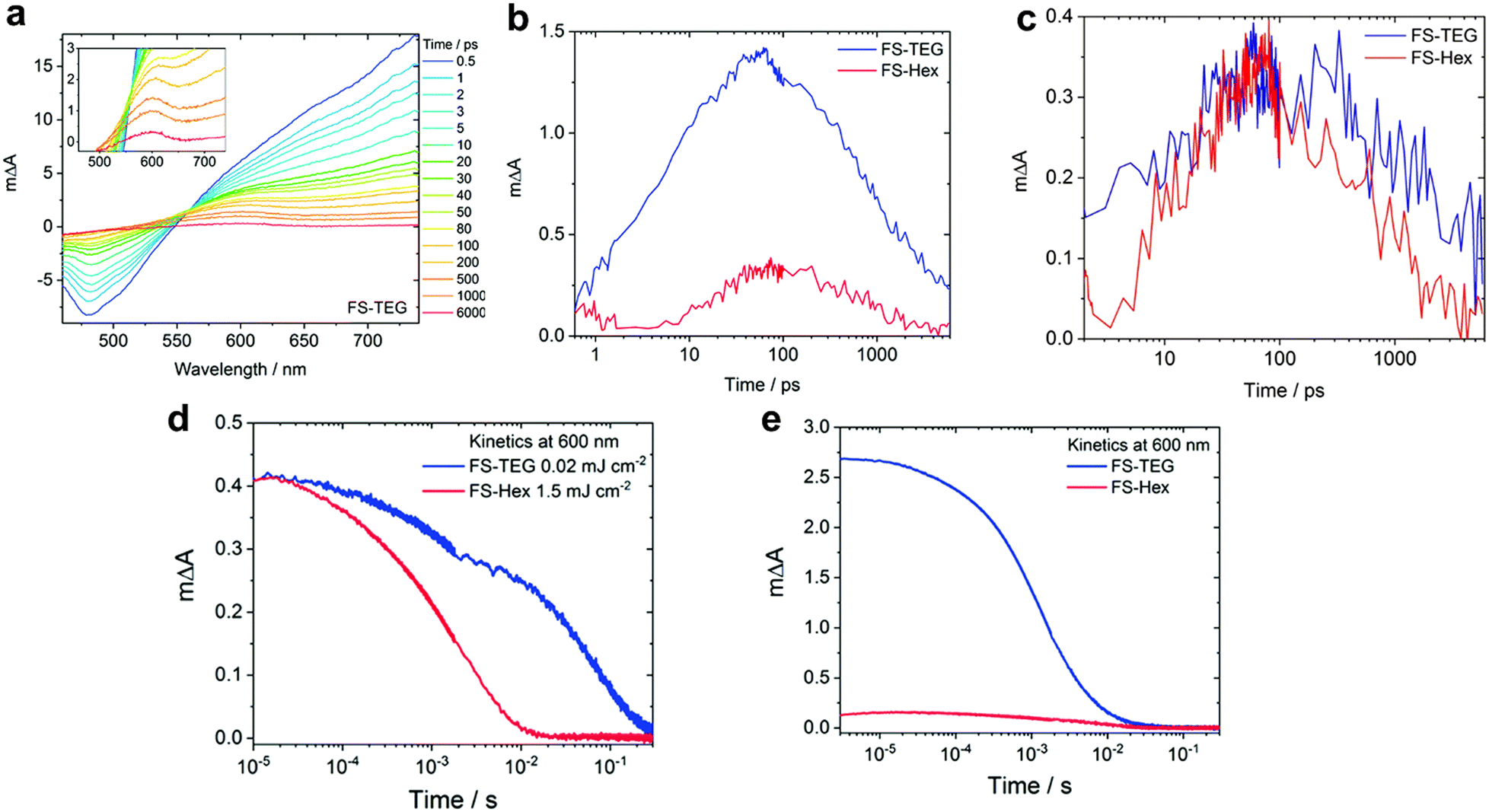 | ||
| Fig. 6 (a) fs-TA spectra of the FS-TEG dispersion in H2O/TEA/MeOH using 420-nm excitation. (b) Deconvoluted transient absorption kinetics for FS-Hex and FS-TEG dispersions probed at 600 nm in picoseconds. Both spectra were measured at the same fluence of 87 μJ cm−2. (c) Comparison of negative polaron absorption kinetics at 600 nm of the FS-TEG and FS-Hex dispersions excited at different fluences, where the maximum exciton densities corresponding to 725 nm are identical (FS-TEG at 102 μJ cm−2 and FS-Hex at 13 μJ cm−2). (d) μs-TA spectrum kinetics at 600 nm, obtained from the FS-TEG and FS-Hex dispersions in the H2O/TEA/MeOH solution excited at different fluences (0.02 mJ cm−2 for FS-TEG and 1.5 mJ cm−2 for FS-Hex). (e) Kinetics of μs-TAS at 600 nm, normalized by the maximum 750 nm signal when irradiated at the same fluence (0.92 mJ cm−2). Reproduced with permission.91 Copyright 2022, The Royal Society of Chemistry. | ||
Considering timescales ranging from microseconds to seconds, which are crucial for the proton reduction reaction, both FS-TEG and FS-Hex exhibited similar polaron amplitudes following excitation at different fluences (Fig. 6d). However, the polaron signal lifetime was notably extended for FS-TEG compared with that of FS-Hex, suggesting an inherently longer polaron lifetime in FS-TEG during the timescale relevant to proton reduction, even when their electron densities were equivalent. To quantitatively evaluate the polaron formation efficiency, the kinetic profiles of the 600-nm peak were normalised against the maximum 750-nm signal, which corresponds to exciton state absorption. Under identical excitation conditions, the signal intensity for FS-TEG significantly increased during the microsecond regime, indicating a more efficient exciton-to-polaron conversion (Fig. 6e). Consequently, the presence of a polar sidechain enhances the affinity and permeability of the photocatalyst for water, thereby increasing the number of available electrons for proton reduction reactions through the stabilised lifetime of the photogenerated charges.
Following a similar approach, in 2022, Kosco et al. integrated polar oligoethylene glycol sidechains into an IDTBT backbone to investigate the impact of water permeability on the photocatalytic charge dynamics of a BT-based polymer.92 Through the comparative picosecond-scale TAS analysis of glycolated (gIDTBT) and non-glycolated polymers, it was confirmed that gIDTBT exhibited positive absorption peaks at approximately 1050 nm, indicating the presence of free charges without the need for additional cocatalysts or sacrificial electron donors (Fig. 7a). Deconvolution of the absorption kinetics at this wavelength revealed a rapid signal increase within 100 ps, followed by decay with a lifetime exceeding 1 ns (Fig. 7b). This demonstrated the prompt formation of charges in the glycolated polymer nanoparticles, albeit insufficient for oxidising ascorbic acid, owing to the nanosecond-scale lifetime relatively shorter than oxidation reaction rate of ascorbic acid. Conversely, the binary polymer nanoparticles composed of gIDTBT and OIDTBR exhibited enhanced charge dynamics characterised by ultrafast charge formation and prolonged charge lifetimes (Fig. 7c). Notably, these dynamics surpassed those observed for nanoparticles lacking glycol sidechains (Fig. 7d). These findings demonstrate the role of polar sidechains in releasing the Coulombic attraction between charges and elongating their lifetimes. Furthermore, fluence-dependent absorption kinetics indicated suppressed geminate charge recombination in gIDTBT-based nanoparticle photocatalysts (Fig. 7c).93 The observed enhancements in charge separation and extended charge lifetimes in gIDTBT were attributed not solely to the glycol itself but rather to increased water permeability within aqueous dispersions. This facilitated free charge generation and prevented geminate charge recombination, ultimately leading to a 30-fold increase in the HER compared with those of the non-glycolated nanoparticles in the blended system with OIDTBR. These findings highlight the critical role of the polar sidechains in enhancing the performance of BT-based polymer photocatalysts, emphasising their potential for efficient charge separation and utilisation in HERs.94
 | ||
| Fig. 7 (a) TA spectra of the gIDTBT dispersion. (b) Deconvoluted transient absorption kinetics probed at 750 nm. Transient absorption kinetics at different fluences of (c) gIDTBT:OIDTBR and (d) IDTBT:OIDTBR. All transient absorption data were measured under 650 nm excitation using a 5 μJ cm−2 fluence. Reproduced with permission.92 Copyright 2021, Wiley-VCH publication. | ||
Therefore, the polar sidechain facilitates free-charge generation by improving exciton separation and prevents geminate recombination. Consequently, the HER was significantly enhanced by a factor of 30 compared with that of the non-glycolated nanoparticles in the blended system with OIDTBR. Previous studies have reported that the glycol sidechain is characterised by a slow dielectric response that does not directly reduce the electron–hole binding energy, which suggests that the observed effects are not solely attributable to the glycol itself. Instead, the increased permeability to water in an aqueous dispersion environment appears to be the primary driving factor.95
Establishing a close interaction between water and photocatalysts is a promising strategy for overcoming the Eb and extending the charge lifetime. The high dielectric-constant nature of water molecules is crucial for screening the Coulombic attraction between the electrons and holes of photogenerated excitons at the interface. These insights provide valuable guidance for enhancing the water affinity in the design of high-performance photocatalysts.96
4.3. Interfacial engineering for water uptake
