
The synergistic effect of dry air and surfactants enables water to be a promising green solvent for stable and efficient perovskite solar cells†
Yanrui
Zhang‡
a,
Lixia
Ren‡
a,
Peng
Zhai
*a,
Jingjing
Xin
a,
Jiarong
Wu
a,
Qi
Zhang
a,
Xin
Chen
a,
Kui
Zhao
 a,
Lu
Zhang
a and
Shengzhong
(Frank) Liu
*abc
a,
Lu
Zhang
a and
Shengzhong
(Frank) Liu
*abc
aKey Laboratory for Applied Surface and Colloid Chemistry, National Ministry of Education, Shaanxi Engineering Lab for Advanced Energy Technology, School of Materials Science and Engineering, Shaanxi Normal University, Xi’an 710119, China. E-mail: zhaipeng@snnu.edu.cn; szliu@dicp.ac.cn
bDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China
cCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 18th November 2023
Abstract
In recent years, perovskite has emerged as a prominent mineral to compete with all other existing PV materials. However, its fabrication typically involves the use of toxic organic solvents. A desirable solution would be to replace these harmful solvents with the genial water. Unfortunately, the high surface tension of water often results in the growth of Pb(NO3)2 crystals resembling islands, leading to morphological imperfections in the final perovskite film. In this study, the detailed Pb(NO3)2 crystallization process under different relative humidity is examined. It is found that moisture in the ambient air plays a pivotal role in the spreading dynamics of aqueous inks and the nucleation rate of Pb(NO3)2. Potassium oleate (PO), a long-alkyl chain anionic surfactant, is therefore designed to reduce the surface tension of water. Through the synergistic effect of dry air and oleate anion, a high-quality, pinhole-free conformal perovskite film with enlarged grain size can be readily obtained. Meanwhile, crystallinity regulation of Pb(NO3)2 by PO is proven to boost the further crystallization of PbI2 and improve the formation kinetics of perovskite. Consequently, PO treated PSCs exhibit a power conversion efficiency (PCE) of 24.14% (0.09 cm2), one of the highest PCEs among all water-processed PSCs. More importantly, benefitting from the wetting-controlled strategy, a PCE of 22.09% is achieved for the first time on a large area (1 cm2). The bare device without any protection shows outstanding stability under continuous thermal stress and light illumination. This triumph is expected to shed light on the scalable production of PSCs using a minimal quantity of harmful organic solvents.
Broader contextThe toxicity of traditional solvents used for the preparation of perovskites is a major challenge in the large-scale commercial manufacturing of perovskite solar cells (PSCs). Fully replacing toxic solvents with nonhazardous solvents is one of the key strategies for their green, safe, and scalable production. For this purpose, preparation of perovskites from an aqueous lead nitrate ink (Pb(NO3)2/H2O) is considered to be the most desirable and promising protocol. However, due to the high surface tension of water, Pb(NO3)2 usually grows into island-like crystals, and morphological imperfections inevitably occur in perovskites, severely degrading their performance and limiting the expansion of the PSC size. In this study, we develop surface engineering to prepare a compact and uniform perovskite film with a large area. To accomplish this, we first scrutinize the crystallization process of Pb(NO3)2 at different relative humidities (RHs). Then, a long-alkyl chain anionic surfactant (potassium oleate) is carefully introduced to reduce the surface tension of water. By taking advantage of the synergistic effect of dry air and oleate anion, a PCE of 22.09% is achieved for the first time on a large area (1 cm2), demonstrating the advantages and great potential for large-area PSC production using green water solvent. |
1. Introduction
The power conversion efficiency (PCE) of perovskite solar cells (PSCs) has improved from 3.8% to 26.1% in the past decade, demonstrating their commercial viability.1–3 However, almost all solvent systems implemented for the preparation of perovskites are toxic to the environment. Vapor deposition without the use of liquid solution is preferred for large-area perovskite fabrication. The device performance of PSCs prepared by this technique is catching up and comparable with that of devices prepared using conventional toxic solution-based methods.4 From the perspective of green synthesis of perovskites by solution deposition, water is regarded as the most promising solvent. In 2015, Wei et al. first invented a low-toxicity aqueous lead nitrate ink (Pb(NO3)2/H2O) to prepare MAPbI3 PSCs via sequential deposition. However, due to the perovskite functional layer's poor coverage, the device PCE was poor (only 12.58%).5 Typically, preparation of perovskite crystals by the Pb(NO3)2/H2O protocol consists of a two-step reaction: (i) the quick reaction of Pb(NO3)2 with MAI to form PbI2, and (ii) the slow transformation of PbI2 into MAPbI3 by reacting with MAI.6 By understanding and regulating the kinetics of intermediate ion exchange among NO3− and mixed cations/anions, the perovskite conversion was enhanced through compositional engineering.7 Seed-promoted aqueous inks were developed to accelerate crystal growth, enabling a printed PSC with a PCE of 16.53%.8 It is worth noting that Pb(NO3)2 is one of the raw materials for synthesizing PbI2. Whether in the laboratory research stage or at the mass production scale, the aqueous Pb(NO3)2 precursor has been confirmed to achieve an enormous cost reduction over conventional PbI2 organic solutions.9Given the use of water as the solvent, the water-processed perovskite is most suitable for preparation under ambient air conditions. There are three major challenges in improving PCE. Firstly, water has a higher surface tension (72.8 mN m−1) compared with the DMF (25.7 mN m−1), and the topography of Pb(NO3)2 is island-like rather than a continuous film. With the expansion of the area, morphological imperfections inevitably occur in perovskites, which will aggravate the surface charge recombination. Secondly, TiO2 is a typical self-cleaning material owing to its photocatalytic properties.10,11 When TiO2 surfaces in ambient environments are irradiated with ultraviolet light, they become hydrophilic, but they slowly revert to hydrophobic in the dark. The stark transition in surface wettability caused by moisture will have a profound impact on film deposition.12 Thirdly, the perovskite conversion based on the Pb(NO3)2/H2O protocol is too slow relative to the toxic PbI2/DMF system, which will lead to the notorious Ostwald ripening effect and incomplete transformation.13 Therefore, it is urgent to better manage the crystallization of Pb(NO3)2 and optimize the formation kinetics of perovskite in order to enhance both the efficiency and stability of water-processed PSCs.
Recently, we have developed a light modulation strategy to activate the perovskite nucleation and heal the pinhole defects, reaching a high average PCE of ∼22.51%.14 However, if light is used in mass production, it will increase equipment cost and energy consumption. In this study, we pioneered the development of a wetting-controlled strategy to prepare a compact and uniform perovskite film with a large area. We first scrutinize the crystallization of Pb(NO3)2 at different relative humidities (RHs). It is revealed that compared with humid air (RH ∼ 50%), dry air (RH ∼ 10%) has a stronger promotion effect on the nucleation rate than the growth rate, and it also maintains a superwetting solid surface, which will facilitate more effective spreading of aqueous inks. Potassium oleate is further introduced to reduce the surface tension of water from 72.77 to 25.30 mN m−1. Through the synergistic effect of dry air and oleate anion, a pinhole-free conformal perovskite capping layer can be easily realized. PO not only physically constructs a glue-like scaffold to eliminate the residual stresses, but also promotes orientated crystallization of α-phase FA-rich perovskites and passivates trap defects. With these optimizations, water-processed PSCs yield a PCE of 24.14% in a small area (0.09 cm2) and 22.09% in a large area (1 cm2), demonstrating the advantages and great potential of this method for large-area PSC production from aqueous inks. The bare device without any protection exhibits outstanding stability under the conditions of ambient dark storage for 2100 hours, continuous heating for 800 hours at 85 °C and light exposure in a N2-filled glovebox under continuous 1 sun illumination.
