
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic combination of active Pt species and light-driven photothermal catalysis for highly efficient toluene oxidation†
Meng
Zhang‡
a,
Ying
Zhang‡
ac,
Qianglong
Xu
ac,
Xiaolan
Li
a,
Jing
Chen
 *bc and
Hongpeng
Jia
*ac
*bc and
Hongpeng
Jia
*ac
aXiamen Key Laboratory of Materials for Gaseous Pollutant Control, Institute of Urban Environment, Chinese Academy of Sciences, Xiamen 361021, China. E-mail: hpjia@iue.ac.cn
bState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 19th August 2024
Abstract
Energy shortage and environmental pollution problems force us to find low-energy consumption methods to deal with volatile organic compounds (VOCs). In this work, Pt-P25 was synthesized through a simple wet impregnation method and calcination method. The study showed that different calcination temperatures and calcination atmospheres affected the activation state of Pt species, showing significant differences in the photothermal catalytic toluene oxidation reaction efficiency. The Pt-P25-800N sample activated in a nitrogen atmosphere at 800 °C exhibits better activity compared to other samples, achieving a toluene conversion rate of 95% and mineralization rate of 65% under a light intensity of 400 mW cm−2. Characterization results demonstrate that low-valent Pt species are positively correlated with toluene oxidation activity and play a major role in the reaction. The ultraviolet (UV), visible (vis) and infrared (IR) components in the spectrum all contribute to the toluene oxidation process. The catalytic bed is heated to the required temperature mainly through thermal effects, thus overcoming the reaction energy barrier. The traditional photocatalytic process over TiO2 also plays an auxiliary enhancement role. Due to the efficient conversion capability of the active sites, Pt-P25-800N achieves long-term stability of at least 50 hours under low light intensity input and water vapor conditions, accompanied by minimal accumulation of intermediate products. The above results reveal that the comprehensive effect between active Pt species and photothermal catalysis jointly achieves efficient degradation of VOCs and alleviates the energy and environmental crisis.
1. Introduction
Volatile organic compounds (VOCs) represent prevalent organic solvents utilized across various industries, and their volatilization poses significant risks to human health and contributes to environmental pollution.1,2 Catalytic oxidation technology emerges as a prominent, energy-efficient method for VOC removal, wherein the introduction of catalysts notably reduces the requisite temperature for the oxidation reaction.3,4 Extensive research endeavors have been directed towards catalyst design, yet the high-temperature conditions indispensable for catalytic oxidation persist as a limiting factor. The prevailing energy infrastructure predominantly relies on fossil fuel combustion owing to its accessibility and stability. However, the concurrent energy and environmental challenges necessitate the exploration of novel technological paradigms to surmount existing constraints. Photocatalytic oxidation works at ambient temperature, relying on semiconductor catalysts, such TiO2,5,6 ZnO (ref. 7 and 8) and g-C3N4,9 but usually faces low quantum efficiency and facile recombination of charge carriers. Photothermal catalysis technology harnesses full spectrum of solar irradiance to drive catalytic reactions, offering not only thermally stable reaction conditions but also leveraging high-energy photons induced by a photocatalytic process to augment reaction kinetics.10,11 Central to its efficacy is the formulation of robust photothermal catalytic systems alongside a comprehensive elucidation of the underlying catalytic reaction mechanisms. Compared to other technologies such as plasma12,13 and microwave14 for process intensification, photothermal catalysis technology only exploit the solar spectrum, achieving the exploration of sustainable energy.TiO2 is a commonly used semiconductor photocatalytic material due to its appropriate bandgap and electric potential location of valence and conduction bands.15,16 However, its bandgap width is large and charge carriers are prone to recombination. And in the catalytic oxidation reaction of VOCs, TiO2 is also a carrier with stable thermal and chemical properties, but it faces the problem of easy carbon deposition and deactivation.17,18 Therefore, it is a reasonable idea to apply TiO2 in photothermal catalytic reactions through rational regulation. However, the large band gap of TiO2 inhibits its spectral absorption efficiency. Theoretically, loading precious metals, such as Au, Ag, Pt and Pd, can not only enhance spectral absorption, but also improve VOC oxidation activity, attributed to their plasmonic resonance effect. For example, the utilization of TiO2 supports with distinct crystalline phases for Pt loading leads to varying degrees of interfacial interaction, influencing the Pt species state and redox properties and ultimately impacting the catalytic oxidation efficacy of chlorine-containing VOCs.19 The reconstruction phenomena of Pt nanoparticles on anatase and rutile surfaces contribute favorably to the hydrogenation and oxidation functionalities of the respective catalysts.20 These results demonstrate the feasibility of Pt supported on a TiO2 support for efficient catalytic degradation of toluene. However, the activation parameters governing noble metals remain insufficiently examined, and their utilization in photothermal catalysis has not undergone systematic investigation.
In this work, the influence of different calcination temperatures and atmospheres have been studied on the state of the Pt nanoparticles on the P25 support. Pt-P25 catalysts are used in photothermal catalytic toluene oxidation reactions. The photocatalytic reaction process over P25 and the thermal catalytic reaction process on the Pt particles are further explored. High-resolution transmission electron microscopy (HR-TEM), X-ray photoelectron spectroscopy (XPS), and O2-TPD are applied to investigate the state of the Pt species. Following toluene oxidation, Pt nanoparticles exhibit a state of carbon accumulation. This prompts further investigation into the process and mechanism underlying the catalytic toluene oxidation reaction mediated by Pt-P25.
2. Experimental
Reagents
Titanium dioxide-P25 with a particle size of 25 nm was purchased from Macklin Inc. Chloroplatinic acid hexahydrate (H2PtCl6·6H2O) was bought from Shaanxi Kaida Chemical Co., Ltd and prepared into a solution for further use. Rutile TiO2 with a particle size of 40 nm was purchased from Aladdin Company. γ-Al2O3 and CeO2 were obtained from Sinopharm Chemical Reagent Co., Ltd.Catalysts synthetic procedures
All Pt-P25 samples were synthesized by a wet impregnation method. Usually, 1 g H2PtCl6 was dissolved in ultrapure water and diluted to 100 mL in a volumetric flask to form a 3.8 gPt L−1 H2PtCl6 solution, as H2PtCl6 is easily hydrolyzed, and not convenient for weighing each time. Firstly, 3.98 mL of H2PtCl6 solution was added into 50 mL deionized water, and then 1.5 g P25 was dispersed in the solution, in which the theoretical Pt weight content is approximately 1%. The mixture was stirred at room temperature and 300 r min−1 for 1 hour and Pt4+ ions were absorbed onto the P25 support. Subsequently, the slurry was stirred until dry and moved into an oven at 80 °C. The solids were collected and ground in a mortar. Then the samples were calcined under different temperatures and reaction atmospheres. According to the activation conditions, the catalysts are named Pt-P25-800N, Pt-P25-800A, Pt-P25-400N, Pt-P25-400A, in which 800 and 400 represent the calcination temperature, and N and A indicate nitrogen and air atmospheres. To clarify the role of Pt nanoparticles, P25, Pt–CeO2 and Pt–Al2O3 were treated at 800 °C under a nitrogen atmosphere for comparison, named as P25-800N, Pt–CeO2-800N and Pt–Al2O3-800N.Catalyst characterization
The catalysts were characterized by powder X-ray diffraction (XRD), N2 adsorption–desorption measurements, X-ray fluorescence (XRF), high-resolution transmission electron microscopy (HR-TEM), X-ray photoelectron spectroscopy (XPS), chemisorption, thermal gravimetric analysis (TGA), elemental analysis and in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). The specific experimental details are presented in the ESI.†Photothermal catalytic toluene oxidation test
The reaction device for the photothermal catalytic toluene oxidation activity test was the same as the previous device.21 The reaction is carried out in a homemade cylindrical stainless-steel reactor with a piece of quartz glass on the top for sealing, as exhibited in Fig. S1.† Light enters the reactor through the quartz glass to the catalytic bed. In order to fully expose to light, the catalyst bed for the photothermal catalytic reaction is designed to be different from the thermal catalytic reaction bed. 20 mg of catalyst powder was added to the solvent and dispersed by sonication. The catalyst is then evenly coated on the glass fiber membrane through a filtration device, and the thickness of the catalyst is less than 1 mm. The glass fiber membrane coated with the catalyst was then placed in an oven at 80 °C to dry before use.Toluene/air mixed gas in the buffer tank has been prepared by bubbling method, with a flow rate of 20 mL min−1 and a concentration of 400 ppm. The reaction gas enters from the bottom of the catalytic bed, flows out from above after passing through the catalytic bed, and finally goes through a gas chromatograph equipped with two flame ionization detectors (FIDs) to determine the concentration. A simulated sunlight xenon lamp (PLS-SXE300+, Beijing Perfectlight Technology Co., Ltd.) is used to power the catalytic reaction, and the light source is the only energy source for the photothermal catalytic reaction. The light intensity is controlled by changing the photocurrent intensity or the distance between the light source and the catalysts. By adding optical filters, the light irradiation of different wavelength bands can be controlled to provide energy for the catalytic reaction. The thermal catalytic reaction was carried out in an electrically heated furnace. The temperature of the catalytic bed was tested with a K-type thermocouple and recorded with a temperature recorder. The catalyst membrane was placed in the middle of the reactor. After turning on the lights, the catalyst bed quickly heats up and the layer temperature remains stable, and then toluene/air mixture reactants are cut into the reactor for the reaction. Usually gas–solid phase heterogeneous reactions go through the processes of diffusion, adsorption activation and reaction, so performing the adsorption process first is beneficial to the progress of the reaction. However, due to the rapid temperature rise of the catalyst bed under light conditions, a large number of adsorbed reaction gas molecules will be absorbed, which affects the judgment of catalytic activity. Therefore, in the photothermal catalytic reaction, light is first introduced to stabilize the temperature of the catalytic bed before the reactants cut into the reactor. The toluene conversion rate and CO2 mineralization rate are calculated based on the concentrations of toluene and CO2 before and after the reaction. The calculation formula is as follows:


3. Results and discussion
Characterization of the catalyst structure
As depicted in Fig. 1a, the predominant crystal phase structure of TiO2 within P25 is identified as anatase, and the crystal phase configuration remains unaltered subsequent to calcination at 400 °C in nitrogen and air atmosphere.19 Upon elevation of the calcination temperature to 800 °C, TiO2 undergoes a phase transition to rutile, concomitantly with notable agglomeration of Pt, as evidenced by pronounced peaks at 39.8° in X-ray diffraction (XRD) analysis.22,23 Consequently, temperature emerges as the primary determinant influencing the crystalline configuration of the catalyst. Given that gas–solid phase heterogeneous catalytic reactions entail diffusion and adsorption–desorption phenomena within catalyst pores, N2 adsorption–desorption isotherms serve as a diagnostic tool for characterizing the pore structure and specific surface area of the catalysts. As illustrated in Fig. 1b, N2 adsorption–desorption isotherms for Pt-P25, Pt-P25-400A, and Pt-P25-400N exhibit typical IV isotherms with H3 hysteresis loops, suggesting the existence of a mesoporous structure.24 The hysteresis loop of the sample subjected to high-temperature calcination (800 °C) nearly vanishes, indicative of pore structure degradation owing to the elevated calcination temperature. Specific surface area measurements, as presented in Table 1, indicate a slight increase in the specific surface area for Pt-P25-400A (92.63 m2 g−1) and Pt-P25-400N (88.29 m2 g−1) compared to P25 (82.68 m2 g−1). Notably, calcination at 400 °C induces a marginal augmentation in the specific surface area, whereas high-temperature calcination leads to a substantial decline for Pt-P25-800A (9.77 m2 g−1) and Pt-P25-800N (11.16 m2 g−1) due to pore structure collapse induced by thermal treatment. The accurate Pt weight content over the calcined Pt-P25 samples were tested by XRF, and the results are recorded in Table 1, which are around 1.33%, a little higher than the theoretical content (1%). This might be due to the oxygen loss during migration of the metal species.25,26 | ||
| Fig. 1 (a) XRD patterns and (b) N2 adsorption–desorption isotherms of Pt-P25, Pt-P25-400A, Pt-P25-400N, Pt-P25-800A and Pt-P25-800N. | ||
| Catalysts | BET (m2 g−1) | Pore volume (cm3 g−1) | Pore diameter (nm) | Pt0/Pt2+ | Oads/Olatt | Pt content (%) |
|---|---|---|---|---|---|---|
| Pt-P25-800N | 11.16 | 0.02 | 2.97 | 1.48 | 0.2 | 1.33 |
| Pt-P25-800A | 9.77 | 0.02 | 2.97 | 0.89 | 0.25 | 1.29 |
| Pt-P25-400N | 88.3 | 0.28 | 15.5 | 0.24 | 0.19 | 1.33 |
| Pt-P25-400A | 92.6 | 0.3 | 15.6 | 0 | 0.18 | 1.37 |
| P25 | 82.7 | 0.37 | 3.71 | — | 0.2 | — |
As evidenced in Fig. 2, Pt particle agglomeration is absent in samples subjected to calcination at 400 °C in air, contrasting with the conspicuous presence of significantly agglomerated large particles in samples calcined under alternative conditions. Notably, particles in samples subjected to calcination in a nitrogen (N2) atmosphere display uniform distribution on the substrate subsequent to particle reassembly, whereas Pt particles combusted in air at 800 °C exhibit notable aggregation, forming prominent large particles subsequent to migration. Besides, CO-diffuse reflectance infrared Fourier transform spectroscopy (CO-DRIFT) results (Fig. S2†) demonstrate that the dispersion degree of Pt subjected to low-temperature calcination (400 °C) surpasses that of Pt subjected to high-temperature calcination (800 °C).