The hydrogen evolution rate can be effectively increased by modifying polymers itself, while methods of increasing catalyst efficiency without significantly modifying the polymer itself, such as post treatment, have also been reported as effective means of engineering the interface between catalysts and reaction media. In addition, the establishment of a well-defined semiconductor/electrolyte interface in the photocatalytic systems within the reaction containers has proven to be important. The formation of a close interface facilitates rapid photo-induced charge transfer to the sacrificial agent via reductive/oxidative quenching, resulting in the generation of free charges with extended lifetimes ranging from microseconds to milliseconds. The suppression of charge recombination is essential for efficient photocatalytic reactions. However, because most organic semiconductors are inherently hydrophobic, optimising the size of the dispersed particles is also a strategy for enhancing the contact between the photocatalyst and water. This also provides a solution to the short-exciton diffusion length problem by reducing the distance over which excitons migrate to the active sites, while simultaneously increasing the interface area with the reactant solution.In 2016, Tian's group employed the nanoemulsion method to fabricate nanosized F8BT polymer dots.81 This process involves ultrasonicating a THF solution containing a PS-PEG-COOH surfactant and F8BT in a distilled water bath, followed by Ar gas blowing to eliminate THF and filtration. The resulting nanosized particles were stabilised by a surfactant, which prevented agglomeration. Although it is much larger than the commonly discussed exciton diffusion length, this approach resulted in a product that delivered a five-orders-of-magnitude enhancement in the HER compared with the results achieved with the bulk polymer photocatalyst. Although the post-nanoemulsion method is favourable for the fabrication of polymer nanoparticles, its feasibility depends on the solubility of the polymer in an organic solvent. However, insoluble polymers cannot be processed into nanoparticles by this method. For P10, nanoparticles were prepared using Pd-catalysed emulsion polymerisation.97 In this method, an aqueous solution of sodium dodecyl sulphate surfactant and Na2CO3 were added to an organic solution of monomers and Pd(PPh3)4, followed by sonication and heating. The resulting dispersed P10 nanoparticles, with an average hydrodynamic particle size of 160 nm, exhibited a HER of 60 mmol h−1 g−1 under visible light, along with a high AQY of 20.4% at 420 nm. Notably, although surfactants stabilise polymer nanoparticles and prevent their aggregation in aqueous solutions, they can hinder the approach of water molecules and charge transfer to the reactants because of their insulating properties. Therefore, Chou et al. introduced conjugated oligomeric polyelectrolytes as surfactants.98 This innovation improved the HER compared with the results achieved with polymer dots when using PS-PEG-COOH. They showed that the improvement was caused by the increased wettability, which also facilitated Förster resonance energy transfer from the polyelectrolyte to the polymer photocatalyst, as proved by the spectra overlap between them and the enhanced and accelerated PL quenching of the polymer.
In addition, Chung's group, introduced photocrosslinkers into a polymer to exploit the morphology of the catalysts. To overcome the physical swelling of the polymer dots in the aqueous phase, which could be a major impediment to hydrogen evolution, post-crosslinking was introduced. They introduced bis(fluorophenyl azide) for physical fixation of the synthesised polymer dots to increase the hydrogen evolution rate and achieve a high AQY (11.024 mmol h−1 g−1, 0.8%).99 Furthermore, Chung et al. introduced a glycol-based bridge chain crosslinker to directly increase the permeability of water molecules and achieve a 2.5-fold increased hydrogen evolution rate. In the nanoemulsion method, there is a solubility condition in which the polymer must be dissolved in THF, whereas introducing a PEG-based crosslinker can easily tune the water permeability once the polymer emulsion is formed. However, because research is still in its early stages, more detailed research on the related dynamics needs to be conducted.
4.4. Effect of cocatalysts
As discussed previously, cocatalysts are essential elements for producing solar fuel. In addition, for generation of different types of solar fuels, different types of cocatalysts can be introduced, for which Pd and Pt metals are commonly used for syngas production.100 Cocatalysts play a crucial role in providing active sites for photocatalysts and reducing their overpotentials. In photocatalytic systems, charge transfer from the semiconductor to cocatalyst occurs on timescales ranging from picoseconds to nanoseconds or, in some cases, from late femtoseconds to early microseconds. Notably, the precise effect of the cocatalyst on the photocatalytic process, particularly the role of residual Pd resulting from the Suzuki–Miyaura coupling condensation, is yet to be fully investigated. For example, in the F8BT photocatalyst, the correlation between the HER and Pd content is intriguing because while Pd removal at concentrations below 1 ppm resulted in a negligible photocatalyst activity, the HER was rapidly saturated at approximately 100 ppm of Pd. This result suggests that even trace amounts of Pd quantities significantly influence the performance of photocatalysts.101 For in-depth insights into the role of Pd as a cocatalyst, McCulloch and Durrant's research groups tracked the behaviour of charge carriers in F8BT and P10 nanoparticles, aiming to investigate the differing Pd dependencies of these photocatalysts using transient spectroscopy (Fig. 8).102 Their results revealed that while the photocatalytic activity of F8BT was saturated at 200 ppm of Pd, the HER of P10 reached saturation at a Pd concentration of 17000 ppm. The analysis of the Pd-dependent TCSPC data for F8BT revealed that 54% of the photogenerated electrons in F8BT were quenched by Pd within 10 ns (Fig. 8a, left). Furthermore, 10% of these electrons were rapidly quenched within 100 fs, as determined using fs-TAS (Fig. 8b). Importantly, the reduced Pd species would likely act as recombination centres for the F8BT holes, consequently diminishing the photocatalytic efficiency. This electron quenching by Pd outperformed the irreversible exciton quenching by sacrificial donors, which generally occurs on a picosecond to nanosecond timescale. This implies that the electron transfer to Pd, even at low Pd concentrations (36–196 ppm), dominates the quenching process, leading to electron localisation on Pd, which is a phenomenon confirmed by microsecond PIAS combined with spectroelectrochemistry (SEC) (Fig. 8c).
