
Rejuvenating propylene carbonate-based electrolytes by regulating the coordinated structure toward all-climate potassium-ion batteries†
Zixing
Wang
a,
Kang
Luo
a,
Jian-Fang
Wu
a,
Peng
Gao
a,
Kexuan
Wang
b,
Shi
Chen
 b,
Jian
Tu
c,
Xiulin
Fan
d and
Jilei
Liu
*a
b,
Jian
Tu
c,
Xiulin
Fan
d and
Jilei
Liu
*a
aSchool of Materials Science and Engineering, Hunan Joint International Laboratory of Advanced Materials and Technology of Clean Energy, Hunan Province Key Laboratory for Advanced Carbon Materials and Applied Technology, Hunan University, Changsha 410082, China. E-mail: liujilei@hnu.edu.cn
bJoint Key Laboratory of the Ministry of Education, Institute of Applied Physics and Materials Engineering, University of Macau, Avenida da Universidade, Taipa, Macau 999078, China
cLI-FUN Technology Corporation Limited, Zhuzhou 412000, Hunan, China
dState Key Laboratory of Silicon Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, Zhejiang, China
First published on 22nd November 2023
Abstract
Potassium-ion batteries (PIBs) suffer from a restricted desolvation process, unstable interfaces and severe capacity deterioration at extreme temperatures, which hinders their application as an alternative technology to lithium-ion batteries. Herein, by regulating the ion–solvent-coordinated structure, substantial ion-pairs and aggregates are formed while the solvent-separated ion pairs are reduced, enabling a KF-rich interface and a low desolvation barrier to improve wide-temperature performance. Consequently, the designed propylene carbonate-based electrolyte rejuvenates graphite anodes, delivering a high reversible capacity of ∼220 mA h g−1 without attenuation at 0–60 °C. Moreover, graphite‖K–FeHCFe full cells can fully restore their original capacity at 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C and 25°C even after operation at 60°C and −20°C. Remarkably, the full cells also exhibit a high room-temperature capacity retention of ∼63.7 mA h g−1 (66.6%) at −20 °C and achieve stable cycling over 1000 cycles at 0 °C (capacity retention of 92.1%), 25 °C (85.7%) and 60 °C (85.3%). This work rejuvenates propylene carbonates in graphite-based all-climate potassium-ion batteries, thereby promoting the development of a low-cost electrochemical energy storage system.
°C and 25°C even after operation at 60°C and −20°C. Remarkably, the full cells also exhibit a high room-temperature capacity retention of ∼63.7 mA h g−1 (66.6%) at −20 °C and achieve stable cycling over 1000 cycles at 0 °C (capacity retention of 92.1%), 25 °C (85.7%) and 60 °C (85.3%). This work rejuvenates propylene carbonates in graphite-based all-climate potassium-ion batteries, thereby promoting the development of a low-cost electrochemical energy storage system.
Broader contextLarge-scale energy storage systems are critical to address the intermittent supply and unbalanced energy distribution. Nevertheless, lithium-ion batteries (LIBs) have failed to meet the requirements because of the scarcity of lithium and high price for manufacturing. Therefore, new secondary batteries are in high demand to replace LIBs. The development of low-cost potassium-ion batteries (PIBs) has attracted extensive attention recently due to the abundant potassium resources, low redox potential of K+/K, smaller Stokes’ radius of K+ and reversible K+ de-/intercalation in graphite, which makes it an ideal alternative to LIBs. However, the practical operation of PIBs at extreme temperatures suffers from slow reaction kinetics and severe capacity deterioration. Among various methods to increase the temperature tolerance, optimal design of the electrolyte formulations evolves as a crucial part of widening the operating temperature range of PIBs at this stage, while the in-depth understanding of the electrolytes and the effects on the electrochemical performance of PIBs under extreme conditions remain unclear. Therefore, it is of great significance to systematically study the temperature tolerance of PIB electrolytes and further promote the development of low-cost electrochemical energy storage technology in the field of large-scale energy storage systems, polar scientific investigation and other low temperature conditions. |
Introduction
Potassium-ion batteries (PIBs) are considered potential alternatives to lithium-ion batteries (LIBs) due to the abundant terrestrial resources (2.09 wt%) of K and reversible intercalation/de-intercalation of K ions into graphite.1–7 Importantly, PIBs exhibit good rate performance potential for the sake of the small Stokes’ radius of K+ (3.6 Å in propylene carbonate (PC)), and high theoretical energy density because of the low redox potential (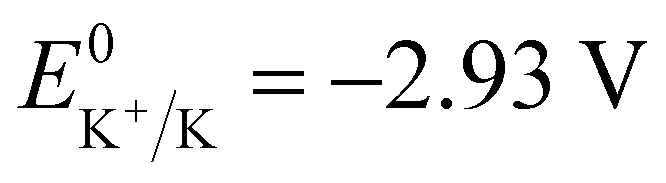 ).1,8,9 Except for the electrochemical performance, cost and safety, which have been improved considerably, the operating temperature of PIBs should also be expanded to adapt to the extreme conditions, such as cold winter or hot summer. At low temperatures, the batteries suffer from slow chemical and electrochemical reaction kinetics, difficulty in charging and rapid capacity decay.10–12 And the problems of severe decomposition of electrolytes, unstable solid electrolyte interfaces (SEI) and thermal runaway always exist when the batteries are operated at high temperatures.13,14 Therefore, it is necessary to understand the failure mechanism of PIBs employed in wide temperature ranges, which will guide the improvements of their electrochemical performance, extend their large-scale application, and further promote their commercialization.
).1,8,9 Except for the electrochemical performance, cost and safety, which have been improved considerably, the operating temperature of PIBs should also be expanded to adapt to the extreme conditions, such as cold winter or hot summer. At low temperatures, the batteries suffer from slow chemical and electrochemical reaction kinetics, difficulty in charging and rapid capacity decay.10–12 And the problems of severe decomposition of electrolytes, unstable solid electrolyte interfaces (SEI) and thermal runaway always exist when the batteries are operated at high temperatures.13,14 Therefore, it is necessary to understand the failure mechanism of PIBs employed in wide temperature ranges, which will guide the improvements of their electrochemical performance, extend their large-scale application, and further promote their commercialization.
The fundamental approaches to widen the operating temperature of batteries focus on improving the intrinsic properties of the batteries, such as electrodes,14–16 electrolytes17–19 and electrode–electrolyte interfaces.20–23 Currently, remarkable progresses are made to mitigate the impact of high or low temperatures on PIB performance by designing electrodes with a suitable structure (e.g., interlayer spacing and particle sizes)14,15 and excellent physical and chemical properties (e.g., thermal stability and conductivity).16,24 For example, Teng et al. reported robust high-temperature PIBs enabled by a carboxyl functional group energy storage mechanism and the p-phthalic acid electrodes, which displayed an excellent stability of 86% capacity retention after 160 cycles at 62.5 °C.14 Shui et al. reported the fabrication of reduced graphene oxide (RGO) anodes with surface defects, high electrical conductivity and large interlayer spacing for PIBs, which presented a capacity of 171 mA h g−1 at −40 °C.15 However, as an indispensable component in cells,25–28 the in-depth understanding of the electrolyte and its effects on electrochemical performance of PIBs under extreme conditions remain unclear. Therefore, optimal electrolyte design is crucial in widening the operating temperature range of PIBs.
Although widely adopted in commercial electrolytes, the narrow liquid range (36.4 °C), high viscosity, and low anodic stability of ethylene carbonate (EC) based electrolytes, substantially limit their electrochemical performance and hence confine the application scenarios of batteries in a narrow temperature range (i.e., −20 to 60 °C).28–30 Alternatively, propylene carbonate (PC) has been a widely used solvent since the early development of lithium metal batteries because of the wide liquid temperature range (−49 to 240 °C), strong solvation capability, and high anodic stability.22,29,30 However, PC is incompatible with the graphite anode, because of the continuous decomposition of PC and the incomplete solid electrolyte interphase (SEI) film on the surface of the graphite anode, resulting in formidable solvent co-intercalation and catastrophic exfoliation of graphite layers.22,29,31 At present, the key to solving this thorny problem is to form a stable SEI on graphite, such as using fluorinated solvents22 or developing highly concentrated electrolytes,29,32 localized highly concentrated electrolytes.33 Therefore, it is of great significance to systematically study the temperature tolerance of PC-based electrolytes and further rejuvenate the application of PC in all-climate electrolytes.