 | ||
| Fig. 2 HRTEM images and mapping of the Pt element over (a) Pt-P25-800N, (b) Pt-P25-800A, (c) Pt-P25-400N and (d) Pt-P25-400A. | ||
While the crystal phase structure and specific surface area of TiO2 are notably influenced by calcination conditions, the binding energy position of the Ti element remains unchanged (Fig. 3a). In the Ti 2p spectra, Ti ions consistently manifest in the oxidation state of +4 across all examined samples. Conversely, the binding energy of the Pt element undergoes considerable variation. In Fig. 3b, Pt 4f mainly exhibited Pt0 and Pt2+, and the ratios of Pt0/Pt2+ of the catalysts are recorded in Table 1. With increasing calcination temperature, there is a notable augmentation in the abundance of low-valent Pt species. This phenomenon is attributed to the substantial disparity in electronegativity between Pt (2.28) and Ti (1.54).27 Furthermore, as reported in a previous study, Pt has demonstrated an inclination towards maintaining a reduced valency, which can be ascribed to the electronic stabilization arising from the robust interaction between Pt and P25.19Fig. 3c reveals the deconvolution of the O 2p spectrum into adsorbed oxygen and lattice oxygen species, localized at 531.5 and 529.9 eV, respectively.28,29 The ratios of adsorbed oxygen to lattice oxygen of the samples are detailed in Table 1. Notably, the proportion of adsorbed oxygen remains relatively consistent for samples calcined at 400 °C. However, upon elevation of the calcination temperature to 800 °C, there is a significant increase in the proportion of adsorbed oxygen. This phenomenon is attributed to alterations in the Pt species state, thereby enhancing the adsorption and activation of O2 molecules.30 The heightened presence of adsorbed oxygen species serves as indicative evidence of the catalyst's augmented capacity for reactant activation, thereby conferring advantageous implications for the catalytic oxidation reaction.31,32
 | ||
| Fig. 3 XPS spectra of (a) Ti, (b) Pt and (c) O, and (d) O2-TPD signals over the samples. | ||
The O2-TPD findings depicted in Fig. 3d reveal a discernible pattern in the desorption temperature and sequence of oxygen species, delineated as follows: physically adsorbed oxygen (<200 °C), chemically adsorbed oxygen and superficial lattice oxygen (200–400 °C), and bulk lattice oxygen (400–800 °C).33,34 The TiO2 spectrum exhibits two desorption peaks at 316 and 442 °C, both attributed to surface lattice oxygen. Upon introduction and activation of Pt species through calcination at 400 °C, there is a notable elevation in the desorption temperature of lattice oxygen. This phenomenon is ascribed to the higher electronegativity of Pt, enhancing oxygen affinity and promoting desorption.35,36 However, with an increase in activation temperature to 800 °C, the treatment process results in the removal of adsorbed and lattice oxygen from the catalyst surface, consequently yielding minimal desorption signals for any oxygen species. These findings suggest that oxygen species in Pt-P25-800A and Pt-P25-800N primarily originate from the adsorption activation of O2 molecules at active sites, rather than participation of lattice oxygen. Moreover, oxygen species swiftly undergo conversion post-adsorption and dissociation on Pt without a prolonged residence time. Notably, the desorption temperature of oxygen species significantly surpasses the actual reaction temperature, indicating solely the reactivity of oxygen species on the catalyst. Thus, the abundance of oxygen species on Pt-P25-800A and Pt-P25-800N does not constitute a pivotal determinant of catalytic reaction activity.
Photothermal catalytic toluene oxidation performance evaluation
Photothermal catalytic oxidation of toluene was investigated over the samples under irradiation intensities of 282 and 400 mW cm−2, respectively. Compared to traditional thermal catalysis supplied by electric heating, photothermal catalysis offers the advantage of rapidly reaching high temperatures.37 Therefore, before introducing the reactants, the catalyst layers were pre-exposed to light irradiation until they stabilize at the desired temperature. During this process, the catalyst structures remain unchanged, as presented in Fig. S3.† As depicted in Fig. 4, rapid absorption and conversion of toluene were observed over P25 upon light exposure. However, a decrease in toluene conversion over time was noted, attributed to the accumulation of carbonaceous intermediates. After 120 minutes, toluene conversion declined from 92% to approximately 21% (Fig. 4a), underscoring the inadequacy of reactive active sites on pure TiO2. Even upon Pt loading onto P25, Pt-P25-400A exhibited negligible enhancement in activity, suggesting incomplete activation of Pt species under the conditions of 400 °C and air atmosphere. In contrast, Pt-P25-400N achieved toluene conversion of 96% and CO2 mineralization of 57% under light irradiation at an intensity of 400 mW cm−2 (Fig. 4a and b). Moreover, an increase in the abundance of low-valent Pt species upon calcination at 800 °C led to higher activity over Pt-P25-800A and Pt-P25-800N. Based on the aforementioned structural characterization analysis, it can be inferred that low-valent Pt species serve as active sites for the toluene oxidation reaction. Under an irradiation intensity of 400 mW cm−2, Pt-P25-800A and Pt-P25-800N exhibited toluene conversion of 95%, and CO2 mineralization of 64% and 65%, respectively. However, at a reduced light intensity of 282 mW cm−2, noticeable differences in catalytic activity were observed among the samples, following the trend: Pt-P25-800N > Pt-P25-800A > Pt-P25-400N > Pt-P25-400A ≈ P25-400N (Fig. 4c and d). In addition, further reduction in light intensity resulted in decreased catalytic activity over Pt-P25-800N with toluene conversion of 23% and CO2 yield of 11% under 236 mW cm−2, albeit remaining stable within a two-hour timeframe (Fig. S4†). Conversely, the catalytic activity of P25 remained largely consistent under varying light intensities, indicating predominant photocatalytic activity on pure TiO2. However, incomplete conversion of toluene and limited deep mineralization were attributed to rapid charge carrier recombination. According to XRD results (Fig. 1a), the TiO2 support within Pt-P25 samples subjected to calcination at 800 °C exhibits the rutile phase. Consequently, the catalytic performance of the Pt-loaded catalyst on the rutile phase was systematically investigated, with the results presented in Fig. S5.† Comparatively, Pt-rutile TiO2 samples subjected to calcination under air or nitrogen atmospheres exhibited comparatively lower catalytic activity, underscoring the significance of utilizing P25 as a carrier material. It is conceivable that the phase transition phenomenon of P25 during high-temperature calcination engenders a unique interaction with Pt species, thereby endowing Pt-P25-800A and Pt-P25-800N configurations with enhanced catalytic efficacy. Besides, the low performance of P25-800N evidence that Pt species are the main active sites. In addition, Pt was loaded onto reducible oxide CeO2 and non-reducible oxide Al2O3, respectively. The active state of Pt species in Pt-Al2O3-800N might be similar to Pt-P25-800N, thus exhibiting high performance. However, Pt–CeO2-800N didn't show much conversion effect for toluene, suggesting that Pt species was not fully activated over CeO2 or ruined during CeO2 structural deformation. Light irradiation only affects the adsorption and activation of reactants during the reaction, but has no effect on the catalyst structure itself, as revealed in Fig. S6.† | ||
| Fig. 4 Toluene conversion and CO2 mineralization of the catalysts under light irradiation with light intensity of (a and b) 400 and (c and d) 282 mW cm−2, and (e) catalytic stability of Pt-P25-800N. | ||
Catalyst assessment typically encompasses not only activity and selectivity but also stability under operational conditions. Consequently, the catalyst's resilience against carbon deposition and water vapor was examined under low-temperature and water vapor environments, respectively. As illustrated in Fig. 4e, the favored catalyst, Pt-P25-800N, demonstrated sustained stability for a minimum of 50 hours, exhibiting resistance to deactivation by intermediate products and water vapor generated during low-temperature reactions. However, under water vapor conditions, the catalyst's mineralization rate exhibited a decrease, presumably attributable to water vapor occupying active sites and impeding the conversion of intermediate products.37,38 Upon cessation of the water vapor supply, the catalytic activity reverted to its original level, indicative of reversible or non-chemically irreversible catalyst behavior.
Photothermal catalytic reaction process
Given that higher light intensity provides energy, the catalyst can utilize either thermal energy within the spectrum or high-energy photons. Consequently, it is imperative to delve into the source of catalytic activity. The light absorption capability of the material governs the efficiency of light energy utilization by the catalyst. Hence, the light absorption performance of the catalyst was initially scrutinized through UV-vis-IR diffuse reflectance spectroscopy, with the results depicted in Fig. 5a. P25 exhibits a robust photoresponse to wavelengths below 400 nm due to the intrinsic electron transitions in the bandgap (O2p → Ti3d), yet demonstrates minimal absorption in the visible and infrared spectra.39,40 Following the introduction of Pt, the catalyst's light absorption in the visible and infrared regions undergoes significant enhancement, attributed to plasmonic effect.41 This augmentation in light intensity absorption demonstrates a positive correlation with the catalyst bed's reaction temperature under identical light intensities (Fig. 5b), implying that the introduction of Pt and its activation method impact its photoresponse. Notably, the augmentation in low-valent Pt species in samples treated at 800 °C leads to reduced light absorption performance in the UV region. Considering that the infrared region primarily induces local thermal effects on the catalytic material, a catalyst bed exhibiting strong absorption in the infrared region may attain higher temperatures. Under light intensities of 280 and 400 mW cm−2, the temperature of the catalytic layer over the samples is illustrated in Fig. 5b, with a sequence approximately as follows: Pt-P25-800N ≈ Pt-P25-800A > Pt-P25-400N > Pt-P25-400A > P25-400N. After the absorption of spectral energy by the catalysts, non-radiative heat generated by charge carrier recombination and molecular vibration induced by infrared thermal effect collectively elevate the temperature of the catalytic bed.42,43 Upon exposure to light irradiation, the catalytic bed layers demonstrate rapid heating, followed by prompt cooling upon cessation of light irradiation, as evidenced in Fig. S7.† This underscores a significant advantage of photothermal catalysis over thermal catalysis, namely localized directional heating and rapid thermal cycling. Modulating the light intensity alters the input energy, consequently affecting the catalytic bed temperature and catalytic activities.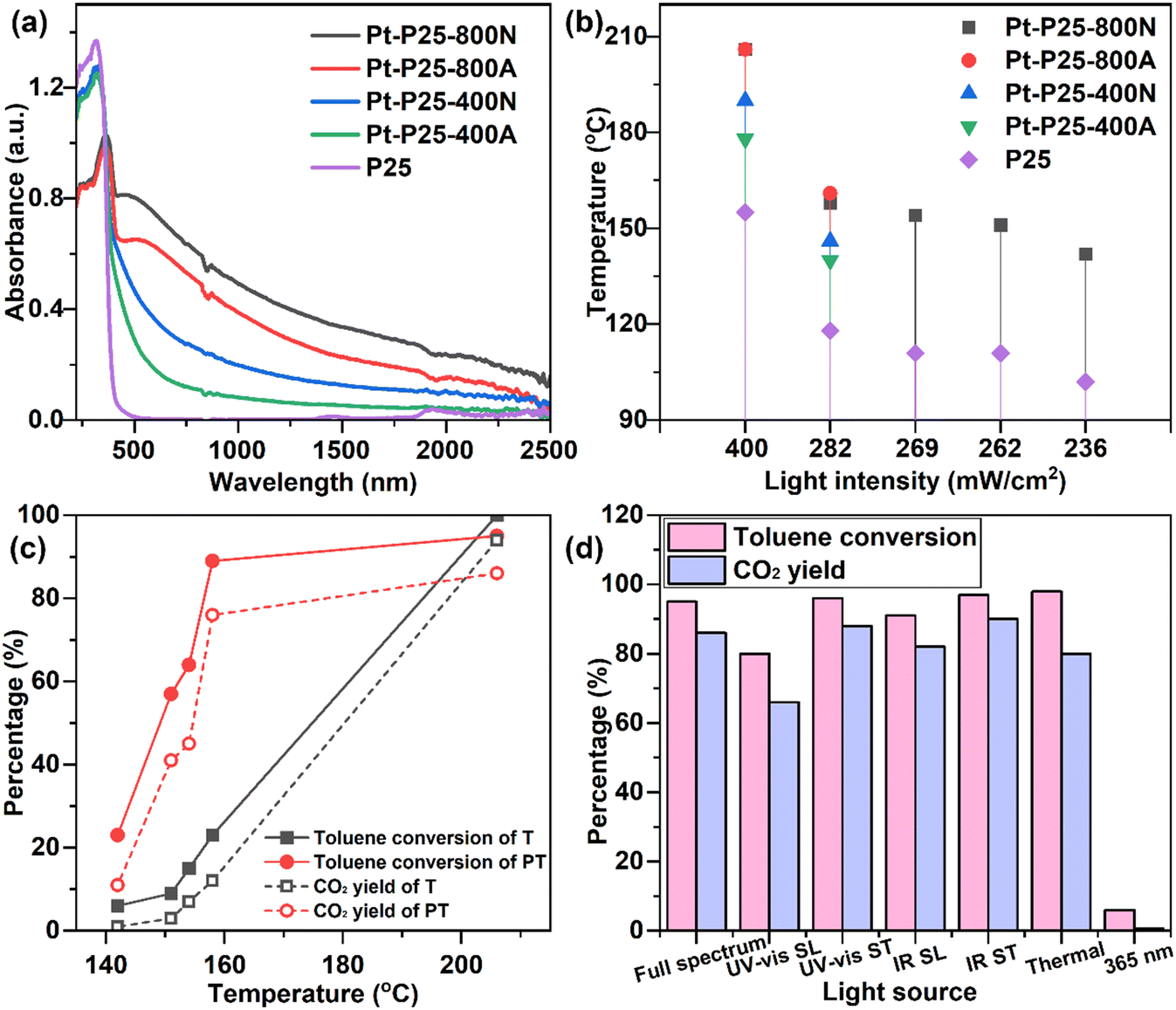 | ||
| Fig. 5 (a) UV-vis-IR DRS spectra and (b) catalyst bed layer temperature over the samples under various light intensities, (c) catalytic performance comparison of thermal catalysis (T) and photothermal catalysis (PT) over Pt-P25-800N, and (d) catalytic performance comparison under light irradiation of full spectrum, UV-vis with the same light intensity (UV-vis SL), UV-vis with the same bed layer temperature (UV-vis ST), IR with the same light intensity (IR SL), IR with the same bed layer temperature (IR ST), 365 nm, and thermal conditions without irradiation. | ||
Exclusively light energy served as the energy source to drive the catalytic reaction in this investigation. The UV component, characterized by shorter wavelengths within the full spectrum, possesses higher energy, thereby exciting the TiO2 semiconductor to generate electrons and holes. These charge carriers subsequently engage with oxygen and water molecules, leading to the production of potent oxidizing oxygen species, which facilitate the oxidation of the toluene reactant. Concurrently, non-radiative heat generated by charge carrier recombination, coupled with the molecular thermal vibration effects induced by the infrared spectrum, collectively contribute to heating the catalyst bed. Upon comparing the catalytic performances of photothermal catalysis and thermal catalytic degradation of toluene at an identical temperature, it was observed that photothermal catalytic activity over Pt-P25-800N surpassed the thermal catalytic activity (Fig. 5c), indicating the involvement of both thermal and light energies within the spectrum. The electric heating furnace, driven by electrical power, offers advantages in terms of uniformity and stability, thus thermal catalysis demonstrates a toluene conversion efficiency approaching 100% under elevated temperature conditions. This catalytic efficacy can similarly be attained under photothermal catalytic conditions.
Manipulating different wavelength bands of light to supply energy for the catalytic reaction revealed that UV-enhanced toluene degradation activity exhibited the highest efficacy at equivalent temperatures, underscoring the auxiliary role of traditional photocatalysis in enhancing the catalytic oxidation process. The catalytic performance under varying light sources is depicted in Fig. 5d, with the respective light intensities and temperatures delineated in Table S1.† Under similar UV illumination conditions, the absence of the thermal effect of infrared light precluded the catalytic bed from reaching elevated temperatures, resulting in diminished catalytic activity, thereby emphasizing the indispensability of high temperatures in the toluene oxidation reaction. Under infrared spectrum conditions with identical light intensity (400 mW cm−2), the temperature of the catalyst bed layer is merely 176 °C, which is lower compared to the temperature attained under full spectrum conditions. Consequently, the catalytic activity is marginally reduced compared to that observed under the full spectrum. Upon achieving the same catalyst bed temperature, the infrared spectrum intensity surpasses that of the full spectrum. The extensive heating and heat retention facilitated by the infrared lamp result in heightened catalytic activity of the catalyst relative to the conditions under the full spectrum. However, under the 365 nm spectral conditions, Pt-P25-800N exhibited solely a toluene conversion rate of 6% and a CO2 mineralization rate of 0.7%, underscoring the necessity of elevated temperatures for facilitating the toluene oxidation reaction on the catalyst. Furthermore, this underscores the presence of a photocatalytic process within the photothermal catalytic reaction, albeit with a minimal contributory effect.
Production of carbon deposits and toluene oxidation reaction pathways
To examine carbon deposition on the catalysts, all samples underwent exposure to a light intensity of 280 mW cm−2 for a duration of 10 hours. As illustrated in Fig. S8,† visible darkening of the catalyst layers of Pt-P25, Pt-P25-400A, and Pt-P25-400N was observed, indicative of carbon deposits. Conversely, the color change of Pt-P25-800A and Pt-P25-800N was less discernible, potentially attributable to the inherent darker color of the catalyst bed itself. Additionally, the substantial quantity of active sites on Pt-P25-800A and Pt-P25-800N may facilitate rapid conversion of reactants, thereby reducing the deposition of intermediate products. Notably, such carbon deposits can be alleviated under light conditions. As demonstrated in Fig. S9a,† upon cessation of the toluene reaction gas, the catalyst undergoes gradual color restoration, accompanied by a decline in bed temperature, affirming the low reactivity of the TiO2 reaction sites and their resistance to chemical poisoning. The P25 sample exhibited no discernible toluene conversion, amounting to less than 2%, until the temperature reached 155 °C. At an irradiance level of 236 °C, the initial temperature of the catalyst bed remained at 66 °C prior to the introduction of toluene. Subsequent to the introduction of toluene, the temperature of the catalyst bed increased to 102 °C. Following a two-hour duration, the sample displayed a toluene conversion rate of 18% and a CO2 mineralization rate of 3% (Fig. S9b†). These findings suggest that the photocatalytic mechanism predominates in the P25 sample, and further imply that temperature augmentation amplifies the efficacy of the photocatalytic process. A similar scenario is also observed in the case of the light-colored CeO2 sample (Fig. S10†). Thermogravimetric and elemental analyses were conducted to assess carbon deposition content in post-reaction samples. As depicted in Fig. 6, the weight loss below 400 °C primarily stemmed from carbon deposits, particularly conspicuous on Pt-P25, Pt-P25-400A, and Pt-P25-400N. Among these, Pt-P25-400A exhibited the highest carbon deposition content, suggesting that Pt modulates the reactant adsorption process. However, incomplete activation impedes further conversion of intermediate products, leading to their deposition on the catalyst. Elemental analysis results in Fig. 6f indicate elevated carbon content in Pt-P25, Pt-P25-400A, and Pt-P25-400N compared to Pt-P25-800A and Pt-P25-800N. Consequently, active sites on Pt-P25-800A and Pt-P25-800N exhibit a higher frequency of intermediate product conversion, effectively mitigating intermediate product deposition. | ||
| Fig. 6 TG analysis of (a) P25, (b) Pt-P25-400A, (c) Pt-P25-400N, (d) Pt-P25-800A, and (e) Pt-P25-800N, and (f) elemental analysis. | ||
Moreover, the reaction mechanism underlying toluene oxidation on the selected catalyst, Pt-P25-800N, was investigated via in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), as shown in Fig. 7a. Initially, toluene was introduced to be adsorbed on Pt-P25-800N for a duration of 30 minutes, primarily yielding an adsorption peak corresponding to toluene. Vibrational peaks observed at 3075, 3030, and 2877 cm−1 represent methyl group vibrations linked to the benzene ring of toluene, while the vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond in the benzene ring typically manifests at 1495 cm−1.44,45 Subsequently, upon elevating the temperature to 158 °C, a peak at 1596 cm−1 indicative of benzaldehyde vibration and a vibration at 1413 cm−1 corresponding to benzoate were observed. Further temperature escalation to 206 and 230 °C resulted in enhanced peaks corresponding to acid anhydride at 1852 and 1786 cm−1, respectively, corroborating deep conversion of intermediate products.46 Given that the reaction was conducted in a continuous flow of toluene, the adsorption peak of toluene persisted on the catalyst surface throughout the reaction. These findings delineate the conversion pathway of toluene on Pt-P25-800N as follows: toluene, benzaldehyde, benzoic acid, acid anhydride, CO2, and water (Fig. 7b). Based on the aforementioned analysis, it can be inferred that the predominant mechanism on Pt-P25-800N involves the activation of reactants facilitated by high temperatures, while the photocatalytic process on P25 contributes to the enhancement of the catalytic reaction kinetics. Notably, the thermal catalytic process is predominantly facilitated by the presence of low-valent Pt species, serving as the primary active sites.
C double bond in the benzene ring typically manifests at 1495 cm−1.44,45 Subsequently, upon elevating the temperature to 158 °C, a peak at 1596 cm−1 indicative of benzaldehyde vibration and a vibration at 1413 cm−1 corresponding to benzoate were observed. Further temperature escalation to 206 and 230 °C resulted in enhanced peaks corresponding to acid anhydride at 1852 and 1786 cm−1, respectively, corroborating deep conversion of intermediate products.46 Given that the reaction was conducted in a continuous flow of toluene, the adsorption peak of toluene persisted on the catalyst surface throughout the reaction. These findings delineate the conversion pathway of toluene on Pt-P25-800N as follows: toluene, benzaldehyde, benzoic acid, acid anhydride, CO2, and water (Fig. 7b). Based on the aforementioned analysis, it can be inferred that the predominant mechanism on Pt-P25-800N involves the activation of reactants facilitated by high temperatures, while the photocatalytic process on P25 contributes to the enhancement of the catalytic reaction kinetics. Notably, the thermal catalytic process is predominantly facilitated by the presence of low-valent Pt species, serving as the primary active sites.
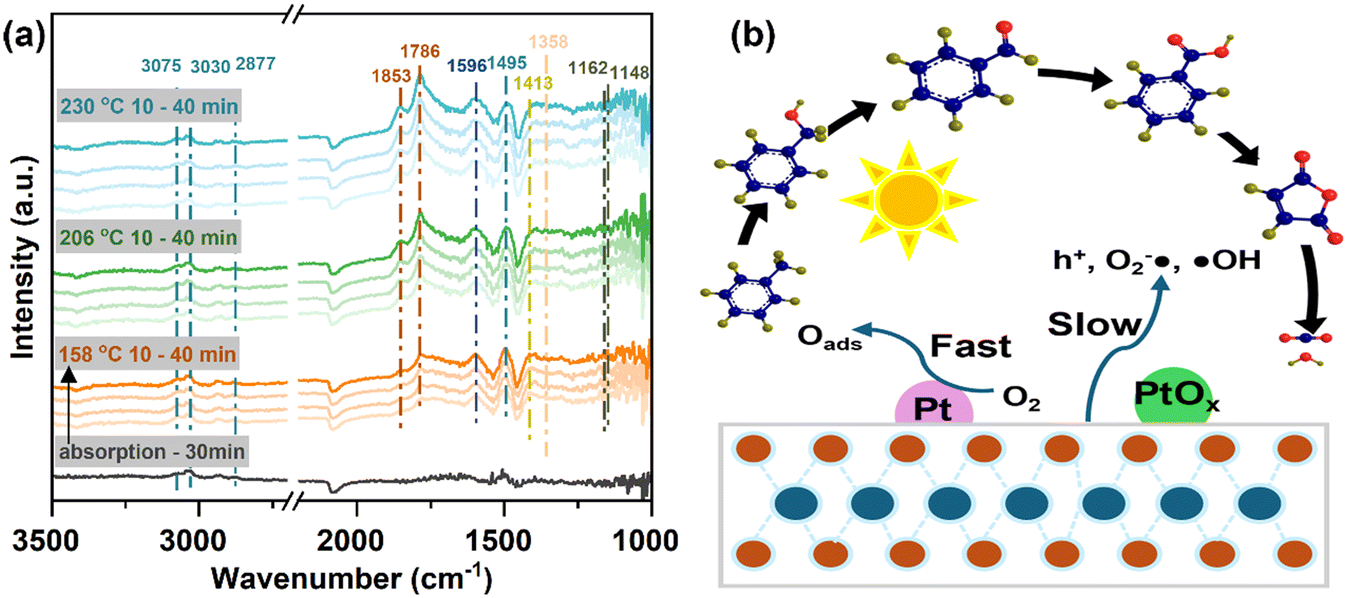 | ||
| Fig. 7 (a) In situ DRIFTS for the detection of reaction intermediates, and (b) toluene oxidation reaction pathways over Pt-P25-800N. | ||
4. Conclusions
In this study, the conventional wet impregnation technique was employed for the synthesis of the Pt-P25 catalyst, with a focus on assessing the influence of various calcination temperatures and atmospheres on the photothermal catalytic oxidation of toluene. Despite the adverse effects observed at an elevated calcination temperature of 800 °C, such as structural pore collapse, Pt particle aggregation, and diminished oxygen adsorption and activation capabilities, heightened catalytic activity persisted, underscoring the superior Pt species activation. Elevated calcination temperatures contribute to an augmentation of low-valent Pt species within Pt-P25, with their abundance positively correlating with catalytic efficacy, thereby highlighting the critical role of low-valent Pt species in the toluene oxidation process. Specifically, under light irradiation with an intensity of 400 mW cm−2, Pt-P25-800N demonstrated a remarkable 95% toluene conversion rate alongside 65% CO2 mineralization, exhibiting commendable stability and resistance to carbon deposition over a span of 50 hours. The catalytic system efficiently harnesses spectral energy to elevate the catalytic bed temperature to the requisite level, thereby furnishing the energy for the catalytic reaction, with photon-mediated mechanisms augmenting this process.Data availability
The authors confirm that the data supporting the findings of this study are available within the manuscript and its ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Natural Science Foundation of China (No. 22376193, 22176187); the STS Program Supporting Project of Fujian Province & CAS [2023T3070].References
- M. Simayi, Y. Shi, Z. Xi, J. Ren, G. Hini and S. Xie, Sci. Total Environ., 2022, 826, 153994 CrossRef CAS PubMed.
- W. Wu, T.-M. Fu, S. R. Arnold, D. V. Spracklen, A. Zhang, W. Tao, X. Wang, Y. Hou, J. Mo, J. Chen, Y. Li, X. Feng, H. Lin, Z. Huang, J. Zheng, H. Shen, L. Zhu, C. Wang, J. Ye and X. Yang, Environ. Sci. Technol., 2024, 58, 5430–5441 CrossRef CAS PubMed.
- X. Wu, R. Han, Q. Liu, Y. Su, S. Lu, L. Yang, C. Song, N. Ji, D. Ma and X. Lu, Catal. Sci. Technol., 2021, 11, 5374–5387 RSC.
- Y. Guo, M. Wen, G. Li and T. An, Appl. Catal., B, 2021, 281, 119447 CrossRef CAS.
- D. S. Selishchev, T. N. Filippov, M. N. Lyulyukin and D. V. Kozlov, Chem. Eng. J., 2019, 370, 1440–1449 CrossRef CAS.
- H. Mao, M. Xu, S. Li, Y. Ren, Y. Zhao, J. Yu, Q. Zhang, W. Zhao, G. Zhang, L. Yan, Z. Xu and Z. Bian, ACS ES&T Eng., 2023, 3, 1851–1863 Search PubMed.
- Anisotropic Au-ZnO photocatalyst for the visible-light expanded oxidation of n-hexane - ScienceDirect, https://www.sciencedirect.com/science/article/pii/S0920586120301851?via%3Dihub, (accessed August 7, 2024).
- Y. Zhang, G. Zhao, Z. Chen, H. Lian, L. Gan and M. Pan, J. Environ. Chem. Eng., 2022, 10, 108690 CrossRef CAS.
- J. Zhou, D. Li, W. Zhao, B. Jing, Z. Ao and T. An, ACS Appl. Mater. Interfaces, 2021, 13, 23843–23852 CrossRef CAS PubMed.
- Y. Yang, S. Zhao, F. Bi, J. Chen, Y. Li, L. Cui, J. Xu and X. Zhang, Cell Rep. Phys. Sci., 2022, 3, 101011 CrossRef CAS.
- Y. Feng, L. Dai, Z. Wang, Y. Peng, E. Duan, Y. Liu, L. Jing, X. Wang, A. Rastegarpanah, H. Dai and J. Deng, Environ. Sci. Technol., 2022, 56, 8722–8732 CrossRef CAS PubMed.
- A. Mohammad Gholipour, N. Rahemi, S. Allahyari and E. Ghareshabani, Top. Catal., 2017, 60, 934–943 CrossRef CAS.
- Z. Bo, M. Cao, H. Zhang, Y. Wang, J. Yan, K. Cen, K. Ostrikov and X. Tu, Chem. Eng. J., 2024, 481, 148399 CrossRef CAS.