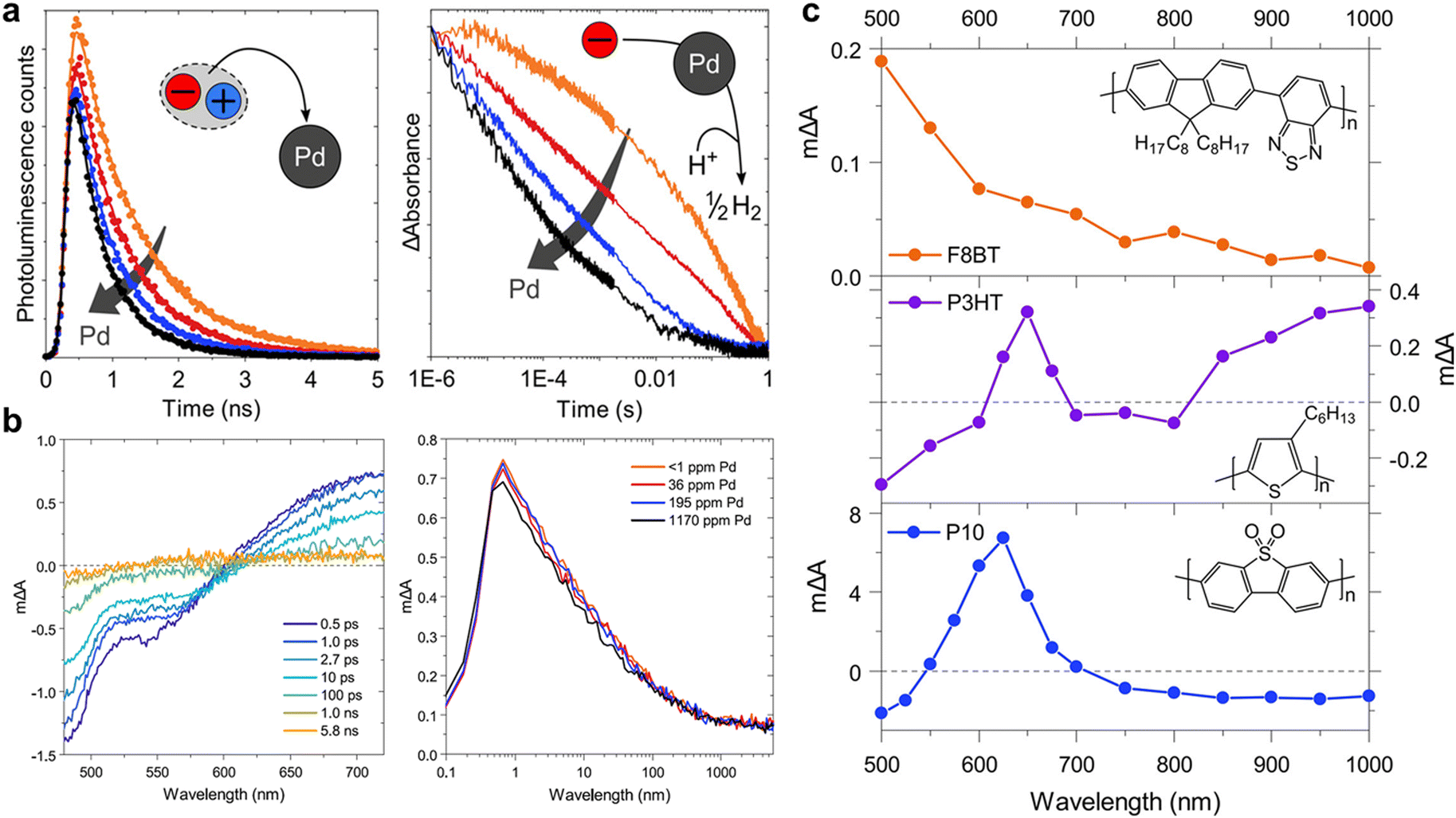 | ||
| Fig. 8 (a) PL decays of F8BT probed at 545 nm and excited using 467 nm at different Pd concentrations in a 30 vol% diethylamine (DEA) aqueous solution (left), and negative polaron transient absorption kinetics at 700 nm probe of the P10 dispersion in the H2O/MeOH/TEA mixture, as a function of the Pd catalyst concentration employed in the synthesis, excited under 355 nm using fluence of 0.32 mJ cm−2 (right). (b) fs-TA spectra of F8BT in H2O (excitation wavelength: 460 nm, fluence: 16.5 μJ cm−2), and kinetics at 700 nm for different Pd concentrations. (c) PIAS of F8BT in a 30 vol% DEA/H2O mixture (excitation: 465 nm, 25.6 mW cm−2), P3HT in the H2O/MeOH/TEA mixture (excitation: 365 nm, 7.1 mW cm−2), and P10 in the H2O/MeOH/TEA mixture (excitation: 365 nm, 12.5 mW cm−2) pulsed for several seconds. Reproduced with permission.102 Copyright 2020, ACS publication. | ||
In contrast, for P10, hole quenching by the sacrificial donor occurred on a subpicosecond timescale, whereas electron quenching by Pd occurred on a microsecond scale (Fig. 8a, right). Consequently, in P10, electrons accumulate as polymer-bound polaron species, indicating that electron transfer to Pd is a limiting process that impedes photocatalytic hydrogen evolution. Although rapid electron transfer to Pd was observed in F8BT, its AQY, which reached 0.1% at 1170 ppm of Pd (340 nm), was two orders of magnitude lower than that of P10 (11.7% at 4000 ppm of Pd, 420 nm). This significant difference can be attributed to the considerably higher number of long-lived charges observed in P10. For this reason, polymers synthesised by the same method might have similar residual Pd concentrations, and because Pd generally exceeds the saturation concentration, it can be said that rate determination by residual Pd is nearly negligible. These findings underline the importance of multifaceted consideration including not only charge transfer to Pd but also reductive quenching through the sacrificial agent to optimise the overall photocatalytic performance. Although it is recognised that sacrificial agents have a very large impact on hydrogen production efficiency, few studies have focused on changing the reaction media conditions, such as the sacrificial agent type, amount, and pH.103 Although it is difficult to accurately analyse the behaviour of atoms inside a polymer catalyst owing to the irregularity of the polymers, analysing the microscopic behaviour of hydrogen atoms while changing various conditions remains an important task for the future.
4.5. Multicomponent photocatalysis
Heterojunction formation with proper band alignment is a well-known strategy for facile photogenerated charge separation and migration. At the interface of a heterojunction, photogenerated electrons and holes can be dissociated into spatially separated domains, facilitating charge separation. Thereafter, these charges participate in reduction and oxidation reactions, respectively. Type-II heterojunction photocatalytic systems, featuring staggered band-edge of electron rich species (D; donor) and electron deficient species (A; acceptor) are widely employed configurations for multicomponent photocatalysts. The effectiveness of type-II heterojunction has been verified in numerous studies involving inorganic,104 organic,105 and hybrid composites with increased charge transfer and separation (Fig. 9a).106 Notably, in this system the separated electrons tend to transfer to the LUMO with a less negative reduction potential, whereas the separated holes move to the HOMO, which has a relatively low oxidation potential. These charge transfers reduce the thermodynamic driving force for photocatalytic reaction, as well as limit the utilizing materials with narrow band gaps that can absorb energy in a broad spectrum exceeding the NIR region for overall water splitting. An artificial Z-scheme photocatalyst mimicking a natural photosynthetic system, in which charge flow between photoexcited components is mediated by a redox mediator (redox mediated Z-scheme) or a built-in electric field at the direct junction (direct Z-scheme), regulates charge recombination between holes in HOMO of D and electrons in LUMO of A (Fig. 9b). Therefore, a narrow-band gap semiconductor can be employed because there is no need to straddle the water redox potential. Despite the advantages of Z-scheme photocatalytic systems, it is difficult to ensure proper contact between two photocatalysts or redox mediators to achieve the Z-scheme configuration. Therefore, it is necessary to investigate the factors that determine what kind of junction would be formed.107,108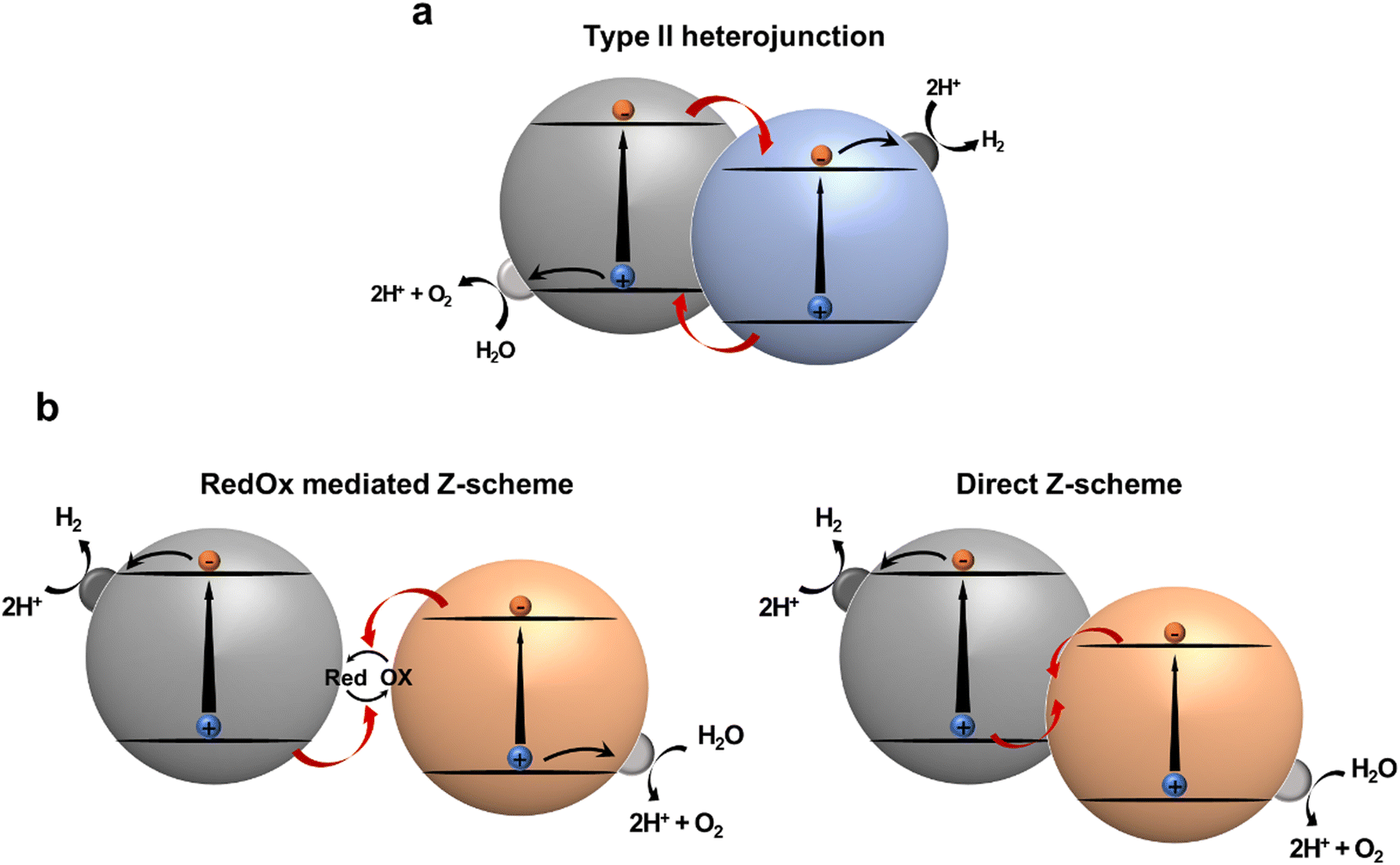 | ||
| Fig. 9 Schematic illustration of (a) type II heterojunction photocatalysts and (b) Z-scheme photocatalysts. | ||
In organic semiconductors, particularly those incorporating linear conjugated polymers, heterojunction formation with small-molecule acceptors with complementary absorption is a powerful approach for resolving the issues of strong exciton binding energy and short diffusion length, as well as enhancing charge dissociation efficiency. In such blend systems, D and A are intimately mixed to form an interface where excitons can undergo quenching via either energy or charge transfer pathways. The efficiency of charge dissociation at the D/A interface, a process that competes with the geminate recombination decaying to the ground state (S0) is influenced by several factors including energetic alignment, local mobility, D/A nanomorphology, internal electric field, and charge transfer state lifetime.109,110
