
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrocyclic complexes of Fe(III) with mixed hydroxypropyl and phenolate or amide pendants as T1 MRI probes†
Elizabeth A.
Kras
 ,
Roy
Cineus
,
Matthew R.
Crawley
and
Janet R.
Morrow
*
,
Roy
Cineus
,
Matthew R.
Crawley
and
Janet R.
Morrow
*
Department of Chemistry, University at Buffalo, the State University of New York, Amherst, NY 14260, USA. E-mail: jmorrow@buffalo.edu
First published on 1st February 2024
Abstract
High-spin Fe(III) complexes of 1,4,7-triazacyclononane (TACN) with mixed oxygen donor pendants including hydroxypropyl, phenolate or amide groups are prepared for study as T1 MRI probes. Complexes with two hydroxypropyl pendants and either amide (Fe(TOAB)) or phenolate (Fe(PTOB)) groups are compared to an analog with three hydroxypropyl groups (Fe(NOHP)), in order to study the effect of the third pendant on the coordination sphere as probed by solution chemistry, relaxivity and structural studies. Solution studies show that Fe(PTOB) has two ionizations with the phenol pendant deprotonating with a pKa of 1.7 and a hydroxypropyl pendent with pKa of 6.3. The X-ray crystal structure of [Fe(PTOB)]Br2 features a six-coordinate complex with two bound hydroxypropyl groups, and a phenolate in a distorted octahedral geometry. The Fe(TOAB) complex has a single deprotonation, assigned to a hydroxypropyl group with a pKa value of 7.0. Both complexes are stabilized as high-spin Fe(III) in solution as shown by their effective magnetic moments and Fe(III)/Fe(II) redox potentials of −390 mV and −780 mV versus NHE at pH 7 and 25 °C for Fe(TOAB) and Fe(PTOB) respectively. Both Fe(PTOB) and Fe(TOAB) are kinetically inert to dissociation under a variety of challenges including phosphate/carbonate buffer, one equivalent of ZnCl2, two equivalents of transferrin or 100 mM HCl, or at basic pH values over 24 h at 37 °C. The r1 relaxivity of Fe(TOAB) at 1.4 T, pH 7.4 and 33 °C is relatively low at 0.6 mM−1 s−1 whereas the r1 relaxivity of Fe(PTOB) is more substantial and shows an increase of 2.5 fold to 2.5 mM−1 s−1 at acidic pH. The increase in relaxivity at acidic pH is attributed to protonation of the phenolate group to provide an additional pathway for proton relaxation.
Introduction
Coordination complexes of Mn(II) and Fe(III) are under development as examples of transition metal-based T1 MRI probes to provide alternatives to clinically relevant Gd(III) MRI contrast agents. While good progress has been made with Mn(II) complexes that have increased stability and relaxivity,1,2 Fe(III) complexes have been less studied.2,3 However, the development of Fe(III) MRI probes has received more attention over the past few years.4–11 A motivation for the development of iron MRI probes is to take advantage of the biochemical mechanisms in animals as well as humans to sequester and store excess iron.12–14 However, ligands designed for Fe(III) chelation need to take into account the unique chemistry of the highly Lewis acidic and small-sized Fe(III) ion.2,3,5 The strong Lewis acidity of the Fe(III) center promotes deprotonation of bound waters at relatively low pKa values to form terminal hydroxides or bridging oxides.15–18 Ready ionization of other ligand groups bound to Fe(III) can further complicate solution chemistry.19,20 Further considerations as outlined in recent reviews involve controlling the oxidation and spin state of iron complexes under biological conditions.3,8Our development of Fe(III) complexes of 1,4,7-triazacyclononane (TACN) as T1 MRI probes has involved a search for pendant groups that produce a stabilized high spin Fe(III) center under physiological conditions.3,20–23 Notably, the TACN macrocycle may stabilize either divalent or trivalent Fe complexes, based on the type of pendant group.3,24 When all three pendant groups are neutral nitrogen donors such as pyridines or neutral oxygen donors such as amide groups, a divalent Fe(II) center is stabilized as shown by redox potentials that are >800 mV versus NHE.25 TACN ligands with neutral five-membered nitrogen heterocyclic pendants such as imidazole produce Fe complexes with intermediate potentials of approximately 330 mV at neutral pH.19,26 In contrast, anionic oxygen donor groups such as phosphonates,23,27 hydroxyalkyls,21 or phenols28 on TACN produce iron complexes with redox potentials that range from −330 mV to −1.2 V versus NHE, signifying stabilization of the trivalent state.
The importance of maintaining the trivalent oxidation state under biological conditions has been discussed in the development of MRI probes.3,8,24 In extracellular space, the most likely one electron reductant for Fe(III) is ascorbate with a standard redox potential of 0.28 V for the Asc˙, H+/HAsc− couple which is closer to 0.10 V under physiological conditions.29 Thus iron(III) complexes with negative redox potentials are desirable to avoid reduction to Fe(II). In terms of spin state, pendants containing oxygen donors including carboxylates, hydroxypropyls or phosphonates generally produce high-spin Fe(III) complexes,23 whereas heterocyclic groups appended to TACN may produce low spin Fe(III) complexes.26,30 Fe(III) complexes of TACN with two pyridine pendants may also be low spin.6
Other considerations common to contrast agents include requirements of good solubility (>5 mM) and kinetic inertness and/or thermodynamic stability towards loss of metal ion.31,32 For example, the stability and kinetic inertness of Gd(III) contrast agents have been the subject of many reviews.31,33 There are fewer examples of studies of the kinetic inertness and stability of Fe(III) MRI probes under physiological conditions.3 Recent studies in our laboratory showed that Fe(III) complexes of linear pentadentate chelates have good stability but are much less kinetically inert towards dissociation than analogous macrocyclic complexes such as those studied here34 as well as recent examples with tetra-azamacrocyclic complexes.35,36 However, Fe(III) complexes of hexadentate linear chelates are both thermodynamically stable and kinetically inert to loss of iron.4
Another major challenge is to identify the most important contributions to proton relaxivity in high-spin Fe(III) T1 MRI probes.8 We have compared Fe(III) macrocyclic complexes containing an inner-sphere water to analogous Fe(III) macrocyclic complexes that lack an inner-sphere water. The former have pentadentate macrocycles whereas the latter have hexadentate macrocyclic ligands with three pendants.20,21 As expected, Fe(III) complexes that lack an inner-sphere water have lower relaxivity than analogous complexes that do contain an inner-sphere water molecule, given the important role of an inner-sphere water in proton relaxation.20 However, a comparison of Fe(III) coordination complexes which lack an inner-sphere water but have different coordinating pendants show that relaxivity ranges from 0.6 to 1.5 mM−1 s−1 at 1.4 T, 33 °C. The hydroxypropyl complex has the highest relaxivity, followed by the phosphonate, and the carboxylate complex showed the lowest relaxivity of the three. These relaxivity differences are based, in part, on distinct second-sphere water interactions as shown by pH-dependent relaxivity studies and variable temperature 17O NMR spectroscopy.23
Whereas Fe(III) complexes that lack an inner-sphere water have lower r1 relaxivity compared to analogs that contain an inner-sphere water, such Fe(III) complexes can still be effective T1 probes if multiple Fe(III) centers are linked together. Multicenter Fe(III) probes may be part of a dinuclear complex,22 or part of a self-assembled cage structure.37 The design of multinuclear Fe(III) complexes is one approach towards more effective T1 MRI probes.
Another challenge in the development of MRI probes is the incorporation of pendant groups that can be readily functionalized. Pendant groups that can be modified for attachment of the metal ion complex to recognition agents used for targeting tissue38 or for incorporation into multinuclear complexes of nanoparticles would be useful.39,40 Moreover, pendant groups that can be easily modified to enhance relaxivity through modulating water or proton exchange,41 or to increase solubility or modulate pharmacokinetic clearance42 is important in MRI probe development. Our choice of amide or phenolate groups reflects their potential as readily functionalized pendants. Here we study the effect of an amide or a phenolate pendant on the aqueous solution properties of Fe(III)-based TACN complexes including ligand ionization, solubility, redox potentials, and kinetic inertness. Also of interest is the modulation of the proton relaxivity in the Fe(III) complexes by the third pendant as mediated by second-sphere or proton exchange. Fe(III) complexes with mixed pendant groups are compared to the complex Fe(NOHP) that contains a single type of pendant (Scheme 1).
 | ||
| Scheme 1 Fe(III) complexes drawn in the expected protonation state at pH 7.4. | ||
Results and discussion
Ligand synthesis
The synthetic schemes for PTOB and TOAB differ by the order of addition of the unique pendant group. If the unique pendant group can be added to protected TACN first followed by deprotection and addition of hydroxypropyl groups, the synthesis is considerably shortened. The PTOB ligand was prepared this way (Scheme 2). However, pendants that are sensitive to the strongly acidic or basic conditions used in the deprotection strategy cannot be added directly to the protected TACN. Sensitive pendant groups such as functionalized amides are best added to a ligand with two hydroxypropyl groups on TACN (DACO), as shown in Scheme S1.† | ||
| Scheme 2 Synthetic procedure for PTOB ligand. | ||
While the synthesis of DACO has been reported,20 a modified synthetic procedure that adds the nitro-benzyl group instead of the benzyl group to TACN prior to catalytic hydrogenation was developed and used in the synthesis of the ligands reported here. The newly reported procedure takes three to four days less than the original procedure and has an increased yield of 59%. One adjustment that was necessary for using the nitro-benzyl group was the addition of acetic acid to the hydrogenation solution to prevent reduction of the nitro group to an amino group, rather than the nitro-benzyl group being removed from the macrocycle. For TOAB, the dibenzylamide group was added to DACO via a single step alkylation reaction (Scheme S2†).40
To prepare PTOB, the coordinating pendant was added first and the hydroxypropyl groups were added second. This route succeeds in part because the methylsulfonyl protecting group on the phenol remains intact through the harsh acid deprotection of the TACN moiety. The hydroxypropyl groups were added while the phenol was still protected to prevent the alkylation of the phenol by the propylene oxide. After the hydroxypropyl pendants were added, the phenol was deprotected using hydroxide.
Fe(III) complex synthesis
The two iron complexes were prepared by similar methods, with slight variations for each. Fe(TOAB) was successfully synthesized in aqueous solution with ferrous bromide while maintaining a pH of 5.5 to 6.5 and a temperature of 50 °C.40 Fe(PTOB) was prepared by using either ferrous chloride or ferrous bromide in ethanol at 60 to 70 °C for 16 to 18 hours. Solutions were exposed to air to allow for oxidation of the ferrous center to ferric iron once bound to the ligands.Structural studies
The atomic connectivity of [Fe(PTOB)]Br2 was determined by single-crystal X-ray diffraction (Fig. 1 and Table S1†). The iron center was found to be six-coordinate with three nitrogen donors from the TACN ring, and three oxygen donors from the TACN pendant groups. Additionally, there were two outersphere Br− counterions which were positionally disordered with Cl− anions. Hydrogen atoms for O2 and O3 were located in the difference map and placed at calculated positions. The iron center can therefore best be described as Fe(III) with disordered Br−/Cl− counterions in a ratio of 1.5/0.5, respectively, and a single phenoxide (O1) arm of the macrocyclic ligand maintain charge neutrality. Hydrogen bonding-type interactions were observed between the hydroxy group protons (H2 and H3) and the nearby (∼2.2 Å) Br−/Cl− groups (see Fig. S31†). The coordination environment is distorted pseudo-octahedral, selected bond lengths and angles are tabulated in Tables S2 and S3.† Surveying the Fe(III) coordination sphere bond lengths reveals the Fe1–O1 distance is ca. 0.2 Å shorter than the Fe–O distances for the protonated alcohols (O2 and O3), further supporting that O1 is indeed deprotonated. | ||
| Fig. 1 Thermal ellipsoid plot of complex cation of [Fe(PTOB)]Br2 at the 50% probability level. H-atoms and outersphere Br− counterions have been omitted for clarity. | ||
Fe(III) complex characterization
The two Fe(III) complexes were characterized by NMR spectroscopy including by Evans method of susceptibility. Fe(TOAB) had an effective magnetic moment of 6.1 ± 0.2 whereas Fe(PTOB) showed an effective magnetic moment of 6.2 ± 0.3. These values are characteristic of high spin Fe(III) complexes and are similar to previously reported high spin Fe(III) contrast agents.20,21 However, high spin Fe(II) centers in TACN ligands may have magnetic moments in this range as well.43,44 In order to further differentiate between high spin Fe(II) and Fe(III), the 1H NMR spectra were collected. High spin Fe(II) complexes of TACN macrocycles typically show relatively sharp paramagnetically shifted proton resonances,43 whereas the 1H NMR spectra of Fe(III) complexes have proton resonances broadened into the baseline due to increased relaxivity properties. The 1H NMR spectra of Fe(TOAB) and Fe(PTOB) showed an absence of proton resonances which, along with relaxivity properties described below, further supports a high spin Fe(III) oxidation state.Aqueous solubility is an important requirement in the design of contrast agents as MRI studies in mice require solutions of at least 5 mM to deliver 50 μmol kg−1 by tail vein. Many Gd(III) based contrast agents have aqueous solubilities of 100 mM or greater.33 Whereas Fe(NOHP) has barely sufficient solubility (6 mM) for animal studies,23 Fe(PTOB) was soluble to at least 15 mM. At neutral pH, Fe(TOAB) was soluble up to 20 mM (with meglumine present), although some precipitation was observed at neutral pH in the presence of HEPES buffer. However, the solubility of this complex over a wide pH range was more limited.
A potential challenge of using amide pendants, such as in Fe(TOAB), is the hydrolysis of the amide to a carboxylate group (Scheme S6†). This is often acid catalyzed, and while most of the intended studies will be done at neutral pH, Fe(III) is a strong Lewis acid that may catalyze the reaction. Since there is a 178 mass unit different between these two species, mass spectrometry was used to determine if hydrolysis occurred over time when Fe(TOAB) was dissolved in water. Fe(TOAB) was studied at pH 4, pH 7, and pH 9 over the course of two weeks. For all pH conditions, no evidence of the hydrolysis product nor dissociation of complex to give free ligand was observed after two weeks at room temperature (∼25 °C). The studies carried out at pH 7 and 9 are shown in Fig. S7 and S8.† In comparison, metal catalyzed amide hydrolysis was examined in a recent study of an analogous Ga(III) based TACN complex. Ga(III) is another highly Lewis acidic metal ion with properties similar to those of Fe(III).45 The Ga(III) center was similarly unable to catalyze the rapid hydrolysis of the amide pendant at room temperature over several hours, although hydrolysis at higher temperatures was observed. However, this study featured a monosubstituted amide with adjacent serine which was proposed to accelerate amide hydrolysis through a N, O acyl shift mechanism.45
In order to determine the speciation of the Fe(III) complexes as a function of pH, spectrophotometric titrations were carried out. Fe(PTOB) showed changes in the LMCT band at 555 nm at acidic pH values (Fig. 2 and Fig. S9†). This change in absorbance is consistent with deprotonation of the phenol group as the pH is increased. The pKa value of 1.7 is lower than that of Fe(III) complexes containing functionalized phenol or hydroxypyridine pendants of linear chelates which were 2.3 and 3.7, respectively.34 In the Fe(PTOB) complex studied here, it is interesting to note that even at very acidic pH values, the LMCT band is maintained, although shifted by 140 nm from the LMCT which is present at pH 3.5 to pH 11. The presence of the LMCT band suggests that the protonated phenol is coordinated even at these very acidic pH values. The second ionization with a pKa value of 6.3 is assigned to one of the hydroxypropyl groups (Fig. S10†). This value is slightly lower than that of other Fe(III) complexes with two hydroxypropyl groups and a water ligand, suggesting that the bound phenolate pendant facilitates ionization of a hydroxypropyl group.20,21 The Fe(TOAB) complex has a single ionization with a pKa value of 7.0 that is assigned to deprotonation of one of the hydroxypropyl groups (Scheme 3 and Fig. S11†). This pKa value is similar to that of the hydroxypropyl group of Fe(NOHP).21
 | ||
| Fig. 2 Spectrophotometric titration of Fe(PTOB) as a function of pH (top). Titration showing isosbestic point at 500 nm along with a plot of the change in absorbance and fit to a single pKa value of 1.7 (bottom). All solutions contained 20 mM HEPES buffer, 100 mM NaCl, 200 μM Fe(PTOB) and were adjusted to the indicated pH value using HCl or NaOH. | ||
 | ||
| Scheme 3 Fe(III) complexes and their ionizations in aqueous solutions. | ||
The electrochemistry of the two iron complexes was studied to determine the effect of the phenolate or amide pendant group on the Fe(III)/Fe(II) reduction potential. Fe(TOAB) shows a quasi-reversible wave at 260 mV versus NHE by cyclic voltammetry when recorded at pH 3.5. This peak shifts to −390 versus NHE at neutral pH (Fig. S12 and S13†). The shift in the potential is consistent with the ionization of a hydroxypropyl group to give an anionic pendant that stabilizes the Fe(III) center. The electrochemistry of Fe(PTOB) was studied in acetonitrile to accommodate the more negative redox potential expected for this complex. For example, an Fe(III) complex of TACN with one phenolate and two carboxylate pendants has a redox potential of −450 mV vs. NHE46 whereas complexes with three phenolate groups have redox potentials of −1.2 V vs. NHE.47 For Fe(PTOB) studies, the reduction potentials are referenced versus the ferrocene/ferrocenium couple (Fc/Fc+), and converted to versus NHE.48 The cyclic voltammogram of FePTOB (Fig. S14†) shows a quasi-reversible reduction potential of −1.4 V vs. Fc/Fc+ (−780 mV vs. NHE), which is assigned to the Fe(III)/Fe(II) redox couple. The negative reduction potential of Fe(PTOB) suggests stabilization of the trivalent state, and supports the fact that Fe(PTOB) is unlikely to be reduced when injected in vivo. In addition, an oxidation wave at 0.5 V is assigned to a ligand-centered redox process associated with the coordinated phenolate.49,50
Fe(III) complex dissociation studies
Kinetic inertness of metal complexes towards dissociation is one of the most important characteristics to consider in the development of contrast agents including those of Gd(III) and Mn(II).8,31,51 Notably, thermodynamic stability may not be as important for a metal ion complex that is highly kinetically inert towards metal ion dissociation. For example, previous studies have shown that Fe(III) complexes of TACN with hydroxypropyl pendants, such as Fe(NOHP), lack thermodynamic stability yet are highly resistant to loss of metal ion even in strong acid or with competing ligands.20,23 Since it is such an important characteristic of MRI probes, kinetic inertness of these complexes was tested under a few different conditions (Table 1).Kinetic inertness in HEPES buffer is an important parameter to test for new MRI contrast agents as HEPES buffer is frequently used for phantoms as well as in other solution studies with the complexes. HEPES buffer is often used in initial studies as an example of a relatively weakly binding buffer, prior to the use of phosphate buffered saline (PBS) of phosphate/carbonate mixtures with physiologically relevant concentrations of these anions. Fe(TOAB) showed a slight change in absorbance over 24 hours of 11–22% (Fig. S16†), that was attributed to precipitation corresponding to the appearance of an orange solid upon addition of HEPES buffer. Due to this, meglumine was tested as an alternative to produce solutions that had a pH of 7 and remained biologically compatible. Meglumine has been used to solubilize Fe(III) complexes21 and is a component of many gadolinium contrast agent formulations.32 Fe(TOAB) was found to be kinetically inert in pH 7, meglumine adjusted solutions, therefore, meglumine was used in UV-vis, relaxivity, and cyclic voltammetry studies to adjust the pH of the solutions to 7. In the case of Fe(PTOB), there was minimal change in absorbance over 24 hours in the presence of only HEPES buffer and therefore HEPES buffer was used in all solution studies. Fe(TOAB) and Fe(PTOB) were found to be kinetically inert in the presence of biologically relevant anions (carbonate and phosphate) as well as in the presence of a molar equivalent of zinc(II) chloride. However, Fe(TOAB) dissociated up to 17.5% in the presence of 100 mM acid after 24 hours (Fig. S15†) and Fe(PTOB) dissociated up to 10% under the same conditions (Fig. S20†). These data suggest that the Fe(III) macrocyclic complexes studied here are more inert towards trans-metalation by Zn(II) than Mn(II) or Gd(III) contrast agents with linear chelates,42 but less inert than macrocyclic complexes of Gd(III) such as Gd(DOTA).52
Further studies of the kinetic inertness of the Fe(PTOB) complex explored incubation at basic pH, and 37 °C over a period of 24 hours. These studies showed no change in the intensity of the LMCT band at 430 nm, suggesting that the complex was inert towards dissociation at pH 9 (Fig. S24†). Moreover, addition of an equivalent of the strongly chelating ligand, EDTA, at pH 7.4, 37 °C produced no discernable dissociation of Fe(PTOB) over 24 hours (Fig. S25†). Finally, addition of 1–2 equivalents of ascorbate (50–100 μM) as a potential reducing agent to solutions of Fe(PTOB) followed by incubation for 24 hours at 37 °C produced no substantial change in the intensity of the LMCT band of Fe(PTOB) (Fig. S26 and S27†). The inability of ascorbate anion to reduce Fe(PTOB) is consistent with the very negative redox potential of the complex which signifies a highly stabilized Fe(III) center.
Although iron is the most abundant transition metal in the human body, maintaining the homeostasis of the iron content in the body is extremely important,53–55 which is one reason why kinetic inertness is a consideration when designing new iron-based contrast agents. Due to its abundance in the human body, there are proteins to bind, transport, and store free iron.55 The major protein carrier for iron throughout the body is transferrin,56–59 which has a strong affinity for iron (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kcond = 20.7 and 19.4 for the two Fe(III) binding sites of human transferrin at pH 7.4).58,60 Due to its abundance (30 μM in the blood plasma),57,60 transferrin could sequester dissociated iron from an injected iron complex if the affinity of the complex for iron is lower than that of transferrin.12 This can be studied by UV-vis as the iron-transferrin complex has a strong absorbance at 465 nm (ε = 4950 M−1 cm−1).59,60
Kcond = 20.7 and 19.4 for the two Fe(III) binding sites of human transferrin at pH 7.4).58,60 Due to its abundance (30 μM in the blood plasma),57,60 transferrin could sequester dissociated iron from an injected iron complex if the affinity of the complex for iron is lower than that of transferrin.12 This can be studied by UV-vis as the iron-transferrin complex has a strong absorbance at 465 nm (ε = 4950 M−1 cm−1).59,60
Using a procedure based on transferrin competition studies with iron MRI contrast agents,4,12 the kinetic inertness of our complexes to transferrin was tested (Table 1). Fe(NOHP), Fe(PTOB) and Fe(TOAB) were incubated with two molar equivalents of apo-transferrin at pH 7 and 37 °C and monitored for 24 hours. Fe(NOHP), Fe(TOAB), and Fe(PTOB) showed 4.3%, 6%, and 7% increase in the absorbance at 465 nm, respectively, consistent with very small amounts of iron transchelation to transferrin over these 24 hours (Fig. S28–S30†). This is comparable with the previously reported Fe-PyC3A which showed <3% transchelation under the same conditions.4 By contrast, an Fe(III) complex with a linear triamine chelate and pendant phenolates lost iron rapidly to transferrin with a half-life of 15–20 minutes.34 This data shows that the macrocyclic ligands or ligands with rigid backbones are required to prevent loss of Fe(III) to transferrin.
Variable temperature 17O NMR studies
The Fe(III) complexes studied here are likely to be coordinatively saturated and lack a coordination site for an inner-sphere water. Indeed, Fe(NOHP) is six-coordinate with no inner-sphere water21 and the solid state structure of Fe(PTOB), as studied here, shows no bound solvent molecules. However, we wondered whether Fe(PTOB) would accommodate an inner-sphere water in solution. The most common method to study whether a paramagnetic metal ion complex has a bound, rapidly exchanging inner-sphere water is through 17O NMR spectroscopy.52,61,62 Methods initially developed for Mn(II) complexes, have employed 17O water resonance line broadening (transverse r2 relaxivity) studies as a function of metal complex concentration and temperature to estimate the number of inner-sphere waters.63 This method has been applied to Fe(III) complexes as well.20,21 The transverse relaxation rate constants (1/T2o or R2o) estimated from 17O line broadening for our complexes were compared to standard Fe(III) complexes that have a bound water, such as Fe(CDTA), and ones that lack a water, such as Fe(DTPA).64 The comparison aids in the assessment of whether there is an inner-sphere water that undergoes exchange on the 17O NMR timescale. At neutral pH, the data collected for Fe(PTOB) shows a transverse r2o (R2o normalized to concentration) that is similar to that of Fe(DTPA) and much lower than that of Fe(CDTA) as shown in Fig. 3. This is similar to reports of Fe(NOTP) and Fe(NOHP)23 and supports the lack of an exchangeable inner-sphere water ligand at neutral pH. | ||
| Fig. 3 Comparison of 17O NMR transverse relaxivity (r2o) for Fe(DTPA), Fe(CDTA), and Fe(PTOB) at pH 6.5–7.2, HEPES buffer, as a function of temperature. | ||
Fe(III) complex proton relaxivity
The mechanism of proton relaxation of paramagnetic metal ion complexes has been described for Fe(III) complexes.7,8,65 Briefly, relaxivity is parsed into contributions from inner-sphere or directly bound waters (rIS1), second-sphere relaxivity (rSS1) for water molecules in close proximity and perhaps bound to ligands, and outer-sphere water (rOS1) for closely diffusing water. These are related to the number of water molecules involved (q), the lifetime of the inner-sphere or second-sphere water interactions with the paramagnetic complex (τm), as well as the relaxation time of the bound water (T1m) as shown in eqn (1). The outer-sphere contribution (rOS1) arises from water molecules that do not have a specific lifetime and interaction associated with the contrast agent. Inner-sphere waters are relatively well defined from the standpoint of number, distance, and orientation with respect to the metal center, whereas second-sphere waters (q′ and τm′) are more difficult to assess.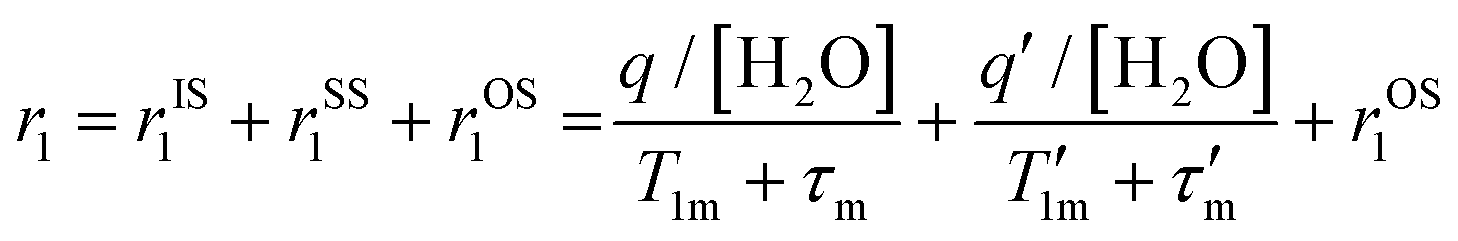 | (1) |
The T1 and T2 water proton relaxation times were measured in the absence and presence of human serum albumin (HSA at 35 mg mL−1) and the relaxivity values for the iron complexes, Fe(TOAB) and Fe(PTOB) are given in Table 2 along with values for Fe(NOHP) for comparison. Measurements were made at pH 7.2 by monitoring the T1 or T2 water proton relaxation times of solutions containing the complex over the concentration range of 50 μM to 1.00 mM for the Fe(III) complex. Fe(TOAB) solutions had meglumine present to adjust the pH and maintain solubility, and did not contain HEPES buffer.
The macrocyclic Fe(III) complexes studied here have proton relaxation values characteristic of Fe(III) complexes that lack an inner-sphere water.20,21,23 However, Fe(TOAB) has decreased relaxivity compared to Fe(NOHP) or Fe(PTOB). A rationale for the unexpected decrease is a change in the second-sphere water contribution to relaxivity by disruption of the coordination sphere by the hydrophobic benzyl amide group. Strong second-sphere water interactions are thought to contribute to the relatively high proton relaxation values for Fe(NOHP) in comparison to other closed coordination sphere complexes.21 The Fe(PTOB) complex shows intermediate relaxivity, higher than that of Fe(TOAB) but lower than that of Fe(NOHP). In comparison, analogous Fe(III) complexes with the TACN framework, two hydroxypropyl groups and a bound water show relaxivities of 2–2.3 mM−1 s−1 at 37 °C and 4.7 T.20,21 Other mononuclear Fe(III) complexes with an exchangeable inner-sphere water ligands and CDTA frameworks have values of 1.9 to 2.4 mM−1 s−1 at intermediate field strengths (3–4.7 T).4,11 In comparison, Gd(DOTA) (DOTAREM) has a r1 relaxivity of 2.8 mM−1 s−1 at 4.7 T, 37 °C as tabulated in a review of the relaxivities of Gd(III) contrast agents.3,66
Relaxivity for the complexes was studied in the presence of 0.6 mM human serum albumin (HSA), to simulate conditions in the blood. As previously noted, Fe(NOHP) shows little to no change in relaxivity when HSA is in solution, consistent with little binding to the serum protein.23 In contrast, Fe(PTOB) shows a moderate 40% increase in the presence of HSA and Fe(TOAB) exhibits an increase of 29% in the presence of HSA, consistent with weak binding to the serum protein.
The r1 relaxivity was further examined as a function of pH for the Fe(PTOB) complex. These studies were enabled by the good solubility of the complex as well as large degree of inertness to loss of iron over a large pH range including highly acidic pH values. In contrast, Fe(III) complexes with phenolate pendants on a linear chelate dissociated at acidic pH and could not be studied over the full pH range.34 The inertness to dissociation of the macrocyclic complex allowed for the study of changes in water proton relaxation that might occur upon protonation of the phenol group. As shown in Fig. 4, the proton relaxivity stays nearly constant from pH 4 to 10. This constancy is observed despite the ionization of one of the hydroxypropyl groups at near neutral pH which might be expected to modulate the relaxivity through a change in second-sphere water interactions or proton exchange. In comparison, Fe(NOHP) also shows a flat pH-relaxivity profile despite a hydroxypropyl group deprotonation at near neutral pH.23 However, there is a dramatic increase in relaxivity at pH values less than 4. This increase is attributed to the formation of a phenol group and the corresponding contribution of the OH phenol group to proton relaxation. Notably, the UV-vis data suggest that the phenol group remains bound to the Fe(III) center even at very acidic pH values as shown by the presence of the LMCT band (Fig. 2). We propose that the increase in relaxivity at acidic pH is due to a contribution from proton exchange of the OH on the phenol pendent or, alternatively, to enhanced second-sphere water interactions at acidic pH values.
 | ||
| Fig. 4 The pH dependence of relaxivity of Fe(PTOB) at 0.10 M NaCl, 50 mM HEPES, 33 °C. The solutions were adjusted to the reported pH using HCl and NaOH. | ||
Conclusions
Macrocyclic complexes of Fe(III) with mixed oxygen donor pendants show promise as MRI probes. The amide or phenolate pendants on TACN in combination with hydroxypropyl pendants support the formation of high-spin Fe(III) complexes at neutral pH. While both complexes have stabilized Fe(III) centers, the redox potential of the Fe(PTOB) is more negative than that of Fe(TOAB), consistent with highly stabilized trivalent iron found in Fe(III) TACN complexes with phenolate pendants.28,46 Our past studies showed that hydroxypropyl groups promote effective second-sphere interactions resulting in higher relaxivity than other pendant groups as shown for Fe(NOHP).23 However, replacing a single hydroxypropyl pendant with an amide or phenolate resulted in complexes with lower r1 relaxivity. The low relaxivity of Fe(TOAB) is attributed to the disruption of the second-sphere coordination environment, possibly involving the hydrophobic aryl groups on the amide. Other factors that need to be further studied include changes in the electronic relaxation time of the Fe(III) center as might be attributed to a less highly symmetric coordination sphere.8 Still, it is quite surprising that a single substitution of a hydroxypropyl group for an amide decreases the relaxivity to such an extent. Fe(PTOB) complex had r1 relaxivity, at 1.0 mM−1 s−1 at 1.4 T, pH 7.4, 33 °C which is closer to that of Fe(NOHP) (Table 2). This data suggests that the phenolate pendent does not disrupt the second-sphere water interactions as effectively as does the amide. Interestingly, the relaxivity of the Fe(PTOB) complex increased by 2.5-fold as the phenolate pendant is protonated at acidic pH. A coordinated phenol group is supported by the presence of a LMCT band at these acidic pH values. The protonated phenolate may contribute to water proton relaxation through a proton exchange mechanism.41 This suggests an approach to modulate the relaxivity of Fe(III) complexes containing phenolate pendants. However, the pKa of the bound phenol group must be closer to neutral pH for this to have an effect in vivo.Both Fe(TOAB) and Fe(PTOB) are promising complexes for further functionalization. For example, we have reported Fe(III) complexes with functionalized phenolate groups that show modulated solubility in aqueous solution and affect the pKa of ancillary ligands.34 In another example, an amphiphilic analog of Fe(TOAB) was incorporated into the bilayer of a liposome to give a r1 relaxivity that was increased by 4-fold per iron compared to the free small molecule complex.40 Incorporation of the mononuclear small molecule iron complexes into a liposome or micelle could be a means to increase the relaxivity of these highly inert, but low relaxivity TACN-complexes that lack an inner-sphere water. Future efforts to increase the relaxivity of Fe(III) contrast agents will include scaffolds that link together multiple iron centers. Towards this goal, Fe(TOAB) and Fe(PTOB) are inert to transchelation in the presence of apo-transferrin, the major iron binding protein in the blood. Thus, mixed pendant complexes of TACN that retain their high degree of kinetic inertness show promise for the development of Fe(III) MRI probes.
Author contributions
E. A. K.: synthesis, data collection and manuscript preparation. R. C.: synthesis and data collection; M. R. C.: crystallography; J. R. M.: manuscript preparation and editing.Conflicts of interest
J. R. M. is cofounder of Ferric Contrast, a company that develops iron-based M. R. I. contrast agents.Acknowledgements
J. R. M. thanks the NSF (CHE-2004135) for support. R. C. acknowledges support by the National Institute of Health, National Institute of General Medical Science Award (R25 GM095459) to University at Buffalo. The authors would like to thank the Chemistry Instrument Center (CIC), University at Buffalo. This work utilized an ICP-MS that was purchased with funding from a NSF Major Research Instrumentation Program (NSF CHE-0959565), a Bruker 500 MHz NMR (NSF CHE-2018160) and a Rigaku XtaLAB Synergy-S diffractometer (NSF CHE-2216151).References
- M. Botta, F. Carniato, D. Esteban-Gómez, C. Platas-Iglesias and L. Tei, Mn(II) compounds as an alternative to Gd-based MRI probes, Future Med. Chem., 2019, 11(12), 1461–1483 CrossRef CAS PubMed.
- A. Gupta, P. Caravan, W. S. Price, C. Platas-Iglesias and E. M. Gale, Applications for transition-metal chemistry in contrast-enhanced magnetic resonance imaging, Inorg. Chem., 2020, 59(10), 6648–6678 CrossRef CAS PubMed.
- E. A. Kras, E. M. Snyder, G. E. Sokolow and J. R. Morrow, Distinct Coordination Chemistry of Fe(III)-Based MRI Probes, Acc. Chem. Res., 2022, 55(10), 1435–1444 CrossRef CAS PubMed.
- H. Wang, V. C. Jordan, I. A. Ramsay, M. Sojoodi, B. C. Fuchs, K. K. Tanabe, P. Caravan and E. M. Gale, Molecular magnetic resonance imaging using a redox-active iron complex, J. Am. Chem. Soc., 2019, 141(14), 5916–5925 CrossRef CAS PubMed.
- N. Kuznik and M. Wyskocka, Iron(III) Contrast Agent Candidates for MRI: a Survey of the Structure-Effect Relationship in the Last 15 Years of Studies, Eur. J. Inorg. Chem., 2016,(4), 445–458 CrossRef CAS.
- J. Salaam, T. Fogeron, G. Pilet, R. Bolbos, C. Bucher, L. Khrouz and J. Hasserodt, Unprecedented Relaxivity Gap in pH-Responsive Fe(III) -Based MRI Probes, Angew. Chem., Int. Ed., 2023, 62(7), e202212782 CrossRef CAS PubMed.
- Z. Baranyai, F. Carniato, A. Nucera, D. Horvath, L. Tei, C. Platas-Iglesias and M. Botta, Defining the conditions for the development of the emerging class of Fe(III)-based MRI contrast agents, Chem. Sci., 2021, 12, 11138–11145 RSC.
- R. Uzal-Varela, F. Lucio-Martinez, A. Nucera, M. Botta, D. Esteban-Gomez, L. Valencia, A. Rodriguez-Rodriguez and C. Platas-Iglesias, A systematic investigation of the NMR relaxation properties of Fe(III)-EDTA derivatives and their potential as MRI contrast agents, Inorg. Chem. Front., 2023, 10(5), 1633–1649 RSC.
- A. Nucera, F. Carniato, Z. Baranyai, C. Platas-Iglesias and M. Botta, Characterization of the Fe(III)-Tiron System in Solution through an Integrated Approach Combining NMR Relaxometric, Thermodynamic, Kinetic, and Computational Data, Inorg. Chem., 2023, 62(10), 4272–4283 CrossRef CAS PubMed.
- P. Boehm-Sturm, A. Haeckel, R. Hauptmann, S. Mueller, C. K. Kuhl and E. A. Schellenberger, Low-molecular-weight iron chelates may be an alternative to gadolinium-based contrast agents for T1-weighted contrast-enhanced MR imaging, Radiology, 2018, 286(2), 537–546 CrossRef PubMed.
- J. Xie, A. Haeckel, R. Hauptmann, I. P. Ray, C. Limberg, N. Kulak, B. Hamm and E. Schellenberger, Iron(III)-tCDTA derivatives as MRI contrast agents: Increased T-1 relaxivities at higher magnetic field strength and pH sensing, Magn. Reson. Med., 2021, 85(6), 3370–3382 CrossRef CAS PubMed.
- A. Sargun, A. L. Fisher, A. S. Wolock, S. Phillips, M. Sojoodi, S. Khanna, J. L. Babitt and E. M. Gale, A Rationally Designed Complex Replenishes the Transferrin Iron Pool Directly and with High Specificity, J. Am. Chem. Soc., 2023, 145(12), 6871–6879 CrossRef CAS PubMed.
- E. C. Theil, Ferritin: The Protein Nanocage and Iron Biomineral in Health and in Disease, Inorg. Chem., 2013, 52(21), 12223–12233 CrossRef CAS PubMed.
- E. L. MacKenzie, K. Iwasaki and Y. Tsuji, Intracellular iron transport and storage: from molecular mechanisms to health implications, Antioxid. Redox Signal., 2008, 10(6), 997–1030 CrossRef CAS PubMed.
- H. Wang, A. Wong, L. C. Lewis, G. R. Nemeth, V. C. Jordan, J. W. Bacon, P. Caravan, H. S. Shafaat and E. M. Gale, Rational Ligand Design Enables pH Control over Aqueous Iron Magnetostructural Dynamics and Relaxometric Properties, Inorg. Chem., 2020, 59(23), 17712–17721 CrossRef CAS PubMed.
- H. Wang, M. B. Cleary, L. C. Lewis, J. W. Bacon, P. Caravan, H. S. Shafaat and E. M. Gale, Enzyme Control Over Ferric Iron Magnetostructural Properties, Angew. Chem., Int. Ed., 2022, 61(3), e20211401 Search PubMed.
- T. Schneppensieper, S. Seibig, A. Zahl, P. Tregloan and R. van Eldik, Influence of chelate effects on the water-exchange mechanism of polyaminecarboxylate complexes of iron(III), Inorg. Chem., 2001, 40(15), 3670–3676 CrossRef CAS PubMed.
- A. Brausam, J. Maigut, R. Meier, P. A. Szilágyi, H.-J. R. Buschmann, W. Massa, Z. Homonnay and R. van Eldik, Detailed spectroscopic, thermodynamic, and kinetic studies on the protolytic equilibria of FeIIIcydta and the activation of hydrogen peroxide, Inorg. Chem., 2009, 48(16), 7864–7884 CrossRef CAS PubMed.
- P. B. Tsitovich, A. M. Kosswattaarachchi, M. R. Crawley, T. Y. Tittiris, T. R. Cook and J. R. Morrow, An Fe-III Azamacrocyclic Complex as a pH-Tunable Catholyte and Anolyte for Redox-Flow Battery Applications, Chem. – Eur. J., 2017, 23(61), 15327–15331 CrossRef CAS PubMed.
- E. M. Snyder, D. Asik, S. M. Abozeid, A. Burgio, G. Bateman, S. G. Turowski, J. A. Spernyak and J. R. Morrow, A Class of FeIII Macrocyclic Complexes with Alcohol Donor Groups as Effective T1 MRI Contrast Agents, Angew. Chem., 2020, 132(6), 2435–2440 CrossRef.
- D. Asik, R. Smolinski, S. M. Abozeid, T. B. Mitchell, S. G. Turowski, J. A. Spernyak and J. R. Morrow, Modulating the properties of Fe(III) macrocyclic MRI contrast agents by appending sulfonate or hydroxyl groups, Molecules, 2020, 25(10), 2291 CrossRef CAS PubMed.
- D. Asik, S. M. Abozeid, S. G. Turowski, J. A. Spernyak and J. R. Morrow, Dinuclear Fe(III) Hydroxypropyl-Appended Macrocyclic Complexes as MRI Probes, Inorg. Chem., 2021, 60(12), 8651–8664 CrossRef CAS PubMed.
- E. A. Kras, S. M. Abozeid, W. Eduardo, J. A. Spernyak and J. R. Morrow, Comparison of phosphonate, hydroxypropyl and carboxylate pendants in Fe(III) macrocyclic complexes as MRI contrast agents, J. Inorg. Biochem., 2021, 225, 111594 CrossRef CAS PubMed.
- J. R. Morrow, J. J. Raymond, M. S. I. Chowdhury and P. R. Sahoo, Redox-Responsive MRI Probes Based on First-Row Transition-Metal Complexes, Inorg. Chem., 2022, 61(37), 14487–14499 CrossRef CAS PubMed.
- S. J. Dorazio, P. B. Tsitovich, S. A. Gardina and J. R. Morrow, The reactivity of macrocyclic Fe(II) paraCEST MRI contrast agents towards biologically relevant anions, cations, oxygen or peroxide, J. Inorg. Biochem., 2012, 117, 212–219 CrossRef CAS PubMed.
- P. B. Tsitovich, F. Gendron, A. Y. Nazarenko, B. N. Livesay, A. P. Lopez, M. P. Shores, J. Autschbach and J. R. Morrow, Low-Spin Fe(III) Macrocyclic Complexes of Imidazole-Appended 1,4,7-Triazacyclononane as Paramagnetic Probes, Inorg. Chem., 2018, 57(14), 8364–8374 CrossRef CAS PubMed.
- E. Kras, A Comparison of Oxygen-Donor Pendant Groups for Iron(III) Macrocyclic T1 MRI Probes, Ph.D. thesis, University at Buffalo, the State University of New York, 2023 Search PubMed.
- M. D. Snodin, L. Ould-Moussa, U. Wallmann, S. Lecomte, V. Bachler, E. Bill, H. Hummel, T. Weyhermuller, P. Hildebrandt and K. Wieghardt, The molecular and electronic structure of octahedral tris(phenolato)iron(III) complexes and their phenoxyl radical analogues: A Mossbauer and resonance Raman spectroscopic study, Chem. – Eur. J., 1999, 5(9), 2554–2565 CrossRef CAS.
- W. H. Koppenol and R. H. Hider, Iron and redox cycling. Do's and don'ts, Free Radical Biol. Med., 2019, 133, 3–10 CrossRef CAS PubMed.
- J. Wang, C. Gondrand, F. Touti and J. Hasserodt, A pair of highly biotolerated diamagnetic and paramagnetic iron(II) complexes displaying electroneutrality, Dalton Trans., 2015, 44(35), 15391–15395 RSC.
- A. Merbach, L. Helm and E. Toth, in The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, Wiley & sons, 2nd edn, 2013 Search PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers, Chem. Rev., 2019, 119(2), 957–1057 CrossRef CAS PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Gadolinium(III) chelates as MRI contrast agents: structure, dynamics, and applications, Chem. Rev., 1999, 99(9), 2293–2352 CrossRef CAS PubMed.
- R. Cineus, S. M. Abozeid, G. E. Sokolow, J. A. Spernyak and J. R. Morrow, Fe(III) T1 MRI probes containing phenolate or hydroxypyridine-appended triamine chelates and a coordination site for bound water, Inorg. Chem., 2023, 62, 16513–16522 CrossRef CAS PubMed.
- S. Karbalaei, A. Franke, A. Jordan, C. Rose, P. R. Pokkuluri, R. J. Beyers, A. Zahl, I. Ivanovic-Burmazovic and C. R. Goldsmith, A Highly Water- and Air-Stable Iron-Containing MRI Contrast Agent Sensor for H2 O2, Chem. – Eur. J., 2022, 28(46), e202201179 CrossRef CAS PubMed.
- R. T. Kadakia, R. T. Ryan, D. J. Cooke and E. L. Que, An Fe complex for (19)F magnetic resonance-based reversible redox sensing and multicolor imaging, Chem. Sci., 2023, 14(19), 5099–5105 RSC.
- G. E. Sokolow, M. R. Crawley, D. R. Morphet, D. Asik, J. A. Spernyak, A. J. R. McGray, T. R. Cook and J. R. Morrow, Metal-Organic Polyhedron with Four Fe(III) Centers Producing Enhanced T1 Magnetic Resonance Imaging Contrast in Tumors, Inorg. Chem., 2022, 61(5), 2603–2611 CrossRef CAS PubMed.
- J. L. Major and T. J. Meade, Bioresponsive, cell-penetrating, and multimeric MR contrast agents, Acc. Chem. Res., 2009, 42(7), 893–903 CrossRef CAS PubMed.
- S. M. Abozeid, M. S. I. Chowdhury, D. Asik, J. A. Spernyak and J. R. Morrow, Liposomal Fe(III) Macrocyclic Complexes with Hydroxypropyl Pendants as MRI Probes, ACS Appl. Bio Mater., 2021, 4(11), 7951–7960 CrossRef CAS PubMed.
- M. S. I. Chowdhury, E. A. Kras, J. C. Pinti, J. A. Spernyak and J. R. Morrow, Liposomal MRI probes containing encapsulated or amphiphilic Fe(III) coordination complexes, Biomater. Sci., 2023, 11, 5942–5954 RSC.
- S. Aime and Z. Baranyai, How the catalysis of the prototropic exchange affects the properties of lanthanide(III) complexes in their applications as MRI contrast agents, Inorg. Chim. Acta, 2022, 532, 120730 CrossRef CAS.
- E. M. Gale, I. P. Atanasova, F. Blasi, I. Ay and P. Caravan, A manganese alternative to gadolinium for MRI contrast, J. Am. Chem. Soc., 2015, 137(49), 15548–15557 CrossRef CAS PubMed.
- P. B. Tsitovich, J. M. Cox, J. B. Benedict and J. R. Morrow, Six-coordinate Iron(II) and Cobalt(II) paraSHIFT Agents for Measuring Temperature by Magnetic Resonance Spectroscopy, Inorg. Chem., 2016, 55, 700–716 CrossRef CAS PubMed.
- P. B. Tsitovich, J. M. Cox, J. A. Spernyak and J. R. Morrow, Gear Up for a pH Shift: A Responsive Iron(II) 2-Amino-6-picolylAppended Macrocyclic paraCEST Agent That Protonates at a Pendent Group, Inorg. Chem., 2016, 55(22), 12001–12010 CrossRef CAS PubMed.
- D. Smilowicz, S. Eisenberg, R. LaForest, J. Whetter, A. Hariharan, J. Bordenca, C. J. Johnson and E. Boros, Metal-Mediated, Autolytic Amide Bond Cleavage: A Strategy for the Selective, Metal Complexation-Catalyzed, Controlled Release of Metallodrugs, J. Am. Chem. Soc., 2023, 145(29), 16261–16270 CrossRef CAS PubMed.
- C. Stockheim, L. Hoster, T. Weyhermuller, K. Wieghardt and B. Nuber, First-row transition-metal complexes of mixed ‘pendant-arm’ derivatives of 1,4,7-triazacyclononane containing phenolate and carboxylate functional groups, J. Chem. Soc., Dalton Trans., 1996,(23), 4409–4416 RSC.
- B. Adam, E. Bill, E. Bothe, B. Goerdt, G. Haselhorst, K. Hildenbrand, A. Sokolowski, S. Steenken, T. Weyhermuller and K. Wieghardt, Phenoxyl radical complexes of gallium, scandium, iron and manganese, Chem. – Eur. J., 1997, 3(2), 308–319 CrossRef CAS PubMed.
- V. V. Pavlishchuk and A. W. Addison, Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C, Inorg. Chim. Acta, 2000, 298(1), 97–102 CrossRef CAS.
- C. Stockheim, L. Hoster, T. Weyhermüller, K. Wieghardt and B. Nuber, First-row transition-metal complexes of mixed ‘pendant-arm’ derivatives of 1,4,7-triazacyclononane containing phenolate and carboxylate functional groups, J. Chem. Soc., Dalton Trans., 1996, 4409–4416(23), 1 Search PubMed.
- B. Adam, E. Bill, E. Bothe, B. Goerdt, G. Haselhorst, K. Hildenbrand, A. Sokolowski, S. Steenken, T. Weyhermüller and K. Wieghardt, Phenoxyl Radical Complexes of Gallium, Scandium, Iron and Manganese, Chem. – Eur. J., 1997, 3(2), 308–319 CrossRef CAS PubMed.
- J. Wahsner, E. M. Gale, A. Rodriguez-Rodriguez and P. Caravan, Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers, Chem. Rev., 2019, 119(2), 957–1057 CrossRef CAS PubMed.
- V. C. Pierre and M. J. Allen, Contrast Agents for MRI: Experimental Methods, Royal Society of Chemistry, 2018 Search PubMed.
- E. C. Theil, T. Tosha and R. K. Beherat, Solving Biology's Iron Chemistry Problem with Ferritin Protein Nanocages, Acc. Chem. Res., 2016, 49(5), 784–791 CrossRef CAS PubMed.
- D. Berg and H. Hochstrasser, Iron metabolism in Parkinsonian syndromes, Mov. Disord., 2006, 21(9), 1299–1310 CrossRef PubMed.
- A. C. Chua, R. M. Graham, D. Trinder and J. K. Olynyk, The regulation of cellular iron metabolism, Crit. Rev. Clin. Lab. Sci., 2007, 44(5–6), 413–459 CrossRef CAS PubMed.
- D. H. Hamilton, I. Turcot, A. Stintzi and K. N. Raymond, Large cooperativity in the removal of iron from transferrin at physiological temperature and chloride ion concentration, J. Biol. Inorg. Chem., 2004, 9(8), 936–944 CrossRef CAS PubMed.
- K. Gkouvatsos, G. Papanikolaou and K. Pantopoulos, Regulation of iron transport and the role of transferrin, Biochim. Biophys. Acta, 2012, 1820(3), 188–202 CrossRef CAS PubMed.
- P. Aisen, A. Leibman and J. Zweier, Stoichiometric and site characteristics of the binding of iron to human transferrin, J. Biol. Chem., 1978, 253(6), 1930–1937 CrossRef CAS PubMed.
- D. H. Hamilton, E. E. Battin, A. Lawhon and J. L. Brumaghim, Using Proteins in a Bioinorganic Laboratory Experiment: Iron Loading and Removal from Transferrin, J. Chem. Educ., 2009, 86(8), 969 CrossRef.
- H. Wang, V. C. Jordan, I. A. Ramsay, M. Sojoodi, B. C. Fuchs, K. K. Tanabe, P. Caravan and E. M. Gale, Molecular Magnetic Resonance Imaging Using a Redox-Active Iron Complex, J. Am. Chem. Soc., 2019, 141(14), 5916–5925 CrossRef CAS PubMed.
- K. Djanashvili and J. A. Peters, How to determine the number of inner-sphere water molecules in Lanthanide(III) complexes by 17O NMR spectroscopy. A technical note, Contrast Media Mol. Imaging, 2007, 2(2), 67–71 CrossRef CAS PubMed.
- J. A. Peters, K. Djanashvili, C. F. G. C. Geraldes and C. Platas-iglesias, Structure, Dynamics, and Computational Studies of Lanthanide-Based Contrast Agents, in The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, 2013, pp. 209–276 Search PubMed.
- E. M. Gale, J. Zhu and P. Caravan, Direct Measurement of the Mn(II) Hydration State in Metal Complexes and Metalloproteins through 17O NMR Line Widths, J. Am. Chem. Soc., 2013, 135(49), 18600–18608 CrossRef CAS PubMed.
- A. Brausam, J. Maigut, R. Meier, P. A. Szilágyi, H.-J. Buschmann, W. Massa, Z. Homonnay and R. van Eldik, Detailed spectroscopic, thermodynamic, and kinetic studies on the protolytic equilibria of Fe(III)cydta and the activation of hydrogen peroxide, Inorg. Chem., 2009, 48(16), 7864–7884 CrossRef CAS PubMed.
- I. Bertini, F. Capozzi, C. Luchinat and Z. C. Xia, Nuclear and Electron Relaxation of Fe(OH2)63+, J. Phys. Chem., 1993, 97(6), 1134–1137 CrossRef CAS.
- M. Rohrer, H. Bauer, J. Mintorovitch, M. Requardt and H. J. Weinmann, Comparison of magnetic properties of MRI contrast media solutions at different magnetic field strengths, Invest. Radiol., 2005, 40(11), 715–724 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2309236. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt04013e |
| This journal is © The Royal Society of Chemistry 2024 |