- A. Li, X. Cong, H. Qin, W. Xu, X. Zhang, W. Wang and F. Guo, New J. Chem., 2023, 47, 3910–3920 RSC.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- X. Sun, Z. Feng, S. Wang, Q.-N. Wang, P. Zhang, R. Li and C. Li, ACS Catal., 2024, 14, 5356–5365 CrossRef CAS.
- Q. Li, C. Wu, K. Wang, X. Wang, X. Chen, W. Dai and X. Fu, Catal. Sci. Technol., 2022, 12, 237–249 RSC.
- F. He, U. Muliane, S. Weon and W. Choi, Appl. Catal., B, 2020, 275, 119145 CrossRef CAS.
- N. Zhang, X. Li, Y. Guo, Y. Guo, Q. Dai, L. Wang and W. Zhan, Environ. Sci. Technol., 2023, 57, 7086–7096 CrossRef CAS PubMed.
- X. Zhang, Z. Li, W. Pei, G. Li, W. Liu, P. Du, Z. Wang, Z. Qin, H. Qi, X. Liu, S. Zhou, J. Zhao, B. Yang and W. Shen, ACS Catal., 2022, 12, 3634–3643 CrossRef CAS.
- M. Zhang, H. Gao, J. Chen, E. A. Elimian and H. Jia, Appl. Catal., B, 2022, 307, 121208 CrossRef CAS.
- J. Guan, Y. Zan, R. Shao, J. Niu, M. Dou, B. Zhu, Z. Zhang and F. Wang, Small, 2020, 16, 2005048 CrossRef CAS PubMed.
- H. Matsui, A. Shoji, C. Chen, X. Zhao, T. Uruga and M. Tada, Catal. Sci. Technol., 2024, 14, 1501–1511 RSC.
- Y. Cao, G. Yuan, Y. Guo, X. Hu, G. Fang and S. Wang, Appl. Surf. Sci., 2022, 600, 154169 CrossRef CAS.
- H. Kang, L. Zhu, S. Li, S. Yu, Y. Niu, B. Zhang, W. Chu, X. Liu, S. Perathoner, G. Centi and Y. Liu, Nat. Catal., 2023, 6, 1062–1072 CrossRef CAS.
- H. Wang, R. Shimogawa, L. Zhang, L. Ma, S. N. Ehrlich, N. Marinkovic, Y. Li and A. I. Frenkel, Commun. Chem., 2023, 6, 1–7 CrossRef PubMed.
- H. Hojo, M. Gondo, S. Yoshizaki and H. Einaga, Nano Lett., 2022, 22, 145–150 CrossRef CAS PubMed.
- C. Feng, X. Liu, T. Zhu, Y. Hu and M. Tian, Appl. Catal., A, 2021, 622, 118218 CrossRef CAS.
- C. Wang, Y. Li, C. Zhang, X. Chen, C. Liu, W. Weng, W. Shan and H. He, Appl. Catal., B, 2021, 282, 119540 CrossRef CAS.
- W. Tan, S. Xie, D. Le, W. Diao, M. Wang, K.-B. Low, D. Austin, S. Hong, F. Gao, L. Dong, L. Ma, S. N. Ehrlich, T. S. Rahman and F. Liu, Nat. Commun., 2022, 13, 7070 CrossRef CAS PubMed.
- S. Wu, H. Liu, Z. Huang, H. Xu and W. Shen, Appl. Catal., B, 2022, 312, 121387 CrossRef CAS.
- G. Yu, Y. Wang, H. Cao, H. Zhao and Y. Xie, Environ. Sci. Technol., 2020, 54, 5931–5946 CrossRef CAS PubMed.
- Y. Ma, L. Wang, J. Ma, H. Wang, C. Zhang, H. Deng and H. He, ACS Catal., 2021, 11, 6614–6625 CrossRef CAS.
- S. Chen, H. Li, Y. Hao, R. Chen and T. Chen, Catal. Sci. Technol., 2020, 10, 1941–1951 RSC.
- X.-N. Li, Z. Yuan, J.-H. Meng, Z.-Y. Li and S.-G. He, J. Phys. Chem. C, 2015, 119, 15414–15420 CrossRef CAS.
- J. Zhang, X. Qin, X. Chu, M. Chen, X. Chen, J. Chen, H. He and C. Zhang, Environ. Sci. Technol., 2021, 55, 16687–16698 CrossRef CAS PubMed.
- M. Zhang, G. Li, Q. Li, J. Chen, E. A. Elimian, H. Jia and H. He, Environ. Sci. Technol., 2023, 57, 4286–4297 CrossRef CAS PubMed.
- R. Gui, Q. Yan, T. Xue, Y. Gao, Y. Li, T. Zhu and Q. Wang, J. Hazard. Mater., 2022, 439, 129665 CrossRef CAS PubMed.
- Z. Xie, J. Chen, Y. Chen, T. Wang, X. Jiang, Y. Xie and C.-Z. Lu, Appl. Surf. Sci., 2022, 579, 152003 CrossRef CAS.
- J. M. Monteagudo, A. Durán, M. R. Martínez and I. San Martín, Chem. Eng. J., 2020, 380, 122410 CrossRef CAS.
- L.-Y. Lin, S. Kavadiya, X. He, W.-N. Wang, B. B. Karakocak, Y.-C. Lin, M. Y. Berezin and P. Biswas, Chem. Eng. J., 2020, 389, 123450 CrossRef CAS.
- C. Song, Z. Wang, Z. Yin, D. Xiao and D. Ma, Chem Catal., 2022, 2, 52–83 CrossRef CAS.
- D. Mateo, J. Luis Cerrillo, S. Durini and J. Gascon, Chem. Soc. Rev., 2021, 50, 2173–2210 RSC.
- Q. Zhao, Y. Zhang, G. He, J. Ma, L. Wang and H. He, Environ. Sci. Technol., 2024, 58, 9361–9369 CrossRef CAS PubMed.
- M. Xiao, X. Yu, Y. Guo and M. Ge, Environ. Sci. Technol., 2022, 56, 1376–1385 CrossRef CAS PubMed.
- Q. Yu, C. Li, J. Zhao, X. Liu, L. Huang, Y. Zhu, K. Yang, Z. Zhang, D. Ma, Y. Zhang and Q. Huang, Appl. Catal., B, 2023, 327, 122441 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00773e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |