DOI:
10.1039/D4DT00394B
(Paper)
Dalton Trans., 2024,
53, 9874-9886
Synthesis and theoretical study of a mixed-ligand indium(III) complex for fabrication of β-In2S3 thin films via chemical vapor deposition†
Received
8th February 2024
, Accepted 16th May 2024
First published on 17th May 2024
Abstract
Two new heteroleptic indium aminothiolate compounds [InClSC2H4N(Me)SC2H4]3[1] and [InSC2H4N(Me)SC2H4(C8H5F3NO)] [2] were synthesized by in situ salt metathesis reaction involving indium trichloride, aminothiol, and N,O-β-heteroarylalkenol ligands. The complexes were subsequently purified and thoroughly characterized by nuclear magnetic resonance (NMR) analysis, elemental studies, mass spectroscopy, and X-ray diffraction single crystal analysis that showed a trigonal bipyramidal coordination of In(III) in both complexes. Thermogravimetric analysis of [1] revealed a multistep decomposition pathway and the formation of In2S3 at 350 °C, which differed from the pattern of [2] due to the lower thermal stability of [1]. Compound [2] exhibited a three-step decomposition process, resulting in the formation of In2S3 at 300 °C. The Chemical Vapor Deposition (CVD) experiment involving compound [2] was conducted on the FTO substrate, resulting in the production of singular-phase In2S3 deposits. A comprehensive characterization of these deposits, including crystal structure analysis via X-ray diffraction (XRD), and surface topography examination through scanning electron microscopy (SEM) has been completed. The presence of In–S units was also supported by the Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), and energy dispersive spectroscopy (EDS) of the as-deposited films. Moreover, the electronic structure and thermal properties of compound [2] were investigated through DFT calculations. Electron density localization analysis revealed that the highest occupied molecular orbital (HOMO) exhibited dense concentration at the aminothiolate moiety of the complex, while the lowest unoccupied molecular orbital (LUMO) predominantly resided at the N,O-β-heteroarylalkenolate ligand. Furthermore, our computational investigation has validated the formation of indium sulfide by elucidating an intermediate state, effectively identified through EI-MS analysis, as one of the plausible pathways for obtaining In2S3. This intermediate state comprises the aminothiolate ligand (LNS) coordinated with indium metal.
Introduction
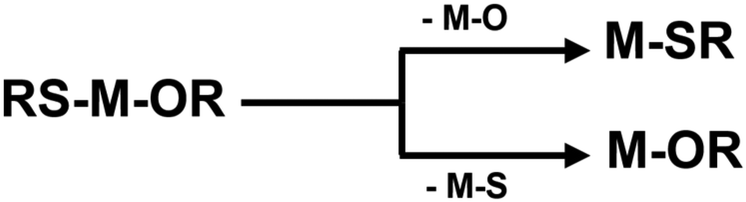
Indium sulfide (In2S3) is an n-type semiconductor with different phases such as α, β, and γ. Among these, β-In2S3, characterized by a narrow band gap of 2.0–2.3 eV shows an interesting defect structure, high stability, and exceptional optical properties. Its notably high photosensitivity suggests the potential for excellent photocatalytic activity,1 photoelectric sensitivity,2 supercarrier mobility,3 efficient CO2 reduction activity,4 and use as photoanodes for water splitting.5 The influence of preorganized metal–ligand units in metal–organic precursors is evident in various functional coatings produced via chemical vapor deposition (CVD) and atomic layer deposition (ALD).6 Specifically designed molecular compounds, achieved through judicious selection of ligand and co-ligand units, play a crucial role in determining the chemical composition, phase, structure, and morphology of the final material.7 By adjusting the chemical configuration of the precursor molecule, the physicochemical properties of the precursors such as volatility or thermal stability can be tailored to meet the requirements of the gas phase process; however, our mechanistic understanding of the conversion of the molecular units into solid-state film remains limited, thus calling for integrated experimental and modeling approaches. While thin film deposition of metal oxides by the transformation of precursors containing preformed metal–oxygen units is extensively reported,8 studies on bond disruption for chalcogens (S and Se) are limited. Metal complexes incorporating sulfur ligands have been extensively studied; however, limited quantitative information is available on the strengths of the metal–ligand bonds.9,10 Metal–ligand (M–S(Se)–R) interactions play a deterministic role in the formation of metal sulfides and selenides.11,12 Gas-phase chemistry offers a unique opportunity to study the intrinsic reactivity of chemical species.13 The metal–thiolate bonds can be strongly covalent, and involve different orbital interactions.14 Moreover, understanding the chemistry behind nanostructure formation in nanomaterials is one of the important and interesting challenges in gas phase chemistry. Precursors, chemicals and associated chemical properties play a critical role in controlling the structural and morphological properties of nanomaterials.15 Therefore, it is important to study the thermodynamics and thermochemistry of dissociation and elimination (Scheme 1) reactions of In2S3-precursors in the gas phase. Several known single source precursors of indium based complexes such as indium thiolates,16,17 dithiocarboxylates,18 dithiocarbamates,19 dithioimidodiphosphinates,20 dithiobiurets,21 xanthates,22 aminothiolates,23 organyl and silylated chalcogenoethers24,25etc. are well established. Despite their extensive experimental investigations, the thermodynamic and kinetic properties, the detailed mechanism of the deposition process, and possible intermediates of many gas-phase In2S3-precursors are unknown. By employing theoretical calculations, many properties of precursors can be predicted with a good degree of precision. These include the metal–ligand bond strength, the thermolysis energy and barrier, the energy of chelation, hydrolysis, and formation processes, as well as crucial thermodynamic parameters like enthalpies, entropies, and Gibbs free energy of dissociation in the gas phase. Furthermore, insights into their molecular orbital properties, which are related to their stabilities and reactivities can be achieved.26
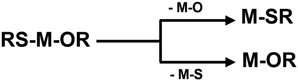 |
| | Scheme 1 Schematic of M–O and M–S (metal–sulfur and metal–oxygen) bond break reaction. | |
Herein, we present a study on the gas phase ligand dissociation and thermal stability of a newly synthesized heteroleptic indium aminothiolate complex [2], which incorporates a chelating N,O-β-heteroarylalkenolate ligand in the coordination sphere. The dissociation energies of In–S, In–O and In–N bonds within the heteroleptic complex that we referred to as ligand dissociation energy (LDE) were successfully investigated using DFT calculations to assess their strengths. The as-synthesized complex [2] demonstrates excellent structural stability and maintains intact transport in the gas phase, minimizing premature decomposition before deposition onto the substrate during the chemical vapour deposition process, ultimately achieving singular-phase In2S3.27
Results and discussion
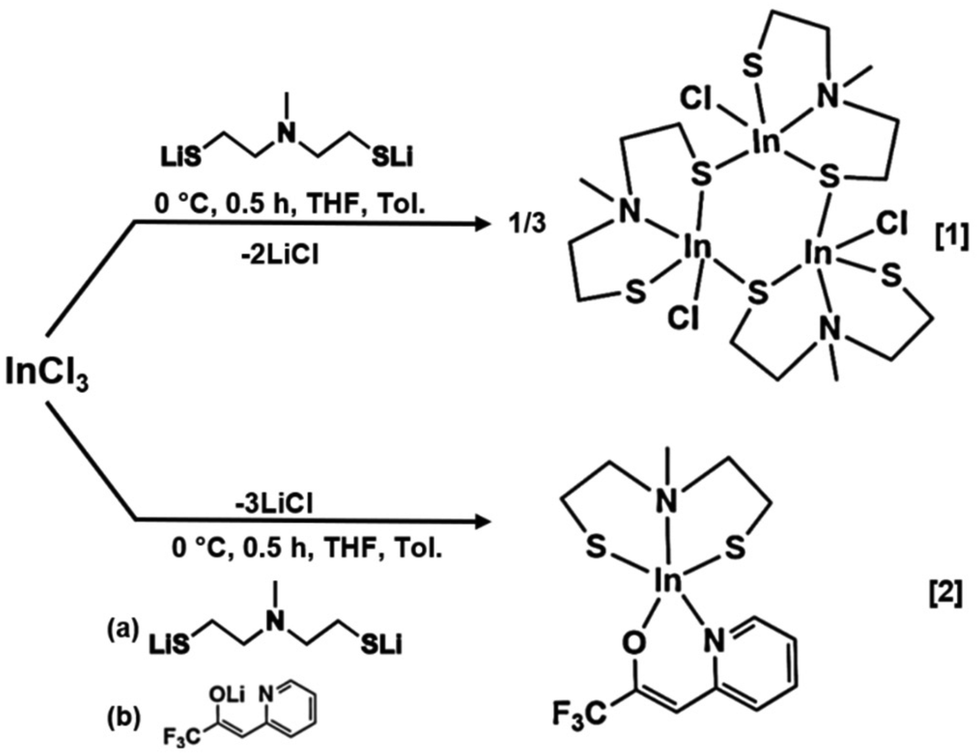
Synthesis of [InClSC2H4N(Me)SC2H4]3[1] and [InSC2H4N(Me)SC2H4(C8H5F3NO)] [2]
The aminothiol28 and β-heteroarylalkenol29 ligands used in this work were synthesized following the procedures described in our research group's publications. The salt metathesis of InCl3 is a well-established synthetic route for obtaining indium complexes, achieved through an acid–base reaction triggered by the stronger chelating ligands.30 The heteroleptic complexes were synthesized by in situ addition of the lithiated ligands to the metal chloride, resulting in the precipitation of LiCl (Scheme 2). Complexes [1] and [2] were extracted using chloro-benzene and chloroform as solvents, respectively, followed by additional purification through pentane washing. This process yielded a white and slightly yellow powder of [1] and [2] respectively. Compound [2] was further purified through sublimation at 100 °C under reduced pressure (10−3 mbar) resulting in the attainment of the final, slightly yellow complex [2]. Both isolated heteroleptic complexes, [1] and [2], were found to be stable under ambient conditions and were easily elucidated in solution by NMR spectral analysis.
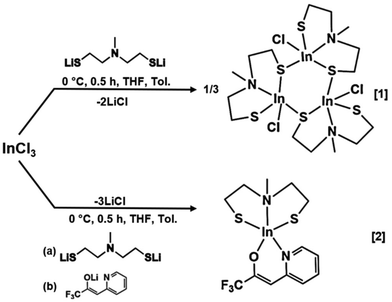 |
| | Scheme 2 Proposed scheme for the synthesis of [InClSC2H4N(Me)SC2H4]3[1] and [InSC2H4N(Me)SC2H4(C8H5F3NO)] [2]. | |
Characterization of [1] and [2]
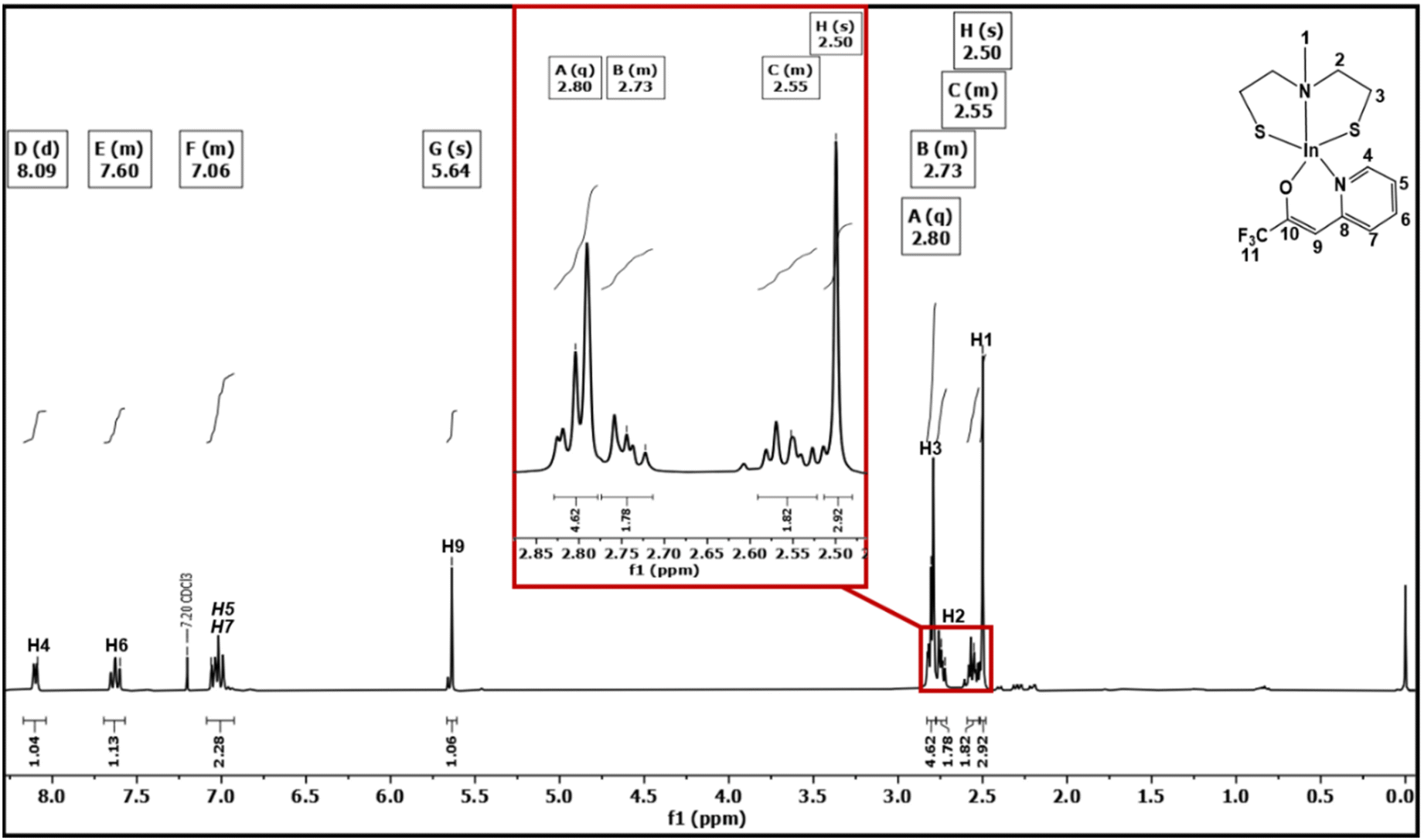
Different signals were detected in the 1H-NMR of [1] at 2.29 and 2.68 ppm with an integration ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8, respectively (Fig. S4†), which, in conjunction with the 13C NMR data, suggests the presence of a monomeric complex of [1] in solution. The singlet observed at 2.29 ppm was assigned to the methyl group of the bridging nitrogen while the broad multiplets at 2.68 ppm were assigned to both the nitrogen-bound and sulfur-bound methylene groups. The 13C NMR spectrum of [1] (Fig. S5†) showed three distinctive signals at 23.13, 42.74 and 58.40 ppm. The 1H NMR data of [2] showed an integration ratio of 3:2:2:4:1:2:1:1 (Fig. 1). The multiplets at 7.06, 7.60, and 8.09 ppm correspond to the protons of the pyridinyl part of the N,O-β-heteroarylalkenolate ligand framework, while the vinylic proton displayed a sharp singlet at 5.64 ppm. The singlet observed at 2.50 ppm was assigned to the methyl group of the bridging nitrogen. The multiplets at 2.55 and 2.73 ppm were assigned to the nitrogen-bound methylene group of both ethanethiolato chains. As noticed, the protons of the nitrogen-bound methylene group of [2] were split into two different signals due to their different positions (endo- or exo-position within the same ring conformation), rendering them chemically non-equivalent. These protons were easily identified using 1H–13C HMQC (Fig. S7†) and 1H–13C HMBC (Fig. S8†) correlation spectral analyses. This effect was also observed for complexes of the types [M(SC2H4N(Me)C2H4S)2] (M = Mo or W) and [SnCl2(SeC2H4N(Me)C2H4Se)] containing a similar aminothiolate ligand.28 Multiplets observed at 2.80 ppm were assigned to the saturated methylene group directly bound to sulfur. The 13C NMR spectrum of [2] displayed notable signals at 23.13, 43.12, 58.86, 94.61, 118.37, 120.64, 124.88, 139.61, 146.24, 156.98 and 157.33 ppm (Fig. S6†). In the 19F NMR spectrum (Fig. S9†) of [2], a singlet was observed at −74.21 ppm, corresponding to the CF3 moiety of the N,O-β-heteroarylalkenolate ligand, which was found to be shifted upfield, when compared to the free ligand (−74.99 ppm, Fig. S3†),31 which is in agreement with previous investigations.32 The unambiguous assignment of proton and carbon chemical shifts using 1H and 13C NMR enabled the prediction of structural units in [2] through 1H–13C HMQC and 1H–13C HMBC correlations (Fig. S7 and S8†).
:8, respectively (Fig. S4†), which, in conjunction with the 13C NMR data, suggests the presence of a monomeric complex of [1] in solution. The singlet observed at 2.29 ppm was assigned to the methyl group of the bridging nitrogen while the broad multiplets at 2.68 ppm were assigned to both the nitrogen-bound and sulfur-bound methylene groups. The 13C NMR spectrum of [1] (Fig. S5†) showed three distinctive signals at 23.13, 42.74 and 58.40 ppm. The 1H NMR data of [2] showed an integration ratio of 3:2:2:4:1:2:1:1 (Fig. 1). The multiplets at 7.06, 7.60, and 8.09 ppm correspond to the protons of the pyridinyl part of the N,O-β-heteroarylalkenolate ligand framework, while the vinylic proton displayed a sharp singlet at 5.64 ppm. The singlet observed at 2.50 ppm was assigned to the methyl group of the bridging nitrogen. The multiplets at 2.55 and 2.73 ppm were assigned to the nitrogen-bound methylene group of both ethanethiolato chains. As noticed, the protons of the nitrogen-bound methylene group of [2] were split into two different signals due to their different positions (endo- or exo-position within the same ring conformation), rendering them chemically non-equivalent. These protons were easily identified using 1H–13C HMQC (Fig. S7†) and 1H–13C HMBC (Fig. S8†) correlation spectral analyses. This effect was also observed for complexes of the types [M(SC2H4N(Me)C2H4S)2] (M = Mo or W) and [SnCl2(SeC2H4N(Me)C2H4Se)] containing a similar aminothiolate ligand.28 Multiplets observed at 2.80 ppm were assigned to the saturated methylene group directly bound to sulfur. The 13C NMR spectrum of [2] displayed notable signals at 23.13, 43.12, 58.86, 94.61, 118.37, 120.64, 124.88, 139.61, 146.24, 156.98 and 157.33 ppm (Fig. S6†). In the 19F NMR spectrum (Fig. S9†) of [2], a singlet was observed at −74.21 ppm, corresponding to the CF3 moiety of the N,O-β-heteroarylalkenolate ligand, which was found to be shifted upfield, when compared to the free ligand (−74.99 ppm, Fig. S3†),31 which is in agreement with previous investigations.32 The unambiguous assignment of proton and carbon chemical shifts using 1H and 13C NMR enabled the prediction of structural units in [2] through 1H–13C HMQC and 1H–13C HMBC correlations (Fig. S7 and S8†).
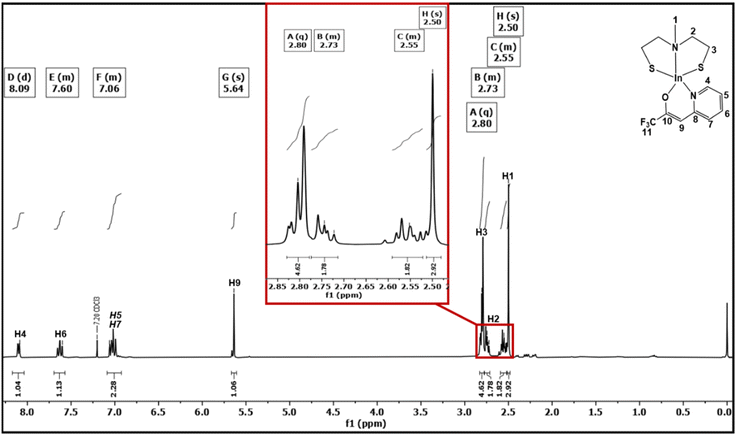 |
| | Fig. 1
1H-NMR spectra of compound [2] recorded in CDCl3. | |
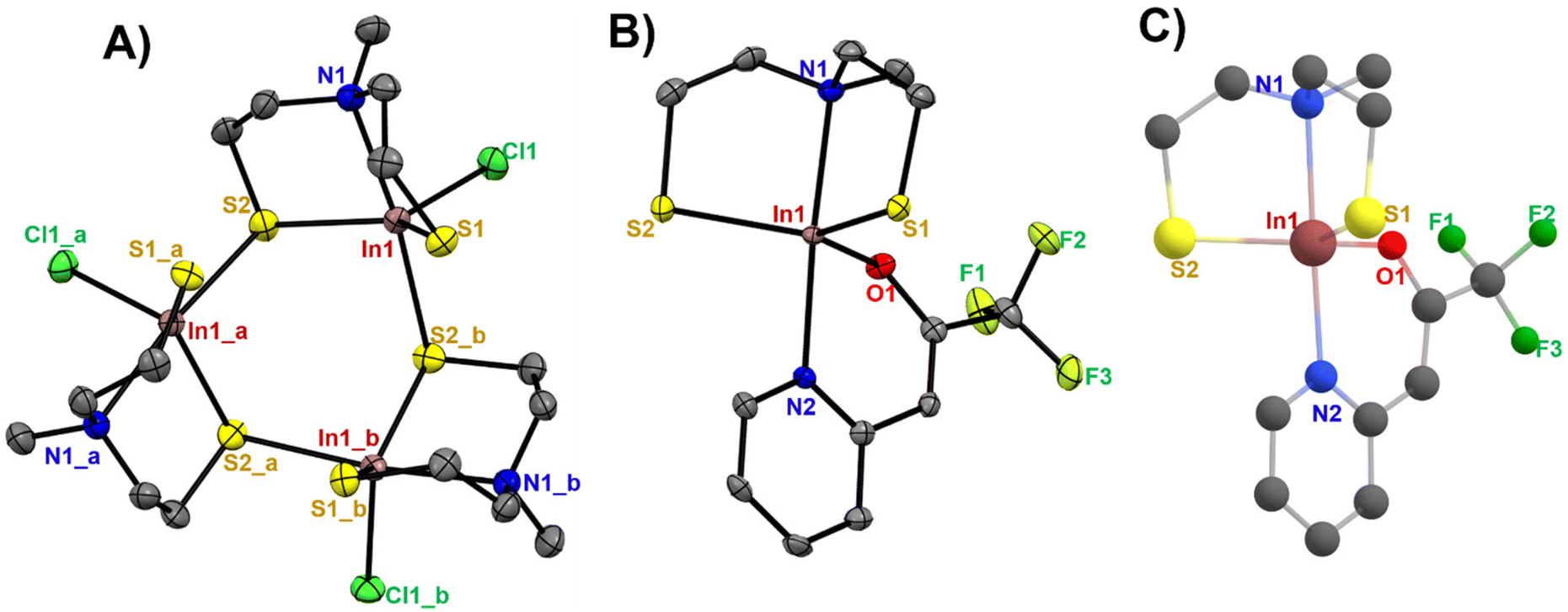
Suitable crystals for single-crystal X-ray diffraction measurements were grown from a concentrated solution of [1] and [2] in chloro-benzene and chloroform, respectively. Selected cell parameters of these complexes are listed in Table 1. The single crystal X-ray analysis of [1] and [2] displayed a distorted trigonal bipyramidal coordination around the metal centre (Fig. 2A–C). The crystallographic analysis of complex [1] shows a trinuclear structure containing three crystallographically related indium centres, in which three aminothiolate ligand units coordinate each central indium atom in a tridentate fashion through two thiolate groups and the nitrogen atom present in the bridging amine group of the aminothiolate ligand (the centroid of this cyclic trimer is located on a crystallographically imposed 3-fold axis). The equatorial positions were occupied by two sulfur atoms of the thiolate ligand and a terminal chlorine atom, while the axial positions were occupied by a nitrogen atom present in the bridging amine group and one of the bridging sulfur atoms. The molecular structure of [2] showed a trigonal–bipyramidal coordination sphere for the indium ion, where the trigonal basal plane is formed by two sulfur atoms of the dianionic thiolate ligand, and the oxygen atom of the N,O-β-heteroarylalkenolate ligand while the apical positions are occupied by the nitrogen atom present in the bridging amine group of the dianionic thiolate ligand and one nitrogen atom of the heteroarylalkenolate ligand. The In–S bond distances (Table S1†) in [1] were found to be in the range of 2.404(1)–2.676(2) Å, while the bond lengths of the bridging sulfur atoms in In1–S2 and In1_a-S2 were found to be longer at 2.485(1) Å and 2.676(2) Å, respectively, than the terminal In1–S1 bond length at 2.404(1) Å. These values correspond well to similar reported values of dimeric, trimeric and oligomeric organoindium compounds.33–37 The In–N bond length (2.393(5) Å) and In–Cl bond length (2.415(1) Å) in [1] are both within the reported ranges.35,38–41 Notably, the three dependent indium sub-units in [1] and their corresponding atoms display uniform bond lengths and angles. For instance, the bond lengths between In1–Cl1, In1_a-Cl1_a, and In1_b-Cl1_b (2.4150(10) Å) are consistent, and the bond angles of In1–S2–In1_a, In1_a-S2_a-In1_b, and In1_b-S2_b-In1 are 111.10(4)°. For compound [2], the In–S bond distances were observed to be 2.4197(9) for In1–S1 and 2.4288(9) Å for In1–S2, with a calculated value of 2.4398–2.4408 Å (Table 2).
