
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBimetallic CPM-37(Ni,Fe) metal–organic framework: enhanced porosity, stability and tunable composition†‡
Soheil
Abdpour
,
Marcus N. A.
Fetzer
,
Robert
Oestreich
 ,
Thi Hai Yen
Beglau
,
István
Boldog
* and
Christoph
Janiak
*
,
Thi Hai Yen
Beglau
,
István
Boldog
* and
Christoph
Janiak
*
Institut für Anorganische Chemie und Strukturchemie, Heinrich-Heine-Universität Düsseldorf, 40204 Düsseldorf, Germany. E-mail: boldogi@hhu.de; Janiak@hhu.de
First published on 16th January 2024
Abstract
A newly synthesized series of bimetallic CPM-37(Ni,Fe) metal–organic frameworks with different iron content (Ni/Fe ≈ 2, 1, 0.5, named CPM-37(Ni2Fe), CPM-37(NiFe) and CPM-37(NiFe2)) demonstrated high N2-based specific SBET surface areas of 2039, 1955, and 2378 m2 g−1 for CPM-37(Ni2Fe), CPM-37(NiFe), and CPM-37(NiFe2), having much higher values compared to the monometallic CPM-37(Ni) and CPM-37(Fe) with 87 and 368 m2 g−1 only. It is rationalized that the mixed-metal nature of the materials increases the structural robustness due to the better charge balance at the coordination bonded cluster, which opens interesting application-oriented possibilities for mixed-metal CPM-37 and other less-stable MOFs. In this work, the CPM-37-derived α,β-Ni(OH)2, γ-NiO(OH), and, plausibly, γ-FeO(OH) phases obtained via decomposition in the alkaline medium demonstrated a potent electrocatalytic activity in the oxygen evolution reaction (OER). The ratio Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe ≈ 2 from CPM-37(Ni2Fe) showed the best OER activity with a small overpotential of 290 mV at 50 mA cm−2, low Tafel slope of 39 mV dec−1, and more stable OER performance compared to RuO2 after 20 h chronopotentiometry at 50 mA cm−2.
:Fe ≈ 2 from CPM-37(Ni2Fe) showed the best OER activity with a small overpotential of 290 mV at 50 mA cm−2, low Tafel slope of 39 mV dec−1, and more stable OER performance compared to RuO2 after 20 h chronopotentiometry at 50 mA cm−2.
Introduction
Metal–organic frameworks (MOFs) are known for their tuneability stimulating both the fundamental and the applied research dimensions. The variability of MOFs, which are predominantly crystalline porous coordination polymers (PCPs), are usually, but not exclusively, associated with the tailorability of the organic ligand. The high-surface area, the variable size-, shape-, and nature of the pore surface are excellent prerequisites for applications such as gas storage and separation, catalysis, as well as energy conversion.1An interesting aspect of MOF-tuneability is the mixed-metal MOF approach towards functional materials,2,3 with such recent representative reported examples as luminescence-based temperature-sensing in a mixed lanthanide [(CH3)2NH2][(Eux/Tb1−x)(biphenyl-3,3′,5,5′-tetracarboxylate)] complex,4 benzene oxidation by a mixed-metal MOF-derived CeO2-Cr2O3 catalyst, and CO2 reduction to MeOH by a MOF-74(Cu, Zn) catalyst,5 water stability and/or adsorption tuning in UiO-66 and MOF-808(Zr,Ce),6 MOF-74(Mg, M) M = Co, Ni,7,8 HKUST-1(Cu, M) M = Ca, Mg, Co, Zn,9 as well as, proton conductance in MOF-808(Zr,Ce).10 Noteworthy, an increased stability of mixed-metal MOFs is sometimes emphasized.2,3 For example Mg-MOF-74 gains water stability by incorporation of Ni2+ or Co2+.7,8 Bimetallic Ce/Zr-UiO-66 and Ce/Zr-MOF-808 have higher thermal stability and acid resistance.6
In the absolute majority of published cases, the metal distribution corresponds to a solid solution, i.e. it is governed statistically. However, the non-contiguous coordination clusters, which are often constituting the secondary building units, are potentially well-suited for a structurally defined mixed-metal composition. The latter is equivalent to a uniform distribution of the metals on the nano-level. Electrode materials, which are represented by or derived from MOFs, are particularly interesting objects, due to the synergy of uniformly distributed two or more metal ion types. Current research also investigates electrode materials that rely on solid-solution type MOFs, e.g. the [Cd4CuO(calix[4]resorcinarene)(H2O)4]·4DMF·5H2O for electrocatalytic oxidation of uric acid,11 mixed-metal MOF derived CeO2-Cr2O3 oxides for catalytic benzene oxidation,12 and others, mentioned below.
Our interest in this contribution also focuses on electrocatalysts for the oxygen-evolution reaction (OER) as a test area for optimization of mixed-metal MOF-based synergy. The focus is highly topical in the context of hydrogen economy, as electrochemical water splitting is practically the only readily available means to generate green hydrogen in large quantities.13
During the past decades, nickel-based materials—as a cost-effective alternative to the benchmark iridium and ruthenium oxides (IrO2 and RuO2)—including sulfides,14 oxides,15 phosphides,16 selenides,17 metal oxide/(oxy)hydroxides,18 received significant attention for developing high-performance anodic electrocatalysts. The potential porosity of the electrode materials alleviates diffusion limitations and increases the apparent surface area, which was proven to be beneficial for improving the electrocatalytic performance.19 Hence, MOFs are among promising candidates for precatalysts of electrode materials.20
Interestingly, the decomposition of MOFs during the OER in an alkaline aqueous medium sometimes leads to in situ formed, structured (also seemingly hierarchically porous) stable residues, typically represented by metal hydroxides/oxides.21 In this case, the MOF is regarded as a ‘precatalyst’, which influences the nature of the catalyst actually formed during the electrochemical process.22,23 This ‘conversion’ approach, which does not demand any special additional treatment, such as an energetically demanding pyrolysis, is particularly attractive, even if there is no general reliable way to predict the activity of the catalyst from the exact nature- and treatment method of the precatalyst. Some examples of mixed-metal MOF-derived materials were also reported recently, e.g. the MOF-derived NixCo3−xO4 spinels for enhanced oxygen evolution,24 or Fe/Co MOF derived electrocatalysts for water splitting.25,26 It is worth mentioning again that the MOF precursors represent solid solutions with statistical distribution of metals, as discussed above.
In this work, the highly porous CPM-37 MOF platform – which is a permanently porous derivative of the well-known flexible MIL-88 MOF with a ‘pore-space-partitioning’ trigonal structure ligand – was studied in the context of increasing its permanent stability using bimetallic CPM-37(Ni,Fe) compound class extensions of the known CPM-37(Ni), featuring low stability.27 A series of bimetallic CPM-37(Ni,Fe) represented by CPM-37(Ni2Fe), CPM-37(NiFe), and CPM-37(NiFe2) with different molar ratios between nickel and iron were synthesized for the first time (Fig. 1a–c and Fig. S16‡). The novel mixed-metal CPM-37(Ni,Fe) MOFs with enhanced stability compared to the single-metal analogues due to the stabilization of certain metal ratios in the cluster were evaluated regarding the synergy between iron and nickel for the intended use as an electrode precatalyst (or precursor) material for the OER reaction. The selection of the nickel and iron metal pair for the bimetallic CPM-37s was stipulated by the known synergy of Ni and Fe in OER catalysts;28,29 see Table S14‡ for a short survey of materials, considered as relevant benchmarks in the context of this work.
 | ||
| Fig. 1 (a) Synthesis of CPM-37(Ni). (b) Network level representation of the CPM-37(Ni) structure with the green edges representing the MIL-88 sub-topology with the space-partitioning nodes and edges shown in violet. (c) Structure of CPM-37(Ni2Fe) with polyhedral representation of the Ni atom with the octahedral {NiO5N} environment. (d) Expected stable mixed-valence metal-based CPM-37 variants with the MIII metal content in the range of one to two ions per cluster with the general formula of the material given. (e and f) Conceived instable CPM-37 variants with too much or too scarce amount of MIII content compared to the expected optimum. | ||
Experimental
Materials and methods
All chemicals were purchased from commercial vendors and used as received: nickel nitrate hexahydrate, Ni(NO3)2·6H2O (98.5%, Merck GmbH); iron sulfate heptahydrate, FeSO4·7H2O (ACROS GmbH); biphenyl-4,4′-dicarboxylic acid, H2BPDCA (98%, Abcr GmbH); N,N-dimethylformamide, DMF (p.a., Fisher chemical GmbH); N-methyl-2-pyrrolidone, NMP (p.a., Fisher Scientific); dichloromethane, CH2Cl2 (p.a., Fisher Chemical GmbH); potassium hydroxide, KOH (1 mol L−1, Roth), hydrazine monohydrate N2H4·H2O (Thermo Scientific), polyvinylidene fluoride, PVDF (CAS: 24937-79-9, Sigma Aldrich); nickel foam, NF (99.5%, average porosity 95.2%, relative average density 4.8%, thickness 1.6 mm, produced by Recemat BV, Cell Material Engineering), and conductive Carbon Black (type Vulcan XC-72R, Fuelcellstore, product code 590106-1). Ultrapure water was produced using a Sartorius Arium Mini water purifier.Powder X-ray diffraction (PXRD) analysis was carried out at ambient temperature on a Rigaku Miniflex 600 powder diffractometer (Rigaku, Japan) using Cu Kα1,2 radiation with average λ = 1.5406 Å (40 kV, 15 mA, 600 W) and a flat silicon low background sample holder in the 2θ range of 5°–100°. Scanning electron microscopy (SEM) was used with a Jeol JSM-6510LV QSEM Advanced electron microscope equipped with a Bruker Xflash 410 silicon drift detector for energy-dispersive X-ray (EDX) spectroscopy to determine the morphology and establish the metal ratios in the samples. Independent quantification of the metal content in the samples was performed using a Perkin-Elmer PinaAcle 900T atomic absorption spectrometer. The Microtrac MRB Belsorp MAX II analyzer was used to record the nitrogen adsorption isotherms of the samples at 77 K. The solvent-exchanged samples were degassed at 60 °C and ∼5 × 10–2 mbar for 16 h before the gas adsorption measurements (the details on the solvent exchange with CH2Cl2 see in section 2.3, ESI‡). Thermogravimetry analysis (TGA) was conducted using a Netzsch TG 209 F3 Tarsus instrument with a 5 K min−1 heating rate using nitrogen as a carrier gas. Fourier transform infrared spectroscopy (FT-IR) measurements were conducted by a Bruker TENSOR 37 IR spectrometer in the range of 4000–400 cm−1. X-ray photoelectron spectroscopy (XPS) data were collected using a ULVAC-PHI Versa Probe II microfocus X-ray photoelectron spectrometer. The spectra were recorded using a polychromatic aluminum Kα X-ray source (1486.8 eV) and referenced to the carbon 1s orbital with a binding energy of 284.8 eV. The XPS spectra were processed using the CasaXPS 2.3.19PR1.0 software.
