
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and luminescent properties of hetero-bimetallic and hetero-trimetallic Ru(II)/Au(I) or Ir(III)/Au(I) complexes†
Richard C.
Knighton‡
 and
Simon J. A.
Pope
*
and
Simon J. A.
Pope
*
School of Chemistry, Cardiff University, Main Building, Cardiff, CF10 3AT, UK. E-mail: popesj@cardiff.ac.uk; Tel: +44 (0)29-20879316
First published on 6th February 2024
Abstract
A series of Ru(II) and Ir(III) based photoluminescent complexes were synthesised that incorporate an ancillary 2,2′-bipyridine ligand adorned with either one or two pendant N-methyl imidazolium groups. These complexes have been fully characterised by an array of spectroscopic and analytical techniques. One Ir(III) example was unequivocally structurally characterised in the solid state using single crystal X-ray diffraction confirming the proposed formulation and coordination sphere. These complexes were then transformed into their heterometallic, Au(I)-containing, analogues in two steps to yield either bi- or trimetallic complexes that integrate {Au(PPh3)}+ units. X-ray diffraction was used to corroborate the solid state structure of the hetero bimetallic complex, based upon a Ru(II)–Au(I) species. The heterometallic complexes all displayed red photoluminescent features (λem = 616–629 nm) that were consistent with the parent Ru(II) or Ir(III) lumophores in each case. The modulation of the emission from the Ru(II)–Au(I) complexes was much more strongly evident than for the Ir(III)–Au(I) analogues, which is ascribed to the inherent differences in the specific triplet excited state character of the emitting states within each heterometallic species.
Introduction
Interest in discrete, molecular, heterometallic complex species has been driven by a number of research disciplines including cooperative1 photocatalysis,2 biological imaging3 and medicinal applications,4 including for cancer therapy.5 Our interests, represented herein, are focussed upon the investigation of the photoluminescent properties of heterometallic complexes that integrate Au(I) complex moieties. Of particular relevance to this aim are the limited reports on mixed-metal, luminescent Ru(II)/Au(I) complexes that have been described previously. For example, Knoll et al. reported a heterobimetallic Ru(II)/Au(I) system, combining the {Ru(bipy)3}2+ photoactive unit with a pendant {(R3P)AuCl} moiety, which showed visible light mediated catalytic activity.6 Similarly, structurally related bimetallic Ru(II)/Au(I) complexes have been developed for photoredox catalysis, wherein the combination of the metals provided an enhancement in catalytic performance.7 Heterobimetallic Ru(II)/Au(I) have also been reported for biological studies,8 including a bifunctional system where the Au(I) portion (possessing a structure closely related to auranofin) showed anti-cancer activity while the {Ru(bipy)3}2+ unit of the molecule was utilised as a lumophore in cell imaging, thus providing a means of tracking the agent within live cells through triplet metal-to-ligand charge transfer (3MRuLCT) emission.9 In fact heterometallic complexes, in general, have been identified as very promising candidates for theranostic applications.10 The therapeutic potential of histidine targeting Ru(II)/Au(I) bimetallics (albeit with a non-emissive Ru(II) constituent) has also been investigated by Dyson and co-workers.11The approaches to ligand design for Ru(II)/Au(I) bimetallic species must consider the different types of donor suitable for the Au(I) moiety, particularly if biological applications are of interest. A photoactive {Ru(bipy)3}2+ unit has been covalently linked to a pendant N-heterocyclic carbene (NHC) ligand which was then coordinated to {Au(I)Cl}. The study showed that the resultant bimetallic compounds can exploit the 3MLCT luminescence of the Ru(II) component which again allows cell imaging studies.12
In contrast to several reports of luminescent Re(I)/Au(I) hetero organometallics,3,13 the first example of which was published in 2004,14 mixed Ir(III)/Au(I) complexes are quite rare. Notably, a recent report described the potential of peptide conjugates of Ir(III)/Au(I) bimetallics as prospective theranostic agents, wherein the {Ir(C^N)2(N^N)}+ portion of the species provided the visible luminescence essential for cell imaging studies. The study proposed that the precise nature of the Au(I)–peptide bond could play a crucial role in the biological behaviour, and intracellular distribution, of the bimetallic complex.15 Another example investigated how a bis(diphenylphosphino)methanide can be utilised as a bridging ligand between Ir(III) and Au(I) yielding luminescent, biologically active dimetallics.16 One other example shows how benzene dithiolate can be used as an anciallary ligand on Ir(III) and support Au(I) coordination.17 A number of other reports describe Ir/Au dimetallics, but for dual catalysis rather than photophysical applications.18
Motivated by these recent studies, the aims of the current work were therefore to investigate the syntheses and spectroscopic properties of heterobi- and heterotrimetallic complexes that combine either Ru(II)/Au(I) or Ir(III)/Au(I) constituents. The comparison of the two systems allowed an investigation of the influence of the cationic Au(I) fragment upon the photophysical properties of the parent Ru(II) or Ir(III) photoactive units.
Results and discussion
Synthesis and characterisation of the monometallic complexes
The mono-functionalised N-methyl imidazolium ligand (3-([2,2′-bipyridin]-5-ylmethyl)-1-methyl-1H-imidazol-3-ium chloride; bipy-Im1) was synthesised according to previous methods,19 while the bi-functionalised analogue (3,3′-([2,2′-bipyridine]-5,5′-diylbis(methylene))bis(1-methyl-1H-imidazol-3-ium) dichloride; bipy-Im2) was obtained from the reaction of 5,5′-bischloromethyl-2,2′-bipyridine and N-methylimidazole.20,21 The coordination chemistry of these ligands with Ru(II) to yield [Ru(bipy)2(bipy-Im1)][PF6]3 and [Ru(bipy)2(bipy-Im1)][PF6]4 (Scheme 1), the latter previously reported by the authors.18 | ||
| Scheme 1 Structures of the N-methyl imidazolium functionalised monometallic Ru(II) and Ir(III) complexes (black) synthesised in this study, together with relevant reference compounds (grey). | ||
The imidazolium functionalized Ir(III) complexes (Scheme 1) were synthesised from Ir(III) species containing 2-phenylquinoxaline-based ligands that we have previously shown to be very effective cyclometalating agents for Ir(III).22 Specifically, 2,6,7-trimethyl-3-phenylquinoxaline (hereafter abbreviated to Me3quinox) was selected as the cyclometalating ligand for Ir(III). The dimetallic precursor [Ir(Me3quinox)2(μ2-Cl)]2, which possesses sufficient solubility to permit characterisation via1H NMR spectroscopy, was converted to the bis-acetonitrile adduct [Ir(Me3quinox)2(MeCN)]PF6 (Fig. S2 and S3, ESI†). Subsequent reaction with either bipy-Im1 or bipy-Im2 was achieved under ambient conditions. This approach yielded two Ir(III) complexes, [Ir(Me3quinox)(bipy-Im1)][PF6]2 and [Ir(Me3quinox)(bipy-Im2)][PF6]3 (Scheme 1) that were isolated and fully characterised. Firstly, high resolution mass spectrometry (HRMS) confirmed the formation of the proposed complexes with appropriate m/z values consistent with the overall charges in each case (Fig. S7 and S10, ESI†).
NMR spectral data was employed to confirm the formation of [Ir(Me3quinox)(bipy-Im1)][PF6]2 and [Ir(Me3quinox)(bipy-Im2)][PF6]3. The presence of different methyl groups within the ligand structures, provide rich information in the aliphatic regions of the 1H NMR spectra. For comparison, the precursor [Ir(Me3quinox)2(MeCN)2]PF6 was also analysed and revealed three distinct methyl environments between 2.5–3.2 ppm associated with the cyclometalated ligand. Upon formation of [Ir(Me3quinox)(bipy-Im2)][PF6]3 the three methyl resonances of the C^N ligand, highlighting the symmetrical nature of the bipyridine ligand, became more different, appearing at 1.72–3.24 ppm (Fig. 1). An additional singlet at 3.89 ppm, associated with the equivalent imidazolium methyl groups, was also observed. The methylene CH2 group, that links the imidazolium to the bipyridine, showed a coupling pattern consistent with diastereotopic protons, indicative of restricted rotation of the imidazolium group(s). For [Ir(Me3quinox)(bipy-Im1)][PF6]2 the introduction of an unsymmetrical auxiliary ligand correspondingly led to the appearance of subtle inequivalence across the methyl resonances (1.72–3.24 ppm) of the quinoxaline ligand. Again a singlet at 3.89 ppm was observed for the imidazolium methyl group, and a diastereotopic pattern for the CH2 linker were also present. All relevant NMR spectral data is presented in the ESI (Fig. S4–S10†).
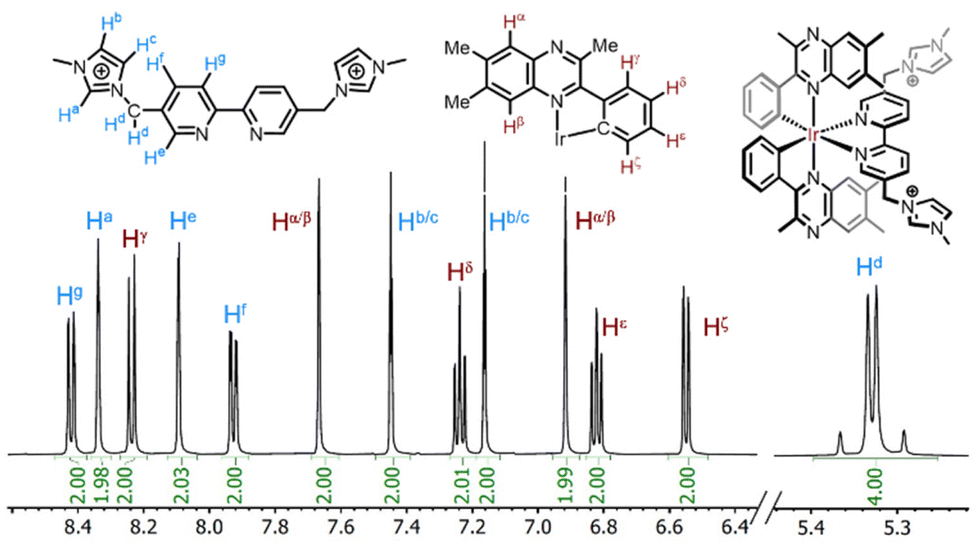 | ||
| Fig. 1 1H NMR spectrum (500 MHz, 293 K, CD3CN; expansion of aromatic signals shown above) of [Ir(Me3quinox)(bipy-Im2)][PF6]3. The diastereotopic CH2 resonance is noted at 5.37–5.29 ppm. | ||
Single crystal X-ray diffraction
Single crystals suitable for X-ray crystallographic studies were obtained for the [Ir(Me3quinox)2(MeCN)2]BF4 precursor and [Ir(Me3quinox)(bipy-Im2)][PF6]3. Crystals of the bis-acetonitrile precursor were obtained by liquid diffusion of Et2O into an MeCN solution, while crystals of [Ir(Me3quinox)(bipy-Im2)][PF6]3 were obtained by liquid diffusion of pentane into a CH2Cl2/MeOH (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution. Both structures (Fig. 2, 3 and ESI Fig. S28–S30 and S32–S34†) showed the expected formulations and ligand configurations within the complexes. In each case the cyclometalating ligands are coordinated in a way which imposes a mutually cis arrangement for the Ir–C bonds. In [Ir(Me3quinox)2(MeCN)2]PF6 the coordinated solvent molecules are mutually cis as expected. For [Ir(Me3quinox)(bipy-Im2)][PF6]3 the imidazolium moieties are positioned away from the cationic metal centre. The bond lengths and bond angles that describe the distorted octahedral coordination sphere are closely comparable with little variance induced by the addition of cationic imidazolium units (crystal structure metrics can be found in the ESI Tables S1–S3†). In fact, these structural parameters are very closely comparable to previous relevant examples,23 including those based upon this type of 2-phenylquinoxaline cyclometalating ligand at Ir(III).24
:1) solution. Both structures (Fig. 2, 3 and ESI Fig. S28–S30 and S32–S34†) showed the expected formulations and ligand configurations within the complexes. In each case the cyclometalating ligands are coordinated in a way which imposes a mutually cis arrangement for the Ir–C bonds. In [Ir(Me3quinox)2(MeCN)2]PF6 the coordinated solvent molecules are mutually cis as expected. For [Ir(Me3quinox)(bipy-Im2)][PF6]3 the imidazolium moieties are positioned away from the cationic metal centre. The bond lengths and bond angles that describe the distorted octahedral coordination sphere are closely comparable with little variance induced by the addition of cationic imidazolium units (crystal structure metrics can be found in the ESI Tables S1–S3†). In fact, these structural parameters are very closely comparable to previous relevant examples,23 including those based upon this type of 2-phenylquinoxaline cyclometalating ligand at Ir(III).24
 | ||
| Fig. 2 Single crystal X-ray diffraction structure of complex [Ir(Me3quinox)2(MeCN)2]BF4 (H-atoms and counter anion omitted for clarity; ellipsoids plotted at the 50% probability level). CCDC no. 2271292.† | ||
 | ||
| Fig. 3 Single crystal X-ray diffraction structure of complex [Ir(Me3quinox)2(bipy-Im2)][PF6]3 (H-atoms and counter anions omitted for clarity; ellipsoids plotted at the 50% probability level). CCDC no. 2271291.† | ||
Reactions with AuCl(PPh3): synthesis and characterisation of the heterometallic complexes
Four luminescent cationic complexes, [Ir(Me3quinox)(bipy-Im1)][PF6]2, [Ir(Me3quinox)(bipy-Im2)][PF6]3 and the related Ru(II) species [Ru(bipy)2(bipy-Im1)][PF6]2 and [Ru(bipy)2(bipy-Im2)][PF6]3, each possessing the pendant N-methyl imidazolium moieties, were therefore available for investigation as reagents for the formation of heterometallic complexes with Au(I). From a synthetic perspective, the following strategy was adopted and shown to be compatible with both the Ru(II) and Ir(III) systems. The imidazolium appended complexes were reacted with Ag2O in MeCN to yield the Ag(I)–NHC intermediate(s), which was then immediately reacted with AuCl(PPh3)25 at room temperature in DCM, via a transmetallation approach, to successfully yield the four heterometallic species [Ir–Au][PF6]2, [Ir–Au2][PF6]3, [Ru–Au][PF6]3 and [Ru–Au2][PF6]4 (structures shown in Scheme 2). Further purification, where necessary, was achieved using column chromatography (details provided in the Experimental section). AuCl(PPh3) was chosen as the preferred Au(I) source as it introduces the highly lipophilic triphenylphosphine moiety that aids solubility, provides a convenient 31P NMR spectroscopic handle that allows the direct interrogation of the Au(I) coordination sphere within the hetero-bi- and hetero-trimetallic products, and maintains the same overall charge of the products when compared to the precursors. | ||
| Scheme 2 Structures of the heterometallic complexes synthesised in this study. | ||
Key NMR spectral data to support the coordination of the NHC donor to Au(I) was provided, firstly, through the 31P NMR data which showed a single downfield peak ca. +40 ppm in each case, which is consistent with previous reports on mixed-ligand [Au(NHC)PPh3]+ species,26 and a characteristic septet associated with hexafluorophosphate counter ion (Fig. 4 and Fig. S11–S26†). Secondly, the 13C{1H} NMR spectra showed a downfield signal attributed to the coordinated carbon atom of the NHC ligand at around 188 ppm; this resonance appeared as a doublet with 2JPC coupling (128 Hz in all cases), which is therefore consistent with coordination to the {Au–PPh3} moiety.
 | ||
| Fig. 4 (a) 31P{1H} NMR spectrum (202 MHz, 293 K, (CD3)2CO) of [Ir–Au][PF6]2. (b) HRMS comparing the experimental (bottom) and theoretical isotope pattern for [Ir–Au][PF6]2. | ||
Comparison of the 13C carbenic resonances and 31P shifts (Fig. 5) reveals only a small covariance between the sets of [M − Au]n+ and [M − Au2]n+ complexes. Larger deviations were observed comparing Ru to Ir, indicating that changes in the respective spectra of the {(NHC)AuPPh3} moiety are probably determined by the overall charge of the Ru/Ir fragment.
 | ||
| Fig. 5 (a) 31P{1H} NMR (202 MHz, 293 K, (CD3)2CO) spectra and (b) carbene region of 13C{1H} NMR (126 MHz, 293 K, (CD3)2CO) spectra of the family of [M − Aux]n+ complexes. The latter shows the 2JPC coupling (ca. 128 Hz). | ||
The 1H NMR data showed well-resolved signals that supported the formation of the complexes, albeit with the triphenylphosphine protons superimposed with the other aromatic resonances. In the trimetallic cases of [Ir–Au2][PF6]3 and [Ru–Au2][PF6]4 the NMR spectral data confirmed the time-averaged equivalence of both Au–NHC units. Conversion to the Au–NHC analogues resulted in a downfield shift (5–5.5 ppm), and a broader ppm range, in the appearance of the diastereotopic signals for the methylene CH2 group. For [Ir–Au][PF6]2 and [Ru–Au][PF6]3 the unsymmetrical nature of the complexes resulted in a similar pattern of signals as observed for the Ir/Ru precursors. Excellent HRMS data was obtained in all cases (see inset Fig. 4, and Fig. S14, S18, S22, S26†) consistent with the appropriate charge of the complex cation and also the isotopic distribution associated Ru or Ir.
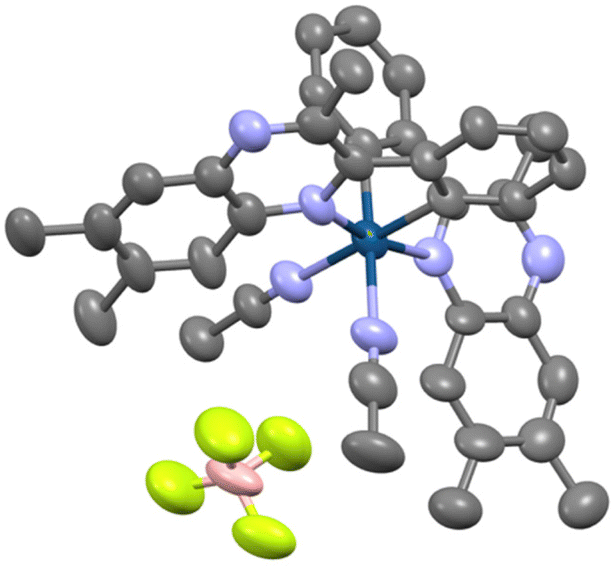
Single crystals of [Ru–Au][PF6]3 suitable for X-ray crystallographic studies were obtained from a MeOH/MeCN solution of the complex. The structure (Fig. 6 and ESI Table S5†) showed the anticipated molecular arrangement predicted by solution state spectroscopies. The coordination sphere of the Au(I) atom is pseudo linear (177.94(12)°) with both Au–P (2.2884(9) Å) and Au–C (2.034(3) Å) bond lengths that are closely comparable to previously reported structure on a [(PPh3)Au(NHC)]+ complex (Au–P = 2.274 Å; Au–C = 2.034 Å; ∠P–Au–C = 176.96°).27 The structure of [Ru–Au][PF6]3 showed no evidence of intermolecular aurophilic interactions, presumably due to the significant steric bulk that surrounds each Au(I) and the inherent electrostatic repulsion of the overall +3 charge. The coordination sphere of Ru(II) is as expected for a polypyridine species, and the bond lengths and angles show only a small difference compared to monometallic Ru(II) 2,2′-bipyridine imidazolium species (Table S5†).21
 | ||
| Fig. 6 Single crystal X-ray diffraction structure of complex [Ru–Au][PF6]3 (H-atoms and counter anions omitted for clarity; ellipsoids plotted at the 50% probability level). CCDC no. 2295692.† | ||
Electronic properties
The UV-vis absorption properties were assessed on dilute aerated MeCN solutions using a range of wavelengths to address both ligand-centred and metal-to-ligand charge transfer (MLCT) type transitions. Firstly, the parent Ru(II) and Ir(III) monometallic species were measured and showed, in each case, the expected collection of intense (ε > 10000 M−1 cm−1) ligand based bands in the UV region, attributed to different spin-allowed (S0 → Sn) π → π* transitions that belong to the different aromatic ligands (Fig. S36†). In addition, the visible region was characterised by a typical MLCT absorption for the Ru(II) species at ca. 451 nm, which is directly comparable to the benchmark species, [Ru(bipy)3]2+.28 The monometallic Ir(III) species display a broader (with the potential for spin–forbidden absorptions, S0 → Tn, arising from the increased spin orbit coupling of Ir), but weaker absorption in the visible region, which, as noted previously, is likely to comprise different CT characters, but maintain a strong MLCT contribution to the absorption.
The spectroscopic data showed that conversion to the corresponding heterometallic species [M–Aun] resulted in absorption spectra that were very subtly modulated, but nonetheless retained the key features identified for the parent monometallic species in each case (Fig. 7). Of course, while the imidazolium unit becomes a neutral NHC donor upon coordination, the cationic {Au–PPh3} fragment ensures that the overall charges of the respective heterometallic species are the same as the relevant monometallic precursors.
 | ||
| Fig. 7 Absorption spectra for the family of heterometallic M–Au complexes (293 K, aerated MeCN, 10−5 M). | ||
Comparison of the absorption spectra of [Ru–Au][PF6]3 and [Ru–Au2][PF6]4 show broadly similar spectra (Fig. 8), but with minor deviations in the low energy shoulder of the 1MLCT band and a bathochromic tendency in the ligand-centred π → π* transition at ca. 290 nm for [Ru–Au2][PF6]4. The analogous [Ir–Au][PF6]2 and [Ir–Au2][PF6]3 complexes possess spectra that are, again, comparable to one another, but distinct from the [Ru–Aun] species. Notably, the [Ir–Aun] complexes possess a broader absorption in the visible region which extends >500 nm and is attributed to spin-allowed MLCT/LLCT/ILCT contributions noted previously for 2-phenylquinoxaline complexes of Ir(III).19 A strong absorption band at 350–400 nm is likely to be due to a combination of ligand-centred and CT transitions.
 | ||
| Fig. 8 Normalised excitation and emission spectra for the polycationic iridium complexes (293 K, aerated MeCN, 10−5 M). | ||
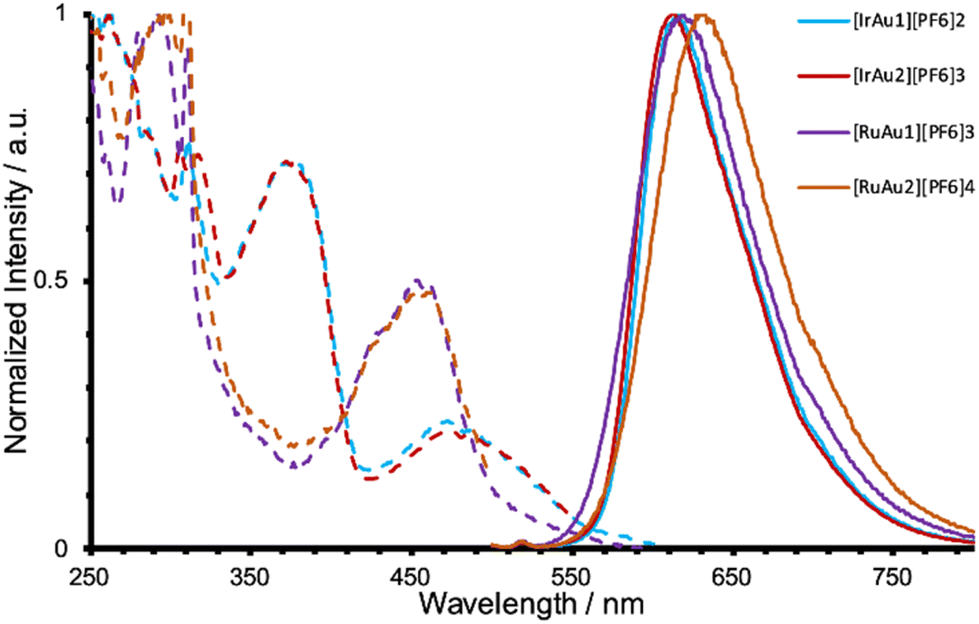
The photophysical data of the synthesised complexes were ascertained at room temperature in dilute aerated MeCN solution and are contextualised with the parameters for the known reference compounds, [Ru(bipy)3][PF6]2 and [Ir(Me3quinox)2(bipy)]PF6 (Table 1). The monometallic cationic complexes were firstly assessed, each demonstrating a broad featureless emission in the red part of the visible region. The Ru(II) species showed a characteristic 3MLCT emission band which is progressively bathochromically shifted from 610 nm to 633 nm to 654 nm upon the addition of N-methyl imidazolium groups to the bipy ligand. As noted previously, this implies a modest stabilisation of the 3MLCT excited state upon addition of the cationic fragments.
| Complexa | λ MLCT (M−1 cm−1)/nm | λ em /nm | τ obs /ns | Φ /% |
|---|---|---|---|---|
| a All measurements obtained in aerated MeCN at 293 K. b 1 × 10−5 M. c λ ex = 400 nm. d Observed lifetime, λex = 295 nm. e Using [Ru(bipy)3][PF6]2 in aerated MeCN as a reference (Φ = 0.018),29 errors are estimated at 15%. | ||||
| [Ir(Me3quinox)2(bipy)]PF6 | 474 (4800) | 617 | 450 | 5.1 |
| [Ir(Me3quin)2(bipy-Im1)][PF6]2 | 466 (5700) | 619 | 350 | 6.2 |
| [Ir(Me3quin)2(bipy-Im2)][PF6]3 | 468 (5500) | 619 | 378 | 5.6 |
| [Ir–Au][PF6]2 | 472 (4900) | 616 | 379 | 9.7 |
| [Ir–Au2][PF6]3 | 473 (5400) | 613 | 494 | 9.3 |
| [Ru(bipy)3][PF6]2 | 450 (15300) |
610 | 157 | 1.8 |
| [Ru(bipy)2(bipy-Im1)][PF6]3 | 450 (14000) |
633 | 218 | 1.7 |
| [Ru(bipy)2(bipy-Im2)][PF6]4 | 452 (13100) |
654 | 236 | 1.4 |
| [Ru–Au][PF6]3 | 451 (12800) |
619 | 235 | 2.7 |
| [Ru–Au2][PF6]4 | 451 (12100) |
629 | 325 | 3.0 |
In contrast, [Ir(Me3quin)2(bipy-Im1)][PF6]2 and [Ir(Me3quin)2(bipy-Im2)][PF6]3 possess superimposable emission profiles (Fig. 8) which are also closely comparable to the benchmark species, [Ir(Me3quinox)2(bipy)]PF6: the increase in overall charge does not strongly modulate the emission properties. In contrast to the Ru(II) examples discussed above, this reflects the nature of the emitting excited emitting state on the Ir(III) complexes, the locale of which are centred on the 2-phenylquinoxaline(s) (i.e. the bipy-Im ligand is not involved in the important LUMOs that govern emission from the Ir(III) species). The associated excitation spectra of the monometallic Ir(III) complexes are very similar and reminiscent of the corresponding UV-vis data.
Time-resolved measurements showed that in all cases the observed lifetimes fall in the range 218–378 ns, supporting the triplet assignment to the emitting states, which is clearly consistent with the values for benchmark species. Finally the quantum yield measurements showed that the monometallic species are broadly in line with relevant comparators, and that the Ir(III) species are approximately four times more emissive than the Ru(II) analogues.
Addition of the Au(I) moieties to the cationic complexes resulted in notable changes to the photophysical properties of the Ru(II) and Ir(III) emissive moieties. For [Ru–Au][PF6]3 and [Ru–Au2][PF6]4, the emission wavelengths showed clear hypsochromic shifts, accompanied by a lengthening of the observed lifetime and increase in quantum yield. This modulation appeared to be most pronounced for the trimetallic species, [Ru–Au2][PF6]4. In comparison, [Ir–Au][PF6]2 and [Ir–Au2][PF6]3 retain the emission characteristics of the parent monometallic species with little deviation in λem, although a notable enhancement in quantum yield to ca. 10%, was noted for both [Ir–Au][PF6]2 and [Ir–Au2][PF6]3. We ascribe this increase in quantum yield to the possible shielding effects via the triphenylphosphine moieties, and thus decreased non-radiative quenching. Taken together, the photophysical data shows that the inherent visible phosphorescence of the cationic Ru(II) and Ir(III) precursors is retained after functionalisation with the {Au–PPh3} fragments (Fig. 9). Furthermore, the Ru(II) systems show a more pronounced modulation in spectral properties which lead to a blue-shift in the emission maxima and corresponding changes to the lifetime and quantum yield that are consistent with the energy gap law. Again, these different observations relate to the specific character of the excited emitting states in each case. For the Ru(II) complexes the cationic imidazolium units clearly stabilise the 3MLCT state. Further functionalisation, in the guise of Au(I) coordination, probably dissipates the positive charge across the NHC/Au/PPh3 moiety which subtly destabilises the energy of the 3MLCT leading to a hypsochromic shift.
 | ||
| Fig. 9 Normalized excitation and emission spectra for the family of bimetallic complexes (293 K, aerated MeCN, 10−5 M). | ||
Cyclic voltammetry was also conducted on the complexes to provide some insight into the redox characteristics. The electrochemical properties were explored using cyclic voltammetry in MeCN solution at 1 mM concentration, using [nBu4N][PF6] as the supporting electrolyte (0.25 M) and the Fc/Fc+ redox couple as a reference. For the monometallic Ir complexes clear non-reversible oxidation and reduction waves were observed (Table S4 and Fig. S38†). The oxidation potentials corresponding to the Ir3+/4+ couple were observed at +0.98 V and +0.99 V for [Ir(Me3quin)2(bipy-Im1)][PF6]2 and [Ir(Me3quin)2(bipy-Im2)][PF6]3 respectively. For the heterobimetallic Ir–Au species oxidation waves were shifted to slightly higher potential, reflecting the presence of a second cationic gold centre. For the Ru–Au compounds typical quasi-reversible oxidation waves were observed at +0.93 V and +0.94 V for [Ru–Au][PF6]3 and [Ru–Au2][PF6]4 respectively. The reduction processes for all studied complexes were complicated by many non-reversible processes which precluded meaningful analysis.
Conclusions
The work has shown that 2,2′-bipyridine ligands adorned with N-methyl imidazolium units can be successfully utilised to build luminescent, heterometallic complexes. Our studies have shown that functionalised polypyridyl Ru(II) and cyclometalated Ir(III) complexes can be used to form stable heterometallic species with Au(I). Photophysical studies showed that these new heterometallic complexes are luminescent in the red region with greater efficiency of emission demonstrated in the Ir(III) series of compounds. As highlighted in the introduction, interest in the development of heterometallic complexes has been driven by a range of potential applications. For the complexes presented herein, we perceive genuine prospects within the field of optical cell imaging agents and, potentially, theranostics. The heterometallic compounds combine good luminescent properties under aerated conditions that are highly desirable in confocal fluorescence microscopy (good Stokes’ shifts, long wavelength emission), with a [Au(NHC)PR3] fragment which are known to be biologically active. Our future work will therefore consider these attributes in a range of different contexts and seek to establish the biological potential of these new complexes.Experimental
General considerations
All reagents and solvents were commercially available and were used without further purification if not stated otherwise. For the measurement of 1H, 31P{1H}, and 13C{1H} APT NMR spectra on a Bruker Fourier300 (300 MHz), Bruker AVANCE HD III equipped with a BFFO SmartProbe™ (400 MHz) or Bruker AVANCE III HD with BBO Prodigy CryoProbe (500 MHz) was used. The obtained chemical shifts δ are reported in ppm and are referenced to the residual solvent signal. Spin–spin coupling constants J are given in Hz. 31P and 13C (APT) spectra are proton decoupled unless otherwise stated.Mass spectra were obtained by the staff at Cardiff University. High resolution mass spectral (HRMS) data were obtained on a Waters MALDI-TOF mx at Cardiff University. Elemental analyses were obtained using the service at London Metropolitan University. IR spectra were obtained from a Shimadzu IR-Affinity-1S FTIR. Reference to spectroscopic data are given for known compounds. UV-Vis studies were performed on a Shimadzu UV-1800 spectrophotometer as MeCN solutions (1.0 × 10−5 or 2.5 × 10−6 M). Photophysical data were obtained on a Jobin Yvon–Horiba Fluorolog spectrometer fitted with a JY TBX picosecond photodetection module as MeCN solutions. The pulsed source was a nano-LED configured for 295 nm output operating at 1 MHz. Luminescence lifetime profiles were obtained using the Jobin Yvon–Horiba FluoroHub single photon counting module and the data fits yielded the lifetime values using the provided DAS6 deconvolution software. Quantum yield measurements were obtained on aerated MeCN solutions of the complexes and determined using comparative actinometry using [Ru(bipy)3](PF6)2 in aerated MeCN as a standard (Φ = 0.018).20
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 7.93 (s, 4H, Ar), 7.35 (s, 4H, Ar), 6.77 (app. tt, Japp = 7.6, 4H, Ar), 6.23 (app. tt, Japp = 7.6, 4H, Ar), 5.65 (d, 3JHH = 7.6, 4H, Ar), 3.21 (s, 12H, ArC3), 2.57 (s, 12H, ArC3), 1.56 (s, 12H, ArC3). ESI-MS m/z calcd for [C68H60ClIr2N8]+ 1409.3916 found 1409.3844, corresponding to [M − Cl]+. IR (ATR): νmax 3050, 2968, 2916, 1576, 1521, 1447, 1346, 1319, 1022, 983, 696, 626, 656 cm−1.
