DOI:
10.1039/D4DD00135D
(Communication)
Digital Discovery, 2024,
3, 2417-2423
Spectra to structure: contrastive learning framework for library ranking and generating molecular structures for infrared spectra†
Received
17th May 2024
, Accepted 15th October 2024
First published on 17th October 2024
Abstract
Inferring complete molecular structure from infrared (IR) spectra is a challenging task. In this work, we propose SMEN (Spectra and Molecule Encoder Network), a framework for scoring molecules against given IR spectra. The proposed framework uses contrastive optimization to obtain similar embedding for a molecule and its spectra. For this study, we consider the QM9 dataset with molecules consisting of less than 9 heavy atoms and obtain simulated spectra. Using the proposed method, we can rank the molecules using embedding similarity and obtain a Top 1 accuracy of ∼81%, Top 3 accuracy of ∼96%, and Top 10 accuracy of ∼99% on the evaluation set. We extend SMEN to build a generative transformer for a direct molecule prediction from IR spectra. The proposed method can significantly help molecule library ranking tasks and aid the problem of inferring molecular structures from spectra.
1 Introduction
Spectroscopy is a branch of science that studies the absorption and emission of electromagnetic radiation by molecules and matter. Infrared (IR) spectroscopy produces an infrared spectrum – a graph of infrared radiation absorbance or transmittance across a frequency range. The IR spectrum is rich with molecular information, which spectroscopists can use to interpret the presence of specific molecular substructures or functional groups by assessing the peaks. Computer-assisted structure elucidation (CASE) algorithms showcase promising capabilities in generating possible molecular structures given spectroscopy data.1 CASE algorithms are predominantly used on NMR data due to the large availability of NMR data. CASE algorithms are not complete on their own, and a synthetic chemist’s intervention may be required. So far, no concrete computational solutions exist for obtaining molecular structures from IR spectra. While several elucidating methods exist for other spectra forms like NMR, the usage of such techniques for IR spectra is still limited.
Although capable conditional molecule generation methods have been developed, there is limited success in the spectral domain. In literature, researchers have proposed a novel framework that uses Monte Carlo tree search (MCTS) and graph convolution network (GCN) to build a molecule iteratively given an NMR spectrum.2 The majority of the machine learning-based structure elucidation methods that have been developed are based on NMR spectra, such as cross-modal retrieval methods for compound identification using NMR data,3 molecule ranking using a convolution neural network for featurizing spectra followed by molecular graph generation algorithms4 and molecular structure prediction from IR and NMR spectra using Markov Decision Process (MDP) and Monte-Carlo Tree Search (MCTS).5 Jonas and Kuhn6 provided a machine-learning method for NMR shift predictions and uncertainty modelling for efficient structure elucidation tasks. Subsequently, Kuhn et al.7 propose a convolutional neural network (CNN) based approach for substructure elucidation on mixtures with just training on pure compounds.
Fessenden and Györgyi8 proposed one of the early methods for IR-based structure elucidation that used a simple two-layer neural network for functional group absence/presence classification. Subsequently, various traditional methods were employed for such structure elucidation tasks. Hemmer and Gasteiger9 proposed a network for generating structure codes for spectra and use the similarity of the structure codes with a library of codes for 3D structure elucidation. Wang et al.10 showcased that support vector machines (SVMs) are capable of functional group classification on 16 different functional groups. Nalla et al.11 leveraged the use of domain-specific information and a rule-based method (obtained using expert knowledge) for machine learning-aided structure elucidation using K-Nearest Neighbors (KNNs), Random Forest Classifiers (RFCs), Multi-Layer Perceptrons (MLPs) and SVMs. One of the recent methods involved the usage of the CNN network for functional group classification.12 All these results highlighted the potential of the usage of machine learning methods for automatic structure elucidation. Current literature suggests that most of these methods are limited to substructure elucidation. Alberts et al.13 proposed transformer encoder–decoder architecture for molecular structure elucidation. Ellis et al.14 proposed a Q-Learning-based approach to build a molecule step-by-step from the IR spectrum.