2. Results and discussion
In theory, when the wettability between the deposited material and the substrate is weak, the monomer–monomer interaction will become stronger compared to the monomer–substrate interaction, and the nuclei will form separately and grow into ‘islands’ on a substrate, which is called the Volmer–Weber growth mode.15 As observed in Fig. 1(a) and Fig. S1 (ESI†), all Pb(NO3)2 films grown under different RHs follow this growth mode and present an island-like morphology on mesoporous TiO2 substrates. As the humidity increases from 10% to 50% during spin coating, the obtained films show an increase in grain size and a decrease in the crystal density. The gap between islands gets larger and the surface coverage becomes worse across the substrate. The transmittance spectra of Pb(NO3)2 films are shown in Fig. S2 (ESI†). Pb(NO3)2 deposited under RH ∼ 50% (hereinafter referred to as Pb(NO3)2-50) displays higher transparency below 600 nm than that of Pb(NO3)2-10, because the large gaps between islands allow the shorter wavelength light to pass through. Long wavelength light scattering occurs on the rough Pb(NO3)2-50 surface with large crystals, which will affect the incident light harvesting above 600 nm. The processes of nucleation and growth can be illustrated using the LaMer diagram, which is divided into three stages, i.e., regions I (pre-nucleation), II (nucleation), and III (growth), respectively.16 In the first step, the monomeric species are initially accumulated in solution. In the second step (minimum or critical supersaturation), the monomer concentration reaches a critical level (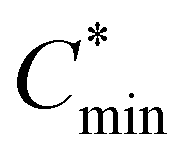 ) for nucleation and subsequently, the nucleation occurs albeit at a very slow rate. In the third step (maximum supersaturation), the nucleation is dramatically accelerated as the monomer concentration reaches the maximum level (
) for nucleation and subsequently, the nucleation occurs albeit at a very slow rate. In the third step (maximum supersaturation), the nucleation is dramatically accelerated as the monomer concentration reaches the maximum level (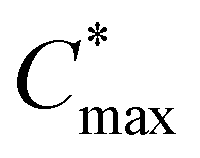 ). At the spin-coating stage, with fast evaporation of water the Pb(NO3)2/H2O precursor rapidly exceeds the solubility level (Cs) and supersaturation status in sequence, and then begins to nucleate. An apparent nucleation with a boosted density is generated for Pb(NO3)2-10, while the nucleation density of Pb(NO3)2-50 is much lower due to the adsorption of excess water vapor from humid air, which prevents the precursor film from reaching
). At the spin-coating stage, with fast evaporation of water the Pb(NO3)2/H2O precursor rapidly exceeds the solubility level (Cs) and supersaturation status in sequence, and then begins to nucleate. An apparent nucleation with a boosted density is generated for Pb(NO3)2-10, while the nucleation density of Pb(NO3)2-50 is much lower due to the adsorption of excess water vapor from humid air, which prevents the precursor film from reaching 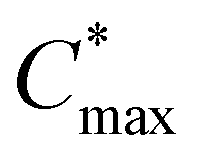 . According to the Burton–Cabrera–Frank (BCF) theory, the number of crystallites per unit area (N), which is inversely proportional to the grain size, can be expressed as:
. According to the Burton–Cabrera–Frank (BCF) theory, the number of crystallites per unit area (N), which is inversely proportional to the grain size, can be expressed as: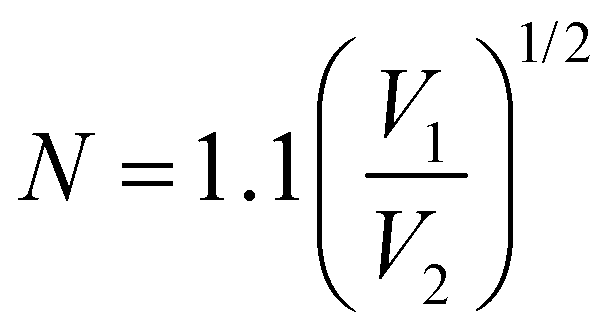 | (1) |
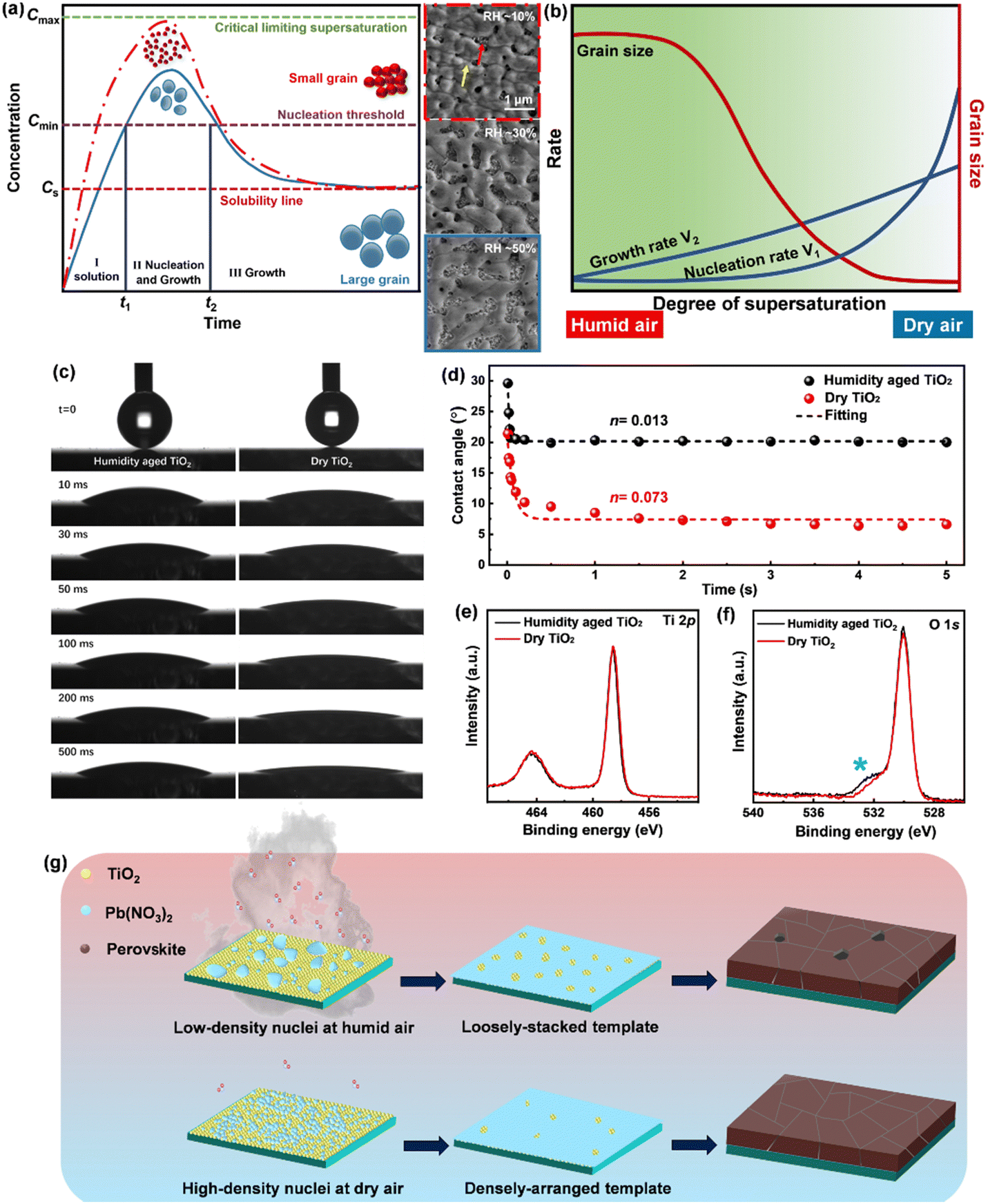 | ||
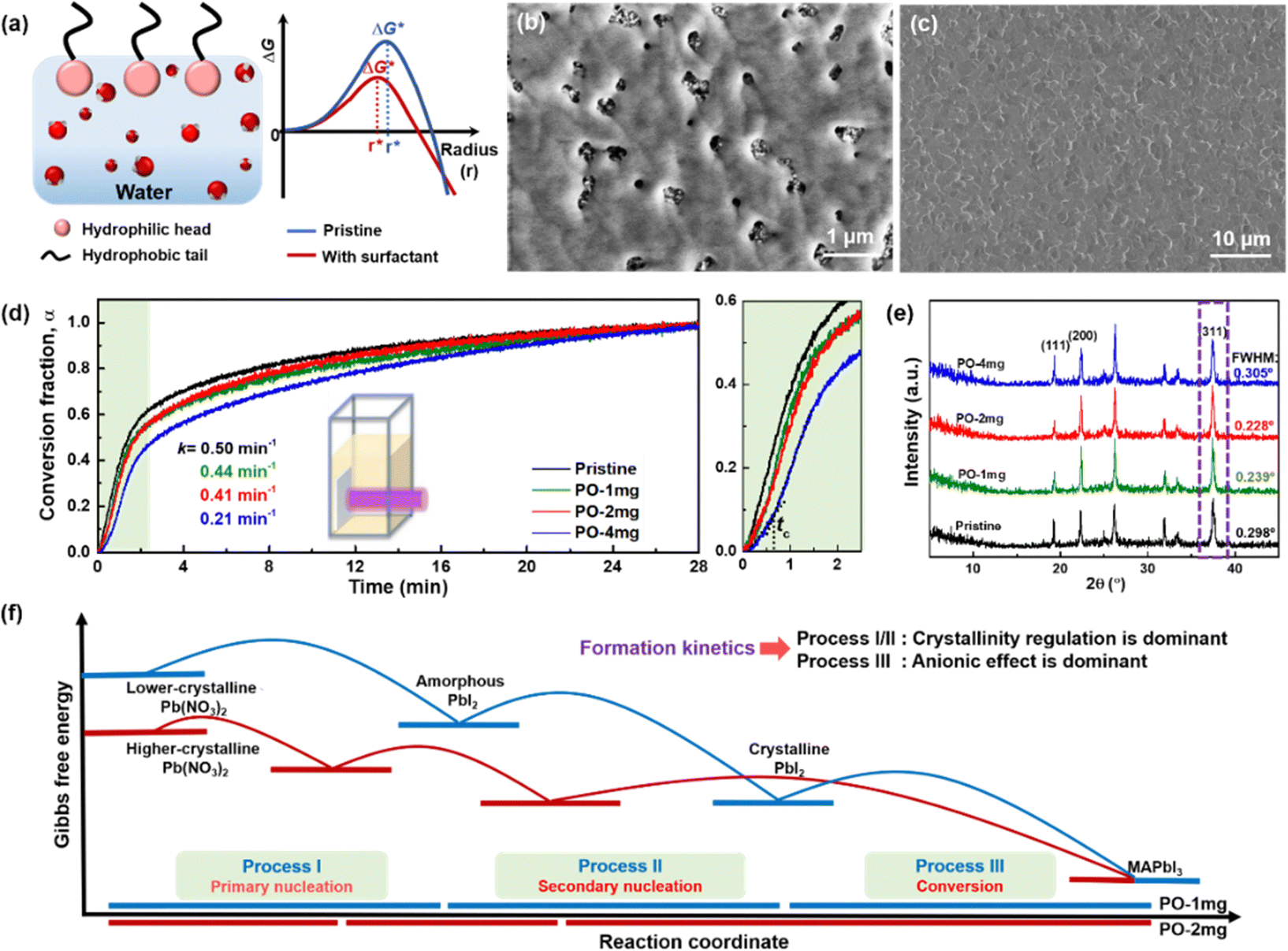
| Fig. 1 (a) The classical LaMer model (left) to explain the difference of nucleation and growth of the aqueous Pb(NO3)2 precursor film for two cases: spin coating under dry air conditions (red dash dotted line) and spin coating under humid air conditions (blue line). SEM images of Pb(NO3)2 films prepared under different RH conditions (right). The red and yellow arrows refer to the mp-TiO2 substrate and Pb(NO3)2 film, respectively. (b) The nucleation and growth rates (blue line as marked separately) as a function of the supersaturation degree, and the corresponding grain size (red line). (c) Dynamics of liquid spreading on various TiO2 substrates. Selected images showing the spreading of water droplets at early stages after coming into contact with a humidity-aged TiO2 and a dry TiO2 surface. The static contact angles for the TiO2 and the surfaces are 20.0° and 6.5°, respectively. (d) Contact angle variation as a function of time for various TiO2 substrates. XPS spectra of various TiO2 substrates: (e) Ti 2p and (f) O 1s. (g) Schematic drawing of the crystallization under different RH conditions. The densely-arranged Pb(NO3)2 template is conducive to forming the pinhole free perovskite. | ||
Complete wetting and spreading have significance for inking, where enhanced spreading is desired. The spreading behavior of water droplets in contact with TiO2 substrates is studied through dynamic contact angle (θ) measurements. Fig. 1(c) and Fig. S3a (ESI†) show the initial spreading stage, time-dependent behavior of θ for water and PEDOT-PSS droplets deposited on TiO2, respectively. The speed of the droplet spreading for drops of similar volume is visibly faster on the dry TiO2 (fresh annealed, with less water vapor adsorption) than on the humidity aged one (stored at RH ∼ 50% for 30 min, with more water vapor adsorption). The spreading of a liquid on a solid surface is generally described by the Tanner power law,18,19
| θ ∝ (t0 + t)−n | (2.) |
γlv![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ = γsv − γsl, cosθ = γsv − γsl, | (3) |
The surface free energy of TiO2 substrates is determined using the Owens and Wendt model.20 This method considers γs as a sum of two components such that:
| γs = γds + γps, | (4) |
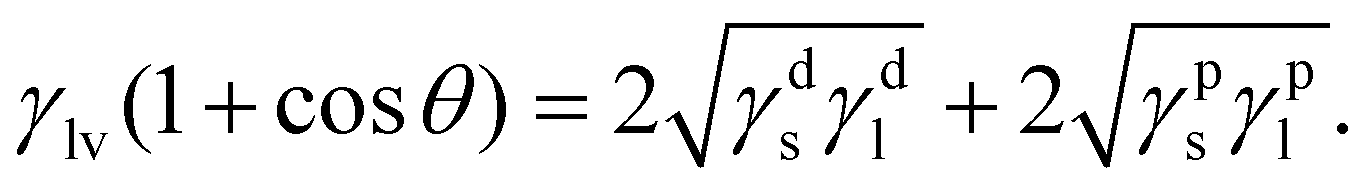 | (5) |
Compared with the humidity aged TiO2, dry TiO2 exhibits higher values of the surface free energy (291.83 mJ m−2) and polar component (179.75 mJ m−2), indicating its better wettability (listed in Table S1, ESI†). X-ray photoemission spectroscopy (XPS) analysis is used to map out the elemental composition of these two TiO2 substrates. In Fig. 1(e), the Ti 2p XPS spectra are assigned to Ti 2p3/2 and 2p1/2 peaks centered at binding energies of 458.55 and 464.50 eV, which are typical for the Ti4+–O bonds in TiO2.21 As seen from the O 1s spectra (in Fig. 1(f)), a pronounced shoulder peak appears in the humidity aged TiO2, which is attributed to the absorption of excess carboxylates from water vapor in humid air.12 We correlate the de-wetting of the TiO2 surface with the spontaneous formation of mixed formate/acetate layers, which will transform the initially hydrophilic surface into a less wettable surface. Deposition in dry air can maintain the superwetting properties of the TiO2 surface, which will allow for more effective spreading of aqueous inks and enhance the surface coverage of Pb(NO3)2.
Due to the necessity of NO3− removal, spin-coating organic amine salt solutions onto the Pb(NO3)2 film is unsuitable, although it is common in conventional PbI2/DMF systems. We have developed an effective fabrication method that first converts Pb(NO3)2 into MA-based perovskite by multiple-cycle dipping. Then, FAI/MAI/MACl/IPA solution is deposited on it, and an FA-rich perovskite is formed through FA–MA cation intermixing during annealing (in Video S1, ESI†).22 Fig. S5 and Video S2 (ESI†) show the gradual color change after immersing a series of Pb(NO3)2 samples in MAX/IPA solution (X = I/Cl, MAI/MACl = 4:1). All Pb(NO3)2 films turns brown immediately after immersion in MAX/IPA. A more rapid and pronounced color change per second can be observed in Pb(NO3)2-50. During the growth of nuclei, Pb(NO3)2 nanocrystals infiltrated in the TiO2 scaffold (the uncovered area) tend to first crystallize by virtue of their small radius of curvature. Fast color change is an indicator of poor coverage in Pb(NO3)2. In addition, the conversion from PbI2 to MAPbI3 induces almost double the volume expansion,23 the densely arranged Pb(NO3)2-10 can form high-density PbI2 crystals, which will compensate for the original voids during the immersion and obtain perovskite films free of pinholes (in Fig. S6, ESI†). In contrast, pinhole defects are observed in Pb(NO3)2-30 and -50 based perovskites due to their large gap, which will provide a path for localized electrical shunting between the electron- and hole-transporting layers.24 The crystallization process is illustrated schematically in Fig. 1(g). We conclude that strict humidity control (RH < 10%) should be a prerequisite for preparing perovskite films from aqueous inks, because the formation of optimal density/size of the nuclei and the improvement of surface coverage are crucial to obtaining high-quality perovskite films in ambient air.