Electrochemical experiments
The nickel foam (NF), used as a support material for the electrode, was prepared as follows. A flat NF pad was accurately cut into 1 × 1 cm pieces. The NF pieces were washed with ultrasonic assistance using acetone (10 min, followed by drying in air) and cold (0 °C, intermediary ice-water bath) 2 mol L−1 HCl for 5 min to remove the surface nickel oxide (the very slow dissolution of nickel metal in cold HCl was implicitly employed). The treatment was followed by rinsing with ultra-pure water, further ultrasonication in ultra-pure water for 10 min, and finally in absolute EtOH for 10 min. The washed nickel foam pieces were then vacuum-oven dried for 15 min and used freshly as a substrate for the precatalyst-containing slurry.The electrochemical measurements were carried out at room temperature using a three-electrode cell setup of a Gamry Interface 1010E Potentiostat. A Pt foil- and a reversible hydrogen electrode (RHE) were used as a counter and a reference electrode, respectively. Nickel foam (NF) coated by the investigated electrode material was used as a working electrode (see below for a single exception). The coating was performed using a slurry in N-methyl-2-pyrrolidone (NMP), containing 5 mg precatalyst, 0.6 mg carbon black and 0.6 mg polyvinylidene fluoride (PVDF). The slurry was carefully distributed on one side of the NF piece, and then the precatalyst-coated NF was dried at 60 °C for 12 h in a vacuum oven. Before conducting the electrochemical measurement, the 1 mol L−1 KOH electrolyte was freed from oxygen by bubbling an N2 gas stream through the solution, which was maintained during the measurement. The RuO2 electrode as a benchmark was prepared using the same method. The Faradaic efficiency (FE) was calculated according to the method described in section 2.11.1, ESI‡.
The linear sweep voltammetry (LSV) at 5 mV s−1 was conducted to determine the electrocatalytic performance of the as-prepared samples. Before that, cyclic voltammetry was performed at 100 mV s−1 (20 cycles) between 1–1.7 V vs. RHE to stabilize the catalyst performance. All of the LSV polarization curves were corrected by iR compensation.30 The electrochemical impedance spectroscopy (EIS) was performed to determine the charge transfer resistance of the as-prepared samples in the frequency range of 1–100 kHz at 1.5 V vs. RHE. The stability of selected electrocatalysts was evaluated by chronopotentiometry at a fixed current density of 50 mA cm−2 for 20 h.
The tests regarding the conversion of CPM-37 in an aqueous alkaline medium without applying current were done by soaking ∼30 mg of a sample in ∼ 5 mL of 1 mol L−1 aqueous KOH for 20 h at room temperature. The obtained solid residue was separated by centrifugation and washed with 2 × 5 mL of water.
For post-mortem analysis after OER of the material derived from CPM-37(Ni2Fe), a special Ni-support was prepared from a standard piece of nickel foam (see above). The foam was compressed to form a foil-like support, which was then loaded with the slurry of 5 mg precatalyst and 0.6 mg polyvinylidene fluoride using the standard approach described above, however without the addition of carbon black. This procedure decreased the conductivity as well as the contact between the support surface and the deposited electrode material, and changed the properties of the electrode. However, it allowed an easy separation of the deposited electrode material after the OER, which was the sole goal of the procedure. The specially prepared electrode was subjected to OER in the form of standard 20-hour chronopotentiometry (see above). The converted electrode material was removed by sonication directly in the electrochemical cell and separated by centrifugation in a quantity, sufficient for small-scale PXRD and XPS analyses (this method ensured virtually no admixture of nickel metal and a decreased carbon content).
Synthesis of the N,N′,N′′-tris-(pyrid-4-yl)-trimesamide (TPAMA) ligand
The TPAMA ligand was synthesized according to the literature procedure31 with slight modifications (ESI, Fig. S1‡).Synthesis of CPM-37(Ni), CPM-37(Fe), and the bimetallic CPM-37(Ni,Fe) materials
CPM-37(Ni), with the idealized framework formula of [Ni3(OH)(BPDCA)3(TPAMA)] (Fig. 1a and Fig. S20‡), was synthesized according to the procedure reported by Feng et al. with some modifications.27 In a typical experiment, 0.15 mmol (43.6 mg) of Ni(NO3)2·6H2O, 0.15 mmol (36.33 mg) of H2BPDCA, and 0.05 mmol (21.92 mg) of TPAMA were dissolved in 12 mL of DMF within a thick wall 20 mL crew cap glass vial. After stirring the mixture for two hours, the sealed glass vial was placed in a preheated oven and kept at 120 °C for three days. The formed pale green hexagonal crystals of the product were isolated by gravity filtration and washed four times with DMF (20 mL per washing). The different iron-containing CPM-37(Ni,Fe) materials and CPM-37(Fe) were synthesized using the same procedure, except employing different molar ratios of nickel and iron, while keeping the combined concentration of the metal ions constant (0.15 mmol in 12 mL of DMF). The bimetallic CPM-37(Ni,Fe) series were synthesized using 0.1 mmol (29.1 mg) Ni(NO3)2·6H2O and 0.05 mmol (14 mg) FeSO4·7H2O; 0.075 mmol (21.8 mg) Ni(NO3)2·6H2O and 0.075 mmol (20.8 mg) FeSO4·7H2O; 0.05 mmol (14.5 mg) Ni(NO3)2·6H2O and 0.1 mmol (27.8 mg) FeSO4·7H2O for CPM-37(Ni2Fe), CPM-37(NiFe), and CPM-37(NiFe2), respectively. The CPM-37(Fe) was synthesized using 0.15 mmol (47 mg) FeSO4·7H2O. The concentrations (and hence also the molar ratios) of H2BPDCA and TPAMA were kept the same for all syntheses. The yield of the products was as follows: 30 mg for CPM-37(Ni), 60 mg for CPM-37(Ni2Fe), 65 mg for CPM-37(NiFe), 63 mg CPM-37(NiFe2), and 58 mg for CPM-37(Fe).Results and discussion
Synthesis and characterization of the CPM-37 materials
The synthesis of the MOF materials was carried out under the same low-temperature solvothermal conditions as reported for the single already known representative, the prototypal CPM-37(Ni).27 The latter was crystallizing as well-formed rods or small blocks, in the latter case with a tendency to adapt the shape of hexagonal platelets, consistent with the reported P63/mmc crystallographic symmetry (Fig. S5‡). The morphology of the crystals depended on the selected conditions; the conditions reported here led mostly to the latter case of the small platelets. The identity of the two morphologies to the published phase was proven either by cell measurements for selected crystals using single crystal XRD and PXRD techniques. The new CPM-37(Ni,Fe) and CPM-37(Fe) were only obtained in a form of microcrystalline powders. All attempts to obtain them in single crystalline form by variation of temperature and solvent, using N,N-diethylformamide and N,N-dimethylacetamide as DMF-analogues, were not successful. It is worth noting the good tuneability of the metal ratio in the mixed-metal CPM-37(Ni,Fe) materials by varying the ratio of the reactant salts. The 2:1, 1:1, and 0.5:1 Ni:Fe reactant molar ratios yielded similar experimentally verified metal ratios in the products (see the AAS analysis below).
The PXRD patterns of CPM-37(Ni), CPM-37(Fe), and the bimetallic CPM-37(Ni,Fe) compounds are very similar, featuring the first five intensive peaks at 6.4°, 7.8°, 8.5°, 11.5°, and 12.5°, Fig. 2a. The experimental PXRDs correspond very well to the simulated pattern based on the single crystal XRD structure of CPM-37(Ni) reported by the Feng group,27 confirming the isostructural nature of the compound family. An important difference is the apparently lower crystallinity of CPM-37(Ni), which features broader peaks compared to the bimetallic CPM-37(Ni,Fe) compounds and CPM-37(Fe). The CPM-37(Ni) is less stable and the deterioration was seemingly aggravated by the prolonged multiple washings with DMF (4 × 20 mL). In the original report of the CPM-37(Ni) synthesis,27 the experimental PXRD pattern and N2-sorption measurement have not been reported, so the data regarding the relatively low observed stability and non-robust permanent porosity of CPM-37(Ni) (87 m2 g−1) are new and somewhat unexpected. The activation involving solvent exchange with CH2Cl2 and degassing at 60 °C for 16 h under vacuum (∼5 × 10–2 mbar) was mild, yet, it evidently was an appreciable stress factor in terms of the material's permanent porosity. While CPM-37(Fe) (368 m2 g−1) demonstrated similar low-stability issues after degassing, the same treatment of the bimetallic CPM-37(Ni,Fe) samples did not compromise the stability according to the PXRD (Fig. S2‡) and N2-adsorption data. The surface areas of the three bimetallic CPM-37(Ni,Fe) samples were 2039, 1955 and 2378 m2 g−1 for CPM-37(Ni2Fe), CPM-37(NiFe), and CPM-37(NiFe2), respectively.
