
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMLstructureMining: a machine learning tool for structure identification from X-ray pair distribution functions†
Emil T. S.
Kjær
 a,
Andy S.
Anker
a,
Andrea
Kirsch
a,
Joakim
Lajer
a,
Olivia
Aalling-Frederiksen
a,
Simon J. L.
Billinge
*b and
Kirsten M. Ø.
Jensen
*a
a,
Andy S.
Anker
a,
Andrea
Kirsch
a,
Joakim
Lajer
a,
Olivia
Aalling-Frederiksen
a,
Simon J. L.
Billinge
*b and
Kirsten M. Ø.
Jensen
*a
aDepartment of Chemistry and Nano-Science Center, University of Copenhagen, 2100 Copenhagen Ø, Denmark. E-mail: kirsten@chem.ku.dk
bDepartment of Applied Physics and Applied Mathematics Science, Columbia University, New York, NY 10027, USA. E-mail: sb2896@columbia.edu
First published on 27th March 2024
Abstract
Synchrotron X-ray techniques are essential for studies of the intrinsic relationship between synthesis, structure, and properties of materials. Modern synchrotrons can produce up to 1 petabyte of data per day. Such amounts of data can speed up materials development, but also comes with a staggering growth in workload, as the data generated must be stored and analyzed. We present an approach for quickly identifying an atomic structure model from pair distribution function (PDF) data from (nano)crystalline materials. Our model, MLstructureMining, uses a tree-based machine learning (ML) classifier. MLstructureMining has been trained to classify chemical structures from a PDF and gives a top-3 accuracy of 99% on simulated PDFs not seen during training, with a total of 6062 possible classes. We also demonstrate that MLstructureMining can identify the chemical structure from experimental PDFs from nanoparticles of CoFe2O4 and CeO2, and we show how it can be used to treat an in situ PDF series collected during Bi2Fe4O9 formation. Additionally, we show how MLstructureMining can be used in combination with the well-known methods, principal component analysis (PCA) and non-negative matrix factorization (NMF) to analyze data from in situ experiments. MLstructureMining thus allows for real-time structure characterization by screening vast quantities of crystallographic information files in seconds.
Introduction
Pair distribution function (PDF) analysis of total scattering data is widely used to characterize the atomic structure of materials. A PDF is obtained by Fourier transforming corrected and normalized total scattering data, and as both diffuse scattering and Bragg diffraction is included in the Fourier transform, PDF analysis can be used for characterization of bulk crystalline materials, as well as nanoscale1–3 and disordered materials4,5 with only local range structural order. As a PDF can be interpreted as a histogram of interatomic distances, some structural information can be obtained from simple, model free analysis. However, to analyze PDFs quantitatively, structure modelling is required. In the modelling process, a structure model is first identified from which a PDF can be calculated. The structural parameters in the model are then refined until a good agreement between the experimental and calculated PDF is obtained. Such PDF refinements are often performed based on crystallographic structures that can be obtained from structural databases. After the user identifies a promising structure candidate, PDF refinement is performed using dedicated software such as PDFgui,6 DiffPy-CMI,7 DISCUS8 or Topas.9With millions of potential structure candidates present in databases, identifying a structure model for refinement can be challenging and time consuming, and often involves manually browsing through possible candidates. Automated screening methods to identify candidate starting structures have begun to appear, for example, the structureMining10 app at https://PDFitc.org11 to find crystal structural candidates given a measured PDF of a well ordered material, or the clusterMining12 algorithm for screening large numbers of models of close-packed metallic nanoparticles. Whilst quick, these tools require the user inputting prior chemical information and are not fully automated.10,12–14 Here we explore the use of machine learning (ML) to accelerate and automate this process for the case of crystal structure model screening. ML has been successfully employed for various tasks in crystallography and structural analysis, for example, for isolating unique signals from in situ PDF series,15,16 suggesting space groups17 and identifying structures ab initio from PDF data.14,18,19
For this task, we have developed a tree-based ML classifier named MLstructureMining, which has been trained to identify crystal structures from PDFs. MLstructureMining works by matching experimental PDFs with simulated PDFs from a structure catalogue such that structural information can be extracted. MLstructureMining's speed allows fast analysis of e.g., in situ and operando data, and could potentially be used for real-time structure characterization during such experiments. MLstructureMining has been trained on PDFs simulated from 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 833 crystallographic structures obtained from the Crystallography Open Database (COD)20 that contain at least one transition metal, post-transition metal, lanthanide, or actinide, and can only contain one or a combination of O, H or S besides the metals. When using MLstructureMining, a PDF is given as input, while the output is a ranked list of suggested structures, whose simulated PDF matches with the input PDF. To reduce the number of possible structure suggestions, the list contains structure classes, where the CIFs that results in very similar simulated PDFs are bundled together into one class. We first show that MLstructureMining obtains a top-3 accuracy of 99% on simulated PDFs not seen during training, with a total of 6062 possible classes. We then demonstrate that MLstructureMining can be used to identify the chemical structure from experimental PDFs obtained from metal oxide nanoparticles of different sizes. Lastly, we show how MLstructureMining can be used in combination with the well-known methods principal component analysis (PCA) and non-negative matrix factorization (NMF) to analyze a large PDF dataset obtained from an in situ experiment on the formation of Bi2Fe4O9.
833 crystallographic structures obtained from the Crystallography Open Database (COD)20 that contain at least one transition metal, post-transition metal, lanthanide, or actinide, and can only contain one or a combination of O, H or S besides the metals. When using MLstructureMining, a PDF is given as input, while the output is a ranked list of suggested structures, whose simulated PDF matches with the input PDF. To reduce the number of possible structure suggestions, the list contains structure classes, where the CIFs that results in very similar simulated PDFs are bundled together into one class. We first show that MLstructureMining obtains a top-3 accuracy of 99% on simulated PDFs not seen during training, with a total of 6062 possible classes. We then demonstrate that MLstructureMining can be used to identify the chemical structure from experimental PDFs obtained from metal oxide nanoparticles of different sizes. Lastly, we show how MLstructureMining can be used in combination with the well-known methods principal component analysis (PCA) and non-negative matrix factorization (NMF) to analyze a large PDF dataset obtained from an in situ experiment on the formation of Bi2Fe4O9.
Method
Data preparation of structures and PDFs
The use of MLstructureMining is shown schematically in Fig. 1. The classification task performed by MLstructureMining can be thought of as PDF matching. When used, MLstructureMining compares the input PDF to PDFs simulated from structure models in a structure catalogue. The best matching PDFs can then be found through SoftMax scores as described below. Having identified the best matching PDF, structural models (space group, unit cell, composition and atomic fractional coordinates) can be inferred from knowing which structure/structures were used for simulating each PDF.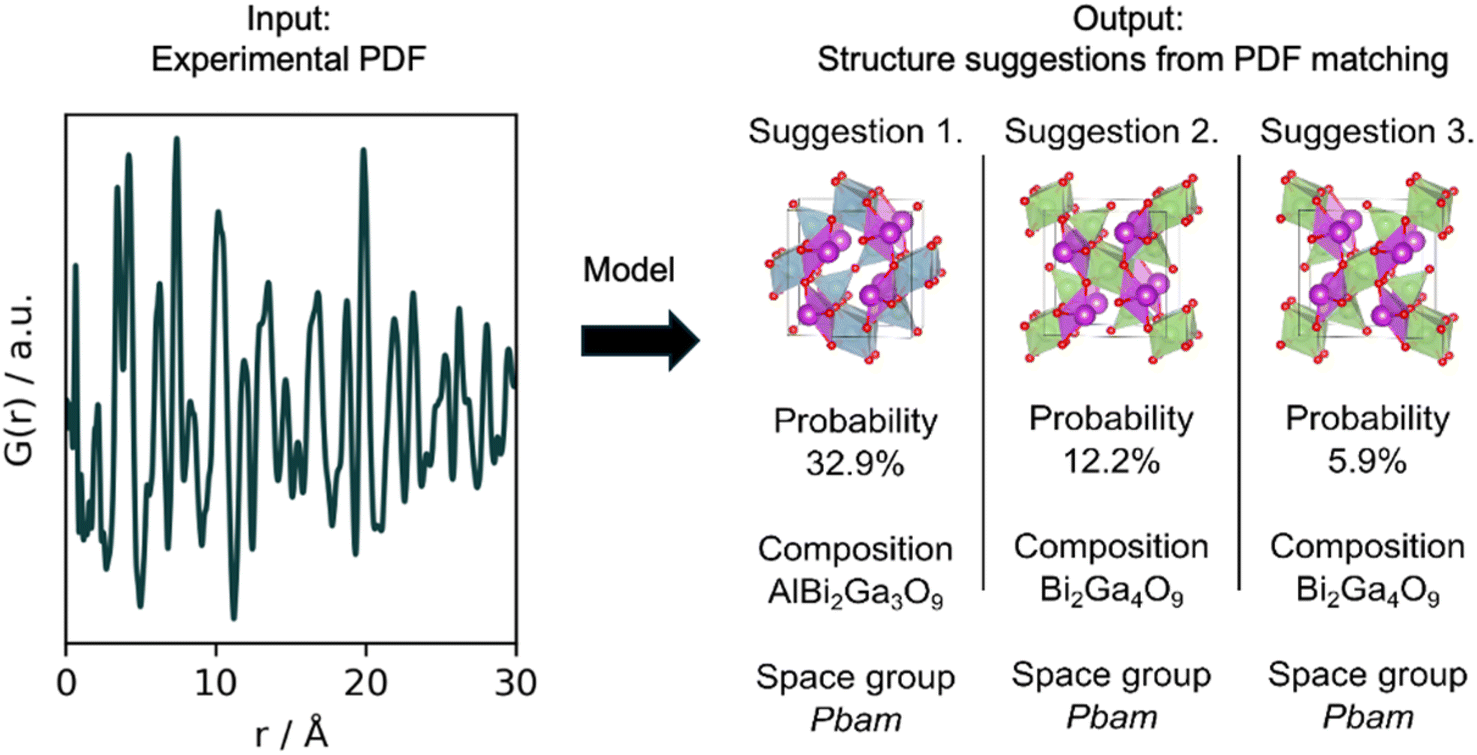 | ||
| Fig. 1 Inference process of MLstructureMining. This schematic illustrates the inference process utilized by MLstructureMining, where a Pair Distribution Function (PDF) is inputted, and the tool subsequently proposes viable structure candidates through PDF matching. The output is a prioritized list of PDFs matching the input PDF, which identifies potential structural candidates, from which the composition, space group and unit cell are directly derived. | ||
MLstructureMining was trained on simulated PDFs from crystal structure models obtained from the Crystallographic Open Database (COD).20,21 The PDFs were simulated using DiffPy-CMI7 and the simulation parameters mimic typical experimental PDFs as shown in Section B in the ESI.†
The structure models, represented as Crystallographic Information Files (CIFs) were downloaded from COD on the 24th of January 2023. Only structure models containing at least one transition metal, post-transition metal, lanthanide, or actinide and either O, H or S were included. This query resulted in a total of 10833 crystal structures. However, some of these structure models are almost identical and contain similar structural information and thus result in highly similar simulated PDFs. As described below, we therefore determine the similarity between different structures and PDFs and bundle structures resulting in similar PDFs together. This results in a ‘structure catalogue’ containing structure models and corresponding PDFs.
To determine PDF similarity, we use the Pearson Correlation Coefficient (PCC)22 as defined in Section C in the ESI.† We calculate the PCC between simulated PDFs from all pairs of structures in our dataset. If two PDFs have a PCC equal to or above 0.95, then the structures are considered similar, and they will be referred to as the same entity in the structure catalog. After this step, the total number of unique structures with simulated PDFs in the structure catalog was reduced to a total of 6062. Identifying and grouping structurally similar entries within our structure catalog reduces the potential concerns regarding the similarity among the PDFs and the skewed representation of different structure types within COD. Given the inevitability of some degree of similarity between PDFs from distinct structures, we conducted comprehensive testing using zeroth-order optimization (ZOO) and various experimental datasets, including in situ PDF data from which we extracted NMF components. This was done to ensure that our model was truly learning to generalize rather than merely overfitting.
We have chosen to set the PCC threshold to 0.95, however, this is an arbitrary value and can be configured after need. By increasing the value of the PCC threshold less structures will be grouped together, hence increasing the overall structural similarity within each entity of the structure catalog. The PCC threshold was determined by looking at various tungsten oxide structures, as they contain similar structural building blocks but with various defects and oxygen disorder. After comparing different structures, 0.95 was determined to be a suitable threshold as it allows for some oxygen disorder but not for new structural peaks within the PDF.
For each entity in the structure catalog, we simulate 100 PDFs with various unit cell dimensions and isotropic atomic displacement parameters (Uiso) chosen using Latin hypercube sampling.23 The unit cell parameter of a, b and c were varied taking into account space group symmetry constraints. The unit cell parameters were varied with ±4%, and the Uiso values were varied from 0.005 Å2 to 0.025 Å2. All Uiso values are set to the same value independent of the atom type. The simulation parameters for the instrumental parameters (Qmin, Qmax, Qdamp) mimic typical experimental PDFs as shown in Section B in the ESI.† The PDFs are simulated from 0 Å to 30 Å with a step size of 0.1 Å, which due to the Shannon–Nyquist sampling theorem24 is a sufficiently small step size for PDFs generated with up to Qmax of 31.4 Å−1.25
We use XGBoost as our ML model for the classification task in MLstructureMining,26 as gradient tree boosting has proven to provide state-of-the-art results on classification benchmarks.27 To train, validate and test MLstructureMining, the simulated PDFs were split into a training, validation and test set with the ratios of 80%, 10% and 10%. We ensure equal representation of each structure in the training, validation and test split. Hence, 80 PDFs of each structure were used for training, 10 for validation and 10 for testing. MLstructureMining's hyperparameters was optimized using Bayesian optimization,28 and the best model was selected from the validation score. Incorporating Latin hypercube sampling was done to minimize the similarities in the training, validation and testing split by systematically sampling across the simulation parameter space. All hyperparameters are shown in Section B in the ESI.† After hyperparameter optimization, MLstructureMining obtains an accuracy of 91% for structure suggestion and a top-3 accuracy of 99%, both are determined from predictions on the test set and with a total of 6062 possible classes. To test the robustness of MLstructureMining, we deploy ZOO from the Adversarial Robustness Toolbox (ART)29 library to perform adversarial attacks. These attacks indicate that MLstructureMining is well regularized and robust as it obtains an accuracy of 89% and a top-3 accuracy of 97%, with a total of 6062 possible classes. For the ZOO attacks the test data was used. Further explanation can be found in Section B in the ESI.†
MLstructureMining outputs the SoftMax score for each class which can provides and indication of its prediction certainty. We note here that using the SoftMax confidence as a proxy for uncertainty has proven to not be exact.30 Other methods can be used to better estimate uncertainties such as bootstrapping31,32 and ensemble methods,33 but these are beyond the scope of this article.
MLstructureMining has been implemented as a Python package with a command line interface. The implementation makes it possible to install the library through a wheel file with pip, which will automatically install all missing dependencies. Additionally, MLstructureMining has been implemented as a Hugging Face application, see the ‘Code availability’ section. The only input required for MLstructureMining is a PDF file or a directory of PDF files. Here, the PDF should be given with r in the units of Å, with an r-range from minimum 0 Å to 30 Å. After providing the path to the PDF data, MLstructureMining proposes structural candidates for all data that can be found within the provided path.
Results and discussion
Introduction of the four experimental PDFs
We demonstrate the capabilities of MLstructureMining on four experimental PDFs, which have been obtained from different samples with varying experimental parameters. The four PDFs are shown in Fig. 2, while their synthesis and data collection can be found in Section D in the ESI.† Example 1 is an experimental PDF obtained from CoFe2O4 nanoparticles with the spinel structure (purple, Fig. 2). The experimental PDF was obtained with a Qmax of 17.5 Å−1 compared to the training data with a Qmax of 25 Å−1. The CoFe2O4 nanoparticles are ca. 17 nm large and are crystalline with no significant structural disorder. This makes them a suitable first trial for MLstructureMining, as the PDF closely resemblances the training data. Examples 2 and 3 are experimental PDFs from ultra-small CeO2 nanoparticles with the fluorite structure (red, Fig. 2) and tungsten oxide nanoparticles with a structure related to W5O14 (blue, Fig. 2). As seen from the PDFs, both samples have crystalline domains of approximately 20 Å. The small particle sizes means that the two experimental PDFs are outside MLstructureMining's training distribution. Additionally, the tungsten oxide PDF (example 3) is obtained with a significantly lower Qmax (15 Å−1) than that of the training data (25 Å−1), and the sample shows a high degree of structural disorder.3,13 Such disorder causes peak broadening and asymmetric peaks due to peak overlap, which adds an extra layer of complexity to analyzing the PDF.13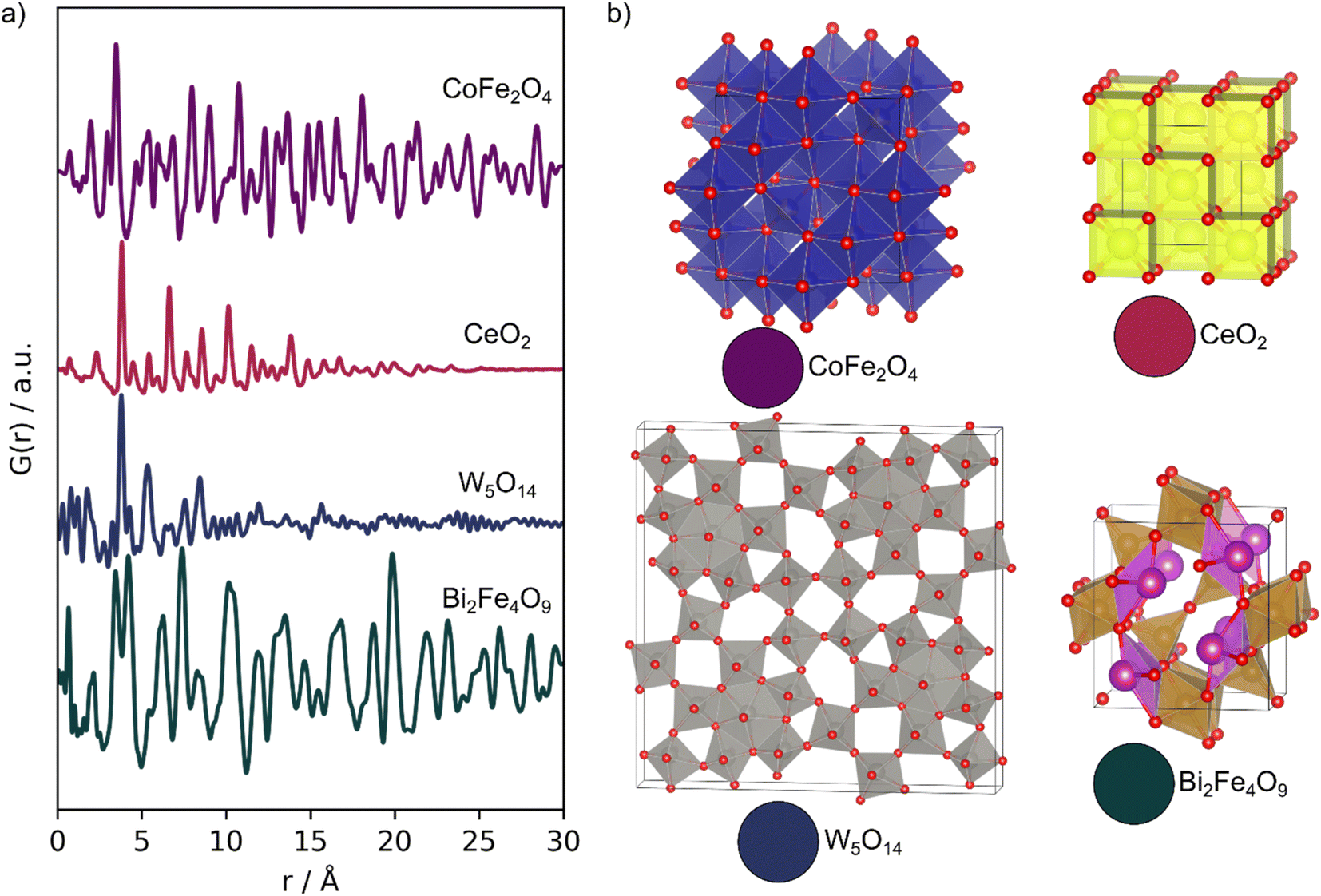 | ||
| Fig. 2 Experimental PDFs. (a) Experimental PDFs obtained from; CoFe2O4 (purple), CeO2 (red), W5O14 (blue) and mullite Bi2Fe4O9 (green). (b) The expected structures of the four samples. | ||
Example 4 is an in situ PDF dataset collected during the formation of multiferroic Bi2Fe4O9 nanoparticles (green, Fig. 2). In this experiment, an amorphous precursor powder was heated at 700 °C for 1 h, while scattering patterns were collected every 5 seconds to follow its transformation into the crystalline product. With this example, we investigate MLstructureMining capabilities to analyze larger amounts of data.
Example 1: identifying the crystal structure with MLstructureMining on an experimental PDF obtained from CoFe2O4
We start out by using MLstructureMining to analyze the experimental PDF obtained from CoFe2O4 nanoparticles, shown in purple in Fig. 2. The experimental parameters used to obtain the PDF, e.g. Qmin of 1.6 Å−1 and Qmax of 17.5 Å−1 resembles the data used to train MLstructureMining. The crystallinity and size of the nanoparticles furthermore means that the PDF shows structural information beyond 30 Å, as was also the case for the training data simulated for crystalline materials. However, the nanoparticle size still results in a slight damping of the PDF peaks at higher r. Such damping is not seen in the training data as all PDFs are simulated for crystals with infinite size. Real-space Rietveld refinement using the CoFe2O4 spinel structure are shown in Section E in the ESI.† The refinement shows that the size is approximately 17 nm.The experimental PDF is directly given as input for MLstructureMining, which performs structure identification as described above. The top-5 structure suggestions for the CoFe2O4 PDF are shown in Table 1. Within this list of predicted structures, only spinel structure types are proposed. Only the chromite structure differs in space group as it has a tetragonal distortion.34 All structures belong to the family of spinel structures, where the oxygens are arranged in an fcc structure, and metal ions occupy octahedral and tetrahedral sites.35
| Rank | Composition | Space group | Probability [%] | R wp [%] | COD ID |
|---|---|---|---|---|---|
| 1 | Cr2NiO4 | I41/amd | 34.4 | 16.9 | 1536758 |
| 2 | Co2.28Cu0.72O4 |
Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m m |
9.2 | 17.7 | 1537073 |
| 3 | Ga2NiO4 |
Fdm |
7.7 | 17.3 | 1541403 |
| 4 | Co3O4 |
Fdm |
4.9 | 49.7 | 5910031 |
| 5 | Cd0.75Fe2O4Zn0.25 |
Fdm |
3.3 | 27.5 | 1539596 |
To check the structure suggestions, real-space Rietveld refinements were performed using each of the top-3 structures. Here, the scale factor, cell (a, b, c, α, β and γ if allowed by symmetry), the particle size (psize), δ2 and an isotropic Uiso were refined. The Uiso values were refined to take the same value for all atoms. The structure suggestions along with their fits to the experimental PDF are shown in Fig. 3. We see that the fits using the top-3 suggestions are almost identical and as the Rwp values range from 16.9% to 17.7%. The second and third structural starting models used have slightly different lattice parameters, which has caused them to not be bundled together in the structure catalog as their PCC is below 0.95. However, when the structures are refined to the PDF, almost identical results are obtained.
 | ||
| Fig. 3 Real-space Rietveld refinements of the top-3 structures suggestions from MLstructureMining for the experimental PDF obtained from CoFe2O4. (a) 1st prediction; Cr2NiO4 (COD ID: 1536758), (b) 2nd prediction; Co2.28CuO.72O4 (COD ID: 1537073) and (c) 3rd prediction; Ga2NiO4 (COD ID: 1541403). All fit parameters are provided in Section F in the ESI.† | ||
From the fits shown in Fig. 3 we can conclude that MLstructureMining found suitable structural candidates to describe the experimental PDF as it suggests all structures of the spinel type. However, MLstructureMining does not suggest a structure with the correct composition of containing Co and Fe. This relates to the almost indistinguishable X-ray scattering factors of Cr (24 e−), Fe (26 e−) and Co (27 e−), Ni (28 e−), Cu (29 e−) and Ga (31 e−), and illustrates that MLstructureMining and X-ray PDF in general cannot stand alone for complete structural and chemical composition. We do not see this as a significant limitation of the method. In many cases, the chemical composition will be known from e.g., the synthesis or other analysis methods. PDF is then used for structure characterization, and here, MLstructureMining clearly shows that the CoFe2O4 particles have the spinel structure.
To further benchmark the capabilities of MLstructureMining, we compare its results with those of an existing structure finding tool, namely structureMining,10 which is available as a web service at https://PDFitc.org.36 We provide structureMining with the PDF and the composition of structure we want it to search through, see Section G in the ESI.† structureMining requires the user to input information about elements that are present in the target material, although ‘wild-cards’ for elements can also be used where appropriate. Here we used ‘Fe–O’ as composition query, which searches through all stoichiometries of iron oxide. This query yielded a total of 151 structures. The top five structures returned by structureMining all have the Fe3O4 composition with Rwp values between 17% to 18%. MLstructureMining and structureMining thus yielded similar results as spinel type structures were returned in both cases. Both approaches perform well, but there are differences in their use. Firstly, chemical composition information does not need to be submitted for the ML model to work making it convenient to use, though it will only work on compounds similar to those it is trained on, i.e., oxides, sulphides and hydrides of transition metal, post-transition metal, lanthanide, or actinide, compounds (∼11000 structures). structureMining, on the other hand, can be asked to mine from any compositional subset of the entire database of ∼400000 structures specified by the user. Looser compositional queries can be given to structureMining (for example, “Fe–O–*” would search for all structures in the database that contain Fe, O and any other element) but the method becomes very slow if the query is too broad. MLstructureMining always returns results rapidly.
Example 2 and 3: identifying the structure of CeO2 and W5O14 nanoparticles
We now proceed to more challenging experimental PDFs from nanoparticles of CeO2 and W5O14, which are less similar to the PDFs that MLstructureMining has been trained on. Both PDFs only show peaks to approximately 20 Å. Since MLstructureMining has not been trained on PDFs of nanoparticles but on simulated PDFs of ideal, infinitive crystals, these examples let us test MLstructureMining's capabilities to extrapolate outside of its learned training distribution.The top-5 structure suggestions for the experimental PDF from CeO2 nanoparticles are shown in Section H in the ESI.† MLstructureMining's SoftMax output for its top-3 suggestions are 41.7%, 7.2%, and 3.0%. This suggests that MLstructureMining finds suitable structures even though the experimental PDF is outside of the training distribution. Fig. 4 shows real-space Rietveld refinements using the three best structures. Four out of five structures show promising resemblance to the baseline structure of CeO2 as all structures are fluorite related. In top-3 the first and second suggested structures result in a low Rwp, (16.5% and 17.3%) which indicates high structural agreement with the experimental PDF. In the fluorite structure, the metal ions are arranged in an fcc lattice, while the anions occupy the octahedral sites. Suggestion two and three are both fluorite-structured doped uranium oxides, and they are thus closely related to the expected CeO2 structure. MLstructureMining's first suggestion is a rhombohedral RIII phase where La and U layers alternate along [111]c.37 Prediction two deviates from the classical fluorite structure, as it takes the velikite structure.38 Compared to the fluorite structure, the velikite structure misses every second O and the remaining oxygens are replaced with S. Nevertheless, the metal atoms are the same position as the fluorite structure.
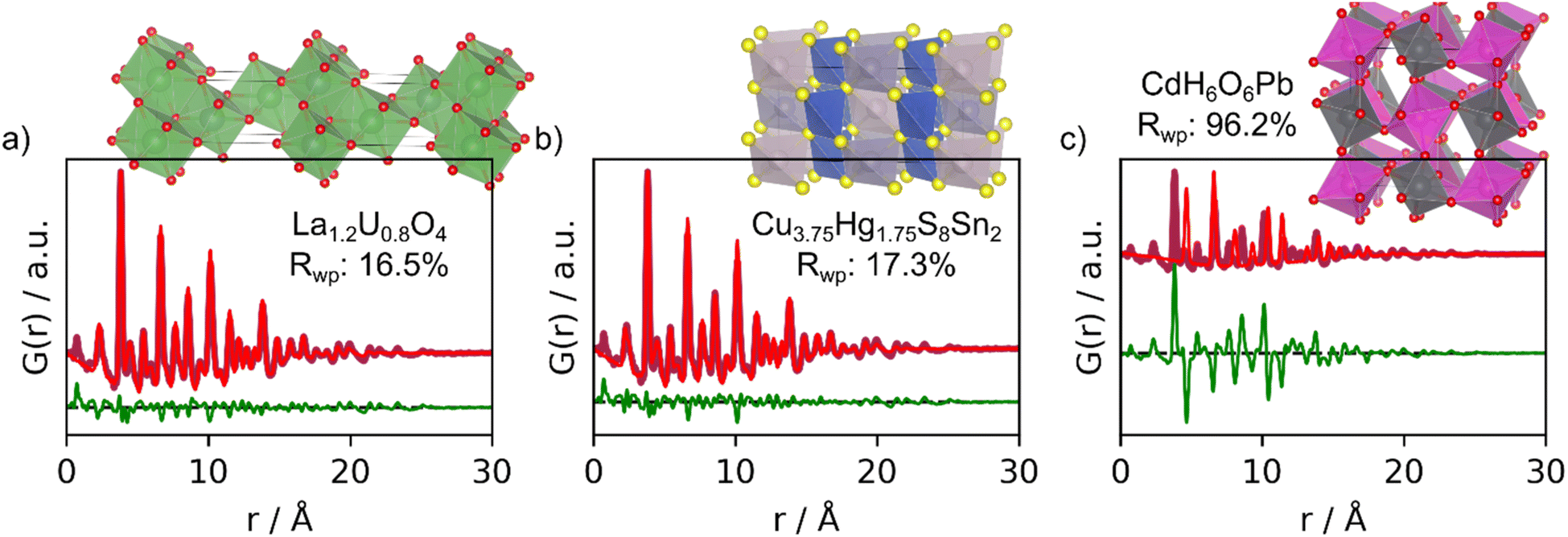 | ||
| Fig. 4 Real-space Rietveld refinements of the top-3 structure suggestions for the experimental PDF obtained from CeO2. (a) 1st prediction; fluorite La1.2U0.8O4 (COD ID: 1006067), (b) 2nd prediction; velikite Cu3.75Hg1.75S8Sn2 (COD ID: 1527617) and (c) 3rd prediction; CdH6O6Pb (COD ID: 1527729). All fit parameters are given in Section F in the ESI.† | ||
Again, MLstructureMining does not suggest structures with the correct chemical composition. Instead of Ce, it suggests a La/U-based oxide, with a structure with higher electron density on the metal sites compared to CeO2. If comparing simulated PDFs of CeO2 and La1.2U0.8O4 (Section I in the ESI†). It is seen that the higher electron density results in a slight change in ratio between the PDF peak intensities. In La1.2U0.8O4, metal–metal PDF peaks are relatively more intense than oxygen–oxygen and oxygen–metal peaks compared to CeO2. Interestingly, the Rwp values of the fit with suggestion 1 (16.5%) is lower than the fit with the expected CeO2 structure (17.32%, Section E in the ESI†), i.e., the La/U-based oxide gives a better description of the experimental data. This difference in fit quality could relate to oxygen vacancies or oxygen disorder in our CeO2 nanoparticles, however, further analysis of this effect is outside the scope of the paper.
Considering structure suggestions with unexpected chemical compositions may thus provide additional information on the sample in play. However, it is also possible to introduce chemical constrains which means that MLstructureMining only returns structures with relevant chemical composition or space groups. If we eliminate all proposed structures that do not include Ce, then we obtain a range of different cerium oxides in our top-5 prediction. Interestingly, MLstructureMining's first cerium oxide suggestion is a zirconium doped fluorite structure in spacegroup P42/nmc. Performing a real-space Rietveld refinement using the proposed structure obtain an Rwp of 17.9%, which is comparable with the baseline CeO2 structure. However, since Ce and Zr has a large difference in electron density (18 e−), removing the Zr doping, thus making it a pure CeO2 structure provide an Rwp of 16.1%. Fit parameters can be found in Section J in the ESI.† This underlines the value of having a tool for being able to rapidly screen through thousands of structures, while being able to apply structural constraints to the output.
