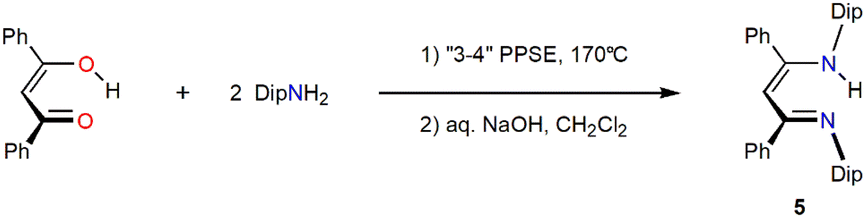DOI:
10.1039/D4DT01298D
(Paper)
Dalton Trans., 2024,
53, 9887-9895
Alkyl backbone variations in common β-diketiminate ligands and applications to N-heterocyclic silylene chemistry†‡
Received
2nd May 2024
, Accepted 20th May 2024
First published on 21st May 2024
Abstract
We report the extension of the common β-diketimine proligand class, RArnacnacH (HC(RCNAr)2H), where R is an alkyl group such as Et or iPr, plus Ph, and Ar is a sterically demanding aryl substituent such as Dip = 2,6-diispropylphenyl, Dep = 2,6-diethylphenyl, Mes = 2,4,6-trimethylphenyl or mesityl, Xyl = 2,6-dimethylphenyl, via one-pot condensation procedures. When a condensation reaction is carried out using the chemical dehydrating agent PPSE (polyphosphoric acid trimethylsilylester), β-diketiminate phosphorus(V) products such as (iPrMesnacnac)PO2 can also be obtained, which can be converted to the respective proligand iPrMesnacnacH via alkaline hydrolysis. The RArnacnacH proligands can be converted to their alkali metal complexes with common methods and we have found that deprotonation of iPrDipnacnacH is significantly more sluggish than that of related β-diketimines with smaller backbone alkyl groups. The basicity of the RArnacnac− anions can play a role in the success of their salt metathesis chemistry and we have prepared and structurally characterised the EtDipnacnac-derived silicon(II) compounds (EtDipnacnac)SiBr and (EtDipnacnac’)Si, where EtDipnacnac’ is the deprotonated variant MeCHC(NDip)CHC(NDip)Et.
1. Introduction
β-Diketiminate (BDI) ligands, or “nacnac's” (Fig. 1) from the acetylacetone (acacH) analogy, are a highly popular ligand class that has been employed in complexes from all parts of the periodic table and for a wide variety of applications.1–16 Their general ease of synthesis, including sterically demanding variants, and their generally robust nature has made these ligands a go-to choice where N,N′-chelating monoanionic spectator ligands are desirable. For example, these ligands have been successfully and widely employed for a range of reactive low oxidation state metal complexes from across the periodic table.2,9,10,12 Their structure allows the tuning of steric and electronic factors,1,12 for example via varying the R and Ar groups in the symmetric variants shown in Fig. 1A. Regarding electronic factors, even fully fluorinated β-diketiminates have been prepared and assessed.17,18 In many fields and applications where steric factors are a dominant influence, e.g., in s-block chemistry,8–10 variation of predominantly the N-aryl groups, but also the substituents (R) in the ligand backbone (see Fig. 1A and B) have been focused on. In some cases, small steric changes can effect large differences on product outcomes.19 Most nacnac variants in the literature employ the readily available parent “acac” backbone fragment, and these are typically cheap and easy to synthesise. Although nacnac's generally serve as robust monoanionic spectator ligands, the backbone unit can engage in various side reactions owing to the electrophilic, nucleophilic and CH acidic properties of various parts of the acac-derived backbone, see Fig. 1B.5 In addition, other decomposition reactions are possible, including the extrusion of an ArN (e.g. DipN) unit from the ligand scaffold.5 A range of metal coordination modes other than the most common N,N′-chelating one (Fig. 1B) can be observed,1,5 not unrelated to the structures of the main tautomers of RArnacnacH species (Fig. 1C). The delocalised free β-diketiminate unit can form various isomers (E/Z) that are accessible within a low range of energies.20 Ligand backbone units that allow wider electronic delocalisation, e.g., those with aryl groups, can induce some non-innocent ligand behaviour and are prone to reduction to a radical dianionic ligand unit in some instances.1,4,5 Overall, it is surprising that only few studies have placed more emphasis on the ligand backbone substitution.4,5 Other common backbone modifications are, for example, tBu2-backbone substituted systems, such as tBuDipnacnacH.21 These bulky substituents provide a significant steric effect and also have an electronic impact on the nature of the ligand system.
 |
| | Fig. 1
RArnacnacH species. | |
A common method for the synthesis of nacnac ligands is a simple condensation reaction between a β-diketone and two equivalents of anilines or primary amines,1,21–24 although for some examples, the activation of the diketone via a Meerwein salt has been employed.1 For the construction of bulky backbone variants like tBuDipnacnacH, tBuMesnacnacH21 and PhDipnacnacH,25 however, two ArN-containing halves have to be synthesised followed by linking these in a C–C bond formation reaction to build the backbone unit in a multistep approach. Aside from hydrocarbyl groups for backbone substituents (R), introduction of secondary amino groups in these positions affords bisamidines that act as electronically modified, electron-rich “N-nacnac” ligands.26,27 We have very recently introduced a one-pot condensation method to prepare iPrDipnacnacH28 and this work expands on this backbone modification. We also consider EtArnacnacH proligands which have been sporadically mentioned in the literature,29–32 but in some cases without much synthetic or spectroscopic detail.