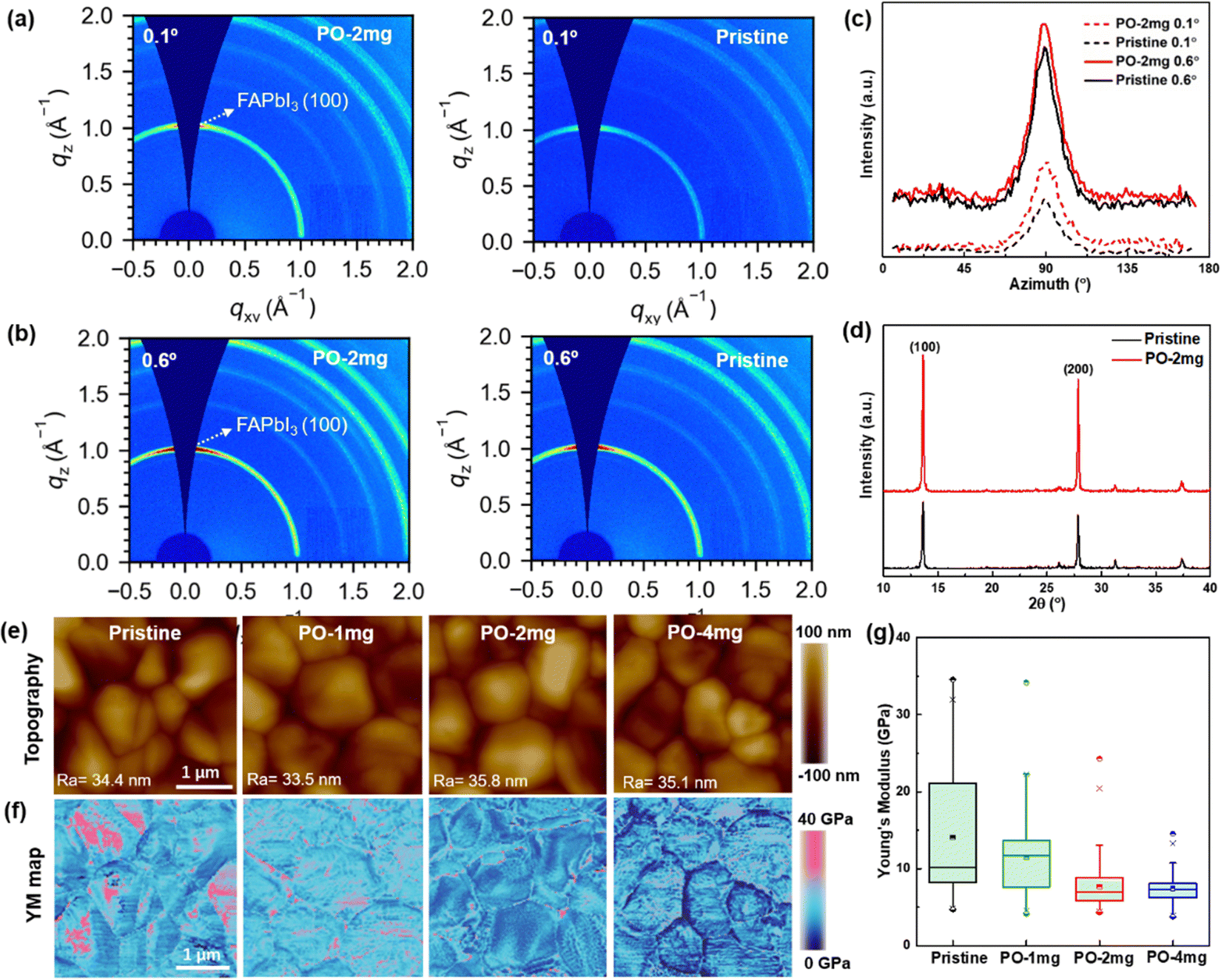
In principle, wetting is favored by low interfacial free energy, high solid surface free energy and low liquid surface free energy. Long-alkyl chain surfactants have been regarded as effective additives to change the rheological properties of the inks and improve the quality of perovskite films.25,26 In view of their molecular structure, the hydrophobic groups are generally alkyl chains or cycloalkanes containing 8–18 carbon atoms. We have systematically screened the effect of long-chain surfactants according to the number and length of their alkyl chains and concluded that increasing the alkyl-chain length is benign to the performance.27 The longer the alkyl chain, the higher the value of the electrostatic potential (ESP). Bonding strength will enhance along with the increase of the maximum value of ESP. Based on these results, we select potassium oleate (C18H33KO2), comprising a long hydrocarbon organic endowed with a carboxyl functionality (R-COO−) and a small inorganic cation (K+) as a surfactant and passivating agent (shown in Fig. 2(a)). The electrostatic potential distribution (shown in Fig. S7a, ESI†) indicates that the PO molecule containing electron-rich R-COO− can interplay with undercoordinated Pb2+ through electrostatic coupling and thus passivate toward the iodide vacancy. The long tail also can form a hydrophobic net to protect the perovskite film from moisture attachment. The K+ cation is reported to play a passivating role at both the grain boundaries (GBs) and the interface.28 As observed in Fig. S7b (ESI†), the critical micellar concentration (CMC) is determined to be 0.3 mg mL−1 and micelles are expected to form in the bulk solution.29 On one hand, oleate anion can orient hydrophilic carboxyl groups towards the aqueous phase and hydrophobic long alkyl chains towards the air phase. These molecules build a compact homogeneous adsorption layer on the surface, effectively reducing the surface tension of the solution.30 On the other hand, the micelles in the bulk solution will passivate defects at the GBs of the perovskite. Fig. S7c (ESI†) reveals that PO incorporation has little effect on viscosity and will not alter the fluidity of the solution. Via this wetting-controlled strategy, the growth of Pb(NO3)2 changes from island-like to a nearly continuous film (shown in Fig. 2(b)). The gaps between the islands are further reduced, and the uniform distribution of micro-sized voids (shown in Fig. S8, ESI†) is conducive to the diffusion and reaction of MAX. Here, we discuss the general effects of surfactants on the nucleation of crystals in accordance with the classical thermodynamic theory.31 The crystal nucleation rate (V1) is described as:
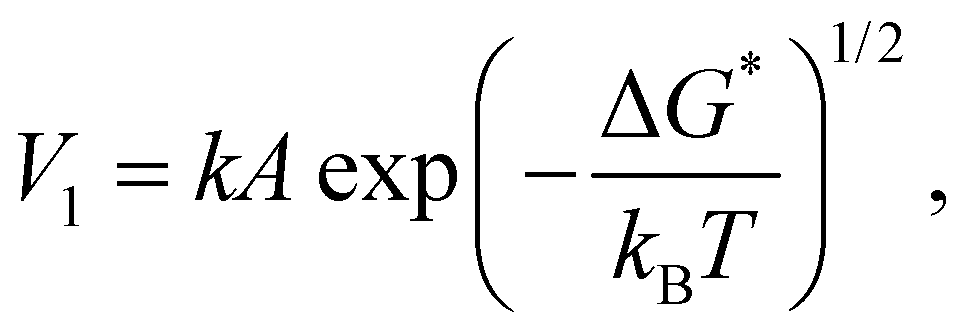 | (6) |
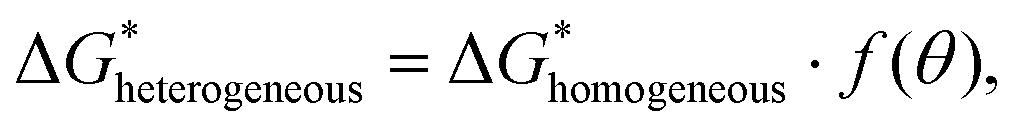 | (7) |
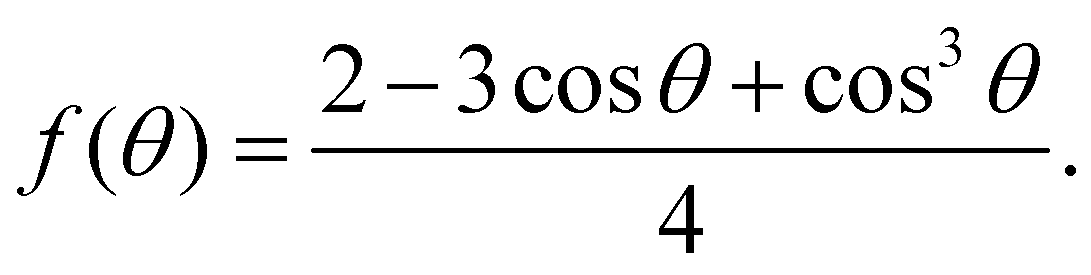 | (8) |
 | ||
| Fig. 2 (a) Schematic illustration of PO adsorption layer on the water surface (left) and graph of the functional relationship between ΔG and cluster radius r (right). SEM images of (b) PO-2mg treated Pb(NO3)2 film and (c) the resulting perovskite. (d) In situ UV-Vis absorption kinetic data of various Pb(NO3)2 substrates dipped in MAX/IPA solution at room temperature shown as the conversion fraction (α). (e) XRD spectra of the Pb(NO3)2 films w/o adding PO. (f) Schematic of the perovskite conversion process in the PO treated Pb(NO3)2 films, with Gibbs free energy shown as a function of the reaction coordinate. | ||
To track the formation kinetics of the perovskite, we conduct in situ UV-Vis absorption measurements. As depicted in Fig. 2(d), the absorbance of the perovskite at 700 nm is monitored during conversion, in which Pb(NO3)2 films are dipped in MAX/IPA solution.32 The ratio of the absorbance at time t to that after complete conversion is defined as the conversion fraction [α(t); 0 ≤ α(t) ≤ 1], representing the fraction of perovskite formed from the Pb(NO3)2 at time t. Compared with the pristine Pb(NO3)2, it takes longer for the sample with a surfactant to convert into perovskite. In the enlarged area (green color), there is a three-stage growth process in the Pb(NO3)2 with PO treatment (hereafter denoted as PO-1mg, -2mg and -4mg): first it shows that the absorbance gradually increases until the kink point (tc), while above tc, the rate of increase is accelerated rapidly, and then the increase rate slows down until the complete transformation is reached. The mild conversion in the initial stage (<40 s) is a consequence of the anionic extraction effect, because R-COO− can extract MA+ from the perovskite nuclei and destroy the perovskite lattice at the beginning of nucleation.22 After the anionic effect is weakened (above tc), more “stable” perovskites will re-crystallize at a rapid rate. The relatively slow increase of absorbance in the later stage is due to the light-scattering effect from large perovskite crystals induced by the Ostwald ripening effect.7 On the opposite, the conversion of pristine Pb(NO3)2 shows a typical two-stage growth process without the interruption of the anionic effect. The kinetic data can be fitted by means of nonlinear regression according to Avrami models:
| 1− α = exp(−ktn) | (9) |
As observed in the X-ray diffraction (XRD) spectra in Fig. 2(e), the main peak (311) of PO-2mg shows the lowest full-width at half-maximum (FWHM) value, implying that moderate introduction of surfactants enhances crystallinity, while excessive introduction reduces crystallinity. Subsequently, as seen in Fig. S11 (ESI†), the PbI2 intermediate created from PO-2mg in the incubation still shows higher crystallinity than its counterparts. We have revealed that the reaction starts with the transformation of low-crystalline PbI2 from Pb(NO3)2 (named process I), followed by secondary nucleation of PbI2 (named process II), and finally the conversion of the high-crystalline PbI2 proceeds via the intercalation of MAX to form the perovskite (named process III).14 Crystallinity regulation by surfactant can form higher-crystallinity Pb(NO3)2 with lower Gibbs free energy, which will boost the faster completion of process I/II.33 For the entire reaction, although the anionic effect hinders the process III, the acceleration of process I/II compensates for it (shown in Fig. 2(f)). As a result, the k value of PO-2mg does not decrease significantly relative to PO-1mg. However, excessive introduction (PO-4mg) drastically retards the conversion and requires an extended immersion time. Hence, the PO-2mg treated perovskite is selected as the target due to its optimal morphology and formation kinetics.
In Fig. S12a (ESI†), the XPS binding energy peaks of Pb 4f obviously shift to lower positions after PO incorporation, indicating the formation of chemical bonding between undercoordinated Pb2+ and R-COO−. We assume that some of the R-COO− ions induce the extraction effect, while the rest coordinate with Pb2+ and passivate the iodide vacancy, thus reducing the trap states within the material's surface and GBs. In Fig. S12b (ESI†), the O 1s of PO treated film can be divided into two single peaks, which are attributed to O2− (532.44 eV) and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif)
![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) (533.77 eV). Time-of-flight secondary-ion mass spectrometry (ToF-SIMS) technique confirms that the spatial distribution of PO preferentially accumulates on the surface (in Fig. S13, ESI†). The surface aggregation of PO is also underpinned by the enhancement of the water contact angle from 52.9° to 65.8° in the example of PO-2mg. (in Fig. S14, ESI†). As seen in the Fourier-transform infrared (FT-IR) spectra (in Fig. S15, ESI†), the exclusive stretching vibration (νs(COO−)) at 1400 cm−1 refers to the feature of oleate ligand in perovskite treated with PO.34
(533.77 eV). Time-of-flight secondary-ion mass spectrometry (ToF-SIMS) technique confirms that the spatial distribution of PO preferentially accumulates on the surface (in Fig. S13, ESI†). The surface aggregation of PO is also underpinned by the enhancement of the water contact angle from 52.9° to 65.8° in the example of PO-2mg. (in Fig. S14, ESI†). As seen in the Fourier-transform infrared (FT-IR) spectra (in Fig. S15, ESI†), the exclusive stretching vibration (νs(COO−)) at 1400 cm−1 refers to the feature of oleate ligand in perovskite treated with PO.34
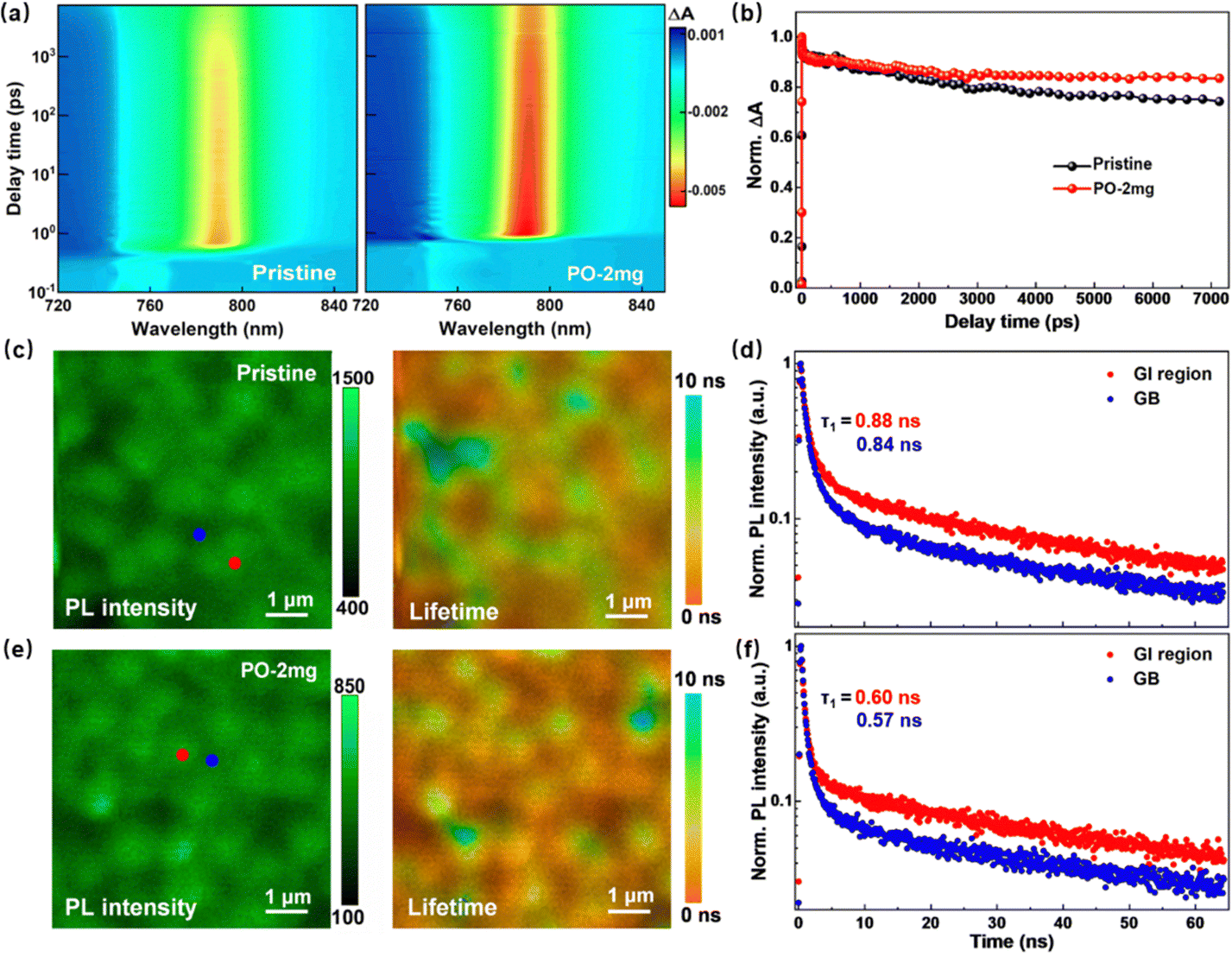
Grazing incident wide-angle X-ray scattering (GIWAXS) measurements with different incident angles (i.e., 0.1° for surface region and 0.6° for bulk domain) are carried out to probe the crystal orientation of the perovskite films. As shown in Fig. 3(a) and (b), for the pristine and the PO treated perovskite, strong scattering rings are observed in both films, which is indicative of the polycrystalline nature of the perovskite films. Both the surface and bulk of PO treated film display the strong scattering signals at qxy = 1 and 2 Å−1, which correspond to the (100) and (200) crystal plane family orientations of α-FAPbI3. By radially integrating intensity along the ring at qxy = 1 Å−1, the PO treated film presents a relatively strong peak with an azimuth angle of 90° (in Fig. 3(c)), suggesting a higher preferential orientation of perovskite crystals compared to the pristine film.35 XRD spectra in Fig. 3(d) display that the diffraction intensities of the (100) and (200) peaks for the PO treated film are enhanced. As expected, the alkyl chains attached to perovskites regulate the growth rates of different crystal planes and eventually promote the formation of films dominated by the (100) orientation.36 Peak force quantitative nanomechanical atomic force microscopy (PFQNM-AFM) is employed to investigate the surface morphologies and corresponding mechanical properties of the perovskite films (shown in Fig. 3(e) and (f)). The modulus map shows a strong correlation with the grain structure and reveals substantial variations in Young's Modulus (YM) across the sample. YM within the grain intragranular (GI) regions (pink color) is higher than that region near GBs (blue color). When increasing the amount of PO, it is observed in Fig. 3(g) that the average YM decreases from 14.10 GPa to 7.67 GPa. High-resolution transmission electron microscopy (HRTEM) results confirm that the PO amorphous layer aggregates at the GBs of perovskite crystals (in Fig. S16, ESI†). Due to the van der Waals force of long alkyl chains, grains can be wrapped within them. Long-alkyl chains are linked with each other, physically connecting adjacent grains together to produce a softening effect.37 The tight contact between grains contributes to maintaining the film integrity under stress. Furthermore, to study the residual strain in the perovskite film, grazing incident X-ray diffraction (GIXRD) is performed using the classical 2θ − sin2(ψ) method, where θ and ψ represent the diffraction and tilt angles, respectively.38,39 For the pristine sample in Fig. S17a (ESI†), the (012) diffraction peaks shift to lower angles as ψ increases from 10° to 50°, indicating gradual expansion of the lattice. This means that the pristine perovskite bears tensile stress along the in-plane direction. After the modulation of PO (in Fig. S17b, ESI†), the shift of the characteristic peaks significantly decreased, implying that the residual stress was largely released. To evaluate the lattice strain relaxation more accurately, the slopes of the fitting linear are calculated by linear fitting of the 2θ − sin2(ψ) curves. The fitting curves of the slope decrease from 0.099 to 0.018, indicating that PO effectively relieves the residual strain in the perovskite film (in Fig. S17c, ESI†). Preferred orientated crystallization and surface softening effect can help inhibit lattice expansion and mitigate lattice distortion. Hence, the residual strain is relaxed.
