
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination-triggered redox activity of early and late lanthanide calix[4]arene complexes†
Yushu
Jiao
 a,
Sergio
Sanz
*b,
Lucie
Koláčná
c,
Jan
van Leusen
a,
Natalya V.
Izarova
a,
Sidra
Sarwar
a,
Jiří
Ludvík
*c and
Paul
Kögerler
*ab
a,
Sergio
Sanz
*b,
Lucie
Koláčná
c,
Jan
van Leusen
a,
Natalya V.
Izarova
a,
Sidra
Sarwar
a,
Jiří
Ludvík
*c and
Paul
Kögerler
*ab
aInstitute of Inorganic Chemistry, RWTH Aachen University, 52056 Aachen, Germany. E-mail: paul.koegerler@ac.rwth-aachen.de
bPeter Grünberg Institute, Electronic Properties (PGI-6), Forschungszentrum Jülich, 52425 Jülich, Germany. E-mail: s.calvo@fz-juelich.de
cJ. Heyrovský Institute of Physical Chemistry AS CR, Department of Molecular Electrochemistry and Catalysis, Dolejškova 2155/3, 182 23 Prague 8, Czech Republic. E-mail: jiri.ludvik@jh-inst.cas.cz
First published on 23rd February 2024
Abstract
The methylation of p-tert-butylcalix[4]arene in the distal 1,3-phenolic sites provides H2L = {p-tert-butylcalix[4](OMe)2(OH)2arene}. This unit acts as a rigid coordinating ligand to early and late lanthanide metal ions, enabling the construction of two families of mononuclear compounds featuring (N(nBu)4)[LnIIIL(acac)2]·CH3CN (Ln = Pr (1), Nd (2), Ho (3), and Er (4)) and (N(nBu)4)2[LnIIIL{Mo5O13(OMe)4(NO)}]·CH2Cl2 (Ln = Nd (5) and Er (6)). The metal ions adopt distorted bicapped trigonal prismatic coordination environments, resulting in slow relaxation of the magnetization for 4. These compounds exhibit reversible redox waves at positive potentials, centered within the calix[4]arene ligand, representing a new type of calix[n]arene-based electrochemical activity induced by coordination to the metal centers.
Introduction
Calix[4]arenes (Fig. 1) are typically cone-shaped molecules constructed by the repetition of four phenolic units linked by methylene bridges.1 The organic skeleton is not electrochemically active, presenting an irreversible oxidation wave at high potentials.2 However, their functionalization with external redox centers (e.g. ferrocene,3 quinone,4 bipyridine, TTF,5 and nitro6 groups) facilitates electrochemical investigation and potential exploitation in various fields, such as sensors, catalysis, ion selectivity, energy storage devices, and molecular electronics. Furthermore, the electrochemical activity is specifically localized within these newly introduced redox-active groups, and it is influenced by the surrounding structural environment and susceptible to alterations in response to external stimuli.7 | ||
| Fig. 1 (a) Representation of calix[4]arene (C[4]) and p-tert-butylcalix[4]arene (TBC[4]), (b) p-tert-butylcalix[4](OMe)2(OH)2arene (H2L) originating from bis-methylation at two distal positions of TBC[4]. | ||
Another important approach is the utilization of its hydroxy groups at the lower rim for complexation with lanthanide metal centers. While calix[4]arenes, and more specifically p-tert-butylcalix[4]arene (TBC[4]), are ideal to construct polynuclear complexes with lanthanide (Ln) metal ions8 (with 42 entries for polynuclear and 6 for mononuclear calix[4]arene-based structures with Ln ions in the Cambridge Structural Database), the bis-methylation at the distal positions provides a slightly different ligand, i.e. p-tert-butylcalix[4](OMe)2(OH)2 arene (referred to hereafter as H2L; Fig. 1b), which favors the construction of monometallic Ln-based complexes.9 The design of compounds with single lanthanide metal ions has been specifically investigated in the design of single-ion magnets (SIMs) due to their large intrinsic magnetic anisotropy and the possibility to change the alignment of the anisotropy axis via judicious choice of the ligands.10 These molecules have been suggested for potential applications in molecular spintronics and quantum technologies, where specific symmetries such as D4d, D5h, and D6d are proposed in the design of high-performance LnIII-SIMs.11 In 2013, Zuo et al. reported a seven-coordinate dysprosium ion encapsulated between an H2L and a Kläui-type tripodal ligand displaying field-induced (dc = 900 Oe) SIM behavior with Ueff = 73.7 K and τ0 = 9.1 × 10−9 s.12 Recently we investigated the use of H2L in combination with acetylacetonate (acac) and polyoxopentamolybdate to obtain (N(nBu)4)[TbIIIL(acac)2] and (N(nBu)4)2[DyIIIL{Mo5O13(OMe)4(NO)}], where the LnIII ions are eight-coordinate in distorted square-antiprismatic coordination geometries (D4d symmetry), presenting slow relaxation of the magnetization.13
Early and late Ln ions also show potential for constructing SIMs, although they are the subject of significantly fewer studies. Herein, we have expanded our previous work to both early and late Ln metals featuring (N(nBu)4)[LnIIIL(acac)2]·CH3CN (acac = acetylacetonate; Ln = Pr (1), Nd (2), Ho (3), and Er (4)) and (N(nBu)4)2[LnIIIL{Mo5O13(OMe)4(NO)}]·CH2Cl2 (Ln = Nd (5) and Er (6)). While our previous reports concentrated on the magnetic properties resulting from coordination to Gd, Tb, and Dy ions, this study expands upon our prior investigations to offer a comprehensive understanding of the redox activity facilitated by metal coordination. The studied compounds display two reversible or quasi-reversible redox waves at positive potentials localized within the L2− ligand. Given that the employed lanthanide centers are not redox-active, our investigations demonstrate that this unprecedented electrochemical activity on the calix[4]arene ligand is induced by the coordination of lanthanide metal centers.