2. Results and discussion
Ligand synthesis
Installing two ethyl groups into the backbone unit, i.e., forming EtArnacnacH proligands, can be achieved by simply modifying a common acid-catalysed condensation reaction22,32 according to Scheme 1. This requires stoichiometric para-toluenesulfonic acid hydrate (pTsOH·H2O), an arene solvent such as toluene and heating under reflux with the use of a Dean–Stark trap to remove water from the condensation reaction system. For sterically more demanding Ar = Dip the use of xylene for up to two days has been advantageous. We recommend carrying out reactions that require high temperatures or long reaction times under nitrogen atmosphere to limit side-reactions. A typical alkaline workup with aqueous sodium carbonate solution and dichloromethane, followed by treatment with methanol afforded crystalline crops of EtDipnacnacH 1a (65%), EtDepnacnacH 1b (48%), EtMesnacnacH 1c (72%), and EtXylnacnacH 1d (75%) in moderate to good isolated yields and higher in situ yields.
 |
| | Scheme 1 Synthesis of proligands 1. | |
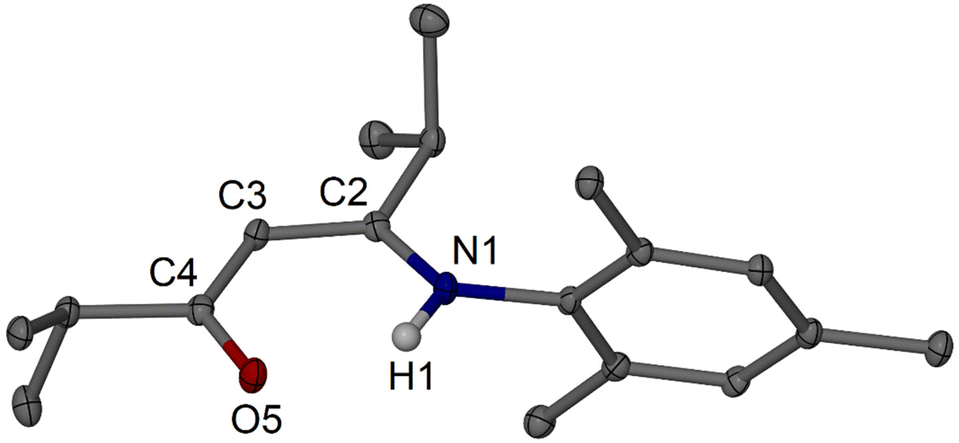
Moving to isopropyl-substituted targets, i.e., to iPrArnacnacH, we first tested simple condensation protocols for the synthesis of the N-mesityl substituted system, i.e., to afford iPrMesnacnacH 2c. Reactions of two equivalents of 2,4,6-trimethylaniline (mesidine, MesNH2), one equivalent of pTsOH·H2O, in refluxing xylene (mixture of isomers, boiling range. ca. 136–140 °C) with a Dean–Stark trap under nitrogen atmosphere for 16 h afforded predominantly the monosubstituted β-ketoimine species iPrC(NHMes)CHC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)iPr 3c as a colourless crystalline material after workup (Fig. 2). The crude product from this reaction did show NMR resonances of low integration for the desired iPrMesnacnacH 2c, and thus a more forcing reaction setup with longer reaction times was employed using 4.5 equivalents of MesNH2, 2 equivalents of pTsOH·H2O, and reflux in xylene under nitrogen for 6–7 days. This was followed by an alkaline aqueous work-up and distilling off of excess MesNH2 under vacuum to afford crude 2c as a light-brown oil. The crude product contained small quantities of crystalline iPrC(NHMes)CHC(O)iPr 3c as a side product which could be separated off via column chromatography to afford 2c in 47% isolated yield (Scheme 2). In one instance, compound 2c crystallised after standing at room temperature for months to a few large colourless crystals, and was structurally characterised (Fig. 3).
O)iPr 3c as a colourless crystalline material after workup (Fig. 2). The crude product from this reaction did show NMR resonances of low integration for the desired iPrMesnacnacH 2c, and thus a more forcing reaction setup with longer reaction times was employed using 4.5 equivalents of MesNH2, 2 equivalents of pTsOH·H2O, and reflux in xylene under nitrogen for 6–7 days. This was followed by an alkaline aqueous work-up and distilling off of excess MesNH2 under vacuum to afford crude 2c as a light-brown oil. The crude product contained small quantities of crystalline iPrC(NHMes)CHC(O)iPr 3c as a side product which could be separated off via column chromatography to afford 2c in 47% isolated yield (Scheme 2). In one instance, compound 2c crystallised after standing at room temperature for months to a few large colourless crystals, and was structurally characterised (Fig. 3).
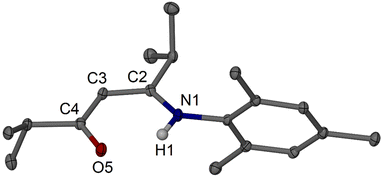 |
| | Fig. 2 Molecular structure of iPrC(NHMes)CHC(O)iPr 3c (30% thermal ellipsoids). Hydrogen atoms except H1 are omitted for clarity. Selected bond lengths (Å) and angles (°): O5–C4 1.247(2), N1–C2 1.343(2), N1–C6 1.438(2), C2–C3 1.386(3), C3–C4 1.420(3); N1–C2–C3 120.49(17), C2–N1–C6 127.07(16), C2–C3–C4 123.50(17), O5–C4–C3 123.34(17). | |
 |
| | Scheme 2 Synthesis of iPrMesnacnacH 2c and 3c. | |
 |
| | Fig. 3 Molecular structure of iPrMesnacnacH, 2c (30% thermal ellipsoids). Hydrogen atoms except H1 are omitted for clarity. Selected bond lengths (Å) and angles (°): N1–C2 1.354(2), N5–C4 1.299(2), C2–C3 1.373(3), C3–C4 1.438(3); C2–N1–C6 127.68(17), N1–C2–C3 119.33(18), C2–C3–C4 125.27(18), N5–C4–C3 119.34(17). | |
It is evident that the method leading to 2c would be difficult to translate to derivatives with sterically more demanding aryl groups and led us to investigate chemical dehydrating agents with the aim of a one-step condensation protocol. For this, we explored the related powerful acidic dehydrating agent polyphosphoric acid trimethylsilylester, PPSE.33,34 PPSE is prepared by adding phosphorus pentoxide to hexamethyldisiloxane in dichloromethane, followed by a reflux period and removal of all volatiles. The honey-like residue is then treated with the respective substituted aniline and diketone (2,6-dimethyl-3,5-heptanedione) and heated to 170 °C for approximately 16–24 hours. For workup, the reaction mixture is cooled to only ca. 90–95 °C to prevent it from solidifying, which impedes the neutralisation, and is slowly and carefully (caution!) treated with aqueous sodium hydroxide (NaOH) solution, that can be introduced via the reflux condenser, to quench the mixture and bring it to a basic pH value. After further cooling, extraction with dichloromethane yielded the crude products that can in most cases be precipitated using methanol. This afforded iPrDipnacnacH 2a (86%), iPrDepnacnacH 2b (53%), and iPrMesnacnacH 2c (51%) in good (2a, 2b) or moderate (2c) isolate yields (Scheme 3). The yield for 2a has been slightly optimised from our initial report28 using this method by gradually lowering the stoichiometric amount of PPSE for the synthesis, and similar optimisations could be envisaged for the preparations of 2b and 2c. For the synthesis of iPrMesnacnacH 2c, another main product was produced alongside 2c, which was characterised as the β-diketiminate phosphorus(V) oxide (iPrMesnacnac)PO24c in approximately 29% yield (Scheme 3). Thus, 2c and 4c were often afforded in approximately similar quantities. The compounds can be easily separated by adding n-hexane to the crude product which precipitates 4c, see Fig. 4 for the molecular structure, and yielded highly soluble 2c. Workups from syntheses of iPrDepnacnacH 2b also contained varying quantities of (iPrDepnacnac)PO24b, according to NMR spectroscopy that were difficult to separate by extracting into different solvents or by fractional crystallisation. No related species was found alongside the synthesis of iPrDipnacnacH 2a. Compound 4c is air stable and a rare β-diketiminate phosphorus compound that is N,N′-chelated, vide infra. We eventually found that stirring (iPrMesnacnac)PO24c in an aqueous sodium hydroxide solution and dichloromethane mixture, i.e., similar to the workup conditions, deprotects 4c to afford the proligand 2c, which can thus be obtained in high yield overall. It appears that the β-diketiminate phosphorus(V) oxide species (iPrArnacnac)PO24 are most stable for the smaller aryl groups (Mes) whereas the bulkier ones (Dip, Dep) more readily hydrolyse under the workup conditions. Proligand 1a could also be prepared using the PPSE method, but without optimising the conditions, we found no improvement in yield compared to the standard condensation reaction (Scheme 1).
 |
| | Scheme 3 Synthesis of iPrArnacnacH 2 and (iPrArnacnac)PO24. | |
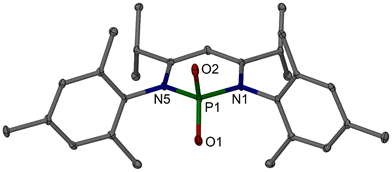 |
| | Fig. 4 Molecular structure of (iPrMesnacnac)PO2, 4c (30% thermal ellipsoids). Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): P1–O1 1.4712(16), P1–O2 1.4720(17), P1–N5 1.7518(17), P1–N1 1.7548(17), N1–C2 1.356(3), N5–C4 1.356(3), C2–C3 1.387(3), C3–C4 1.390(3); O1–P1–O2 121.67(10), N5–P1–N1 98.47(8), O1–P1–N1 108.54(9), O2–P1–N1 108.50(9), O1–P1–N5 109.55(9), O2–P1–N5 107.62(9). | |
To investigate how widely PPSE can be used for the one-step condensation synthesis of bulky β-diketimine proligands, we used the same method that successfully forms compound 2, but using 2,2,6,6-tetramethyl-3,5-heptanedione instead of 2,6-dimethyl-3,5-heptanedione as the diketone with 2,6-diisopropylaniline to attempt the synthesis of tBuDipnacnacH21 in a one-step procedure. This, however, afforded a mixture of products after workup, did not yield detected quantities of tBuDipnacnacH, and the conditions were deemed unsuitable to convert this bulkier diketone. The method was, however, suitable to convert dibenzoylmethane (1,3-diphenylpropane-1,3-dione) with 2,6-diisopropylaniline, DipNH2, to the known proligand PhDipnacnacH 5,25 in 56% isolated yield (unoptimised, Scheme 4). To test if the method is suitable to easily install bulky tertiary alkyl groups into the N-positions of a β-diketimine proligand, a reaction between 2,6-dimethyl-3,5-heptanedione, two equivalents of 1-adamantylamine (1-aminoadamantane) and the respective quantities of PPSE under the usual conditions was conducted. After workup, however, a product mixture was obtained that contained large quantities of 1-aminoadamantane and this system was not further studied.
 |
| | Scheme 4 Synthesis of PhDipnacnacH 5. | |
The β-diketimine proligands EtDipnacnacH, 1a (Fig. S61†), EtDepnacnacH, 1b (Fig. S62†), EtXylnacnacH, 1d (Fig. S63†), iPrDepnacnacH, 2b (Fig. S64†), and iPrMesnacnacH, 2c (Fig. 3), were structurally characterised and show the expected overall structures and geometrical features, see the ESI† for details. Across the structures, the level of localised versus delocalised bonding in the conjugated backbone unit that can be observed varies and is comparable to previously characterised examples, e.g.2a,28 but some disorder or poorly ordered features are often present and thus these structures are not discussed in detail. The mesityl compounds iPrC(NHMes)CHC(O)iPr 3c, iPrMesnacnacH 2c, and (iPrMesnacnac)PO24c (Fig. 1–3) are shown here with representative bond lengths and angles. Some localisation of bonds of the ligand backbone unit can be inferred from the structure of 2c, the well-defined tautomeric form of 3c is observed, and the geometrical features are unremarkable. Similarly, the NMR spectroscopic data of compounds 1, 2, and 3c are as expected. The three discussed RDipnacnacH proligands, for R = Me, Et, iPr, were exclusively observed by NMR spectroscopy in solution in their tautomeric enamine form, i.e., with a highly downfield shifted NH1H NMR resonance near δ 12 ppm. The tBuDipnacnacH proligand, on the other hand, shows the dominant diimine form in solution with some percentage present in the enamine form (Fig. 1C).21 This may be explained by considering the basicity of the deprotonated fragments, RDipnacnac−. In these, the HOMO is typically predominantly associated with the p-orbital on the (delocalised) backbone CH unit1,18 (“enamine form”) which is likely destabilised by the positive inductive effect of the electron-rich tert-butyl groups on the neighbouring iminyl groups. Thus, this tautomer may become less stable, the CH backbone unit becomes the preferred position for protonation and the diimine form of the proligand becomes energetically favourable.
Compound 4c shows the phosphorus centre to be N,N′-chelated as part of a six-membered ring with PO bonds (P1–O1 1.4712(16), P1–O2 1.4720(17)) that are of similar lengths to those found in a few related species.35–37 The N–P–N angle (98.47(8)°) is narrow due to the constraints of the heterocycle and, accordingly, the O–P–O angle (121.67(10)°) is quite obtuse. The introduction of phosphorus centres into a classical β-diketiminate system38–45 is relatively rare and often met with unexpected product outcomes displaying different ligand-P connectivities, for example, when targeted via salt metathesis of β-diketiminate alkali metal complexes with phosphorus(III) halides. Often, the phosphorus centre forms a heterocycle with the β-diketiminate involving an activated backbone methyl group or simply bonds to the γ-carbon. N,N′-bound β-diketiminate phosphorus systems are rare42,43 and both control and prediction of the product type can be difficult. Thus, the convenient one-step formation of 4c offers a new synthetic entry route.
Proligand deprotonation: conversion to alkali metal complexes
Alkali metal complexes of sterically demanding β-diketiminate ligands20,21,46–52 are a common synthon in β-diketiminate chemistry because they are generally easy to obtain and undergo wide salt metathesis chemistry to coordinate the ligand entity to a wide range of elements.1–16 s-block organometallics such as n-butyllithium and benzyl potassium are routinely employed to deprotonate β-diketimines to alkali metal complexes in coordinating or hydrocarbon solvents. These general methods can also be used for the RArnacnacH compounds described herein (Scheme 5), and more details are provided on reactions of Dip-containing variants EtDipnacnacH 1a and iPrDipnacnacH 2a. As widely performed, lithiations of 1a and 2a, and the other presented proligands, with n-butyllithium can be generally carried out in hydrocarbon solvents (toluene, benzene, n-hexane), coordinating solvents (THF, diethylether) or solvent mixtures. Reactions of 1a or 2a, respectively, with benzyl potassium are typically carried out by stirring a suspension of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture in toluene overnight. Often, alkali metal bis(trimethylsilyl)amide complexes, [M{N(SiMe3)2}], M = Li, Na, K, have been used for this type of conversion as well. We have studied the reaction between RDipnacnacH and one equivalent of [Na{N(SiMe3)2}] in deuterated benzene on a small scale followed by 1H NMR spectroscopy (Scheme 6 and Fig. S53, S55, S56†). MeDipnacnacH reacts slowly with [Na{N(SiMe3)2}] at room temperature (e.g., ca. 40% conversion after 16 hours) or rapidly at 60 °C. EtDipnacnacH 1a shows a slower reaction with [Na{N(SiMe3)2}] at room temperature and only slowly converts to [(EtDipnacnac)Na] 7b over several hours at 60 °C, e.g., ca. 60% conversion after 5 hours and full conversion overnight. For the same reaction with iPrDipnacnacH 2a, however, we find only very sluggish conversion at 60 °C and this system required prolonged heating, for example, around five days to afford full conversion to [(iPrDipnacnac)Na] 7a. We next investigated the reaction between RDipnacnacH and ten equivalents of [K{N(SiMe3)2}] in deuterated benzene more quantitatively under pseudo-first order reaction conditions and found rate constants of k′MeDip = 3.0 × 10−2 min−1 at room temperature, k′1a = 7.6 × 10−3 min−1 at room temperature, and k′2a = 2.5 × 10−3 min−1 at 60 °C (Fig. S57–S60†). These differences highlight the significant effect of the ligand backbone, where MeDipnacnacH is deprotonated approximately four times faster than 1a, and 1a is deprotonated approximately three times faster at room temperature compared with 2a at 60 °C. In stoichiometric reactions, 2a can be more quickly deprotonated with the smaller and stronger amide base [Li(NEt2)] in deuterated benzene to [(iPrDipnacnac)Li] 6a after heating to 60 °C for two days. Thus, the acidity of all the studied proligands RDipnacnacH, R = Me, Et, iPr, is higher than that of HN(SiMe3)2 (pKa value 25.8).53 It is tempting to suggest that in addition to there being different kinetic barriers for the deprotonation of RDipnacnacH with [Na{N(SiMe3)2}], likely for steric reasons, the lower acidity of 2a in the series also leads to the markedly slower deprotonation reaction at 60 °C and a more basic and less thermodynamically favoured sodium complex (7a) when compared with reactions of MeDipnacnacH and 1a. As part of this study, we generally found that EtArnacnac-compounds show a higher solubility in aromatic or aliphatic solvents compared with iPrArnacnac-compounds and this can play a role in their further transformations.
:1 mixture in toluene overnight. Often, alkali metal bis(trimethylsilyl)amide complexes, [M{N(SiMe3)2}], M = Li, Na, K, have been used for this type of conversion as well. We have studied the reaction between RDipnacnacH and one equivalent of [Na{N(SiMe3)2}] in deuterated benzene on a small scale followed by 1H NMR spectroscopy (Scheme 6 and Fig. S53, S55, S56†). MeDipnacnacH reacts slowly with [Na{N(SiMe3)2}] at room temperature (e.g., ca. 40% conversion after 16 hours) or rapidly at 60 °C. EtDipnacnacH 1a shows a slower reaction with [Na{N(SiMe3)2}] at room temperature and only slowly converts to [(EtDipnacnac)Na] 7b over several hours at 60 °C, e.g., ca. 60% conversion after 5 hours and full conversion overnight. For the same reaction with iPrDipnacnacH 2a, however, we find only very sluggish conversion at 60 °C and this system required prolonged heating, for example, around five days to afford full conversion to [(iPrDipnacnac)Na] 7a. We next investigated the reaction between RDipnacnacH and ten equivalents of [K{N(SiMe3)2}] in deuterated benzene more quantitatively under pseudo-first order reaction conditions and found rate constants of k′MeDip = 3.0 × 10−2 min−1 at room temperature, k′1a = 7.6 × 10−3 min−1 at room temperature, and k′2a = 2.5 × 10−3 min−1 at 60 °C (Fig. S57–S60†). These differences highlight the significant effect of the ligand backbone, where MeDipnacnacH is deprotonated approximately four times faster than 1a, and 1a is deprotonated approximately three times faster at room temperature compared with 2a at 60 °C. In stoichiometric reactions, 2a can be more quickly deprotonated with the smaller and stronger amide base [Li(NEt2)] in deuterated benzene to [(iPrDipnacnac)Li] 6a after heating to 60 °C for two days. Thus, the acidity of all the studied proligands RDipnacnacH, R = Me, Et, iPr, is higher than that of HN(SiMe3)2 (pKa value 25.8).53 It is tempting to suggest that in addition to there being different kinetic barriers for the deprotonation of RDipnacnacH with [Na{N(SiMe3)2}], likely for steric reasons, the lower acidity of 2a in the series also leads to the markedly slower deprotonation reaction at 60 °C and a more basic and less thermodynamically favoured sodium complex (7a) when compared with reactions of MeDipnacnacH and 1a. As part of this study, we generally found that EtArnacnac-compounds show a higher solubility in aromatic or aliphatic solvents compared with iPrArnacnac-compounds and this can play a role in their further transformations.
 |
| | Scheme 5 Formation of β-diketiminate alkali metal complexes. | |
 |
| | Scheme 6 Formation of β-diketiminate alkali metal complexes via alkali metal bis(trimethylsilyl)amides. | |
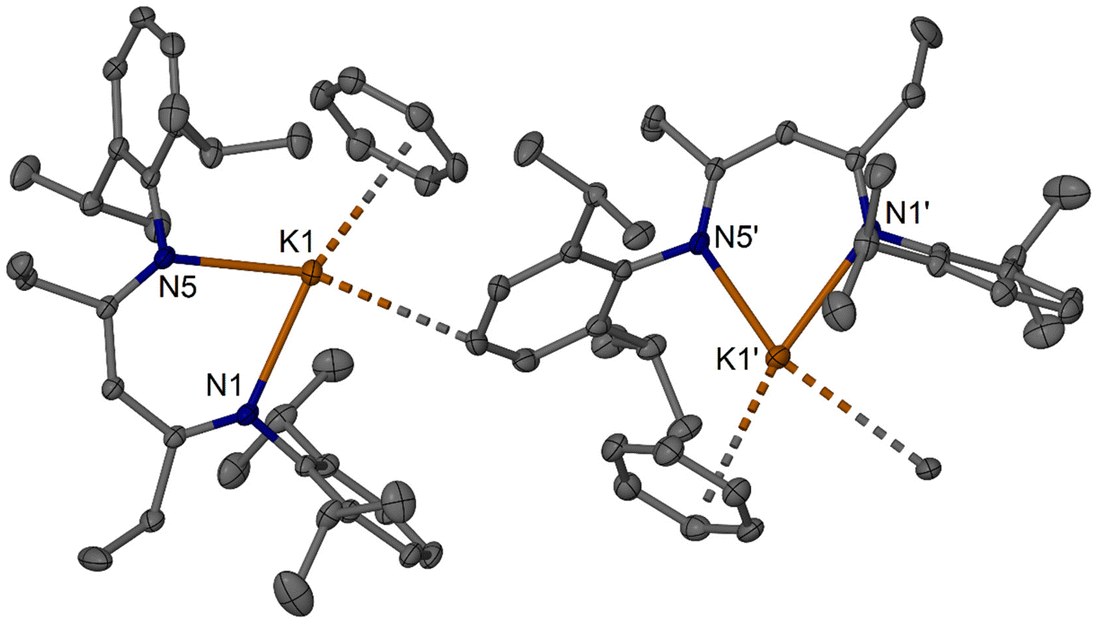
As part of the study, NMR data was acquired for a series of species, see Scheme 5, and the molecular structures of [(iPrDipnacnac)Li] 6a (Fig. 5), [(iPrDipnacnac)Li(OEt2)] 6a(OEt2) (Fig. S71†), and [(EtDipnacnac)K(C6H6)] 8b(C6H6) (Fig. 6), were structurally characterised. Complex 6a crystallised as a weakly-interacting dimer, highly similar to the structure of [(MeDipnacnac)Li]. For the latter, a weakly-bound dodecameric isomer has been characterised as well.23 The molecular structure of 6a(OEt2) also mimics the previously characterised [(MeDipnacnac)Li(OEt2)].23 In complex 8b(C6H6), the K+ ion is N,N′-chelated by the nacnac ligand and approximately η4-coordinated by a benzene molecule and shows a contact to a para-CH unit of a Dip-substituent from a neighbouring molecule leading to weak one-dimensional contacts in the lattice. In benzene-free [(MeDipnacnac)K], the “completion” of the potassium coordination sphere is achieved by K η5-coordination to a neighbouring Dip-substituent.46,47
 |
| | Fig. 5 Molecular structure of [(iPrDipnacnac)Li] 6a (30% thermal ellipsoids). Hydrogen atoms are omitted for clarity, and weak Li⋯C interactions are shown with dashed bonds. Selected bond lengths (Å) and angles (°): N1–Li1 1.886(4), N5–Li1 1.893(4), Li1⋯C28′ 3.199(5); N1–Li1–N5 100.2(2). | |
 |
| | Fig. 6 Molecular structure of [(EtDipnacnac)K(C6H6)] 8b(C6H6) (30% thermal ellipsoids). Hydrogen atoms are omitted for clarity, and weak K⋯C interactions are shown with dashed bonds. Selected bond lengths (Å) and angles (°): K1–N1 2.6979(12), K1–N5 2.6613(12), K1⋯H25′ 2.831, K1⋯C25′ 3.2747(16); N5–K1–N1 70.67(3). | |
Synthesis of β-diketiminate-derived silylene compounds
A case where the facile activation of the β-diketiminate ligand backbone plays a crucial role in product stability and compound reactivity is in low oxidation state silicon chemistry.54–59 Driess and co-workers have introduced a reactive two-coordinate silylene based on the dideprotonated β-diketimine MeDipnacnacH, i.e., (MeDipnacnac’)Si, where MeDipnacnac’ is H2CC(NDip)CHC(NDip)Me.60,61 Related to this, the groups of Driess62 and Aldridge63 have recently introduced and studied the chemistry of rare β-diketiminate chlorosilylene species and ligand backbone modifications were instrumental in accessing the desired target compounds.