The application of D/A heterojunctions in hydrogen evolution photocatalysis was pioneered by Kosco et al. They fabricated nanoparticles using the polymer donor, PTB7-Th, and the acceptor, EH-IDTBR (previously designed for organic photovoltaics (OPV) active materials), employing SDS or TEBS as a stabilizing surfactant. Thereafter, they investigated the effect of the blend morphology (the core–shell for SDS and bulk heterojunction for TEBS).111 Notably, the enhanced photocatalytic activity observed with TEBS suggested that the internal D/A junctions accelerated charge separation and enhanced charge extraction to the Pt cocatalyst, primarily due to the increased exposure of the EH-IDTBR component. The distinct morphologies are attributed to the different interactions between the surfactants and polymers. The aromatic ring of TEBS interacts with EH-IDTBR, reducing the difference between interfacial tension of PTB7-Th and EH-IDTBR (chloroform phase) with aqueous phase, hindering core–shell formation. More recently, McCulloch's group investigated the formation of long-lived charges in PM6/PCBM and PM6/Y6 nanoparticles with core–shell and intermixed structures respectively.112 Their results showed that the D/A blend system with an enhanced type-II junction facilitated charge separation and the formation of long-lived charges. Most strikingly, PIAS measurements under quasi-steady-state (several seconds) irradiation indicated that the photoexcitation of PM6/PCBM nanoparticles resulted in the accumulation of ∼600 PM6+ species per nanoparticle, with lifetimes of milliseconds to seconds. The amplitude of long-lived charges was further enhanced by the addition of Pt cocatalysts because of the suppression of charge recombination. The long-lived charges rapidly decayed after the addition of ascorbic acid as a sacrificial agent, indicating fast charge extraction from the nanoparticles to the sacrificial agent. As a result, the relatively larger signals of the long-lived charges in the PM6/PCBM nanoparticles (HER of 73.7 mmol h−1 g−1) compared with those of PM6/Y6 (HER of 43.9 mmol h−1 g−1) were directly contributed to their photocatalytic performance.
These observations may deviate from a trend in OPV. For instance, fullerene-based electron acceptors exhibit lower photoconversion efficiency compared to Y6 because of short exciton diffusion length (∼5 nm) and limited absorption ability in the visible and NIR region.113 Moreover the higher photocatalytic performance, achieved with PM6/PCBM nanoparticle featuring core–shell structures, indicates that various complementary factors, such as reaction solution, cocatalyst, and micro- and macro-phase morphology, should be considered in particulate photocatalysis.
To further improve the comprehension of the charge-carrier behavior of type-II heterojunction nanoparticles prepared by nanoemulsification method, Tian's group systematically investigated the charge dynamics of nanoparticles with various ratios of PFODTBT (D) and ITIC (A) via steady-state and transient spectroscopy.114 They found that when the A fraction was low, charge separation process occurred by sequential energy transfer from D* to A and charge transfer from A* to D. Conversely, the one-step electron and hole transfer became a dominant pathway when the A fraction exceeded 62 wt% (Fig. 10a). Through the TRPL decay of pure D nanoparticles, red shift of PL which indicating the energetic disorder was observed (Fig. 10b). However, this phenomenon was absent in binary D/A nanoparticles with 51 wt% of A, exhibiting that they are not subjected to energetic disorder (Fig. 10c). Additionally, ultrafast charge generation within 200 fs was demonstrated by a rapid increase in positive absorption at 475 nm, corresponding to the overlap of two oppositely charged species in the TAS spectra of binary nanoparticles with 62 wt% of A (Fig. 10d). These fast charge separation and energetic disorder-independent charge carriers are contributed to the fast electron and hole transfer between D and A interfaces in nanosized particles and the suppressed charge recombination. Indeed, in nanoparticles exciton diffusion is confined within highly mixed D/A morphology, resulting in fast exciton dissociation. However, it has been revealed that at low A concentration the large interfacial area serves as nongeminate recombination site, leading to charge carrier loss, via a significant excitation light intensity dependent TA decay of A˙−. In contrast, the recombination rate decreased at high concentrations of A more than 62 wt%, attributed to the aggregation of A and the separated phases of the D and A. These results highlight the pivotal role of morphologies in driving the observed dynamics.
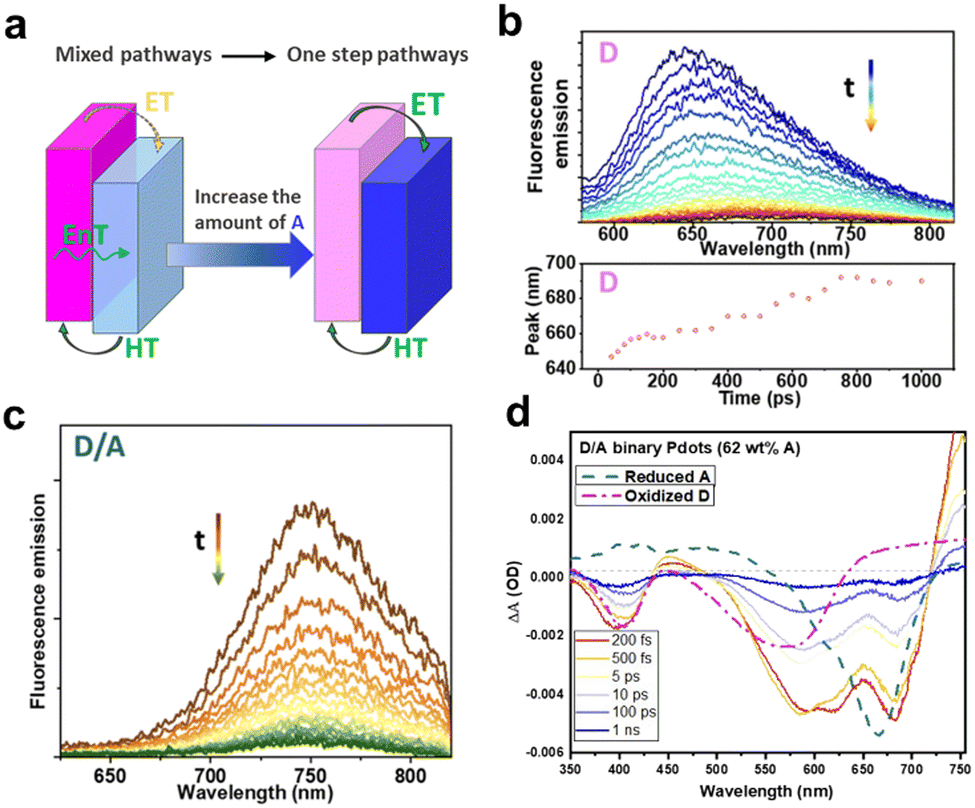 | ||
| Fig. 10 (a) Schematic of the charge pathway in binary nanoparticles. Time-resolved PL spectra of (b) D and (c) D/A binary nanoparticles with 51 wt% A, excited at 550 nm. (d) Transient absorption spectra of D/A binary nanoparticles (62 wt% A) under excitation at 550 nm using fluence of 140 μW. The dashed line indicates the reduced A and oxidized D, obtained from the spectroelectrochemical spectra. Reproduced with permission.114 Copyright 2023, Royal Society of Chemistry. | ||
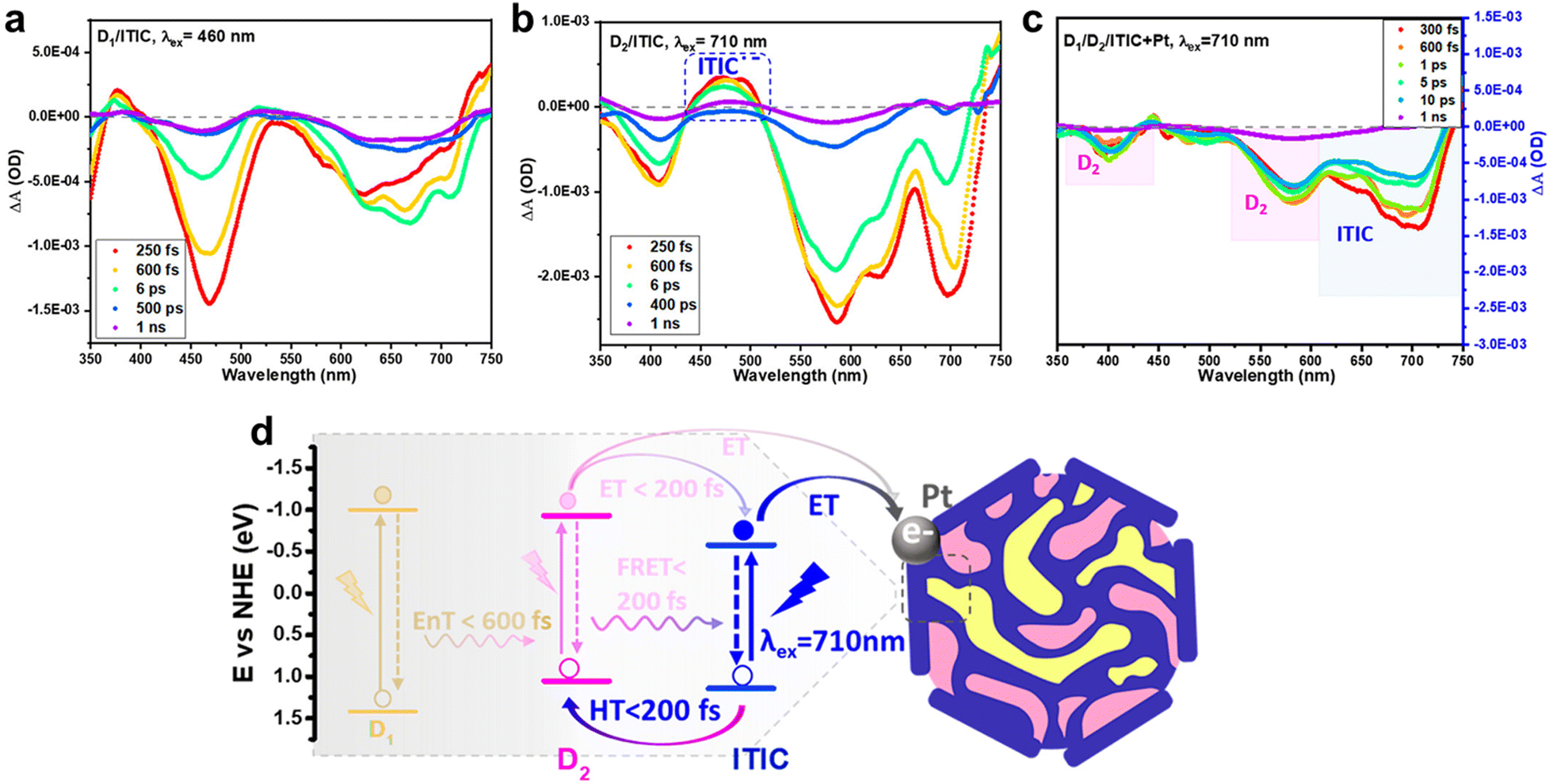
Furthermore, Tian's group explored the charge behavior in a ternary photocatalytic system consisting of two donors and one acceptor which has panchromatic light absorption via TAS, accompanied by complementary steady-state spectroscopy analysis such as PL and SEC.115 In the fs-TAS of the D1:ITIC blend, upon excitation of D1 at 460 nm, the GSB of ITIC (635–700 nm) emerged alongside D1 GSB (Fig. 11a). Together with PL quenching measurement, the rapid rise in ITIC GSB within 430 fs can be interpreted as energy transfer from D1 to ITIC. In the D2/ITIC TAS data pulsed at the optical band gap of ITIC (710 nm), the positive absorption at 440 and 510 nm was indicative of the reduced ITIC, which corresponds to the SEC data. Further, negative absorption signals of D2˙+ polaron GSB emerged with rise times of 120 and 180 fs, respectively (Fig. 11b). This demonstrated the hole transfer from the excited ITIC to D2, even with a low energy offset. These rapid energy and charge transfers were attributed to the close contact among the components and the optimal morphology of the ternary polymer nanoparticles. In addition, when Pt cocatalysts were deposited, selectively reduced ITIC˙− negative polaron and ITIC GSB signals indicated that the electron transfer from ITIC to Pt was the dominant charge extraction pathway in this system (Fig. 11c and d). The above results indicated that panchromatic band absorption, sequential charge transfer among three components, and fast charge extraction to Pt from the ITIC are the significant factors contributing to the high performance of multicomponent photocatalysts.