Results and discussion
Structure descriptions
A two-step synthetic strategy (Fig. 2) was applied to obtain the hybrid complex compounds (N(nBu)4)2[LnIIIL{Mo5O13(OMe)4(NO)}]. The first step involved the synthesis of a series of (N(nBu)4)[LnIIIL(acac)2] complexes by reacting (N(nBu)4)(acac), LnIIICl3 and, H2L in dry toluene at reflux for 6 h under Schlenk conditions (Fig. 2, center). These complexes served as good starting materials for a further metathesis reaction with the monolacunary Lindqvist-type pentamolybdate14 [Mo5O13(OMe)4(NO)(Na(MeOH))]2− to obtain the targeted [LnIIIL{Mo5O13(OMe)4(NO)}]2− complexes (Fig. 2, right). The six complexes crystallize in the monoclinic system and structure solution was performed in the C2/c space group. The SCXRD data reveal that complexes within families 1–4 and 5, 6 are structurally analogous, we therefore only discuss the structural description of the representative erbium derivatives 4 and 6.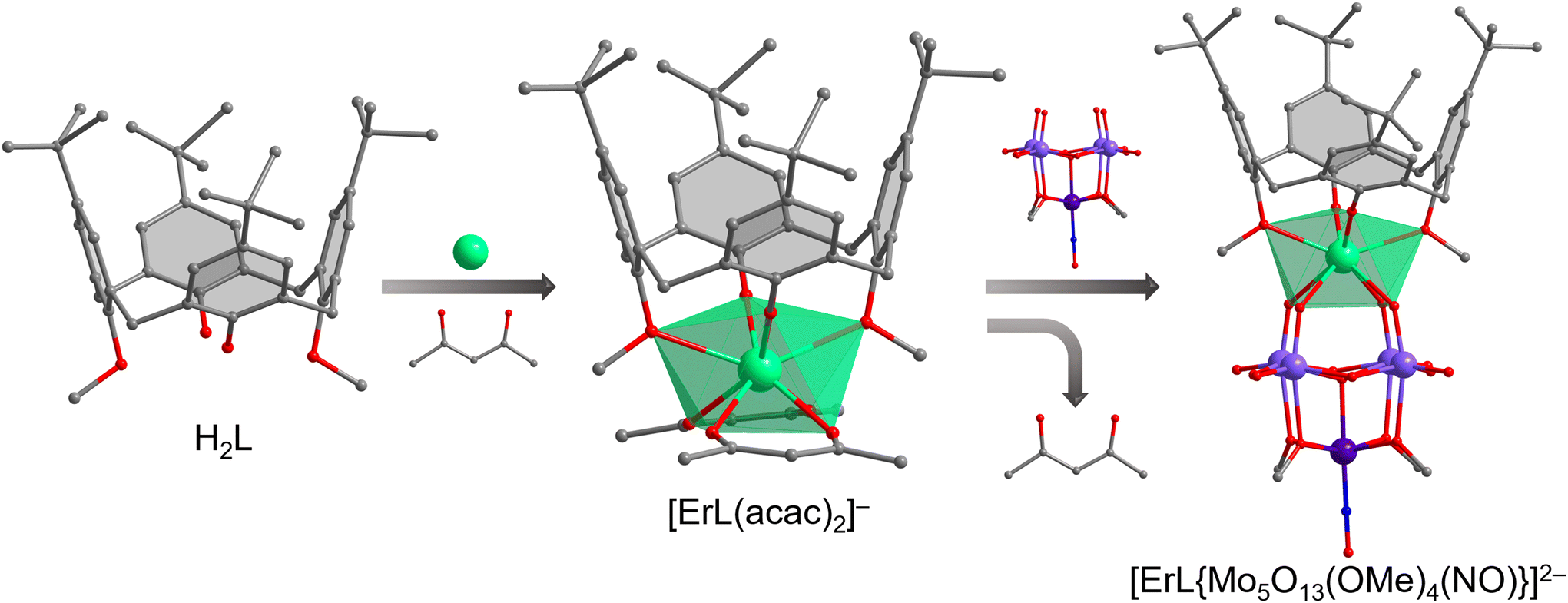 | ||
Fig. 2 The combination of acac and H2L ligands under basic conditions yields [ErIIIL(acac)2]−. A metathesis reaction of [ErIIIL(acac)2]− with [Mo5O13(OMe)4(NO)Na(MeOH)]2− in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry provides [ErIIIL{Mo5O13(OMe)4(NO)}]2−. In both structures, ErIII resides in a distorted bicapped trigonal prismatic ErIIIO8 environment. Color code: Er: green, O: red, N: blue, MoIV: purple, MoNO: dark purple, C: gray. N(nBu)4+, solvents and H atoms are omitted for clarity. :1 stoichiometry provides [ErIIIL{Mo5O13(OMe)4(NO)}]2−. In both structures, ErIII resides in a distorted bicapped trigonal prismatic ErIIIO8 environment. Color code: Er: green, O: red, N: blue, MoIV: purple, MoNO: dark purple, C: gray. N(nBu)4+, solvents and H atoms are omitted for clarity. | ||
Colorless rod-like single crystals of 4 were obtained by slow diffusion of CH3CN into a concentrated THF solution. The H2L ligand is fully deprotonated (L2−) and coordinates to the erbium metal ion as a tetradentate ligand through the lower-rim O sites (ErIII–OPh: 2.147(4) Å; ErIII–OMePh: 2.600(4) Å). The L2− does not adopt the typical bowl shape (C4v) but a pinched conformation (C2v) where the two methylated aromatic rings are nearly coplanar and the two phenoxide rings splay (Fig. 3). This conformation creates an O4 pocket defined by two edge-sharing triangular units (O1/O1/O2, Fig. 3a) with a dihedral angle of 24.0°. The further coordination of two acac ligands (ErIII–O: 2.350(4) and 2.364(4) Å) completes the distorted bicapped trigonal prismatic O8 environment of the ErIII center (Fig. S1†).
 | ||
| Fig. 3 Top view of the (a) [ErIIIL(acac)2]− and (b) [ErIIIL{Mo5O13(OMe)4(NO)}]2− displaying the typical pinched conformation of the L2− ligand and its C2v symmetry. Color code: Er = green, O = red, MoIV = purple, and C = gray. N(nBu)4+ and H-atoms are omitted for clarity. | ||
Brown rod-like single crystals of 6 were grown by slow evaporation of a concentrated solution of the complex in dichloromethane/n-hexane (1/3, v/v) for one week. The monolacunary Lindqvist pentamolybdate [Mo5O13(OMe)4(NO)]3− grows from a central oxygen atom (μ5-Oc) bridging to four equatorial MoVI ions (MoVI–Oc: 2.296(5)–2.364(5) Å) and one axial [Mo(NO)]3+ unit (Mo–Oc: 2.109(5) Å). On the outside, four μ2-Ob connect the central MoVI ions (MoVI–Ob: 1.900(5)–1.923(5) Å). The link between the central MoVI ions and the axial [Mo(NO)]3+ unit is completed by four μ2-OOMe methoxy groups (MoVI–OOMe: 2.249(5)–2.282(5) Å; Moaxial–OOMe: 1.994(5)–2.012(5) Å). The remaining coordination sites are constituted of four terminal oxo groups (MoVI–Ot: 1.693(5)–1.701(5) Å) and one linear nitrosyl group (Moaxial–NNO: 1.755(7) Å). The lacunary Mo5 provides four reactive oxygen atoms (MoVI–O: 1.741(5)–1.755(5) Å) in a nearly O4 square (O⋯O: 2.754(7)–2.848(7) Å) to coordinate with the [ErIIIL]+ unit. The calix[4]arene adopts the same pinched conformation as in complex 4, with a 24.3° dihedral angle between the two adjacent triangles O1/O3/O2 and O1/O3/O4 (Fig. 3b) formed by the lower-rim O4 oxygen atoms (O⋯O: 2.835(7)–2.968(6) Å). The two tetradentate ligands generate a distorted bicapped trigonal prismatic ErIIIO8 environment (Fig. S1;† ErIII–OPOM: 2.408(5)–2.479(5) Å; ErIII–OPh: 2.107(5) and 2.120(5) Å; ErIII-OMePh: 2.518(4) and 2.552(5) Å).
In the crystal lattice, complexes 4 and 6 (Fig. S2 and S3†) pack in an antiparallel bilayer array arrangement in which the molecules interdigitate, with the presence of N(nBu4)+ countercations in the interstitial space for charge balance. The shortest ErIII–ErIII distance is 11.204 Å (4) and 13.155 Å (6), and the closest intermolecular contacts between molecules are mediated by CH3(tBu)–CH3(acac) and CH3(tBu)–CH(Ph) interactions at 3.643 and 3.744 Å, respectively (4) and NO–CH3(POM) and CH3(POM)–CH3(POM) interactions at 3.855 and 4.194 Å, respectively (6).
ESI-HMRS, FT-IR, UV-Vis, and TGA experiments
The ESI-HRMS spectra in the negative-ion mode of 1–6 (Fig. S8–S11†) show fragmentations related to the loss of N(nBu4)+ countercations. In 1–4, the presence of only a main ion is observed, assigned to [M − N(nBu4)]− (M = N(nBu)4)[LnIIIL(acac)2] in the m/z region of 1013–1041. Complexes 5 and 6 present two different fragmentations, the singly [M − N(nBu)4]− (m/z region of 1904–1926) and the doubly charged [M − 2N(nBu)4]2− (m/z region of 830–842) ions, where (M = N(nBu)4)2[LnIIIL{Mo5O13(OMe)4(NO)}]. The isotopic distributions of the calculated species in 1–6 perfectly match the experimental values. In the FT-IR spectra of 1–6 (Fig. S12–S14†), the most relevant bands are related to vibrations associated with ν(C–H) ∼3000–2800 cm−1 (s), overlapping vibrations of ν(arC–C)/δ(CH2)/δas(CH3) ∼1480–1410 cm−1 (vs), δs(CH3) ∼1332 cm−1 (m), ν(C–O) ∼1210 cm−1 (w). The strong stretching band appearing at ∼1597 cm−1 in 1–4 is assigned to C–Odelocalized in the acac ligand. In 5 and 6, specific groups of the POM show vibrations related to ν(N–O) ∼1622 cm−1 (m), ν(C–O) ∼1037 cm−1 (m), ν(Mo–Ot) ∼927–838 cm−1 (s), ν(Mo–O–Mo) ∼680 cm−1 (vs). The TGA results for 1–4 (Fig. S15 and S16†) reveal thermal stability up to ∼160 °C for the complexes, characterized by a slight decline in mass given the loss of crystallization solvent within the calixarene cavities. Above this temperature, the complexes start to decompose. However, complexes 5 and 6 are thermally stable up to ∼280 °C (Fig. S17†). The electronic absorption spectra of 1–4 (Fig. S18 and S19†) in CH3CN solutions display two intense absorption bands at ∼282 and 258 nm, corresponding to the typical π–π* electronic transitions centered on the phenolic rings of the L2− ligand. Upon substitution of the acac by the {Mo5} ligand in 5 and 6, the bands display a small red shift to ∼310 (5) and 258 nm (6), in agreement with the color change from colorless in complexes 1–4 to brown in 5, 6. In addition, a shoulder band at ∼258 nm can be attributed to the n–π* electron transition in the [Mo5O13(OMe)4(NO)]3− unit.Magnetic studies
The magnetic properties of complexes 1–6 at static fields are shown in Fig. 4a as χmT vs. T at 0.1 T and Mmvs. B at 2.0 K and B = 0.1–5.0 T. At 290 K, the χmT values of 1.51 (1), 1.50 (2), 13.55 (3), 11.10 (4), 1.50 (5), and 11.21 cm3 K mol−1 (6) are within the expected ranges15 for the respective isolated LnIII centers: 1.45–1.62 (1, PrIII), 1.45–1.53 (2 and 5, NdIII), 13.26–13.78 (3, HoIII), and 11.05–11.28 cm3 K mol−1 (4 and 6, ErIII). χmT values remain nearly constant down to 100 K (1, 2 and 5) and 150 K (3, 4 and 6). Subsequently, complexes 1, 2 and 5 exhibit a faster decrease to minima values of 0.07 (1), 0.48 (2) and 0.67 cm3 K mol−1 (5) at 2.0 K. While 3, 4 and 6 show a progressive decrease to 12.58 (3), 8.36 (4) and 9.36 (6) in the temperature range of 150–25 K, wherefrom they approach 8.13 (3), 6.06 (4) and 5.83 (6) at 2.0 K, respectively. Their behavior at lower temperatures (<25 K) is due to the thermal depopulation of the mJ energy sublevels of the ground terms (3H4 for PrIII, 4I9/2 for NdIII, 5I8 for HoIII, and 4I15/2 for ErIII) arising from the spin–orbit coupling, interelectronic repulsion and ligand field. At 2 K, the molar magnetization Mmvs. applied field B (Fig. 4a, inset) of 1–6 show an approximately linear dependence in the range of 0–5 T. The Mm values are 0.32 (1), 1.2 (2), 6.5 (3), 4.6 (4), 1.4 (5), and 5.0NAμB (6) at 2 K and 5 T. They do not reach the saturation point (PrIII 3.2, NdIII 5.7, HoIII 10.0, and ErIII 9.0NAμB) due to the measurement of ground/powdered samples, i.e. the determination of the mean value of randomly oriented crystallites consisting of magnetically anisotropic centers.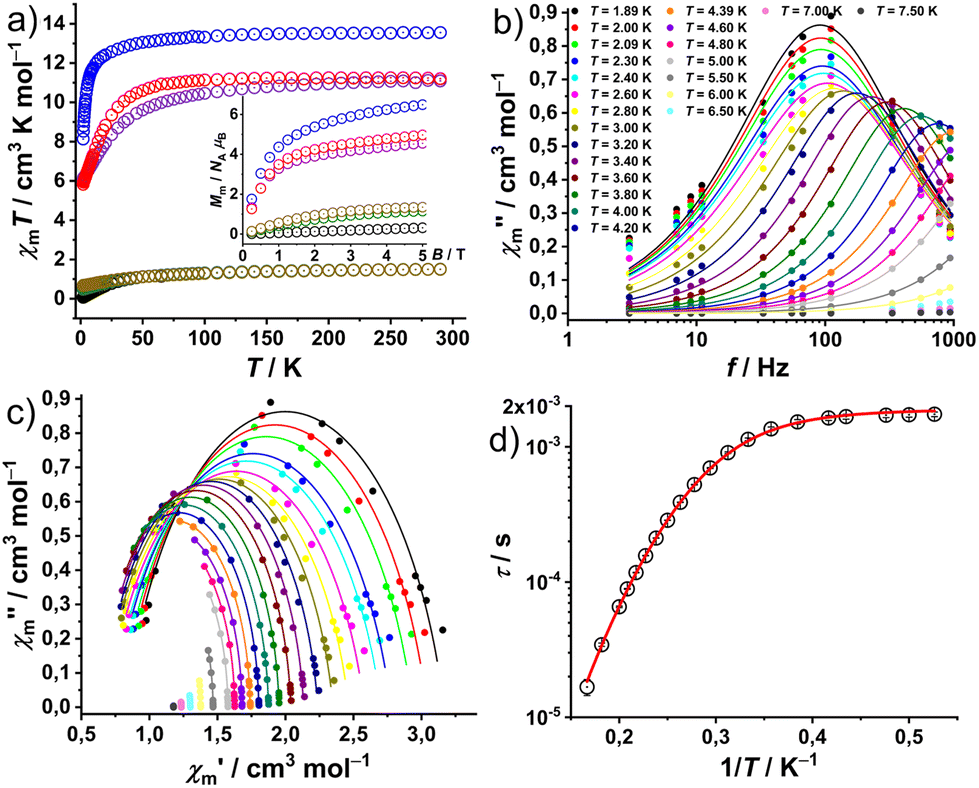 | ||
| Fig. 4 (a) Susceptibility data for complexes 1–6: χmT vs. T at 0.1 (circles: 1 black, 2 green, 3 blue, 4 purple, 5 yellow, and 6 red) and Mmvs. B at 2.0 K (inset; same colour code as in 4a). (b) Out-of-phase molar magnetic susceptibility χ′′mvs. frequency f for 4; (c) Cole–Cole plot of 4 in the range 1.89–7.5 K at a static bias field of 300 Oe (circles: data, lines: fits to a generalised Debye expression). (d) Plot of relaxation time τ vs. T−1 (circles) for 4; the solid red line represents a fit considering quantum tunnelling of magnetisation and Raman relaxation processes. | ||
In complex 4 out-of-phase (χ′′m) signals were detected at a static bias field, with more pronounced curvature observed at 300 Oe in the Cole–Cole plot of the out-of-phase (χ′′m) vs. the in-phase magnetic susceptibility (χ′m) data (Fig. 4c). The simultaneous fitting of a generalized Debye expression16 to the χ′mvs. f (Fig. S20†) and χ′′mvs. f data, yields the solid lines shown in Fig. 4b, c and the relaxation times τ with the distribution α = 0.095 ± 0.082, indicating the presence of multiple relaxation pathways. The best reproduction of the Arrhenius plot τ vs. 1/T data, as shown in Fig. 4d, required considering a field-independent contribution of the quantum tunneling of magnetization (QTM) and a Raman slow relaxation process, as found for analog compounds before,13a and as expressed in the formula τ−1 = B + CTn. The best-fit yields a QTM with B = (529 ± 10) s−1 and, a constant C = (0.143 ± 0.008) s−1 K−n and an exponent n = 7.2 ± 0.1 for the Raman process. An exponent of 7 is well-known from literature and first reported by Kronig.17a It describes the typical Raman process by spin-two-phonon interaction for which the phonon energy is larger than the splitting of ground and excited state.17b
Electrochemical studies
The electrochemical experiments of 1–6 (cyclic voltammetry – CV and rotating disk voltammetry – RDV) were recorded with GC-disk electrodes (stationary and rotating) in dry DMF solutions. The results of oxidation and reduction potentials are summarized in Table S5† and the voltammograms are presented in Fig. 5 and S24.† At positive potentials, the [LnIIIL(acac)2]− family displays two reversible oxidation steps (Fig. 5a and c), with potential differences of 200–270 mV between them. The first oxidation step in 1–4 appears at a potential range of 0.33–0.34 V, showing full reversibility with a peak separation of about 70 mV and a comparable cathodic/anodic peak current ratio. The second oxidation step in 1 and 2 occurs at approximately 60–70 mV less positive than for 3 and 4, and the cathodic counter peak is nearly absent, whereas in 3 and 4 the reversibility is still evident. The decrease of reversibility (quasireversibility) of the second oxidation step in 1–4 is caused by a combination of slower heterogeneous kinetics on the electrode (manifested by anodic/cathodic peak separation of ∼160 mV) and by a relatively fast follow-up reaction (displayed by nearly absent cathodic counterpeak at the standard scan rate of 200 mV s−1). At negative potentials, no reduction behavior is observed. We postulate that the two (quasi)reversible redox couples represent the formation of the “phenoxy radical” in the calix[4]arene unit, which is further oxidized to “cyclohexadienone” in the second step (Fig. 5). Such electrochemical transformations related to these species have been previously reported in p-substituted phenolic molecules.18 Due to the absence of electronic communication between the two phenolic units within the C2v symmetrical calix[4]arene, the oxidation reactions within each phenolic unit occur simultaneously, resulting in overlapping of the anodic peaks/waves. | ||
| Fig. 5 (a) Cyclic voltammograms (200 mV s−1) of oxidation of 1–4 (1 light green; 2 dark green; 3 pink; 4 purple). (b) RD-voltammograms (1000 rpm) of oxidation of 1–4. (c) Cyclic voltammograms (200 mV s−1) of oxidation of 5 (orange) and 6 (blue). (d) RD-voltammograms (1000 rpm) of oxidation of 5 and 6. (e) Proposed oxidation mechanism. | ||
CV and RDV of the [LnIIIL{Mo5O13(OMe)4(NO)}]2− family (Fig. 5b and d) exhibit two fully reversible one-electron oxidation processes at 0.53 ± 10 mV and 0.78 ± 20 mV (CV-peak separation about 60–80 mV and comparable cathodic/anodic peak current ratio). The observed potential difference of approximately 260 mV between the two reversible oxidation steps in 5 and 6 is in good agreement with the range of values found in 1–4. This alignment suggests that 5 and 6 exhibit the same oxidation processes, with only a shift by about 200 mV towards more positive values, attributed to the slightly more intensive electron-withdrawing effect of the [Mo5O13(OMe)4(NO)]3− unit comparing to the acac ligands. At negative potentials, four reduction signals can be observed in 5 and 6 (Fig. S25†), probably related to the reduction and re-oxidation of four MoIV and one MoII ions in [Mo5O13(OMe)4(NO)]3−, given the non-innocent nature of this ligand.
By comparing the oxidation processes in 1–6, we found that the first anodic potential in 1–4 (0.33 V), and 5 and 6 (0.53 V) are identical. This observation indicates that the type of lanthanides has a small influence on the electronic properties of the phenoxy radical. However, in the second oxidation step, early lanthanides Pr (1) and Nd (2, 5) complexes are oxidized at less positive potentials than late lanthanides Ho (3) and Er (4, 6) by approximately 60 mV. Here, the subtle yet consistent shift in the oxidation potentials is affected by the coordination to the lanthanide metals, which agrees with the different nature of the bond as depicted in Fig. 5e, right.
Conclusions
In summary, we expand our study on the two previously reported families of monometallic lanthanide complexes to early and late lanthanide metals featuring (N(nBu)4)[LnIIIL(acac)2]·CH3CN (Ln = Pr (1), Nd (2), Ho (3), and Er (4)) and (N(nBu)4)2[LnIIIL{Mo5O13(OMe)4(NO)}]·CH2Cl2 (Ln = Nd (5) and Er (6)). The erbium complex (4) shows typical SIM behavior in the presence of a bias field, where different relaxation processes are active. The analysis of the magnetization reversal indicates both QTM and Raman processes. Given that uncoordinated H2L is not redox-active by itself, the coordination to the lanthanide metal ions triggers a new type of electroactivity in the calix[4]arene ligand without the necessity of its derivatization with redox-active groups (e.g. ferrocene, nitro, bipyridine, cobaltocenium). The redox waves observed by cyclic voltammetry are reminiscent of the ones obtained with the tetrathiafulvalene (TTF) ligand and derived compounds. This finding paves the way for future endeavors in the creation of new families of calix[n]arene-based monometallic lanthanide complexes, in which to explore the intrinsic electrochemical activity and to study shifts in the redox activity associated with their host/guest chemistry ability of the calix[4]arene unit.Experimental section
All reagents (99.9%) used in this study were obtained from commercial sources. High-grade dry solvents were obtained using a MBRAUN MB-SPS 800 solvent purification system. (N(nBu)4)[LnIIIL(acac)2]·CH3CN and (N(nBu)4)2[LnIIIL{Mo5O13(OMe)4(NO)}]·CH2Cl2 were synthesized following our previous work.13Synthesis of (N(nBu)4)[LnIIIL(acac)2]·CH3CN
Acetylacetone (0.20 mL, 2.0 mmol) and N(nBu4)OH (1.30 mL from a 40 wt% in MeOH, 2.0 mmol) were dissolved in 40 mL of MeOH and stirred at 60 °C for two hours. The solvent was then evaporated to dryness resulting in a yellow oil of N(nBu4)(acac). The obtained product, LnIIICl3 (74.72 mg for PrCl3, 135.64 mg for HoCl3, 125.29 mg for NdCl3, 136.80 mg for ErCl3, 0.50 mmol), and H2L (338.49 mg, 0.50 mmol) were dissolved in 50 mL of dry toluene under Ar and stirred at 135 °C for six hours. The resulting solution was cooled, filtered, evaporated to dryness, and washed with CH2Cl2 to obtain a white powder. Colorless single crystals were obtained by slow diffusion of CH3CN in a concentrated solution of THF containing the product. Yield (235 mg, 39% for 1); (277 mg, 42% for 2); (260 mg, 40% for 3), and (240 mg, 36% for 4).Synthesis of (N(nBu)4)2[LnIIIL{Mo5O13(OMe)4(NO)}]·CH2Cl2
(N(nBu)4)[LnIIIL(acac)2]·CH3CN (0.1 mmol) and (N(nBu)4)2[Mo5O13(OMe)4(NO)(Na(MeOH))] (113.52 mg, 0.1 mmol) were dissolved in 50 mL of dry CH3CN and stirred under reflux for 12 hours. The resulting solution was then filtered and evaporated to yield a brown oil. Consecutive re-dissolution, filtration, and evaporation of this crude in CH2Cl2, CH3CN, and THF yielded a brown solid free of unreacted starting materials and by-products. This solid was washed with 500 mL Et2O, filtered, and evaporated to afford a brown solid. Brown single crystals were obtained by slow diffusion of hexane in a concentrated solution of CH2Cl2 containing the product. Yield (39 mg, 18% for 5) and (33 mg, 15% for 6).
Crystal
data for (1) (CCDC 2299497†): C74H111N2O8Pr, Mr = 1297.55 g mol−1, colorless plate, 0.07 × 0.10 × 0.27 mm3, monoclinic, space group C2/c, a = 23.746(5) Å, b = 16.124(3) Å, c = 21.064(4) Å, α = 90°, β = 120.16(3)°, γ = 90°, V = 6973(3) Å3, Z = 4, STOE STADIVARI diffractometer, MoKα radiation (λ = 0.71073 Å), T = 100 K, 6124 reflections collected, 36655 unique (Rint = 0.0828), 4056 observed (I > 2σ(I)). Final GooF = 1.018, R1 = 0.0611 (I > 2σ(I)) and wR2 = 0.1612 (all data).
Crystal data for (2) (CCDC 2299498†): C74H111N2O8Ho, Mr = 1321.57 g mol−1, colorless plate, 0.05 × 0.05 × 0.20 mm3, monoclinic, space group C2/c, a = 23.831(5) Å, b = 16.057(3) Å, c = 21.071(4) Å, α = 90°, β = 120.42(3)°, γ = 90°, V = 6953(3) Å3, Z = 4, STOE STADIVARI diffractometer, MoKα radiation (λ = 0.71073 Å), T = 100 K, 6136 reflections collected, 36272 unique (Rint = 0.0697), 5110 observed (I > 2σ(I)). Final GooF = 1.043, R1 = 0.0581 (I > 2σ(I)) and wR2 = 0.1486 (all data).
Crystal data for (3) (CCDC 2299499†): C74H111N2O8Nd, Mr = 1300.88 g mol−1, colorless plate, 0.05 × 0.11 × 0.32 mm3, monoclinic, space group C2/c, a = 23.771(5) Å, b = 16.146(3) Å, c = 21.069(4) Å, α = 90°, β = 120.26(3)°, γ = 90°, V = 6984(3) Å3, Z = 4, STOE STADIVARI diffractometer, MoKα radiation (λ = 0.71073 Å), T = 100 K, 6166 reflections collected, 35449 unique (Rint = 0.1083), 4033 observed (I > 2σ(I)). Final GooF = 1.035, R1 = 0.0697 (I > 2σ(I)) and wR2 = 0.1848 (all data).
Crystal data for (4) (CCDC 2299500†): C74H111N2O8Er, Mr = 1323.90 g mol−1, colorless plate, 0.075 × 0.21 × 0.30 mm3, monoclinic, space group C2/c, a = 23.790(5) Å, b = 16.055(3) Å, c = 21.038(4) Å, α = 90°, β = 120.40(3)°, γ = 90°, V = 6931(3) Å3, Z = 4, STOE STADIVARI diffractometer, MoKα radiation (λ = 0.71073 Å), T = 100 K, 6114 reflections collected, 32953 unique (Rint = 0.0657), 5309 observed (I > 2σ(I)). Final GooF = 1.047, R1 = 0.0579 (I > 2σ(I)) and wR2 = 0.1487 (all data).
Crystal data for (5) (CCDC 2299501†): C83H144N3O22Cl2Mo5Nd, Mr = 2230.84 g mol−1, brown needle, 0.10 × 0.14 × 0.41 mm3, monoclinic, space group C2/c, a = 67.598(14) Å, b = 12.541(3) Å, c = 23.425(5) Å, α = 90°, β = 103.49(3)°, γ = 90°, V = 19311(7) Å3, Z = 8, STOE STADIVARI diffractometer, MoKα radiation (λ = 0.71073 Å), T = 100 K, 16875 reflections collected, 87889 unique (Rint = 0.0652), 13308 observed (I > 2σ(I)). Final GooF = 1.025, R1 = 0.0490 (I > 2σ(I)) and wR2 = 0.1430 (all data).
Crystal data for (6) (CCDC 2299502†): C83H144N3O22Cl2Mo5Er, Mr = 2253.86 g mol−1, brown needle, 0.07 × 0.20 × 0.33 mm3, monoclinic, space group C2/c, a = 66.701(13) Å, b = 12.581(3) Å, c = 23.523(5) Å, α = 90°, β = 103.25(3)°, γ = 90°, V = 19311(7) Å3, Z = 8, STOE STADIVARI diffractometer, MoKα radiation (λ = 0.71073 Å), T = 100 K, 16897 reflections collected, 85513unique (Rint = 0.0898), 11966 observed (I > 2σ(I)). Final GooF = 1.005, R1 = 0.0660 (I > 2σ(I)) and wR2 = 0.1833 (all data).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Chinese Scholarship Council (CSC) and the Punjab Educational Endowment Fund (PEEF) for Dr Yushu Jiao and Dr Sidra Sarwar's scholarships. J. Ludvík and L. Koláčná are grateful to the grant GAČR 230646 S and to the institutional support RVO 61388955.References
-
C. D. Gutsche, Calixarenes 2001, Kluwer Academic Publishers, 2001 Search PubMed
.
- B. C. M. A. Ashwin, R. K. Chitumalla, A. H. A. Baby, J. Jang and P. M. Mareeswaran, J. Inclusion Phenom. Macrocyclic Chem., 2018, 90, 51–60 CrossRef CAS
-
(a) P. D. Beer and A. D. Keefe, J. Chem. Soc., Dalton Trans., 1990, 3675–3682 RSC
-
(a) M. Gómez-Kaifer, P. A. Reddy, C. D. Gutsche and L. Echegoyen, J. Am. Chem. Soc., 1994, 116, 3580–3587 CrossRef
-
(a) B. T. Zhao, M. J. Blesa, N. Mercier, F. Le Derf and M. Salle, J. Org. Chem., 2005, 70, 6254–6257 CrossRef CAS PubMed
-
(a) A. Liška, P. Vojtíšek, A. J. Fry and J. Ludvík, J. Org. Chem., 2013, 78, 10651–10656 CrossRef PubMed
- T. D. Chung and H. Kim, J. Inclusion Phenom., 1998, 32, 179–193 CrossRef CAS
-
(a) G. Guillemot, B. Castellano, T. Prange, E. Solari and C. Floriani, Inorg. Chem., 2007, 46, 5152–5154 CrossRef CAS PubMed
- J. Gottfriedsen, R. Hagner, M. Spoida and Y. Suchorski, Eur. J. Inorg. Chem., 2007, 2288–2295 CrossRef CAS
-
(a) N. Ishikawa, M. Sugita, T. Okubo, N. Tanaka, T. Iino and Y. Kaizu, Inorg. Chem., 2003, 42, 2440–2446 CrossRef CAS PubMed
- J. L. Liu, Y. C. Chen and M. L. Tong, Chem. Soc. Rev., 2018, 47, 2431–2453 RSC
- F. Gao, L. Cui, Y. Song, Y. Z. Li and J. L. Zuo, Inorg. Chem., 2013, 53, 562–567 CrossRef PubMed
-
(a) Y. Jiao, S. Sarwar, S. Sanz, J. van Leusen, N. V. Izarova, C. L. Campbell, E. K. Brechin, S. J. Dalgarno and P. Kögerler, Dalton Trans., 2021, 50, 9648–9654 RSC
- A. Proust, P. Gouzerh and F. Robert, Inorg. Chem., 1993, 32, 5291–5298 CrossRef CAS
-
H. Lueken, Magnetochemie, Teubner Verlag, Stuttgart, 1999 Search PubMed
- K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 10, 98–105 CrossRef
-
(a) R. de L. Kronig, Physica, 1939, 6, 33–43 CrossRef
-
(a) J. A. Richards, P. E. Whitson and D. H. Evans, J. Electroanal. Chem., 1975, 63, 311–327 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and experimental data related to the discussion of results. CCDC 2299497–2299502. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03828a |
| This journal is © The Royal Society of Chemistry 2024 |