In this work, we have developed a PC-based electrolyte using only thermally stable PC and potassium bis(fluorosulfonyl)amide (KFSI) for sustainable PIBs featuring all-climate adaptability. The highly concentrated electrolyte prepared by regulating the ion–solvent-coordinated structure can effectively alleviate the challenges of slow charge–discharge kinetics at low temperatures, and insufficient interfacial stability at high temperatures. It was found that the highly concentrated electrolyte weakened the affinity between PC and K+, formed a more favorable solvation structure of contact ion pair (CIP) and aggregate (AGG), promoted the desolvation on the graphite surface, and formed a robust anion-derived KF-rich interface, thereby improving the electrochemical stability of the graphite anode. Furthermore, the PC-based electrolyte exhibited enhanced electrochemical performance when used with Prussian white (K–FeHCFe) cathodes. As a result, the graphite‖K half-cell with 4.0 M PC–KFSI delivered a high reversible capacity of ∼220 mA h g−1 without attenuation for more than 6 months at 0 °C and maintained stable cycling even at elevated temperatures up to 60 °C. Moreover, the graphite‖K–FeHCFe full cells with this designed electrolyte showed excellent long-term cycling stability (up to 1000 cycles) and variable temperature reversibility (–20 to 60 °C), demonstrating the great practical feasibility of regulating the solvation structure for PC-based electrolytes operating under all-climate conditions.
Results and discussion
Rational design of the PC-based electrolyte
The graphite‖K half-cells with commercial electrolytes (0.8 M KPF6 in EC/DEC) displayed rapid capacity fading at high temperatures and difficulty in charging at low temperatures, as shown in Fig. 1(a) and Fig. S1 (ESI†). Specifically, at 60 °C, the capacity decreased from 217 to 144 mA h g−1 for the first five cycles. And the commercial electrolyte showed a low capacity of ∼70 mA h g−1 at 0 °C. These poor performances were attributed to the high melting point (36.4 °C), poor thermal stability of EC and limited solubility of KPF6 (∼0.9 mol kg−1)34 (Fig. 1(b)). Therefore, new electrolyte formulations are highly desirable to widen the temperature range of PIBs. PC was widely used as a co-solvent in low-temperature electrolytes due to its low melting point (−49 °C) and weak temperature dependence of the SEI.22,35–38 However, the poor compatibility of PC with graphite anodes hindered its application. The highly concentrated electrolytes have attracted much attention because the unique solvation structure formed a favorable interface that can reversibly intercalate K+ in the graphite anode. Nevertheless, the commonly used KPF6 showed limited solubility, so we chose the KFSI electrolyte with higher ionic conductivity (7.2 mS cm−1) and higher solubility (7.5 mol kg−1) (Fig. 1(b)). Consequently, we selected the combination of thermally stable PC and highly soluble KFSI to rejuvenate the application of PC in all-climate electrolytes by regulating the ion–solvent-coordinated structure.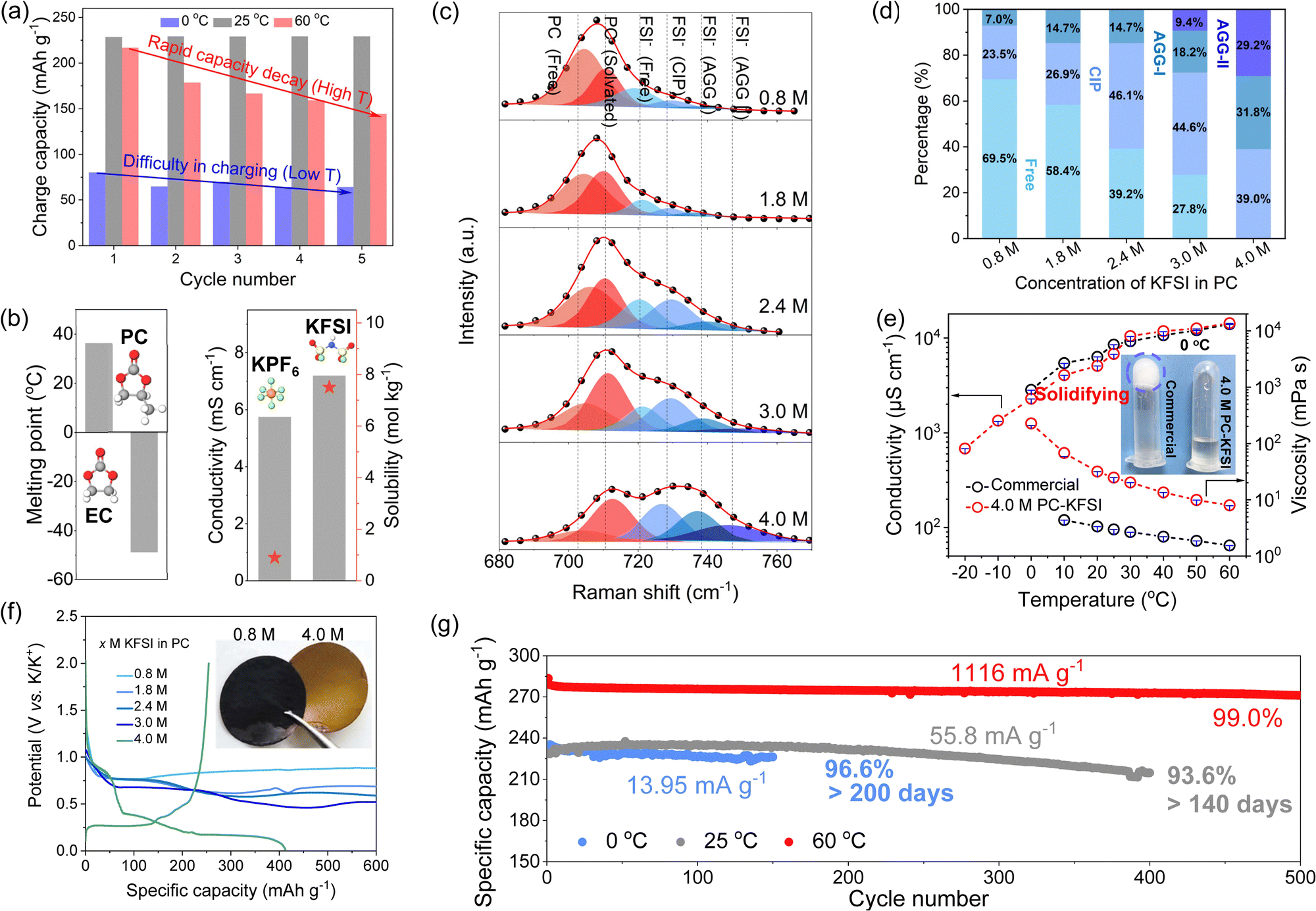 | ||
| Fig. 1 Rational design of a PC-based electrolyte and its electrochemical performance. (a) The charge capacity of initial five cycles of Gr‖K half cells in commercial electrolytes at 0 °C, 25 °C and 60 °C with a current density of 13.95 mA g−1. (b) The comparison of physical and chemical properties of PC and EC, KFSI and KPF6. (c) Raman spectra of KFSI-PC electrolytes in the region from 680 to 770 cm−1. Black plots and red solid lines represented the original spectra and the fitting results, respectively. Curve fitting was performed with Gaussian–Lorentzian functions. (d) Distribution of the K+ solvates for different concentrations of KFSI in PC electrolytes from Raman spectra. (e) Ionic conductivities and viscosities of different electrolytes at different temperatures. Inset showed a digital image of the electrolytes after being stored at 0 °C for 1 h. (f) Initial charge–discharge curves of the graphite anode in the PC electrolyte with different concentrations. (g) Cycling performance of K-Gr cells at 0 °C, 25 °C and 60 °C. | ||
The solvation structures of different concentrations of KFSI in PC electrolyte systems were studied by Raman vibrational spectroscopy in a frequency range from 680 to 770 cm−1, where the S–N–S vibration of the FSI− anion and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration of PC could be observed (Fig. 1(c) and Fig. S2, ESI†). The CO band of PC could be divided into two peaks: the peak at ∼705 cm−1 referred to the free PC, and the another peak at ∼712 cm−1 was associated with the K+ coordinated PC.39 Obviously, the coordination number of PC decreased with increasing salt concentration (Fig. S3, ESI†), suggesting that a small number of PC molecules participated in the solvation structure of K+. The bands at ∼720 cm−1, ∼729 cm−1, ∼737 cm−1, and ∼749 cm−1 were attributed to free FSI− anions, contact ion pairs (CIP, FSI− coordinating to one K+), aggregates-I (AGG-I, FSI− coordinating to two K+), and aggregates-II (AGG-II, FSI− coordinating to more than two K+), respectively.40–42 As shown in Fig. 1(d), the distribution of solvation structures as a function of electrolyte concentration was acquired from fitting results of the Raman spectra. In the dilute electrolyte, K+ was mainly solvated and shielded by solvents, and the FSI− anion existed in the form of free anions. With the further increase of the KFSI content, owing to the lack of sufficient PC molecules to separate K+ cations and FSI− anions, the charge shielding effect of K solvation weakened, and the cation–anion interaction became stronger, resulting in the formation of aggregates (AGGs) by the coordination of more than two cations to one anion. Specifically, the ratio of AGGs increased from 7% to 61% with increasing concentration, while the FSI− anion signal was almost absent. Compared with commercial dilute electrolytes, the highly concentrated electrolytes were composed of ion–solvent-coordinated structures (coordinated solvent PC molecules and FSI− anions), and there were almost no free solvent molecules and anions, which can effectively solve the problem of crystallization at low temperatures and evaporation/decomposition at high temperatures, so that the battery could work in a wide temperature range.