 | ||
| Fig. 11 Transient absorption spectra of the (a) D1/ITIC, (b) D2/ITIC, and (c) ternary D1/D2/ITIC nanoparticles without with Pt cocatalyst, at an excitation wavelength of 710 nm with fluence of 80 μW. (d) Schematic of the charge carrier pathways in the photocatalytic process involving ternary nanoparticles. Reproduced with permission.115 Copyright 2021, ACS publication. | ||
Current studies implied that in multicomponent particulate photocatalysts, constructing appropriate D and A phase morphologies that provide interfaces for charge dissociation while maintaining pure domains for charge transport channels is important for generating long-lived charges. Simultaneously, for facile charge extraction, suitable contact with the cocatalysts and surrounding environment is required.
5. Summary and outlook
Organic photocatalysts with tuneable structures and properties are promising energy platforms for hydrogen evolution. High photocatalytic performance begins with the design of materials that efficiently absorb light across a broad spectrum, including the visible and NIR regions,116 which can be achieved by systematically tuning the building blocks of conjugated polymers. The photogenerated charges in photocatalysts can be divided into three processes: (1) light absorption and exciton generation, (2) exciton diffusion and charge transport to photocatalytically active sites, and (3) charge transfer to the reactant for photocatalytic redox reactions. Understanding the correlation between the molecular structure, charge carrier dynamics, and photocatalytic performance is crucial for developing rational design principles for organic photocatalysts. However, the complex charge behaviour of organic photocatalytic systems remains a significant challenge to completely understanding. This review provides a comprehensive summary of the design strategies for organic photocatalysis, focusing on charge carrier dynamics in linear conjugated polymer-based photocatalysts employing time-resolved analyses, such as TAS and TRPL.In organic semiconductors, photogenerated charges undergo various recombinations within nanoseconds owing to their strong exciton binding energies and inherent energetic disorder. This leads to low charge separation efficiency, which directly affects the photocatalytic performance. To address this issue, effective strategies such as introducing D–A alternating copolymers, planar backbones, and π-linkers with elongated conjugation have emerged to enhance charge separation and migration. Another method involves the use of D/A blends to achieve efficient light absorption across a wide spectrum and facile charge separation. Most organic moieties employed in multicomponent photocatalysis were initially designed for use in OPVs. Owing to the distinct differences in both the operational contexts and attributes required for OPVs and photocatalysis, a tailored design approach specifically for photocatalysis is essential. Furthermore, manipulating the photocatalyst–aqueous solution interface through sidechain and backbone modifications has proven to be feasible. Increasing hydrophilicity and water permeability can stabilise free charges and extend their lifetimes. Considering the limited exciton diffusion lengths and absence of driving forces for photoinduced electron–hole dissociation in organic photocatalysts, shortening the distance for charge migration to the interface and enabling rapid charge transfer are important. Organic semiconductors offer better processability and tunability than inorganic materials, allowing various forms conducive to mass and charge transport pathways.
Although this review primarily focuses on sacrificial-condition photocatalytic hydrogen evolution, advancing photocatalytic OWS for practical artificial photosynthesis applications is essential. To date, only one single-component linear polymer photocatalyst, namely P10, with Ir and Pd dual cocatalysts has been reported for OWS.117 However, the solar-to-hydrogen conversion efficiency remains moderate in OWS. A significant challenge for organic semiconductor-based photocatalysts is the large overpotential associated with the four-electron reaction required for oxygen evolution. In addition, the thermodynamically favoured recombination of charge carriers during OWS further hampers the efficiency. Addressing these obstacles will necessitate innovative approaches, such as the implementation of Z-scheme systems or precise integration of cocatalysts within organic photocatalysts. Potential solutions include spatially separating the HER from the water oxidation reaction, employing selective cocatalysts for either proton reduction or water oxidation,118,119 and utilising alternative oxidation substrates, such as photoreforming.120,121
Furthermore, the resultant product and conversion efficiency of the photocatalytic reactions can be modulated by adjusting factors such as the surrounding conditions; characteristics of the reactant binding sites, including the steric effect and hydrophilicity; number of transferred charges; and choice of cocatalysts. Therefore, further investigations and multifaceted adjustments must be implemented to connect these factors and redox couples, leading to the production of valuable chemicals beyond hydrogen gas.