), 8.13 (dd, 3JHH = 8.2, 4JHH = 1.3, 2H, Ar), 7.96 (s, 2H, Ar), 7.03 (ddd, 3JHH = 8.3, 3JHH = 7.2, 4JHH = 1.3, 2H, Ar), 6.72 (d app. t, Japp = 7.8, 4JHH = 1.3, 2H, Ar), 6.11 (dd, 3JHH = 7.8, 4JHH = 1.2, 2H, Ar), 3.21 (s, 6H, ArC3), 2.57 (s, 6H, ArC3), 2.53 (s, 6H, ArC3), 1.96 (s, 6H, ArC3). 13C NMR spectrum (126 MHz, 293 K, CD3CN) 164.6 (s, Ar), 152.4 (s, Ar), 147.4 (s, Ar), 146.7 (s, Ar), 143.1 (s, Ar), 142.6 (s, Ar), 140.4 (s, Ar), 140.0 (s, Ar), 133.9 (s, Ar), 131.0 (s, Ar), 130.6 (s, Ar), 128.5 (s, Ar), 127.0 (s, Ar), 124.0 (s, Ar), 27.3 (s, Ar
), 7.93 (s, 4H, Ar), 7.35 (s, 4H, Ar), 6.77 (app. tt, Japp = 7.6, 4H, Ar), 6.23 (app. tt, Japp = 7.6, 4H, Ar), 5.65 (d, 3JHH = 7.6, 4H, Ar), 3.21 (s, 12H, ArC3), 2.57 (s, 12H, ArC3), 1.56 (s, 12H, ArC3). ESI-MS m/z calcd for [C68H60ClIr2N8]+ 1409.3916 found 1409.3844, corresponding to [M − Cl]+. IR (ATR): νmax 3050, 2968, 2916, 1576, 1521, 1447, 1346, 1319, 1022, 983, 696, 626, 656 cm−1.
), 8.13 (dd, 3JHH = 8.2, 4JHH = 1.3, 2H, Ar), 7.96 (s, 2H, Ar), 7.03 (ddd, 3JHH = 8.3, 3JHH = 7.2, 4JHH = 1.3, 2H, Ar), 6.72 (d app. t, Japp = 7.8, 4JHH = 1.3, 2H, Ar), 6.11 (dd, 3JHH = 7.8, 4JHH = 1.2, 2H, Ar), 3.21 (s, 6H, ArC3), 2.57 (s, 6H, ArC3), 2.53 (s, 6H, ArC3), 1.96 (s, 6H, ArC3). 13C NMR spectrum (126 MHz, 293 K, CD3CN) 164.6 (s, Ar), 152.4 (s, Ar), 147.4 (s, Ar), 146.7 (s, Ar), 143.1 (s, Ar), 142.6 (s, Ar), 140.4 (s, Ar), 140.0 (s, Ar), 133.9 (s, Ar), 131.0 (s, Ar), 130.6 (s, Ar), 128.5 (s, Ar), 127.0 (s, Ar), 124.0 (s, Ar), 27.3 (s, Ar![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3), 20.8 (s, ArH3), 20.0 (s, ArH3). ESI-MS m/z calcd for [C34H30IrN4]+ 687.2101 found 687.2101, corresponding to [M − 2MeCN − PF6]+. IR (ATR): νmax 3053, 2992, 2287, 1577, 1560, 1344, 1215, 991, 831, 729, 555 cm−1.
:1, 20 mL) and stirred at ambient temperature for 18 hours. The solvent was removed in vacuo and the crude material purified by column chromatography (SiO2; MeCN/H2O/sat. KNO3(aq); 14:2:1). The organic solvent was removed in vacuo and the product precipitated via the addition of 0.1 M NH4PF6(aq) (5 mL). The solid was filtered to give the title compound as a red solid (244 mg, 0.199 mmol, 86%). 1H NMR spectrum (500 MHz, 293 K, CD3CN) 8.43 (dd, 3JHH = 8.3, 4JHH = 1.2, 2H, Ar), 8.35 (s, 1H, Ar), 8.29 (ddd, 3JHH = 5.6, 4JHH = 1.6, 5JHH = 0.8, 1H, Ar), 8.15 (d, 3JHH = 8.5, 1H, Ar), 8.15–8.11 (m, 2H, Ar), 7.98 (td, 3JHH = 7.9, 4JHH = 1.6, 1H, Ar), 7.65 (s, 1H, Ar), 7.61 (s, 1H, Ar), 7.59 (ddd, 3JHH = 7.6, 3JHH = 5.4, 4JHH = 1.2, 1H, Ar), 7.45 (app. t, Japp = 1.8, 1H, Im), 7.29–7.15 (m, 3H, Ar + Im), 7.03 (s, 1H, Ar), 6.93 (s, 1H, Ar), 6.87–6.77 (m, 2H, Ar), 6.69 (dd, 3JHH = 7.8, 4JHH = 1.3, 1H, Ar), 6.54 (dd, 3JHH = 7.8, 4JHH = 1.3, 1H, Ar), 5.39–5.27 (m, 2H, bipyC2Im), 3.89 (s, 3H, ImC3), 3.27 (s, 3H, ArC3), 3.24 (s, 3H, ArC3), 2.28 (s, 6H, ArC3), 1.73 (s, 3H, ArC3), 1.72 (s, 3H, ArC3). 13C NMR spectrum (126 MHz, 293 K, CD3CN) 163.9 (s, Ar), 163.8 (s, Ar), 156.8 (s, Ar), 155.4 (s, Ar), 153.2 (s, Ar), 153.2 (s, Ar), 152.9 (s, Ar), 152.4 (s, Ar), 149.2 (s, Ar), 147.8 (s, Ar), 145.9 (s, Ar), 145.7 (s, Ar), 142.6 (s, Ar), 142.4 (s, Ar), 142.0 (s, Ar), 141.9 (s, Ar), 140.9 (s, Ar), 140.5 (s, Ar), 140.4 (s, Ar), 140.2 (s, Ar), 139.7 (s, Ar), 139.3 (s, Ar), 136.0 (s, Ar), 135.6 (s, Ar), 135.5 (s, Ar), 135.5 (s, Ar), 131.8 (s, Ar), 131.7 (s, Ar), 131.2 (s, Ar), 131.1 (s, Ar), 129.1 (s, Ar), 129.1 (s, Ar), 125.7 (s, Ar), 125.5 (s, Ar), 125.3 (s, Ar), 124.7 (s, Ar), 124.7 (s, Ar), 124.1 (s, Ar), 124.0 (s, Ar), 123.3 (s, Ar), 50.1 (s, ArH2Im), 37.3 (s, ImH3), 28.1 (s, ArH3), 27.9 (s, ArH3), 20.3 (s, ArH3), 19.9 (s, ArH3), 19.7 (s, ArH3), 19.6 (s, ArH3). ESI-MS m/z calcd for [C49H45IrN8]2+ 469.1700 found 469.1715, corresponding to [M − 2PF6]2+. IR (ATR): νmax 3165, 3119, 3050, 1602, 1577, 1560, 1523, 1444, 1400, 1344, 1309, 1217, 1165, 759, 738, 555 cm−1. Elemental analysis found, C 47.94%, H 2.89%, N 8.74%; expected, C 47.92%, H 3.69%, N 9.12% for C49H45F12IrN8P2.
:1, 30 mL) and stirred at ambient temperature for 18 hours. The solvent was removed in vacuo and the crude material purified by column chromatography (SiO2; MeCN/H2O/sat. KNO3(aq); 14:2:1). The organic solvent was removed in vacuo and the product precipitated via the addition of 0.1 M NH4PF6(aq) (5 mL). The solid was filtered to give the title compound as a red solid (367 mg, 0.249 mmol, 75%). 1H NMR spectrum (500 MHz, 293 K, CD3CN) 8.42 (dd, 3JHH = 8.3, 4JHH = 1.2, 2H, Ar), 8.34 (s, 2H, Im), 8.24 (d, 3JHH = 8.5, 2H, Ar), 8.09 (dd, 4JHH = 2.2, 5JHH = 0.7, 2H, Ar), 7.93 (dd, 3JHH = 8.5, 4JHH = 2.1, 2H, Ar), 7.67 (s, 2H, Ar), 7.45 (app. t, Japp = 1.8, 2H, Im), 7.24 (ddd, 3JHH = 8.3, 3JHH = 7.2, 4JHH = 1.3, 2H, Ar), 7.16 (app. t, Japp = 1.9, 2H, Im), 6.92 (s, 2H, Ar), 6.82 (ddd, 3JHH = 7.8, 3JHH = 7.2, 4JHH = 1.2, 2H, Ar), 6.55 (dd, 3JHH = 7.8, 4JHH = 1.2, 2H, Ar), 5.40–5.25 (m, 4H, ArC2Im), 3.89 (s, 6H, ImC3), 3.24 (s, 6H, ArC3), 2.31 (s, 6H, ArC3), 1.72 (s, 6H, ArC3). 13C NMR spectrum (126 MHz, 293 K, CD3CN) 163.7 (s, Ar), 156.0 (s, Ar), 152.9 (s, Ar), 152.3 (s, Ar), 148.0 (s, Ar), 145.6 (s, Ar), 142.7 (s, Ar), 142.1 (s, Ar), 140.7 (s, Ar), 140.4 (s, Ar), 139.4 (s, Ar), 137.5 (s, Ar), 136.0 (s, Ar), 135.6 (s, Ar), 131.8 (s, Ar), 131.3 (s, Ar), 129.2 (s, Ar), 125.9 (s, Ar), 125.8 (s, Ar), 124.7 (s, Ar), 124.2 (s, Ar), 123.7 (s, Ar), 50.0 (s, ArH2Im), 37.3 (s, ImH3), 28.2 (s, ArH3), 20.4 (s, ArH3), 19.6 (s, ArH3). ESI-MS m/z calcd for [C54H52F6IrN10P]2+ 589.1826 found 589.1822, corresponding to [M − 2PF6]+. IR (ATR): νmax 3165, 3119, 3055, 2922, 2851, 1578, 1560, 1525, 1483, 1400, 1369, 1344, 1321, 1217, 1165, 829, 759, 740, 669, 624, 556 cm−1. Elemental analysis found, C 43.21%, H 3.75%, N 9.13%; expected, C 43.64%, H 3.66%, N 9.42% for C54H52F18IrN10P3·H2O.
:2:1). The organic solvent was removed in vacuo and the product precipitated via the addition of 0.1 M NH4PF6(aq) (5 mL). The solid was filtered to give the title compound as an orange solid (37 mg, 23.7 μmol, 52%). 1H NMR spectrum (500 MHz, 293 K, (CD3)2CO) 8.85–8.81 (m, 1H, Ar), 8.79–8.75 (m, 2H, Ar), 8.74–8.68 (m, 2H, Ar), 8.61 (dt, 3JHH = 8.1, 4JHH = 1.2, 1H, Ar), 8.30–8.21 (m, 2H, Ar), 8.21–8.13 (m, 4H, Ar), 7.99 (dddd, 3JHH = 8.7, 3JHH = 5.6, 4JHH = 1.5, 5JHH = 0.7, 2H, Ar), 7.95 (tt, 3JHH = 5.5, 4JHH = 1.1, 2H), 7.96–7.92 (m, 2H, Ar), 7.89 (ddd, 3JHH = 5.6, 4JHH = 1.5, 5JHH = 0.7, 1H, Ar), 7.76–7.71 (m, 1H, Ar), 7.71–7.65 (m, 3H, Ar), 7.64–7.55 (m, 8H, Ar), 7.54–7.45 (m, 9H, Ar), 7.41–7.34 (m, 2H, Ar + (NHC)), 7.32–7.30 (m, 1H, (NHC)), 7.27 (ddd, 3JHH = 7.7, 3JHH = 5.6, 4JHH = 1.3 Hz, 1H, Ar), 5.79–5.63 (m, 2H, ArC2Im), 4.14 (s, 3H, ImC3). 13C NMR spectrum (126 MHz, 293 K, (CD3)2CO) 187.6 (d, 1JPC = 127, P–), 157.9 (s, Ar), 157.9 (s, Ar), 157.8 (s, Ar), 157.7 (s, Ar), 157.6 (s, Ar), 157.4 (s, Ar), 152.8 (s, Ar), 152.7 (s, Ar), 152.6 (s, Ar), 152.5 (s, Ar), 149.2 (s, Ar), 139.2 (s, Ar), 139.2 (s, Ar), 139.0 (s, Ar), 139.0 (s, Ar), 138.9 (s, Ar), 138.8 (s, Ar), 137.3 (s, Ar), 135.0 (d, 2JPC = 14, P–CH–), 133.4 (d, 4JPC = 3, P–CH–), 130.7 (d, 3JPC = 12, P–CH–CH–), 129.3 (s, Ar), 129.0 (s, Ar), 128.9 (s, Ar), 128.8 (s, Ar), 128.7 (s, Ar), 125.6 (s, Ar), 125.3 (s, Ar), 125.0 (s, Ar), 125.0 (s, Ar), 124.9 (s, Ar), 124.9 (s, Ar), 124.2 (s, Ar), 51.7 (s, ArH2(NHC)), 39.0 ((NHC)H3). 31P NMR (202 MHz, 293 K, (CD3)2CO) 40.4 (s,
H3), 20.8 (s, ArH3), 20.0 (s, ArH3). ESI-MS m/z calcd for [C34H30IrN4]+ 687.2101 found 687.2101, corresponding to [M − 2MeCN − PF6]+. IR (ATR): νmax 3053, 2992, 2287, 1577, 1560, 1344, 1215, 991, 831, 729, 555 cm−1.
:1, 20 mL) and stirred at ambient temperature for 18 hours. The solvent was removed in vacuo and the crude material purified by column chromatography (SiO2; MeCN/H2O/sat. KNO3(aq); 14:2:1). The organic solvent was removed in vacuo and the product precipitated via the addition of 0.1 M NH4PF6(aq) (5 mL). The solid was filtered to give the title compound as a red solid (244 mg, 0.199 mmol, 86%). 1H NMR spectrum (500 MHz, 293 K, CD3CN) 8.43 (dd, 3JHH = 8.3, 4JHH = 1.2, 2H, Ar), 8.35 (s, 1H, Ar), 8.29 (ddd, 3JHH = 5.6, 4JHH = 1.6, 5JHH = 0.8, 1H, Ar), 8.15 (d, 3JHH = 8.5, 1H, Ar), 8.15–8.11 (m, 2H, Ar), 7.98 (td, 3JHH = 7.9, 4JHH = 1.6, 1H, Ar), 7.65 (s, 1H, Ar), 7.61 (s, 1H, Ar), 7.59 (ddd, 3JHH = 7.6, 3JHH = 5.4, 4JHH = 1.2, 1H, Ar), 7.45 (app. t, Japp = 1.8, 1H, Im), 7.29–7.15 (m, 3H, Ar + Im), 7.03 (s, 1H, Ar), 6.93 (s, 1H, Ar), 6.87–6.77 (m, 2H, Ar), 6.69 (dd, 3JHH = 7.8, 4JHH = 1.3, 1H, Ar), 6.54 (dd, 3JHH = 7.8, 4JHH = 1.3, 1H, Ar), 5.39–5.27 (m, 2H, bipyC2Im), 3.89 (s, 3H, ImC3), 3.27 (s, 3H, ArC3), 3.24 (s, 3H, ArC3), 2.28 (s, 6H, ArC3), 1.73 (s, 3H, ArC3), 1.72 (s, 3H, ArC3). 13C NMR spectrum (126 MHz, 293 K, CD3CN) 163.9 (s, Ar), 163.8 (s, Ar), 156.8 (s, Ar), 155.4 (s, Ar), 153.2 (s, Ar), 153.2 (s, Ar), 152.9 (s, Ar), 152.4 (s, Ar), 149.2 (s, Ar), 147.8 (s, Ar), 145.9 (s, Ar), 145.7 (s, Ar), 142.6 (s, Ar), 142.4 (s, Ar), 142.0 (s, Ar), 141.9 (s, Ar), 140.9 (s, Ar), 140.5 (s, Ar), 140.4 (s, Ar), 140.2 (s, Ar), 139.7 (s, Ar), 139.3 (s, Ar), 136.0 (s, Ar), 135.6 (s, Ar), 135.5 (s, Ar), 135.5 (s, Ar), 131.8 (s, Ar), 131.7 (s, Ar), 131.2 (s, Ar), 131.1 (s, Ar), 129.1 (s, Ar), 129.1 (s, Ar), 125.7 (s, Ar), 125.5 (s, Ar), 125.3 (s, Ar), 124.7 (s, Ar), 124.7 (s, Ar), 124.1 (s, Ar), 124.0 (s, Ar), 123.3 (s, Ar), 50.1 (s, ArH2Im), 37.3 (s, ImH3), 28.1 (s, ArH3), 27.9 (s, ArH3), 20.3 (s, ArH3), 19.9 (s, ArH3), 19.7 (s, ArH3), 19.6 (s, ArH3). ESI-MS m/z calcd for [C49H45IrN8]2+ 469.1700 found 469.1715, corresponding to [M − 2PF6]2+. IR (ATR): νmax 3165, 3119, 3050, 1602, 1577, 1560, 1523, 1444, 1400, 1344, 1309, 1217, 1165, 759, 738, 555 cm−1. Elemental analysis found, C 47.94%, H 2.89%, N 8.74%; expected, C 47.92%, H 3.69%, N 9.12% for C49H45F12IrN8P2.
:1, 30 mL) and stirred at ambient temperature for 18 hours. The solvent was removed in vacuo and the crude material purified by column chromatography (SiO2; MeCN/H2O/sat. KNO3(aq); 14:2:1). The organic solvent was removed in vacuo and the product precipitated via the addition of 0.1 M NH4PF6(aq) (5 mL). The solid was filtered to give the title compound as a red solid (367 mg, 0.249 mmol, 75%). 1H NMR spectrum (500 MHz, 293 K, CD3CN) 8.42 (dd, 3JHH = 8.3, 4JHH = 1.2, 2H, Ar), 8.34 (s, 2H, Im), 8.24 (d, 3JHH = 8.5, 2H, Ar), 8.09 (dd, 4JHH = 2.2, 5JHH = 0.7, 2H, Ar), 7.93 (dd, 3JHH = 8.5, 4JHH = 2.1, 2H, Ar), 7.67 (s, 2H, Ar), 7.45 (app. t, Japp = 1.8, 2H, Im), 7.24 (ddd, 3JHH = 8.3, 3JHH = 7.2, 4JHH = 1.3, 2H, Ar), 7.16 (app. t, Japp = 1.9, 2H, Im), 6.92 (s, 2H, Ar), 6.82 (ddd, 3JHH = 7.8, 3JHH = 7.2, 4JHH = 1.2, 2H, Ar), 6.55 (dd, 3JHH = 7.8, 4JHH = 1.2, 2H, Ar), 5.40–5.25 (m, 4H, ArC2Im), 3.89 (s, 6H, ImC3), 3.24 (s, 6H, ArC3), 2.31 (s, 6H, ArC3), 1.72 (s, 6H, ArC3). 13C NMR spectrum (126 MHz, 293 K, CD3CN) 163.7 (s, Ar), 156.0 (s, Ar), 152.9 (s, Ar), 152.3 (s, Ar), 148.0 (s, Ar), 145.6 (s, Ar), 142.7 (s, Ar), 142.1 (s, Ar), 140.7 (s, Ar), 140.4 (s, Ar), 139.4 (s, Ar), 137.5 (s, Ar), 136.0 (s, Ar), 135.6 (s, Ar), 131.8 (s, Ar), 131.3 (s, Ar), 129.2 (s, Ar), 125.9 (s, Ar), 125.8 (s, Ar), 124.7 (s, Ar), 124.2 (s, Ar), 123.7 (s, Ar), 50.0 (s, ArH2Im), 37.3 (s, ImH3), 28.2 (s, ArH3), 20.4 (s, ArH3), 19.6 (s, ArH3). ESI-MS m/z calcd for [C54H52F6IrN10P]2+ 589.1826 found 589.1822, corresponding to [M − 2PF6]+. IR (ATR): νmax 3165, 3119, 3055, 2922, 2851, 1578, 1560, 1525, 1483, 1400, 1369, 1344, 1321, 1217, 1165, 829, 759, 740, 669, 624, 556 cm−1. Elemental analysis found, C 43.21%, H 3.75%, N 9.13%; expected, C 43.64%, H 3.66%, N 9.42% for C54H52F18IrN10P3·H2O.
:2:1). The organic solvent was removed in vacuo and the product precipitated via the addition of 0.1 M NH4PF6(aq) (5 mL). The solid was filtered to give the title compound as an orange solid (37 mg, 23.7 μmol, 52%). 1H NMR spectrum (500 MHz, 293 K, (CD3)2CO) 8.85–8.81 (m, 1H, Ar), 8.79–8.75 (m, 2H, Ar), 8.74–8.68 (m, 2H, Ar), 8.61 (dt, 3JHH = 8.1, 4JHH = 1.2, 1H, Ar), 8.30–8.21 (m, 2H, Ar), 8.21–8.13 (m, 4H, Ar), 7.99 (dddd, 3JHH = 8.7, 3JHH = 5.6, 4JHH = 1.5, 5JHH = 0.7, 2H, Ar), 7.95 (tt, 3JHH = 5.5, 4JHH = 1.1, 2H), 7.96–7.92 (m, 2H, Ar), 7.89 (ddd, 3JHH = 5.6, 4JHH = 1.5, 5JHH = 0.7, 1H, Ar), 7.76–7.71 (m, 1H, Ar), 7.71–7.65 (m, 3H, Ar), 7.64–7.55 (m, 8H, Ar), 7.54–7.45 (m, 9H, Ar), 7.41–7.34 (m, 2H, Ar + (NHC)), 7.32–7.30 (m, 1H, (NHC)), 7.27 (ddd, 3JHH = 7.7, 3JHH = 5.6, 4JHH = 1.3 Hz, 1H, Ar), 5.79–5.63 (m, 2H, ArC2Im), 4.14 (s, 3H, ImC3). 13C NMR spectrum (126 MHz, 293 K, (CD3)2CO) 187.6 (d, 1JPC = 127, P–), 157.9 (s, Ar), 157.9 (s, Ar), 157.8 (s, Ar), 157.7 (s, Ar), 157.6 (s, Ar), 157.4 (s, Ar), 152.8 (s, Ar), 152.7 (s, Ar), 152.6 (s, Ar), 152.5 (s, Ar), 149.2 (s, Ar), 139.2 (s, Ar), 139.2 (s, Ar), 139.0 (s, Ar), 139.0 (s, Ar), 138.9 (s, Ar), 138.8 (s, Ar), 137.3 (s, Ar), 135.0 (d, 2JPC = 14, P–CH–), 133.4 (d, 4JPC = 3, P–CH–), 130.7 (d, 3JPC = 12, P–CH–CH–), 129.3 (s, Ar), 129.0 (s, Ar), 128.9 (s, Ar), 128.8 (s, Ar), 128.7 (s, Ar), 125.6 (s, Ar), 125.3 (s, Ar), 125.0 (s, Ar), 125.0 (s, Ar), 124.9 (s, Ar), 124.9 (s, Ar), 124.2 (s, Ar), 51.7 (s, ArH2(NHC)), 39.0 ((NHC)H3). 31P NMR (202 MHz, 293 K, (CD3)2CO) 40.4 (s, ![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) Ph3), 144.3 (sept., 1JPF = 708, F6). ESI-MS m/z calcd for [C53H45AuF12N8P3Ru]+ 1412.1511 found 1412.1570, corresponding to [M − PF6]+. IR (ATR): νmax 3169, 2922, 2376, 2345, 2322, 1466, 1439, 1400, 1344, 1318, 1244, 1217, 1165, 1101, 997, 839, 759, 741, 692, 556 cm−1.
:2:1). The organic solvent was removed in vacuo and the product precipitated via the addition of 0.1 M NH4PF6(aq) (5 mL). The solid was filtered to give the title compound as an orange solid (122 mg, 54.1 μmol, 73%). 1H NMR spectrum (500 MHz, 293 K, (CD3)2CO) 8.74 (d, 3JHH = 8.4, 2H, Ar), 8.70 (dt, 3JHH = 8.2, 4JHH = 1.1, 2H, Ar), 8.58 (dt, 3JHH = 8.2, 4JHH = 1.1, 2H, Ar), 8.26 (app. td, 3Japp = 7.9, 4JHH = 1.5, 2H, Ar), 8.21 (dd, 3JHH = 8.5, 4JHH = 2.0, 2H, Ar), 7.87–7.82 (m, 4H, Ar), 7.69–7.41 (m, 36H, Ar), 7.34 (dd, 3JHH = 1.9, 4JHH = 1.0 Hz, 2H, (NHC)), 7.25–7.20 (m, 2H, (NHC)), 7.10 (ddd, 3JHH = 7.7, 5.6, 4JHH = 1.3, 2H, Ar), 5.82–5.59 (m, 4H, ArC2(NHC)), 4.15 (s, 6H, (NHC)C3). 13C NMR spectrum (126 MHz, 293 K, (CD3)2CO) 187.6 (d, 1JPC = 127, P–), 157.7 (s, Ar), 157.4 (s, Ar), 156.9 (s, Ar), 152.89 (s, Ar), 152.6 (s, Ar), 149.2 (s, Ar), 139.3 (s, Ar), 138.8 (s, Ar), 137.5 (s, Ar), 135.0 (d, 2JPC = 14, P–C–H), 133.4 (d, 4JPC = 2, P–C–CH–CH–H), 130.7 (d, 3JPC = 12, P–C–CH–H), 129.3 (s, Ar), 128.9 (s, Ar), 128.8 (s, Ar), 125.2 (s, Ar), 125.0 (s, Ar), 125.0 (s, Ar), 124.8 (s, Ar), 124.8 (s, Ar), 124.3 (s, Ar), 51.7 (s, ArH2(NHC)), 39.0 (s, (NHC)H3). 31P NMR (202 MHz, 293 K, (CD3)2CO) 40.4 (s, Ph3), 144.3 (sept., 1JPF = 708, F6). ESI-MS m/z calcd for [C76H66Au2F12N10P4Ru]2+ 983.1313 found 983.1312, corresponding to [M − 2PF6]2+. IR (ATR): νmax 3169, 2376, 2347, 2322, 1466, 1437, 1398, 1101, 839, 731, 712, 691, 556 cm−1. Elemental analysis found, C 39.99%, H 2.90%, N 6.21%; expected, C 40.67%, H 3.19%, N 6.16% for C77H72Au2F24N10P6Ru.
:2:1). The organic solvent was removed in vacuo and the product precipitated via the addition of 0.1 M NH4PF6(aq) (5 mL). The solid was filtered to give the title compound as an orange solid (55 mg, 32.6 μmol, 67%). 1H NMR spectrum (500 MHz, 293 K, (CD3)2CO) 8.52 (ddd, 3JHH = 5.5, 4JHH = 1.6, 5JHH = 0.7, 1H, Ar), 8.45 (ddd, 3JHH = 8.4, 5.0, 4JHH = 1.2, 2H, Ar), 8.38 (dt, 3JHH = 8.2, 4JHH = 1.1, 1H, Ar), 8.27 (t, J = 1.7 Hz, 2H), 8.29–8.24 (m, 2H, Ar), 8.20 (app. td, 3JHH = 7.9, 4JHH = 1.6, 1H, Ar), 8.09 (dd, 3JHH = 1.9, 4JHH = 0.9, 1H, Ar), 7.84 (ddd, 3JHH = 7.6, 5.5, 4JHH = 1.2, 1H, Ar), 7.79 (dd, J = 1.9, 0.8 Hz, 1H), 7.67–7.57 (m, 5H, Ar), 7.51–7.43 (m, 12H, Ar), 7.24–7.16 (m, 3H, Ar), 6.97 (s, 1H, Ar), 6.81–6.76 (m, 2H, Ar), 6.71 (ddd, 3JHH = 7.8, 7.1, 4JHH = 1.2, 1H, Ar), 6.51 (dd, 3JHH = 7.7, 4JHH = 1.3, 1H, Ar), 5.88 (d, 2JHH = 16.6, 1H, ArCH(NHC)), 5.76 (d, J = 16.6 Hz, 1H, ArCH(NHC)), 4.31 (s, 3H, (NHC)C3), 3.32 (s, 3H, ArC3), 3.26 (s, 3H, ArC3), 2.15 (s, 3H, ArC3), 2.12 (s, 3H, ArC3), 1.79 (s, 3H, ArC3), 1.64 (s, 3H, ArC3). 13C NMR spectrum (126 MHz, 293 K, (CD3)2CO) 187.8 (d, 1JPC = 127, P–), 163.9 (s, Ar), 163.3 (s, Ar), 156.4 (s, Ar), 155.5 (s, Ar), 154.1 (s, Ar), 152.9 (s, Ar), 152.4 (s, Ar), 152.4 (s, Ar), 149.0 (s, Ar), 146.8 (s, Ar), 145.8 (s, Ar), 145.3 (s, Ar), 142.4 (s, Ar), 142.1 (s, Ar), 141.8 (s, Ar), 141.5 (s, Ar), 141.2 (s, Ar), 140.3 (s, Ar), 140.3 (s, Ar), 140.2 (s, Ar), 139.6 (s, Ar), 139.1 (s, Ar), 138.6 (s, Ar), 136.0 (s, Ar), 135.2 (s, Ar), 135.0 (d, 2JPC = 14, P–C–H), 133.3 (d, 4JPC = 3, P–C–CH–CH–H), 131.6 (s, Ar), 131.4 (s, Ar), 131.1 (s, Ar), 130.6 (d, 3JPC = 12, P–C–CH–H), 129.8 (s, Ar), 129.3 (s, Ar), 129.3 (s, Ar), 129.2 (s, Ar), 128.9 (s, Ar), 125.8 (s, Ar), 125.6 (s, Ar), 125.2 (s, Ar), 124.9 (s, Ar), 124.4 (s, Ar), 123.9 (s, Ar), 123.8 (s, Ar), 123.7 (s, Ar), 51.86 (s, ArC2(NHC)), 39.2 (s, ArH3), 29.9 (s, ArH3), 29.8 (s, ArH3), 29.6 (s, ArH3), 28.5 (s, ArH3), 27.7 (s, ArH3), 20.5 (s, ArH3), 19.9 (s, ArH3), 19.5 (s, ArH3), 19.5 (s, ArH3). 31P NMR (202 MHz, 293 K, (CD3)2CO) 40.2 (s, Ph3), 144.3 (sept., 1JPF = 708, F6). ESI-MS m/z calcd for [C67H59AuF6IrN8P2]+ 1541.3542 found 1541.3612, corresponding to [M − PF6]+. IR (ATR): νmax 3169, 3050, 2918, 2851, 1578, 1524, 1508, 1452, 1437, 1398, 1373, 1344, 1319, 1217, 1199, 1165, 1101, 993, 829, 741, 712, 692, 628, 556 cm−1. Elemental analysis found, C 46.81%, H 3.00%, N 6.51%; expected, C 47.19%, H 3.66%, N 6.57% for C67H60AuF12IrN8P3·H2O.
:2:1). The organic solvent was removed in vacuo and the product precipitated via the addition of 0.1 M NH4PF6(aq) (5 mL). The solid was filtered to give the title compound as an orange solid (72 mg, 29.9 μmol, 44%). 1H NMR spectrum (500 MHz, 293 K, (CD3)2CO) 8.41 (dd, 3JHH = 8.4, 4JHH = 1.2, 2H, Ar), 8.30 (d, 4JHH = 2.1 Hz, 2H), 8.26 (dd, 3JHH = 8.6, 4JHH = 0.7, 2H, Ar), 8.03 (d, 4JHH = 2.0, 2H, Ar), 7.76 (dd, 4JHH = 1.9, 5JHH = 0.9, 2H, Ar), 7.66–7.61 (m, 6H, Ar), 7.53–7.45 (m, 28H, Ar), 7.42–7.40 (m, 4H, Ar), 7.19–7.15 (m, 2H, Ar), 7.01 (s, 2H, Ar), 6.69–6.66 (m, 2H, Ar), 6.57 (dd, 3JHH = 7.8, 4JHH = 1.3, 2H, Ar), 5.87 (d, 2JHH = 16.7, 2H, ArCH), 5.76 (d, 4JHH = 16.7, 2H, ArCH), 4.29 (s, 6H, ArCH2(NHC)), 3.24 (s, 6H, ArC3), 1.96 (s, 6H, ArC3), 1.62 (s, 6H, ArC3). 13C NMR spectrum (126 MHz, 293 K, (CD3)2CO) 187.8 (d, 1JPC = 127, P–), 163.2 (s, Ar), 155.5 (s, Ar), 153.1 (s, Ar), 152.4 (s, Ar), 146.8 (s, Ar), 145.3 (s, Ar), 142.4 (s, Ar), 141.7 (s, Ar), 140.5 (s, Ar), 140.3 (s, Ar), 139.1 (s, Ar), 139.0 (s, Ar), 135.3 (s, Ar), 135.0 (d, 2JPC = 14, P–C–H), 133.3 (d, 4JPC = 2, P–C–CH–CH–H), 131.4 (s, Ar), 130.6 (d, 3JPC = 12, P–C–CH–H), 129.3 (s, Ar), 129.3 (s, Ar), 128.8 (s, Ar), 125.7 (s, Ar), 124.6 (s, Ar), 124.0 (s, Ar), 51.8 (s, ArH2(NHC)), 39.2 (s, ArH3), 20.6 (s, ArH3), 19.4 (s, ArH3). 31P NMR (202 MHz, 293 K, (CD3)2CO) 40.2 (s, Ph3), 144.3 (sept., 1JPF = 708, F6). ESI-MS m/z calcd for [C90H81Au2F6IrN10P3]2+ 1047.23 found 1047.24, corresponding to [M − 2PF6]2+. IR (ATR): νmax 3169, 3055, 2378, 2347, 1703, 1609, 1570, 1524, 1508, 1481, 1452, 1436, 1396, 1373, 1319, 1217, 1200, 1134, 1101, 995, 827, 741, 712, 691, 556 cm−1. Elemental analysis found, C 45.07%, H 2.74%, N 5.55%; expected, C 45.49%, H 3.61%, N 5.83% for C91H86Au2F18IrN10P5.
Ph3), 144.3 (sept., 1JPF = 708, F6). ESI-MS m/z calcd for [C53H45AuF12N8P3Ru]+ 1412.1511 found 1412.1570, corresponding to [M − PF6]+. IR (ATR): νmax 3169, 2922, 2376, 2345, 2322, 1466, 1439, 1400, 1344, 1318, 1244, 1217, 1165, 1101, 997, 839, 759, 741, 692, 556 cm−1.
:2:1). The organic solvent was removed in vacuo and the product precipitated via the addition of 0.1 M NH4PF6(aq) (5 mL). The solid was filtered to give the title compound as an orange solid (122 mg, 54.1 μmol, 73%). 1H NMR spectrum (500 MHz, 293 K, (CD3)2CO) 8.74 (d, 3JHH = 8.4, 2H, Ar), 8.70 (dt, 3JHH = 8.2, 4JHH = 1.1, 2H, Ar), 8.58 (dt, 3JHH = 8.2, 4JHH = 1.1, 2H, Ar), 8.26 (app. td, 3Japp = 7.9, 4JHH = 1.5, 2H, Ar), 8.21 (dd, 3JHH = 8.5, 4JHH = 2.0, 2H, Ar), 7.87–7.82 (m, 4H, Ar), 7.69–7.41 (m, 36H, Ar), 7.34 (dd, 3JHH = 1.9, 4JHH = 1.0 Hz, 2H, (NHC)), 7.25–7.20 (m, 2H, (NHC)), 7.10 (ddd, 3JHH = 7.7, 5.6, 4JHH = 1.3, 2H, Ar), 5.82–5.59 (m, 4H, ArC2(NHC)), 4.15 (s, 6H, (NHC)C3). 13C NMR spectrum (126 MHz, 293 K, (CD3)2CO) 187.6 (d, 1JPC = 127, P–), 157.7 (s, Ar), 157.4 (s, Ar), 156.9 (s, Ar), 152.89 (s, Ar), 152.6 (s, Ar), 149.2 (s, Ar), 139.3 (s, Ar), 138.8 (s, Ar), 137.5 (s, Ar), 135.0 (d, 2JPC = 14, P–C–H), 133.4 (d, 4JPC = 2, P–C–CH–CH–H), 130.7 (d, 3JPC = 12, P–C–CH–H), 129.3 (s, Ar), 128.9 (s, Ar), 128.8 (s, Ar), 125.2 (s, Ar), 125.0 (s, Ar), 125.0 (s, Ar), 124.8 (s, Ar), 124.8 (s, Ar), 124.3 (s, Ar), 51.7 (s, ArH2(NHC)), 39.0 (s, (NHC)H3). 31P NMR (202 MHz, 293 K, (CD3)2CO) 40.4 (s, Ph3), 144.3 (sept., 1JPF = 708, F6). ESI-MS m/z calcd for [C76H66Au2F12N10P4Ru]2+ 983.1313 found 983.1312, corresponding to [M − 2PF6]2+. IR (ATR): νmax 3169, 2376, 2347, 2322, 1466, 1437, 1398, 1101, 839, 731, 712, 691, 556 cm−1. Elemental analysis found, C 39.99%, H 2.90%, N 6.21%; expected, C 40.67%, H 3.19%, N 6.16% for C77H72Au2F24N10P6Ru.
:2:1). The organic solvent was removed in vacuo and the product precipitated via the addition of 0.1 M NH4PF6(aq) (5 mL). The solid was filtered to give the title compound as an orange solid (55 mg, 32.6 μmol, 67%). 1H NMR spectrum (500 MHz, 293 K, (CD3)2CO) 8.52 (ddd, 3JHH = 5.5, 4JHH = 1.6, 5JHH = 0.7, 1H, Ar), 8.45 (ddd, 3JHH = 8.4, 5.0, 4JHH = 1.2, 2H, Ar), 8.38 (dt, 3JHH = 8.2, 4JHH = 1.1, 1H, Ar), 8.27 (t, J = 1.7 Hz, 2H), 8.29–8.24 (m, 2H, Ar), 8.20 (app. td, 3JHH = 7.9, 4JHH = 1.6, 1H, Ar), 8.09 (dd, 3JHH = 1.9, 4JHH = 0.9, 1H, Ar), 7.84 (ddd, 3JHH = 7.6, 5.5, 4JHH = 1.2, 1H, Ar), 7.79 (dd, J = 1.9, 0.8 Hz, 1H), 7.67–7.57 (m, 5H, Ar), 7.51–7.43 (m, 12H, Ar), 7.24–7.16 (m, 3H, Ar), 6.97 (s, 1H, Ar), 6.81–6.76 (m, 2H, Ar), 6.71 (ddd, 3JHH = 7.8, 7.1, 4JHH = 1.2, 1H, Ar), 6.51 (dd, 3JHH = 7.7, 4JHH = 1.3, 1H, Ar), 5.88 (d, 2JHH = 16.6, 1H, ArCH(NHC)), 5.76 (d, J = 16.6 Hz, 1H, ArCH(NHC)), 4.31 (s, 3H, (NHC)C3), 3.32 (s, 3H, ArC3), 3.26 (s, 3H, ArC3), 2.15 (s, 3H, ArC3), 2.12 (s, 3H, ArC3), 1.79 (s, 3H, ArC3), 1.64 (s, 3H, ArC3). 13C NMR spectrum (126 MHz, 293 K, (CD3)2CO) 187.8 (d, 1JPC = 127, P–), 163.9 (s, Ar), 163.3 (s, Ar), 156.4 (s, Ar), 155.5 (s, Ar), 154.1 (s, Ar), 152.9 (s, Ar), 152.4 (s, Ar), 152.4 (s, Ar), 149.0 (s, Ar), 146.8 (s, Ar), 145.8 (s, Ar), 145.3 (s, Ar), 142.4 (s, Ar), 142.1 (s, Ar), 141.8 (s, Ar), 141.5 (s, Ar), 141.2 (s, Ar), 140.3 (s, Ar), 140.3 (s, Ar), 140.2 (s, Ar), 139.6 (s, Ar), 139.1 (s, Ar), 138.6 (s, Ar), 136.0 (s, Ar), 135.2 (s, Ar), 135.0 (d, 2JPC = 14, P–C–H), 133.3 (d, 4JPC = 3, P–C–CH–CH–H), 131.6 (s, Ar), 131.4 (s, Ar), 131.1 (s, Ar), 130.6 (d, 3JPC = 12, P–C–CH–H), 129.8 (s, Ar), 129.3 (s, Ar), 129.3 (s, Ar), 129.2 (s, Ar), 128.9 (s, Ar), 125.8 (s, Ar), 125.6 (s, Ar), 125.2 (s, Ar), 124.9 (s, Ar), 124.4 (s, Ar), 123.9 (s, Ar), 123.8 (s, Ar), 123.7 (s, Ar), 51.86 (s, ArC2(NHC)), 39.2 (s, ArH3), 29.9 (s, ArH3), 29.8 (s, ArH3), 29.6 (s, ArH3), 28.5 (s, ArH3), 27.7 (s, ArH3), 20.5 (s, ArH3), 19.9 (s, ArH3), 19.5 (s, ArH3), 19.5 (s, ArH3). 31P NMR (202 MHz, 293 K, (CD3)2CO) 40.2 (s, Ph3), 144.3 (sept., 1JPF = 708, F6). ESI-MS m/z calcd for [C67H59AuF6IrN8P2]+ 1541.3542 found 1541.3612, corresponding to [M − PF6]+. IR (ATR): νmax 3169, 3050, 2918, 2851, 1578, 1524, 1508, 1452, 1437, 1398, 1373, 1344, 1319, 1217, 1199, 1165, 1101, 993, 829, 741, 712, 692, 628, 556 cm−1. Elemental analysis found, C 46.81%, H 3.00%, N 6.51%; expected, C 47.19%, H 3.66%, N 6.57% for C67H60AuF12IrN8P3·H2O.
:2:1). The organic solvent was removed in vacuo and the product precipitated via the addition of 0.1 M NH4PF6(aq) (5 mL). The solid was filtered to give the title compound as an orange solid (72 mg, 29.9 μmol, 44%). 1H NMR spectrum (500 MHz, 293 K, (CD3)2CO) 8.41 (dd, 3JHH = 8.4, 4JHH = 1.2, 2H, Ar), 8.30 (d, 4JHH = 2.1 Hz, 2H), 8.26 (dd, 3JHH = 8.6, 4JHH = 0.7, 2H, Ar), 8.03 (d, 4JHH = 2.0, 2H, Ar), 7.76 (dd, 4JHH = 1.9, 5JHH = 0.9, 2H, Ar), 7.66–7.61 (m, 6H, Ar), 7.53–7.45 (m, 28H, Ar), 7.42–7.40 (m, 4H, Ar), 7.19–7.15 (m, 2H, Ar), 7.01 (s, 2H, Ar), 6.69–6.66 (m, 2H, Ar), 6.57 (dd, 3JHH = 7.8, 4JHH = 1.3, 2H, Ar), 5.87 (d, 2JHH = 16.7, 2H, ArCH), 5.76 (d, 4JHH = 16.7, 2H, ArCH), 4.29 (s, 6H, ArCH2(NHC)), 3.24 (s, 6H, ArC3), 1.96 (s, 6H, ArC3), 1.62 (s, 6H, ArC3). 13C NMR spectrum (126 MHz, 293 K, (CD3)2CO) 187.8 (d, 1JPC = 127, P–), 163.2 (s, Ar), 155.5 (s, Ar), 153.1 (s, Ar), 152.4 (s, Ar), 146.8 (s, Ar), 145.3 (s, Ar), 142.4 (s, Ar), 141.7 (s, Ar), 140.5 (s, Ar), 140.3 (s, Ar), 139.1 (s, Ar), 139.0 (s, Ar), 135.3 (s, Ar), 135.0 (d, 2JPC = 14, P–C–H), 133.3 (d, 4JPC = 2, P–C–CH–CH–H), 131.4 (s, Ar), 130.6 (d, 3JPC = 12, P–C–CH–H), 129.3 (s, Ar), 129.3 (s, Ar), 128.8 (s, Ar), 125.7 (s, Ar), 124.6 (s, Ar), 124.0 (s, Ar), 51.8 (s, ArH2(NHC)), 39.2 (s, ArH3), 20.6 (s, ArH3), 19.4 (s, ArH3). 31P NMR (202 MHz, 293 K, (CD3)2CO) 40.2 (s, Ph3), 144.3 (sept., 1JPF = 708, F6). ESI-MS m/z calcd for [C90H81Au2F6IrN10P3]2+ 1047.23 found 1047.24, corresponding to [M − 2PF6]2+. IR (ATR): νmax 3169, 3055, 2378, 2347, 1703, 1609, 1570, 1524, 1508, 1481, 1452, 1436, 1396, 1373, 1319, 1217, 1200, 1134, 1101, 995, 827, 741, 712, 691, 556 cm−1. Elemental analysis found, C 45.07%, H 2.74%, N 5.55%; expected, C 45.49%, H 3.61%, N 5.83% for C91H86Au2F18IrN10P5.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Leverhulme Trust is thanked for funding (RPG-2021-003). We acknowledge the use of the EPSRC funded Physical Sciences Data-science Service hosted by the University of Southampton and STFC under grant no. EP/S020357/1. Dr B. Kariuki is also thanked for the acquisition of X-ray diffraction data at Cardiff University. We thank the EPSRC UK National Crystallography Service at the University of Southampton for the collection of the crystallographic data for [Ir(Me3quinox)2(MeCN)2]BF4.Notes and references
- M. H. Prosenc, M. M. Kappes and G. Niedner-Schatteburg, Chem. – Eur. J., 2021, 27, 15019 CrossRef PubMed.
- For example: D. Liu, M. Zhang, H.-H. Huang, Q. Feng, C. Su, A. Mo, J.-W. Wang, Z. Qi, X. Zhang, L. Jiang and Z. Chen, ACS Sustainable Chem. Eng., 2021, 9, 28 Search PubMed.
- For example: M. Redrado, V. Fernandez-Moreira and M. C. Gimeno, ChemMedChem, 2021, 16, 932 CrossRef CAS PubMed; V. Fernandez-Moreira, I. Marzo and M. C. Gimeno, Chem. Sci., 2014, 5, 4434 RSC.
- A. Jain, Coord. Chem. Rev., 2019, 401, 213067 CrossRef CAS; Z. Huang and J. J. Wilson, Eur. J. Inorg. Chem., 2021, 1312 CrossRef.
- L. Ma, L. Li and G. Zhu, Inorg. Chem. Front., 2022, 9, 2424 RSC; A. Jain, Coord. Chem. Rev., 2019, 401, 213067 CrossRef CAS.
- D. M. Knoll, C. Zippel, Z. Hassan, M. Nieger, P. Weis, M. M. Kappes and S. Brase, Dalton Trans., 2019, 48, 177704 RSC.
- L. Bayer, B. S. Birenheide, F. Kramer, S. Lebedkin and F. Breher, Chem. – Eur. J., 2022, 28, e202201856 CrossRef CAS PubMed.
- R. T. Mertens, S. Gukathasan, A. S. Arojojoye, C. Olelewe and S. G. Awuah, Chem. Rev., 2023, 123, 6612 CrossRef CAS PubMed.
- M. Wenzel, A. de Almeida, E. Bigaeva, P. Kavanagh, M. Picquet, P. Le Gendre, E. Bodio and A. Casini, Inorg. Chem., 2016, 55, 2544 CrossRef CAS PubMed.
- V. Fernandez-Moreira and M. C. Gimeno, Chem. – Eur. J., 2018, 24, 3345 CrossRef CAS PubMed.
- L. K. Batchelor, D. Ortiz and P. J. Dyson, Inorg. Chem., 2019, 58, 2501 CrossRef CAS PubMed.
- L. Boselli, M. Carraz, S. Mazeres, L. Paloque, G. Gonzalez, F. Benoit-Vical, A. Valentin, C. Hemmert and H. Gornitzka, Organometallics, 2015, 34, 1046 CrossRef CAS.
- V. Fernández-Moreira, I. Marzo and M. C. Gimeno, Chem. Sci., 2014, 5, 4434 RSC; A. Luengo, V. Fernández-Moreira, I. Marzo and M. C. Gimeno, Inorg. Chem., 2017, 56, 15159 CrossRef CAS PubMed; A. Luengo, V. Fernández-Moreira, I. Marzo and M. C. Gimeno, Organometallics, 2018, 37, 3993 CrossRef; A. Luengo, M. Redrado, I. Marzo, V. Fernández-Moreira and M. C. Gimeno, Inorg. Chem., 2020, 59, 8960 CrossRef PubMed; A. Luengo, I. Marzo, V. Fernández-Moreira and M. C. Gimeno, Appl. Organomet. Chem., 2022, e6661 CrossRef.
- Y. Yamamoto, M. Shiotsuka and S. Onaka, J. Organomet. Chem., 2004, 689, 2905 CrossRef CAS.
- A. Luengo, I. Marzo, M. Reback, I. M. Daubit, V. Fernandez-Moreira, N. Metzler-Nolte and M. C. Gimeno, Chem. – Eur. J., 2020, 26, 12158 CrossRef CAS PubMed.
- M. Redrado, A. Benedi, I. Marzo, A. L. Garcia-Otin, V. Fernandez-Morieira and M. C. Gimeno, Chem. – Eur. J., 2021, 27, 9885 CrossRef CAS PubMed.
- V. H. Nguyen, R. S. H. Khoo and J. H. K. Yip, Inorg. Chem., 2015, 54, 2264 CrossRef CAS PubMed.
- S. Sabeter, J. A. Mata and E. Peris, Chem. – Eur. J., 2012, 18, 6380 CrossRef CAS PubMed; M. Bohmer, F. Kampert, T. T. Y. Tan, G. Guisado-Barrios, E. Peris and F. E. Hahn, Organometallics, 2018, 37, 4092 CrossRef; G. M. Brown, M. R. J. Elsegood, A. J. Lake, N. M. Sanchez-Ballester, M. B. Smith, T. S. Varley and K. Blann, Eur. J. Inorg. Chem., 2007, 1405 CrossRef; C. S. Brinke and F. E. Hahn, Eur. J. Inorg. Chem., 2015, 3227 CrossRef; R. C. Nishad, S. Kumar and A. Rit, Organometallics, 2021, 40, 915 CrossRef; S. J. Coles, P. N. Horton, P. Kimber, W. T. Klooster, P. Liu, F. Plasser, M. B. Smith and G. J. Tizzard, Chem. Commun., 2022, 58, 5598 RSC.
- L. J. Charbonnière, N. Weibel and R. F. Ziessel, J. Org. Chem., 2002, 67, 3933 CrossRef CAS PubMed; F. Forato, A. Belhboub, J. Monot, M. Petit, R. Benoit, V. Sarou-Kanian, F. Fayon, D. Jacquemin, C. Queffelec and B. Bujoli, Chem. – Eur. J., 2018, 24, 2457 CrossRef PubMed; S. G. Telfer, G. Bernardinelli and A. F. Williams, Dalton Trans., 2003, 435 RSC.
- A crystal structure has been reported: S. Park, S.-H. Moon, T. H. Kim and K.-M. Park, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, E67, o2392 CrossRef PubMed.
- R. C. Knighton, J. M. Beames and S. J. A. Pope, Inorg. Chem., 2023, 62, 19446 CrossRef CAS PubMed.
- K. A. Phillips, T. M. Stonelake, K. Chen, Y. Hou, J. Zhao, S. J. Coles, P. N. Horton, S. J. Keane, E. C. Stokes, I. A. Fallis, A. J. Hallett, S. P. O'Kell, J. B. Beames and S. J. A. Pope, Chem. – Eur. J., 2018, 24, 8577 CrossRef CAS PubMed; A. H. Day, M. H. Übler, H. L. Best, E. Lloyd-Evans, R. J. Mart, I. A. Fallis, R. K. Allemann, E. A. H. Al-Wattar, N. I. Keymer, N. J. Buurma and S. J. A. Pope, Chem. Sci., 2020, 11, 1599 RSC; T. M. Stonelake, K. A. Phillips, H. Y. Otaif, Z. C. Edwardson, P. N. Horton, S. J. Coles, J. M. Beames and S. J. A. Pope, Inorg. Chem., 2020, 59, 2266 CrossRef PubMed; S. A. Fitzgerald, H. Y. Otaif, C. E. Elgar, N. Sawicka, P. N. Horton, S. J. Coles, J. M. Beames and S. J. A. Pope, Inorg. Chem., 2021, 60, 15467 CrossRef PubMed.
- C. E. Elgar, H. Y. Otaif, J. M. Beames, P. N. Horton, S. J. Coles, A. J. Hallett, S. P. O'Kell and S. J. A. Pope, Eur. J. Inorg. Chem., 2023, e202300102 CrossRef CAS.
- F.-M. Hwang, H.-Y. Chen, P.-S. Chen, C.-S. Liu, Y. Chi, C.-F. Shu, F.-I. Wu, P.-T. Chou, S.-M. Peng and G.-H. Lee, Inorg. Chem., 2005, 44, 1344 CrossRef CAS PubMed.
- T. Tamai, K. Fujiwara, S. Higashimae, A. Nomoto and A. Ogawa, Org. Lett., 2016, 18, 2114 CrossRef CAS PubMed.
- M. V. Baker, P. J. Bernhard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, J. Organomet. Chem., 2005, 690, 5625 CrossRef CAS.
- D. Curran, H. Muller-Bunz, S. I. Bar, R. Schobert, X. Zhu and M. Tacke, Molecules, 2020, 25, 3474 CrossRef CAS PubMed.
- V. Balzani, P. Ceroni, A. Credi and M. Venturi, Coord. Chem. Rev., 2021, 433, 213758 CrossRef CAS; M. K. De Armond and M. L. Myrick, Acc. Chem. Res., 1989, 22, 364 CrossRef.
- H. Ishida, S. Tobita, Y. Hasegawa, R. Katoh and K. Nozaki, Coord. Chem. Rev., 2010, 254, 2449 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional spectral data and details associated with X-ray diffraction. CCDC 2271291, 2271292 and 2295692. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03690a |
| ‡ Current address: School of Chemistry, University of Southampton, Southampton, SO17 1BJ, UK. |
| This journal is © The Royal Society of Chemistry 2024 |