O vibration of PC could be observed (Fig. 1(c) and Fig. S2, ESI†). The CO band of PC could be divided into two peaks: the peak at ∼705 cm−1 referred to the free PC, and the another peak at ∼712 cm−1 was associated with the K+ coordinated PC.39 Obviously, the coordination number of PC decreased with increasing salt concentration (Fig. S3, ESI†), suggesting that a small number of PC molecules participated in the solvation structure of K+. The bands at ∼720 cm−1, ∼729 cm−1, ∼737 cm−1, and ∼749 cm−1 were attributed to free FSI− anions, contact ion pairs (CIP, FSI− coordinating to one K+), aggregates-I (AGG-I, FSI− coordinating to two K+), and aggregates-II (AGG-II, FSI− coordinating to more than two K+), respectively.40–42 As shown in Fig. 1(d), the distribution of solvation structures as a function of electrolyte concentration was acquired from fitting results of the Raman spectra. In the dilute electrolyte, K+ was mainly solvated and shielded by solvents, and the FSI− anion existed in the form of free anions. With the further increase of the KFSI content, owing to the lack of sufficient PC molecules to separate K+ cations and FSI− anions, the charge shielding effect of K solvation weakened, and the cation–anion interaction became stronger, resulting in the formation of aggregates (AGGs) by the coordination of more than two cations to one anion. Specifically, the ratio of AGGs increased from 7% to 61% with increasing concentration, while the FSI− anion signal was almost absent. Compared with commercial dilute electrolytes, the highly concentrated electrolytes were composed of ion–solvent-coordinated structures (coordinated solvent PC molecules and FSI− anions), and there were almost no free solvent molecules and anions, which can effectively solve the problem of crystallization at low temperatures and evaporation/decomposition at high temperatures, so that the battery could work in a wide temperature range.
Obviously, the introduction of an ion–solvent-coordinated structure in the electrolyte system will change its physical and electrochemical properties. The ionic conductivities and viscosities from −20 to 60 °C were compared in Fig. 1(e). For the commercial electrolytes, we could not measure the true ionic conductivities below 0 °C due to the inevitable solidification, which was solidified at 0 °C from the inset of Fig. 1(e). However, the highly concentrated electrolytes can still remain completely in liquid state after storage at 0 °C for 1 h, which was attributed to the absence of the free solvent in the highly concentrated electrolytes that made them behave like ionic liquids. The results of differential scanning calorimetry (DSC) indicated that no exothermic or endothermic peak was observed for the 4.0 M PC–KFSI electrolyte, suggesting that it can maintain a fully liquid state within the measuring temperature range from −80 to 20 °C, while the commercial electrolyte showed obvious exothermic peaks at 8 °C during cooling (Fig. S4, ESI†). Although the viscosity of the highly concentrated electrolyte was nearly one order of magnitude higher than that of the commercial electrolyte, which required further optimization of the formulation (localized highly concentrated electrolytes, weak-solvation electrolytes), the highly concentrated electrolyte still preserved a high conductivity of ∼680 μS cm−1 even at −20 °C, which was beneficial for building all-climate PIBs.
Traditional PC-based electrolytes would form an incomplete SEI film on the surface of the graphite anode, resulting in co-intercalation and further exfoliation of graphite, so we firstly investigated the compatibility of the graphite anode with the designed PC-based electrolyte rich in AGG solvation structures. The electrochemical performances of graphite in PC-based electrolytes with different concentrations were compared as shown in Fig. 1(f). The graphite‖K cells failed to work at low concentrations (<4.0 M) and displayed long irreversible discharge plateaus that decreased with the increased molarity, corresponding to the continuous co-intercalation and decomposition of PC. When the molarity reached 4.0 M, the reversible charging capacity was up to ∼250 mA h g−1, indicating an obvious KC8 species (inset of Fig. 1(f)), verifying the effectiveness of highly concentrated electrolyte for improving electrochemical reversibility of graphite in PC electrolyte. To investigate the morphologies of graphite (Fig. S5, ESI†), SEM images were recorded using a SEM equipped with a vacuum transfer accessory. It is clear that the Gr electrode in 0.8 M PC–KFSI (∼80 μm) was much thicker than that in 4.0 M PC–KFSI (∼25 μm). Furthermore, good cycling and rate performance in a wide temperature range (0 °C to 60 °C) was clearly demonstrated (Fig. 1(g) and Fig. S6, ESI†). Among them, the graphite‖K half-cell with 4.0 M PC–KFSI exhibited a reversible capacity of ∼235 mA h g−1 and a retention of 93.6% after 400 cycles at 25 °C, thus outperforming the cells with the commercial electrolyte and other reported works remarkably (Table S1, ESI†).5,31,43–51 Even at 0 °C, the graphite‖K half-cell with 4.0 M PC–KFSI still delivered a high reversible capacity of ∼220 mA h g−1 without attenuation for more than 6 months. In contrast, the cell showed a limited capacity of ∼70 mA h g−1 with rapid deterioration in commercial electrolytes originating from its solidification (Fig. S1, ESI†). Moreover, the graphite‖K half-cell maintained cycling stably in highly concentrated electrolytes even at elevated temperatures up to 60 °C, regardless of low or high current densities (Fig. S7, ESI†), superior to the commercial electrolytes. Simultaneously, the symmetric K‖K and K‖Cu half cells based on the highly concentrated electrolyte could operate stably under varied temperatures (Fig. S8, ESI†), further demonstrating the practical feasibility of regulating the solvation structure for PC-based electrolytes.