 | ||
| Fig. 3 Two-dimensional GIWAXS patterns of (a) PO treated film and (b) pristine film at different incident angles. (c) Radially integrated intensity plots along the ring at q = Å−1, assigned to the perovskite (100) plane for both the PO treated film and pristine film. (d) XRD spectra of the PO treated film and pristine film. (e) AFM images of the PO treated film and pristine film. (f) PFQNM-AFM mapping of perovskite thin films. Young's modulus maps of perovskite film with surfactant treatment. The colored scale bar represents the magnitude of modules. (g) The box plot of the YM values across the whole image. | ||
As seen in Fig. S18 (ESI†), pristine perovskite and PO treated films display very similar UV-vis absorption spectra with an identical optical absorption edge around 800 nm, indicating that the PO does not affect the intrinsic optical properties of perovskite materials. To evaluate the carrier transfer and recombination dynamics of the films, femtosecond (fs) transient absorption (TA) spectroscopy is performed. Pump excitation induces an absorption change (ΔA) related to ground-state bleaching (GSB). The 2D TA spectra in Fig. 4(a) show that perovskite films with and without PO treatment present distinct GSB peaks at approximately 790 nm, and the photoinduced carrier dynamics can be revealed through the GSB peaks. Fig. 4(b) shows the normalized decay kinetics of the corresponding GSB peaks. The PO treated perovskite film shows a much slower decay, representing a much longer lifetime of photo-induced carriers.40,41 We further perform the fluorescence lifetime imaging microscopy (FLIM) characterization to explore the charge transfer kinetics of perovskite films based on the structure of FTO glass/TiO2/perovskite.42,43 As seen in Fig. 4(c), although there are some PL heterogeneities between different grains, probably due to the size effects on the FLIM, the PL inside the grain is more homogeneous after PO incorporation. We can readily isolate emission from within a grain (red circle) compared to at a GB (blue circle), and conclude the fast early time component (τ1) to the lifetime is likely due to rapid nonradiative recombination at the GB. Moreover, the incorporation of PO can quench the PL intensity/lifetime throughout the whole thin films, suggesting more sufficient electron transport from the perovskite layer to the TiO2 layer.44 Space-charge-limited-current (SCLC) analysis based on an electron-only device with the structure FTO/TiO2/perovskite/PCBM/Ag is tested to quantitatively obtain the density of trap states in these perovskite films.45,46 As per Fig. S19 (ESI†), the trap-filled limit voltages (VTFL) are determined as 0.68 V and 0.36 V, and the corresponding densities of trap states are calculated as 1.09 × 1016 cm−3 for the pristine sample and 5.77 × 1015 cm−3 for the PO treated sample.
 | ||
| Fig. 4 (a) TAS spectra of pristine and PO-2mg treated perovskites. (b) Temporal dynamics of bleach signals of these two films. Bulk fluorescence intensity/lifetime imaging of the (c) pristine perovskite and (e) PO-treated perovskite films on FTO/TiO2 substrates. Lifetime plots of the two locations indicated in the PL maps (c) and (e) for the different films: (d) pristine perovskite and (f) PO-treated perovskite. | ||
We fabricate n–i–p devices with the water-processed perovskite and the statistical PCE values for the devices with different amounts of PO in perovskite are summarized in Fig. S20 and Table S2 (ESI†). Clearly, the incorporation of PO leads to an average PCE enhancement from 22.40% (pristine, the control) to 23.61% (PO-2mg, the target), whereas a further increase of PO leads to PCE degradation. Fig. 5(a) shows the J–V curves of the champion devices and the key photovoltaic parameters are summarized in the inset. Compared to the pristine sample, PO-1mg treated perovskite exhibits higher PCE (23.69%), but due to insufficient introduction, its PCE is slightly lower than that of the PO-2mg sample (24.14%). The PCE enhancement is ascribed to the optimal formation kinetics, as well as the passivation and morphology repair effects of PO molecules. Excessive PbI2 residues are detected in the PO-4mg treated perovskite film (shown in Fig. S21, ESI†), which causes electrical insulation and reduces device performance. In Fig. 5(b), the target PSC displays a boosted stabilized power output (SPO) of 23.84% (biased at 1.00 V) compared with an SPO of 22.71% (biased at 0.96 V) for the control. Fig. 5(c) provides the external quantum efficiency (EQE) spectrum and integrated JSC of the corresponding devices. The target device exhibits a higher integrated JSC of 24.83 mA cm−2 than that of the control device (24.65 mA cm−2), which is consistent with the J–V curves. As shown in Fig. 5(d), benefitting from the densely arranged Pb(NO3)2 template, the target PSC achieves a PCE of 22.09% on a large area (1 cm2), while the poor PCE of the control (16.34%) is mainly attributed to the presence of pinhole defects as shown in Fig. S22 (ESI†). When expanding the area, morphological imperfections are almost inevitable and are recognized as a performance limiting factor. To our knowledge, this is the first reported PCE with an area of 1 cm2 for water-processed PSCs. We provide a performance comparison in Fig. S23 (ESI†) between the state-of-the-art green fabrication methods. Considering the low carbon emissions and low cost of the entire process, the Pb(NO3)2/H2O protocol has significant advantages. Meanwhile, the ideality factor (n) is measured to examine the effect of PO treatment on the Shockley–Read–Hall recombination of the device, as shown in Fig. 5(e). The slope of VOCversus the natural logarithm of light intensity for the target device (1.38 kBT q−1) is smaller than that of the control device (1.48kBTq−1), indicating a suppressed trap-assisted charge recombination through PO treatment.47 In Fig. 5(f), we calculate the impact of charge transport losses and non-radiative recombination losses on FF. The maximum FF (FFmax) is estimated by the following equation without considering charge transport losses:48,49
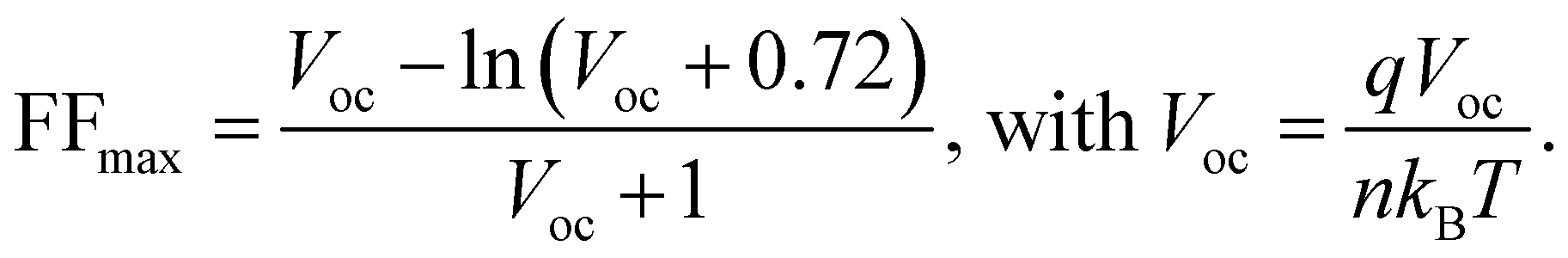 | (10) |
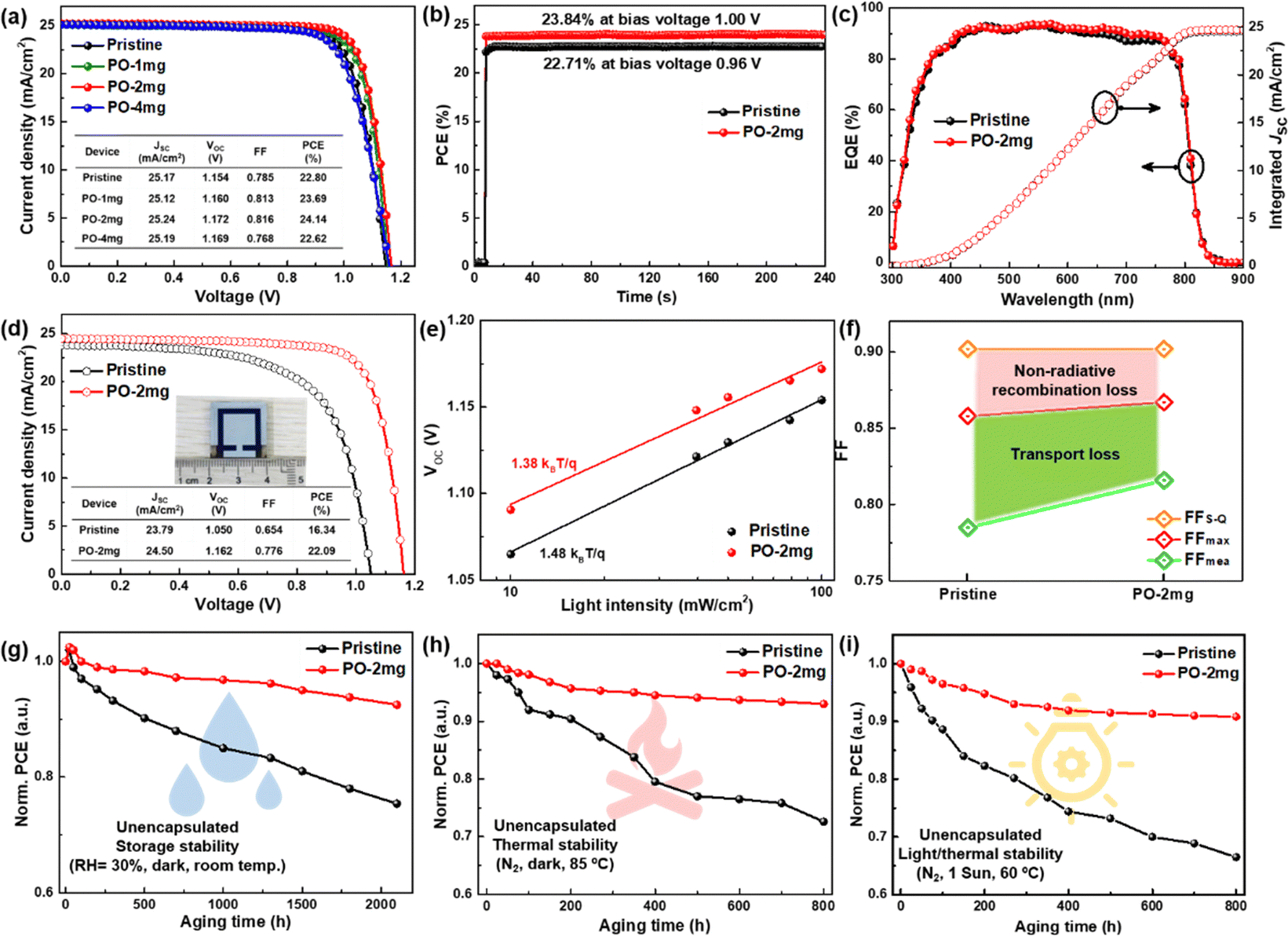 | ||
| Fig. 5 (a) J–V curves of the champion PSCs. (b) The MPP of PSCs in ambient air under AM1.5G illumination. (c) EQE spectra and integrated current density values for devices based on pristine and PO-2mg treated perovskites. (d) J–V curves of the PSCs with a large area (1 cm2). (e) VOC as a function of light intensity of various perovskite devices. (f) Device FFS-Q compared to FFmea and FFmax. Stability measurement of solar cells. (g) Stability trails of the photovoltaic parameters of pristine and PO treated devices (average relative humidity of 30% and room temperature of ∼25 °C). (h) Thermal stability test at 85 °C for 800 hours under a N2 atmosphere. It is noted that the hole transporting material Spiro-OMeTAD used for the thermal stability test is replaced by poly[bis(4-phenyl)(2,4,6-trimethylphenyl)-amine] (PTAA). (i) Light soaking stability under illumination of 100 mW cm−2. The unencapsulated PSCs are placed in a N2-filled glove box under one sun irradiation (60 °C) under open circuit conditions. | ||
The calculated FFmax for the control device is 0.858, while the calculated FFmax for the target device is 0.867. For the control device, the difference between FFmax and the measured FF (FFmea) and between FFmax and FFS-Q is 0.073 and 0.044, respectively. However, the differences between the target devices becomes smaller, which are 0.051 and 0.035, respectively. The improvement of the FF originates from defect passivation and preferential crystallization, which can suppress non-radiative recombination and minimize charge transfer losses.
The stability of unencapsulated devices has been tested at a temperature of 25 °C and humidity of 30% under an air environment for 2100 h. As shown in Fig. 5(g), the PCE of the target device maintains 92.5% of the original PCE after 90 days. By contrast, after obvious degradation the control device only retains 75.4% original PCE. The thermal stability of the PSCs is investigated by continuous heating at 85 °C for 800 hours, and the results are displayed in Fig. 5(h). The target device has much better thermal stability, and the device maintains 93.1% of its original efficiency while the control device only maintains 72.6%. To further explore the coupled aging stressors (thermal/light stability, 1 sun/60 °C), the PCE for different devices is constantly monitored as a function of the aging time. As shown in Fig. 5(i), the PCE of the target device can maintain 90.8% original PCE after 800 h illumination. Nonetheless, the control device gives relatively limited light stability, only keeping 66.5% of the original PCE after 800 h illumination. The unencapsulated devices are tested at the maximum power point (MPP) under full-sun illumination under a N2 atmosphere to explore their operational stability (in Fig. S24, ESI†). The target device maintains 95.2% of its maximum efficiency after 800 h, while the control device degrades to 80.3% of its initial performance after 460 h. Generally, the generation of I2 under illumination has also been identified as one of the critical factors for the degradation.50,51 The iodine loss from perovskite can be tracked by placing the film samples in a solvent with low polarity and testing the UV-vis absorption spectra of the extractions.52,53 The pristine film and PO treated film are immersed in toluene at 80 °C under 1 sun illumination. As shown in Fig. S25 (ESI†), the pristine perovskite films release substantial molecular iodine within 24 hours revealed by the rise of absorbance at around 500 nm while the PO treated perovskite films generate less I2 in the extraction. The slower release of I2 implies that interface/GB modification by PO substantially reduces ion migration and suppresses I2 formation.
3. Conclusion
By examining the crystallization process of Pb(NO3)2 under different RH conditions, we figure out that dry air has a stronger promotion effect on the nucleation rate, and boosts the spreading dynamics of aqueous inks. Potassium oleate as a long-alkyl chain anionic surfactant is introduced to reduce the surface tension of water. By adopting a wetting-controlled strategy, a pinhole-free conformal perovskite film with a large area can be readily achieved. These results indicate that PO can not only form a soft framework around the perovskite grains to reduce the film residual stress, but also coordinate with the unsaturated Pb2+ and induce preferential crystallization. The device processed in dry air achieves a PCE of 24.14% in a small area (0.09 cm2) and 22.09% in a large area (1 cm2). It is expected that this wetting strategy will promote the large-scale production of perovskites using the Pb(NO3)2/H2O protocol.Author contributions
P. Z. conceived the idea. S. L. supervised the entire team and the project. L. R. and P. Z. designed the experiments. Y. Z. fabricated and optimized all the devices and samples for measurements. Q. Z., J. X. and J. W. conducted the XRD and UV-vis spectra and characterization of PSCs. X. C. and K. Z. conducted the GIWAX measurements. P. Z. and L. R. conducted PL, TRPL and EQE measurements. L. Z. carried out TAS/FLIM measurements and analyzed the data. Y. Z. conducted SEM measurements. P. Z. and L. R. wrote the first draft of the paper. All authors contributed to the discussion and review of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the National Key Research Program of China (2016YFA0202403), the Changjiang Scholar and Innovative Research Team (IRT_14R33), the Overseas Talent Recruitment Project (B14041), the 111 Project (B21005), the National Natural Science Foundation of China (51702263), the Key Research and Development Program of Shaanxi (program no. 2022LL-JB-08), and the Natural Science Basic Research Plan in Shaanxi Province of China (2020JQ-144, 2022JQ-683). The authors acknowledge that GIWAXS data were obtained from BL14B1 of the Shanghai Synchrotron Radiation Facility (SSRF), China.References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- Y. Zhao, F. Ma, Z. Qu, S. Yu, T. Shen, H.-X. Deng, X. Chu, X. Peng, Y. Yuan, X. Zhang and J. You, Science, 2022, 531–534 CrossRef CAS PubMed.
- I. Kosta, H. Grande and R. Tena-Zaera, Electrochim. Acta, 2017, 246, 1193–1199 CrossRef CAS.
- L. Tan, J. Zhou, X. Zhao, S. Wang, M. Li, C. Jiang, H. Li, Y. Zhang, Y. Ye, W. Tress, L. Ding, M. Gratzel and C. Yi, Adv. Mater., 2023, 35, e2205027 CrossRef PubMed.
- T. Y. Hsieh, T. C. Wei, K. L. Wu, M. Ikegami and T. Miyasaka, Chem. Commun., 2015, 51, 13294 RSC.
- T. Y. Hsieh, T. S. Su, M. Ikegami, T. C. Wei and T. Miyasaka, Mater. Today Energy, 2019, 14, 100125 CrossRef.
- P. Zhai, T. S. Su, T. Y. Hsieh, W. Y. Wang, L. X. Ren, J. Y. Guo and T. C. Wei, Nano Energy, 2019, 65, 104036 CrossRef CAS.
- B. Zhang, Y. Xiao, Y. Liu, X. Yu, Z. Ku, W. Li, J. Xiao, F. Long, Y. Cheng and Y. Peng, ACS Sustainable Chem. Eng., 2022, 10, 5225–5232 CrossRef CAS.
- Y. C. Teng, T. S. Su, S. Lan, A. F. Musa and T. C. Wei, Nanomaterials, 2022, 12, 3783 CrossRef CAS PubMed.
- J. T. Yates, Surf. Sci., 2009, 603, 1605–1612 CrossRef CAS.
- K. Liu, M. Cao, A. Fujishima and L. Jiang, Chem. Rev., 2014, 114, 10044–10094 CrossRef CAS.
- J. Balajka, M. A. Hines, W. J. I. DeBenedetti, M. Komora, J. Pavelec, M. Schmid and U. Diebold, Science, 2018, 361, 786–789 CrossRef CAS.
- T. Y. Hsieh, M. Pylnev, E. Palomares and T. C. Wei, Adv. Funct. Mater., 2020, 30, 1909644 CrossRef CAS.
- P. Zhai, L. X. Ren, S. Q. Li, L. Zhang, D. Li and S. Z. Liu, Matter, 2022, 5, 4450–4466 CrossRef CAS.