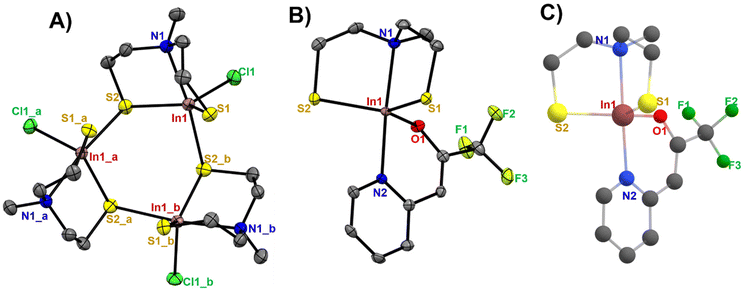 |
| | Fig. 2 The ORTEP drawing of (A) [1] (symmetry operations for In1_a: −x + y, 1 − x, z and In1_b: 1 − y, 1 + x − y, z), (B) [2] with thermal ellipsoids drawn at a 50% probability level with hydrogen atoms omitted for clarity and (C) DFT-optimized molecular structure of [2] (B3lyp/6-31G(d,p)/LanL2DZ level of theory). | |
Table 1 Comparison of selected cell parameters of [1] and [2]
| Compound |
[1]
|
[2]
|
| Empirical formula |
C15H33Cl3In3N3S6 |
C13H16F3InN2OS2 |
| Formula weight |
898.61 |
452.22 |
| Crystal system |
Trigonal |
Orthorhombic |
| Space group |
R![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pbca
|
|
T (K) |
100(2) |
100(2) |
|
a/Å |
16.6411(5) |
11.1065(3) |
|
b/Å |
16.6411(5) |
13.0043(5) |
|
c/Å |
19.4879(9) |
22.9345(7) |
|
α/° |
90 |
90 |
|
β/° |
90 |
90 |
|
γ/° |
120 |
90 |
| Volume/Å3 |
4673.7(4) |
3312.48(19) |
|
Z
|
6 |
8 |
|
ρ
calc g cm−3 |
1.916 |
1.814 |
|
F(000) |
2628.0 |
1792.0 |
| Independent reflections |
2613 [Rint = 0.0590, Rsigma = 0.0260] |
3937 [Rint = 0.0676, Rsigma = 0.0387] |
| Goodness of fit |
1.095 |
1.048 |
| Final R indexes [I> ≧ 2σ(I)] |
R
1 = 0.0376, wR2 = 0.0957 |
R
1 = 0.0333, wR2 = 0.0691 |
| Final R indexes [all data] |
R
1 = 0.0437, wR2 = 0.0995 |
R
1 = 0.0463, wR2 = 0.0759 |
| Largest diff. peak/hole/e Å−3 |
1.86/−0.73 |
0.57/−0.99 |
Table 2 Experimental and DFT-calculated geometries of compound [2]
| |
XRD |
DFTa |
|
DFT calculations on the B3lyp/6-31G(d,p)/LanL2DZ level of theory.
τ
5 = 1 corresponds to ideal trigonal bipyramidal geometry and τ5 = 0 corresponds to the ideal square pyramidal coordination geometry.42
|
| Bond lengths (Å) |
| In1–S1 |
2.4197(9) |
2.4408 |
| In1–S2 |
2.4288(9) |
2.4398 |
| In1–O1 |
2.089(2) |
2.030 |
| In1–N1 |
2.340(3) |
2.404 |
| In1–N2 |
2.268(3) |
2.282 |
| Bond angles (°) |
| S2–In1–S1 |
127.98(3) |
125.13 |
| O1–In1–S1 |
115.37(7) |
116.36 |
| O1–In1–S2 |
115.79(7) |
117.21 |
| N1–In1–S2 |
85.67(7) |
84.87 |
| N1–In1–S1 |
85.23(8) |
84.78 |
| N1–In1–O1 |
90.28(10) |
89.33 |
| N2–In1–O1 |
84.87(10) |
87.17 |
| N2–In1–S1 |
97.57(7) |
98.14 |
| N2–In1–S2 |
95.70(7) |
95.40 |
| N2–In1–N1 |
174.98(10) |
176.14 |
|
τ
5
|
0.78 |
0.85 |
Similarly, the In–N bond distance between the central indium atom and the bridging nitrogen atom of the thiolate ligand was found to be 2.340(3) Å for In1–N1 (cal. 2.404 Å), while the distance between the central indium atom and the nitrogen atom of the N,O-β heteroarylalkenolate ligand was found to be 2.268(3) Å for In1–N2, with a calculated value of 2.282 Å (Table 2). These values aligned with reported values.23,43–47 However, the slight bond elongation observed at 2.340(3) Å for In1–N1 reflects the weakening of the bond due to the greater donor strength of the thiolate ligand, as reported for similar complexes of the aminothiolate ligand.48 The observed and calculated bond lengths of In1–O1 were found to be 2.089(2) and 2.030 Å, respectively. The In1–O1 bond length of 2.089(2) Å is notably shorter compared to those observed in complexes with higher coordination numbers, such as [(SOOS)In(py)(NO3)], [(SNNS)In(OAc)], [(NNS2)In(NO3)], [(NNS2)In(OAc)], and [(SNNSPr)In(OAc)] complexes with In–O bond lengths in the range of 2.162(4)–2.851(4) Å.33 The equatorial positions of both compounds [1] (S2–In–S1) and [2] (S2–In1–S1) exhibited deviations from the ideal equatorial geometry of 120°, which are manifest in the distorted bond angles of 131.40(4) and [(127.98(3)°) (cal. 125.13°)], respectively. Additionally, the axial bond angles 171.64(9)° (N1–In1–S6) of [1] and 174.98(10)° (N1–In1–N2) (cal. 176.14°) of [2] deviated from linearity. Since oxygen (3.44) and chlorine (3.16) exhibit greater electronegativity than sulfur (2.58), the observed coordination geometry is distorted from the ideal trigonal-planar arrangement of 120° in both compounds [1] and [2]. This distortion will lead to wider S–In–S angles and smaller O–In–S or Cl–In–S angles due to the differing spatial demand of electron lone pairs present in S/O–In or S/Cl–In bonds. The distortion in [2] could have also contributed to the slight widening of the bite angle (84.87(10)°).41
The analysis of intermolecular stacking interactions reveals interaction between the hydrogen atom of the pyridine ring and the sulfur atom in adjacent molecules (H⋯S), with a stacking distance of 2.939 Å. Additionally, distinct interactions are observed between fluorine atoms in adjacent molecules (F⋯F), with a separation of 2.877 Å (Fig. S10†). These interactions may have contributed to the minute deviations in the In–S bond lengths observed in [2].
Vapor phase and thermal characterization studies
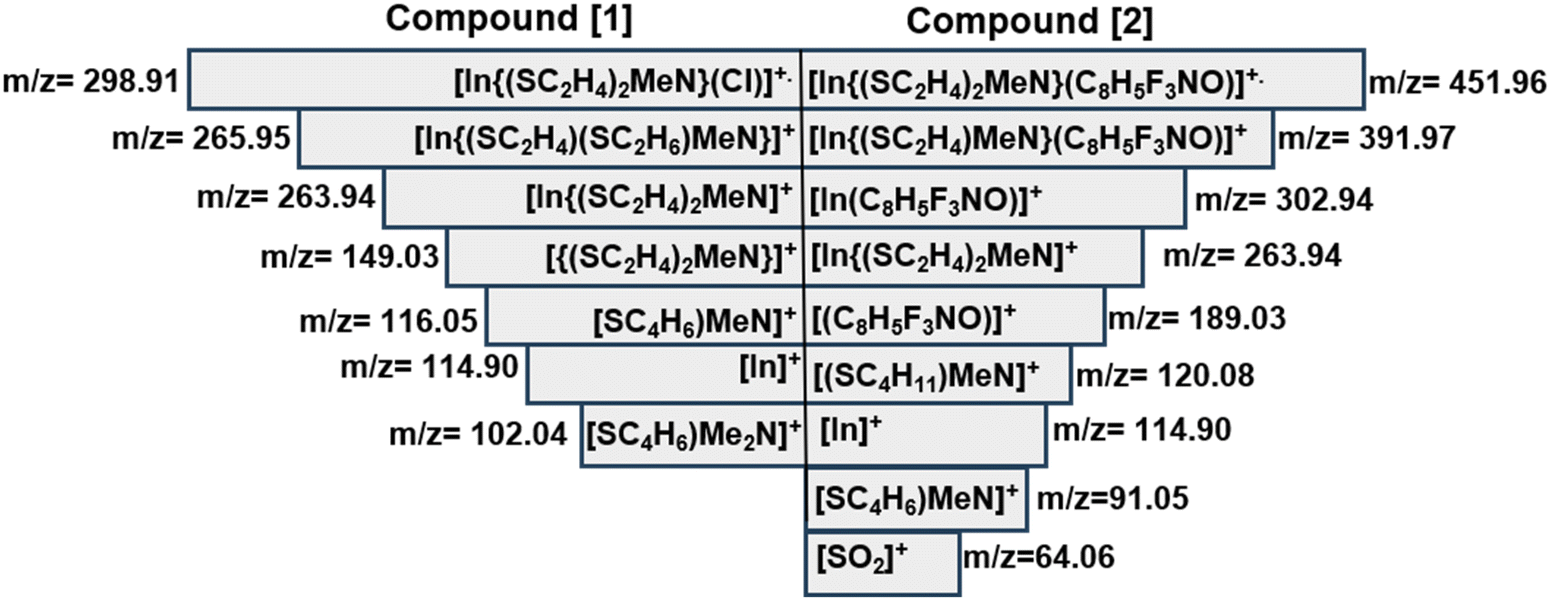
The electron ionization mass spectra of [1] and [2] recorded at an ionization energy of 70 eV revealed distinct radical cations (Fig. S12 and S13†). These were verified through comparative analysis of the calculated and measured isotopic patterns of the signals. Mass spectrometric analysis of [1] identified the molecular peak [M]˙+ (m/z = 298.91). Subsequent fragmentation of the terminal Cl− group generates the fragment [In{(SC2H4)MeN}]+ (m/z = 263.94). The remaining signals could conclusively be assigned to the aminothiolate ligand species and the indium metal (Scheme 3). For compound [2], the molecular ion peak [M]˙+ was observed at m/z = 451.96 [In{(SC2H4)2MeN}(C8H5F3NO)]˙+, with an intensity of 5%. The base peak dominated the spectra, with intensities reaching 100% at m/z = 114.9, possibly resulting from the loss of both ligands.
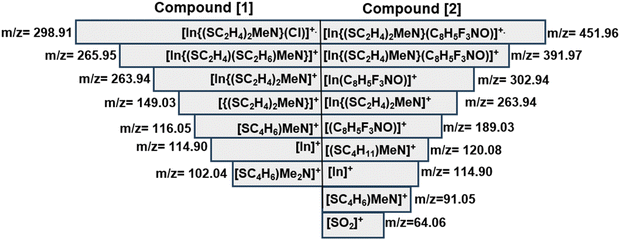 |
| | Scheme 3 Fragment ions of [1] and [2] detected by EI-MS. | |
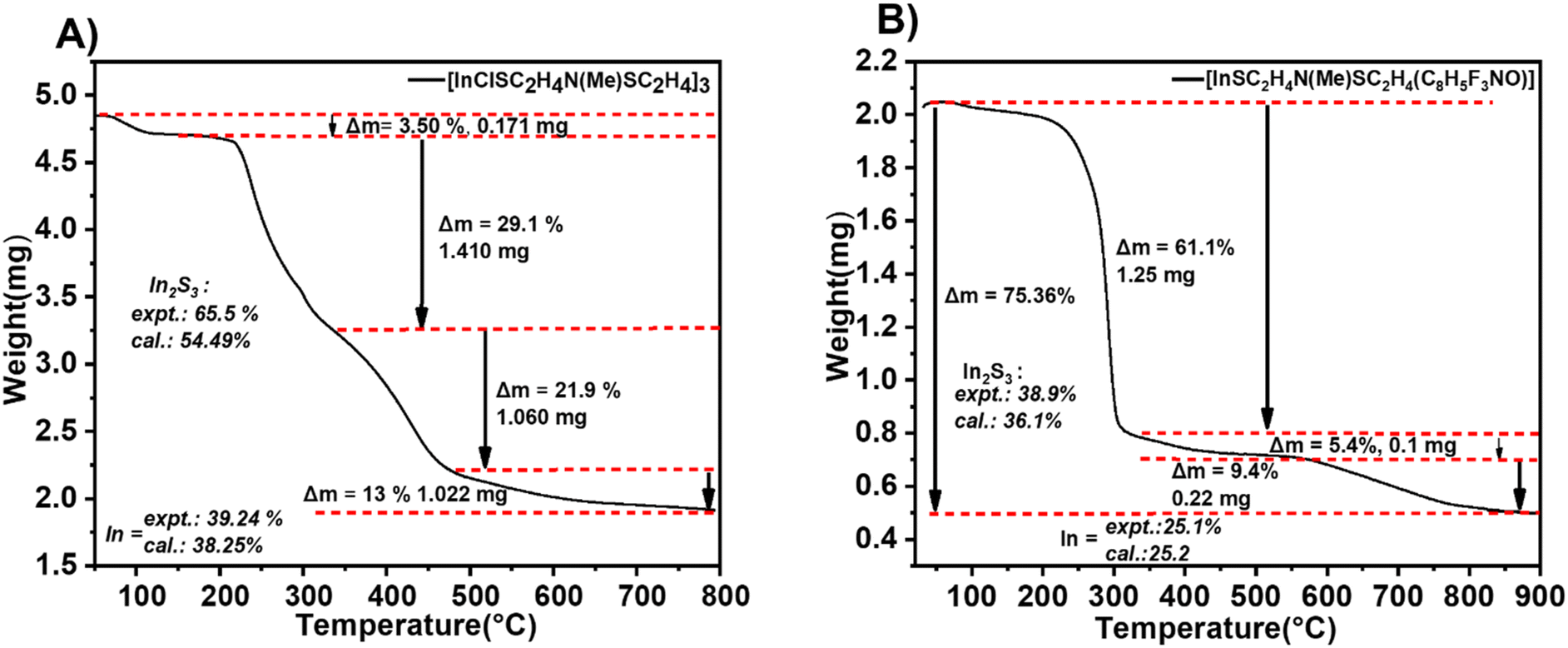
Apart from the molecular ion peak, fragments at m/z = 391.97 [In{(SC2H4)MeN}(C8H5F3NO)]˙+, m/z = 302.94 [In(C8H5F3NO)]+, m/z = 263.94 [In{(SC2H4)2MeN}]+ and m/z = 189.04 [(C8H5F3NO)]˙+ further demonstrate the fragmentation pathway of [2]. Interestingly, m/z = 64.06 [SO2]+ further demonstrates the oxidation of In2S3 to In2O3 which precedes the facile reduction of In2O3 to metallic indium in the presence of the carbon residue. The remaining signals could be assigned to the fragments of the aminothiolate ligand. Thermogravimetric analysis of the heteroleptic compound [1] conducted under continuous N2(g) (Fig. 3A) exhibited a multi-step decomposition process as a result of the lower thermal stability of [1] as compared to that of [2] (Fig. 3B). Initially, a minor mass loss occurring within the temperature range of 100–250 °C, corresponding to 3.5% sample weight, is attributed to the loss of chlorine from the molecular structure of the complex [1]. Subsequently, a substantial mass loss of 29.1% is observed around 350 °C, indicating the onset of In2S3 formation and possible emergence of intermediates (InSCl), which is a likely scenario in the gas phase fragmentation of such bulky molecules. It is plausible that the ligand moieties coordinated to the indium centre can attach to other volatile fragments (e.g. InCl or RInCl) upon thermolysis-driven fragmentation, resulting in new intermediates. This could also explain the disparities observed between the experimental (65.5%) and calculated data (54.49%). This decomposition is followed by another significant mass loss of approximately 21.9%, observed at 500 °C, corresponding to subsequent oxidation to In2O3. Further heating led to a consequent mass loss of 13% at 800 °C, showing the reduction of In2O3 to metallic indium facilitated by the presence of carbon. The formation of metallic indium at 800 °C was also evident in the mass spectral analysis above (Scheme 3). Thermogravimetric analysis of [2] exhibited a three-step decomposition process. The first decomposition mass loss occurred at 300 °C, with a mass loss of 61.1%, corresponding to the formation of In2S3, which remained stable until 550 °C, displaying a plateau indicative of the formation of the In2S3 material. The residual mass at 38.9% for In2S3 could be seen to be significantly higher than the theoretical value of 36.1% due to carbon and fluoride residues in the ligand. Further heating from 550 to 900 °C resulted in the final weight loss of 9.4%, assuming the formation of metallic indium at a 25.1% experimental value (25.2% calculated value), which agrees with the mass spectral analysis results of [2]. Similar reduction of chalcogenide complexes to metallic elements has been observed by Mishra et al.25
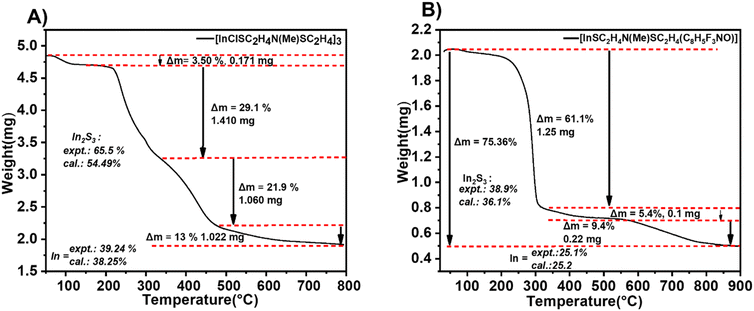 |
| | Fig. 3 Thermographic analysis plot of (A) compound [1] and (B) compound [2] at a heating rate of 10 °C min−1. | |
Chemical vapor deposition of compound [2]
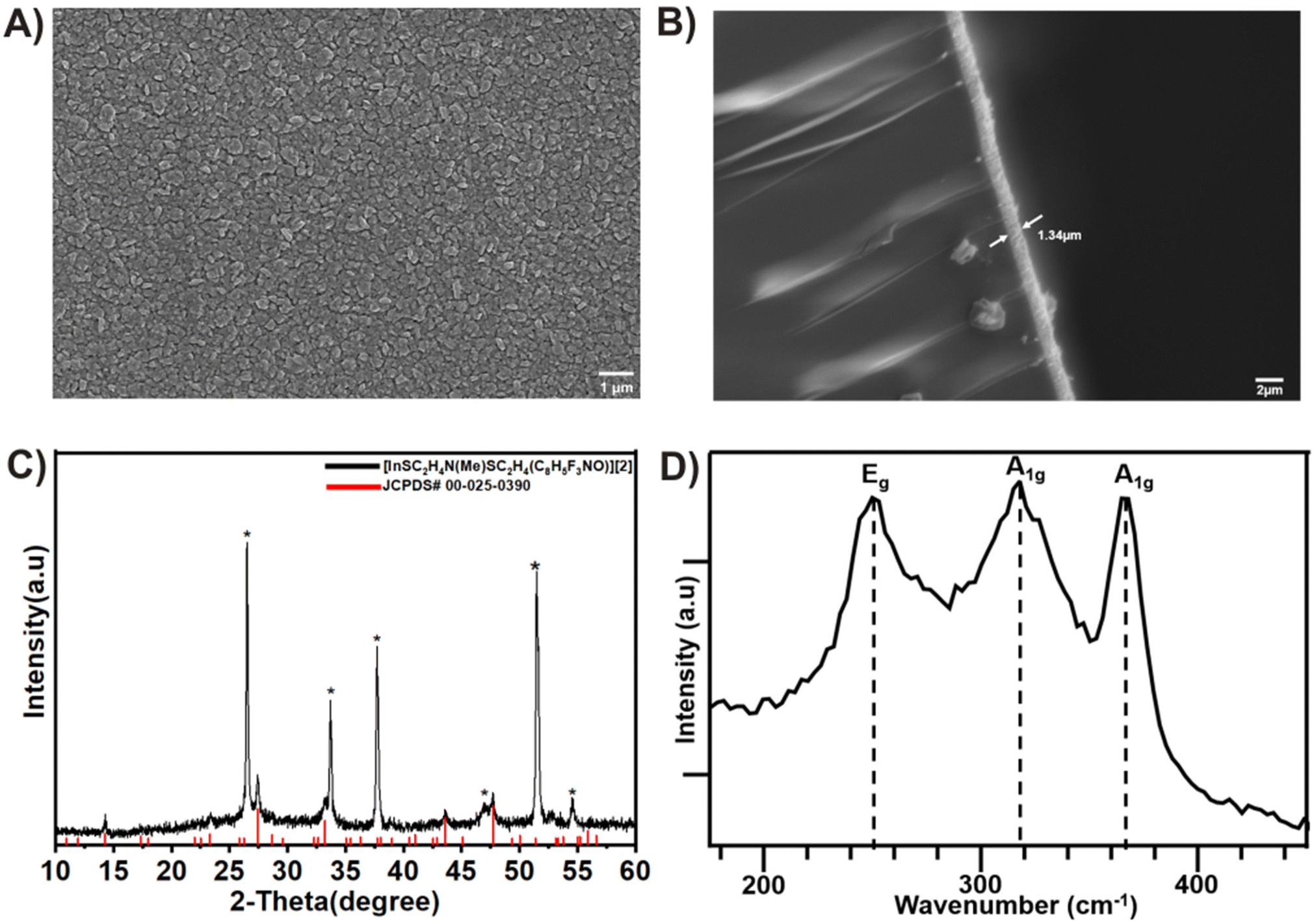
The chemical vapor deposition (CVD) experiments of [2] on an FTO substrate at a substrate temperature of 300 °C resulted in crystalline β-In2S3 films.49 Following the CVD process, the FTO substrate was uniformly covered with In2S3 grains after 30 minutes of deposition time. The SEM analyses (Fig. 4A) of the deposited film revealed that the In2S3 films were composed of nanocrystalline grains with an average size of 384 nm, evenly distributed throughout. The films exhibited good coverage, homogeneous morphology, and strong adhesion to the substrate, as depicted in the cross-sectional analysis (Fig. 4B). X-ray diffraction peaks (Fig. 4C) were identifiable and attributed to the tetragonal structure of the In2S3 film (JCPDS# 00-025-0390).50 This observation was further corroborated through energy dispersive X-ray spectroscopy (EDS) analysis, which revealed a S:In stoichiometric ratio of 1.6 (Fig. S14–S17†). The Raman spectra of the as-deposited thin films, recorded in the wavenumber range of 100–1000 cm−1 at room temperature, exhibited distinct peaks at 247 cm−1, 319 cm−1, and 365 cm−1 (Fig. 4D), corresponding to tetragonal β-In2S3.51
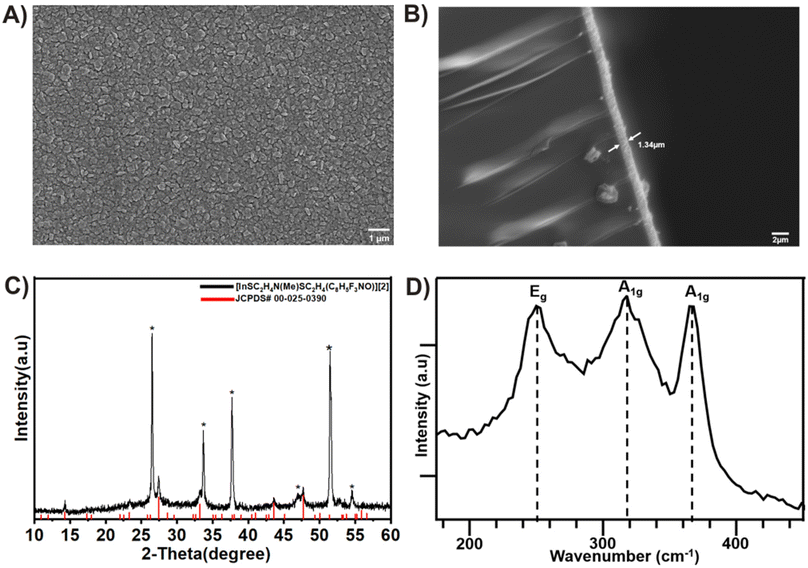 |
| | Fig. 4 (A) In-plane scanning electron microscopy (SEM). (B) The cross-sectional image of the β-In2S3 film deposited on FTO. (C) The XRD pattern of the deposited β-In2S3 thin film at 300 °C. The peaks identified by asterisks represent the FTO signals, and (D) Raman spectrum of the deposited film of In2S3 from compound [2]. | |
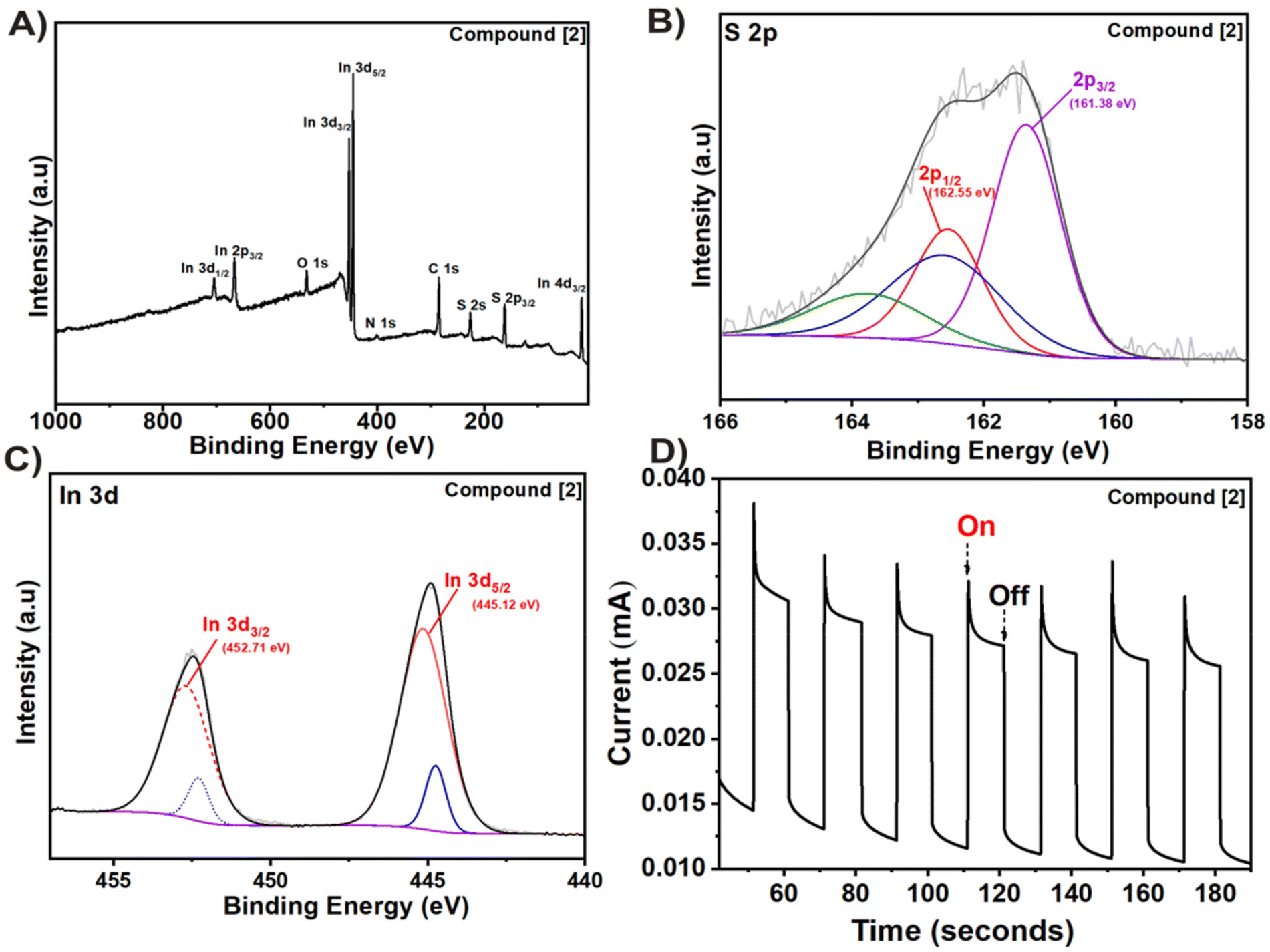
For further analysis of the In2S3 thin film via X-ray photoelectron spectroscopy (XPS), a small sample was gently removed from the FTO substrate. The survey spectrum showed the presence of elements including indium (In), sulfur (S), carbon (C), and oxygen (O). It is worth noting that the carbon signal is likely attributed to residual ligands adsorbed onto the film (Fig. 5A). This is possible due to lower decomposition temperature as residual carbon decomposes generally at higher temperatures (>500 °C).11 The presence of oxygen may be inevitably caused by sample handling contamination or surface oxidation during or after the CVD experiment, respectively.
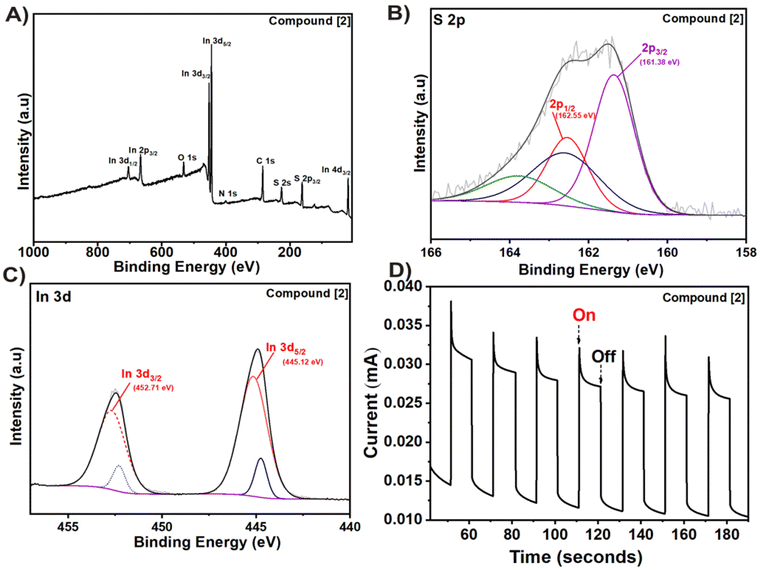 |
| | Fig. 5 (A) XPS survey, (B) S 2p, (C) In 3d high resolution spectra and (D) transient photoresponse of the deposited In2S3 film from compound [2]. | |
The atomic ratio of S:In was estimated to be 1.65 from the survey spectrum confirming the presence of In2S3 (Table S2†). In the 2p spectrum (Fig. 5B), two peaks at 161.38 eV and 162.55 eV could be seen corresponding to S 2p3/2 and S 2p1/2, respectively.52 Two strong photoelectron peaks at binding energies of 445.12 and 452.71 eV are assigned to 3d5/2 and In 3d3/2 transitions, respectively, and a spin–orbit splitting of 7.6 eV (ref. 53) (Fig. 5C). The high-resolution O 1s spectra showed the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O and M–O bonds in the In2S3 film (Fig. S18A†), while the remaining ligand fragments present on the surface could be observed in the high-resolution C 1s spectra (Fig. S18B†).
O, C–O and M–O bonds in the In2S3 film (Fig. S18A†), while the remaining ligand fragments present on the surface could be observed in the high-resolution C 1s spectra (Fig. S18B†).
The photocurrent response and chronoamperometric I–t curve was recorded under periodic on–off solar illumination (100 mW cm−2) with respect to time (Fig. 5D). Noticeably, the photoanode showed instantaneous, repetitive, and stable photoresponses indicating its potential in photoelectrochemical applications. The current spike observed in the transient photoresponse is possibly caused by charge carrier recombination.54
Density functional theory (DFT) calculations
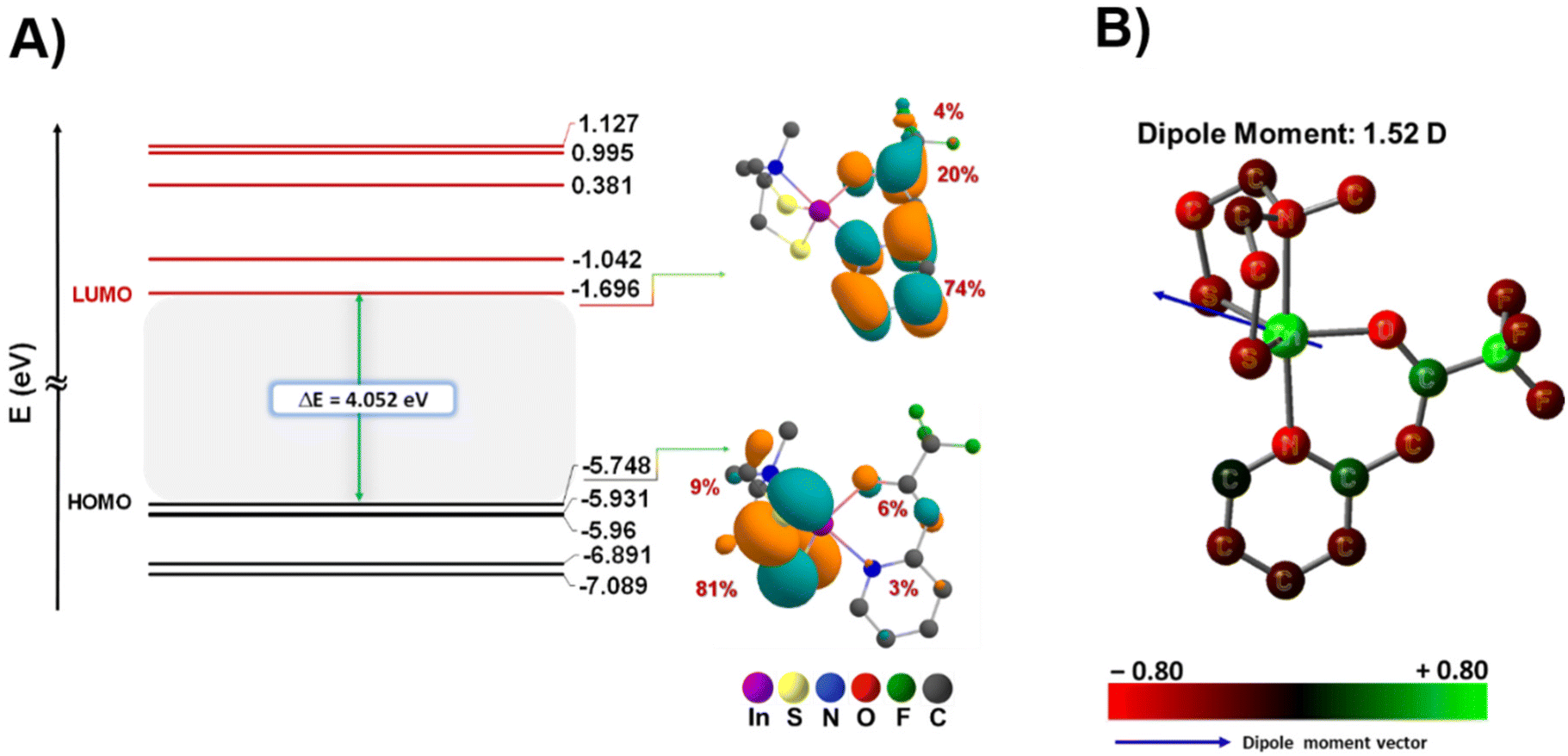
To enhance our comprehension of the electronic structure and thermal characteristics of the precursor, density functional theory (DFT) calculations were conducted to optimize the molecular structure of compound [2] (Fig. 2C), and ascertain the bond dissociation energy between the ligands and the metal centre (Fig. 7). Table S3† displays the Cartesian coordinates of the optimized structure of the complex. The bond lengths and bond angles, obtained through both theoretical calculations and crystallographic analysis, are presented in Table 2. Notably, as the geometry index τ5 = 1 corresponds to an ideal trigonal bipyramidal coordination geometry,42 the calculated and experimentally determined τ5 value (0.85 and 0.78, respectively) for the complex is in good agreement, providing compelling evidence for the distorted trigonal bipyramidal geometry adopted by the complex (Table 2). These results reveal a consistent correspondence between the datasets derived from theoretical predictions and the values experimentally determined.
Furthermore, the FT-IR spectroscopy results of the experimental values for the complex were compared with the calculated values, as depicted in Fig. S19,† revealing a consistent correlation. This congruence further supports the robustness of our theoretical framework in accurately describing the structural attributes of the investigated complex. The graphical representation in Fig. 6 illustrates that the energies associated with the HOMO (−5.748 eV) and LUMO (−1.696 eV) are characterized by negative values, indicating the stability within the complex.55 Additionally, the observed band gap (known as the energy difference between the HOMO and LUMO) magnitude aligns well with the existing literature, further affirming the appropriateness of our theoretical framework.56 Through DFT calculations, we elucidated the localization of electron density within the frontier orbitals (Fig. 6 and Table S4†). Our analysis reveals that in the highest occupied molecular orbital (HOMO), approximately 90% of electron density is concentrated on the aminothiolate ligand (LNS), with two sulfur atoms accounting for 81% and the methyl group of the amine contributing 9%. Conversely, in the lowest unoccupied molecular orbital (LUMO), around 98% of the electron density is localized on the N,O-β-heteroarylalkenolate ligand (LNO), distributed across the aromatic ring (74%), carbonyl group (20%), and fluorinated groups (4%) as illustrated in Fig. 6A. Remarkably, the contribution of the metal centre to both the HOMO and LUMO is negligible. This distinctive localization pattern of the HOMO and LUMO supports the assignment of complex 2 as a donor–acceptor LL'CT complex, where the LNS ligand exhibits donor characteristics while the LNO ligand demonstrates acceptor characteristics. These results underscore the presence of ligand-to-ligand charge transfer, with a donor–bridge–acceptor pattern, particularly from ligand LNS to ligand LNO, mediated by indium as the metal centre (Fig. S11†).57,58
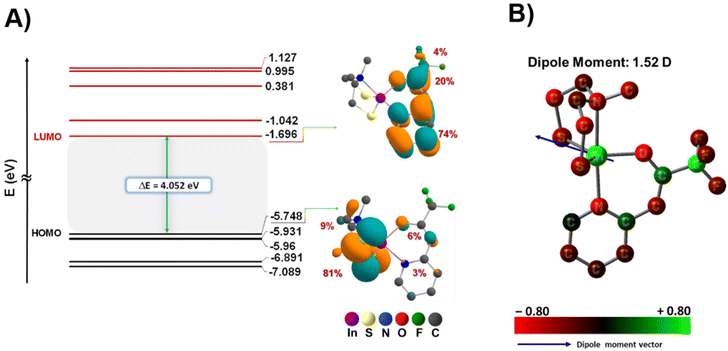 |
| | Fig. 6 (A) Frontier Kohn–Sham orbital diagram for complex [2], and electron density distributions of frontier orbitals, HOMO and LUMO states and (B) structures of atomic charges of complex [2] (B3lyp/6-31G(d,p)/LanL2DZ level of theory). | |
To determine the ligand dissociation energy, we have taken into account both homolytic and heterolytic cleavage of metal–ligand bonds. The plausible reaction pathways for ligand dissociations of compound [2] are displayed in Fig. 7. In context of the specified pathway, we performed optimization and frequency calculations on all fragments utilizing the same level of theoretical analysis in the gas phase. Additionally, single-point calculations were performed and the thermal correction derived from frequency calculation was incorporated into the single-point energy, enhancing the accuracy of the estimates for total energy, enthalpy and Gibbs free energy. The theoretical ligand dissociation energy for each ligand, characterized by one comprising three bonds (LNS) with indium and the other (LNO) featuring two bonds, was calculated as the enthalpy changes from both the enthalpy of the complex and the produced fragments at 298.15 K and 1 atm for each reaction pathway. This calculation was performed according to the following formula (eqn (1)).
| | | LDE = ∑H298.15Fragments − H298.15Complex | (1) |
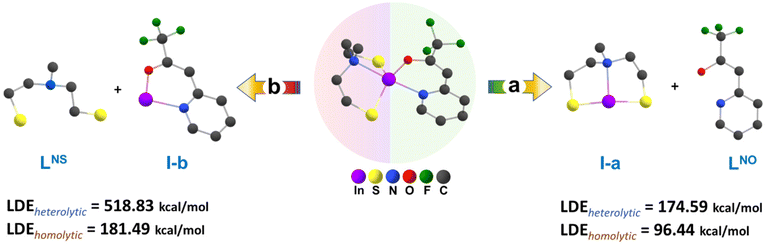 |
| | Fig. 7 Model reactions for metal–ligand dissociation of compound [2] at the B3lyp/6-31G(d,p)/LanL2DZ and M06/6-311G(d,p)/LanL2DZ levels of theory. | |
In Fig. 7, the calculated bond dissociation energies, referred to as ligand dissociation energy (LDE), revealed distinctive values for the aminothiolate and N,O-β-heteroarylalkenolate ligands. Notably, for both homolytic and heterolytic cleavages, the energy required for the cleavage of N,O-β-heteroarylalkenolate ligands (LNO) is lower than that for the thiolate ligand (LNS) (Fig. 7 and Table 3). These results indicate that, during the chemical vapor deposition process of the complex, the thermodynamic stability of In bonds with the LNS ligand is greater than with the LNO ligand. Consequently, the N,O-β-heteroarylalkenolate ligand, with lower LDE values, is more prone to preferential dissociation. It is anticipated that the formation of an intermediate species containing indium with the aminothiolate ligand (depicted as I-a in Fig. 7) will be more probable, demonstrating heightened thermal stability compared to that of the alternative species represented as I-b. It is noteworthy that the mentioned intermediate (I-a) has been successfully detected in EI-MS analysis, as depicted in Scheme 3 and Fig. S13,† with a mass-to-charge ratio (m/z) of 263.94.
Table 3 Calculated energy changes, and changes in enthalpy and Gibbs free energy for ligand–metal dissociation at the B3lyp/6-31G(d,p)/LanL2DZ and M06/6-311G(d,p)/LanL2DZ levels of theory
| Cleavage type |
Pathway a |
Pathway b |
| Homolytic |
Heterolytic |
Homolytic |
Heterolytic |
|
ΔE = ∑EFragments − EComplex.
ΔH = ∑H298.15Fragments − H298.15Complex.
ΔG = ∑G298.15Fragments − G298.15Complex.
|
| Fragment specification (Charge/Spin) |
I-a
(0/2) |
I-a
(1/1) |
I-b
(0/3) |
I-b
(2/1) |
|
LNO
(0/2) |
LNO
(−1/1) |
LNS
(0/3) |
LNS
(−2/1) |
| |
| ΔEa (kcal mol−1) |
95.85 |
173.99 |
180.90 |
518.24 |
| ΔHb (kcal mol−1) |
96.44 |
174.59 |
181.49 |
518.83 |
| ΔGc (kcal mol−1) |
80.67 |
161.47 |
164.59 |
500.15 |
Conclusions
Two novel heteroleptic indium(III) complexes, [1] and [2], were successfully synthesized using aminothiol and β-heteroarylalkenol as chelating ligands. Compared to compound [1], which could not be successfully applied in the CVD process, the heteroleptic compound [2] exhibited a predictable decomposition pattern and potential for molecular preorganisation, enhancing the predictability of materials synthesis through both experimental and theoretical outcomes. Both the mass spectral analysis and thermogravimetric analysis (TGA) of [2] revealed a distinct sequential breakdown of the two distinct ligands within varying temperature ranges. This observation underscores the importance of tailoring precursor design to incorporate ligands with varying bonding strengths. To elucidate the experimental results, further investigation into the dissociation of ligands in compound [2] was conducted by DFT calculations. Specifically, we investigated one of the plausible pathways leading to the formation of In2S3, which included an intermediate of indium metal with ligand LNS. According to our results, in both homolytic and heterolytic cleavages, the energy required for the cleavage of the LNS ligand is greater than that for the LNO ligand. This suggests that the indium bond with the LNS ligand is more thermodynamically stable than that with the LNO ligand. This perspective strengthens the probability of In2S3 formation as a consequence of the decomposition of the aforementioned intermediate.
Experimental section
All manipulations were performed under a nitrogen atmosphere in the stock-line. Solvents were dried with conventional methods and stored under a N2 atmosphere with molecular sieves in Schlenk flasks. InCl3 was dried under vacuum for 1 h before use, and aminothiol and N,O-β-heteroarylalkenol ligands were synthesized according to Mathur et al.28,29 All other reagents were procured commercially from Aldrich and used without further purification. NMR spectra were recorded using a Bruker Avance II 300 or Avance II+ 600 spectrometer equipped with a double resonance (BBFO) 5 mm observe probe head with z-gradient coil (Bruker, Rheinhausen, Germany), operating at 1H: 300.1 MHz; 600 MHz and 19F: 282.4 MHz. All 2D NMR experiments were performed using standard pulse sequences from the Bruker pulse program library. NMR spectra were plotted using the Bruker TopSpin 3.2 software. Elemental analyses were carried out on a HEKAtech CHNS Euro EA 3000. Mass spectra was recorded on a Finnigan MAT 95 (EI, 70 eV) in m/z (intensity in %). TG analysis was performed using a TG/DSC STA 6000 apparatus with nitrogen gas and a heating rate of 10 °C min−1. Powder X-ray diffraction was performed on a STOE diffractometer with a STADI MP system and either Mo Kα (λ = 0.71 Å) or Cu Kα radiation (λ = 1.54 Å). FT-IR spectra were measured using a Platinum ATR spectrometer with samples analyzed using OPUS software. The Varian 50 Scan UV-visible photometer was utilized to perform measurements for UV-visible absorption. Quartz glass cuvettes with a length of 1 cm were employed for the purpose, and the spectra was subjected to baseline correction. The MOCVD tests were carried out in a cold-wall CVD reactor with cooling traps, an internal pressure sensor, and external temperature sensors at low-pressure settings, and the CVD chamber consisted of a quartz glass tube fitted with an inductively heated graphite holder on which the substrate (FTO) was mounted.12 The deposited film, Pt coil and Hg/HgO are taken as the working (anode), counter (cathode) and reference electrodes. The molecular precursor was introduced to the reactor through a glass flange by applying dynamic vacuum (10−3 mbar) and heating the precursor reservoir to the desired temperature. No carrier or reactive gases were used. Raman spectra were recorded with an alpha300R Confocal Raman Microscope with a WITec UHTS 300 spectrometer with a 532 nm laser. The samples’ microstructures were examined using field-emission scanning electron microscopy (SEM, Zeiss Sigma VP 300 RIESE). Appropriate single crystals were cooled to 100(2) K, mounted on a MiTiGen Microloom™ and attached to the goniometer head of an SC-XRD Bruker D8 VENTURE KAPPA with a microfocus sealed tube using a multilayer mirror as a monochromator and a Bruker OHOTON III detector. The diffractometer was equipped with an Oxford Cryostream 800 low-temperature apparatus and Mo Kα radiation (λ = 0.71073 Å) was used. APEX2 was used to process the photographs after the entire dataset was captured. The crystal structure was solved with SHELXT 2018/2 using Olex2,59–61 and refined based on F2 (SHELXL-2018/3)60,62,63 using Olex261 and SHELX programs. Editing of CIFs and construction of tables and bond lengths and angles were achieved using Olex2. For the photoresponse measurement, a conventional three-electrode device on an electrochemical workstation (SP-200 Potentiostat) was employed. The obtained samples on FTO substrates with a fixed coating area of 1.0 cm × 1.0 cm were used as the working electrodes. Ag/AgCl (3 M KCl) and Pt wire acted as the reference and counter electrodes. The light source was an LED solar simulator (LSH-7320 ABA) and the illumination intensity was calibrated to 100 mW cm–2. Mixed NaSO3/Na2S (0.35 M/0.25 M, pH = 12.5) aqueous solution was used as the electrolyte.
Synthesis of [1]
To a solution of the aminothiol ligand (0.5 g, 3.30 mmol) in toluene (15 mL) at 0 °C, 2.64 mL (6.60 mmol) of n-BuLi was added dropwise and stirred for 30 minutes, after which 0.730 g (3.30 mmol) of InCl3, activated in THF (15 mL), was added in situ and stirred for 30 minutes under ambient conditions to obtain a cloudy solution. The solvents (toluene and THF) were completely removed under vacuum to obtain a white solid [1] that was extracted in a following step with chlorobenzene, using four aliquots of 10 mL each. Following this, the solution of chlorobenzene containing the dissolved product was reduced to 20 mL by removing a part under vacuum. Subsequently, n-pentane was added until a complete precipitation of the product was achieved, leaving a clear supernatant. After gently decanting the solvent, the resulting solid was additionally washed with n-pentane and ultimately dried under vacuum to obtain [1] as a white powder. Suitable crystals were obtained by slow evaporation using dichlorobenzene as solvent. Yield: 85% (0.84 g, 2.28 mmol). 1H NMR (600 MHz, DMSO) δ 2.76–2.59 (m, 8H), 2.29 (s, 3H). 13C NMR (101 MHz, DMSO) δ 58.40, 42.74, 23.13. EI-MS (70 eV, 50–220 °C): m/z (intensity) = m/z = 298.91 [In{(SC2H4)2MeN}(Cl)]+˙, m/z = 265.95 [In{(SC2H4)(SC2H6)MeN}]+, m/z = 263.94 [In{(SC2H4)2MeN}], m/z = 149.03 [{(SC2H4)2MeN}]+, m/z = 116.05 [(SC4H6)MeN]+, m/z = 114.90 [In]+, m/z = 102.04 [(SC4H6)Me2N]+. Elemental analysis C5H11ClInNS2: calculated, C, 20.05; H, 3.70; N, 4.68; S, 21.41. Found C, 20.01; H, 3.51; N, 4.64; S, 21.37.
Synthesis of [2]
To a solution of aminothiol ligand (0.5 g, 3.30 mmol) in toluene (15 mL) at 0 °C, 2.64 mL (6.60 mmol) of n-BuLi was added dropwise and stirred for 30 minutes, after which 0.730 g (3.30 mmol) of InCl3, activated in THF (15 mL), was added in situ and stirred for 30 minutes. After 30 minutes, a solution of deprotonated O-β-heteroarylalkenol ligand (0.644 g, 3.30 mmol, deprotonated by the same method as stated above) in THF (15 mL) was added in situ to the reaction mixture and stirred for 30 minutes under ambient conditions to obtain a slightly yellow solution. Work-up was done in the same way as in the synthesis of [1] using chloroform and n-pentane as solvents, which yielded compound [2] in the form of a slightly yellow powder. Suitable crystals were obtained by slow evaporation using chloroform as solvent. Yield: 86% (1.2 g, 2.65 mmol). 1H NMR (300 MHz, CDCl3) δ 8.09 (d, J = 5.5, 1.5 Hz, H-4), 7.60 (m, J = 7.8, 1.8 Hz, H-6), 7.06 (m, H-5, H-7), 5.64 (s, H-9), 2.89–2.78 (m, H-3, H-2), 2.50 (s, H-1). 13C NMR (75 MHz, CDCl3) δ 157.33 (C10), 156.98 (C8), 146.24 (C4), 139.61 (C6), 124.88 (C7), 120.68 (C5), 118.37 (C11), 94.61 (C9), 58.86 (C2), 43.12 (C1), 23.13 (C3). 19F NMR (282 MHz, CDCl3) δ −74.21. EI-MS (70 eV, 50–150 °C): m/z = 451.96 [In{(SC2H4)2MeN}(C8H5F3NO)]+, 391.97 [In{(SC2H4)MeN}(C8H5F3NO)]+, m/z = 302.94 [In(C8H5F3NO)]+, m/z = 263.93 [In{(SC2H4)2MeN}]+, m/z = 189.04 [(C8H5F3NO)]+, m/z = 120.08[(SC4H11)MeN]+, m/z = 114.90 [In]+, m/z = 91.05 [(SC4H6)MeN]+, m/z = 64.06 [SO2]. Elemental analysis (C13H16F3InN2OS2): calculated, C, 34.53; H, 3.57; N, 6.19; S, 14.18. Found, C, 35.15; H, 3.72; N, 6.28; S, 14.98.
Computational methods
All calculations were performed via density functional theory (DFT) using Gaussian 16, revision C.01, to investigate the geometry, electronic structure and stability of the compounds.64 The molecular structure optimization and frequency calculations of the all compounds in the gas phase were carried out using the B3LYP functional65,66 in conjunction with the 6-31G(d,p) basis set for light atoms (C, N, O, F, and H), and the quasi-relativistic LanL2DZ basis set for indium. The absence of imaginary frequencies in the calculated geometries indicates the optimized geometries as energetic minima. The Cartesian coordinates of the optimized structure and calculated compositions (%) of frontier molecular orbitals in the ground state (S0) for complex [2] are included in the ESI (Tables S3 and S4†). Single-point energy calculations were conducted on the optimized structure using the M06 functional. In the case of indium, both geometry optimization and single-point energy calculations were carried out with the same basis set (LanL2DZ). However, for the light atoms, a larger basis set (6-311G(d,p)) was employed. For the complex and all its fragments, the total energy, enthalpies, and Gibbs free energy corrections obtained from frequency calculations were incorporated into the calculated single-point energies. The visualization of the molecular structure and molecular orbitals was accomplished through the utilization of GaussView (version 6)67 and Chemcraft.68
Conflicts of interest
There are no conflicts of interests to declare.
Acknowledgements
The authors gratefully acknowledge the University of Cologne and the German Science Foundation (DFG) for providing the infrastructural and financial support. The PhD fellowship, provided by DAAD, to Mr Chijioke Kingsley Amadi wih funding number 91729650 and travel grant obtained through the Research Training Group TIDE (IRTG 2591 – Template-Designed Organic Electronics) are gratefully acknowledged.
References
- H. Xu, H. Chen, S. Chen, K. Wang and X. Wang, Int. J. Hydrogen Energy, 2021, 46, 32445–32454 CrossRef CAS.
- J. Hou, S. Cao, Y. Sun, Y. Wu, F. Liang, Z. Lin and L. Sun, Adv. Energy Mater., 2018, 8, 1701114 CrossRef.
- W. Huang, L. Gan, H. Yang, N. Zhou, R. Wang, W. Wu, H. Li, Y. Ma, H. Zeng and T. Zhai, Adv. Funct. Mater., 2017, 27, 1702448 CrossRef.
- A. G. Alhamzani, T. A. Yousef, M. M. Abou-Krisha, K. Y. Kumar, M. K. Prashanth, L. Parashuram, B. Hun Jeon and M. S. Raghu, Chemosphere, 2023, 322, 138235 CrossRef CAS PubMed.
- B. R. Lee and H. W. Jang, Electron. Mater. Lett., 2021, 17, 119–135 CrossRef CAS.
-
S. Mathur, A. P. Singh, A. R. Müller, T. Leuning, T. Lehnen and H. Shen, J. Ceram. Sci. Technol, John Wiley & Sons, Ltd, 1st edn, 2011 Search PubMed.
- L. Jürgensen, M. Frank, D. Graf, I. Gessner, T. Fischer, K. Welter, W. Jägermann and S. Mathur, Z. Phys. Chem., 2020, 234, 911–924 CrossRef.
- S. Mathur and T. Ruegamer, Int. J. Appl. Ceram. Technol., 2011, 8, 1050–1058 CrossRef CAS.
- X. Cao, D. Heidelberg, J. Ciupka and M. Dolg, Inorg. Chem., 2010, 49, 10307–10315 CrossRef CAS PubMed.
- M. D. Pluth and Z. J. Tonzetich, Chem. Soc. Rev., 2020, 49, 4070–4134 RSC.
- F. Hartl, V. Brune, S. Lüggert, C. Hegemann, D. van Gerven, M. Wilhelm, S. Ji, H. Choi and S. Mathur, Inorg. Chem., 2023, 62, 6274–6287 CrossRef CAS PubMed.
- U. Atamtürk, E. Jung, T. Fischer and S. Mathur, Chem. Mater., 2022, 34, 7344–7356 CrossRef.
- E. I. Solomon, S. I. Gorelsky and A. Dey, J. Comput. Chem., 2006, 27, 1415–1428 CrossRef CAS PubMed.
- L. Capron, W. Y. Feng, C. Lifshitz, B. L. Tjelta and P. B. Armentrout, J. Phys. Chem., 1996, 100, 16571–16576 CrossRef CAS.
- S. B. Bandgar, M. M. Vadiyar, Y.-C. Ling, J.-Y. Chang, S.-H. Han, A. V. Ghule and S. S. Kolekar, ACS Appl. Energy Mater., 2018, 1, 638–648 CrossRef CAS.
- D. P. Dutta, G. Sharma, A. K. Tyagi and S. K. Kulshreshtha, Mater. Sci. Eng., B, 2007, 138, 60–64 CrossRef CAS.
- M. Lazell, P. O'Brien, D. J. Otway and J.-H. Park, Dalton Trans., 2000, 4479–4486 RSC.
- T. C. Deivaraj, M. Lin, K. P. Loh, M. Yeadon and J. J. Vittal, J. Mater. Chem., 2003, 13, 1149–1155 RSC.
- G. Hogarth and D. C. Onwudiwe, Inorganics, 2021, 9, 70 CrossRef CAS.
- J. Waters, D. Crouch, J. Raftery and P. O'Brien, Chem. Mater., 2004, 16, 3289–3298 CrossRef CAS.
- K. Ramasamy, M. A. Malik, P. O'Brien and J. Raftery, Dalton Trans., 2010, 39, 1460–1463 RSC.
- F. Biegger, C. Rameshan, A. K. Opitz, J. Noll, T. Haunold, H. Lang and S. Barth, New J. Chem., 2016, 40, 6962–6969 RSC.
- J. H. Park, T. M. Chung, B. K. Park and C. G. Kim, Inorg. Chim. Acta, 2020, 505, 119504 CrossRef CAS.
- S. Mishra, Chem. Commun., 2022, 58, 10136–10153 RSC.
- S. Gahlot, B. Purohit, E. Jeanneau and S. Mishra, Chem. – Eur. J., 2021, 27, 10826–10832 CrossRef CAS PubMed.
- A. D. Martínez-Iniesta, A. Morelos-Gómez, E. Muñoz-Sandoval and F. López-Urías, RSC Adv., 2021, 11, 2793–2803 RSC.
- V. Brune, C. Hegemann, M. Wilhelm, N. Ates and S. Mathur, Z. Anorg. Allg. Chem., 2022, 648, e202200049 CrossRef CAS.
- V. Brune, C. Hegemann and S. Mathur, Inorg. Chem., 2019, 58, 9922–9934 CrossRef CAS PubMed.
- L. Czympiel, J. Pfrommer, W. Tyrra, M. Schäfer and S. Mathur, Inorg. Chem., 2015, 54, 25–37 CrossRef CAS PubMed.
- J. Więcławik and A. Chrobok, Molecules, 2023, 28, 1955 CrossRef PubMed.
- L. Jürgensen, D. Höll, M. Frank, T. Ludwig, D. Graf, A. K. Schmidt-Verma, A. Raauf, I. Gessner and S. Mathur, Dalton Trans., 2020, 49, 13317–13325 RSC.
- L. Brückmann, W. Tyrra, S. Stucky and S. Mathur, Inorg. Chem., 2012, 51(1), 536–542 CrossRef PubMed.
- T. S. Anderson, G. G. Briand, R. Brüning, A. Decken, M. J. Margeson, H. M. Pickard and E. E. Trevors, Polyhedron, 2017, 135, 101–108 CrossRef CAS.
- B. Yearwood, S. U. Ghazi, M. J. Heeg, N. Richardson and J. P. Oliver, Organometallics, 2000, 19, 865–871 CrossRef CAS.
- G. G. Briand, B. F. T. Cooper, D. B. S. MacDonald, C. D. Martin and G. Schatte, Inorg. Chem., 2006, 45, 8423–8429 CrossRef CAS PubMed.
- F. Salsi, M. Roca Jungfer, A. Hagenbach and U. Abram, Eur. J. Inorg. Chem., 2020, 2020, 1222–1229 CrossRef CAS.
- J. S. Casas, M. S. García-Tasende and J. Sordo, Coord. Chem. Rev., 2000, 209, 197–261 CrossRef CAS.
- R. D. Adams, B. Captain and Q. F. Zhang, Z. Anorg. Allg. Chem., 2007, 633, 2187–2190 CrossRef CAS.
- K. R. Margulieux, C. Sun, L. N. Zakharov, A. W. Holland and J. J. Pak, Inorg. Chem., 2010, 49, 3959–3961 CrossRef CAS PubMed.
- S. Abram, C. Maichle-Mössmer and U. Abram, Polyhedron, 1997, 16, 2291–2298 CrossRef CAS.
- K. Kumar, R. K. Sahani, S. Garai and S. Bhattacharya, Dalton Trans., 2023, 52, 17499–17513 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, Dalton Trans., 1984, 7, 1349–1356 RSC.
- M. Risto, T. Chivers and J. Konu, Dalton Trans., 2011, 40, 8238–8246 RSC.
- K. Kumar, P. Tiwari, S. Moharana, R. Kant and S. Bhattacharya, J. Mol. Struct., 2022, 1260, 132801 CrossRef CAS.
- C. Stamou, Z. G. Lada, C. T. Chasapis, D. Papaioannou, P. Dechambenoit and S. P. Perlepes, Dalton Trans., 2022, 51, 15771–15782 RSC.
- R. C. F. Leitao, F. Silva, G. H. Ribeiro, I. C. Santos, J. F. Guerreiro, F. Mendes, A. A. Batista, F. R. Pavan, P. I. da S. Maia, A. Paulo and V. M. Deflon, J. Inorg. Biochem., 2023, 240, 112091 CrossRef CAS PubMed.
- P. Mpofu, P. Rouf, N. J. O'Brien, U. Forsberg and H. Pedersen, Dalton Trans., 2022, 51, 4712–4719 RSC.
- V. Brune, N. Raydan, A. Sutorius, F. Hartl, B. Purohit, S. Gahlot, P. Bargiela, L. Burel, M. Wilhelm, C. Hegemann, U. Atamtürk, S. Mathur and S. Mishra, Dalton Trans., 2021, 50, 17346–17360 RSC.
- S. Rasool, K. Saritha, K. T. R. Reddy, M. S. Tivanov, V. F. Gremenok, S. P. Zimin, A. S. Pipkova, L. A. Mazaletskiy and I. I. Amirov, Mater. Res. Express, 2020, 7, 016431 CrossRef CAS.
- L. Wang, X. Lin, A. Ennaoui, C. Wolf, M. Lux-Steiner and R. Klenk, EPJ Photovoltaics, 2016, 7, 70303 CrossRef.
- B. Hemanth Kumar, S. Shaji and M. C. Santhosh Kumar, J. Mater. Sci.: Mater. Electron., 2019, 30, 17986–17998 CrossRef CAS.
- S. Kumar and T. T. John, Mater. Chem. Phys., 2022, 286, 126222 CrossRef CAS.
- A. Nayak, L. Seungwon, Y. Sohn and D. Pradhan, CrystEngComm, 2014, 15, 8064–8072 RSC.
- Z. Peng, Y. Su, I. Ennaji, A. Khojastehnezhad and M. Siaj, Chem. Eng. J., 2023, 477, 147082 CrossRef CAS.
- S. Parvarinezhad, M. Salehi, M. Kubicki and R. Eshaghi Malekshah, Appl. Organomet. Chem., 2022, 36, e6836 CrossRef CAS PubMed.
- J. R. Diniz, J. R. Correa, D. F. S. Machado, S. L. M. Soares, C. C. Gatto, H. C. B. de Oliveira, P. E. N. de Souza, M. O. Rodrigues and B. A. D. Neto, J. Lumin., 2020, 225, 117391 CrossRef CAS.
- L. Z. Fu, P. He, J.-W. Wang, F. Ma, C. Liu, G. Chen and X.-Y. Yi, Chem. Commun., 2024, 60, 1595–1598 RSC.
- L. A. Cameron, J. W. Ziller and A. F. Heyduk, Chem. Sci., 2016, 7, 1807–1814 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
-
G. M. Sheldrick, SHELXL-2018, Universität Göttingen, Göttingen, Germany, 2018 Search PubMed.
- V. V. V. Hugo, H. S. M. Alejandro, V. S. A. María, R. H. María, L. R. M. Antonio, P. O. M. Guadalupe, M. G. M. Antonio, A. H. Fernando, A. Víctor, C. A. Diego and Á. Enrique, Comput. Chem., 2018, 7, 1–26 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
-
R. Dennington, T. A. Keith and J. M. Millam, GaussView, version 6.0. 16, Semichem Inc., Shawnee Mission, KS, USA, 2016, pp. 143–150.
-
G. A. Zhurko, Chemcraft: https://www.chemcraftprog.com. (accessed February 2, 2024).
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Touraj
Karimpour
a,
Touraj
Karimpour