Interphase chemistry
For dilute electrolytes, SEI was mainly formed by the decomposition of the solvents, while substantial ion-pairs and aggregates were formed in highly concentrated electrolytes, which showed a higher energy level of the lowest unoccupied molecular orbital, leading to better reduction stability of solvent molecules. Moreover, such a solvation structure also promoted the formation of anion-derived SEI, further protecting the solvent molecule from reductive decomposition.10 The potassiation/depotassiation potential profiles maintained a similar shape as the temperature increased from 0 to 60 °C, indicating the presence of similar electrochemical reactions. However, the plateaus of SEI formation were different between commercial electrolytes and highly concentrated electrolytes, and the plateaus advanced with increasing temperatures (Fig. S9, ESI†), which manifested differences in the formation mechanism and components of SEI. According to the intensity evolution in the deconvoluted spectra of C1s and F1s from depth-profiling XPS experiments (Fig. S10–S13, ESI† and Fig. 2(a)), it was concluded that the outer layer of SEI was mainly composed of rich organic species (ethylene oxide oligomers and potassium ethylene dicarbonate),52,53 which were formed by the decomposition of solvents; the inner layer was mainly KF-rich species, which arose from the decomposition of the anion in designed solvation structures. It could also be found that the intensity changes of the high-concentration electrolyte at different temperatures were similar, indicating the excellent thermal stability of the special SEI formed in highly concentrated electrolytes. However, the strength of the SEI formed in the commercial electrolyte changed significantly in both C1s and F1s profiles with increasing temperature (Fig. 2(a)). Generally, the intensity of the bulk Gr peak (C–C/C–H, ∼285.0 eV) in the C1s spectra gradually increased as the sputtering progressed due to the removal of the surface SEI. The areal intensity ratios of the bulk Gr peak in the entire C1s region (279.0–293.0 eV) of the highly concentrated electrolyte samples during sputtering were significantly smaller than that of the commercial electrolyte (Fig. 2(b)), indicating that the SEI formed in a highly concentrated electrolyte was more robust or thicker. This might be attributed to the KF-rich species derived from the anion in highly concentrated electrolytes, while in the commercial electrolyte the SEI was mainly composed of solvent-derived organic components as further proven by the KF contents in Fig. 2(c). Compared with organic components, the KF showed lower activation energy, higher high-voltage stability, lower solubility in electrolytes and lower electronic conductivity, which was expected to improve the overall strength of SEI, reduce the specific resistance and improve cycling stability.10,54 The difference in XPS results indicated that the 4.0 M PC–KFSI rich in ion–solvent-coordinated structures leads to the formation of a robust and KF-rich SEI on the Gr anode surface, which can reduce the decomposition of PC solvents and improve the stability under extreme conditions.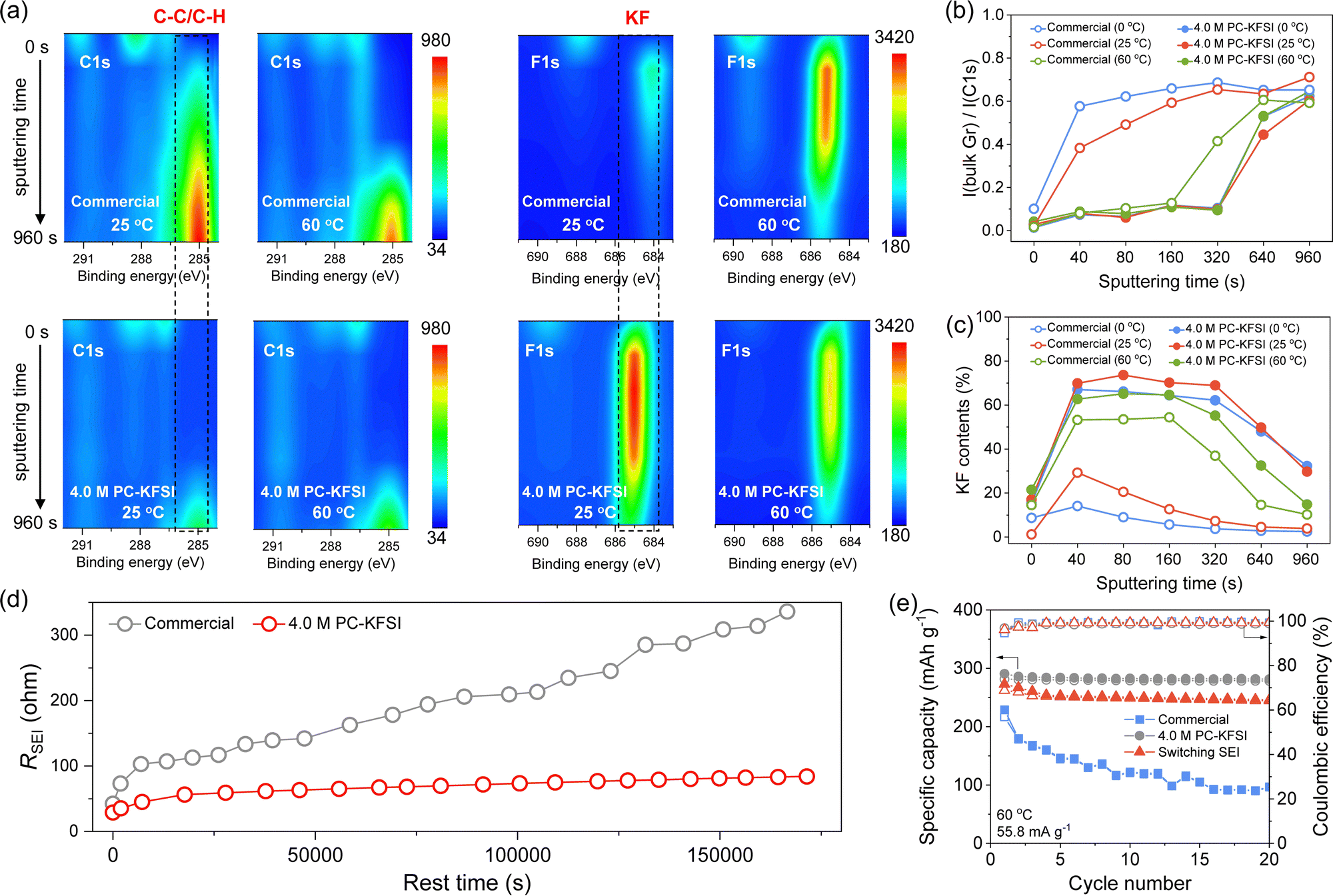 | ||
| Fig. 2 SEI layer compositions and stability of the PC-based electrolyte. (a) Contour plots of the XPS C1s and F1s of the pre-cycled graphite electrodes with 4.0 M PC–KFSI and commercial electrolytes at 25 and 60 °C. (b) Areal intensity ratio of the C1s peak at ∼285.0 eV over the entire C 1s region. (c) KF contents changes in SEI at 0, 25 and 60 °C. (d) The interface resistance RSEI as a function of the rest time at 60 °C. (e) The cycle performance of graphite anodes reassembled in different electrolytes at 60 °C. | ||
Furthermore, in order to study the stability of SEI, the impedance of graphite‖K half-cells was monitored at the open-circuit potential for ∼20 h after being discharged up to a pre-defined potential. The impedance plot clearly showed that the resistance of the SEI formed in the commercial electrolyte increased significantly during the resting period, while the impedance of the highly concentrated electrolyte was relatively small and stable (Fig. S14, ESI†). We plotted the resistance against rest time after collecting all RSEI values (Fig. 2(d)), and it can be clearly observed that the RSEI values of the highly concentrated electrolyte were lower than 100 ohms even after resting for nearly 50 hours at 60 °C, ensuring stable operation of the cells in a high-temperature environment. To verify the effectiveness of SEI derived from highly concentrated electrolytes for enhancing the all-climate stability of graphite, the exchange experiments were designed (Fig. S15, ESI†). Specifically, the graphite anode was precycled in 4.0 M PC–KFSI to form a specific SEI (Gr@SEI), then taking out Gr@SEI to reassemble the half-cell with the commercial electrolyte. Inspiringly, the Gr@SEI‖K cell exhibited cycling performance as stable as that of highly concentrated electrolytes at 60 °C (Fig. 2(e)), significantly better than that of commercial electrolytes. The results showed that the KF-rich SEI obtained in highly concentrated electrolytes was beneficial for improving the high-temperature stability of commercial electrolytes, further verifying the superiority of this ion–solvent-coordinated structure. However, low-temperature performance was not improved by switching SEI, suggesting that SEI played a dominant role in high-temperature stability, while low temperature might be attributed to other factors.