The organic photocatalysts discussed in this review are promising candidates for the industrialisation of future energy solutions. Organic semiconductors offer a variety of structural combinations, enabling precise synthesis tailored to industrial requirements, such as toxic substances realised during synthesis and ease of decomposition without placing a burden on the environment.122–125 For example, Cooper's group recently utilised machine-assisted synthesis to rapidly screen thousands of photocatalytic organic materials and identify optimal structures, reaction conditions, and combinations.126,127 This approach promises significant advances in industrial applications and can be used in a variety of other applications because of its compatibility with various robotic and computer systems.
For these future energy applications, the stability of organic photocatalysts remains a critical issue. Recent research has shown that a low dielectric constant in polymers can lead to the destruction of the conjugated structure through the accumulation of free electrons within the polymer matrix.128,129 To ensure sufficient stability, it is essential to prevent internal charge accumulation by enabling efficient charge migration to the active site. This can be achieved using strategies such as Z-scheme systems, heterojunctions, charge collection via cocatalysts, and hybridisation with inorganic semiconductors. Thus, the stability and efficiency can be improved through the integration of polymer properties and interdisciplinary approaches.
Although still in the early stages of research, recent studies combining polymer catalysts with biological applications, such as artificial photosynthesis, have made undergone remarkable progress.103,130 This interdisciplinary approach highlights the versatility of polymers across various disciplines and presents ongoing challenges for polymer scientists to integrate these materials into diverse applications, requiring continuous innovation and adaptation.
Although linear polymer photocatalysts are still in their infancy, their tuneable structures and optoelectronic properties have immense potential. To establish organic semiconductors as versatile platforms, research efforts should focus on providing in-depth insights into the relationships among the structure, charge dynamics, and performance of these catalysts using complementary analytical tools.
Author contributions
J. C. and W. J. contributed equally to this work. J. C. initiated and wrote the manuscript. W. J. reviewed and edited the manuscript. S. G.-C. and J. R. D. reviewed the manuscript. T. P. and H. C. reviewed, edited, and supervised the manuscript.Data availability
Data for this article, including figures are available at https://doi.org/10.1039/D4EE01808G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work supported by National Research Foundation of Korea (NRF) grants funded by Ministry of Science and ICT (MSIT) (no. 2021R1A5A1084921, 2021R1A2C3004420 and RS-2023-00213920).References
- P. K. Ozili and E. Ozen, The Impact of Climate Change and Sustainability Standards on the Insurance Market, Wiley, 2023, pp. 439–454 Search PubMed.
- Q. Wang, C. Pornrungroj, S. Linley and E. Reisner, Nat. Energy, 2021, 7, 13–24 CrossRef.
- F. Vilela, K. Zhang and M. Antonietti, Energy Environ. Sci., 2012, 5, 7819–7832 RSC.
- S. Y. Son, K. Choi, J. Lee, H. I. Kim, T. Park and M. Kim, Chem. Mater., 2022, 34, 4856–4864 CrossRef CAS.
- H. Cha, G. Y. Lee, Y. Fu, Y. J. Kim, C. E. Park and T. Park, Adv. Energy Mater., 2013, 3, 1018–1024 CrossRef CAS.
- S. Lim, G. Lee, S. Han, J. Kim, S. Yun, J. Lim, Y.-J. Pu, M. J. Ko, T. Park and J. Choi, ACS Energy Lett., 2021, 6, 2229–2237 CrossRef CAS.
- D. Choi, H. Kim, Y. Bae, S. Lim and T. Park, ACS Energy Lett., 2024, 9, 2633–2658 CrossRef CAS.
- H. Lim, I. Jeong, J. Choi, G. Shin, J. Kim, T.-H. Kim and T. Park, Appl. Surf. Sci., 2023, 610, 155601 CrossRef CAS.
- H. Lim, G. H. Han, D. H. Lee, G. Shin, J. Choi, S. H. Ahn and T. Park, Small, 2024, 2400031 CrossRef PubMed.
- J. Kosco, F. Moruzzi, B. Willner and I. McCulloch, Adv. Energy Mater., 2020, 10, 2001935 CrossRef CAS.
- J. Jia, L. C. Seitz, J. D. Benck, Y. Huo, Y. Chen, J. W. Ng, T. Bilir, J. S. Harris and T. F. Jaramillo, Nat. Commun., 2016, 7, 13237 CrossRef CAS PubMed.
- F. Urbain, V. Smirnov, J.-P. Becker, A. Lambertz, F. Yang, J. Ziegler, B. Kaiser, W. Jaegermann, U. Rau and F. Finger, Energy Environ. Sci., 2016, 9, 145–154 RSC.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- V. Andrei, G. M. Ucoski, C. Pornrungroj, C. Uswachoke, Q. Wang, D. S. Achilleos, H. Kasap, K. P. Sokol, R. A. Jagt, H. Lu, T. Lawson, A. Wagner, S. D. Pike, D. S. Wright, R. L. Z. Hoye, J. L. MacManus-Driscoll, H. J. Joyce, R. H. Friend and E. Reisner, Nature, 2022, 608, 518–522 CrossRef CAS PubMed.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- Y. Ma, L. Lin, T. Takata, T. Hisatomi and K. Domen, Phys. Chem. Chem. Phys., 2023, 25, 6586–6601 RSC.
- K. Kranthiraja, H. Kim, J. Lee, U. K. Aryal, S. S. Reddy, R. D. Gayathri, T. Gokulnath and S.-H. Jin, Macromol. Res., 2023, 31, 897–905 CrossRef CAS.
- J.-W. Ha, J. G. Jung, D. H. Ryu, S. Lee, C. E. Song, B. Lim, Y. J. Jung, J. M. Park and D.-H. Hwang, Macromol. Res., 2023, 31, 25–31 CrossRef CAS.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- S. A. Ansari, M. M. Khan, M. O. Ansari and M. H. Cho, New J. Chem., 2016, 40, 3000–3009 RSC.
- Y. Zhang, Y. Hu, J. Zhao, E. Park, Y. Jin, Q. Liu and W. Zhang, J. Mater. Chem. A, 2019, 7, 16364–16371 RSC.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- Y. Zhang, Y. Li, X. Xin, Y. Wang, P. Guo, R. Wang, B. Wang, W. Huang, A. J. Sobrido and X. Li, Nat. Energy, 2023, 8, 504–514 CrossRef CAS.
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS PubMed.
- K. Ding, B. Chen, Y. Li, Y. Zhang and Z. Chen, J. Mater. Chem. A, 2014, 2, 8294–8303 RSC.
- G. Li and K. A. Gray, Chem. Phys., 2007, 339, 173–187 CrossRef CAS.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474–475 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- A. Irshad, M. H. Arshed, H. H. Somaily, H. Sabeeh, A. Noor ul, I. Ayman, M. F. Warsi and I. Shakir, Macromol. Res., 2023, 31, 91–104 CrossRef CAS.
- S. Kuecken, A. Acharjya, L. Zhi, M. Schwarze, R. Schomacker and A. Thomas, Chem. Commun., 2017, 53, 5854–5857 RSC.
- J. Xie, Z. Fang and H. Wang, Polymers, 2022, 14, 1363 CrossRef CAS PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- C. Li, J. Liu, H. Li, K. Wu, J. Wang and Q. Yang, Nat. Commun., 2022, 13, 2357 CrossRef CAS PubMed.
- M. G. Schwab, M. Hamburger, X. Feng, J. Shu, H. W. Spiess, X. Wang, M. Antonietti and K. Müllen, Chem. Commun., 2010, 46, 8932–8934 RSC.
- C. Han, S. Xiang, S. Jin, C. Zhang and J.-X. Jiang, ACS Catal., 2023, 13, 204–212 CrossRef CAS.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- M. V. Pavliuk, S. Wrede, A. Liu, A. Brnovic, S. Wang, M. Axelsson and H. Tian, Chem. Soc. Rev., 2022, 51, 6909–6935 RSC.
- S. Matsuoka, T. Kohzuki, A. Nakamura, C. Pac and S. Yanagida, J. Chem. Soc., Chem. Commun., 1991, 580–581, 10.1039/C39910000580.
- X. Yuan, C. Wang, L. Vallan, A. T. Bui, G. Jonusauskas, N. D. McClenaghan, C. Grazon, S. Lacomme, C. Brochon, H. Remita, G. Hadziioannou and E. Cloutet, Adv. Funct. Mater., 2023, 33, 2211730 CrossRef CAS.
- O. P. Dimitriev, Chem. Rev., 2022, 122, 8487–8593 CrossRef CAS PubMed.
- H. Wang, S. Jin, X. Zhang and Y. Xie, Angew. Chem., Int. Ed., 2020, 59, 22828–22839 CrossRef CAS PubMed.
- H. Wang, W. Liu, X. He, P. Zhang, X. Zhang and Y. Xie, J. Am. Chem. Soc., 2020, 142, 14007–14022 CrossRef CAS PubMed.
- M. Yu, W. Zhang, Z. Guo, Y. Wu and W. Zhu, Angew. Chem., Int. Ed., 2021, 60, 15590–15597 CrossRef CAS PubMed.
- Z. A. Lan, G. Zhang, X. Chen, Y. Zhang, K. A. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 10236–10240 CrossRef CAS PubMed.
- E. Medina, C. Sandoval-Pauker, P. Salvador and B. Pinter, Inorg. Chem., 2022, 61, 18923–18933 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- P. Drude, Ann. Phys., 1900, 306, 566–613 CrossRef.
- H. F. Haneef, A. M. Zeidell and O. D. Jurchescu, J. Mater. Chem. C, 2020, 8, 759–787 RSC.
- J. Rivnay, S. C. B. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488–5519 CrossRef CAS PubMed.
- S. Fratini, M. Nikolka, A. Salleo, G. Schweicher and H. Sirringhaus, Nat. Mater., 2020, 19, 491–502 CrossRef CAS PubMed.
- S. Giannini and J. Blumberger, Acc. Chem. Res., 2022, 55, 819–830 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- Y. Li, L. Yang, H. He, L. Sun, H. Wang, X. Fang, Y. Zhao, D. Zheng, Y. Qi, Z. Li and W. Deng, Nat. Commun., 2022, 13, 1355 CrossRef CAS PubMed.
- C. Ding, J. Shi, Z. Wang and C. Li, ACS Catal., 2017, 7, 675–688 CrossRef CAS.
- X. Yang, C. Du, R. Liu, J. Xie and D. Wang, J. Catal., 2013, 304, 86–91 CrossRef CAS.
- Z. Hu, X. Zhang, Q. Yin, X. Liu, X.-F. Jiang, Z. Chen, X. Yang, F. Huang and Y. Cao, Nano Energy, 2019, 60, 775–783 CrossRef CAS.
- H. Lim, J.-Y. Jeong, D. H. Lee, S.-W. Myeong, G. Shin, D. Choi, W. B. Kim, S. M. Choi and T. Park, J. Mater. Chem. A, 2023, 11, 25938–25944 RSC.
- D. Kim, H. Choi, W. Jung, C. Kim, E. Y. Park, S. Kim, N. J. Jeon, S. Song and T. Park, Energy Environ. Sci., 2023, 16, 2045–2055 RSC.
- K. Choi, D. H. Lee, W. Jung, S. Kim, S. H. Kim, D. Lee, S. Song and T. Park, Adv. Funct. Mater., 2022, 32, 2112027 CrossRef CAS.
- H. U. Kim, T. Kim, C. Kim, M. Kim and T. Park, Adv. Funct. Mater., 2023, 33, 2208082 CrossRef CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, 2006 Search PubMed.
- M. Jones and G. D. Scholes, J. Mater. Chem., 2010, 20, 3533–3538 RSC.
- R. Berera, R. van Grondelle and J. T. Kennis, Photosynth. Res., 2009, 101, 105–118 CrossRef CAS PubMed.
- I. H. van Stokkum, D. S. Larsen and R. Van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- J. Zhang, B. Zhu, L. Zhang and J. Yu, Chem. Commun., 2023, 59, 688–699 RSC.
- C. O’Rourke and A. Mills, Chem. Commun., 2021, 57, 1242–1245 RSC.
- H. L. Tan, F. F. Abdi and Y. H. Ng, Chem. Soc. Rev., 2019, 48, 1255–1271 RSC.
- H. S. Lee, H. G. Song, H. Jung, M. H. Kim, C. Cho, J.-Y. Lee, S. Park, H. J. Son, H.-J. Yun and S.-K. Kwon, Macromolecules, 2016, 49, 7844–7856 CrossRef CAS.
- S. A. Hillman, R. S. Sprick, D. Pearce, D. J. Woods, W.-Y. Sit, X. Shi, A. I. Cooper, J. R. Durrant and J. Nelson, J. Am. Chem. Soc., 2022, 144, 19382–19395 CrossRef CAS PubMed.
- C.-L. Chang, W.-C. Lin, L.-Y. Ting, C.-H. Shih, S.-Y. Chen, T.-F. Huang, H. Tateno, J. Jayakumar, W.-Y. Jao and C.-W. Tai, Nat. Commun., 2022, 13, 5460 CrossRef CAS PubMed.
- C.-L. Chang, T.-F. Huang, W.-C. Lin, L.-Y. Ting, C.-H. Shih, Y.-H. Chen, J.-J. Liu, Y.-T. Lin, Y.-T. Tseng, Y.-H. Wu, Y.-E. Sun, M. H. Elsayed, C.-W. Chen, C.-H. Yu and H.-H. Chou, Adv. Energy Mater., 2023, 13, 2300986 CrossRef CAS.
- C. Dai, S. Xu, W. Liu, X. Gong, M. Panahandeh-Fard, Z. Liu, D. Zhang, C. Xue, K. P. Loh and B. Liu, Small, 2018, 14, 1801839 CrossRef PubMed.
- L. Hao, R. Shen, S. Chen, W. Bi, L. Wang, G. Liang, P. Zhang and X. Li, J. Mater. Chem. A, 2022, 10, 24064–24072 RSC.
- L. Liu, M. A. Kochman, W. Zhao, M. A. Zwijnenburg and R. S. Sprick, Chem. Commun., 2022, 58, 10639–10642 RSC.
- C. Li, H. Xu, H. Xiong, S. Xia, X. Peng, F. Xu and X. Chen, Adv. Funct. Mater., 2024, 2405539 CrossRef.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef CAS PubMed.
- P. B. Pati, G. Damas, L. Tian, D. L. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 10, 1372–1376 RSC.
- M. Axelsson, C. F. N. Marchiori, P. Huang, C. M. Araujo and H. Tian, J. Am. Chem. Soc., 2021, 143, 21229–21233 CrossRef CAS PubMed.
- S. An, K.-J. Jeong, S. Z. Hassan, G. Ham, S. Kang, J. Lee, H. Ma, J. Kwon, S. Y. Jeong, J. Yang, H. Y. Woo, H.-H. Cho, H. Cha, C. Y. Son and D. S. Chung, Adv. Sci., 2024, 2309786 CrossRef CAS PubMed.
- C. Ru, Y. Wang, P. Chen, Y. Zhang, X. Wu, C. Gong, H. Zhao, J. Wu and X. Pan, Small, 2023, 19, 2302384 CrossRef CAS PubMed.
- P. Chen, F. Ji, D. Ma, Y. Xie, X. Wu, M. Zhang, C. Ru, L. Zhou, J. Wu and X. Pan, J. Mater. Chem. A, 2023, 11, 21146–21152 RSC.
- W.-C. Lin, C.-L. Chang, C.-H. Shih, W.-C. Lin, Z. Yu Lai, J.-W. Chang, L.-Y. Ting, T.-F. Huang, Y.-E. Sun, H.-Y. Huang, Y.-T. Lin, J.-J. Liu, Y.-H. Wu, Y.-T. Tseng, Y.-R. Zhuang, B.-H. Li, A.-C. Su, C.-H. Yu, C.-W. Chen, K.-H. Lin, U.-S. Jeng and H.-H. Chou, Small, 2023, 19, 2302682 CrossRef CAS PubMed.
- W.-C. Lin, Y.-H. Wu, Y.-E. Sun, M. M. Elsenety, W.-C. Lin, J.-C. Yen, H.-K. Hsu, B.-H. Chen, H.-Y. Huang, C.-A. Chang, T.-F. Huang, Y.-R. Zhuang, Y.-T. Tseng, K.-H. Lin, S.-D. Yang, C.-H. Yu and H.-H. Chou, Angew. Chem., Int. Ed., 2024, e202407702 CAS.
- Y. Xiang, X. Wang, L. Rao, P. Wang, D. Huang, X. Ding, X. Zhang, S. Wang, H. Chen and Y. Zhu, ACS Energy Lett., 2018, 3, 2544–2549 CrossRef CAS.
- S. An, Z. Wu, H. Jeong, J. Lee, S. Y. Jeong, W. Lee, S. Kim, J. W. Han, J. Lim, H. Cha, H. Y. Woo and D. S. Chung, Small, 2023, 19, 2204905 CrossRef CAS PubMed.
- D. J. Woods, S. A. Hillman, D. Pearce, L. Wilbraham, L. Q. Flagg, W. Duffy, I. McCulloch, J. R. Durrant, A. A. Guilbert and M. A. Zwijnenburg, Energy Environ. Sci., 2020, 13, 1843–1855 RSC.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, W. Zhang, M. Moser, R. Sheelamanthula, L. Zhao, B. Willner, T. C. Hidalgo and H. Faber, Adv. Mater., 2022, 34, 2105007 CrossRef CAS PubMed.
- I. Montanari, A. F. Nogueira, J. Nelson, J. R. Durrant, C. Winder, M. A. Loi, N. S. Sariciftci and C. Brabec, Appl. Phys. Lett., 2002, 81, 3001–3003 CrossRef CAS.
- T. M. Clarke, F. C. Jamieson and J. R. Durrant, J. Phys. Chem. C, 2009, 113, 20934–20941 CrossRef CAS.
- A. A. Mohapatra, Y. Dong, P. Boregowda, A. Mohanty, A. Sadhanala, X. Jiao, A. Narayan, C. R. McNeill, J. R. Durrant and S. Patil, J. Phys. Chem. C, 2021, 125, 6886–6896 CrossRef CAS.
- E. McQueen, Y. Bai and R. S. Sprick, Nanomaterials, 2022, 12, 4299 CrossRef CAS PubMed.
- C. M. Aitchison, R. S. Sprick and A. I. Cooper, J. Mater. Chem. A, 2019, 7, 2490–2496 RSC.
- M. H. Elsayed, M. Abdellah, Y.-H. Hung, J. Jayakumar, L.-Y. Ting, A. M. Elewa, C.-L. Chang, W.-C. Lin, K.-L. Wang and M. Abdel-Hafiez, ACS Appl. Mater. Interfaces, 2021, 13, 56554–56565 CrossRef CAS PubMed.
- S. An, S. Z. Hassan, J.-W. Jung, H. Cha, C.-H. Cho and D. S. Chung, Small Methods, 2022, 6, 2200010 CrossRef CAS PubMed.
- Z. Fu, A. Vogel, M. A. Zwijnenburg, A. I. Cooper and R. S. Sprick, J. Mater. Chem. A, 2021, 9, 4291–4296 RSC.
- J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, Adv. Energy Mater., 2018, 8, 1802181 CrossRef.
- M. Sachs, H. Cha, J. Kosco, C. M. Aitchison, L. Francas, S. Corby, C. L. Chiang, A. A. Wilson, R. Godin, A. Fahey-Williams, A. I. Cooper, R. S. Sprick, I. McCulloch and J. R. Durrant, J. Am. Chem. Soc., 2020, 142, 14574–14587 CrossRef CAS PubMed.
- M. V. Pavliuk, M. Lorenzi, D. R. Morado, L. Gedda, S. Wrede, S. H. Mejias, A. Liu, M. Senger, S. Glover, K. Edwards, G. Berggren and H. Tian, J. Am. Chem. Soc., 2022, 144, 13600–13611 CrossRef CAS PubMed.
- X. Ruan, X. Cui, Y. Cui, X. Fan, Z. Li, T. Xie, K. Ba, G. Jia, H. Zhang, L. Zhang, W. Zhang, X. Zhao, J. Leng, S. Jin, D. J. Singh and W. Zheng, Adv. Energy Mater., 2022, 12, 2200298 CrossRef CAS.
- F. Dong, Z. Zhao, T. Xiong, Z. Ni, W. Zhang, Y. Sun and W.-K. Ho, ACS Appl. Mater. Interfaces, 2013, 5, 11392–11401 CrossRef CAS PubMed.
- A. Putta Rangappa, D. Praveen Kumar, K. H. Do, J. Wang, Y. Zhang and T. K. Kim, Adv. Sci., 2023, 10, 2300073 CrossRef CAS PubMed.
- B.-J. Ng, L. K. Putri, X. Y. Kong, Y. W. Teh, P. Pasbakhsh and S.-P. Chai, Adv. Sci., 2020, 7, 1903171 CrossRef CAS PubMed.
- C.-Q. Li, X. Du, S. Jiang, Y. Liu, Z.-L. Niu, Z.-Y. Liu, S.-S. Yi and X.-Z. Yue, Adv. Sci., 2022, 9, 2201773 CrossRef CAS PubMed.
- Y. Dong, H. Cha, H. L. Bristow, J. Lee, A. Kumar, P. S. Tuladhar, I. McCulloch, A. A. Bakulin and J. R. Durrant, J. Am. Chem. Soc., 2021, 143, 7599–7603 CrossRef CAS PubMed.
- M. Kim, S. U. Ryu, S. A. Park, Y.-J. Pu and T. Park, Chem. Sci., 2021, 12, 14004–14023 RSC.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater., 2020, 19, 559–565 CrossRef CAS PubMed.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, T. Fei, Y. Dong, R. Sougrat, G. T. Harrison, Y. Firdaus, R. Sheelamanthula, B. Purushothaman, F. Moruzzi, W. Xu, L. Zhao, A. Basu, S. De Wolf, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Nat. Energy, 2022, 7, 340–351 CrossRef CAS.
- S. D. Dimitrov, Z. Huang, F. Deledalle, C. B. Nielsen, B. C. Schroeder, R. S. Ashraf, S. Shoaee, I. McCulloch and J. R. Durrant, Energy Environ. Sci., 2014, 7, 1037–1043 RSC.
- A. Liu, S. Wang, H. Song, Y. Liu, L. Gedda, K. Edwards, L. Hammarström and H. Tian, Phys. Chem. Chem. Phys., 2023, 25, 2935–2945 RSC.
- A. Liu, L. Gedda, M. Axelsson, M. Pavliuk, K. Edwards, L. Hammarström and H. Tian, J. Am. Chem. Soc., 2021, 143, 2875–2885 CrossRef CAS PubMed.
- S. U. Ryu, D. H. Lee, Z. U. Rehman, J.-C. Lee, H. Lim, G. Shin, C. E. Song and T. Park, Chem. Eng. J., 2024, 485, 149865 CrossRef CAS.
- Y. Bai, C. Li, L. Liu, Y. Yamaguchi, M. Bahri, H. Yang, A. Gardner, M. A. Zwijnenburg, N. D. Browning, A. J. Cowan, A. Kudo, A. I. Cooper and R. S. Sprick, Angew. Chem., Int. Ed., 2022, 61, e202201299 CrossRef CAS PubMed.
- S. Nandy, T. Hisatomi, M. Nakabayashi, H. Li, X. Wang, N. Shibata, T. Takata and K. Domen, Joule, 2023, 7, 1641–1651 CrossRef CAS.
- F. Dionigi, P. C. K. Vesborg, T. Pedersen, O. Hansen, S. Dahl, A. Xiong, K. Maeda, K. Domen and I. Chorkendorff, J. Catal., 2012, 292, 26–31 CrossRef CAS.
- S. Linley and E. Reisner, Adv. Sci., 2023, 10, 2207314 CrossRef CAS PubMed.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- J. Lee, G. W. Kim, M. Kim, S. A. Park and T. Park, Adv. Energy Mater., 2020, 10, 1902662 CrossRef CAS.
- H. I. Kim, J. Lee, M. J. Choi, S. U. Ryu, K. Choi, S. Lee, S. Hoogland, F. P. G. de Arquer, E. H. Sargent and T. Park, Adv. Energy Mater., 2020, 10, 2002084 CrossRef CAS.
- J. Lee, S. A. Sang, S. U. Ryu, D. Chung, T. Park and S. Y. Son, J. Mater. Chem. A, 2020, 8(41), 21455–21473 RSC.
- J. A. Chiong, L. Michalek, A. E. Peña-Alcántara, X. Ji, N. J. Schuster and Z. Bao, J. Mater. Chem. C, 2023, 11, 15205–15214 RSC.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef CAS PubMed.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick and A. I. Cooper, Nature, 2020, 583, 237–241 CrossRef CAS PubMed.
- L. Yao, N. Guijarro, F. Boudoire, Y. Liu, A. Rahmanudin, R. A. Wells, A. Sekar, H.-H. Cho, J.-H. Yum, F. Le Formal and K. Sivula, J. Am. Chem. Soc., 2020, 142, 7795–7802 CrossRef CAS PubMed.
- H.-H. Cho, L. Yao, J.-H. Yum, Y. Liu, F. Boudoire, R. A. Wells, N. Guijarro, A. Sekar and K. Sivula, Nat. Catal., 2021, 4, 431–438 CrossRef CAS.
- Y. Yang, M. A. Zwijnenburg, A. M. Gardner, S. Adamczyk, J. Yang, Y. Sun, Q. Jiang, A. J. Cowan, R. S. Sprick, L.-N. Liu and A. I. Cooper, ACS Nano, 2024, 18, 13484–13495 CrossRef CAS PubMed.
Footnote |
| † Jinhyuk Choi and Wooteak Jung contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |