
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal–metal cooperativity boosts Lewis basicity and reduction properties of the bis(gallanediyl) CyL2Ga2†‡
Christoph
Helling§
 a,
Lotta
Döhler
c,
Oleksandr
Kysliak
c,
Helmar
Görls
c,
Phil
Liebing
c,
Christoph
Wölper
a,
Robert
Kretschmer
*cde and
Stephan
Schulz
*ab
a,
Lotta
Döhler
c,
Oleksandr
Kysliak
c,
Helmar
Görls
c,
Phil
Liebing
c,
Christoph
Wölper
a,
Robert
Kretschmer
*cde and
Stephan
Schulz
*ab
aInstitute of Inorganic Chemistry, University of Duisburg-Essen, 45117 Essen, Germany. E-mail: stephan.schulz@uni-due.de
bCenter for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, 45117 Essen, Germany
cInstitute of Inorganic and Analytical Chemistry (IAAC), Friedrich Schiller University Jena, 07743 Jena, Germany. E-mail: robert.kretschmer@chemie.tu-chemnitz.de
dJena Center for Soft Matter (JCSM), Friedrich Schiller University Jena, 07743 Jena, Germany
eInstitute of Chemistry, Chemnitz University of Technology, 09111 Chemnitz, Germany
First published on 23rd February 2024
Abstract
The interplay of two proximate gallium centres equips the bimetallic complex CyL2Ga2 (1, CyL2 = 1,2-trans-Cy[NC(Me)C(H)C(Me)N(Dip)]2, Dip = 2,6-i-Pr2C6H3) with increased Lewis basicity and higher reducing power compared to the monometallic gallanediyl LGa (2, L = HC[MeCN(Dip)]2) as evidenced by cross-over experiments. Quantum chemical calculations were employed to support the experimental findings.
Introduction
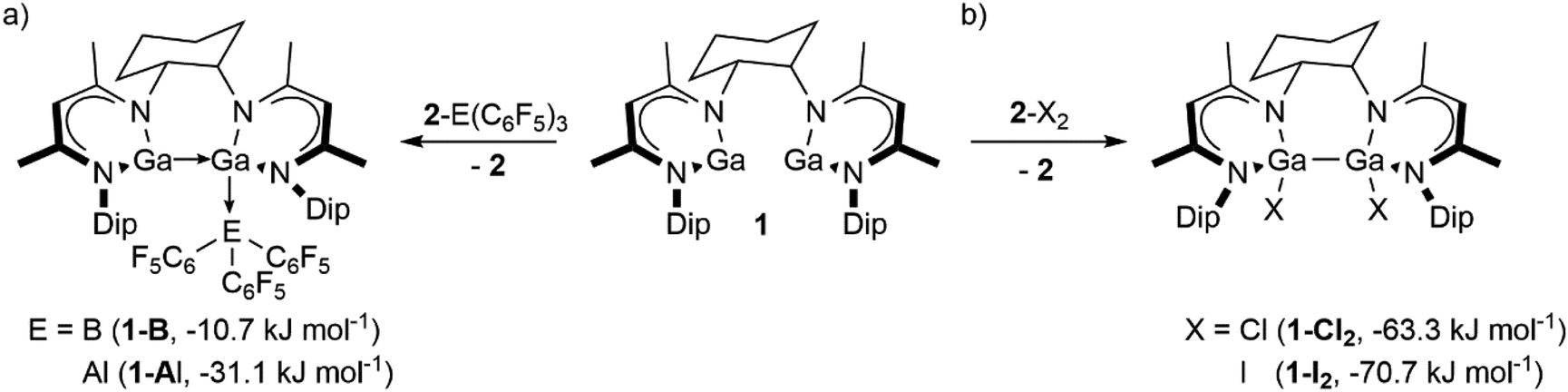
Homobimetallic low-valent main-group metal complexes with metal–metal bonds have attracted increasing attention as their unusual bonding situation and unique frontier molecular orbital arrangements enable transition-metal like reactivity including small molecule activation and catalysis.1 In addition, molecular complexes with only loosely associated or proximate, yet distinct, low-valent main group centres facilitate chemical transformations unavailable with the corresponding monometallic complexes by bimetallic cooperation.2 Unfortunately, metal–metal cooperativity between low-valent main-group metal centres has not yet been studied in full detail due to the scarceness of such compounds. Terphenyl-substituted group 13 diyls [MAr] (M = Al, Ga, In, Tl) often form weakly associated dimetallenes in the solid-state, whereas they typically dissociate into the corresponding monomeric species in solution.3 Power and co-workers demonstrated experimentally and computationally that the reactions of [2,6-Dip2C6H3]Ga (Dip = 2,6-i-Pr2C6H3) towards H2 and ethylene is mediated via the Ga–Ga bonded digallene [{2,6-Dip2C6H3}Ga]2, whereas monomeric [2,6-Trip2-3,5-i-Pr2C6H]Ga (Trip = 2,4,6-i-Pr3C6H2) was found to be unreactive.4Recently, Kretschmer and co-workers reported bis(metallanediyl) complexes X[NC(Me)C(H)C(Me)N(Dip)M]2 (X = linker group, M = Ga, In, Tl), in which two monovalent group 13 metal centres are embedded in a single bis(β-diketiminate) ligand framework.5 Remarkably, the dinuclear GaI complex, CyL2Ga2 (1, CyL2 = 1,2-trans-Cy[NC(Me)C(H)C(Me)N(Dip)]2), exhibits enhanced reactivity in the C–F bond activation of fluoroarenes compared to its monometallic counterpart LGa (2, L = HC[C(Me)N(Dip)]2).5d Furthermore, 1 enables the construction of unusual intermetallic compounds for example with group 13 and 15 compounds.5e Computations revealed that a second isomer of 1, in which the two ambiphilic GaI centres interact by forming an intramolecular donor–acceptor complex (1′, Fig. S30‡), is only slightly higher in energy and should hence be accessible at room temperature.5d The isomer 1′ should feature an increased Lewis basicity compared to 2 as one gallium centre donates electron density to the second and hence increases its donor capabilities. Furthermore, the formation of a Ga–Ga bond upon the oxidative addition should equip 1 with increased reducing power compared to the mononuclear species. Herein, we report the results of cross-over experiments between 1 and donor–acceptor complexes as well as halogenated compounds CyL2Ga2X2 (1-X2; X = Cl 1-Cl2, I 1-I2), undoubtedly proving the enhanced reactivity of 1 compared to 2.
Results and discussion
To prove our hypothesis on the increased Lewis basicity of 1 compared to 2, we first synthesised the Lewis acid–base adducts 1-E(C6F5)3 (E = B 1-B, Al 1-Al) by reaction of 1 with equimolar amounts of E(C6F5)3 (Scheme 1, top). Based on in situ1H, 11B and 19F NMR spectra, 1-B forms quantitatively in solution but could not be isolated due to its fast decomposition during work-up. However, the nature of compound 1-B is established by use of multinuclear (1H, 13C, 11B, 19F) NMR spectroscopy. In contrast, 1-Al forms selectively within 12 h at ambient temperature and was obtained after work-up as a yellow crystalline solid in moderate yield (44%). | ||
| Scheme 1 (Top) Synthesis of 1-B and 1-Al. (Bottom) Molecular structure of 1-Al in the solid-state. Hydrogen atoms, the minor component of the disordered i-Pr group, and co-crystallised benzene molecules were omitted for clarity. Displacement ellipsoids are drawn at 50% probability level. | ||
1-Al is stable in the solid-state under inert gas atmosphere but slowly decomposes in solution to yet unidentified products. The 1H and 13C{1H} NMR spectra of 1-B and 1-Al reflect the asymmetry of the molecules due to the adduct formation, resulting in two distinct sets of resonances for the inequivalent β-diketiminate moieties. The 11B NMR resonance of 1-B (−18.7 ppm) is in a comparable region as observed for 2-B(C6F5)3 (−20.3 ppm) and Cp*Ga–B(C6F5)3 (−17.96a/−19.6 ppm;7 Cp* = C5Me5), hence allowing no conclusions on the donor capability of 1. The 19F NMR spectrum of 1-Al shows the expected three signal pattern, indicating three equivalent C6F5 groups, whereas 1-B features a more complex spectrum containing distinct signals for each fluorine nucleus, potentially due to additional secondary Ga⋯F interactions8 or restricted Ga–B bond rotation. Single crystals of 1-Al were obtained from a saturated benzene solution by storage at room temperature (Scheme 1, bottom). Its molecular solid-state structure confirms the formation of a rare GaI–AlIII Lewis acid–base adduct, of which only three examples have yet been structurally characterised.6b,9 The almost planar six-membered C3N2Ga ring containing Ga2 coordinates to Ga1 of the nearly perpendicularly oriented second gallacycle, which additionally coordinates to Al1 of the Al(C6F5)3 moiety, hence creating a virtually planar coordination environment at Ga2 (sum of bond angles: 358.9(3)°) and distorted tetrahedral coordination geometries at Ga1 and Al1, respectively. The Ga1–Ga2 (2.6109(6) Å) bond is longer than the electron-sharing bonds of CyL2Ga(F)Ga(ArF) (2.4196(5)–2.4520(8) Å)5d and the dative interactions in 2-Ga(C6F5)3 (2.4819(2) Å)6b and Cp*Ga–GaX2Cp* (X = Cl 2.4245(3) Å, I 2.437(2) Å),7 respectively, but resembles the computed Ga–Ga bond length of 1′ (2.67 Å).5d The Ga1–Al1 (2.5494(13) Å) bond length is identical to that of 2-Al(C6F5)3 (2.5482(4) Å),6b consistent with dative Ga2→Ga1 (GaI GaI) and Ga1 Al1 (GaI AlIII) bonding interactions. The sum of the C–Al–C bond angles in 1-Al (323.4(5)°) is smaller than those in Cp*Ga–Al(C6F5)3 (344.6(1)°, 338.3(1)°)9a and 2-Al(C6F5)3 (334.1(2)°),6b suggesting a stronger Ga–Al interaction in accordance with an increased donor strength of 1.10
Next, we conducted cross-over experiments between the known LGa–E(C6F5)3 adducts (2-E(C6F5)3; E = B 2-B, Al 2-Al)6 and CyL2Ga21 (Scheme 2a), which were monitored by 1H NMR spectroscopy (Fig. S19 and S20‡). Addition of 1 to solutions of 2-E(C6F5)3 immediately afforded bright yellow solutions of 1-B and 1-Al, respectively, and compound 2 (Scheme 2a). Despite the relatively small energy gain (vide infra), a variable-temperature (VT) 1H NMR study on the reaction mixture of 1/2-B (Fig. S23‡) showed no indication of a temperature-dependent equilibrium, while the addition of an excess of 2 (Fig. S24‡) did not result in the formation of 2-B(C6F5)3 and liberation of 1, thus supporting an irreversible Lewis acid transfer. The cross-over experiments proved the enhanced Lewis basicity of 1 compared to 2, which is likely induced by the GaI–GaI donor–acceptor interaction. Similar cross-over experiments of CyL2Ga21 with LAl–B(C6F5)3 (3-B, L = HC[MeCN(Dip)]2, Fig. S25‡) showed no conversion even at elevated temperatures, proving the stronger Lewis basic character of LAl (3) compared to 1. However, the significance of the experiment might be biased due to additional Al–F interaction in 3-B.8
 | ||
| Scheme 2 Cross-over experiments for the evaluation of (a) the Lewis basicity and (b) the reduction capabilities of 1 compared to its mononuclear relative 2 along with the calculated Gibbs free energies at the BP86(D3-BJ)/def2-SVP level of theory. | ||
Quantum chemical calculations at the BP86(D3-BJ)/def2-SVP level of theory11 were conducted to gain mechanistic insights and the computational findings support the experimental results. The cross-over reactions of the 1/2-E(C6F5)3 couples are both exergonic, but the gain in energy is more pronounced for the reaction of CyL2Ga21 with 2-Al (−31.1 kJ mol−1) as compared to the reaction with 2-B (−10.7 kJ mol−1). For the 1/2-B(C6F5)3 couple, we could locate an SN2-type transition structure on the potential-energy surface associated with a small barrier of only 13.3 kJ mol−1, Fig. S28,‡ which agrees well with the observed smooth reaction progress. In contrast, the reaction of 1 with 3-B is endergonic by 52.1 kJ mol−1, in-line with its non-occurrence in our experiments.
Since low-valent group 13 β-diketiminate complexes can act as two-electron reductants,12 we studied the reducing capability of 1 by probing the reductive dehalogenation of LGaX2 (2-X2; X = Cl, I) with 1, Scheme 2b. In order to identify the expected reaction products, i.e., CyL2Ga2X2 (1-X2; X = Cl 1-Cl2, I 1-I2), these were independently synthesised by reactions of 2 with InCl3 (1-Cl2, 29%) and AuI (1-I2, 56%), respectively (Scheme S1‡). Compounds 1-Cl2 and 1-I2 are stable in the solid-state and in solution under air- and moisture-free conditions. The 1H and 13C{1H} NMR spectra of 1-Cl2 and 1-I2 each exhibit two distinct sets of resonances for the two β-diketiminate moieties despite the nominally symmetric nature of the molecules, arising from a fixed conformation of the trans-cyclohexylene-bridge caused by the strain in the polycyclic ring system.
Single-crystals of 1-Cl2 and 1-I2 were obtained from saturated solutions in n-hexane at ambient temperature and toluene at 5 °C, respectively (Fig. S23 and S25‡). The halide substituents in the dihalodigallanes adopt syn-periplanar conformations with respect to the Ga–Ga bond (torsion angles X–Ga–Ga–X: 1-Cl2 13.92(2)°, 1-I2 1.64(3)°), which contrasts with non-bridged dihalodigallanes containing β-diketiminate13 and diazabutadienide14 ligands that adopt synclinal and anti-periplanar conformations, respectively. The Ga–Ga and Ga–X bond lengths in 1-Cl2 (Ga–Ga 2.4185(3) Å; Ga–Cl 2.2159(4) Å, 2.2327(3) Å) and 1-I2 (Ga–Ga 2.4615(6) Å; Ga–I 2.6337(6) Å, 2.5779(6) Å) are comparable to those of related dihalodigallanes (Ga–Ga 2.242–2.576 Å; Ga–Cl 2.210–2.218 Å; Ga–I 2.586–2.633 Å)13,14 based on non-bridging ligands and those of CyL2Ga(F)Ga(ArF) (2.4196(5)–2.4520(8) Å),5d indicating covalent GaII–GaII and GaII–X bonding interactions.
The cross-over experiments between 2-X2 and 1 were then monitored by 1H NMR spectroscopy (Scheme 2b and Fig. S21, S22‡). While no reaction occurred at ambient temperature, heating the reaction mixtures to 90 °C for 2 days (1-Cl2) and 70 °C for 5 days (1-I2) resulted in the irreversible consumption of 1 and 2-X2 as well as liberation of 2 and formation of 1-Cl2 and 1-I2, respectively, which were identified by 1H NMR spectroscopy (Fig. S21 and S22‡). 1-Cl2 and 1-I2 are formed together with small amounts of yet unidentified side products, altering the reaction stoichiometry. This likely results from the higher reaction temperatures required to overcome the activation barrier for the halogen transfer imposed by the sterically demanding environments of the Ga centres in 1 and 2. However, the cross-over experiments undoubtedly proved the higher reducing power of binuclear 1 compared to mononuclear 2. The reactions are consistent with one-electron oxidation per Ga centre each in 1 and a two-electron reduction of the Ga centre in 2-X2, while the syn-periplanar arrangement of the halide substituents in 1-Cl2 and 1-I2 supports a suprafacial addition of the halogen atoms to 1.
In contrast, 1 failed to react with LAlI2 (3-I2) even at elevated temperatures (Fig. S26‡), indicating a stronger reduction potential of LAl (3) compared to 1. This reaction may be additionally hampered by the larger steric congestion of the Al centre due to the slightly shorter Al–N and Al–I bond lengths, respectively.15 These findings agree with the computations according to which the dehalogenation of 2-X2 is exergonic by 63.3 (X = Cl) and 70.7 kJ mol−1 (X = I) but endergonic by 53.0 kJ mol−1 in case of 3-I2. We also tried to compute the reaction pathway but despite repeated attempts, we could not locate the respective transition structures. However, we could identify two local minima en route1 + 2-X2 → 1-X2 + 2, Fig. S29,‡ these are a formal encounter complex of 1 and 2-X2 and an intermediate in which one halogen each is bound to 1 and 2, respectively.
Aiming to identify a general preference of compound 1 to form the corresponding GaII–GaII complexes instead of two separate GaIII moieties, we investigated the reactions of compounds 1 and 2 with the aminoxyl radical TEMPO (2,2,6,6-tetramethylpiperidine-N-oxyl, ˙ONR2). Gallanediyl 2 cleanly reacts with two equivalents of TEMPO with oxidation of the GaI centre (Scheme 3) and formation of LGa(TEMPO)2 (2-(TEMPO)2), which was isolated as pale yellow crystals in high yield (78%).
 | ||
| Scheme 3 Synthesis of 2-(TEMPO)2 and 1-(TEMPO)4 (NR2 = N(Me2CCH2)2CH2). | ||
2-(TEMPO)2 features the expected resonances in its 1H and 13C{1H} NMR spectra, and its molecular constitution was confirmed by sc-XRD (Fig. S26‡). Similarly, bis(gallanediyl) 1 reacts with four equivalents of TEMPO to give CyL2Ga2(TEMPO)4 (1-(TEMPO)4) containing two discrete GaIII centres (Scheme 3) in 29% yield; using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometry provided a mixture of 1 and 1-(TEMPO)4. 1-(TEMPO)4 was characterised by sc-XRD (Fig. S27‡), whereas no meaningful NMR spectra could be obtained due to its poor solubility in common organic solvents. The Ga centres in 2-(TEMPO)2 and 1-(TEMPO)4 are distorted tetrahedrally coordinated and feature Ga–O bond lengths (2-(TEMPO)2 1.8505(19) Å, 1.8555(15) Å; 1-(TEMPO)4 1.833(6) Å, 1.850(7) Å) in the expected range. These results show that all four electrons of the two GaI centres in 1 are in principle available for oxidation reactions.
:2 stoichiometry provided a mixture of 1 and 1-(TEMPO)4. 1-(TEMPO)4 was characterised by sc-XRD (Fig. S27‡), whereas no meaningful NMR spectra could be obtained due to its poor solubility in common organic solvents. The Ga centres in 2-(TEMPO)2 and 1-(TEMPO)4 are distorted tetrahedrally coordinated and feature Ga–O bond lengths (2-(TEMPO)2 1.8505(19) Å, 1.8555(15) Å; 1-(TEMPO)4 1.833(6) Å, 1.850(7) Å) in the expected range. These results show that all four electrons of the two GaI centres in 1 are in principle available for oxidation reactions.
Experimental
General procedures
All manipulations were carried out under an atmosphere of purified argon using standard Schlenk and glovebox techniques. Toluene and n-hexane were dried with an MBraun Solvent Purification System, and benzene was distilled from Na/K alloy. Deuterated benzene was dried over activated molecular sieves (4 Å) and degassed prior to use. Starting materials CyL2Ga2 (1),5d LGa (2),16 B(C6F5)3,17 Al(C6F5)3(toluene),18 LGa–B(C6F5)3 (2-B(C6F5)3),6a LGa–Al(C6F5)3 (2-Al(C6F5)3),6b LGaCl2 (2-Cl2),15 and LGaI2 (2-I2)15 were prepared according to literature procedures. AuI, InCl3, and TEMPO were obtained from commercial sources and used as received. NMR spectra (δ in ppm) were recorded using a Bruker Avance DPX 300 (1H 300.1 MHz, 13C{1H} 75.5 MHz, 11B 96.3 MHz, 19F 282.4 MHz), a Bruker Avance Neo 400 (1H 400.1 MHz, 13C{1H} 100.6 MHz, 11B 128.4 MHz, 19F 376.5 MHz), or a Bruker Avance III HD (1H 600.1 MHz, 13C{1H} 150.9 MHz, 11B 192.5 MHz, 19F 564.7 MHz) spectrometer and were referenced to internal C6D5H (1H δ = 7.16, 13C δ = 128.06), external BF3(OEt2) (11B δ = 0.00), or external CFCl3 (19F δ = 0.00) standards. IR spectra were recorded in a glovebox either with an Agilent Cary 630 FTIR or an ALPHA-T FT-IR spectrometer both equipped with a single-reflection ATR sampling module. Microanalyses were performed at the elemental analysis laboratory of the University of Duisburg-Essen. Melting points were measured in wax-sealed glass capillaries under argon atmosphere using a Thermo Scientific 9300 apparatus and are uncorrected.Synthetic procedures
Computations
All geometry optimisations and frequency calculations were performed with the Gaussian16 program package19 in conjunction with the BP86 functional11a,b and def2-SVP basis sets.11c The D3 version of Grimme's dispersion correction with the Becke–Johnson damping function was applied to account for dispersion effects.11d The absence of imaginary frequencies confirmed stationary points as minima. All energies (given in kJ mol−1) are corrected by the (unscaled) zero-point vibrational energy and by thermal energies at 298.15 K.Crystallography
The crystals of 1-Al, 2-Cl2, 2-I2, 1-(TEMPO)4, 2-(TEMPO)2, and S1 were mounted on nylon loops in inert oil. Crystallographic data of 1-Al, 1-Cl2, 2-(TEMPO)2, and S1 were collected on a Bruker D8 Kappa diffractometer with APEX2 detector (Mo-Kα radiation, λ = 0.71073 Å) at 100(2) K. Absorption corrections were performed semi-empirically from equivalent reflections on the basis of multiscans (Bruker AXS APEX2). The intensity data of 2-I2 and 1-(TEMPO)4 were collected on a Nonius KappaCCD diffractometer using graphite-monochromated Mo-Kα radiation. Data were corrected for Lorentz and polarisation effects; absorption was taken into account on a semi-empirical basis using multiple-scans.20 The crystallographic data are summarised in Tables S1 and S2.‡ The structures were solved by direct methods (SHELXS-9721 and SHELXS22) and refined anisotropically by full-matrix least-squares on F2 (SHELXL-2017 and SHELXL-2018).23 Hydrogen atoms were refined using a riding model or rigid methyl groups. In 1-Al, an i-Pr group is disordered over two positions. Its bond lengths and angles were restrained to be equal (SADI) and RIGU restraints were applied to its atoms’ displacement parameters. Due to their close proximity, C35 and C35′ were refined with common displacement parameters (EADP). A benzene molecule is strongly disordered over a centre of inversion. Two alternate positions could be identified of which one is on the centre of inversion. The local symmetry of these positions was ignored in the refinement (negative PART). The bond lengths and angles of all solvent molecules were restrained to be equal (SADI) and the atoms were restrained to lie on a common plane (FLAT). RIGU restraints were applied to the anisotropic displacement parameters. Additionally, for residue 3 ISOR restraints were used. In 1-Cl2, the cyclohexylene ring is disordered over two positions. The displacement of the smaller component could only be refined isotropically and EADP constraints had to be applied to displacement parameters of C11 and C11′ to avoid correlations with the occupancy. In addition, one chlorine atom is disordered over two positions. The Cl–Ga bond lengths of both components were restrained to be equal (SADI). One of the i-Pr groups (C10–C12) of 1-I2 is disordered. The disorder could be resolved to a ratio of 65:35%. The crystal of 1-(TEMPO)4 was treated as a twin by 180° rotation and additionally by inversion. Determination of the twinned cell was performed with CellNow, whereas TWINABS was used for data reduction and absorption correction.24 The two i-Pr groups of 1-(TEMPO)4 are disordered. The disorder could be resolved to ratios of 69:31% (C15–C17) and 54:46% (C18–C20), respectively. On the crystal of 2-(TEMPO)2 there was a small satellite crystal that could not be removed. Treatment as non-merohedral twin revealed its reflections to be very weak (I < 1σ). Including the second component had no influence on the model thus the twinning/satellite could be ignored. The absolute structure of S1 could be determined reliably. Parsons’ quotient method was used to determine the absolute structure parameter x.25
Conclusions
To conclude, the superior basicity and enhanced reducing capability of bimetallic 1 compared to its monometallic counterpart 2 were unambiguously demonstrated by cross-over experiments involving Lewis acid and halogen transfer reactions. The increased reactivity of 1 is attributed to an interaction between the GaI centres. Such synergistic interactions in low-valent bimetallic complexes are expected to play a crucial role in main group element-mediated bond activation processes and catalytic transformations in the future.Author contributions
C. H. conceived the fundamental scheme of the study, executed the experimental work with support by L. D. and O. K., while R. K. performed the quantum chemical calculations. C. W., H. G., and P. L. conducted the sc-XRD measurements and processed the corresponding data. R. K. and S. S. supervised the study. The manuscript was written by C. H, R. K., and S. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (SCHU 1069/23-1, INST 20876/282-1 FUGG, KR4782/3-1, KR4782/2-4), the Chemnitz University of Technology (R. K.), and the Open Access Publication Fund of the University of Duisburg-Essen (S. S.) is acknowledged. We are thankful to S. H. Schreiner for analytic support (IR, melting points) and to the Rechenzentrum of the Friedrich Schiller University Jena for the allocation of computer time.References
-
(a) T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608 CrossRef CAS PubMed
; (b) P. P. Power, Nature, 2010, 463, 171 CrossRef CAS PubMed
-
(a) J. Campos, Nat. Rev. Chem., 2020, 4, 696 CrossRef CAS PubMed
-
(a) J. D. Queen, A. Lehmann, J. C. Fettinger, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2020, 142, 20554 CrossRef CAS PubMed
- C. A. Caputo, J. Koivistoinen, J. Moilanen, J. N. Boynton, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2013, 135, 1952 CrossRef CAS PubMed
-
(a) M. E. Desat, S. Gärtner and R. Kretschmer, Chem. Commun., 2017, 53, 1510 RSC
-
(a) N. J. Hardman, P. P. Power, J. D. Gorden, C. L. B. Macdonald and A. H. Cowley, Chem. Commun., 2001, 1866 RSC
- P. Jutzi, B. Neumann, G. Reumann, L. O. Schebaum and H.-G. Stammler, Organometallics, 2001, 20, 2854 CrossRef CAS
- Z. Yang, X. Ma, R. B. Oswald, H. W. Roesky, H. Zhu, C. Schulzke, K. Starke, M. Baldus, H.-G. Schmidt and M. Noltemeyer, Angew. Chem., Int. Ed., 2005, 44, 7072 CrossRef CAS PubMed
-
(a) J. D. Gordon, C. L. B. MacDonald and A. H. Cowley, Main Group Chem., 2005, 4, 33 CrossRef
-
(a) H. Jacobsen, H. Berke, S. Döring, G. Kehr, G. Erker, R. Fröhlich and O. Meyer, Organometallics, 1999, 18, 1724 CrossRef CAS
-
(a) A. D. Becke, Phys. Rev. A, 1998, 38, 3098 CrossRef PubMed
- M. Zhong, S. Sinhababu and H. W. Roesky, Dalton Trans., 2020, 49, 1351 RSC
- R. Laubenstein, M. Ahrens and T. Braun, Z. Anorg. Allg. Chem., 2017, 643, 1723 CrossRef CAS
-
(a) R. J. Baker, R. D. Farley, C. Jones, D. P. Mills, M. Kloth and D. M. Murphy, Chem. – Eur. J., 2005, 11, 2972 CrossRef CAS PubMed
- M. Stender, B. E. Eichler, N. J. Hardman, P. P. Power, J. Prust, M. Noltemeyer and H. W. Roesky, Inorg. Chem., 2001, 40, 2794 CrossRef CAS PubMed
- N. J. Hardman, B. E. Eichler and P. P. Power, Chem. Commun., 2000, 1991 RSC
- A. G. Massey and A. J. Park, J. Organomet. Chem., 1964, 2, 245 CrossRef CAS
- G. S. Hair, A. H. Cowley, R. A. Jones, B. G. McBurnett and A. Voigt, J. Am. Chem. Soc., 1999, 121, 4922 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
-
(a)
COLLECT, Data Collection Software, Nonius B.V., Netherlands, 1998 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed
-
(a)
G. M. Sheldrick, SHELXL-2014, Program for the Refinement of Crystal Structures; University of Göttingen, Göttingen, Germany, 2014 CrossRef CAS PubMed
-
Bruker, Apex III, CellNow and TWINABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed
-
(a) S. Parsons and H. D. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 2004, 60, s61 Search PubMed
Footnotes |
| † The work is dedicated to Dietmar Stalke on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Spectroscopic (NMR and IR), crystallographic and computational data. CCDC 2128586 (2-(TEMPO)2), 2128587 (1-Al), 2128588 (1-Cl2), 2128594 (S1), 2129461 (1-I2) and 2132253 (1-(TEMPO)4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00172a |
| § Current address: School of Chemistry, Monash University, PO Box 23, Melbourne, VIC, 3800, Australia. |
| This journal is © The Royal Society of Chemistry 2024 |