Kinetics behavior
The desolvation process was considered to be the main interfacial kinetic barrier at low temperature, thus accelerating the desolvation kinetics could effectively improve the low-temperature performance of batteries.20,21,55,56 Limited kinetics induced polarization was typically observed for the battery charged at low temperatures. Therefore, in order to better study the kinetics of batteries at low temperature, three-electrode electrochemical impedance spectroscopy (EIS) was conducted and the distribution of the relaxation time (DRT)57 were further converted to uncovering and separating the contribution of different processes to cell impedance (Fig. S16, ESI†). According to the previous works, two processes (RS and Rct) of the three-electrode cells have been identified and were attributed to the SEI formation and desolvation processes.20 As shown in Fig. 3(a) and (b), the reaction processes of charging and discharging were similar in different temperature ranges for 4.0 MPC–KFSI and commercial electrolytes, except that as the temperature increased, the formation potential of SEI advanced and the overall impedance decreased, consistent with the result of dQ/dV (Fig. S9, ESI†). For the discharging process in highly concentrated electrolytes (Fig. 3(a)), the Rct peak (10−1–102 s) strength increased obviously and the RSEI (10−3–10−1 s) shifted significantly at 1.0–0.5 V, corresponding to SEI formation. At 0.5–0.01 V, the Rct peak intensity decreased significantly with the intercalation of K+. This phenomenon was reversible during the charging process, indicating the stable potassiation/depotassiation behavior.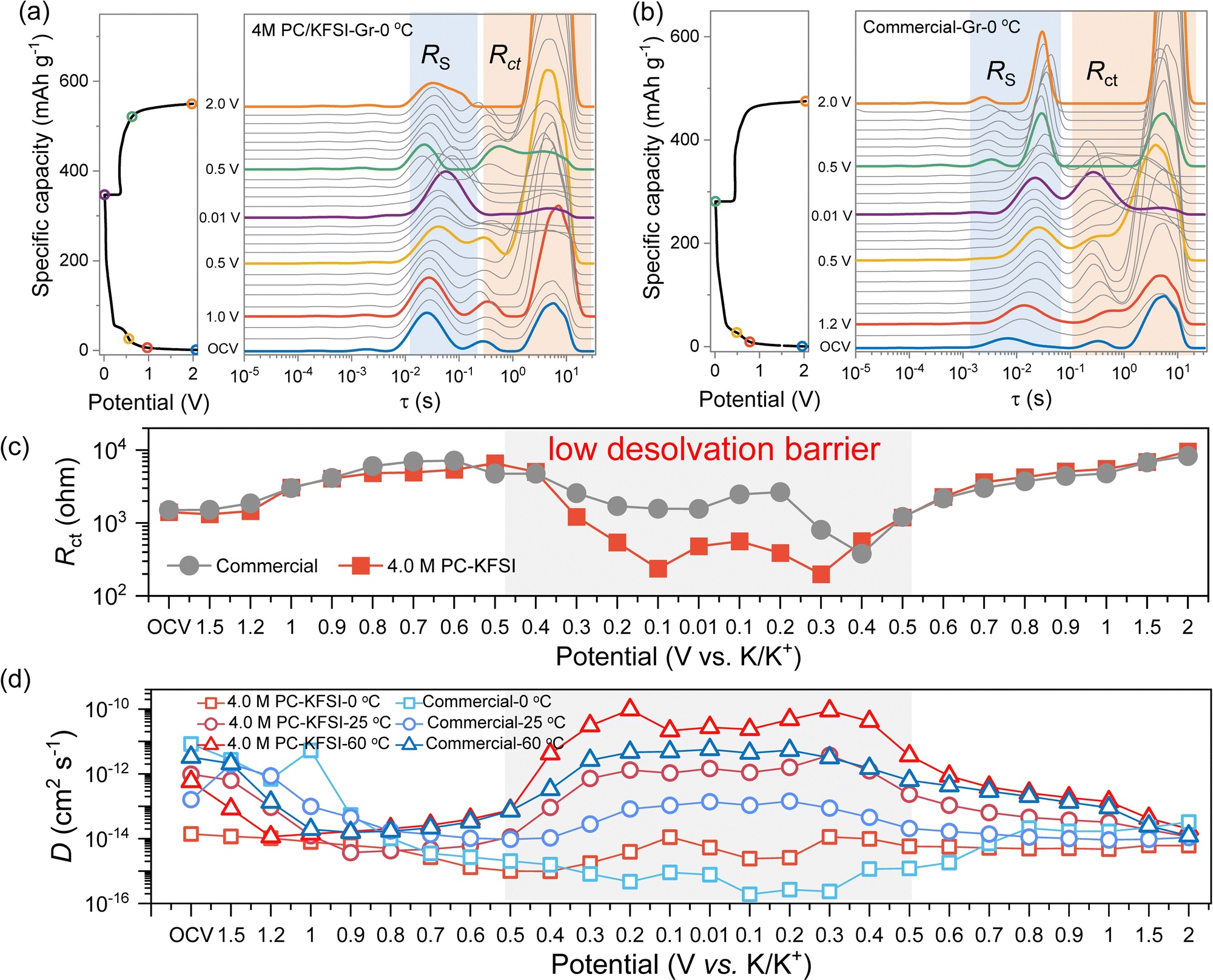 | ||
| Fig. 3 Kinetics behavior of the PC-based electrolyte. In situ EIS during 1st charge–discharge process at 0 °C of the graphite‖K half-cell with (a) 4.0 M PC–KFSI and (b) commercial electrolytes. (c) Fitting impedance versus voltage profiles based on DRT results for Rct. (d) K+ diffusion coefficients at various voltages for graphite‖K half-cells at different temperatures. | ||
It was found that the desolvation and diffusion ability of the highly concentrated electrolyte were significantly better than that of the commercial electrolyte at low temperatures. As shown in Fig. 3(c), the Rct values in highly concentrated electrolytes were smaller than those obtained in commercial electrolytes, which was ascribed to the homogeneous and conductive SEI formed by the CIP- and AGG-rich solvation structures. Based on the Nyquist plots at low frequency, the apparent K+ diffusion coefficients were calculated. As shown in Fig. 3(d), the D values of the highly concentrated electrolyte were an order of magnitude higher than that of the commercial electrolyte in the same temperature range during the process of intercalation and desorption of K+, which was consistent with the results of impedance. Obviously, 4.0 M PC–KFSI with a special ion–solvent coordination structure could not only form a KF-rich SEI interface, improving the high-temperature stability of batteries, but enabled fast desolvation and greatly reduced the interface kinetic barrier, improving low temperature performance.
Electrochemical performance of the graphite‖K–FeHCFe full cell
The highly concentrated electrolytes also showed excellent electrochemical stability under high voltage (Fig. S17, ESI†). Therefore, we carried out long-term charge–discharge cycling tests to check if this electrolyte was compatible with the Prussian white (K–FeHCFe) cathode material (Fig. S18, ESI†), which was the representative high-capacity cathode materials of PIBs because of their three-dimensional open framework structure, high stability, and low cost. As shown in Fig. 4(a), reasonable capacity retentions were achieved over 100 cycles at 0–60 °C for the cells using the 4.0 M PC–KFSI electrolytes, indicating the potential application of this highly concentrated electrolyte at high voltages. While the cells using the commercial electrolyte underwent severe capacity deterioration after 100 cycles (Fig. S19–S21, ESI†). Overall, the graphite anodes and the K–FeHCFe cathodes with 4.0 M PC–KFSI exhibited significantly better long-term cycling stability than those with commercial electrolytes at both low temperatures and high temperatures. The enhanced performance was believed to be associated with weak K+–solvent interactions, and robust anion-derived inorganic-rich interphases. Having confirmed the stable operation of both K–FeHCFe‖K and graphite‖K half-cells under all-climate conditions, 4.0 M PC–KFSI was further applied to graphite‖K–FeHCFe full cells, as diagrammatically shown in Fig. 4(b). According to the charge–discharge profiles, the graphite‖K–FeHCFe full cell exhibited an average potential of 3.45 V and a reversible capacity of 128.1 mA h g−1 at 20 mA g−1. The full-cell also displayed two reversible redox peaks (3.76/3.67 V and 3.4/3.06 V corresponding to the conversion between Fe2+HS/Fe3+HS and Fe2+LS/Fe3+LS), measured by dQ/dV curves (Fig. S22, ESI†). Moreover, a better rate performance could also be obtained in a graphite‖K–FeHCFe full cell with 4.0 M PC–KFSI, exhibiting high capacities of 111.6, 102.9, 94.9, 89.2, 86.4, 82.6 and 73 mA h g−1 at different rates of 50, 100, 200, 300, 400, 500 and 1000 mA g−1, respectively (25 °C, Fig. 4(c)). The graphite‖K–FeHCFe full cell showed a high-rate capability even at 0 °C (∼64 mA h g−1 at 200 mA g−1, Fig. S23a, ESI†) and 60 °C (∼94 mA h g−1 at 3000 mA h g−1, Fig. S23b, ESI†), respectively.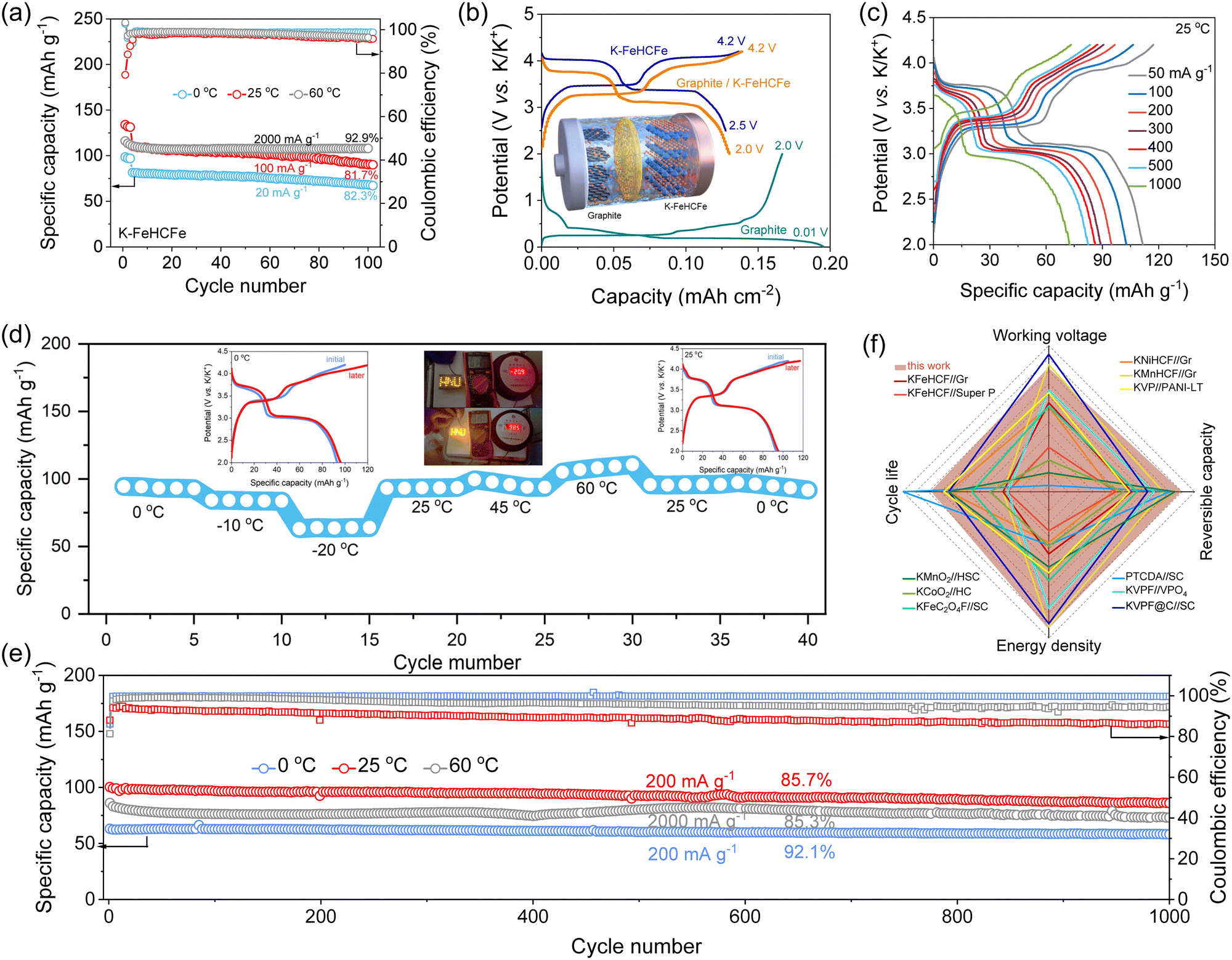 | ||
| Fig. 4 Electrochemical performance of graphite‖K–FeHCFe full cells. (a) Cycling stability of K–FeHCFe‖K profiles at different temperatures. (b) Typical depotassiation/potassiation profiles of the full battery. Inset shows the schematic representation of the working mechanism of the Gr‖K–FeHCFe full cells. (c) Rate performance of Gr‖K–FeHCFe cells at 25 °C. (d) The specific capacity of Gr‖K–FeHCFe cells in the concentrated electrolyte at varied temperatures. (e) The cycling stability of Gr‖K–FeHCFe cells at 0–60 °C in 4.0 M PC–KFSI. (f) Comparisons of the electrochemical performance in this work with those in the literature.6,8,58–68 | ||