Reacting [(iPrDipnacnac)Li] 6a with Roesky, Stalke and co-workers’ N-heterocyclic carbene adduct IDipSiCl2 (IDip = IPr = {HC(NDip)}2C)64 as a SiCl2 source in an attempted salt metathesis reaction in deuterated benzene afforded large quantities of proligand 2a alongside uncoordinated IDip, as judged by 1H NMR spectroscopy. In this reaction, we suspected that the backbone CH on the IDip ligand may be activated via an abnormal carbene-species65–67 with the highly basic ligand system using 6a. Thus, we attempted a similar salt metathesis reaction of 6a using the imidazolinylidene-based N-heterocyclic carbene adduct SIDipSiBr2 (SIDip = SIPr = {H2C(NDip)}2C)68 introduced by Filippou and co-workers. In the latter, the saturated N-heterocyclic carbene backbone appears to be more inert towards deprotonation reactions in general. This reaction, however, also formed proligand 2a and uncoordinated SIPr as part of the reaction mixture. Switching to a similar reaction of the less basic [(EtDipnacnac)Li] 6b with SIDipSiBr2 in deuterated benzene which was monitored by 1H NMR spectroscopy saw the starting materials get consumed and showed the appearance of NMR resonances for a main new β-diketiminate-containing product of anticipated symmetry plus those for uncoordinated SIDip. Performing the reaction on a slightly larger scale at room temperature afforded (EtDipnacnac)SiBr 9 in around 43% isolated yield (Scheme 7), which could also be structurally characterised, see Fig. 7. The overall molecular structure of 9 is similar to those of β-diketiminate chlorosilylenes introduced by Driess62 and Aldridge.63 In 9, the β-diketiminate-Si–Br angle is quite acute; 83.81(10)° for the Br1–Si1⋯C3backbone angle and 98.75(10)° when using the Br1–Si1–(N1⋯N5-midpoint) angle. The Si–Br distance in 9 (2.405(2) Å) is similar to that found in a diiminophosphinate silicon(II) bromide (2.4545(10) Å)69 whereas the one present in a related amidinate silicon(II) bromide70 is too disordered for reliable comparison. In solution, compound 9 shows four doublets and two septets for the protons of the isopropyl groups by 1H NMR spectroscopy in accordance with its expected symmetry showing different environments above and below the nacnacSi-plane. A 29Si{1H} NMR resonance of δ-7.4 ppm was found for compound 9 that lies in the expected range (cf δ-10.7 ppm (ref. 62), δ 2 ppm (ref. 63)).
 |
| | Scheme 7 Synthesis of (EtDipnacnac)SiBr 9. | |
 |
| | Fig. 7 Molecular structure of (EtDipnacnac)SiBr 9 (30% thermal ellipsoids). Hydrogen atoms and the minor component of disorder are omitted for clarity. Selected bond lengths (Å) and angles (°): Br1–Si1 2.405(2), Si1–N1 1.8406(12), Si1–N5 1.8652(12), N1–C2 1.3505(18), C2–C3 1.383(2), C3–C4 1.399(2), N5–C4 1.3323(18); N1–Si1–N5 94.53(6), N1–Si1–Br1 97.20(9), N5–Si1–Br1 94.67(8), C2–C3–C4 125.50(14), Br1–Si1⋯C3 83.81(10), Br1–Si1–(N1⋯N5-midpoint) 98.75(10). | |
To test the robustness of the ligand system, we treated (EtDipnacnac)SiBr 9 with the strong amide base [KN{N(SiMe3)2}] in aromatic solvents at room temperature and the reaction led to formal HBr elimination and formation of (EtDipnacnac’)Si 10, where EtDipnacnac’ is the backbone-deprotonated “divalent” ligand variant MeCHC(NDip)CHC(NDip)Et, i.e., an analogue of Driess’ silylene, plus KBr and HN(SiMe3)2 (Scheme 8). This demonstrates that the backbone positions in EtDipnacnac compounds can still be deprotonated, but likely less easily than those of MeDipnacnac derivatives. Compound 10 (Fig. 8) crystallised with half a molecule in the asymmetric unit and thus the backbone unit is disordered by symmetry and individual bond lengths in that part of the molecule will not be commented on. The silicon(II) centre is part of a planar six-membered ring formed by bonding to both nitrogen atoms of the EtDipnacnac’ ligand. The average Si–N distance in 10 (1.736 Å) is marginally longer than the average Si–N distance in Driess’ silylene (MeDipnacnac’)Si (1.708 Å),60 and as expected significantly shorter than those in the three coordinate silicon(II) compound 9 (1.853 Å mean). There is an absence of any weak long-range Si⋯Si contacts in the solid state structures of both 10 and 9. NMR spectra of 10 are broadly comparable to those of related (MeDipnacnac’)Si showing four doublets and two septets for the protons of the isopropyl groups (1H NMR spectroscopy) as expected for a planar central unit with two different molecular halves plus resonances that support the backbone deprotonation, i.e., a quartet (1 H) at δ 3.84 ppm and an associated doublet (3 H) at δ 1.62 ppm. The 29Si NMR resonance of 10 (δ 87.9 ppm) is very close to that of (MeDipnacnac’)Si (δ 88.4 ppm).60
 |
| | Scheme 8 Synthesis of (EtDipnacnac’)Si 10. | |
 |
| | Fig. 8 Molecular structure of (EtDipnacnac’)Si 10 (30% thermal ellipsoids). Hydrogen atoms and the minor component disorder are omitted for clarity. Selected bond lengths (Å) and angles (°): Si1–N1 1.7358(12), Si1–N1′ 1.7357(12); N1–Si1–N1′ 99.23(8). | |
3. Conclusions
In this work we have presented the synthesis of a range of β-diketimine proligands, RArnacnacH, where the backbone substituents (R) are Et or iPr and the N-aryl groups (Ar) are Dip, Dep, Mes or Xyl. Derivatives with R = Et can be synthesised by a common condensation type protocol, whereas for derivatives with R = iPr, a powerful dehydrating agent, PPSE, was used in a one-pot procedure. The latter also afforded the phosphorus(V) β-diketiminate compound (iPrMesnacnac)PO24c which could be hydrolysed to iPrMesnacnacH 2c under aqueous alkaline conditions and likely presents the initial product formed during the assembly reaction in PPSE before workup. The β-diketimine proligands RArnacnacH can be converted to alkali metal complexes using common reagents such as n-butyllithium and benzyl potassium. Reactions of RDipnacnacH with sodium or potassium bis(trimethylsilyl)amide highlight significant differences in reaction rates for the deprotonation reaction and show one aspect of an influence of the alkyl substitution in the backbone unit. Furthermore, the different basicities and/or minor differences in steric profile of the ligands RDipnacnac− play a role in salt metathesis of their alkali metal complexes. For [(iPrDipnacnac)Li] 6a side-reactions such as proton abstraction were observed likely due to the high basicity, whereas less basic [(EtDipnacnac)Li] 6b could be used in a salt metathesis approach with SIDipSiBr2 to afford the rare β-diketiminate silicon(II) bromide compound (EtDipnacnac)SiBr 9. Nevertheless, despite the protected backbone unit in 9, HBr can be eliminated using [KN{N(SiMe3)2}] to afford the analogue of Driess’ silylene, (EtDipnacnac’)Si 10. Now, a range of backbone R groups including Me, Et, iPr, tBu and Ph can be easily introduced to popular RArnacnacH proligands and all except R = tBu are accessible via a one-step protocol. This allows researchers to choose suitable nacnac- or BDI-ligands from a wider pool based on steric profile, basicity, solubility, and ligand robustness for their choice of application.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We are grateful to the EPSRC DTG (EP/R513337/1), the China Scholarship Council, and the School of Chemistry at the University of St Andrews for support.
References
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3065 CrossRef CAS PubMed.
- Y. C. Tsai, Coord. Chem. Rev., 2012, 256, 722–758 CrossRef CAS.
- E. Glöckler, S. Ghosh and S. Schulz, Polym. Rev., 2023, 63, 478–514 CrossRef.
-
S. Ito and Y. Morimoto, β-Diketiminates as Redox Non-innocent Supporting Ligands in Coordination Chemistry, in Chemical Science of π-Electron Systems, ed. T. Akasaka, et al., Springer, Japan 2016 Search PubMed.
- C. Camp and J. Arnold, Dalton Trans., 2016, 45, 14462–14498 RSC.
- R. Kretschmer, Chem. – Eur. J., 2020, 26, 2099–2119 CrossRef CAS PubMed.
- R. B. Ferreira and L. J. Murray, Acc. Chem. Res., 2019, 52, 447–455 CrossRef CAS PubMed.
- S. P. Sarish, S. Nembenna and H. W. Roesky, Acc. Chem. Res., 2011, 44, 157–170 CrossRef CAS PubMed.
- C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS.
- B. Rösch and S. Harder, Chem. Commun., 2021, 57, 9354–9365 RSC.
- L. S. Vasil'ev, S. V. Baranin and I. V. Zavarzin, Russ. Chem. Bull., Int. Ed., 2017, 66, 1398–1418 CrossRef.
- M. Zhong, S. Sinhababu and H. W. Roesky, Dalton Trans., 2020, 49, 1351–1364 RSC.
- C. Chen, S. M. Bellows and P. L. Holland, Dalton Trans., 2015, 44, 16654–16670 RSC.
- R. L. Webster, Dalton Trans., 2017, 46, 4483–4498 RSC.
- S. Hohloch, B. M. Kriegel, R. G. Bergman and J. Arnold, Dalton Trans., 2016, 45, 15725–15745 RSC.
- D. Zhu and P. H. M. Budzelaar, Dalton Trans., 2013, 42, 11343–11354 RSC.
- K. Huse, C. Wölper and S. Schulz, Eur. J. Inorg. Chem., 2018, 3472–3480 CrossRef CAS.
- K. Huse, H. Weinert, C. Wölper and S. Schulz, Dalton Trans., 2020, 49, 9773–9780 RSC.
- R. Lalrempuia, C. E. Kefalidis, S. J. Bonyhady, B. Schwarze, L. Maron, A. Stasch and C. Jones, J. Am. Chem. Soc., 2015, 137, 8944–8947 CrossRef CAS PubMed.
- H. Videa and A. J. Martínez-Martínez, Dalton Trans., 2023, 52, 13058–13062 RSC.
- P. H. M. Budzelaar, A. B. van Oort and A. G. Orpen, Eur. J. Inorg. Chem., 1998, 1485–1494 CrossRef CAS.
-
D. J. Mindiola, P. L. Holland and T. H. Warren, Complexes of Bulky β-Diketiminate Ligands, in Inorganic Syntheses, ed. T. B. Rauchfuss, John Wiley & Sons, 2010, vol. 35, pp. 1–55 Search PubMed.
- M. Stender, R. J. Wright, B. E. Eichler, J. Prust, M. M. Olmstead, H. W. Roesky and P. P. Power, J. Chem. Soc., Dalton Trans., 2001, 3465–3469 RSC.
- I. El-Zoghbi, A. Ased, P. O. Oguadinma, E. Tchirioua and F. Schaper, Can. J. Chem., 2010, 88, 1040–1045 CrossRef CAS.
- M. Arrowsmith, M. R. Crimmin, M. S. Hill and G. Kociok-Köhn, Dalton Trans., 2013, 42, 9720–9726 RSC.
- D. C. H. Do, A. Keyser, A. V. Protchenko, B. Maitland, I. Pernik, H. Niu, E. L. Kolychev, A. Rit, D. Vidovic, A. Stasch, C. Jones and S. Aldridge, Chem. – Eur. J., 2017, 23, 5830–5841 CrossRef CAS PubMed.
- M. A. Land, B. Huo, K. N. Robertson, K. E. O. Ylijoki, P. T. K. Lee, J. Areephong, D. Vidovic and J. A. C. Clyburne, Dalton Trans., 2018, 47, 10195–10205 RSC.
- S. Burnett, C. Bourne, A. M. Z. Slawin, T. van Mourik and A. Stasch, Angew. Chem., Int. Ed., 2022, 61, e202204472 CrossRef CAS PubMed.
-
M. Gao, J. Wang, H. Liu, J. Ma, X. Cai, C. Li, J. Hu, X. Zhang, X. Li, Z. Zhang, R. Duan, L. Yang and C. Ma, US patent, US2017/0073441A1, 2017 Search PubMed.
- W. Liu, Y. Ding, D. Jin, Q. Shen, B. Yan, X. Ma and Z. Yang, Green Chem., 2019, 21, 3812–3815 RSC.
- M. C. Eaton, B. J. Knight, R. Brahmi, R. B. Ferreira, V. J. Catalano, A. L. Rheingold, I. Ghiviriga and L. J. Murray, J. Org. Chem., 2020, 85, 13579–13588 CrossRef CAS PubMed.
- N. Demidov, M. Grebogi, C. Bourne, A. P. McKay, D. B. Cordes and A. Stasch, Molecules, 2023, 28, 7569 CrossRef CAS PubMed.
- S. E. López, J. Restrepo and J. Salazar, J. Chem. Res., 2007, 497–502 CrossRef.
- K. Yamamoto and H. Watanabe, Chem. Lett., 1982, 1225–1228 CrossRef CAS.
- A. E. Sheshenev, E. V. Boltukhina, A. A. Grishina, I. Cisarova, I. M. Lyapkalo and K. K. (Mimi) Hii, Chem. – Eur. J., 2013, 19, 8136–8143 CrossRef CAS PubMed.
- H. Kalita, W.-Z. Lee and M. Ravikanth, Inorg. Chem., 2014, 53, 9431–9438 CrossRef CAS PubMed.
- A. Hinz, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Chem. – Eur. J., 2014, 20, 14659–14673 CrossRef CAS PubMed.
- P. B. Hitchcock, M. F. Lappert and J. E. Nycz, Chem. Commun., 2003, 1142–1143 RSC.
- P. J. Ragogna, N. Burford, M. D′eon and R. McDonald, Chem. Commun., 2003, 1052–1053 RSC.
- R. Rahm, S. Espenlaub, U. R. Werz and G. Maas, Heteroat. Chem., 2005, 16, 437–446 CrossRef CAS.
- D. Vidovic, Z. Lu, G. Reeske, J. A. Moore and A. H. Cowley, Chem. Commun., 2006, 3501–3503 RSC.
- Z. Lu, G. Reeske, J. A. Moore and A. H. Cowley, Chem. Commun., 2006, 5060–5061 RSC.
- L. A. Lesikar, W. D. Woodul and A. F. Richards, Polyhedron, 2007, 26, 3242–3246 CrossRef CAS.
- Z. Lu, M. Findlater and A. H. Cowley, Chem. Commun., 2008, 184–186 RSC.
- N. D. Reddy, A. Jana, H. W. Roesky, P. P. Samuel and C. Schulzke, Dalton Trans., 2010, 39, 234–238 RSC.
- W. Clegg, E. K. Cope, A. J. Edwards and F. S. Mair, Inorg. Chem., 1998, 37, 2317–2319 CrossRef CAS PubMed.
- A. G. Avent, M. R. Crimmin, M. S. Hill and P. B. Hitchcock, J. Organomet. Chem., 2006, 691, 1242–1250 CrossRef CAS.
- H. Hamaki, N. Takeda, T. Yamasaki, T. Sasamori and N. Tokitoh, J. Organomet. Chem., 2007, 692, 44–54 CrossRef CAS.
- A. J. Wooles, W. Lewis, A. J. Blake and S. T. Liddle, Organometallics, 2013, 32, 5058–5070 CrossRef CAS.
- R. M. Gauld, J. R. Lynch, A. R. Kennedy, J. Barker, J. Reid and R. E. Mulvey, Inorg. Chem., 2021, 60, 6057–6064 CrossRef CAS PubMed.
- B. Rösch, T. X. Gentner, J. Eyselein, J. Langer, H. Elsen and S. Harder, Nature, 2021, 592, 717–721 CrossRef PubMed.
- D. D. L. Jones, S. Watts and C. Jones, Inorganics, 2021, 9, 72 CrossRef CAS.
- R. R. Fraser, T. S. Mansour and S. Savard, J. Org. Chem., 1985, 50, 3232–3234 CrossRef CAS.
- S. Yao, A. Saddington, Y. Xiong and M. Driess, Acc. Chem. Res., 2023, 56, 475–488 CrossRef CAS PubMed.
- Y. Wang and G. H. Robinson, J. Am. Chem. Soc., 2023, 145, 5592–5612 CrossRef CAS PubMed.
- C. Shan, S. Yao and M. Driess, Chem. Soc. Rev., 2020, 49, 6733–6754 RSC.
- S. Fujimori and S. Inoue, Eur. J. Inorg. Chem., 2020, 3131–3142 CrossRef CAS PubMed.
- R. S. Ghadwal, R. Azhakar and H. W. Roesky, Acc. Chem. Res., 2013, 46, 444–456 CrossRef CAS PubMed.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
- M. Driess, S. Yao, M. Brym, C. van Wüllen and D. Lentz, J. Am. Chem. Soc., 2006, 128, 9628–9629 CrossRef CAS PubMed.
- M. Driess, S. Yao, M. Brym and C. van Wüllen, Angew. Chem., Int. Ed., 2006, 45, 6730–6733 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, A. Kostenko and M. Driess, Dalton Trans., 2018, 47, 2152–2155 RSC.
- D. C. H. Do, A. V. Protchenko, M. A. Fuentes, J. Hicks, E. L. Kolychev, P. Vasko and S. Aldridge, Angew. Chem., Int. Ed., 2018, 57, 13907–13911 CrossRef CAS PubMed.
- R. S. Ghadwal, H. W. Roesky, S. Merkel, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 5683–5686 CrossRef CAS PubMed.
- S. C. Sau, P. K. Hota, S. K. Mandal, M. Soleilhavoup and G. Bertrand, Chem. Soc. Rev., 2020, 49, 1233–1252 RSC.
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed.
- S. Würtemberger-Pietsch, U. Radius and T. B. Marder, Dalton Trans., 2016, 45, 5880–5895 RSC.
- A. C. Filippou, O. Chernov and G. Schnakenburg, Chem. – Eur. J., 2011, 17, 13574–13583 CrossRef CAS PubMed.
- S. Takahashi, J. Sekiguchi, K. Nakaya, A. Ishii and N. Nakata, Inorg. Chem., 2022, 61, 7266–7273 CrossRef CAS PubMed.
- H.-X. Yeong, K.-C. Lau, H.-W. Xi, K. H. Lim and C.-W. So, Inorg. Chem., 2010, 49, 371–373 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Alexandra M. Z.
Slawin
,
Alexandra M. Z.
Slawin