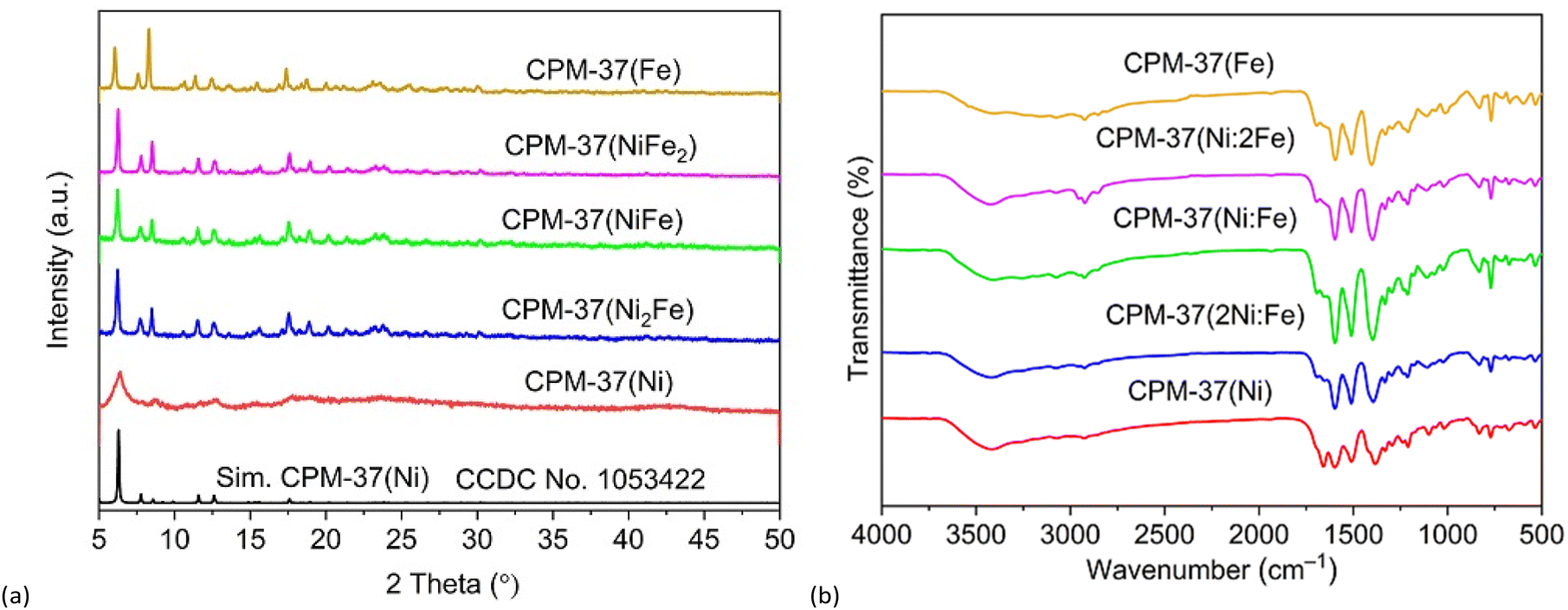 | ||
| Fig. 2 (a) PXRD patterns of the as-prepared samples, (b) FT-IR spectra of the as-prepared samples. | ||
FT-IR spectroscopy was used for accessing the presence of the characteristic functional groups (Fig. 2b). The spectra imply high similarity regarding the chemical composition (the assignments of FT-IR bands of the resulted catalysts are provided in Table S1‡). The broad peak around 3300–3500 cm−1 can be ascribed to stretching and bending vibrations of hydroxyl groups from coordinated and adsorbed water molecules, associated by H-bonds.32,33 The asymmetric vibration of carboxylate at 1598–1513 cm−1 and the respective symmetric counterpart are located at 1398–1330 cm−1. The band at 460–580 cm−1 can be attributed to the metal-oxygen stretching vibrational mode.34
Scanning electron microscopy of the samples is represented in Fig. 3. The three bimetallic samples CPM-37(Ni2Fe), CPM-37(NiFe), and CPM-37(NiFe2) consist of aggregated nano-platelet crystals. The monometallic CPM-37(Ni) features single crystals with a trigonal symmetry (>0.2 mm; Fig. S5‡), while the monometallic CPM-37(Fe) is composed of similar, but significantly smaller plates (Fig. S6g‡). The EDX elemental mapping of CPM-37(Ni2Fe) in Fig. 3e and f reveals a uniform distribution of nickel and iron in the sample. The EDX elemental mapping of CPM-37(NiFe), CPM-37(NiFe2), and CPM-37(Fe) are shown in Fig. S6,‡ the SEM-EDX spectra in Fig. S7‡ (see also Table S3‡).
 | ||
| Fig. 3 SEM images of (a and d) CPM-37(Ni2Fe), (b) CPM-37(NiFe), (c) CPM-37(NiFe2). EDX elemental mappings for (e) Ni and (f) Fe. | ||
Atomic absorption spectroscopy (AAS; see section 2.2 ESI‡) was used to determine the absolute nickel and iron content in the CPM-37(Ni,Fe) samples. EDX analyzes a thin surface layer of approx. 1–2 μm,35 and provides only a relative element content. The two methods are complementary with expected minor discrepancies between them; the results are compared in Table 1.
| Sample | SEM-EDX | AAS | Expected Ni and Fe contents and their molar ratiosa | ||||
|---|---|---|---|---|---|---|---|
| Ni/Fe mol. ratio | Ni wt% | Fe wt% | Ni/Fe mol. ratio | Ni wt% | Fe wt% | Ni/Fe mol. ratio | |
| a For the theoretical Ni and Fe weight % content, the idealized formula [Ni3(OH)(C14H8O4)3(C24H18N6O3)] with the molecular weight of 1352.18 g mol−1 was used in all cases. The slight molecular weight differences due to the different content of the constituting metal ions and counter-anions X were not accounted due to low significance. The expected contents and molar ratios correspond to the molar metal ratios, which were used in the synthesis. | |||||||
| CPM-37(Ni2Fe) | 1.58 | 7.90 | 3.90 | 1.91 | 8.40 | 4.20 | 2.00 |
| CPM-37(NiFe) | 0.96 | 4.80 | 4.90 | 1.11 | 6.40 | 6.10 | 1.00 |
| CPM-37(NiFe2) | 0.45 | 3.56 | 7.10 | 0.54 | 4.20 | 8.20 | 0.50 |
The quantification of the metal content confirmed that the Ni:Fe ratio in the samples is close to their initial ratio in the reaction medium (i.e. there is no strong observed preference for the incorporation of Ni or Fe, which is somewhat unexpected, due to different expected oxidation states of +2 and +3, respectively, and hence different affinities).
The thermogravimetry analysis (TGA) under N2 as a carrier gas was performed on the CPM-37 samples after a solvent exchange with CH2Cl2 (section 2.2 and Fig. S12, ESI‡). Two major weight-loss steps were registered: the first, at 50–350 °C, represents ∼16–24% weight loss resulting from the desorption of the solvent guest molecules (CH2Cl2, water, and residual DMF). Even prolonged exchange does not remove the DMF completely, which means that the found surface areas for the degassed samples are not the optimal values, but rather the best ones, we were able to achieve.36 The second step at 350–550 °C shows a ∼57–66% weight loss, which is attributed to the decomposition (decarboxylation and carbonization) of the constituting organic ligands.37
The porosity of the as-prepared catalysts was studied by N2-adsorption measurement (Fig. 4a). The observed IUPAC Type IB isotherms indicate microporous structures of the materials,38 while the porosities are radically different: the monometallic CPM-37(Ni) and CPM-37(Fe), which are assumed to have lower stability, demonstrate much lower uptakes and Brunauer-Emmett-Teller (BET) surface areas compared to the bimetallic CPM-37(Ni,Fe) materials (Table 2).
 | ||
| Fig. 4 (a) Nitrogen adsorption isotherms of the CPM-37 samples at 77 K (adsorption: filled circles; desorption: empty circles), (b) NLDFT-based pore size distributions for the CPM-37 samples. | ||
The NLDFT-based calculated pore size distribution in Fig. 4b shows a trimodal distribution of the CPM-37(Ni,Fe) materials in the microporous 0.5–2.0 nm pore diameter range, which slightly exceeds the maximum below 1.0 nm for the expected unimodal distribution (the absolute values for the mode-maxima are of low precision, i.e. indicative only, as no matching MOF model for the DFT kernel is available).38 On the contrary, CPM-37(Ni) showed a broad pore size distribution with dominantly represented mesoporous and microporous ranges,38 which is attributed to the partial structural collapse as a result of the solvent exchange and the subsequent degassing procedure. The corresponding total pore volumes of the catalysts derived from N2-adsorption isotherms at 77 K are summarized in Table 2.
X-ray photoelectron spectroscopy (XPS) was conducted to determine the principal question regarding the oxidation state of the metal ions in the synthesized CPM-37, namely in CPM-37(Ni2Fe), which was proven to be the best material for OER, and in the monometallic CPM-37(Fe) and CPM-37(Ni) for comparison. It is important to note that XPS has a relatively shallow penetration depth, approximately one order of magnitude less than EDX.35 Therefore, the elemental composition obtained through XPS analysis (Fig. S13a‡), as shown in Table S6,‡ was considered to have lower precision regarding the average values compared to EDX analysis (the determined distribution of oxidation states features the same surface-specific error, however, it gives a valuable semi-quantitative insight, which is not readily accessible by other means).
The high-resolution Ni 2p spectrum of CPM-37(Ni) and CPM-37(Ni2Fe) (Fig. 5a) reveals a mixed Ni2+/Ni3+ oxidation state for nickel in both the monometallic CPM-37(Ni) and bimetallic CPM-37(Ni2Fe) samples. In the case of the CPM-37(Ni) sample, two distinct peaks observed at 856.0 and 873.6 eV can be attributed to Ni 2p3/2 and Ni 2p1/2 transitions for Ni2+, respectively. The deconvolution of those peaks allows to detect the respective transitions for Ni3+ at 857.5 eV and 875.0 eV as well. The two peaks at 861.8 and 879.7 eV are ascribed to Ni 2p satellites.39–41
 | ||
| Fig. 5 High-resolution XPS spectrum of (a) Ni 2p in CPM-37(Ni) and CPM-37(Ni2Fe), (b) Fe 2p in CPM-37(Fe) and CPM-37(Ni2Fe). | ||
The calculated ratio of Ni2+:Ni3+ for CPM-37(Ni) was 4.9 (Table S10‡), indicating that the dominant oxidation state of nickel is Ni2+, which aligns with the reported [Ni3(OH)(BPDCA)3(TPAMA)] framework formula.27 Interestingly, the deconvolution analysis of the Ni 2p spectrum of CPM-37(Ni2Fe) also confirms the presence of a mixed Ni oxidation state. Specifically, the peaks observed at 855.9 and 873.5 eV were assigned to Ni2+, the peaks at 857.1 and 875 eV to Ni3+. The two satellite peaks are detectable at 861.9 and 880.2 eV.40,41 The calculated ratio of Ni2+:Ni3+ for CPM-37(Ni2Fe) is 2.4 (Table S10‡).
The deconvolution analysis of the Fe 2p high-resolution XPS spectrum consistently indicated only an Fe3+ oxidation state for both CPM-37(Fe) and CPM-37(Ni2Fe) (Fig. 5b). For CPM-37(Fe), two peaks located at 711.5 and 725.1 eV were observed, while for CPM-37(Ni2Fe) there are two peaks centered at 711.9 and 725 eV, both characteristic for Fe3+.42–44 A pair of satellite peaks at 717.9 and 729.8 eV as well as 717.5 and 730.5 eV were observed for CPM-37(Fe) and CPM-37(Ni2Fe), respectively.45 The high-resolution XPS spectrum of Fe 3p also confirmed the nearly only Fe3+ state in both CPM-37(Fe) and CPM-37(Ni2Fe) samples (Fig. S14‡).
An interesting separate point in the question of the composition is the presence of minor amounts of sulfur in the XPS spectra of the investigated iron-containing CPM-37, namely the CPM-37(Fe) and CPM-37(Ni2Fe) (S 2p peaks in Fig. S13a‡), which is attributable to the residual SO42− ions from FeSO4·7H2O, used for their synthesis. At least Li et al. have also reported the incorporation of SO42− in the structure of analogous MOFs and suggested its charge-balancing role.46 We could confirm only a minor role of the sulfate, which amounts to ∼1 mol% of the total metal content (note, that the XPS analysis of sulfur is not precise due to low content, aggravated by the inherent surface nature of the analysis, which could be affected by surface defects and the associated variation of charge balancing counter-anions. Hence the given content is semi-qualitative). It seems that the role of the sulfate is not fundamental and the content depends on the method of preparation. As for the C, O, and N elements, the respective XPS peaks of C 1s, O 1s, and N 1s are shown in Fig. S13b–d,‡ with the corresponding peak assignments listed in Tables S7–S9,‡ respectively.
The proposed final composition is based on the single-crystal structure of the reported CPM-37(Ni) (CCDC 1053422; Fig. 1a–c),27 which is confirmed by us via comparison of the simulated and experimental PXRDs, as well as by SCXRD-based cell-determination. The formerly ascribed composition based on the SCXRD data corresponds to the idealized formula of [(Ni2+)2(Ni3+)(μ3-OH)(BPDCA)3(TPAMA)] implying a Ni2+:Ni3+ ratio of 2:1 (Fig. 1d; stable composition with x = 1). Our XPS data suggest though, a deficiency of Ni3+ compared to this formula, showing a Ni2+:Ni3+ ratio of ∼5:1 (note that the precision of XPS, which is surface-relevant only, is limited, but still, the deflection is too high to be ignored). The Ni3+ oxidation state, normally unstable under the given conditions, is stabilized by its necessary counterbalancing role of the charged μ3-OH central ligand, which in turn stabilizes the cationic environment. If only Ni2+ were present, the role of the central ligand could have been played only by μ3-H2O (Fig. 1f), which is evidently an unstable configuration, where three cations are close to each other without an anionic counterbalance in-between. The observed structural instability of CPM-37(Ni) during direct degassing as well as the results of the XPS suggests, that some, or even a substantial amount of the framework-constituting clusters might indeed be constituted by Ni2+ only. It is reasonable to suggest, that while the freshly prepared CPM-37(Ni) might be closer to the ideal formula given above, a part of Ni3+ could reduce to Ni2+ upon storage, solvent exchange, and degassing (and/or upon strong local heating during the XPS measurement) thereby destabilizing the framework. The general formula in this case could be expressed as [(Ni2+)3−x(Ni3+)x(μ3-OH)x(μ3-OH2)1−x(BPDCA)3(TPAMA)], where x is rather closer to 0.5 than to 1 (Fig. 1f), as in the idealized formula. Degassing at elevated temperatures should further destabilize the structure.
In CPM-37(Ni2Fe) one-third of Ni atoms are substituted by Fe atoms, as confirmed by the found Ni:Fe ≈ 2.0 ratio by the AAS analysis, while the found oxidation states by XPS were Ni2+and Ni3+ with a Ni2+:Ni3+ = 2.4 ratio and Fe3+ only. Thus, the CPM-37(Ni2Fe) is enriched by M3+ ions even more than minimally necessary to charge-balance the central μ3-OH ligand. The framework formula could be expressed as [(Ni2+)3−x(Ni3+∼0.33Fe3+∼0.66)x(μ3-OH)2−x(μ3-O)x–1(BPDCA)3(TPAMA)] with x ≈ 1.5 (the content of the central oxido ligand might be slightly less, namely by ∼z/2, where z∼0.03 is the amount of the SO42− present. The difference is smaller than the precision of the XPS, so it could be neglected, particularly while a part of the sulfate might also be simply trapped/occluded as well). Thus, the composition corresponds to the expected stability range with MII content of x ∈ [1–2] in the three-metal cluster (Fig. 1d). The presence of the Ni3+ is somewhat unexpected, but it is in line with the expected additional stabilizing effect of the charge-balancing central μ3-O ligand instead of the μ3-OH (in other words there is a factor-equilibrium between the added stabilizing effect of the μ3-O ligand and instability of the Ni3+ oxidation state).
CPM-37(Ni2Fe), as well as the other mixed-metal CPM-37(Ni,Fe) compounds investigated in this work demonstrate good stability allowing direct degassing without strong deterioration of the expected high surface areas (in stark contrast to CPM-37(Ni)). In addition, the discussion above allows to suggest, why the CPM-37(Fe) is also less stable than the mixed-metal counterparts (even if to a lesser extent compared to CPM-37(Ni)). Formally, the situation is unexpected as MIL-88(Fe) or MIL-100(Fe) based on the {FeIII3O(OH)} cluster core are stable. The possible reason is that in CPM-37(Fe) (cf.Fig. 1e) there are no free coordination sites at the iron ions, which could accept the additional charge-balancing terminal hydroxido-ligands, unlike in the case of the parent MIL-88(Fe). It is not a fundamental problem, as the charge-balancing counteranions could also be localized in the pores as the bromide ion in NH2-MIL-88D(Fe), according to the crystal structure with KOKKOL CSD code.47 In the current case of CPM-37(Fe) the sulfate ions could play the same role. However, MOFs with cationic frameworks (cf.Fig. 1e) do not receive full Coulombic stabilization and hence are less stable than the analogues with fully charge-compensated frameworks.
To deepen the understanding of the relative stabilities of mixed-metal CPM-37(Ni,Fe) materials in the context of permanent porosity, two further representatives CPM-37(Ni2.5Fe0.5) and CPM-37(Ni0.5Fe2.5) with metal ratios approaching the single-metal compositions were synthesized. The syntheses as well as the solvent exchange and degassing were done exactly the same way as for the other CPM-37(Ni,Fe) materials. It turned out (see section 2.9 for relevant analytics) that CPM-37(Ni2.5Fe0.5) with SBET = 1684 m2 g−1 and actual SEM-based Ni:Fe = 3.9:1 as well as CPM-37(Ni0.5Fe2.5) with SBET = 435 m2 g−1 and Ni:Fe = 1:0.2 are both characterized by decreased surface areas, as expected. However, CPM-37(Ni2.5Fe0.5), which also shows a tendency towards increased iron content compared to the reactant ratio (seemingly a manifestation of the tendency to ensure better charge compensation), still demonstrated significant permanent porosity and is closer to CPM-37(Ni2Fe) in this regard, while CPM-37(Ni0.5Fe2.5) is of relatively low porosity and close to CPM-37(Fe). Aside of slight enrichment of CPM-37(Ni2.5Fe0.5) by iron, the other reason for sufficiently good stabilization is due to the presence of small amounts of NiIII, which was observed for CPM-37(Ni). It is also worth noting that the TGA shows incomplete removal of DMF in the degassed samples, hence higher surface areas might be reached, which would distinguish materials with different compositions better. Nevertheless, the given procedure was the best that we have found (which might be due to the slight stabilizing role of the residual DMF), and the similarity of the activation process allows to suggest with a high probability that the found relative stabilities, with a stability plateau for CPM-37(Ni2Fe), CPM-37(NiFe), and CPM-37(Fe2Ni), is most likely correct.
The observation that the mixed-metal CPM-37 compounds, at least for the examples of Ni and Fe materials, are much more stable than their single-metal peers regarding their high attainable permanent porosity and easy preparation is of high practical importance.
Electrocatalytic oxygen evolution reaction (OER) performance
Among the two electrochemical processes constituting the water splitting, namely the (cathodic) hydrogen evolution reaction (HER) and (anodic) oxygen evolution reaction (OER),48 the latter requires, as a rule, higher overpotentials, and is hence less efficient.49 The optimization of the OER, primarily via finding electrocatalytic materials with low overpotentials at high current densities, is one of the evident challenges.The work focuses on a comparative study, and the success criterion is based on comparison of the CPM-37 materials with the RuO2 benchmark. The OER performance of the samples was evaluated using a three-electrode setup (RHE as a reference and Pt foil as a counter electrode) in 1.0 mol L−1 KOH (pH = 13.3) solution, degassed by continous bubbling of N2 gas. The linear sweep voltammetry (LSV) polarization curve (Fig. 6a), confirmed that the presence of both iron and nickel in the CPM-37 samples remarkably increases the OER performance of the electrocatalyst compared to the monometallic CPM-37(Ni) and CPM-37(Fe) (here and further it is implied that the CPM-37 MOFs are precursor material to the catalytic species actually formed in the alkaline medium).
 | ||
| Fig. 6 (a) OER polarization curves for the converted CPM-37 precatalysts, i.e. derived electrode materials. (b) Overpotentials determined at 50, 100, and 200 mA cm−2 for the derived materials. (c) Tafel plot of the derived materials determined in 1 mol L−1 KOH solution. (d) Nyquist plots for the selected samples at 1.5 V vs. RHE. The Voigt circuit model (black lines) was used to fit the data, where Rs represents the electrolyte resistance, Rp is the resistance associated with the electrode's porosity, and Rct with the charge transfer, respectively. Constant phase elements (CPE) were used to show double-layer capacitance. | ||
Despite promising prerequisites, the usage of MOFs as electrode materials also meets strong inherent obstacles. The two most important ones are the low conductivity (which is to some extent amendable by the use of conductive additives constituting a composite) and generally low stability of MOFs in acidic, but particularly in basic conditions.50,51 However, MOFs have been proven to be at least good precursors for electrode materials.52–54 Pyrolysis (carbonization) of MOFs under an inert atmosphere is the most utilized way to produce electrocatalytic materials from MOFs. The resulting porous carbon materials with much-improved conductivity feature relatively uniformly distributed metal nanoparticles or reduced metal-based species, which could serve as electrocatalytic active sites.20 Typical problems are low yield, poor reproducibility and/or scalability, as well as limitations regarding local uniformity and purity of the ensuing electrode materials.55,56
The OER performance enhancement for the bimetallic CPM-37(Ni,Fe)-derived materials can be attributed to the electrochemical Ni–Fe synergism in alkaline media,57 and the introduction of additional structural vacancies in nickel-based MOFs.58 Among all CPM-37 materials, the bimetallic CPM-37(Ni2Fe) has the best performance regarding the OER reaction – namely, it has the lowest overpotential for current densities above 50 mA cm−2 (Fig. 6a), suggesting a Ni:Fe ratio optimum at ∼2. The advantage increases with current densities, reflected by the lowest Tafel slope of ∼39 mV dec−1 for CPM-37(Ni2Fe) compared to the other CPM-37 materials. At a low current density of 25 mA cm−2, the well-known benchmark material, RuO2, demonstrates a slightly lower overpotential compared to CPM-37(Ni2Fe), namely at 274 vs 278 mV respectively, but already at 50 mA cm−2 the order reversed to be 300 vs 290 mV.
At current densities above 80 mA cm−2, all iron-containing CPM-37 materials outperform the RuO2 benchmark, however, the CPM-37(Fe) is the worst performer. Therefore, the synergistic presence of Ni is important, and the ratio optimization led to an appreciable overpotential decrease with an optimum for CPM-37(Ni2Fe) at >50 mA cm−2 currents (Fig. 6b).39 As it can be seen from Fig. 6a, in the bimetallic CPM-37(Ni,Fe) samples the oxidation of Ni2+ is hindered, which is inferred from a positive shift of the Ni2+/3+ peaks to >1.4 V compared to <1.4 V for CPM-37(Ni).
Tafel analysis was conducted to assess the intrinsic kinetics of the OER process. The resulting Tafel slope, obtained from the plot, is commonly used to semi-quantitatively determine the reaction rate at the electrode-electrolyte interface using the following equation:59
| η = b log(j) + a | (1) |
The calculated Tafel slopes for CPM-37(Ni), CPM-37(Ni2Fe), CPM-37(NiFe), CPM-37(NiFe2), CPM-37(Fe) were, respectively, 67, 39, 44, 47, 49 mV dec−1 against 57 mV dec−1 for RuO2 (Fig. 6c). The lowest Tafel slope for the CPM-37(Ni2Fe)-derived material signifies the lowest energy losses associated with the overpotential. Speaking in kinetic terms, electrons transfer is faster, which improves the performance.60
The electrochemical impedance spectroscopy (EIS) measurement was conducted to find correlations with the catalytic activity trend of the materials.61Fig. 6d represents the Nyquist plots of the selected samples derived from EIS measurements at 1.5 V vs. RHE. The smaller semicircle radius of the plot for the CPM-37(Ni2Fe)-derived material infers lower charge transfer resistance on the electrode-electrolyte interface, which means accelerated kinetics of the OER.62
The Nyquist plots were fitted using a Voigt-type circuit (Fig. 6d) to determine the charge transfer resistance, Rct (Table 3). The lowest (1.45 Ω) and highest (66 Ω) Rct values were observed for the CPM-37(Ni2Fe)- and CPM-37(Ni)-derived materials, respectively. These findings correlate with the observed trend in catalytic activity as evaluated by the linear sweep voltammetry (LSV) polarization curves, indicating that CPM-37(Ni2Fe) showcased superior OER performance in comparison to the other CPM-37-derived materials. The smaller (Rct) for CPM-37(Ni2Fe) (1.45 Ω) signifies higher electrode-electrolyte ion transport speed compared to the analogs, which is a prerequisite for more favorable OER kinetics.63 Furthermore, the catalyst that was derived from CPM-37(Ni2Fe) had a Faradaic efficiency of 84% (section 2.11.1, ESI‡).
| Sample | Overpotential at 50 mA cm−2 (mV) | Tafel slope (mV dec−1) | Charge transfer resistance, Rct (Ω) |
|---|---|---|---|
| CPM-37(Ni) | 369 | 67 | 66.0 |
| CPM-37(Ni2Fe) | 290 | 39 | 1.45 |
| CPM-37(NiFe) | 298 | 44 | 1.66 |
| CPM-37(NiFe2) | 307 | 47 | 3.15 |
| CPM-37(Fe) | 316 | 49 | 5.15 |
| RuO2 | 300 | 57 | 1.23 |
Long-term stability is one of the most important practical performance-defining qualities of an electrocatalyst.64 The stability of the CPM-37(Ni2Fe)-derived material and RuO2 as a reference material was investigated by chronopotentiometric analysis (CP) at a fixed current density of 50 mA cm−2 over 20 h (Fig. 7a). The results of the OER stability test confirmed the superiority of CPM-37(Ni2Fe) compared to RuO2 regarding the long-term performance. The overpotential needed to achieve a current density of 50 mA cm−2 experienced a relatively modest increase, from 290 mV to 304 mV, for the electrode material derived from CPM-37(Ni2Fe). Conversely, the OER performance of RuO2 exhibited a substantial decline after 20 hours, resulting in a significant overpotential rise from 300 mV to 387 mV.
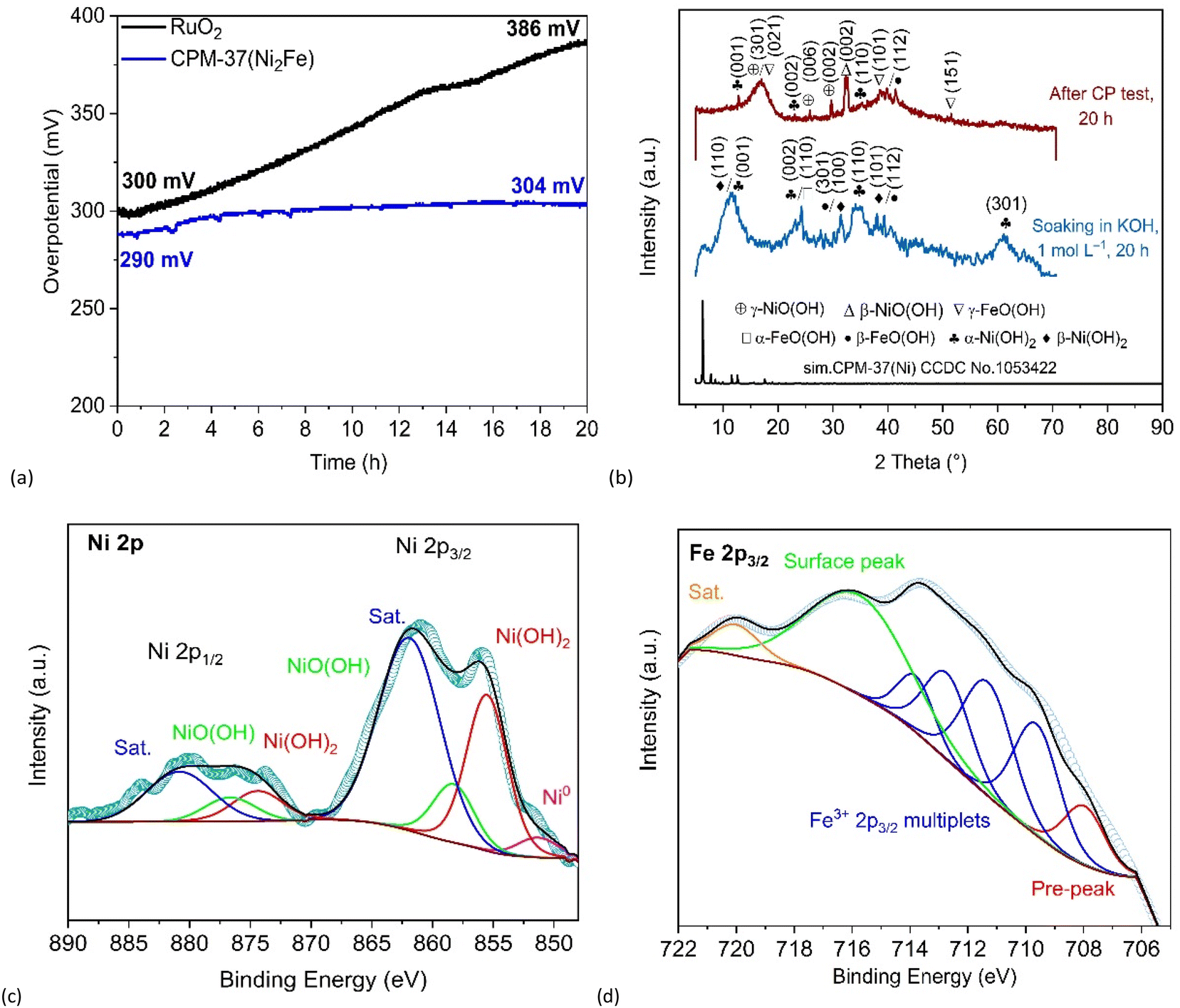 | ||
| Fig. 7 (a) Chronopotentiometry (CP) analysis of CPM-37(Ni2Fe) and RuO2 at 50 mA cm−2 for 20 h. (b) PXRD patterns of CPM-37(Ni2Fe) after CP test in 1 mol L−1 KOH for 20 h, and soaking in 1 mol L−1 KOH for 20 h. (c) High-resolution XPS spectrum of Ni 2p in CPM-37(Ni2Fe) after CP test in 1 mol L−1 KOH for 20 h. (d) High-resolution XPS spectrum of Fe 2p in CPM-37(Ni2Fe) after CP test in 1 mol L−1 KOH for 20 h. | ||
To investigate more in-depth the conversion of the initial CPM-37 materials in aqueous alkali, dedicated tests were performed on larger scales of the material without the involvement of electrochemical processes (this test is not equivalent to the investigation of the actual electrode material after the OER – see below – but it gives an interesting insight to an approximation of its composition). A sample of CPM-37 was soaked in 1 mol L−1 an aqueous KOH for 20 h, and the recovered material was analyzed by means of PXRD analysis (Fig. S3‡). In line with the known low stability of carboxylate MOFs in alkaline aqueous media,65 complete decomposition of the CPM-37 MOFs was observed, together with microscopically witnessed loss of initial morphology (Fig. S8 and Table S4‡). As a result, a mixed-phase residue of metal (oxy)hydroxides was formed (Fig. S3‡). It is worth noting here the purposefulness of the TPAMA ligand choice; unlike the acidic H2BPDCA, the former has relatively low solubility in aqueous alkaline solutions, but it is chemically labile under the given conditions, and the products of hydrolysis are well soluble.
Thus, in the case of CPM-37(Ni2Fe) (Fig. 7b), most interesting in the context of OER, four phases were identified: α-Ni(OH)2 (ICDD 38-0715),66 β-Ni(OH)2 (ICDD 14-0117),67 α-FeO(OH) (ICDD 29-0713),68 and β-FeO(OH) (ICDD 01-080-1770).69 For CMP-37(NiFe) and CPM-37(NiFe2) the same phases, except β-Ni(OH)2, were observed (Fig. S3‡). The conversion of the monometallic CPM-37(Ni) and CPM-37(Fe) led to a mixture of hydroxides, dominantly to β-Ni(OH)2 and β-FeO(OH), respectively (see Fig. S4 and Table S2‡ for the IR spectra of the formed residues and the respective assignments).
It is worth noting that the residues derived from CPM-37(Ni2Fe) in an aqueous alkaline solution without the application of current contain an intermediary quantity of β-Ni(OH)2 compared to its dominant content for CPM-37(Ni)- and near absence for other more iron-rich CPM-37-derived materials. The conversion of the β-phase to the α-Ni(OH)2 might be stimulated by a phenomenon known as ‘interstratification’, caused by the presence of charge-imbalancing Fe3+ cations.70 The latter creates defects by substituting the Ni2+ ions in β-Ni(OH)2, causing an excess of positive charge, which is compensated by anions localized between the β-Ni(OH)2 layers. This arrangement stimulates the formation of α-Ni(OH)2via conversion of the β-Ni(OH)2 phase.71–73 The layered striated morphology with tightly incorporated iron ions, whose synergistic presence is crucial,68 might play a role in increased efficiency. On the other hand, there is a formal contradiction, as α-Ni(OH)2 itself has a higher catalytic activity in OER than β-Ni(OH)2 (according to the Bode model,74 the α-Ni(OH)2 more readily oxidizes to γ-NiO(OH) during the OER reaction, which in turn takes part in the catalytic cycle).75,76 Accordingly, it was suggested that the derived material after the OER could have a different composition.
Therefore, the best-performing actual electrode material derived from CPM-37(Ni2Fe) was also analyzed by means of PXRD, using a sample collected after the application of the current, i.e. after the OER. This type of test is generally cumbersome as it is hard to separate the non-metal part of the electrode material from the nickel foam without introducing nickel metal and/or separating the material non-uniformly. Hence a specially prepared ‘flattened’ Ni electrode was loaded (see the materials and method section), processed under the same conditions as the other samples, and then a simple separation of the deposited non-metal part of the electrode material was achieved by ultrasonication. Such modification of the electrode comes at a cost of higher overpotential due to worse contact with the metal support. Hence, the standard and modified electrodes could be compared only regarding the qualitative composition of the electrode material's outcome. The comparison of the initial metal oxide/hydroxide mixed phase residue (previous tests), and the electrode material after the OER allows to observe the result of the evolution of the material during the process.
The PXRD of the CPM-37(Ni2Fe)-derived material after the OER on the special electrode revealed the presence α-Ni(OH)2 (ICDD 38-0715),66 γ-NiO(OH) (ICCD 06-0075),67 β-FeO(OH) (ICDD 01-080-1770),69 and γ-FeO(OH) (JCPDS no. 76-2301),77 and possibly minor amounts of β-Ni(OH)2 (Fig. 7b and Fig. S10‡). During the OER the latter converted to other Ni-containing phases, while a part of the Ni(II) was oxidized to nickel(III) oxide hydroxide phases.
According to the generally accepted theoretical model, the nickel oxy(hydroxide) species play a crucial role in the oxygen evolution reaction (OER) in alkaline aqueous medium due to the electrocatalytic role of the Ni3+/Ni2+ redox pair. See Table S14‡ for relevant published benchmark materials; the obtained results in this paper are good, however not extraordinary. The main focus here is on the possibility of fine-tuning the performance through the Ni/Fe ratio and on morphological enhancement, which allows better uniformity, at least in principle. γ-NiO(OH) and γ-FeO(OH) are usually identified as synergistic active sites for the OER process.39,78,79 The role of Fe could be at least partially postulated as catalyst for the decomposition of the peroxy-species and, hence, the mixture of nickel and iron tight at the nano-scale is viewed as advantageous. The mechanism of the OER reaction is usually given by eqn (2)–(5),80 where the Σ and Σ+ symbols represent the active centers associated with the Ni2+ and Ni3+ catalyst:
| Σ-OH− → Σ+-OH + e− | (2) |
| Σ+-OH + OH− → Σ+-O˙ + H2O + e− | (3) |
| Σ+-O˙ + OH− → Σ-OOH + e− | (4) |
| Σ+-OOH + OH− → Σ + O2 + H2O + e− | (5) |
The XPS analysis of the CPM-37(Ni2Fe)-derived material recovered from the special electrode after the OER (Fig. S15a‡) shows the presence of both Ni2+ and Ni3+ (Fig. 7c and d and also section 2.8.2, ESI‡). Thus the Ni 2p peaks are found both for Ni2+ (at 855.7 eV for Ni 2p3/2 and 874.8 eV for Ni 2p1/2) and Ni3+ (at 858.7 eV for Ni 2p3/2 and 876.7 eV for Ni 2p1/2), which should be associated with Ni(OH)2 and NiO(OH) phases respectively, also identified by PXRD (Fig. 7b). The two further peaks located at 862.2 and 881.4 eV are attributed to satellite peaks of Ni 2p3/2 and Ni 2p1/2 respectively, while the small peak at 852 eV can be ascribed to metallic nickel, which is produced by Ar sputtering during XPS analysis.39,81
Regarding iron, practically only Fe3+ is expectedly detected in the CPM-37(Ni2Fe)-derived material. Unlike in the case of the Fe in the parent MOF, where the 2p3/2 envelope was fitted by a single peak (which is a strong simplification), it was attempted to make a more precise fit using the four strongest ‘Gupta and Sen’ (GS) multiplet components (the four-peak fitting is a simplification as the two further weaker peaks at ∼714.1 and ∼719.5 are not taken in account because it is hard to fit them reliably. The simplification could underestimate the content of iron up to ∼17%, but due to peak overlap the value tends to be lower). The obtained values 709.7 (32.5%), 711.4 (32.3%), 712.8 (22.7%), and 713.7 eV (12.5%) are fully consistent with the average Fe(III) (except that a small ‘pre-peak’ at 707.4 eV could be interpreted as minor amounts of iron in lower oxidation states formed during Ar sputtering).82 However, the result does not allow to distinguish, for example, α-FeO(OH) and γ-FeO(OH), which are overall the closest candidates. Thus, the reference values for the latter are 710.3 (31.9%), 711.3(32.2%), 712.3 (23.5%), 713.9 (12.2%),82 while the difference from α-FeO(OH) is within 0.2 eV(2.5%) max., and those values are somewhat closer than those are from α-Fe2O3 and β-Fe2O3.83–85 The dominant role of γ-FeO(OH) was rather postulated, based on generally higher stability under similar conditions and the measured low-quality PXRD.
Among the further observed elements, including the evident potassium from the KOH solution, carbon, oxygen, and sulfur, represented by K 2p, C 1s, O 1s, and S 2p bands, the observation of residual sulfur is interesting as it demonstrates the presence of a small amount of SO42− in the CPM-37(Ni2Fe) introduced during the synthesis of the precursor MOF as discussed above. No N 1s signal was detected, confirming that the CPM-37(Ni2Fe) structure underwent complete decomposition during the OER reaction.
Conclusions
A series of newly developed bimetallic CPM-37(Ni,Fe) metal–organic frameworks showed much better stability and surface areas after activation than the monometallic Ni- or Fe-based peers. This observation is easily rationalizable: the low stability of CPM-37(Ni) is due to the relative instability of the Ni3+ state, which should be present to allow the μ3-OH cluster-supporting central ligand. On the other hand, the CPM-37(Fe), featuring exclusively the stable Fe3+ state, should have some of the counter-anions to be localized in the pores. The cationic framework should have comparatively lesser stability. The observation of increased stability of mixed-metal CPM-37(Ni,Fe) coupled with easiness of preparation, high surface areas (∼2000 m2 g−1) and variability of metal content are important for the application-oriented MOF-field and calls for in-depth investigations of similar mixed-metal MOFs (due to the relative novelty of the analogous CPM materials, their prospects are somewhat obscured; it could be indirectly inferred from the absence of reports on permanent porosity of some representatives, e.g. CPM-37(Ni)). In this contribution, the uniform distribution of the two constituent metals down to the nano-scale, stipulated by the increased stability of the constituent coordination-bonded cluster, was inherently employed for the preparation of a nano-structured composite of metal oxides and oxo hydroxides, yielding the core functional part of the electrode material, by hydrolytic decomposition of the parent MOF.Among the electrode materials derived from the CPM-37(Ni,Fe) precursors, the CPM-37(Ni2Fe) material had the highest OER performance, with a low overpotential of 290 mV at 50 mA cm−2, a low charge transfer resistance (Rct) of 1.45 Ω and a low Tafel slope of 39 mV dec−1, implying favorable OER kinetics. This is better than for the benchmark RuO2 material (300 mV overpotential at 50 mA cm−2 but with a significantly higher Tafel slope at 57 mV dec−1). The long-term performance is also better as witnessed by the results of the chronopotentiometry performance (CP) test after 20 h: the CPM-37(Ni2Fe)-derived material with the overpotential increase from 290 to 308 mV outperformed the RuO2 with the overpotential increased from 300 to 386 mV. The comparison of the materials derived by soaking CPM-37(Ni2Fe) in 1 mol L−1 KOH solution and the material derived after the OER reaction showed the formation of γ-NiO(OH) phase during the electrochemical process, supporting the expected electrocatalytic role of Ni3+ species.
In general, the CPM-37(Ni2Fe) derived material, where the Ni:Fe content was optimized, performs comparably or better also in comparison with Ni(OH)2 (in various forms), Fe-doped Ni(OH)2, and other materials derived from bimetallic Ni,Fe-MOF precatalysts (Table S12‡), suggesting the advantages of the highly porous CPM-37(Ni2Fe) precatalyst and the morphology of the derived material, as well as of the optimized Ni/Fe ratio. The low Tafel-slope for the CPM-37(Ni2Fe), which is a prerequisite for the use at high current-densities, demonstrates the room, which still exists for optimization of advanced electrode materials with improved ion transport kinetics based on microporous MOF precursors with adjustable composition, being uniform on the nanoscale.
Author contributions
Conceptualization: S. A (Soheil Abdpour), I. B. (István Boldog); methodology: S. A; validation: S. A; formal analysis: S. A, M. N. A. F. (Marcus. N. A. Fetzer), I. B., R. O. (Robert Oestreich), T. H. Y. B. (Thi Hai Yen Beglau): investigation, S. A; resources: C. J. (Christoph Janiak); writing original draft: S. A; writing – review & editing: I. B., C. J.; supervision: C. J.; project administration: C. J.; funding acquisition, C. J. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Mr Takin Haj Hassani Sohi for single crystal XRD-based unit cell parameter checking for the known CPM-37(Ni) phase and Ms Annette Ricken for the AAS measurements. This research was funded by a joint National Natural Science Foundation of China–Deutsche Forschungsgemeinschaft (NSFC-DFG) project (DFG JA466/39-1) and within the Priority Program SPP 1928/2 COORNETs (C. J. grant Ja466/43-1).References
- Y. Cui, B. Li, H. He, W. Zhou, B. Chen and G. Qian, Acc. Chem. Res., 2016, 49, 483–493 CrossRef CAS PubMed.
- S. Abednatanzi, P. G. Derakhshandeh, H. Depauw, F.-X. Coudert, H. Vrielinck, P. Van Der Voort and K. Leus, Chem. Soc. Rev., 2019, 48, 2535–2565 RSC.
- L. Chen, H.-F. Wang, C. Li and Q. Xu, Chem. Sci., 2020, 11, 5369–5403 RSC.
- Y. Pan, H. Q. Su, E. L. Zhou, H. Z. Yin, K. Z. Shao and Z. M. Su, Dalton Trans., 2019, 48, 3723–3729 RSC.
- T. Stolar, A. Prasnikar, V. Martinez, B. Karadeniz, A. Bjelic, G. Mali, T. Friscic, B. Likozar and K. Uzarevic, ACS Appl. Mater. Interfaces, 2021, 13, 3070–3077 CrossRef CAS PubMed.
- M. Lammert, C. Glissmann and N. Stock, Dalton Trans., 2017, 46, 2425–2429 RSC.
- J. D. Howe, C. R. Morelock, Y. Jiao, K. W. Chapman, K. S. Walton and D. S. Sholl, J. Phys. Chem. C, 2016, 121, 627–635 CrossRef.
- Y. Jiao, C. R. Morelock, N. C. Burtch, W. P. Mounfield, J. T. Hungerford and K. S. Walton, Ind. Eng. Chem. Res., 2015, 54, 12408–12414 CrossRef CAS.
- I. Jahan, M. A. Islam, T. H. Rupam, M. L. Palash and B. B. Saha, Therm. Sci. Eng. Prog., 2022, 35, 101453 CrossRef CAS.
- W. H. Ho, S. C. Li, Y. C. Wang, T. E. Chang, Y. T. Chiang, Y. P. Li and C. W. Kung, ACS Appl. Mater. Interfaces, 2021, 13, 55358–55366 CrossRef CAS PubMed.
- Y.-X. Ma, C. Liu, J.-F. Ma and Y. Zhao, ACS Mater. Lett., 2022, 4, 2522–2527 CrossRef CAS.
- N. Ye, J. Zheng, K. Xie, B. Jiang and S. Zuo, J. Rare Earths, 2023, 41, 889–895 CrossRef CAS.
- X. Wang, H. Xiao, A. Li, Z. Li, S. Liu, Q. Zhang, Y. Gong, L. Zheng, Y. Zhu, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, J. Am. Chem. Soc., 2018, 140, 15336–15341 CrossRef CAS PubMed.
- A. P. Tiwari, Y. Yoon, T. G. Novak, K. S. An and S. Jeon, ACS Appl. Nano Mater., 2019, 2, 5061–5070 CrossRef CAS.
- F. E. S. Oztuna, T. Beyazay and U. Unal, J. Phys. Chem. C, 2019, 123, 28131–28141 CrossRef.
- J. Zhou, C. Huang, Q. Zhou, Y. Xie, L. Yang, L. Yu and Y. Yu, Inorg. Chem., 2022, 61, 9318–9327 CrossRef CAS PubMed.
- S. Abdpour, L. Rademacher, M. N. A. Fetzer, T. H. Y. Beglau and C. Janiak, Solids, 2023, 4, 181–200 CrossRef CAS.
- D. Y. Chung, P. P. Lopes, P. F. B. D. Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Pkovic, D. Strmcnik, Y. Zhu, S. Seifert, S. Lee, V. R. Stamenkovic and N. M. Markovic, Nature, 2020, 5, 222–230 Search PubMed.
- T. H. Y. Beglau, L. Rademacher, R. Oestreich and C. Janiak, Molecules, 2023, 28, 4464 CrossRef CAS PubMed.
- J. Liu, D. Zhu, C. Guo, A. Vasileff and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1700518 CrossRef.
- G. Lee, W. Na, J. Kim, S. Lee and J. Jang, J. Mater. Chem. A, 2019, 7, 17637–17647 RSC.
- L. Gao, Z. Lin, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
- S. Chen, L. Ma, Z. Huang, G. Liang and C. Zhi, Cell Rep., 2022, 3, 100729 CAS.
- D. K. Singha, T. Ping and B. K. Jena, ACS Appl. Energy Mater., 2023, 6, 9963–9974 CrossRef CAS.
- S. Dai, Y. Liu, Y. Mei, J. Hu, K. Wang, Y. Li, N. Jin, X. Wang, H. Luo and W. Li, Dalton Trans., 2022, 51, 15446–15457 RSC.
- Y. Dang, P. Han, Y. Li, Y. Zhang and Y. Zhou, J. Mater. Sci., 2020, 55, 13951–13963 CrossRef CAS.
- X. Zhao, X. Bu, Q. Zhai, H. Tran and P. Feng, J. Am. Chem. Soc., 2015, 137, 1396–1399 CrossRef CAS PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- D. H. Taffa, D. Balkenhohl, M. Amiri and M. Wark, Small Struct., 2022, 4, 2200263 CrossRef.
- S. Anantharaj and S. Noda, J. Mater. Chem. A, 2022, 10, 9348–9354 RSC.
- S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607–2614 CrossRef CAS PubMed.
- P. Maniam and N. Stock, Inorg. Chem., 2011, 50, 5085–5097 CrossRef CAS PubMed.
- L. Sondermann, W. Jiang, M. Shviro, A. Spieß, D. Woschko, L. Rademacher and C. Janiak, Molecules, 2022, 27, 1241 CrossRef CAS PubMed.
- T. Zhang, N. Zhao, J. Li, H. Gong, T. An, F. Zhao and H. Ma, RSC Adv., 2017, 7, 23583–23590 RSC.
- D. Titus, E. J. J. Samuel and S. M. Roopan, Nanoparticle characterization techniques in: Green Synthesis, Characterization and Applications of Nanoparticles, ed. A. K. Shukla and S. Iravani, Elsevier, Amsterdam, 2019, ch. 12, pp. 303–319 Search PubMed.
- W. Zhang, H. Yin, Z. Yu, X. Jia, J. Liang, G. Li, Y. Li and K. Wang, Nanomaterials, 2022, 12, 2062 CrossRef CAS PubMed.
- H. Yang, F. Peng, C. Dang, Y. Wang, D. Hu, X. Zhao, P. Feng and X. Bu, J. Am. Chem. Soc., 2019, 141, 9808–9812 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. R. Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- Y. Jia, Z. Xu, L. Li and S. Lin, Dalton Trans., 2022, 51, 5053–5060 RSC.
- Y. Zhang, J. Wang, L. Ye, M. Zhang and Y. Gong, Dalton Trans., 2021, 50, 4720–4726 RSC.
- Y. Wang, C. Wang, H. Shang, M. Yuan, Z. Wu, J. Li and Y. Du, J. Colloid Interface Sci., 2022, 605, 779–789 CrossRef CAS PubMed.
- F. Li, J. Du, X. Li, J. Shen, Y. Wang, Y. Zhu and L. Sun, Adv. Energy Mater., 2018, 8, 1702598 CrossRef.
- B. Wang, D. Chen, S. Jiao, Q. Zhang, W. Wang, M. Lu, Z. Fang, G. Pang and S. Feng, New J. Chem., 2021, 45, 19646–19650 RSC.
- N. Geng, W. Chen, H. Xu, M. Ding, T. Lin, Q. Wu and L. Zhang, Ultrason. Sonochem., 2021, 72, 105411 CrossRef CAS PubMed.
- Y. Lin, L. Zhao, L. Wang and Y. Gong, Dalton Trans., 2021, 50, 4280–4287 RSC.
- F. L. Li, P. Wang, X. Huang, D. J. Young, H. F. Wang, P. Braunstein and J. P. Lang, Angew. Chem., Int. Ed., 2019, 58, 7051–7056 CrossRef CAS PubMed.
- S. Bauer, C. Serre, T. Devic, P. Horcajada, J. Marrot, G. Férey and N. Stock, Inorg. Chem., 2008, 47, 7568–7576 CrossRef CAS PubMed.
- Z. P. Wu, X. F. Lu, S. Q. Zang and X. W. Lou, Adv. Funct. Mater., 2020, 30, 1910274 CrossRef CAS.
- Q. Zha, F. Yuan, G. Qin and Y. Ni, Inorg. Chem., 2020, 59, 1295–1305 CrossRef CAS PubMed.
- T. Zhang, J. Du, P. Xi and C. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 362–370 CrossRef CAS PubMed.
- J. Huo, Y. Wang, L. Yan, Y. Xue, S. Li, M. Hu, Y. Jiang and Q.-G. Zhai, Nanoscale, 2020, 12, 14514–14523 RSC.
- A. Radwan, H. Jin, D. He and S. Mu, Nanomicro Lett., 2021, 13, 132 CAS.
- L. Yang, G. Zhu, H. Wen, X. Guan, X. Sun, H. Feng, W. Tian, D. Zheng, X. Cheng and Y. Yao, J. Mater. Chem. A, 2019, 7, 8771–8776 RSC.
- F. Sun, G. Wang, Y. Ding, C. Wang, B. Yuan and Y. Lin, Adv. Energy Mater., 2018, 8, 1800584 CrossRef.
- J. Tian, F. Jiang, D. Yuan, L. Zhang, Q. Chen and M. Hong, Angew. Chem., Int. Ed., 2020, 59, 13101–13108 CrossRef CAS PubMed.
- M. K. Sahoo, A. K. Samantara and J. N. Behera, Inorg. Chem., 2020, 59, 12252–12262 CrossRef CAS PubMed.
- D. Li, H. Liu and L. Feng, Energy Fuels, 2020, 34, 13491–13522 CrossRef CAS.
- J. Duan, S. Chen and C. Zhao, Nat. Commun., 2017, 8, 15341 CrossRef CAS PubMed.
- S. Anantharaj, S. R. Ede, K. Karthick, S. S. Sankar, K. Sangeetha, P. E. Karthik and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC.
- S. Öztürk, G. H. Moon, A. Spieß, E. Budiyanto, S. Roitsch, H. Tüysüz and C. Janiak, ChemPlusChem, 2021, 86, 1106–1115 CrossRef PubMed.
- S. Anantharaj and S. Noda, ChemElectroChem, 2020, 7, 2297–2308 CrossRef CAS.
- M. Chauhan, K. P. Reddy, C. S. Gopinath and S. Deka, ACS Catal., 2017, 7, 5871–5879 CrossRef CAS.
- X. Cheng, Z. Pan, C. Lei, Y. Jin, B. Yang, Z. Li, X. Zhang, L. Lei, C. Yuan and Y. Hou, J. Mater. Chem. A, 2019, 7, 965–971 RSC.
- A. Tahira, Z. Hussain, M. Vagin, U. Aftab, M. I. Abro, M. Willander and O. Nur, Catal. Sci. Technol., 2019, 9, 2879–2887 RSC.
- M. Ding, X. Cai and H.-L. Jiang, Chem. Sci., 2019, 10, 10209–10230 RSC.
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Proc. R. Soc. A, 2015, 471, 20140792 CrossRef PubMed.
- Y. F. Yuan, X. H. Xia, J. B. Wu, J. L. Yang, Y. B. Chen and S. Y. Guo, Electrochim. Acta, 2011, 56, 2627–2632 CrossRef CAS.
- X. Zhang, Y. Qiu, W. Zhang, X. Ji and J. Liu, Sustainable Energy Fuels, 2021, 5, 2228–2233 RSC.
- G. Kasparis, A. S. Erdocio, J. M. Tuffnell and N. T. K. Thanh, CrystEngComm, 2019, 21, 1293–1301 RSC.
- S. Krehula, M. Ristić, C. Wu, X. Li, L. Jiang, J. Wang, G. Sun, T. Zhang, M. Perović, M. Bošković, B. Antić, L. Kratofil Krehula, B. Kobzi, S. Kubuki and S. Musić, J. Alloys Compd., 2018, 750, 687–695 CrossRef CAS.
- M. Rajamathi, P. V. Kamath and R. Seshadri, J. Mater. Chem., 2000, 10, 503–506 RSC.
- L. Guerlou-Demourgues, C. Denage and C. Delmas, J. Power Sources, 1994, 52, 269–274 CrossRef CAS.
- K. Lawson, S. P. Wallbridge, A. E. Catling, C. A. Kirk and S. E. Dann, J. Mater. Chem. A, 2023, 11, 789–799 RSC.
- H. Bode, K. Dehmelt and J. Witte, Electrochim. Acta, 1996, 11, 1079–1084 CrossRef.
- J. Yu, S. Pan, Y. Zhang, Q. Liu and B. Li, Front. Mater., 2019, 6, 1–11 CrossRef.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
- J.-Q. Xie, Y.-Q. Ji, J.-H. Kang, J.-L. Sheng, D.-S. Mao, X.-Z. Fu, R. Sun and C.-P. Wong, Energy Environ. Sci., 2019, 12, 194–205 RSC.
- J. Zhao, J.-J. Zhang, Z.-Y. Li and X.-H. Bu, Small, 2020, 16, 2003916 CrossRef CAS PubMed.
- M. K. Adak, L. Mallick, K. Samanta and B. Chakraborty, J. Phys. Chem. C, 2023, 127, 154–168 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 335, 6321 Search PubMed.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Phys. Chem. Chem. Phys., 2012, 14, 2434–2442 RSC.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- R. P. Gupta and S. K. Sen, Phys. Rev. B: Solid State, 1975, 12, 15–19 CrossRef CAS.
- X. Han, Y. Niu, C. Yu, Z. Liu, H. Huang, S. Li, W. Guo, X. Tan and J. Qiu, Nano Energy, 2020, 69, 104367 CrossRef CAS.
- G. T. Vuong, M.-H. Phama and T. O. Do, CrystEngComm, 2013, 15, 9694–9703 RSC.
Footnotes |
| † Dedicated to Prof. Dr Rhett Kempe on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Synthesis and 1H NMR spectrum of TPAMA ligand, additional details regarding the AAS, PXRD, SEM-EDX, TGA and XPS, as well as comparison of the OER performance of different materials. See DOI: https://doi.org/10.1039/d3dt03695b |
| This journal is © The Royal Society of Chemistry 2024 |