The wide-temperature applicability of the graphite‖K–FeHCFe full cells was investigated. Fig. 4(d) showed the electrochemical performance of graphite‖K–FeHCFe full cells with 4.0 M PC–KFSI electrolytes, which first measured at a lower temperature (−20 °C) from 0 °C, then a higher temperature (60 °C), and finally returned to 0 °C. The graphite‖K–FeHCFe full cell with the highly concentrated electrolytes could still exhibit high retention of room-temperature capacity: ∼93.5 mA h g−1 (97.7%), ∼83.8 mA h g−1 (87.6%), ∼63.7 mA h g−1 (66.6%) when the temperature was reduced to 0 °C, −10 °C and −20 °C. For high-temperature application, the graphite‖K–FeHCFe full cells with the highly concentrated electrolytes exhibited good cycle stability and a high capacity of about ∼108 mA h g−1 at 60 °C (Fig. 4(d)). Surprisingly, this full cell can fully restore its original capacity at 0°C and 25°C even after high-temperature operation at 60°C and low-temperature at −20°C, indicating the stability of the robust KF-rich interphases formed by highly concentrated electrolytes under all climates (inset of Fig. 4(d)). The assembled full cell can be used to power an LED panel, even at −20or 100 °C (inset of Fig. 4(d)). According to the high reversible capacity and high working voltage of the graphite‖K–FeHCFe full cell under all-climate conditions, the specific energies were as high as 203.8 Wh kg−1 (0 °C), 245.5 Wh kg−1 (25 °C), and 220.8 Wh kg−1 (60 °C), surpassing most state-of-the-art PIBs (based on the active materials mass, Table S2, ESI†). Note that the graphite‖K–FeHCFe full cells with the highly concentrated electrolytes could run 1000 cycles reversibly despite the aggravated electrode/electrolyte interface degradation at high temperatures and sluggish reaction kinetic at low temperatures, indicating the practicality of highly concentrated electrolytes under all-climate conditions (Fig. 4(e) and Fig. S24, ESI†). More precisely, after 1000cycles, 92.1% of capacity was retained at 0°C, 85.7% at 25°C, and 85.3% at 60°C. As summarized in Fig. 4(f), the highly concentrated electrolytes in this work opened a unique direction for designing all-climate electrolytes without complex formulation. We believe that once electrode modifications and further electrolyte formulation optimization are employed, the cells could operate reliably at both extremely cold (<−20 °C) and hot (>60 °C) temperatures.
Conclusions
In summary, we rejuvenate PC-based electrolytes in all-climate PIBs by regulating the ion–solvent-coordinated structure. As a proof of concept, we obtained highly concentrated electrolytes rich in AGG and CIP solvation structures based on the PC solvent with a wide liquid temperature range and KFSI salt with high ionic conductivity. A rationally designed 4.0 M PC–KFSI electrolyte is able to form highly thermally stable anion-derived KF-rich interphases on the graphite anode and achieve a low desolvation barrier, leading to stable potassiation/depotassiation behaviors under extreme environments. Specifically, the graphite anodes exhibit a reversible capacity of >220 mA h g−1, and maintain 96.6% capacity retention after 200 days at 0 °C, 93.6% after 400 cycles at 25 °C, and 99.0% after 500 cycles at 60 °C. Notably, the highly concentrated 4.0 M PC–KFSI electrolyte has also been demonstrated to enable stable operation up to 1000 cycles of graphite‖K–FeHCFe full cells over a wide temperature range from −20 to 60 °C. This work proposes the regulation of the ion–solvent-coordinated structure to formulate all-climate electrolytes, which undoubtedly opens new avenues for battery systems operated at extreme temperatures.Experimental section/methods
Preparation of electrolytes
Potassium bis(fluorosulfonyl)imide (KFSI) was purchased from Tokyo Chemical Industry and stored in a glove box without further treatment. Propylene carbonate (PC) was provided by Sigma Aldrich and dried using 4 Å molecular sieves for 24 h prior to electrolyte formulation. The electrolyte solutions were prepared in an Ar-filled glove box at room temperature, where different concentrations (0.8–4.0 M) of KFSI were dissolved in PC solution in a volumetric flask, then poured into the vial and stirred with a magnetic stirrer for 12 h. Commercial electrolyte (0.8 M KPF6 in EC-DEC) was directly provided by Dodo Chem (Suzhou, Jiangsu Province, China).Preparation of electrodes
The graphite electrode slurry was prepared by mixing 85 wt% natural graphite powder (Gr, Beijing electric carbon plant), 10 wt% sodium alginate and 5 wt% ketjen black in deionized water, and further stirred with a magnetic stirrer for 12 h. Then the obtained slurry was homogeneously coated on Cu foil and transferred to a vacuum oven at 80 °C for drying and cut into 12 mm or 14 mm discs. The mass loading of the graphite anode is ca. 1.5–2.0 mg cm−2. Similarly, the Prussian white (K–FeHCFe, PW) electrode was prepared by mixing 70 wt% PW (prepared via a typical coprecipitation method), 20 wt% ketjen black, and 10 wt% polyvinylidene fluoride in N-methyl pyrrolidine solvent. The slurry was coated onto the aluminum foil and dried at 110 °C under vacuum for 12 h. The diameter of the cathode is 12 mm, and the loading of each electrode is ca. 1.0–2.0 mg cm−2.Materials characterizations
The morphologies were investigated by scanning electron microscopy using a JSM-7610FPlus SEM equipped with a vacuum transfer accessory, conducted at the KW-ST Lab (https://www.kewei-scitech.com). Differential scanning calorimetry (DSC, NETZSCH DSC204F1) was used to evaluate the thermal behavior of different electrolytes. XRD was examined using a PANalytical Empyrean X-ray diffractometer with Cu Kα radiation. The Branauer–Emmett–Teller (BET) specific surface area (SSA) was determined via nitrogen adsorption–desorption isotherms (JW-BK200C, Beijing JWGB SCI & TECH). The solvation structure was determined by Raman spectroscopy (WITEC alpha-300R) and Fourier transform infrared spectroscopy (Thermo Fisher Scientific, Nicolet iS50). The structure and components of the SEI on the electrodes were characterized using X-ray photoelectron spectroscopy (XPS, ESCALAB 250i) under nitrogen protection, with an argon ion gun (beam energy 2000 eV) to conduct the depth-profiling tests with etching time t = 0, 40, 80, 160, 320 s, 640 s and 960 s.Electrochemical measurements
All the electrochemical tests were conducted by using coin cells (CR2032) assembled in an Ar-filled glove box with oxygen and water contents below 0.01 ppm. Prior to assembling graphite‖K–FeHCFe full cells, the K–FeHCFe cathodes were predepotassiated in half-cells at 20 mA g−1 (25 °C) and graphite anodes were prepotassiated in half-cells at 13.95 mA g−1 (45 °C). Then, the full cells were assembled using depotassiated K–FeHCFe cathodes and potassiated graphite anodes in a 4.0 M PC–KFSI electrolyte with Whatman glass fiber as the separator and the N/P values were set to be 1:1.4. The three-electrode cells were assembled in a 2032-type coin cell by placing the reference electrode (K@CNT) between the working electrode (Gr) and the counter electrode (K@CNT), separated by two pieces of glass fiber. The electrochemical voltage windows of the electrolytes were measured via the linear sweep voltammetry (LSV) method with Pt as the working electrode and potassium slices as counter and reference electrodes. The scan rate of LSV tests was 0.5 mV s−1. The electrochemical performances were determined using the LAND CT2001A battery testing system and the Neware testing instrument (BTS-610). Differential capacity versus potential (dQ/dV) profiles and electrochemical impedance spectra (EIS) were recorded on a Solartron electrochemical workstation. The dQ/dV curves were obtained in a voltage range of 0.01–2.0 V at a scan rate of 13.95 mA g−1 for K‖graphite half-cells and 2.0–4.2 V at a scan rate of 20 mA g−1 for graphite‖K–FeHCFe full cells. The EIS tests were performed in the frequency range of 0.01–106 Hz with an oscillation amplitude of 10 mV.
Author contributions
J. L. and Z. W. conceived the project. J. L., J. W., P. G., J. T. and X. F. supervised the work. Z. W. carried out most of the experiments and wrote the manuscript. K. L. contributed to part of the preparation and characterization of the electrolyte and the electrode. K. W. and S. C. performed depth-profiling XPS measurements. All authors discussed and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2022YFE0206300), the National Natural Science Foundation of China (U21A2081, 22209047, and 22075074), the Hunan Provincial Natural Science Foundation of China (2021JJ40047 and 2020JJ5035) and the Macau Science and Technology Development Fund (file no. 0013/2021/AMJ).Notes and references
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Chem. Rev., 2020, 120, 6358–6466 CrossRef CAS PubMed.
- K. Zhang, Z. Gu, E. Ang, J. Guo, X. Wang, Y. Wang and X. Wu, Mater. Today, 2022, 54, 189–201 CrossRef CAS.
- J. Wang, X. Liu, H. Jia, P. Apostol, X. Guo, F. Lucaccioni, X. Zhang, Q. Zhu, C. Morari, J.-F. Gohy and A. Vlad, ACS Energy Lett., 2022, 7, 668–674 CrossRef CAS.
- W. Zhang and Z. Guo, Natl. Sci. Rev., 2021, 8, nwab043 CrossRef.
- Z. Jian, W. Luo and X. Ji, J. Am. Chem. Soc., 2015, 137, 11566–11569 CrossRef CAS PubMed.
- L. Li, Z. Hu, Y. Lu, C. Wang, Q. Zhang, S. Zhao, J. Peng, K. Zhang, S. L. Chou and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 13050–13056 CrossRef CAS.
- J. Liu, T. Yin, B. Tian, B. Zhang, C. Qian, Z. Wang, L. Zhang, P. Liang, Z. Chen, J. Yan, X. Fan, J. Lin, X. Chen, Y. Huang, K. P. Loh and Z. X. Shen, Adv. Energy Mater., 2019, 9, 1900579 CrossRef.
- L. Deng, J. Qu, X. Niu, J. Liu, J. Zhang, Y. Hong, M. Feng, J. Wang, M. Hu, L. Zeng, Q. Zhang, L. Guo and Y. Zhu, Nat. Commun., 2021, 12, 2167 CrossRef CAS PubMed.
- M. Zhang, P. Bai, X. Ji, J. Yang, C. Wang and Y. Xu, Adv. Mater., 2021, 33, 2003741 CrossRef PubMed.
- J. Xu, J. Zhang, T. P. Pollard, Q. Li, S. Tan, S. Hou, H. Wan, F. Chen, H. He, E. Hu, K. Xu, X.-Q. Yang, O. Borodin and C. Wang, Nature, 2023, 614, 694–700 CrossRef CAS PubMed.
- J. Holoubek, H. Liu, Z. Wu, Y. Yin, X. Xing, G. Cai, S. Yu, H. Zhou, T. A. Pascal, Z. Chen and P. Liu, Nat. Energy, 2021, 6, 303–313 CrossRef CAS.
- N. Zhang, T. Deng, S. Zhang, C. Wang, L. Chen, C. Wang and X. Fan, Adv. Mater., 2021, 34, 2107899 CrossRef.
- J. Hou, M. Yang, D. Wang and J. Zhang, Adv. Energy Mater., 2020, 10, 1904152 CrossRef CAS.
- X. Lu, X. Pan, D. Zhang, Z. Fang, S. Xu, Y. Ma, Q. Liu, G. Shao, D. Fu, J. Teng and W. Yang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2110912118 CrossRef CAS.
- Y. Wang, X. Gao, L. Li, M. Wang, J. Shui and M. Xu, Nano Energy, 2020, 67, e2110912118 Search PubMed.
- J. Wang, X. Guo, P. Apostol, X. Liu, K. Robeyns, L. Gence, C. Morari, J.-F. Gohy and A. Vlad, Energy Environ. Sci., 2022, 15, 3923–3932 RSC.
- J. Holoubek, K. Kim, Y. Yin, Z. Wu, H. Liu, M. Li, A. Chen, H. Gao, G. Cai, T. A. Pascal, P. Liu and Z. Chen, Energy Environ. Sci., 2022, 15, 1647–1658 RSC.
- X. Dong, Z. Guo, Z. Guo, Y. Wang and Y. Xia, Joule, 2018, 2, 902–913 CrossRef CAS.
- X. Fan, X. Ji, L. Chen, J. Chen, T. Deng, F. Han, J. Yue, N. Piao, R. Wang, X. Zhou, X. Xiao, L. Chen and C. Wang, Nat. Energy, 2019, 4, 882–890 CrossRef CAS.
- C. B. Jin, N. Yao, Y. Xiao, J. Xie, Z. Li, X. Chen, B. Q. Li, X. Q. Zhang, J. Q. Huang and Q. Zhang, Adv. Mater., 2022, 35, e2208340 CrossRef PubMed.
- B. Nan, L. Chen, N. D. Rodrigo, O. Borodin, N. Piao, J. Xia, T. Pollard, S. Hou, J. Zhang, X. Ji, J. Xu, X. Zhang, L. Ma, X. He, S. Liu, H. Wan, E. Hu, W. Zhang, K. Xu, X. Q. Yang, B. Lucht and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202205967 CrossRef CAS PubMed.
- M. Qin, M. Liu, Z. Zeng, Q. Wu, Y. Wu, H. Zhang, S. Lei, S. Cheng and J. Xie, Adv. Energy Mater., 2022, 12, 202201801 Search PubMed.
- J. F. Wu, W. Zhou, Z. Wang, W. W. Wang, X. Lan, H. Yan, T. Shi, R. Hu, X. Cui, C. Xu, X. He, B. W. Mao, T. Zhang and J. Liu, Adv. Mater., 2023, 35, e2209833 CrossRef PubMed.
- S. Ding, B. Zhou, C. Chen, Z. Huang, P. Li, S. Wang, G. Cao and M. Zhang, ACS Nano, 2020, 14, 9626–9636 CrossRef CAS PubMed.
- Y. S. Meng, V. Srinivasan and K. Xu, Science, 2022, 378, eabq3750 CrossRef CAS PubMed.
- H. Liang, Z. Gu, X. Zhao, J. Guo, J. Yang, W. Li, B. Li, Z. Liu, Z. Sun, J. Zhang and X. Wu, Sci. Bull., 2022, 67, 1581–1588 CrossRef CAS PubMed.
- M. Li, C. Wang, Z. Chen, K. Xu and J. Lu, Chem. Rev., 2020, 120, 6783–6819 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- X. Liu, X. Shen, H. Li, P. Li, L. Luo, H. Fan, X. Feng, W. Chen, X. Ai, H. Yang and Y. Cao, Adv. Energy Mater., 2021, 11, 2003905 CrossRef CAS.
- L. Xing, X. Zheng, M. Schroeder, J. Alvarado, A. von Wald Cresce, K. Xu, Q. Li and W. Li, Acc. Chem. Res., 2018, 51, 282–289 CrossRef CAS.
- J. Zhao, X. Zou, Y. Zhu, Y. Xu and C. Wang, Adv. Funct. Mater., 2016, 26, 8103–8110 CrossRef CAS.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- S. Lin, H. Hua, P. Lai and J. Zhao, Adv. Energy Mater., 2021, 11, 202101775 Search PubMed.
- Y. Xu, T. Ding, D. Sun, X. Ji and X. Zhou, Adv. Funct. Mater., 2022, 33, 2211290 CrossRef.
- S. Zhang, K. Xu, J. Allen and T. Jow, J. Power Sources, 2002, 110, 216–221 CrossRef CAS.
- L. Fan, Y. Hu, A. M. Rao, J. Zhou, Z. Hou, C. Wang and B. Lu, Small Methods, 2021, 5, e2101131 CrossRef.
- Y. Liu, C. Gao, L. Dai, Q. Deng, L. Wang, J. Luo, S. Liu and N. Hu, Small, 2020, 16, e2004096 CrossRef.
- J. Wang, H. Wang, X. Zang, D. Zhai and F. Kang, Batteries Supercaps, 2020, 4, 554–570 CrossRef.
- Y. Aoki, M. Oda, S. Kojima, T. Ishihama, T. Nagashima, T. Doi and M. Inaba, ACS Appl. Energy Mater., 2022, 5, 1085–1094 CrossRef CAS.
- S. Zhu and J. Chen, Energy Storage Mater., 2022, 44, 48–56 CrossRef.
- T. D. Pham, A. Bin Faheem and K. K. Lee, Small, 2021, 17, e2103375 CrossRef.
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 CrossRef CAS.
- L. Fan, R. Ma, Q. Zhang, X. Jia and B. Lu, Angew. Chem., Int. Ed., 2019, 58, 10500–10505 CrossRef CAS.
- S. Komaba, T. Hasegawa, M. Dahbi and K. Kubota, Electrochem. Commun., 2015, 60, 172–175 CrossRef CAS.
- S. Liu, J. Mao, Q. Zhang, Z. Wang, W. K. Pang, L. Zhang, A. Du, V. Sencadas, W. Zhang and Z. Guo, Angew. Chem., Int. Ed., 2020, 59, 3638–3644 CrossRef CAS PubMed.
- L. Wang, J. Yang, J. Li, T. Chen, S. Chen, Z. Wu, J. Qiu, B. Wang, P. Gao, X. Niu and H. Li, J. Power Sources, 2019, 409, 24–30 CrossRef CAS.
- L. Qin, N. Xiao, J. Zheng, Y. Lei, D. Zhai and Y. Wu, Adv. Energy Mater., 2019, 9, 1902618 CrossRef CAS.
- Y. Lei, D. Han, J. Dong, L. Qin, X. Li, D. Zhai, B. Li, Y. Wu and F. Kang, Energy Storage Mater., 2020, 24, 319–328 CrossRef.
- L. Li, L. Liu, Z. Hu, Y. Lu, Q. Liu, S. Jin, Q. Zhang, S. Zhao and S. L. Chou, Angew. Chem., Int. Ed., 2020, 59, 12917–12924 CrossRef CAS.
- Y. An, H. Fei, G. Zeng, L. Ci, B. Xi, S. Xiong and J. Feng, J. Power Sources, 2018, 378, 66–72 CrossRef CAS.
- X. Li, Y. Lei, L. Qin, D. Han, H. Wang, D. Zhai, B. Li and F. Kang, Carbon, 2021, 172, 200–206 CrossRef CAS.
- S. Ji, J. Li, J. Li, C. Song, S. Wang, K. Wang, K. S. Hui, C. Zha, Y. Zheng, D. A. Dinh, S. Chen, J. Zhang, W. Mai, Z. Tang, Z. Shao and K. N. Hui, Adv. Funct. Mater., 2022, 32, 202200771 Search PubMed.
- J. Zhang, J.-F. Wu, Z. Wang, Y. Mo, W. Zhou, Y. Peng, B. He, K. Xiao, S. Chen, C. Xu and J. Liu, J. Energy Chem., 2022, 71, 344–350 CrossRef CAS.
- S. U. Yoon, H. Kim, H.-J. Jin and Y. S. Yun, Appl. Surf. Sci., 2021, 547, 149193 CrossRef CAS.
- Q. Li, D. Lu, J. Zheng, S. Jiao, L. Luo, C. M. Wang, K. Xu, J. G. Zhang and W. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 42761–42768 CrossRef CAS.
- J. Holoubek, A. Baskin, J. W. Lawson, H. Khemchandani, T. A. Pascal, P. Liu and Z. Chen, J. Phys. Chem. Lett., 2022, 13, 4426–4433 CrossRef CAS.
- Y. Lu, C.-Z. Zhao, J.-Q. Huang and Q. Zhang, Joule, 2022, 6, 1172–1198 CrossRef CAS.
- X. Bie, K. Kubota, T. Hosaka, K. Chihara and S. Komaba, J. Mater. Chem. A, 2017, 5, 4325–4330 RSC.
- Y.-H. Zhu, X. Yang, D. Bao, X.-F. Bie, T. Sun, S. Wang, Y.-S. Jiang, X.-B. Zhang, J.-M. Yan and Q. Jiang, Joule, 2018, 2, 736–746 CrossRef CAS.
- C. Zhang, Y. Xu, M. Zhou, L. Liang, H. Dong, M. Wu, Y. Yang and Y. Lei, Adv. Funct. Mater., 2017, 27, 1604307 CrossRef.
- L. Fan, R. Ma, J. Wang, H. Yang and B. Lu, Adv. Mater., 2018, 30, e1805486 CrossRef PubMed.
- J. Liao, Q. Hu, X. He, J. Mu, J. Wang and C. Chen, J. Power Sources, 2020, 451, 227739 CrossRef CAS.
- J. Liao, X. Zhang, Q. Zhang, Q. Hu, Y. Li, Y. Du, J. Xu, L. Gu and X. Zhou, Nano Lett., 2022, 22, 4933–4940 CrossRef CAS PubMed.
- B. Lin, X. Zhu, L. Fang, X. Liu, S. Li, T. Zhai, L. Xue, Q. Guo, J. Xu and H. Xia, Adv. Mater., 2019, 31, e1900060 CrossRef.
- S. Zhao, Z. Liu, G. Xie, Z. Guo, S. Wang, J. Zhou, X. Xie, B. Sun, S. Guo and G. Wang, Energy Environ. Sci., 2022, 15, 3015–3023 RSC.
- T. Deng, X. Fan, C. Luo, J. Chen, L. Chen, S. Hou, N. Eidson, X. Zhou and C. Wang, Nano Lett., 2018, 18, 1522–1529 CrossRef CAS.
- B. Ji, W. Yao, Y. Zheng, P. Kidkhunthod, X. Zhou, S. Tunmee, S. Sattayaporn, H. M. Cheng, H. He and Y. Tang, Nat. Commun., 2020, 11, 1225 CrossRef CAS.
- J. Liao, C. Chen, Q. Hu, Y. Du, Y. He, Y. Xu, Z. Zhang and X. Zhou, Angew. Chem., Int. Ed., 2021, 60, 25575–25582 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee03340f |
| This journal is © The Royal Society of Chemistry 2024 |