
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnprecedented Mo3S4 cluster-catalyzed radical C–C cross-coupling reactions of aryl alkynes and acrylates†
Juanjo
Mateu-Campos
 a,
Eva
Guillamón
a,
Vicent S.
Safont
a,
Kathrin
Junge
b,
Henrik
Junge
b,
Matthias
Beller
*b and
Rosa
Llusar
*a
a,
Eva
Guillamón
a,
Vicent S.
Safont
a,
Kathrin
Junge
b,
Henrik
Junge
b,
Matthias
Beller
*b and
Rosa
Llusar
*a
aDepartament de Química Física i Analítica, Universitat Jaume I, Av. Sos Baynat s/n, 12071 Castelló de la Plana, Spain. E-mail: rosa.llusar@uji.es
bLeibniz-Institute for Catalysis e.V., Albert-Einstein Straße, 29a, 18059 Rostock, Germany
First published on 30th January 2024
Abstract
A new method for the generation of benzyl radicals from terminal aromatic alkynes has been developed, which allows the direct cross coupling with acrylate derivatives. Our additive-free protocol employs air-stable diamino Mo3S4 cubane-type cluster catalysts in the presence of hydrogen. A sulfur-centered cluster catalysis mechanism for benzyl radical formation is proposed based on catalytic and stoichiometric experiments. The process starts with the cluster hydrogen activation to form a bis(hydrosulfido) [Mo3(μ3-S)(μ-S)(μ-SH)2Cl3(dmen)3]+ intermediate. The reaction of various aromatic terminal alkynes containing different functionalities with a series of acrylates affords the corresponding Giese-type radical addition products.
Introduction
Molybdenum disulfide (MoS2) based materials have long been used in the petrochemical industry for the catalytic hydrodefunctionalization of fossil fuels.1 In the recent past, new applications of these materials have emerged, e.g. hydrodeoxygenation of biomass or fuels,2 conversion of syngas to alcohols3 or production of amines from nitroderivatives.4 On the other hand, the potential of MoS2-based materials as C–C coupling catalysts has also been reported although to a lesser extent. As an example, Ullmann C–C coupling of 2,8-dibromo-dibenzothiophenes on the surface of substoichiometric MoS2 nanosheets has been monitored by scanning tunnelling microscopy.5 Lee et al. have developed a straightforward protocol for C–H arylation to synthesize heteroarene compounds from aryl diazonium salts and furane or pyridine using two-dimensional MoS2 materials.6 Interestingly, this latter methodology can also be applied to achieve various C–C bond formation reactions. Mechanistic investigations pinpoint the MoS2 terminal sulfur atoms as the active sites for this transformation through electrostatic interaction between the nucleophilic sulfur groups and the aryl diazonium cation. Shreds of evidence for the operation of a radical mechanism are also provided; however, there are inherent difficulties to obtain mechanistic information, so the study of model systems is seen as an interesting alternative. In this context, the structural analogies and similar behaviour patterns between MoS2 and Mo3S4 clusters make these trinuclear complexes excellent models capable to emulate the solid state catalyst active sites.7–9Research carried out by some of us has shown that diamino and diimino cuboidal Mo3(μ3-S)(μ-S)3 clusters are efficient catalysts for the hydrogenation of nitroarenes and azobenzene to afford aniline as well as for the semihydrogenation of alkynes.10,11 Based on experimental and computational results, we have identified the sulfur centres as the active sites for these transformations without any direct metal participation.12 In the case of symmetrical alkynes semihydrogenation, the reaction starts with the interaction of one alkyne molecule with two of the bridging sulfurs of the cluster to form a dithiolene adduct. Then, H2 activation occurs at the third bridging sulfur in cooperation with one of the dithiolene carbon atoms. Finally, the alkene molecule is released and the Mo3S4 cluster complex recovered. In an attempt to extend our catalytic protocol to terminal alkynes, in particular phenylacetylene, we observed the formation of the C–C homocoupling products derived from the 1-phenylethyl radical, together with the semi and fully hydrogenated products. The importance of C–C bond formation in the synthesis of essential building blocks in chemistry motivated us to pursue this observation and perform this study.
In the past decades, we have witnessed significant advances in the development of improved methodologies for radical generation utilizing electrochemical,13 thermal catalytic,14 and photocatalytic approaches.15 In general, a catalyst in these reactions is often referred as “smart initiator” because it avoids the use of stoichiometric amounts of reagents.16 Representative examples of catalytic methodologies for the preparation of the 1-phenylethyl radical are given in Fig. 1.13–15,17,18
 | ||
| Fig. 1 Examples of catalytic methods for the generation of the 1-phenylethyl radical. | ||
Complementary to these known works, herein, we present an additive-free green catalytic protocol for the generation of benzyl radicals from phenylacetylene derivatives using diamino molybdenum sulfido cluster catalysts as “smart initiators” in the presence of hydrogen. A sulfur-based reaction mechanism has been tentatively proposed based on cluster reaction monitoring in combination with stoichiometric and catalytic control experiments. This protocol has also been extended to other conjugated alkynes. The catalytic system is applied to Giese-type radical addition reactions employing acrylates as radical acceptors.
Results and discussion
Catalytic model reactions and mechanistic insights
Our initial serendipitous result on benzyl radical generation using molybdenum sulfide cluster catalysts for the hydrogenation of terminal alkynes prompted us to undertake an exploratory study to identify the Mo3S4 active species. As starting point, hydrogenation of phenylacetylene was taken as a model reaction and several Mo3S4 cluster complexes, shown in Fig. 2, were tested. Initial experiments were conducted in CH3CN at 70 °C and 20 bars of H2 which are typical conditions for the semihydrogenation of symmetrical alkynes (see ESI, section 2 for catalytic protocols†).19 Apart from styrene (2a), ethylbenzene (2b), 2,3-diphenylbutanes (racemic and meso 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (2c), (Z)-1,3-diphenyl-1-butene (2d) and 1,3-diphenylbutane (2e) were identified as reaction products. Unlike symmetric alkyne hydrogenation, which is selective towards the semihydrogenation product, the major reaction product in phenylacetylene hydrogenation was the homocoupling product between 1-phenylethyl radicals (2c). Product quantification was performed by gas chromatography using an FID detector (ESI, section 1†). Compounds 2c and 2d are not commercially available and were synthesized and purified according to literature methods (see ESI, section 3 for more details†).20–23 Among the tested trinuclear clusters only the diamino [Mo3S4Cl3(dmen)3]+ (dmen = N,N′-dimethylethylenediamine) (1+) complex is an active catalyst towards the formation of C–C coupling products while no or negligible conversion leading to unidentified products is observed with the corresponding diphosphino, aminophosphino and diimino clusters. In addition, a moderate conversion of 35% is found for the triamino [Mo3S4(tacn)3]4+ (tacn = 1,4,7-triazacyclononane) complex that led to only 15% yield of the semihydrogenation product.
:1) (2c), (Z)-1,3-diphenyl-1-butene (2d) and 1,3-diphenylbutane (2e) were identified as reaction products. Unlike symmetric alkyne hydrogenation, which is selective towards the semihydrogenation product, the major reaction product in phenylacetylene hydrogenation was the homocoupling product between 1-phenylethyl radicals (2c). Product quantification was performed by gas chromatography using an FID detector (ESI, section 1†). Compounds 2c and 2d are not commercially available and were synthesized and purified according to literature methods (see ESI, section 3 for more details†).20–23 Among the tested trinuclear clusters only the diamino [Mo3S4Cl3(dmen)3]+ (dmen = N,N′-dimethylethylenediamine) (1+) complex is an active catalyst towards the formation of C–C coupling products while no or negligible conversion leading to unidentified products is observed with the corresponding diphosphino, aminophosphino and diimino clusters. In addition, a moderate conversion of 35% is found for the triamino [Mo3S4(tacn)3]4+ (tacn = 1,4,7-triazacyclononane) complex that led to only 15% yield of the semihydrogenation product.
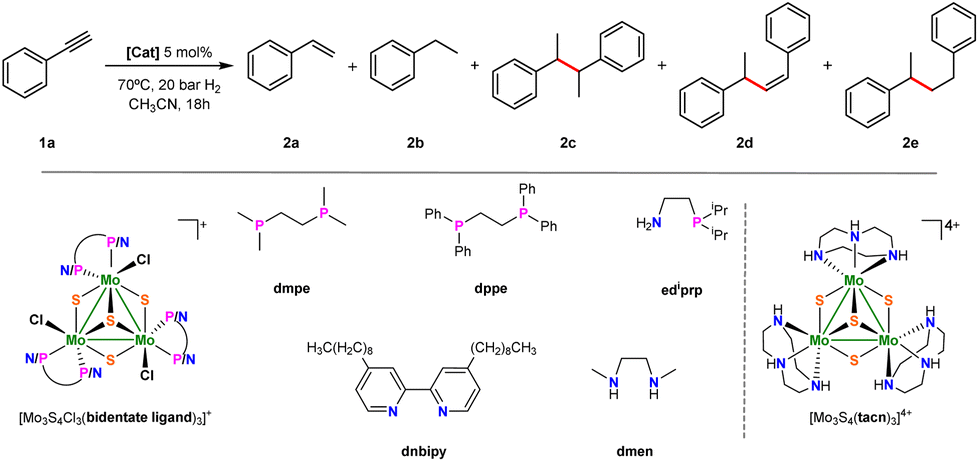 | ||
| Fig. 2 Hydrogenation of phenylacetylene and simplified structures of Mo3S4 clusters tested as catalysts. | ||
Next, we optimized the conditions using the [Mo3S4Cl3(dmen)3]Cl cluster catalyst (solvent, H2 pressure, temperature, and amount of catalyst. See ESI, section 4 for more details†) to enhance the yield of the homocoupling products. Alcohols such as methanol or ethanol are better solvents than acetonitrile towards the obtention of the C–C coupling products (Table S1†). On the other hand, decreasing the H2 pressure from 30 to 10 bars (Table 1, entries 3–5) slightly increased the yield and selectivity of the C–C coupling products (compound 2e is not detected at 10 bars). At lower H2 pressures, 1 to 2 bars (Table 1, entry 2), we did not observe the formation of products 2a to 2e, despite the 35% conversion of phenylacetylene. A 10% conversion of phenylacetylene is found in the absence of hydrogen (Table 1, entry 1). Increasing the temperature from 50 to 90 °C (Table 1, entry 3 and entries 6–9) increased the yield of the C–C coupling products although negligible differences are seen between 80 and 90 °C. Finally, the optimum amount of catalyst was determined to be 5 mol% (Table S2†). To sum up, the optimized conditions leading to 8% of styrene (2a), 10% of ethylbenzene (2b) and 70% of the C–C coupling products (2c–2d) have been determined to be 10 bar of H2 pressure, 80 °C and 5 mol% of the Mo3S4 catalyst in methanol. Other side products are assumed to be formed by poly- or oligomerization of the alkyne or styrene.24,25
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | T (°C) | P H2 (bar) | Conversionb (%) | Yieldb (%) | ||||
| 2a | 2b | 2c | 2d | 2e | ||||
| a Reaction conditions: phenylacetylene (0.1 mmol), catalyst (5 mol%), CH3OH (2 mL), 18 h. b Determined by GC using benzyl benzoate as an internal standard. | ||||||||
| 1 | 70 | 0 | 10 | 0 | 0 | 0 | 0 | 0 |
| 2 | 70 | 1–2 | 35 | 0 | 0 | 0 | 0 | 0 |
| 3 | 70 | 10 | >99 | 9 | 9 | 48 | 15 | 0 |
| 4 | 70 | 20 | >99 | 8 | 17 | 42 | 9 | 4 |
| 5 | 70 | 30 | >99 | 11 | 17 | 34 | 6 | 5 |
| 6 | 50 | 10 | 28 | 4 | 0 | 5 | 2 | 0 |
| 7 | 60 | 10 | 51 | 4 | 1 | 15 | 8 | 0 |
| 8 | 80 | 10 | >99 | 8 | 10 | 52 | 18 | 0 |
| 9 | 90 | 10 | >99 | 7 | 8 | 52 | 16 | 0 |

Recently, some of us have shown that hydrogen activation in symmetrical alkyne semihydrogenation using a diamino Mo3S4 cluster catalyst occurs through a dithiolene adduct intermediate formed upon reacting the cluster unit with the organic substrate.11 In a previous kinetic study on the reaction between [Mo3S4Cl3(dmen)3]+ and phenylacetylene in acetonitrile, we observed the formation of the [3 + 2] cycloaddition product of formula [Mo3(μ3-S)(μ-S)(μ3-SC(Ph)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Ph))Cl3(dmen)3]+, termed as type I adduct, under reversible equilibrium conditions.26 Shibahara et al. have shown that type I adducts can protonate at the carbon position with the concomitant rupture of the dithiolene C–S bond to afford the so called type II adduct.27 This adduct with two bridging sulfides available, can add a second alkyne molecule. In our case, the dithiolene type I adduct does not react with acids and we found no experimental evidence of addition of a second alkyne molecule into the Mo3S4 cluster unit.
C(Ph))Cl3(dmen)3]+, termed as type I adduct, under reversible equilibrium conditions.26 Shibahara et al. have shown that type I adducts can protonate at the carbon position with the concomitant rupture of the dithiolene C–S bond to afford the so called type II adduct.27 This adduct with two bridging sulfides available, can add a second alkyne molecule. In our case, the dithiolene type I adduct does not react with acids and we found no experimental evidence of addition of a second alkyne molecule into the Mo3S4 cluster unit.
To prove the mechanistic assumptions vide supra, we proceeded to isolate the type I cycloaddition product between [Mo3S4Cl3(dmen)3]+ and phenylacetylene. For this purpose we reacted the cluster cation with an excess of the alkyne in acetonitrile (see ESI, section 5 for experimental details†). After phenylacetylene addition, the starting green solution becomes darker and the desired [Mo3(μ3-S)(μ-S)(μ3-SC(Ph)CHS)Cl3(dmen)3]Cl addition product was precipitated with ether. The ESI-MS spectra registered in acetonitrile shows a peak centered at m/z = 889 associated to the type I adduct according to its m/z value and the calculated isotopic pattern (Fig. S1†). Next, we applied the optimized reaction conditions to the hydrogenation of the type I adduct. The product outcome is similar to that of the catalytic hydrogenation of phenylacetylene with some noticeable differences in the final product distribution 2a to 2e. The most remarkable deviations are the increase in the concentration of styrene (2a) from 8 to 26% and the absence of (Z)-1,3-diphenyl-1-butene (2d) in the reaction outcome. Formation of 2d is expected to proceed through C–C coupling between the 1-phenylethyl radical and phenylacetylene. Because this latter compound is not present in the reaction media, product 2d should not be formed.
While the phenylacetylene radical dimerization is scientifically interesting in its own, the cross coupling with other olefins is more valuable from a synthetic point of view. Hence, we decided to investigate the product distribution of the catalytic hydrogenation of phenylacetylene in the presence of acrylonitrile (3a), which is known to act as radical acceptor in Giese-type radical addition reactions.28,29 Reactions were performed with one to ten equivalents of acrylonitrile (Table S3†). Optimum results were obtained using six equivalents of acrylonitrile (Table 2, entry 1). After 18 hours of reaction, styrene (2a) and the desired 4-phenylpentanitrile (4a) were obtained in 10 and 77% yields, respectively. Incidentally, no reaction occurs when phenylacetylene is replaced by styrene, which proves that the 1-phenylethyl radical is not formed by direct styrene hydrogenation. These results suggest the operation of at least two reaction paths, leading to the semihydrogenation product (2a) and to the 1-phenylethyl radical in good agreement with our simplified mechanism proposals represented in Fig. 3b and c (vide infra).
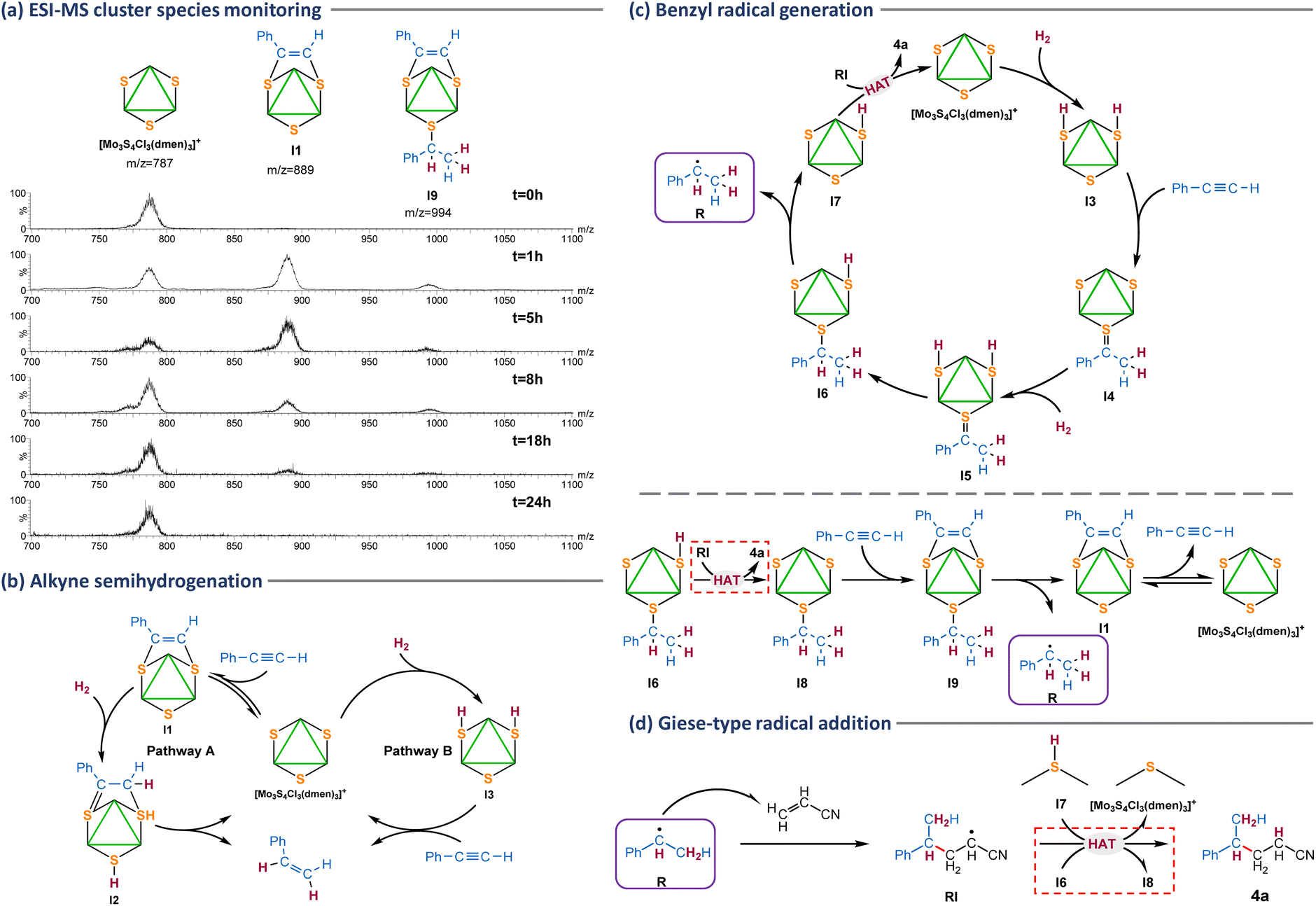 | ||
| Fig. 3 ESI mass spectrum of the catalyst after phenylacetylene hydrogenation at different times. Spectra registered from different batch experiments at 20 V in CH3CN (a). Possible reaction pathways for alkyne semihydrogenation (b). Tentative reaction mechanism for the benzyl radical generation (c). Giese-type radical addition to the activated alkene (d). | ||
|
|
||||
|---|---|---|---|---|
| Entry | Deviation | Conversionb (%) | Yieldb (%) | |
| 4a | 2a | |||
| a Reaction conditions: phenylacetylene (0.1 mmol), acrylonitrile (0.6 mmol), catalyst (5 mol%), 80 °C, H2 pressure (10 bar), CH3OH (2 mL), 18 h. b Determined by GC using benzyl benzoate as an internal standard. The yield in parenthesis corresponds to isolated products. | ||||
| 1 | None | >99 | 77 | 10 |
| 2 | CuCl (5 mol%) | 20 | 0 | 0 |
| 3 | CD3OD | 97 | 71 (68) | 15 |
| 4 | CH3OD | >99 | 61 (56) | 13 |
| 5 | Et3N (5 mol%) | 8 | 3 | 3 |
| 6 | Et3N (15 mol%) | 5 | 0 | 0 |

Cluster evolution during the catalytic hydrogenation of phenylacetylene in the presence of acrylonitrile was monitored from batch experiments at different reaction times (Fig. 3a). Initially, the only peak registered corresponds to the [Mo3S4Cl3(dmen)3]+ (m/z = 787) cluster catalyst. After one hour of reaction, this last signal coexists with a higher intensity peak due to the [Mo3(μ3-S)(μ-S)(μ3-SC(Ph)CHS)Cl3(dmen)3]+ (I1) dithiolene type I adduct (m/z = 889) and a lower intensity peak at m/z = 994 tentatively attributed to [Mo3(μ3-S)(μ-SCH(Ph)(CH3))(μ3-SC(Ph)CHS)Cl3(dmen)3]+ (I9) on the basis of its m/z value and isotopic distribution pattern. The three signals coexist during the catalytic protocol although with different intensity ratios. After eight hours, the signal due to the [Mo3S4Cl3(dmen)3]+ (m/z = 787) cluster catalyst appears as the most intense peak and it remains as the only species at the end of the reaction (t = 24 h). A similar reaction monitoring pattern is found when the reaction is done in an aprotic solvent such as acetonitrile (Fig. S2†). Incidentally, the peak at m/z = 994 tentatively attributed to [Mo3(μ3-S)(μ-SCH(Ph)(CH3))(μ3-SC(Ph)CHS)Cl3(dmen)3]+ (I9) is not observed when reaction monitoring is performed starting from the isolated type I dithiolene (I1) adduct. Thus, we conclude that intermediate I9 can only be formed when there is free alkyne in the reaction media also in agreement with our tentative mechanism proposal shown in Fig. 3c (vide infra).
Several control experiments were then performed to interrogate the system aimed to gain mechanistic knowledge. We have previously shown that catalysis inhibition upon CuCl addition is a clear signature of a sulfur-based reaction mechanism.11,12,19 Complex [Mo3S4Cl3(dmen)3]+ (m/z = 787) reacts with CuCl (Table 2, entry 2) to form a cubane type cluster [Mo3(CuCl)S4Cl3(dmen)3]+ (m/z = 896) in which the three sulfur atoms are blocked by a copper atom precluding the formation of products 4a and 2a. Deuteration studies can also be of great utility to gather mechanistic information about hydrogenation processes. In a very recent study, we were able to prove that (Z)-selective semihydrogenation of phenylacetylene can occur through two competing reaction pathways, named A and B in Fig. 3b.30 Hydrogen activation in pathway A takes place after alkyne insertion into two of the Mo3(μ3-S)(μ-S3) bridging atoms to form I1. Then, H2 activation occurs at the third bridging sulfur atom and one of the dithiolene carbon atoms (I2). On the other hand, activation of H2 in pathway B takes place at two of the bridging atom positions to form a bis(hydrosulfido) intermediate (I3). In the presence of deuterated methanol, pathway B affords the fully deuterated semihydrogenated alkene while only partial deuteration is achieved through pathway A. This is due to the rapid isotope exchange between the I3 S–H protons and the CH3OD deuterium atoms. When the Mo3S4 catalyzed hydrogenative cross coupling of phenylacetylene and acrylonitrile was performed in CD3OD or CH3OD (Table 2, entries 3 and 4), the corresponding fully deuterated coupling product is obtained (see ESI, section 9 and Fig. S3 for experimental details†), which indicates a mechanism going through the I3 intermediate. Interestingly, addition of Et3N (Table 2, entries 5 and 6) inhibits the catalysis suggesting a mechanism for H2 activation in which acidic protons are being generated providing further support on the formation of the bis(hydrosulfido) I3 intermediate. An analogous effect was observed for the catalytic hydrogenation of azobenzene using the diamino [Mo3S4Cl3(dmen)3]+ cluster or for the semihydrogenation of alkynes mediated by the imidazolyl amino [Mo3S4Cl3(ImNH2)3]+ (ImNH2 = 1-methyl-1H-imidazol-2-ylmethanamine) cluster catalysts.12,19
Based on the above results and our previous work, we tentatively propose the simplified mechanism for the benzyl radical production shown in Fig. 3c (top). The reaction begins with the activation of hydrogen by the Mo3S4 cluster to form the bis(hydrosulfido) I3 intermediate as mentioned above. Next, the benzylic carbon of the alkyne is added to the third bridging sulfur atom of the cluster while the two hydrogen atoms of the S–H groups are transferred to the terminal carbon atom of the alkyne to afford [Mo3(μ3-S)(μ-S)(μ3-SC(Ph)CH3)Cl3(dmen)3]+ (I4). Very likely this process takes place in several steps. Further hydrogenation of I4 leads to the bis(hydrosulfido) I5 intermediate which after transferring a hydrogen atom to the unsaturated carbon is transformed into intermediate I6. At this point, the benzyl radical (R) can be released through C–S cleavage to generate the hydrosulfido species I7. Then, the generated nucleophilic radical (R) undergoes a Giese-type addition to acrylonitrile (Fig. 3d) to generate a radical intermediate (RI), that subsequently reacts with the hydrosulfido intermediate I7 to afford the 4a Giese adduct via a hydrogen atom transfer (HAT) process recovering the starting cluster species.31,32 In parallel, the Giese adduct can also be formed via a HAT from I6 to afford the [Mo3(μ3-S)(μ-S)2(μ-SCH(Ph)(CH3))Cl3(dmen)3]+ (I8) intermediate with two bridging sulfurs and one bridging thiolate (shown in Fig. 3c bottom), which upon insertion of a second alkyne affords [Mo3(μ3-S)(μ-SCH(Ph)(CH3))(μ3-SC(Ph)CHS)Cl3(dmen)3]+ (I9) – detected by ESI-MS spectrometry – from which the benzyl radical is released to generate the type I cluster adduct I1, that coexists in equilibrium with the [Mo3S4Cl3(dmen)3]+ cluster catalyst.
Substrate scope of Mo3S4 catalyzed hydrogenative cross coupling reactions
Having developed a simple and green catalytic protocol for benzyl radical generation using a molybdenum sulfide cluster in the presence of hydrogen without the need of additives, we focused our attention on the synthetic utility of our protocol. For that purpose, we explored the tolerance of the reaction system by modifying both, the alkyne substrate and the radical acceptor. First, we investigated the hydrogenation of phenylacetylene (1a) in the presence of several electron-deficient alkenes 3b–3n shown in Scheme S1.† Unfortunately, low conversions were detected by GC-MS analysis in addition to a mixture of products, some of them identified as styrene (2a), the desired Giese-type radical addition product (4) and its corresponding alkene elimination product (5). On the other hand, in the case of electron-deficient 2-ethylhexyl acrylate (3n), the desired cross-coupling product was obtained in acceptable yields.Acrylates are inexpensive feedstock broadly used in polymer industry, but their use in the production of fine chemicals or valuable compounds is also a topic of interest.33 Thus, once 3n acrylate was selected as the appropriate coupling species, we conducted an optimization of the conditions to increase the yield of the respective cross-coupling product. In this case, increasing the hydrogen pressure from 10 to 20 bar at 80 °C we observed an enhancement in the yield of product 4b (Tables S4 and S5†). Next, we modified the amount of catalyst, being 7.5 mol% the optimum amount (Table S6†). Finally, we studied the use of other reductive sources. We selected syngas as an alternative to hydrogen and the use of 20 bar of syngas resulted in a decrease of both, the alkyne conversion, and the C–C coupling product yield. On the other hand, a mixture of formic acid:triethylamine (5:2) was also evaluated. In this last case, the experiment resulted in a slight conversion of the alkyne without the presence of product 4b (Table S7†).
After optimization of the Giese-type reaction conditions to afford acrylates, we studied as coupling partners three more acrylate monomers broadly used in the polymer industry, summarized in Fig. 4. In addition to 2-ethylhexyl acrylate (3n), butyl acrylate (3o) resulted in a suitable acceptor for the benzylic radical. Unfortunately, the system does not tolerate the substitution of the inner position in the alkene since methacrylate 3p was tested and the corresponding C–C product was not detected. Even though benzyl acrylate (3q) was less reactive than 3n and 3o acrylates, the coupling product was isolated in a reasonable yield.
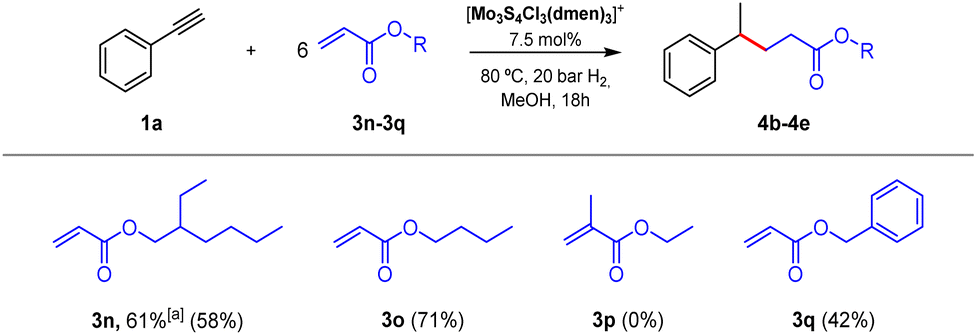 | ||
| Fig. 4 Scope of acrylates. Reaction conditions: phenylacetylene (0.1 mmol), acrylate (0.6 mmol), catalyst (7.5 mol%), 80 °C, H2 pressure (20 bar), CH3OH (2 mL), 18 h. The yield in parenthesis corresponds to isolated products. aDetermined by GC using hexadecane as an internal standard. | ||
Next, we tested different aromatic alkynes to evaluate their tolerance for this methodology (Fig. 5). First, we studied the effect of substituents in the aromatic ring using acrylate 3o as the coupling partner. For the methylated alkynes in para and meta positions (1b–1c), the coupling products were isolated in moderate yields. Meanwhile, when the ortho-methylated alkyne (1d) is used as starting material, a decrease in the yield took place, and the final compound is obtained as a mixture with the alkene-type side product. The system also tolerates phenyl substituent in the para position (1e) to obtain the final product in 59% yield. We also tested the reactivity of a series of halide-containing alkynes (1f–1h), which provided the final products in moderate isolated yields. In addition, alkynes containing electron-rich groups such as 4-OMe and 3-NH2 (1i–1j) were evaluated, and the C–C products were isolated in high yields. In contrast, when alkynes containing electron-withdrawing groups like 4-CF3 and 4-COCH3 (1k–1l) are used, a decrease in the final product yields is observed. This behaviour could be explained by the role of the donor or withdrawing group in stabilizing the radical on the ring.
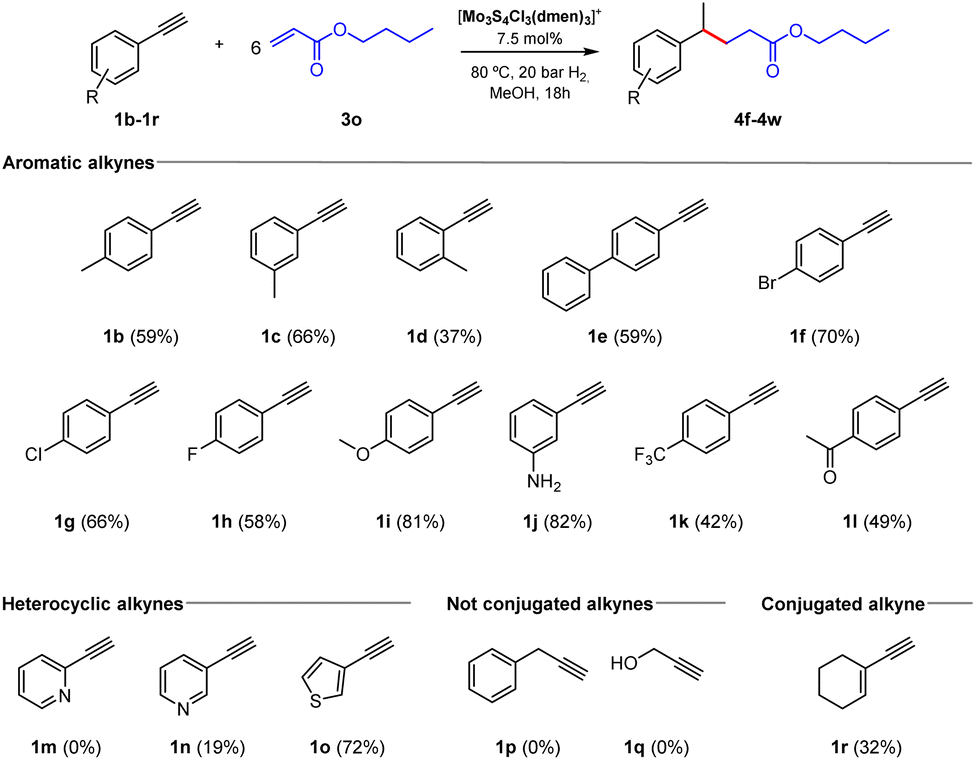 | ||
| Fig. 5 Scope of terminal alkynes. Reaction conditions: alkyne (0.1 mmol), butyl acrylate (0.6 mmol), catalyst (7.5 mol%), 80 °C, H2 pressure (20 bar), CH3OH (2 mL), 18 h. The yield in parenthesis corresponds to isolated products. | ||
Interestingly, this procedure was also extended to alkynes containing heterocycles. Unfortunately, when we used the ortho-substituted pyridine (1m) we were not able to detect the coupling product. Nonetheless, the product was isolated in low yield starting from the meta-substituted pyridine (1n) and the use of 3-ethynylthiophene (1o) resulted in the formation of the C–C coupling product in good yield. Finally, wondering about the need for a conjugated alkyne able to stabilize the generated radical, we investigated a couple of non-conjugated alkynes (1p–1q), but dreadfully we could not detect the formation of the corresponding C–C coupling products. Surprisingly, when we tested the conjugated enyne (1r) we isolated the corresponding coupling product in 32% yield. This experimental evidence supports the requirement of using a conjugated alkyne capable of stabilizing the generated radical intermediates and allowing them to engage in Giese reactions.34,35
Conclusions
A simple and additive-free green protocol for the regioselective cross coupling reaction of aromatic alkynes and olefins has been developed. Key for this transformation is the generation of radicals from terminal aromatic alkynes and molecular hydrogen using an air-stable cuboidal Mo3S4 diamino cluster as smart initiator. The cross-coupling process proceeds via a sulfur-based cluster catalysis mechanism. Our tentative mechanistic proposal for the benzyl radical generation from phenylacetylene starts with the cluster hydrogen activation to form a bis(hydrosulfido) [Mo3(μ3-S)(μ-S)(μ-SH)2Cl3(dmen)3]+ intermediate. The rapid exchange between S–H group hydrogen atoms and CH3OD solvent deuterium atoms results in the formation of deuterated PhĊD-CD2H benzyl radicals. Interestingly, this novel catalytic protocol can be applied to Giese-type radical addition reactions employing different acrylates as radical acceptors. The main advantages of this system include the use of widely available substrates (terminal alkynes and acrylates), as well as the use of inexpensive hydrogen as the reducing agent. Furthermore, the tolerance of this catalytic protocol enables the use of aromatic terminal alkynes containing different functionalities, but also the use of cost-effective acrylate monomers widely used in the polymer industry.Author contributions
R.L. and M. B. directed the project. J.M.C. performed all experiments. E.G. and V.S.S. supervised the catalytic protocol optimization, control and mechanistic experiments. K.J. and H.J. supervised the substrate scope methodology. R.L. and J.M.C. wrote the original manuscript and ESI. M.B. contributed to the final version of the manuscript. E.G., V.S.S., K.J., and H.J reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support of the Spanish Ministerio de Ciencia e Innovación (PID2022-141089NB-I00), Generalitat Valenciana (Grant CIAICO/2021/122) and Universitat Jaume I (UJI-B2021-29 and UJI-B2022-56) is gratefully acknowledged. The authors also thank the Serveis Centrals d'Instrumentació Científica (SCIC) of the Universitat Jaume I for providing us with characterization techniques. Furthermore, we thank Leibniz-Institute for Catalysis for the facilities. K. J., H. J. and M. B. acknowledge financial support from the State of Mecklenburg and Western Pomerania. J. M. C. would like to acknowledge the funding received from the Universitat Jaume I though its Research Stay Grants (E-2022-31) and a predoctoral fellowship.References
- C. Chizallet and P. Raybaud, Catal. Sci. Technol., 2014, 4, 2797–2813 RSC.
- M. Grilc, G. Veryasov, B. Likozar, A. Jesih and J. Levec, Appl. Catal., B, 2015, 163, 467–477 CrossRef CAS.
- J. Hu, L. Yu, J. Deng, Y. Wang, K. Cheng, C. Ma, Q. Zhang, W. Wen, S. Yu, Y. Pan, J. Yang, H. Ma, F. Qi, Y. Wang, Y. Zheng, M. Chen, R. Huang, S. Zhang, Z. Zhao, J. Mao, X. Meng, Q. Ji, G. Hou, X. Han, X. Bao, Y. Wang and D. Deng, Nat. Catal., 2021, 4, 242–250 CrossRef CAS.
- Z. Li, D. Zhang, J. Ma, D. Wang and C. Xie, Mater. Lett., 2018, 213, 350–353 CrossRef CAS.
- Y. Bao, M. Yang, S. J. R. Tan, Y. P. Liu, H. Xu, W. Liu, C. T. Nai, Y. P. Feng, J. Lu and K. P. Loh, J. Am. Chem. Soc., 2016, 138, 14121–14128 CrossRef CAS PubMed.
- E. Hwang, S. M. Lee, S. Bak, H. Hwang, H. M. Kim and H. Lee, Tetrahedron Lett., 2018, 59, 3969–3973 CrossRef CAS.
- M.-L. Grutza, A. Rajagopal, C. Streb and P. Kurz, Sustainable Energy Fuels, 2018, 2, 1893–1904 RSC.
- Y. Yan, B. Xia, Z. Xu and X. Wang, ACS Catal., 2014, 4, 1693–1705 CrossRef CAS.
- J. Al Cheikh, R. Zakari, A. C. Bhosale, A. Villagra, N. Leclerc, S. Floquet, P. C. Ghosh, A. Ranjbari, E. Cadot, P. Millet and L. Assaud, Mater. Adv., 2020, 1, 430–440 RSC.
- E. Pedrajas, I. Sorribes, A. L. Gushchin, Y. A. Laricheva, K. Junge, M. Beller and R. Llusar, ChemCatChem, 2017, 9, 1128–1134 CrossRef CAS.
- A. G. Algarra, E. Guillamón, J. Andrés, M. J. Fernández-Trujillo, E. Pedrajas, J. Á. Pino-Chamorro, R. Llusar and M. G. Basallote, ACS Catal., 2018, 8, 7346–7350 CrossRef CAS.
- E. Guillamón, M. Oliva, J. Andrés, R. Llusar, E. Pedrajas, V. S. Safont, A. G. Algarra and M. G. Basallote, ACS Catal., 2021, 11, 608–614 CrossRef.
- H. Shimakoshi, Z. Luo, K. Tomita and Y. Hisaeda, J. Organomet. Chem., 2017, 839, 71–77 CrossRef CAS.
- H. Jiang, R. Lu, X. Luo, X. Si, J. Xu and F. Lu, Chem. – Eur. J., 2021, 27, 1292–1296 CrossRef CAS PubMed.
- N. Ishida, Y. Masuda, Y. Imamura, K. Yamazaki and M. Murakami, J. Am. Chem. Soc., 2019, 141, 19611–19615 CrossRef CAS PubMed.
- M. Kischkewitz, F. W. Friese and A. Studer, Adv. Synth. Catal., 2020, 362, 2077–2087 CrossRef PubMed.
- H. J. Zhang, F. Su and T. B. Wen, J. Org. Chem., 2015, 80, 11322–11329 CrossRef CAS PubMed.
- X.-L. Su, S.-P. Jiang, L. Ye, G.-X. Xu, J.-J. Chen, Q.-S. Gu, Z.-L. Li and X.-Y. Liu, Tetrahedron, 2021, 89, 132152 CrossRef CAS.
- M. Gutiérrez-Blanco, E. Guillamón, V. S. Safont, A. G. Algarra, M. J. Fernández-Trujillo, K. Junge, M. G. Basallote, R. Llusar and M. Beller, Inorg. Chem. Front., 2023, 10, 1786–1794 RSC.
- A. Lei and X. Zhang, Org. Lett., 2002, 4, 2285–2288 CrossRef CAS PubMed.
- F. D. Greene, M. A. Berwick and J. C. Stowell, J. Am. Chem. Soc., 1970, 92, 867–874 CrossRef CAS.
- D. Müller and A. Alexakis, Chem. – Eur. J., 2013, 19, 15226–15239 CrossRef PubMed.
- V. Krasovskaya, A. Krasovskiy, A. Bhattacharjya and B. H. Lipshutz, Chem. Commun., 2011, 47, 5717–5719 RSC.
- F. Mäsing, X. Wang, H. Nüsse, J. Klingauf and A. Studer, Chem. – Eur. J., 2017, 23, 6014–6018 CrossRef PubMed.
- F. Chen, C. Kreyenschulte, J. Radnik, H. Lund, A.-E. Surkus, K. Junge and M. Beller, ACS Catal., 2017, 7, 1526–1532 CrossRef CAS.
- E. Pedrajas, J. A. Pino-Chamorro, M. Ferrer, M. J. Fernández-Trujillo, R. Llusar, M. Martínez, M. G. Basallote and A. G. Algarra, Int. J. Quantum Chem., 2020, 120, 26353 CrossRef.
- Y. Ide, M. Sasaki, M. Maeyama and T. Shibahara, Inorg. Chem., 2004, 43, 602–612 CrossRef CAS PubMed.
- D. Birnthaler, R. Narobe, E. Lopez-Berguno, C. Haag and B. König, ACS Catal., 2023, 13, 1125–1132 CrossRef CAS.
- Y. Cai, S. Chatterjee and T. Ritter, J. Am. Chem. Soc., 2023, 145, 13542–13548 CrossRef CAS PubMed.
- M. Gutiérrez-Blanco, A. G. Algarra, E. Guillamón, M. J. Fernández-Trujillo, M. Oliva, M. G. Basallote, R. Llusar and V. S. Safont, Inorg. Chem., 2024, 63, 1000–1009 CrossRef PubMed.
- D. M. Kitcatt, S. Nicolle and A.-L. Lee, Chem. Soc. Rev., 2022, 51, 1415–1453 RSC.
- W. Zhou, I. A. Dmitriev and P. Melchiorre, J. Am. Chem. Soc., 2023, 145, 25098–25102 CrossRef CAS PubMed.
- N. Rao, Y.-Z. Li, Y.-C. Luo, Y. Zhang and X. Zhang, ACS Catal., 2023, 13, 4111–4119 CrossRef CAS.
- A. L. G. Kanegusuku and J. L. Roizen, Angew. Chem., Int. Ed., 2021, 60, 21116–21149 CrossRef PubMed.
- T. Drennhaus, D. Leifert, J. Lammert, J. P. Drennhaus, K. Bergander, C. G. Daniliuc and A. Studer, J. Am. Chem. Soc., 2023, 145, 8665–8676 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization and NMR of isolated products. See DOI: https://doi.org/10.1039/d3dt04121b |
| This journal is © The Royal Society of Chemistry 2024 |