CLIP15 provided a contrastive learning approach for effectively connecting two different modalities (images and text) and justified the usage of the embeddings for zero-shot classification. Khandelwal et al.16 showcased the quality of CLIP's representation learning capabilities for vision tasks. In this work, we propose the Spectra and Molecule Encoder Network (SMEN); we take an approach to optimize the contrastive loss between the molecule embeddings and spectra embeddings (Fig. 1). This leads to an accurate representation of molecules and spectra in a high-dimensional latent space, where a molecule and its spectra have high cosine similarity. SMEN achieved the Top 1 accuracy of ∼81%, Top 3 accuracy of ∼96%, and the Top 10 accuracy of ∼99% against test spectra. This showcases the molecule-spectra scoring capabilities of the proposed method. In addition to this, we build a decoder model, SMILES Decoder (SD), that can generate Simplified Molecular Input Line Entry System (SMILES) strings from this high dimensional latent space. Using SD coupled with SMEN (SMEN–SD), we show that we can make one-shot IR-to-molecule predictions with promising accuracies.
 |
| | Fig. 1 SMEN: Spectra and Molecule Encoder Network architecture. (1) Batch of molecules and spectra pairs are featurized using molecule encoder and spectra encoder, respectively. (2) Contrastive optimization for maximizing the diagonal in the product matrix, Si,j indicating the similarity between the ith molecule and jth spectra. (3) Ranking of all the molecules against a target spectra by Si,j. | |
2 Methods
2.1 Dataset
For this study, we have calculated the infrared spectrum for molecules present in the QM9 (ref. 17) Dataset, Version 2.18 The dataset consists of small molecules with less than 9 heavy atoms per molecule. The molecules consist of 5 types of atoms: hydrogen, carbon, oxygen, nitrogen, and fluorine. We computed the IR spectra for each molecule using the Gaussian 09 (ref. 19) toolkit. First, we used B3LYP density functional methods with a 6-31g(2df,p) basis set in the gas phase for geometry optimization and vibrational spectra calculation. This results in line spectra, which were broadened to mimic the actual gas phase spectra using the peak broadening function described in Chemprop-IR20 to mimic the experimental IR. The intensities of the resulting spectra are binned with a bin-width of 2 cm−1 in the spectral range from 400–4000 cm−1 to obtain the final 1801 length representation. An example of a line spectrum and the broadened spectrum for a sample molecule is shown in Fig. 2. Using the above settings, we obtained 121![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 944 chemically valid molecules (out of 133885) and their corresponding spectrum after discarding molecules, which resulted in errors in the Gaussian toolkit.
944 chemically valid molecules (out of 133885) and their corresponding spectrum after discarding molecules, which resulted in errors in the Gaussian toolkit.
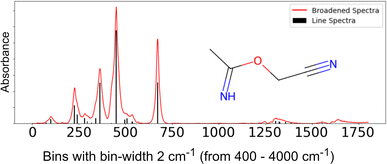 |
| | Fig. 2 Example of spectra obtained after broadening. | |
The idea is to bring two different modalities, IR spectra and 3D molecules, to a unique embedding space such that a molecule and its spectra essentially have extremely similar embeddings. This similarity measure can then be used as a quantitative metric for scoring a library of molecules against spectra or vice versa. A molecule encoder that can learn high-level features relevant to spectral properties and a spectra encoder for a high dimensional representation of the spectra are needed to achieve this. The architectures and the dataset used for meeting the above needs are described in the subsequent sections.
2.2 Spectra encoder
For this study, we split the original spectra into windows or patches similar to splitting an image in vision transformers,21 which is fed to the transformer encoder model, followed by a fully connected network. This is because transformers have shown incredible representation learning capabilities on sequence-based and signal-like data such as text, audio,22 ECG,23 speech, etc. We expect transformers to work incredibly well for IR because of their “attention” mechanisms, which allow these models to prioritize or attend to specific peaks and valleys in the spectrum for efficient structure elucidation, since all regions in the spectra are not equally important for determining the molecular structure. The complete spectra encoder model architecture is showcased in ESI S2.1.† We used a transformer network with 7 attention heads and 5 transformer encoder layers. Further information on implementation details and the model are provided in ESI S2 and S3.†
2.3 Molecule encoder
The molecular data in the QM9 dataset contains the 3D positions of each atom and one-hot node embeddings, which can be used to construct a 3D molecular graph. Chemprop24 showcases the capabilities of Message Passing Neural Networks (MPNNs) for featurizing molecules. Chemprop-IR20 builds upon this for featurizing molecules to predict the spectra, indicating the strength of GNN-based models to capture molecular substructures necessary for correlating to spectral features. Equivariant Graph Neural Networks (EGNNs) are superior to simple 2D GNNs due to their capabilities for encoding 3D structures and spatial composition of atoms, benefitting both from the message passing aspects and 3D constitutional information.25 Additionally, the authors validate EGNN capabilities on the QM9 dataset, which makes it more convenient for our use case. Thus, we believe that EGNNs would be an ideal choice for encoding spectra features. For processing the QM9 molecules and implementation of EGNN, we used the code base provided in https://github.com/vgsatorras/egnn.25 Implementation details are provided in ESI S2 and S3.†
The broad objective is that given a batch of molecules and spectra, the paired molecule encoder and spectra encoder (SMEN) are trained to maximize the cosine similarity of the original pairs and minimize the cosine similarity between the embeddings of non-pairs to obtain a multi-modal embedding space. To achieve this, we optimize the cross entropy loss on the matrix product of both the embeddings for a batch, similar to the approach taken in CLIP26 for images and text. The entire pipeline is shown in Fig. 1. ESI S2 and S3† provide additional information on model architectures and training. Code and data used for this study are available in ESI S1.†
3 Results and discussion
After training the network, given a list of molecules and spectra, we first use SMEN to compute the embeddings for both molecules and spectra. The embeddings obtained are unit normalized (refer to ESI S4† for visualization of embedding space), and cosine similarity of the target spectra embedding with all the molecule embeddings is computed (referred to as score in subsequent sections). The molecules are ranked according to this computed score. If the target molecule exists in top-k ranks, it will contribute to top-k accuracy. We used Chemprop-IR20 on the QM9 dataset as the baseline for comparison (refer S7†). For evaluating the ranking capabilities, we rank the entire QM9 dataset including both train and test set molecules against test spectra (referred to as Full set), and just the test set molecules against test set spectra (referred to as Test set). We report the top-k accuracies in Table 1. The results showcase the potential of the proposed method in scoring molecules against spectra. The top-k scores are very high on the test set (in contrast to the Full set) because, as we keep on increasing the no. of molecules that are screened, the probability that a molecule whose embedding is closer to the target spectra in the embedding space increases (either due to molecule similarity or spectral similarity or an inaccurate embedding). Even if the correct molecule is not ranked the highest, it will likely end up at the top, as indicated by high top-10 accuracies. Sometimes, two not-so-similar molecules may have similar spectra, which can drop the top-k scores for some ranks, as illustrated in Fig. 3. Refer to ESI S5† for more examples. The score alone can act as a good enough metric for quantifying the relationship between a molecule and spectra. The distribution of scores for different ranks of molecules and spectra are provided in Fig. 4.
Table 1 Top K scores for scoring Test set molecules and entire QM9 molecules (Full) against test set spectra
|
|
Top-1 |
Top-2 |
Top-3 |
Top-5 |
Top-10 |
| Test set |
94.1% |
98.8% |
99.3% |
99.6% |
99.8% |
| Full set |
81.4% |
93.0% |
96.1% |
98.1% |
99.3% |
| Chemprop-IR Full set |
74.2% |
83.9% |
87.1% |
89.7% |
91.9% |
 |
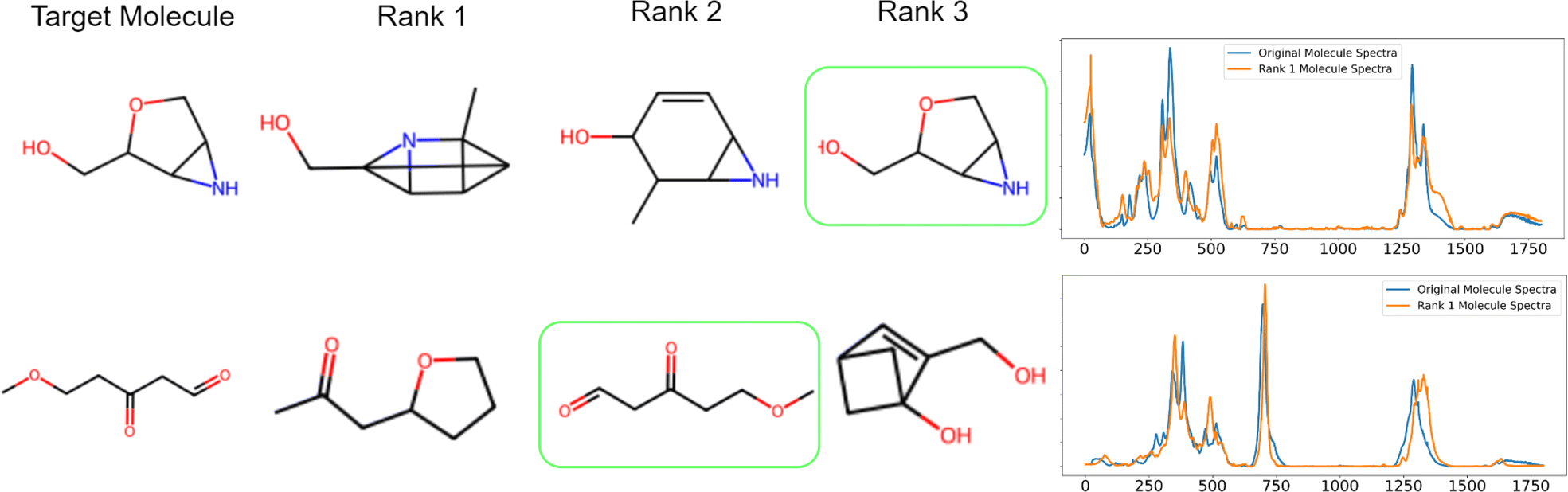
| | Fig. 3 Examples are taken from the Top 3 molecules for two different target spectra. The original molecule and Rank 1 molecule are not very similar (they have similar functional groups C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, R–O–R, OH, and NH) in both examples, but their spectra are similar. This illustrates that the top-ranked molecules can have high spectral similarity with the target molecule spectra even if the ranking is incorrect. O, R–O–R, OH, and NH) in both examples, but their spectra are similar. This illustrates that the top-ranked molecules can have high spectral similarity with the target molecule spectra even if the ranking is incorrect. | |
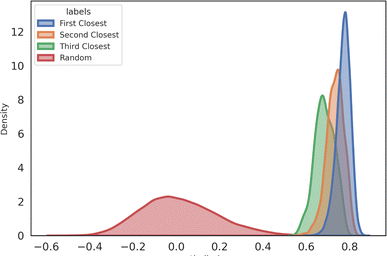 |
| | Fig. 4 Similarity distribution for Top 1, 2, 3 ranked (first, second, and third) pairs and a random sample of an equal number of non-pairs. | |
3.1 Effect of window size
One interesting parameter we use to featurize the IR spectra is patch size. This determines the resolution of the spectral data fed into the transformer model. We experiment with various window sizes for window size w ∈ [3, 5, 7, 9, 11, 13, 15] with a smaller network. Although we do not notice any specific trend in performance, a window length of 7 worked the best for our case. We expected the accuracy to be higher for smaller window lengths due to their higher resolution, but that was not the case. We believe that a window length of 7 here can capture relevant spectral features with sufficient detail (Fig. 5).
 |
| | Fig. 5 Variation of model performance across various window sizes. | |
3.2 Effect of batch size
Several studies have shown the significance of batch size as a parameter for contrastive learning tasks.27 Following the same tradition, we explore the performance of our model across various batch sizes B ∈ [16, 32, 64, 128, 256, 512] with a smaller network as reported in Fig. 6. We clearly see the trend in increasing top-k scores with the increase in batch size. Due to computing resource limitations, we could not explore the effect of an even bigger batch size. However, the figure does suggest that the increase in performance appears to reach a saturation point with the increase in batch size. Such results were expected because the contrastive optimization algorithm heavily depends on batch size; the bigger the batch size, the more accurately the model can contrast them in higher dimensional space.
 |
| | Fig. 6 SMEN's performance across various batch sizes. | |
3.3 Evaluation on NIST quantitative infrared datsaset
Due to lack of enough experimental IR data, we test the SMEN's ranking capabilities on ranking the molecules present in the Quantitative Infrared Dataset https://webbook.nist.gov/chemistry/quant-ir/. We remove all the molecules with atoms other than C, N, O, H and F to have QM9-like molecules. We test SMEN on the remaining 27 molecules. We expected a poor performance due to the differences in experimental and simulated spectra, and also because SMEN has not seen any experimental spectra and has to rank the 27 molecules solely based on the data it learned from simulated data. The results are shown in Fig. S6 and S7.† The results indicate that 7 out of 27 were ranked correctly, 17 out of 27 were ranked in the Top 5. This indicates that although SMEN is not trained on experimental data, to some extent, it is still capable of ranking them. We believe that upon fine-tuning on experimental datasets, this will improve significantly, making SMEN readily usable for real-world examples. Additionally, the current model can generate molecules of all sizes within the range of the QM9 dataset. However, to extend the use cases to large and complex molecules, we could train a larger network on large molecule datasets with even higher-order latent space dimensions, essentially scaling every aspect of the network for complex molecules. An additional improvement or future work to handle large molecules, would be to group various atoms together into a single token.28 For example, the carboxyl [COO] group could be a single token instead of different tokens [C, O, O] (current method). This significantly reduces the complexity of molecules with larger sizes and longer SMILES lengths. We could use an algorithm to find commonly occurring groups of atoms in SMILES strings and create a new token for these groups, increasing the vocabulary size but still keeping the number of tokens required to complete the SMILES smaller. We believe these methods could be used when the molecule space increases exponentially while introducing larger molecules.
3.4 SMILES decoder
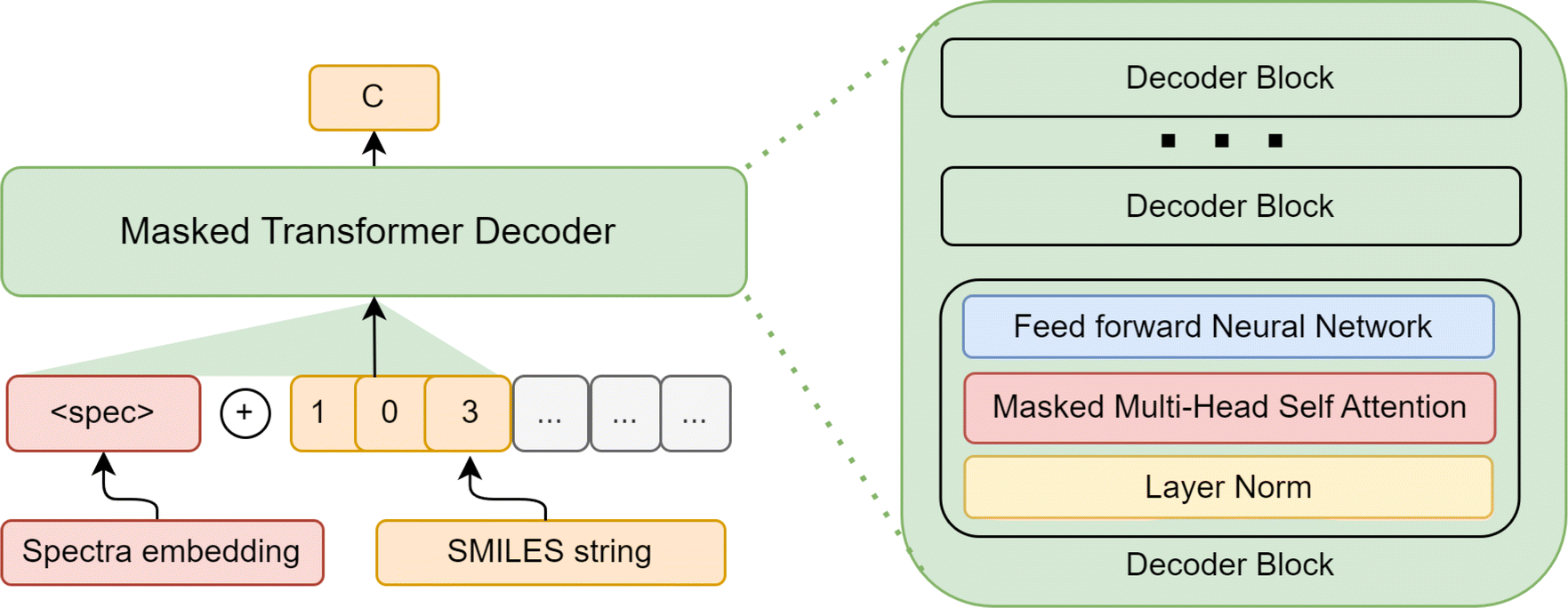
One of the main limitations of both of these methods is that the model can only rank known molecules. One needs to generate novel molecules using another algorithm and then use SMEN for ranking. As an attempt to solve this problem, we built a generative transformer, SMILES Decoder (SD), that can generate a SMILES29 string given an embedding from this shared latent space of molecules and spectra. We take an approach similar to MolGPT,30 where we condition a generative transformer using a pre-trained SMEN framework. For conditioning the transformer on the spectra, we use the spectral embedding obtained by our spectra encoder as the start token and train the transformer to generate subsequent tokens. A simple illustration of SD is shown in Fig. 7. During generation, the first token is predicted using the spectral embedding and is concatenated with the spectral embedding. The next token is predicted from the tokens predicted so far and the spectral embedding. The molecule is sampled till the end token is predicted. Additional architecture information can be found in ESI S2.3.†
 |
| | Fig. 7 SMILES Decoder (SD) used. Given a spectral embedding from the latent space, the embedding is concatenated with SMILES string (embedding 〈spec〉 behaves like the start token), and a Masked Transformer is trained to predict subsequent tokens. | |
We have observed that the trained SMILES decoder module can generate valid molecules 98% of the time. We try greedy decoding (picking the most probable token during sampling) and random sampling (randomly sampling the next token based on the probability distributions) and beam search (keeping track of n beams where each beam is a state of sample SMILES generated so far) for generating SMILES strings. The results suggest that greedy decoding can generate the original molecule with 51% accuracy. As an alternative, we also generate k SMILES samples using the random sampling approach and report the results in Table 2. A few examples are shown in Fig. 8, more in ESI S6.†
Table 2 Percentage of correctly decoded samples using greedy decoding, random sampling with K samples, top beam (target molecule is in the top beam in K beams) and K-beams (target molecule is in one of the K beams)
|
|
K = 1 |
K = 3 |
K = 5 |
| Greedy decoding |
51% |
— |
— |
| Random sampling |
46% |
59% |
63% |
| Top beam |
51% |
56% |
59% |
| K-beams |
51% |
71% |
73% |
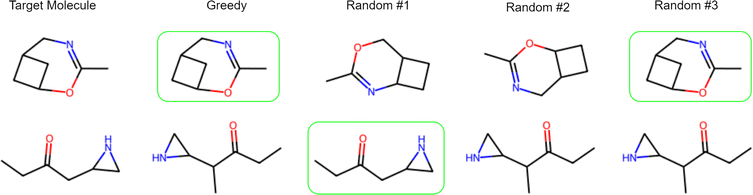 |
| | Fig. 8 Example SMILES generated by the decoder. Correctly generated molecules by the decoder are shown in green. Sometimes, random sampling may generate a target molecule when greedy sampling fails. | |
4 Conclusion
Overall, we see that a simple decoder can be used to extend the applications of SMEN. We can use the SMEN again to rank the molecules the decoder generates. A self-sustaining framework for structure elucidation using IR spectra is proposed. In conclusion, this work proposes a framework SMEN, that uses Equivariant Graph Neural Networks for Molecule encoding and a Transformer for spectra and encodes them to a multi-modal latent space. The contrastive optimization for IR and molecules proposed in this work can generate embeddings that can be used for scoring QM9-like molecules and spectra with high top-k accuracies. We augment this framework with another generative transformer (SD) module that can decode the embedding space to generate molecules with promising accuracy. We believe that our proposed framework will be of significant use for library ranking tasks and direct one-shot molecule prediction using spectra. In the future, one may even use such a pre-trained model for several downstream tasks and to guide other generative models for generating molecules for given spectra.
Data availability
The links to open-sourced code, analysis scripts, and datasets supporting this article have been provided as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank IHub-Data for its support. UDP thanks DST-SERB (CRG/2021/008036) for financial assistance.
Notes and references
- M. Elyashberg and D. Argyropoulos, Magn. Reson. Chem., 2021, 59, 669–690 CrossRef.
- B. Sridharan, S. Mehta, Y. Pathak and U. D. Priyakumar, J. Phys. Chem. Lett., 2022, 13, 4924–4933 CrossRef CAS PubMed.
- Z. Yang, J. Song, M. Yang, L. Yao, J. Zhang, H. Shi, X. Ji, Y. Deng and X. Wang, Anal. Chem., 2021, 93, 16947–16955 CrossRef CAS.
- Z. Huang, M. S. Chen, C. P. Woroch, T. E. Markland and M. W. Kanan, Chem. Sci., 2021, 12, 15329–15338 RSC.
- S. Devata, B. Sridharan, S. Mehta, Y. Pathak, S. Laghuvarapu, G. Varma and U. D. Priyakumar, Digital Discovery, 2024, 3, 818–829 RSC.
- E. Jonas and S. Kuhn, J. Cheminf., 2019, 11, 50 Search PubMed.
- S. Kuhn, E. Tumer, S. Colreavy-Donnelly and R. Moreira Borges, Magn. Reson. Chem., 2022, 60, 1052–1060 CrossRef CAS PubMed.
- R. J. Fessenden and L. Györgyi, J. Chem. Soc., Perkin Trans. 2, 1991, 1755–1762 RSC.
- M. C. Hemmer and J. Gasteiger, Anal. Chim. Acta, 2000, 420, 145–154 CrossRef CAS.
- Z. Wang, X. Feng, J. Liu, M. Lu and M. Li, Microchem. J., 2020, 159, 105395 CrossRef CAS.
-
R. Nalla, R. Pinge, M. Narwaria and B. Chaudhury, Proceedings of the ACM India Joint International Conference on Data Science and Management of Data, Association for Computing Machinery, New York, NY, USA, 2018 Search PubMed.
- G. Jung, S. G. Jung and J. M. Cole, Chem. Sci., 2023, 14, 3600–3609 RSC.
-
M. Alberts and T. Laino and A. C. Vaucher, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-5v27f.
- J. D. Ellis, R. Iqbal and K. Yoshimatsu, IEEE Trans. Artif. Intell., 2024, 5, 634–646 Search PubMed.
-
A. Radford, J. W. Kim, C. Hallacy, A. Ramesh, G. Goh, S. Agarwal, G. Sastry, A. Askell, P. Mishkin, J. Clark, G. Krueger and I. Sutskever, Proceedings of the 38th International Conference on Machine Learning, PMLR, 2021 Search PubMed.
-
A. Khandelwal, L. Weihs, R. Mottaghi and A. Kembhavi, 2022 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), IEEE Computer Society, Los Alamitos, CA, USA, 2022 Search PubMed.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. von Lilienfeld, Sci. Data, 2014, 1, 140022 CrossRef CAS PubMed.
-
R. Ramakrishnan, P. Dral, M. Rupp and O. A. von Lilienfeld, Data for 133885 GDB-9 molecules: Version 2, 2018, https://springernature.figshare.com/articles/dataset/Data_for_6095_constitutional_isomers_of_C7H10O2/1057646 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision E.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- C. McGill, M. Forsuelo, Y. Guan and W. H. Green, J. Chem. Inf. Model., 2021, 61, 2594–2609 CrossRef.
-
A. Dosovitskiy, L. Beyer, A. Kolesnikov, D. Weissenborn, X. Zhai, T. Unterthiner, M. Dehghani, M. Minderer, G. Heigold, S. Gelly, J. Uszkoreit and N. Houlsby, International Conference on Learning Representations, 2021 Search PubMed.
-
B. Elizalde, S. Deshmukh, M. A. Ismail and H. Wang, ICASSP 2023 – 2023 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), 2023 Search PubMed.
- C. Che, P. Zhang, M. Zhu, Y. Qu and B. Jin, BMC Med. Inf. Decis. Making, 2021, 21, 184 CrossRef PubMed.
- E. Heid, K. P. Greenman, Y. Chung, S.-C. Li, D. E. Graff, F. H. Vermeire, H. Wu, W. H. Green and C. J. McGill, J. Chem. Inf. Model., 2024, 64, 9–17 CrossRef PubMed.
-
V. G. Satorras, E. Hoogeboom and M. Welling, Proceedings of the 38th International Conference on Machine Learning, PMLR, 2021 Search PubMed.
-
A. Radford, J. W. Kim, C. Hallacy, A. Ramesh, G. Goh, S. Agarwal, G. Sastry, A. Askell, P. Mishkin, J. Clark, G. Krueger and I. Sutskever, Proceedings of the 38th International Conference on Machine Learning, PMLR, 2021 Search PubMed.
-
T. Chen, S. Kornblith, M. Norouzi and G. Hinton, Proceedings of the 37th International Conference on Machine Learning, PMLR, 2020 Search PubMed.
- X. Li and D. Fourches, J. Chem. Inf. Model., 2021, 61, 1560–1569 CrossRef PubMed.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef.
- V. Bagal, R. Aggarwal, P. K. Vinod and U. D. Priyakumar, J. Chem. Inf. Model., 2022, 62, 2064–2076 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*