We now compare the results from MLstructureMining with those from structureMining. For this query ‘Ce–O’ was provided as composition for structureMining, which yielded a total of 10 structures, see Section G in the ESI.† All the top-4 structures are identical to the CeO2 structure used for the baseline fit, but the best structure has a lowered oxygen occupancy. structureMining thus provides the expected result compared to MLstructureMining.
We continue to challenge MLstructureMining on experimental PDFs that are significantly different from the simulated PDFs that it has been trained on. We now use an experimental PDF with a Qmax of only 15 Å−1. The PDF is obtained from ultra-small tungsten oxide nanoparticles with a large degree of oxygen disorder.13 Neither the low Qmax, the small size, nor oxygen disorder have been taken into account when training MLstructureMining. We have previously analyzed the structure of these tungsten oxide nanoparticles, and shown that the W5O14 structure best describe the PDF.13 However, several other known tungsten oxide structures containing pentagonal columns of [WO6] octahedra can also account for the main PDF peaks, and the unit cells of these structures, including W5O14, are furthermore larger than the nanoparticles. Therefore, using a crystalline model may not be a suitable way to describe their structure. This PDF thus represents an extremely challenging task for MLstructureMining, which it may in fact not be suited for.
The top-3 structures suggested by MLstructureMining can be seen Fig. 5 along with their real-space Rietveld refinements. The SoftMax output for these structures are 1.2%, 1.1% and 1.1%. These values are significantly smaller than those of the suggestions made in example 1–2 (Table 1 and Section H in the ESI†). In this example MLstructureMining predicts an evenly distributed set of output, this behavior indicates that the provided input is outside of the training distribution, which should alert the user about limited success. Suggestions 1 and 2 are metal oxide structures, while suggestion 3 is an alloy. It is evident from the fits that the suggested structures only match the most intense peak located at 3.8 Å (Fig. 5), which corresponds to the distance between tungsten ions in corner-sharing [WO6] octahedra in W5O14. Apart from this, the suggested structures show little structural similarity with tungsten oxide as highlighted by the Rwp values obtained from the real-space Rietveld refinements (66.9%, 67.2% and 61.6%).13 For this PDF, MLstructureMining's suggestions thus seem almost random and are not useful for structural analysis. Fortunately, this can be identified by MLstructureMinings low SoftMax output in its predictions. When using MLstructureMining, the user should thus take note of these values.
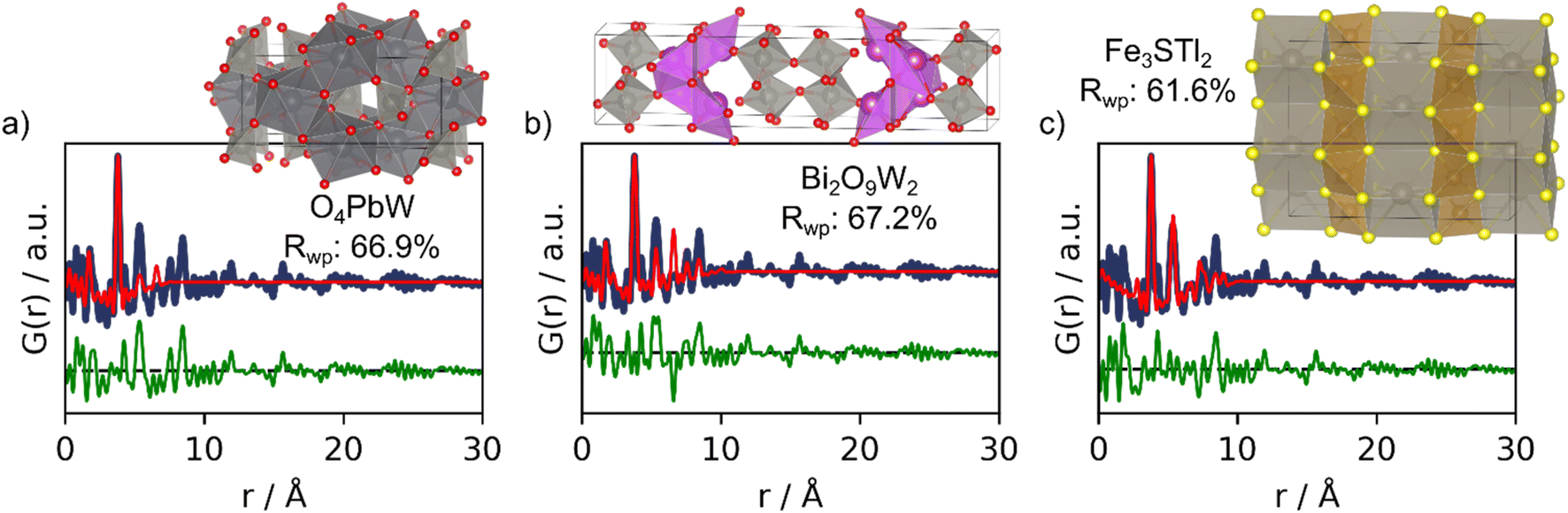 | ||
| Fig. 5 Real-space Rietveld refinements of the-3 three suggestions for the experimental PDF obtained from tungsten oxide nanoparticles. (a) 1st prediction; O4PbW structure (COD ID: 9014025), (b) 2nd prediction; Bi2O9W2 structure (COD ID: 7230340) and (c) 3rd prediction; Fe3S4Tl2 structure (COD ID: 1536855). All fit parameters can be seen in Section F in the ESI.† | ||
Utilizing structureMining for proposing structures for the experimental PDF of W5O14 yielded promising results when using ‘W–O’ as the composition input. This resulted in a total of 25 structures, see Section G in the ESI.† Here, the best and second best structure were W5O14 and W18O49, which is in agreement with prior structure characterization.13 This underlines the usefulness of the structureMining app for cases not suited for MLstructureMining.
Example 4: structure identification from in situ PDFs obtained during the formation of Bi2Fe4O9
To test MLstructureMining's capabilities for larger datasets, we use it to analyze an in situ PDF series collected during the formation of Bi2Fe4O9. In the experiment, we follow the crystallization of an amorphous precursor powder into the crystalline product during heating at 700 °C. The precursor was synthesized by the sol–gel method, which is further described in Section D in the ESI.† During heating, the amorphous precursor transforms into an intermediate crystalline phase before Bi2Fe4O9 forms, as shown in Fig. 6a. The three distinct phases, precursor, intermediate and product, are highlighted in white (Fig. 6a). To get a better overview of the structural changes in the data, we first use a PCC matrix as shown in Fig. 6b. A description of how to obtain the PCC matrix is given in Section C in the ESI† and in previous work.13 The PCC measures the linear relationship between two continuous functions ranging from opposite (−1) to similar (1), and the PCC matrix thus allows us to visually compare the similarity between different PDFs throughout the dataset. The yellow regions of the PCC matrix (Fig. 6b) indicate areas of the in situ PDF series with few structural changes, hence a high PCC value. The transformation of the precursor into the crystalline intermediate is very sudden, which can be seen from the sharp change in PCC value at around 11 minutes (Fig. 6b). In turn, the color gradient is smoother at the transition between the intermediate phase to the product. This indicates that the structural transition occurs slower and that both the intermediate and product might exist at the same time for a certain period.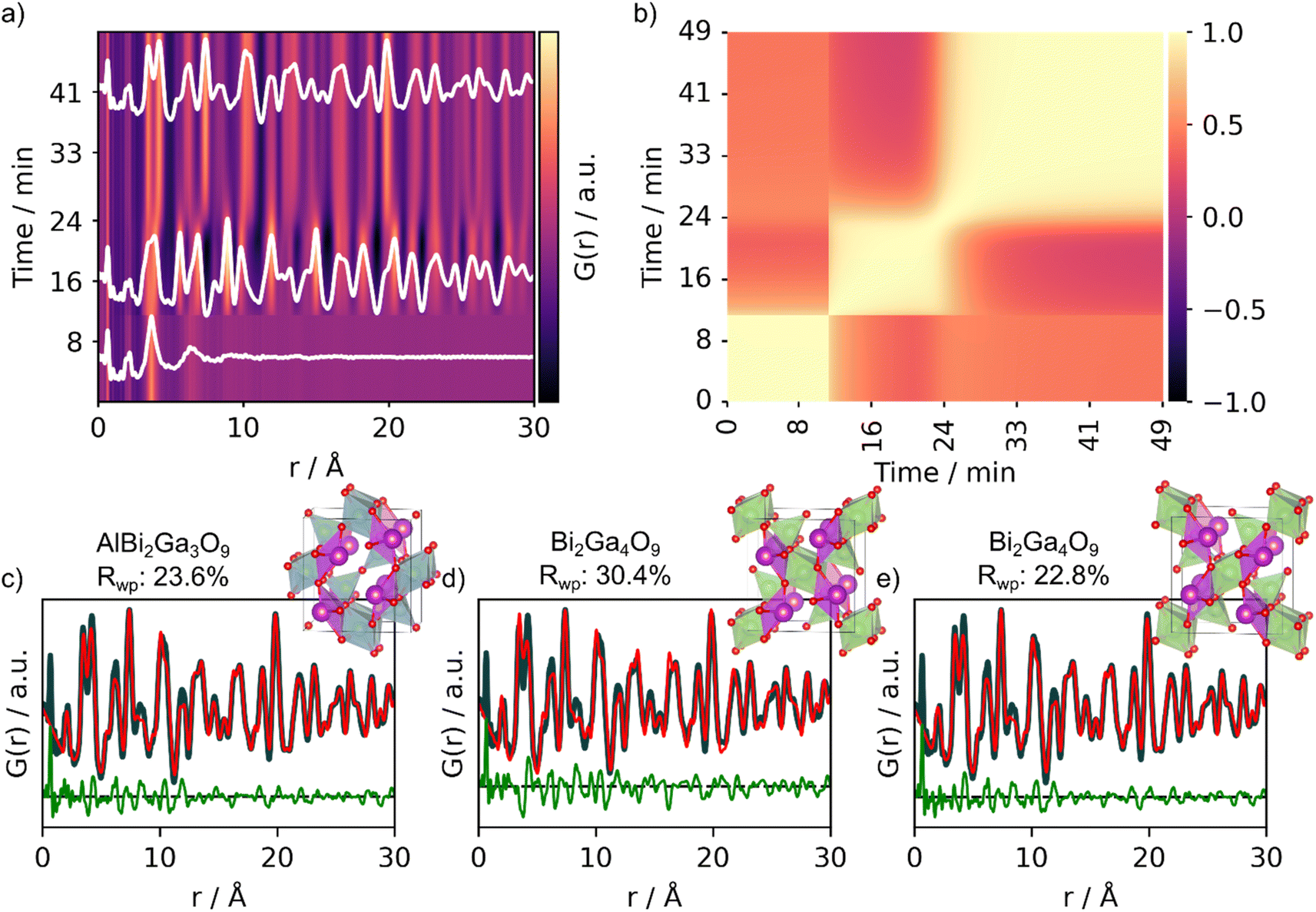 | ||
| Fig. 6 Analysis of the experimental in situ PDFs obtained during the formation of Bi2Fe4O9. (a) Waterfall plot of the PDFs obtained during heating of the amorphous precursor at 700 °C crystallizing into Bi2Fe4O9 and (b) corresponding PCC matrix. MLstructureMining was used for structure prediction on the last frame of the in situ series. The real-space Rietveld refinements of the top-3 predictions of the experimental PDF are shown in (c) AlBi2Ga3O9 (COD ID: 4342599), (d) Bi2Ga4O9 (COD ID: 2104768) and (e) Bi2Ga4O9 (COD ID: 2002314). Fit parameters can be seen in Section F in the ESI.† | ||
After having gained a visual representation of the changes occurring in the in situ PDF series, we use MLstructureMining to suggest structural candidates for the PDFs of the in situ series. The probability for the top-3 structural predictions per PDF for the whole in situ series is plotted in Section K in the ESI.† As finding the structure of the amorphous precursor is currently not within the capabilities of MLstructureMining, we focus on analyzing the structure of the product, while similar analysis for the intermediate can be found in Section L in the ESI.†Fig. 6c and d shows fits of the top-3 structure predictions for the last PDF of the in situ series. The top-5 suggestions are shown in Section H† and the fit parameters are given in Section E in the ESI.† All of the three suggested structures are Bi-based oxides with the mullite-type structure, i.e. Bi2Ga4O9 and Bi2Ga3AlO9. Both of these structures are isostructural with the expected structure Bi2Fe4O9 and differ only in composition.39 All three structures proposed by MLstructureMining provide a suitable fit to the final PDF of the in situ series, which is shown by the low Rwp values (23.6%, 30.4% and 22.68%).
When using structureMining on the experimental PDF of Bi2Fe4O9, three different composition were tested to screen through a larger chemical space, see Section G in the ESI.† Ultimately, structureMining returned the structure of Bi2Fe4O9 and Bi2Ga4O9, which have similar structures to those also proposed by MLstructureMining.
Supervised ML models have successfully been applied to gain structural information from scattering and spectroscopy data.17,18,40–47 However, these methods are limited when dealing with data measured on chemical systems with multiple phases. On the other hand, unsupervised ML models such as PCA and NMF have been employed to identify structural components in scattering and spectroscopy data but these methods do not provide a characterization of the structure.16,44,45,48–51 Here, we demonstrate that by combining supervised and unsupervised ML, it is possible to characterize the structure of data that contains contributions from multiple chemical structures.
PCA and NMF analysis can be employed to both identify the number of phases in a dataset, and for isolating the phases contained within an in situ PDF experiment.16,48,50,52 Here, we use a combination of PCA and NMF to reduce the complexity of the dataset. If the total number of components (here distinct phases in the in situ series) is not known, PCA can be used to propose the number of components needed to fully describe the data by determining the variance explained by each components (Fig. 7a). Once the number of components has been proposed by PCA, NMF can be used to reconstruct the components of the in situ series, in this example going from several hundred PDFs down to a few PDFs.
 | ||
| Fig. 7 PCA, NMF component reconstruction and real space Rietveld refinement of MLstructureMining's top-3 suggestions. (a) Cumulative variance explained by the PCA components. (b) Reconstructed NMF components (orange and red) shown on top of experimental data (blue and purple) together with the difference curve (green) shown below. The real-space Rietveld refinements using the top-3 suggestions to the experimental PDF of the product are shown in (c) AlBi2Ga3O9 (COD ID: 4342599), (d) Bi2O4Pd (COD ID: 2002219) and (e) Bi2Ga4O9 (COD ID: 2104768). Fit parameters can be seen in Section F in the ESI.† | ||
Due to the limited structural information (0–10 Å) from the precursor phase, all PDFs only containing structural information about the precursor was excluded in the PCA and NMF analysis. The PCA and NMF analysis including the entire in situ series is shown in Section M in the ESI.†Fig. 7a shows the cumulative sum of explained variance by each PCA component.53 This information can be used to determine the number of components needed to describe the in situ PDF series. Here, the two first components describe 99% of the variance within the data. Individually, component 1 explains 67% and component 2 explains 32%, while the remaining 1% are either fluctuations in the data caused by noise or small structural changes. Using 3, 4 or 5 components does not improve the description of the data. We therefore chose to compute two components using NMF, as seen in Fig. 7b. We show the reconstructed components on top of the experimental PDFs of the product and intermediate together with their difference curves. NMF component 1 corresponds to the product Bi2Fe4O9, while NMF component 2 corresponds to the intermediate phase appearing during the in situ experiment (Fig. 7b). We then provide NMF component 1 as input for MLstructureMining. From this analysis, we obtain similar results for the top-3 structural candidates as when directly providing MLstructureMining with the last PDF of the in situ series shown in Fig. 6c–e. Fig. 7c–e shows strong agreement between MLstructureMining's first and third suggestions from NMF component 1 and the experimental PDF. It is only the second predictions which differentiates. The fit parameters can be seen in Section E in the ESI† and only show minor differences between the fit parameters obtain from the experimental PDF and NMF component 1.
Applying structureMining on NMF component 1 proposes the same top-5 structures as when used on the experimental PDF of Bi2Fe4O9, see Section G in the ESI.† The consistency of this result further highlights the utilization opportunities of combining PCA with NMF to extract the unique PDF signals from large multiphase in situ experiments.
Conclusion
We have presented a ML tool for identifying structural candidates for PDF refinement called MLstructureMining. MLstructureMining has been trained on PDFs simulated from 10833 crystal structures from the COD. Within a second, it can propose structure candidates for the provided experimental PDF without any additional inputs, and thereby without any bias. Within its top-3 suggestions, MLstructureMining scores an accuracy of 99% on the test data, with a total of 6062 possible classes. MLstructureMining's speed means that it can be used to rapidly analyze large PDF data sets from time- or position resolved experiments.
MLstructureMining was used on four PDFs measured on different instrumental parameters (Section D in the ESI†) and types of chemical systems; crystalline CoFe2O4 nanoparticles, ultrasmall CeO2 and tungsten oxide nanoparticles and an in situ PDF series obtained during the formation of Bi2Fe4O9. For the crystalline CoFe2O4 spinel nanoparticles, MLstructureMining successfully predicts spinel type structures as the most promising structure and automated real-space Rietveld refinements of the top-3 suggestions yield Rwp values of 16.9%, 17.7% and 17.3%. Example 2 demonstrates that MLstructureMining can be applied to experimental PDFs from ultra-small nanoparticles. Here, MLstructureMining suggested several fluorite structures with high structural similarity to the CeO2 structure that the PDF was obtained from. The obtained Rwp values top-2 structure suggestions (16.5% and 17.3%) highlights the structural agreement. MLstructureMining was thus successful even though the experimental PDF shows only little structural coherence, and thus is far from the training PDFs, which are simulated from crystalline materials. In example 3, we demonstrated that the predicted probability scores can be used as a proxy of how trustworthy the MLstructureMining suggestions are. Here, MLstructureMining was used on an extremely challenging experimental PDF with low Qmax obtained from ultra-small tungsten oxide nanoparticles with high degree of oxygen disorder. MLstructureMining's predicted probability scores of top three (1.2%, 1.1% and 1.1%) indicate that no suitable structural models were found. Lastly, in example 4, we demonstrate MLstructureMining's capability to deal with an in situ PDF series and thereby characterize large amounts of data. We furthermore show how a combination of PCA and NMF can reconstruct the unique PDF signals within an in situ PDF series containing multiple components, thus reducing the amount of data from several hundred PDFs to a handful but also enabling supervised ML to identify the structure. The reconstructed NMF components are robust enough for MLstructureMining to analyze and show similar results as directly analyzing experimental PDFs.
MLstructureMining offers the advantage of rapid screening, capable of sifting through thousands of structures in mere seconds without requiring specific compositional input. This feature can be particularly useful when there is a need to broaden the understanding of a synthesized structure. However, if there is a low degree of uncertainty regarding the sample, structureMining offers a more targeted approach. In essence, while MLstructureMining offers speed for the class of materials it is trained on, structureMining provides a deeper, more refined analysis, making them complementary tools in the study of structures.
As shown throughout these four examples presented, MLstructureMining show great generalization capabilities and the SoftMax output show a high correlation with suggestion suitable structures. When MLstructureMining predicts an unevenly distributed set of SoftMax outputs, the predictions have shown to be reliable. Evenly distributed SoftMax outputs indicate little reliability, as in the case for the experimental PDF of W5O14. To evaluate robustness of MLstructuremining, thus ensuring regularized behavior of the model, we deployed ZOO attacks from ART. These demonstrated a modest decrease in the model's top-3 accuracy, from 99% to 97%, indicating a strong generalization capability. We deemed that it was out of the scope for this article to further quantify how far it was possible to push the testing distribution before breakdown of the model was achieved.
MLstructureMining has been implemented as a Python library with a command-line interface and on Hugging Face to ensure easy accessibility to MLstructureMining. MLstructureMining has additionally been installed on the DanMAX beamline at MAXIV in Sweden, and is planned to be implemented in https://PDFitc.org.11 Due to MLstructureMining's easy deployment, fast structure analysis and less biased data analysis capabilities, we expect that MLstructureMining is a new powerful tool for PDF analysis.
Code availability
The code for data preparation and training of MLstructureMining can be found at: https://github.com/EmilSkaaning/MLstructureMining-workflow. MLstructureMining as a Python library can be found at: https://github.com/EmilSkaaning/MLstructureMining. MLstructureMining has been uploaded as a Hugging Face model and can be found at: https://huggingface.co/Ekjaer/MLstructureMining.Data availability
The Python package ‘MLStructureMining’ version 4.1.0 has been used to predict suitable structural models. The link to the code can be found here: https://github.com/EmilSkaaning/MLstructureMining. The code used for the PCA and NMF analysis (extract the number of components and generate NMF features) can be found at: https://github.com/Kabelkim/phase-splitter. The version used was updated on the 13th of March 2022. All crystallographic information files (CIFs) have been downloaded from ‘Crystallography Open Database’ (COD), https://www.crystallography.net/cod/. Data preprocessing and generating Pair Distribution Function (PDF) data was done using the code found at: https://github.com/EmilSkaaning/MLstructureMining-workflow. The version used is the newest version, ‘Merge Pull Request #20’, updated on the 3rd of September 2023.Author contributions
ETSK, ASA and KMØJ conceptualized the project. ETSK, ASA, SJLB, and KMØJ designed the methodology and ETSK, ASA and JL wrote the code. ASA, AK and OAF synthesized the samples and collected the scattering data. KMØJ procured funding and with SJLB supervised the project. All authors were involved with the writing of the paper.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work is part of a project that has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Program (grant agreement No. 804066). We are grateful to the Villum Foundation for financial support through a Villum Young Investigator grant (VKR00015416). Funding from the Danish Ministry of Higher Education and Science through the SMART Lighthouse is gratefully acknowledged. We acknowledge support from the Danish National Research Foundation Center for High Entropy Alloy Catalysis (DNRF 149). AK gratefully acknowledges the Deutsche Forschungsgemeinschaft (DFG, German science foundation) for funding of the project Ki 2427/1-1 (# 429360100). Work in the Billinge group was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award Number DE-SC0019212. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III beamline P02.1 and P21.1. Beamtime was allocated for proposal I-20210486 EC.References
- T. L. Christiansen, S. R. Cooper and K. M. Ø. Jensen, Nanoscale Adv., 2020, 2, 2234–2254 RSC.
- S. J. L. Billinge and I. Levin, Science, 2007, 316, 561–565 CrossRef CAS.
- M. Juelsholt, A. S. Anker, T. L. Christiansen, M. R. V. Jørgensen, I. Kantor, D. R. Sørensen and K. M. Ø. Jensen, Nanoscale, 2021, 13, 20144–20156 RSC.
- S. J. L. Billinge and M. G. Kanatzidis, Chem. Commun., 2004, 749–760, 10.1039/B309577K.
- T. Lindahl Christiansen, E. T. S. Kjaer, A. Kovyakh, M. L. Roderen, M. Hoj, T. Vosch and K. M. O. Jensen, J. Appl. Crystallogr., 2020, 53, 148–158 CrossRef CAS PubMed.
- C. L. Farrow, P. Juhas, J. W. Liu, D. Bryndin, E. S. Božin, J. Bloch, T. Proffen and S. J. L. Billinge, J. Phys.: Condens. Matter, 2007, 19, 335219 CrossRef CAS PubMed.
- P. Juhás, C. L. Farrow, X. Yang, K. R. Knox and S. J. L. Billinge, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 562–568 CrossRef PubMed.
- T. Proffen and R. B. Neder, J. Appl. Crystallogr., 1997, 30, 171–175 CrossRef CAS.
- A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- L. Yang, P. Juhás, M. W. Terban, M. G. Tucker and S. J. Billinge, Acta Crystallogr., Sect. A: Found. Adv., 2020, 76, 395–409 CrossRef CAS PubMed.
- L. Yang, E. A. Culbertson, N. K. Thomas, H. T. Vuong, E. T. S. Kjaer, K. M. O. Jensen, M. G. Tucker and S. J. L. Billinge, Acta Crystallogr., Sect. A: Found. Adv., 2021, 77, 2–6 CrossRef CAS PubMed.
- S. Banerjee, C.-H. Liu, K. M. O. Jensen, P. Juhas, J. D. Lee, M. Tofanelli, C. J. Ackerson, C. B. Murray and S. J. L. Billinge, Acta Crystallogr., Sect. A: Found. Adv., 2020, 76, 24–31 CrossRef CAS PubMed.
- E. T. S. Kjær, O. Aalling-Frederiksen, L. Yang, N. K. Thomas, M. Juelsholt, S. J. L. Billinge and K. M. Ø. Jensen, Chem.: Methods, 2022, 2, e202200034 Search PubMed.
- A. S. Anker, E. T. S. Kjær, M. Juelsholt, T. L. Christiansen, S. L. Skjærvø, M. R. V. Jørgensen, I. Kantor, D. R. Sørensen, S. J. L. Billinge, R. Selvan and K. M. Ø. Jensen, npj Comput. Mater., 2022, 8, 213 CrossRef.
- R. Gu, S. J. L. Billinge and Q. Du, Acta Crystallogr., Sect. A: Found. Adv., 2023, 79, 203–216 CrossRef CAS PubMed.
- H. S. Geddes, H. Blade, J. F. McCabe, L. P. Hughes and A. L. Goodwin, Chem. Commun., 2019, 55, 13346–13349 RSC.
- C.-H. Liu, Y. Tao, D. Hsu, Q. Du and S. J. L. Billinge, Acta Crystallogr., Sect. A: Found. Adv., 2019, 75, 633–643 CrossRef CAS PubMed.
- E. T. S. Kjær, A. S. Anker, M. N. Weng, S. J. L. Billinge, R. Selvan and K. M. Ø. Jensen, Digital Discovery, 2023, 2, 69–80 RSC.
- A. S. Anker, E. T. Kjaer, E. B. Dam, S. J. Billinge, K. M. Jensen and R. Selvan, Proceedings of the 16th International Workshop on Mining and Learning with Graphs (MLG), 2020, DOI:10.26434/chemrxiv.12662222.v1.
- S. Gražulis, D. Chateigner, R. T. Downs, A. Yokochi, M. Quirós, L. Lutterotti, E. Manakova, J. Butkus, P. Moeck and A. Le Bail, J. Appl. Crystallogr., 2009, 42, 726–729 CrossRef PubMed.
- S. R. Hall, F. H. Allen and I. D. Brown, Acta Crystallogr., Sect. A: Found. Adv., 1991, 47, 655–685 CrossRef.
- J. Myers, A. Well and R. Lorch Jr, Research design and statistical analysis Routledge, Routledge, 2010 Search PubMed.
- M. A. Bouhlel, J. Hwang, N. Bartoli, R. Lafage, J. Morlier and J. Martins, Adv. Eng. Software, 2019, 135, 102662 CrossRef.
- C. E. Shannon, Proc. IRE, 1949, 37, 10–21 Search PubMed.
- C. Farrow, M. Shaw, H. Kim, P. Juhás and S. Billinge, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 134105 CrossRef.
- T. Chen and C. Guestrin, Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 2016, vol. 22, pp. 785–794 Search PubMed.
- P. Li, arXiv, 2012, preprint, arXiv:1203.3491, DOI:10.48550/arXiv.1203.3491.
- F. Nogueira, GitHub, 2014, https://github.com/fmfn/BayesianOptimization Search PubMed.
- M.-I. Nicolae, M. Sinn, M. N. Tran, B. Buesser, A. Rawat, M. Wistuba, V. Zantedeschi, N. Baracaldo, B. Chen and H. Ludwig, arXiv, 2018, preprint, arXiv:1807.01069, DOI:10.48550/arXiv.1807.01069.
- T. Pearce, A. Brintrup and J. Zhu, arXiv, 2021, preprint, arXiv:2106.04972, DOI:10.48550/arXiv.2106.04972.
- M. R. Chernick, Bootstrap methods: A guide for practitioners and researchers, John Wiley & Sons, 2011 Search PubMed.
- B. Efron and R. J. Tibshirani, An introduction to the bootstrap, CRC press, 1994 Search PubMed.
- T. G. Dietterich, in Multiple Classifier Systems. MCS 2000. Lecture Notes in Computer Science, 2000, vol. 1857, pp. 1–15 Search PubMed.
- E. Prince, J. Appl. Phys., 2009, 32, S68–S69 CrossRef.
- K. M. Ø. Jensen, H. L. Andersen, C. Tyrsted, E. D. Bøjesen, A.-C. Dippel, N. Lock, S. J. L. Billinge, B. B. Iversen and M. Christensen, ACS Nano, 2014, 8, 10704–10714 CrossRef CAS PubMed.
- L. Yang, E. A. Culbertson, N. K. Thomas, H. T. Vuong, E. T. Kjær, K. Jensen, M. G. Tucker and S. J. Billinge, Acta Crystallogr., Sect. A: Found. Adv., 2021, 77, 2–6 CrossRef CAS PubMed.
- R. M. Rojas, P. Herrero, P. J. G. a. Chain and J. Rodriguez-Carvajal, J. Solid State Chem., 1994, 112, 322–328 CrossRef CAS.
- L. Kaplunnik, E. Pobedimskaya and N. Belov, Sov. Phys. Crystallogr., 1977, 22, 99–100 Search PubMed.
- M. N. Iliev, A. P. Litvinchuk, V. G. Hadjiev, M. M. Gospodinov, V. Skumryev and E. Ressouche, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 024302 CrossRef.
- C. Zheng, K. Mathew, C. Chen, Y. Chen, H. Tang, A. Dozier, J. J. Kas, F. D. Vila, J. J. Rehr and L. F. Piper, npj Comput. Mater., 2018, 4, 12 CrossRef.
- K. T. Butler, M. D. Le, J. Thiyagalingam and T. G. Perring, J. Phys.: Condens. Matter, 2021, 33, 194006 CrossRef CAS PubMed.
- W. B. Park, J. Chung, J. Jung, K. Sohn, S. P. Singh, M. Pyo, N. Shin and K.-S. Sohn, IUCrJ, 2017, 4, 486–494 CrossRef CAS PubMed.
- A. Ziletti, D. Kumar, M. Scheffler and L. M. Ghiringhelli, Nat. Commun., 2018, 9, 2775 CrossRef PubMed.
- Z. Chen, N. Andrejevic, N. C. Drucker, T. Nguyen, R. P. Xian, T. Smidt, Y. Wang, R. Ernstorfer, D. A. Tennant and M. Chan, Chem. Phys. Rev., 2021, 2, 031301 CrossRef.
- Y. Suzuki, H. Hino, T. Hawai, K. Saito, M. Kotsugi and K. Ono, Sci. Rep., 2020, 10, 1–11 CrossRef PubMed.
- J. Kirkpatrick, B. McMorrow, D. H. Turban, A. L. Gaunt, J. S. Spencer, A. G. Matthews, A. Obika, L. Thiry, M. Fortunato and D. Pfau, Science, 2021, 374, 1385–1389 CrossRef CAS PubMed.
- H. Tao, T. Wu, M. Aldeghi, T. C. Wu, A. Aspuru-Guzik and E. Kumacheva, Nat. Rev. Mater., 2021, 6, 701–716 CrossRef.
- C.-H. Liu, C. J. Wright, R. Gu, S. Bandi, A. Wustrow, P. K. Todd, D. O'Nolan, M. L. Beauvais, J. R. Neilson, P. J. Chupas, K. W. Chapman and S. J. L. Billinge, J. Appl. Crystallogr., 2021, 54, 768–775 CrossRef CAS.
- V. Stanev, V. V. Vesselinov, A. G. Kusne, G. Antoszewski, I. Takeuchi and B. S. Alexandrov, npj Comput. Mater., 2018, 4, 43 CrossRef.
- Z. Thatcher, C.-H. Liu, L. Yang, B. C. McBride, G. Thinh Tran, A. Wustrow, M. A. Karlsen, J. R. Neilson, D. B. Ravnsbæk and S. J. Billinge, Acta Crystallogr., Sect. A: Found. Adv., 2022, 78, 242–248 CrossRef CAS PubMed.
- S. Tetef, N. Govind and G. T. Seidler, Phys. Chem. Chem. Phys., 2021, 23, 23586–23601 RSC.
- K. W. Chapman, S. H. Lapidus and P. J. Chupas, J. Appl. Crystallogr., 2015, 48, 1619–1626 CrossRef CAS.
- S. Wold, K. Esbensen and P. Geladi, Chemom. Intell. Lab. Syst., 1987, 2, 37–52 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dd00001c |
| This journal is © The Royal Society of Chemistry 2024 |