- W. A. Dunlap-Shohl, Y. Zhou, N. P. Padture and D. B. Mitzi, Chem. Rev., 2019, 119, 3193–3295 CrossRef CAS PubMed.
- H. L. Hu, M. Singh, X. J. Wan, J. N. Tang, C. W. Chu and G. Li, J. Mater. Chem. A, 2020, 8, 1578–1603 RSC.
- K. Zhang, Z. Wang, G. Wang, J. Wang, Y. Li, W. Qian, S. Zheng, S. Xiao and S. Yang, Nat. Commun., 2020, 11, 1006 CrossRef CAS PubMed.
- V. Zorba, X. B. Chen and S. S. Mao, Appl. Phys. Lett., 2010, 96, 093702 CrossRef.
- G. McHale, N. J. Shirtcliffe, S. Aqil, C. C. Perry and M. I. Newton, Phys. Rev. Lett., 2004, 93, 036102 CrossRef CAS PubMed.
- C. Cioarec, P. Melpignano, N. Gherardi, R. Clergereaux and C. Villeneuve, Langmuir, 2011, 27, 3611–3617 CrossRef CAS PubMed.
- P. Zhai, R. Cheng, L. X. Ren, Y. T. Huang, H. Lee and S. P. Feng, Sol. RRL, 2018, 2, 1800103 CrossRef.
- P. Zhai, L. Ren, Y. Zhang, Z. Xu, Y. Wu, K. Zhao, L. Zhang and S. Liu, Energy Environ. Sci., 2023, 16, 3014–3024 RSC.
- H. A. Harms, N. Tétreault, N. Pellet, M. Bensimon and M. Grätzel, Faraday Discuss., 2014, 176, 251–269 RSC.
- E. Ochoa-Martinez, M. Ochoa, R. D. Ortuso, P. Ferdowsi, R. Carron, A. N. Tiwari, U. Steiner and M. Saliba, ACS Energy Lett., 2021, 6, 2626–2634 CrossRef CAS.
- T. Wang, T. Ye, L. Qiao, W. Kong, F. Zeng, Y. Zhang, R. Sun, L. Zhang, H. Chen, R. Zheng and X. Yang, Sci. China Mater., 2022, 65, 3361–3367 CrossRef CAS.
- W. Zhang, L. He, D. Tang and X. Li, Sol. RRL, 2020, 4, 2000376 CrossRef CAS.
- L. Wang, D. Zheng, Z. Li, B. Farhadi, L. Peng, S. Zhao, Z. Chang, L. Duan, Y. Cao, H. Wang, Y. Tong, M. Du, K. Wang and S. Liu, Matter, 2023, 6, 2987–3005 CrossRef CAS.
- A. Kausar, A. Sattar, C. Xu, S. Zhang, Z. Kang and Y. Zhang, Chem. Soc. Rev., 2021, 50, 2696–2736 RSC.
- Y. Ren, K. Zhang, Z. Lin, X. Wei, M. Xu, X. Huang, H. Chen and S. Yang, Nano-Micro Lett., 2023, 15, 182 CrossRef CAS PubMed.
- Z. Mitrinova, S. Tcholakova, Z. Popova, N. Denkov, B. R. Dasgupta and K. P. Ananthapadmanabhan, Langmuir, 2013, 29, 8255–8265 CrossRef CAS PubMed.
- M. Jung, S. G. Ji, G. Kim and S. I. Seok, Chem. Soc. Rev., 2019, 48, 2011–2038 RSC.
- A. Ummadisingu and M. Gratzel, Sci. Adv., 2018, 4, e1701402 CrossRef PubMed.
- W. Shao, H. Wang, F. Ye, C. Wang, C. Wang, H. Cui, K. Dong, Y. Ge, T. Wang, W. Ke and G. Fang, Energy Environ. Sci., 2023, 16, 252–264 RSC.
- Y. Cui, J. Shi, F. Meng, B. Yu, S. Tan, S. He, C. Tan, Y. Li, H. Wu, Y. Luo, D. Li and Q. Meng, Adv. Mater., 2022, 34, e2205028 CrossRef PubMed.
- X. Huang, F. Cao, S. Zhan, Q. Feng, M. Zhu, Z. Su, X. Gao, J. Yin, J. Li, N. Zheng and B. Wu, Joule, 2023, 7, 1556–1573 CrossRef CAS.
- M. Jung, T. J. Shin, J. Seo, G. Kim and S. I. Seok, Energy Environ. Sci., 2018, 11, 2188–2197 RSC.
- X. Wang, Z. Ying, J. Zheng, X. Li, Z. Zhang, C. Xiao, Y. Chen, M. Wu, Z. Yang, J. Sun, J. R. Xu, J. Sheng, Y. Zeng, X. Yang, G. Xing and J. Ye, Nat. Commun., 2023, 14, 2166 CrossRef CAS PubMed.
- C. Zhu, X. Niu, Y. Fu, N. Li, C. Hu, Y. Chen, X. He, G. Na, P. Liu, H. Zai, Y. Ge, Y. Lu, X. Ke, Y. Bai, S. Yang, P. Chen, Y. Li, M. Sui, L. Zhang, H. Zhou and Q. Chen, Nat. Commun., 2019, 10, 815 CrossRef CAS PubMed.
- J. J. Zhao, Y. H. Deng, H. T. Wei, X. P. Zheng, Z. H. Yu, Y. C. Shao, J. E. Shield and J. S. Huang, Sci. Adv., 2017, 3, eaao5616 CrossRef PubMed.
- W. Xu, Y. Gao, W. Ming, F. He, J. Li, X. H. Zhu, F. Kang, J. Li and G. Wei, Adv. Mater., 2020, 32, e2003965 CrossRef PubMed.
- J. Zhang, B. Che, W. Zhao, Y. Fang, R. Han, Y. Yang, J. Liu, T. Yang, T. Chen, N. Yuan, J. Ding and S. F. Liu, Adv. Mater., 2022, 34, e2202735 CrossRef PubMed.
- M. Q. Wang, W. W. Wu, Y. L. Liu, S. Y. Yuan, D. H. Tian, C. L. Zhang, Z. P. Ma, J. H. Deng, J. H. Chen, Z. Z. Lou, W. Z. Li and J. D. Fan, Adv. Funct. Mater., 2023, 2300700 CrossRef CAS.
- S. Wieghold, J. P. Correa-Baena, L. Nienhaus, S. J. Sun, K. E. Shulenber, Z. Liu, J. S. Tresback, S. S. Shin, M. G. Bawendi and T. Buonassisi, ACS Appl. Energy Mater., 2018, 1, 6801–6808 CrossRef CAS.
- T. Wu, R. J. Zhao, J. M. Qiu, S. H. Wang, X. L. Zhang and Y. Hua, Adv. Funct. Mater., 2022, 32, 2204450 CrossRef CAS.
- D. Yang, X. Zhou, R. X. Yang, Z. Yang, W. Yu, X. L. Wang, C. Li, S. Z. Liu and R. P. H. Chang, Energy Environ. Sci., 2016, 9, 3071–3078 RSC.
- S. Yuan, F. Qian, S. Yang, Y. Cai, Q. Wang, J. Sun, Z. Liu and S. Liu, Adv. Funct. Mater., 2019, 29, 1807850 CrossRef CAS.
- Y. Z. Zhang, Y. J. Wang, L. C. Zhao, X. Y. Yang, C. H. Hou, J. Wu, R. Su, S. Jia, J. J. Shyue, D. Y. Luo, P. Chen, M. T. Yu, Q. Y. Li, L. Li, Q. H. Gong and R. Zhu, Energy Environ. Sci., 2021, 14, 6526–6535 RSC.
- S. Rühle, Sol. Energy, 2016, 130, 139–147 CrossRef.
- Q. Cao, J. B. Yang, T. Wang, Y. K. Li, X. Y. Pu, J. S. Zhao, Y. X. Zhang, H. Zhou, X. Q. Li and X. H. Li, Energy Environ. Sci., 2021, 14, 5406–5415 RSC.
- S. G. Motti, D. Meggiolaro, A. J. Barker, E. Mosconi, C. A. R. Perini, J. M. Ball, M. Gandini, M. Kim, F. De Angelis and A. Petrozza, Nat. Photonics, 2019, 13, 532–539 CrossRef CAS.
- J. Wei, Q. W. Wang, J. D. Huo, F. Gao, Z. Y. Gan, Q. Zhao and H. B. Li, Adv. Energy Mater., 2021, 11, 2002326 CrossRef CAS.
- X. Y. Yang, Y. Ni, Y. Z. Zhang, Y. J. Wang, W. Q. Yang, D. Y. Luo, Y. G. Tu, Q. H. Gong, H. F. Yu and R. Zhu, ACS Energy Lett., 2021, 6, 2404–2412 CrossRef CAS.
- A. Krishna, H. Zhang, Z. Zhou, T. Gallet, M. Dankl, O. Ouellette, F. T. Eickemeyer, F. Fu, S. Sanchez, M. Mensi, S. M. Zakeeruddin, U. Rothlisberger, G. N. Manjunatha Reddy, A. Redinger, M. Gratzel and A. Hagfeldt, Energy Environ. Sci., 2021, 14, 5552–5562 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee02459h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |