
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The chemical nature of SO2 poisoning of Cu-CHA-based SCR catalysts for NOx removal in diesel exhausts†
Anastasia Yu.
Molokova
 a,
Davide
Salusso
a,
Elisa
Borfecchia
b,
Fei
Wen
c,
Stefano
Magliocco‡
b,
Silvia
Bordiga
b,
Ton V. W.
Janssens
*d,
Kirill A.
Lomachenko
*a and
Gloria
Berlier
*b
a,
Davide
Salusso
a,
Elisa
Borfecchia
b,
Fei
Wen
c,
Stefano
Magliocco‡
b,
Silvia
Bordiga
b,
Ton V. W.
Janssens
*d,
Kirill A.
Lomachenko
*a and
Gloria
Berlier
*b
aEuropean Synchrotron Radiation Facility, 71 avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France. E-mail: lomachenko@esrf.fr
bDepartment of Chemistry and NIS Centre, University of Turin, via Giuria 7, 10125 Turin, Italy. E-mail: gloria.berlier@unito.it
cUmicore AG & Co, Rodenbacher Chaussee 4, 63457 Hanau, Germany
dUmicore Denmark ApS, Kogle Allé 1, 2970 Hørsholm, Denmark. E-mail: TonV.W.Janssens@eu.umicore.com
First published on 19th August 2024
Abstract
This study addresses the impact of SO2 exposure on the catalytic performance of a Cu-chabazite-based SCR catalyst, as used in diesel exhausts, to reduce the emission of NOx through the NH3-SCR reaction. The SCR activity is determined by a reaction of NO with the [Cu2II(NH3)4O2]2+ intermediate. The same intermediate is also the most reactive Cu-species towards SO2. We demonstrate here that the reaction with NO at 200 °C is limited after exposure of the [Cu2II(NH3)4O2]2+ complex to SO2 or SO2/O2. Heating the catalyst to 300 °C in NO restores the reaction, albeit at a significantly lower rate. The lower reactivity towards NO indicates that exposure of [Cu2II(NH3)4O2]2+ to SO2 induces changes in the chemistry of Cu in the catalyst. This implies that poisoning of Cu-chabazite catalysts by SO2 is, at least in part, of the chemical nature, and may be not limited to the physical pore blocking.
Introduction
Cu-chabazite (CHA) zeolites are efficient catalysts for the selective catalytic reduction of NOx using ammonia (NH3-SCR) from the exhaust of diesel vehicles, due to their high activity below 300 °C and good hydrothermal stability.1,2 However, the activity of Cu-CHA below 300 °C, which is in the normal operation range for SCR catalysts, is significantly reduced in the presence of SO2, and therefore, the application of Cu-CHA in diesel exhaust after-treatment systems requires ultra-low-sulfur diesel fuel.3,4To understand why SO2 affects the performance of Cu-CHA, it is necessary to understand how SO2 affects the NH3-SCR reaction cycle. The NH3-SCR reaction is a redox cycle, proceeding via a number of Cu-complexes formed by alternating oxidation (CuI → CuII) and reduction (CuII → CuI) steps of Cu, which further involve adsorption and reaction of NO, NH3 and O2 as ligands on the Cu-ions in the CHA zeolite.5–15 Many available studies on the reaction of SO2 with Cu-CHA show that the temperature of the reaction, gas composition and the oxidation state and local environment of Cu play a role in the effect of SO2 on the performance of Cu-CHA.4,16–25 To identify the mechanism of the deactivation of Cu-CHA by SO2, we have shown earlier that the [Cu2II(NH3)4O2]2+ dimeric species are sensitive to SO2.24 The same species are crucial for the performance of the NH3-SCR cycle,11,13,14,26 indicating that the sensitivity of these species to SO2 plays a key role in the deactivation mechanism.
The identification of the mechanism of the reaction between [Cu2II(NH3)4O2]2+ and SO2 is the next necessary step in the study of the SO2 effect on the catalytic performance of Cu-CHA in NH3-SCR. In our recent work,23 we proposed a possible mechanism of the interaction of [Cu2II(NH3)4O2]2+ complexes with SO2 and the structure of the sulfated Cu species forming as a product of the oxidation of SO2 by the [Cu2II(NH3)4O2]2+ complex. We also found that the presence of O2 enhances the formation of sulfated species, which corresponds to the observed increase in the uptake of SO2 by the Cu-CHA catalyst.
According to the theoretical work of Bjerregaard et al.,17 the main reason for the deactivation is the formation of ammonium bisulfate in the zeolite cages, which is formed together with ammonium sulphate. The latter has been observed27 together with sulfuric acid28 and copper and aluminum sulphates.29 The important part of the low-temperature NH3-SCR cycle in Cu-CHA is the activation of a pair of mobile [CuI(NH3)2]+ complexes by O2 to form the active dimeric [Cu2(NH3)4O2]2+ complexes.6,7,11,26 In the model presented by Bjerregaard et al.,17 ammonium bisulfate accumulates in the zeolite and limits the mobility of the [CuI(NH3)2]+ complexes, which hinders the formation of new [Cu2II(NH3)4O2]2+ complexes. If [Cu2II(NH3)4O2]2+ complexes are not formed, the NH3-SCR redox cycle stops. According to this model, deactivation is a consequence of a lower amount of [Cu2II(NH3)4O2]2+ complexes in the catalyst.
In this article, we shed light on whether the observed deactivation of Cu-CHA is entirely due to the lower number of active [Cu2II(NH3)4O2]2+ complexes or a change in the chemistry of the system also plays a role. The interaction of [Cu2II(NH3)4O2]2+ in Cu-CHA with NO is an essential part of the SCR cycle, and the understanding of the effect of SO2 on the performance of Cu-CHA is not possible without revealing how SO2 affects this essential step. The goal of this work is to clarify how the exposure to SO2 alters the reaction between [Cu2II(NH3)4O2]2+ and NO.
Experimental
We apply temperature-programmed reaction with NO, and combine in situ X-ray absorption spectroscopy (XAS) simultaneously with diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) to follow the reaction between [Cu2II(NH3)4O2]2+ complexes in a Cu-CHA NH3-SCR catalyst and NO before and after exposing it to SO2. For the measurements reported here, a Cu-CHA catalyst with a Si/Al ratio of 6.7, containing 3.2 wt% Cu (Cu/Al = 0.24) has been used. To determine the impact of SO2, we first prepare the [Cu2II(NH3)4O2]2+ complexes, expose these to SO2, and then to NO to follow the reaction. The results are then compared to those obtained in a similar measurement without the exposure to SO2.In the NO temperature-programmed reaction, the consumption of NO is followed during heating of the [Cu2II(NH3)4O2]2+ complexes, in NO/N2 from 50 °C to 550 °C to follow the difference in the NO consumption with and without exposure to SO2. The combined XAS + DRIFTS experiment was performed on the BM23 beamline of the European Synchrotron Radiation Facility (ESRF). The [Cu2II(NH3)4O2]2+ complexes, with or without prior treatment in SO2 + O2, were exposed to NO at 200 °C to follow the changes in the oxidation state and local environment of Cu with XAS and redistribution of NH3/NH4+ with DRIFTS. For the sample exposed to SO2 + O2, this was followed by heating to 300 °C in NO.
The experimental details are presented in the ESI.†
Results and discussion
Fig. 1 shows the results for the temperature-programmed reaction between NO and the [Cu2II(NH3)4O2]2+ species with and without prior exposure to SO2. In temperature-programmed reaction with NO, the impact of SO2 on the reaction of NO and the [Cu2II(NH3)4O2]2+ complex becomes visible as a change in the NO consumption as a function of temperature. When the [Cu2II(NH3)4O2]2+ complex is exposed to NO, the NO consumption takes place at around 120 °C. After exposure of the [Cu2II(NH3)4O2]2+ complex to SO2, the NO consumption peak shifts to around 250 °C. The shift indicates that the chemistry of the reaction of NO has changed upon exposure to SO2, such that a higher reaction temperature is required. | ||
| Fig. 1 Temperature-programmed reaction with NO, and NH3 desorption from [Cu2II(NH3)4O2]2+ with (orange) and without (grey) prior exposure to SO2. | ||
We also monitored the desorption of NH3 in this experiment. The desorption peaks for both protocols start at around 300 °C, possibly corresponding to the desorption of NH3 from the Brønsted sites and/or decomposition of the [CuI(NH3)2]+ complex.30 The amount of NH3 desorbed is slightly lower for the “exposed to SO2” experiment, indicating that some NH3 was probably lost during exposure to SO2.
The integrated amounts are presented in the ESI.†
The light-blue curves in Fig. 2a and c correspond to the exposure of the [Cu2II(NH3)4O2]2+ complex to NO at 200 °C. The weak 1s–3d transition at 8977.3 eV characteristic of CuII species disappears, while the 1s–4p transition at 8982.5 eV grows, indicating the reduction to CuI, in agreement with previous results.11,31,32 This reduction is accompanied by the decomposition of the [Cu2II(NH3)4O2]2+ complexes, as indicated by the lower intensity of the first shell in the EXAFS part. The peak in the second shell broadens, which may reveal the presence of a mixture of different Cu species.
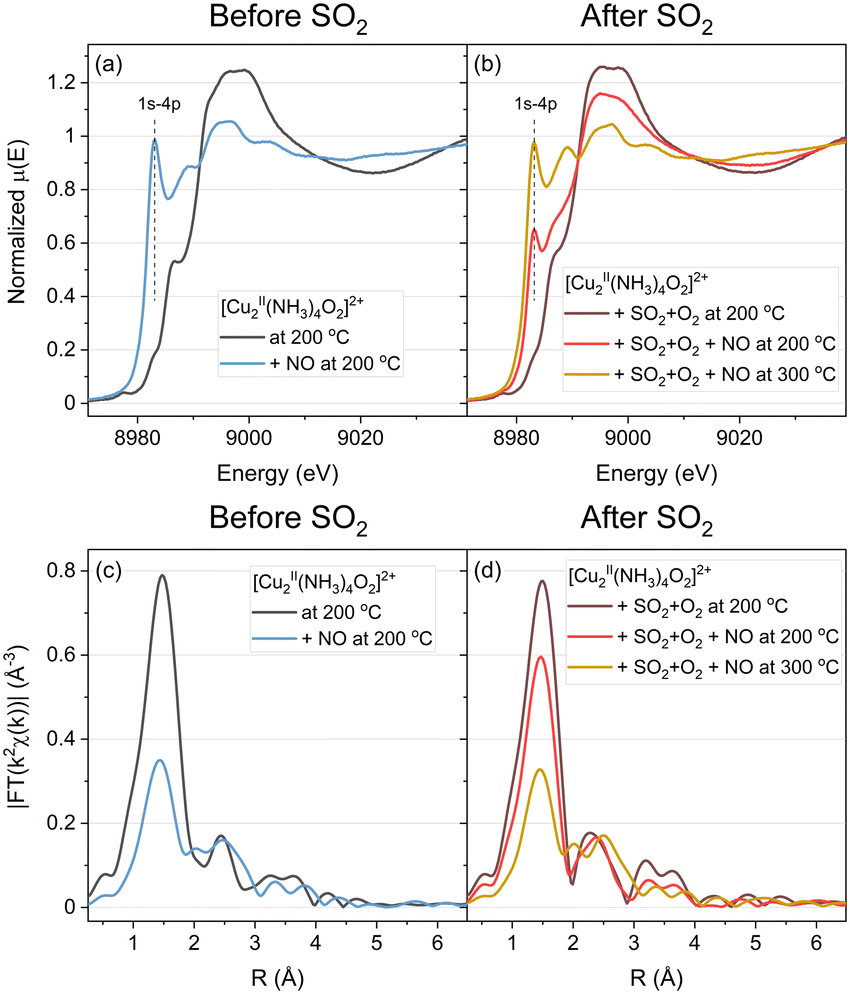 | ||
| Fig. 2 Cu K-edge XANES (a and b) and FT-EXAFS (c and d) spectra collected in situ during the exposure of the [Cu2II(NH3)4O2]2+ species to NO at 200 °C (“not exposed to the SO2” protocol); exposure of [Cu2II(NH3)4O2]2+ to SO2 + O2, then to NO at 200 °C, and at 300 °C (“exposed to the SO2” protocol). | ||
After exposure of the [Cu2II(NH3)4O2]2+ complex to SO2 + O2, a sulfated CuII species is present (brown line in Fig. 2b and d). This species has been rationalized in our previous work as CuIISO4Z, where Z may comprise O from the SO4− group, framework O or NH3.23 Exposure of the catalyst in this state to NO at 200 °C (red line) leads to some reduction of Cu, according to the XANES results, but to a lesser extent as compared to the [Cu2II(NH3)4O2]2+ complexes not exposed to SO2. Both, the increase of the 1s–4p transition at 8982.5 eV and the decrease in the intensity of the first coordination shell in the EXAFS part are weaker, as compared to the fresh [Cu2II(NH3)4O2]2+ complexes, indicating that the reaction with NO is less pronounced. When the catalyst is heated to 300 °C, both XANES and EXAFS features develop further, and the final result becomes similar to the light-blue spectrum of the [Cu2II(NH3)4O2]2+ complex exposed to NO at 200 °C without exposure to SO2.
To quantify the effect of SO2 on the reaction between NO and the [Cu2II(NH3)4O2]2+ complex, we apply a combination of multivariate curve resolution – alternating least squares (MCR-ALS) and linear combination fitting (LCF). This combination is a powerful tool that has been successfully applied to the in situ XANES datasets of Cu-CHA before.23 This approach allows us to resolve the spectra of the unknown Cu-CHA species at different stages of the protocol using multivariate curve resolution (Fig. 3a) and to calculate the concentration profiles of the Cu species from linear combination fitting (Fig. 3b and c). The choice of the references for LCF and the related discussion of errors are explained in the ESI.†
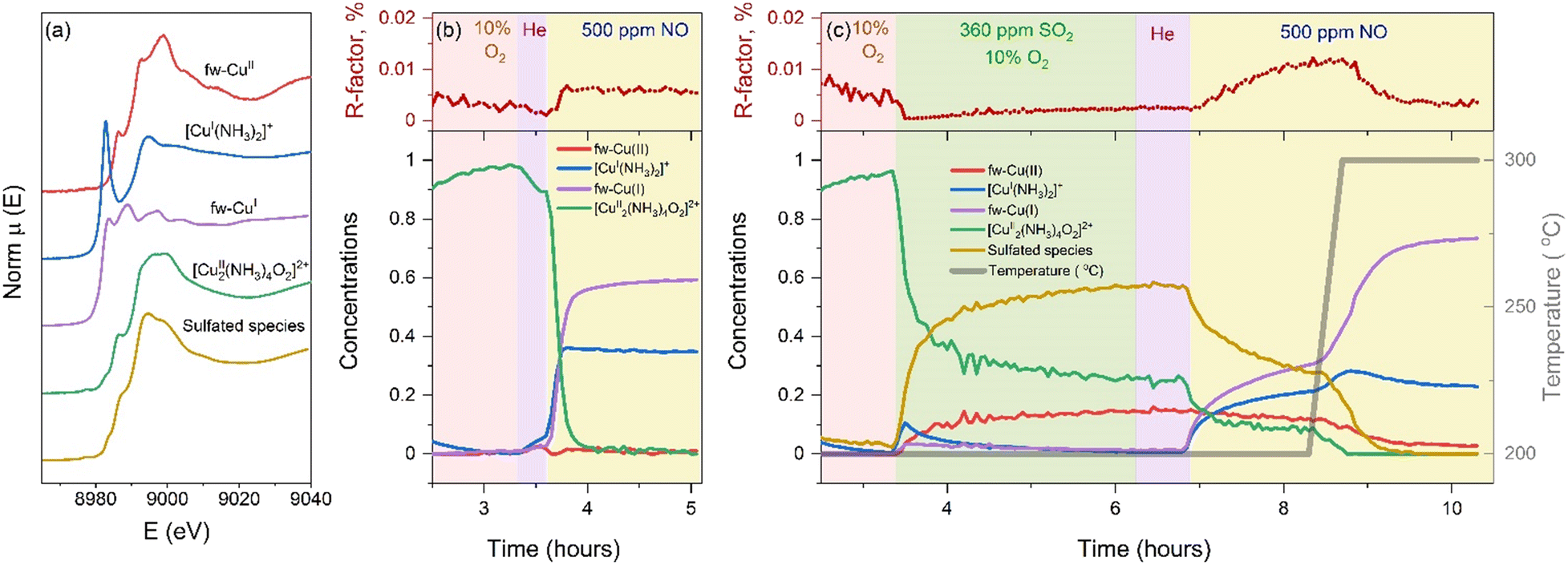 | ||
| Fig. 3 (a) Spectra used as references in the linear combination fit. The spectra of fw-CuII, [CuI(NH3)2]+, and [Cu2II(NH3)4O2]2+ are experimental spectra. The spectra of fw-CuI and the sulfated component were obtained with MCR-ALS. (b and c) Linear combination fit results of the experimental procedures. (b) Exposure of [Cu2II(NH3)4O2]2+ to NO at 200 °C (“not exposed to the SO2” protocol); (c) exposure of [Cu2II(NH3)4O2]2+ to SO2 + O2, then to NO at 200 °C, and then heating in NO to 300 °C (“exposed to the SO2” protocol). Upper panels – R-factors of the fit as a function of time and lower panels – concentration profiles of the reference spectra. | ||
When the [Cu2II(NH3)4O2]2+ complexes are exposed to NO, without exposure to SO2, we see a fast reduction of Cu and the formation of [CuI(NH3)2]+ and framework-coordinated CuI (fw-CuI) species, in agreement with previous reports.11 When we introduce NO at 200 °C after the exposure of [Cu2II(NH3)4O2]2+ to SO2 + O2, (Fig. 3c), we observe a decrease of the concentration of sulfated CuII species (yellow curve) and [Cu2II(NH3)4O2]2+ complexes (green curve).
The concentration of fw-CuII species (red curve), however, remains unchanged. The concentrations of fw-CuI and [CuI(NH3)2]+ (blue and purple curves) increase, but the reaction rate is noticeably lower compared to the rates observed without exposure to SO2.
Upon increasing the temperature to 300 °C, the reaction accelerates and we find a similar fw-CuI and [CuI(NH3)2]+ complex as in the absence of SO2. However, the ratio of the fw-CuI/[CuI(NH3)2]+ complex changes from 60/35 in the absence of SO2 to 73/23 after exposure to SO2 and heating to 300 °C. This explains the difference between the shapes of the blue spectrum in Fig. 2a and the yellow spectrum in Fig. 2b. The concentration of [CuI(NH3)2]+ is lower (23% instead of 35% in the not exposed to SO2), which can be caused by loss of some NH3 from the [Cu2II(NH3)4O2]2+ complexes during the exposure to SO2 + O2 in the previous stage of the protocol, in agreement with the temperature-programmed reaction results in Fig. 1.
The gas composition in the outlet of the cell during the in situ XAS + DRIFTS experiment, measured with a mass spectrometer, corroborates the slower reaction of NO with the [Cu2II(NH3)4O2]2+ complex after SO2 exposure (Fig. 4). When NO reacts with the fresh [Cu2II(NH3)4O2]2+ complexes without SO2, NO is consumed, whereas N2 and H2O form, in agreement with previous reports.11 When the [Cu2II(NH3)4O2]2+ complex is exposed to SO2 + O2 first, the formation of N2 at 200 °C upon exposure to NO is reduced down to 0.36 of the one of the fresh samples, according to the ratio of the corresponding N2 peaks. The main peaks for NO consumption and production of N2 and H2O appear around 250 °C, in good agreement with the observed shift in the temperature-programmed reaction with NO (Fig. 1). Furthermore, we also observe desorption of SO2 at 250 °C, corresponding to the decomposition of the sulfated species, according to the linear combination fit of the XAS data.
 | ||
| Fig. 4 Mass spectrometer data during exposure of the [Cu2II(NH3)4O2]2+ complexes “not exposed to SO2” (top panel) and “exposed to SO2” (bottom panel) to NO. Masses: H2O: 18, NO: 30, N2: 28, and SO2: 48. | ||
Fig. 5a shows in situ DRIFTS spectra after the Kubelka–Munk (KM) transformation, that were obtained simultaneously with the XAS and MS data presented above. The spectrum of the [Cu2II(NH3)4O2]2+ complex (Fig. S4, ESI†) is subtracted from the presented spectra to better highlight the changes upon exposure to NO. In the absence of SO2, exposure of the [Cu2II(NH3)4O2]2+ complexes to NO at 200 °C (light blue line in Fig. 5a) results in the growth of the OH stretching mode (νOH) of Brønsted sites (3590 cm−1),33,34 and the decrease of the broad and complex absorption band related to the νNH of NH3 and NH4+ (2500–3500 cm−1).33,35,36 This indicates some consumption of NH3 from [Cu2II(NH3)4O2]2+ complexes and/or NH4+, leading to restoration of the Brønsted H+ sites.
 | ||
| Fig. 5 (a) In situ difference DRIFTS spectra of the Cu-CHA catalyst during exposure of the [Cu2II(NH3)4O2]2+ complexes to NO at 200 °C (blue), to a mixture of SO2 + O2 (brown) and then to NO at 200 °C (red) and after heating in NO to 300 °C (yellow). The initial spectrum of [Cu2II(NH3)4O2]2+ is subtracted. The presented curves are averages of the last 10 curves of the protocol step, equivalent to a 5-min acquisition time. (b and c) Time evolution of the intensities corresponding to 3590 cm−1 (signal of Brønsted sites) and 3320 cm−1 (signal of NH3/NH4) during: (b) exposure of [Cu2II(NH3)4O2]2+ to NO at 200 ° and (c) exposure of [Cu2II(NH3)4O2]2+ after prior SO2 + O2 treatment at 200 °C, to NO at 200 °C and 200–300 °C. | ||
After exposure of the [Cu2II(NH3)4O2]2+ complexes to SO2 + O2 (red line in Fig. 5a), the band of the Brønsted sites (3590 cm−1) remains unchanged, while the bands in the range 2500–3500 cm−1, corresponding to the νNH of NH3 and NH4+ increase. This could indicate a redistribution of the NH3 ligands in the zeolite, with the formation of NH4+ ions that could exchange fw-Cu sites and/or form ammonium sulphate or bisulfate, as proposed in the literature.17,29,37–39 In this state, exposure to NO at 200 °C does not cause significant changes in the spectrum profile (brown line in Fig. 5a), indicating no reaction, or at most a slow reaction with NO, in agreement with the results from temperature-programmed reaction, XAS, and mass spectrometry as presented above. The subsequent heating to 300 °C in NO (yellow line in Fig. 5a) then results in the release of NH3 from Cu sites or stored on the Brønsted sites. Interestingly, the final state in this case shows a clearly higher intensity for the Brønsted sites (3590 cm−1) and lower intensity for the νNH of NH3 and NH4+ (2500–3500 cm−1), as compared to the not exposed to the SO2 state (blue line). This is in line with the loss of some amount of NH3 from the sample after exposure to SO2 + O2 as evidenced by the XANES linear combination fitting and temperature programmed reaction.
Fig. 5(b and c) presents time evolutions of the intensities in two points: 3590 cm−1 corresponding to the OH stretching mode (νOH) of Brønsted sites and 3320 cm−1 corresponding to the νNH of NH3 and NH4+. By plotting the trends of these two intensities, we can follow the dynamics of the NH3 and NH4+ species in the sample described above during the experimental protocol. The evolutions of the corresponding raw DRIFTS spectra and KM transformed spectra are presented in Fig. S5 and S6 of the ESI.† According to these data, the reaction of NO with the [Cu2II(NH3)4O2]2+ complex is accompanied by an increase in intensity for Brønsted sites, and a decrease in intensity for the νNH of NH3 and NH4+, both without exposure to SO2 and after exposure to SO2. Without exposure to SO2, the reaction is fast at 200 °C. After exposure to SO2, this reaction is very slow at 200 °C and accelerates only after heating to 300 °C.
From the presented results, a reaction of [Cu2II(NH3)4O2]2+ with NO takes place at 150–200 °C, without exposure to SO2. In this reaction, N2 and H2O are formed. Simultaneously, a reduction of CuII to CuI, accompanied by the consumption of NH3 from both Cu and Brønsted sites, consumption of NO and production of N2 and H2O takes place. The reaction stops when the reduction of Cu is complete.
When [Cu2II(NH3)4O2]2+ is exposed to SO2, the reaction with NO at 200 °C is hindered and requires a higher temperature (250–300 °C). The same is true for a reduction of CuII to CuI, consumption of NH3 from Cu and Brønsted sites, consumption of NO and production of N2 and H2O. Furthermore, some desorption of SO2 is observed at around 250 °C. This reaction is also limited by the number of CuII species being able to participate in the reaction. This means that the exposure of the [Cu2II(NH3)4O2]2+ complex to SO2 changes the reaction with NO, as the rate of the observed reaction is lower, compared to the case without SO2 exposure. This is visible on the time evolution curves of the IR bands (Fig. 5b and c) and the concentration profiles resulting from multivariate curve resolution (Fig. 3b and c). Without exposure to SO2, the reaction completes in ∼15 minutes. After exposure to SO2, however, the full transition takes ∼50 minutes, even though the system is clearly above 200 °C the entire time. This indicates that a SO2-induced change in the chemical properties of Cu in Cu-CHA catalysts contributes to the poisoning effect of SO2 on the reaction between Cu-CHA and NO. Because the latter is an important stage in the NH3-SCR reaction cycle, this effect also contributes to the deactivation of Cu-CHA catalysts for NH3-SCR by SO2.
In both procedures, exposed and not exposed to SO2, desorption of NH3 occurs above 300 °C (see NH3 desorption curves in Fig. 1). It is noteworthy that after exposure to SO2, the NH3 desorption peak is slightly smaller due to NH3 loss during SO2 exposure. The linear combination fits of the XAS data are consistent with a loss of NH3 after SO2-exposure, revealing a larger fraction of the Cu bound to the zeolite framework and the lower concentration of the [CuI(NH3)2]+ species in the end of the protocol.
Conclusions
In this work, we monitored the reaction of NO with the [Cu2II(NH3)4O2]2+ complex in a Cu-CHA catalyst for NH3-SCR without and with exposure to SO2, using temperature-programmed reaction with NO and simultaneous in situ X-ray absorption spectroscopy (XAS), diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and mass spectrometry. Since this reaction step occurs in the NH3-SCR reaction cycle after the activation of O2, the effect of SO2 on this reaction reflects the deactivation of Cu-CHA catalysts for NH3-SCR.The reaction of NO with the [Cu2II(NH3)4O2]2+ complex results in the formation of N2 and H2O, a loss of some NH3, formation of some Brønsted acid sites, and a reduction of CuII to CuI. Without exposure of the [Cu2II(NH3)4O2]2+ complex to SO2, the reaction with NO takes place around 120 °C.
When the [Cu2II(NH3)4O2]2+ complex is exposed to SO2, prior to the reaction with NO, the required reaction temperature increases to 250–300 °C. Some desorption of SO2 takes place around 250 °C, while the reaction products and other observed effects are similar to the case without SO2 exposure. However, the fraction of Cu bound to the zeolite framework becomes higher after SO2 exposure, and the formation of Brønsted acid sites, loss of NH3, and the reduction of CuII become significantly slower. From the NH3 desorption profile, less NH3 is stored on Cu, while NH3 stored on Brønsted acid sites is mostly unaffected, in agreement with our previous observation that the SO4− ligands may take the place of some of the NH3 in the coordination sphere of Cu. Our results indicate that SO2 induces changes in the chemical properties of Cu in Cu-CHA catalysts for NH3-SCR, which are, at least in part, responsible for the poisoning of CuCHA catalysts by SO2.
Data availability
ESRF data DOI for the CH-6662 beamtime: https://doi.org/10.15151/ESRF-ES-1049152918.Author contributions
Anastasia Yu. Molokova: investigation, conceptualisation, formal analysis, software, visualization, and writing – original draft; Davide Salusso: investigation and writing – review & editing; Elisa Borfecchia: supervision, conceptualization, and writing – review & editing; Fei Wen: supervision and writing – review & editing; Stefano Magliocco: investigation; Silvia Bordiga: supervision, project administration, and funding acquisition; Ton V. W. Janssens: writing – review & editing, supervision, project administration, and funding acquisition; Kirill A. Lomachenko: investigation, supervision, project administration, and funding acquisition; Gloria Berlier: supervision, project administration, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
ESRF is kindly acknowledged for the provision of beamtime at the BM23 beamline (experiment CH-6662). This project has received funding from the European Union's Horizon 2020 research and innovation Programme under the Marie Skłodowska-Curie grant agreement No. 847439 (InnovaXN). This research acknowledges support from the Project CH4.0 under the MIUR program ‘Dipartimenti di Eccellenza 2023-2027’ (CUP: D13C22003520001).References
- C. K. Lambert, Perspective on SCR NOx control for diesel vehicles, React. Chem. Eng., 2019, 4, 969–974 RSC.
- R. Gounder and A. Moini, Automotive NOx abatement using zeolite-based technologies, React. Chem. Eng., 2019, 4, 966–968 RSC.
- P. S. Hammershoi, Y. Jangjou, W. S. Epling, A. D. Jensen and T. V. W. Janssens, Reversible and irreversible deactivation of Cu-CHA NH3-SCR catalysts by SO2 and SO3, Appl. Catal., B, 2018, 226, 38–45 CrossRef.
- P. S. Hammershoi, A. D. Jensen and T. V. W. Janssens, Impact of SO2-poisoning over the lifetime of a Cu-CHA catalyst for NH3-SCR, Appl. Catal., B, 2018, 238, 104–110 CrossRef.
- L. Chen, T. V. W. Janssens, P. N. R. Vennestrom, J. Jansson, M. Skoglundh and H. Gronbeck, A Complete Multisite Reaction Mechanism for Low-Temperature NH3-SCR over Cu-CHA, ACS Catal., 2020, 10, 5646–5656 CrossRef CAS.
- Y. Feng, X. Wang, T. V. W. Janssens, P. N. R. Vennestrøm, J. Jansson, M. Skoglundh and H. Grönbeck, First-Principles Microkinetic Model for Low-Temperature NH3-Assisted Selective Catalytic Reduction of NO over Cu-CHA, ACS Catal., 2021, 11(23), 14395–14407 CrossRef CAS.
- T. V. W. Janssens, H. Falsig, L. F. Lundegaard, P. N. R. Vennestrom, S. B. Rasmussen, P. G. Moses, F. Giordanino, E. Borfecchia, K. A. Lomachenko, C. Lamberti, S. Bordiga, A. Godiksen, S. Mossin and P. Beato, A Consistent Reaction Scheme for the Selective Catalytic Reduction of Nitrogen Oxides with Ammonia, ACS Catal., 2015, 5, 2832–2845 CrossRef CAS.
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, The Cu-CHA deNOx catalyst in action: temperature-dependent NH3-assisted selective catalytic reduction monitored by operando XAS and XES, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS PubMed.
- R. Millan, P. Cnudde, V. van Speybroeck and M. Boronat, Mobility and Reactivity of Cu+ Species in Cu-CHA Catalysts under NH3-SCR-NOx Reaction Conditions: Insights from AIMD Simulations, JACS Au, 2021, 1, 1778–1787 CrossRef CAS PubMed.
- I. A. Pankin, H. Issa Hamoud, K. A. Lomachenko, S. B. Rasmussen, A. Martini, P. Bazin, V. Valtchev, M. Daturi, C. Lamberti and S. Bordiga, Cu- and Fe-speciation in a composite zeolite catalyst for selective catalytic reduction of NOx: insights from operando XAS, Catal. Sci. Technol., 2021, 11, 846–860 RSC.
- C. Negri, T. Selleri, E. Borfecchia, A. Martini, K. A. Lomachenko, T. V. W. Janssens, M. Cutini, S. Bordiga and G. Berlier, Structure and Reactivity of Oxygen-Bridged Diamino Dicopper(II) Complexes in Cu-Ion-Exchanged Chabazite Catalyst for NH3-Mediated Selective Catalytic Reduction, J. Am. Chem. Soc., 2020, 142, 15884–15896 CrossRef CAS PubMed.
- F. Gao, D. Mei, Y. Wang, J. Szanyi and C. H. Peden, Selective Catalytic Reduction over Cu/SSZ-13: Linking Homo- and Heterogeneous Catalysis, J. Am. Chem. Soc., 2017, 139, 4935–4942 CrossRef CAS PubMed.
- C. Paolucci, I. Khurana, A. A. Parekh, S. C. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-Caballero, A. Yezerets, J. T. Miller, W. N. Delgass, F. H. Ribeiro, W. F. Schneider and R. Gounder, Dynamic multinuclear sites formed by mobilized copper ions in NOx selective catalytic reduction, Science, 2017, 357, 898–903 CrossRef CAS PubMed.
- A. Martini, C. Negri, L. Bugarin, G. Deplano, R. K. Abasabadi, K. A. Lomachenko, T. V. W. Janssens, S. Bordiga, G. Berlier and E. Borfecchia, Assessing the Influence of Zeolite Composition on Oxygen-Bridged Diamino Dicopper(II) Complexes in Cu-CHA DeNOx Catalysts by Machine Learning-Assisted X-ray Absorption Spectroscopy, J. Phys. Chem. Lett., 2022, 13, 6164–6170 CrossRef CAS PubMed.
- C. Negri, A. Martini, G. Deplano, K. A. Lomachenko, T. V. W. Janssens, E. Borfecchia, G. Berlier and S. Bordiga, Investigating the role of Cu-oxo species in Cu-nitrate formation over Cu-CHA catalysts, Phys. Chem. Chem. Phys., 2021, 23, 18322–18337 RSC.
- X. Auvray, M. Arvanitidou, Å. Högström, J. Jansson, S. Fouladvand and L. Olsson, Comparative Study of SO2 and SO2/SO3 Poisoning and Regeneration of Cu/BEA and Cu/SSZ-13 for NH3 SCR, Emiss. Control Sci. Technol., 2021, 7, 232–246 CrossRef CAS.
- J. D. Bjerregaard, M. Votsmeier and H. Grönbeck, Mechanism for SO2 poisoning of Cu-CHA during low temperature NH3-SCR, J. Catal., 2023, 417, 497–506 CrossRef CAS.
- P. S. Hammershoi, P. N. R. Vennestrom, H. Falsig, A. D. Jensen and T. V. W. Janssens, Importance of the Cu oxidation state for the SO2-poisoning of a Cu-SAPO-34 catalyst in the NH3-SCR reaction, Appl. Catal., B, 2018, 236, 377–383 CrossRef.
- Y. Jangjou, Q. Do, Y. T. Gu, L. G. Lim, H. Sun, D. Wang, A. Kumar, J. H. Li, L. C. Grabow and W. S. Epling, Nature of Cu Active Centers in Cu-SSZ-13 and Their Responses to SO2 Exposure, ACS Catal., 2018, 8, 1325–1337 CrossRef CAS.
- X. Jiang, J. Yang, Y. Liang, H. Zhang, Y. Zhou, R. Shan, Q. Liu, W. Liu and L. Yao, In situ DRIFTS and DFT studies of SO2 poisoning over Cu-exchanged X zeolite catalyst for NH3-SCR, J. Environ. Chem. Eng., 2023, 11(3), 109822 CrossRef CAS.
- V. Mesilov, S. Dahlin, S. L. Bergman, P. S. Hammershøi, S. Xi, L. J. Pettersson and S. L. Bernasek, Insights into sulfur poisoning and regeneration of Cu-SSZ-13 catalysts: in situ Cu and S K-edge XAS studies, Catal. Sci. Technol., 2021, 11, 5619–5632 RSC.
- V. Mesilov, L. Pon, S. Dahlin, S. L. Bergman, L. J. Pettersson and S. L. Bernasek, Computational Study of Noble Metal CHA Zeolites: NO Adsorption and Sulfur Resistance, J. Phys. Chem. C, 2022, 126(16), 7022–7035 CrossRef CAS.
- A. Y. Molokova, R. K. Abasabadi, E. Borfecchia, O. Mathon, S. Bordiga, F. Wen, G. Berlier, T. V. W. Janssens and K. A. Lomachenko, Elucidating the reaction mechanism of SO(2) with Cu-CHA catalysts for NH(3)-SCR by X-ray absorption spectroscopy, Chem. Sci., 2023, 14, 11521–11531 RSC.
- A. Y. Molokova, E. Borfecchia, A. Martini, I. A. Pankin, C. Atzori, O. Mathon, S. Bordiga, F. Wen, P. N. R. Vennestrøm, G. Berlier, T. V. W. Janssens and K. A. Lomachenko, SO2 Poisoning of Cu-CHA deNOx Catalyst: The Most Vulnerable Cu Species Identified by X-ray Absorption Spectroscopy, JACS Au, 2022, 2, 787–792 CrossRef CAS PubMed.
- A. J. Shih, I. Khurana, H. Li, J. González, A. Kumar, C. Paolucci, T. M. Lardinois, C. B. Jones, J. D. Albarracin Caballero, K. Kamasamudram, A. Yezerets, W. N. Delgass, J. T. Miller, A. L. Villa, W. F. Schneider, R. Gounder and F. H. Ribeiro, Spectroscopic and kinetic responses of Cu-SSZ-13 to SO2 exposure and implications for NOx selective catalytic reduction, Appl. Catal., A, 2019, 574, 122–131 CrossRef CAS.
- C. Paolucci, J. R. Di Iorio, W. F. Schneider and R. Gounder, Solvation and Mobilization of Copper Active Sites in Zeolites by Ammonia: Consequences for the Catalytic Reduction of Nitrogen Oxides, Acc. Chem. Res., 2020, 53, 1881–1892 CrossRef CAS PubMed.
- V. Mesilov, S. Dahlin, S. L. Bergman, S. Xi, J. Han, L. Olsson, L. J. Pettersson and S. L. Bernasek, Regeneration of sulfur-poisoned Cu-SSZ-13 catalysts: Copper speciation and catalytic performance evaluation, Appl. Catal., B, 2021, 299, 120626 CrossRef CAS.
- J. Du, X. Shi, Y. Shan, G. Xu, Y. Sun, Y. Wang, Y. Yu, W. Shan and H. He, Effects of SO2 on Cu-SSZ-39 catalyst for the selective catalytic reduction of NOx with NH3, Catal. Sci. Technol., 2020, 10, 1256–1263 RSC.
- Y. Qiu, C. Fan, C. Sun, H. Zhu, W. Yi, J. Chen, L. Guo, X. Niu, J. Chen, Y. Peng, T. Zhang and J. Li, New Insight into the In Situ SO2 Poisoning Mechanism over Cu-SSZ-13 for the Selective Catalytic Reduction of NOx with NH3, Catalysts, 2020, 10, 1391 CrossRef CAS.
- L. Chen, T. V. W. Janssens, M. Skoglundh and H. Grönbeck, Interpretation of NH3-TPD Profiles from Cu-CHA Using First-Principles Calculations, Top. Catal., 2018, 62, 93–99 CrossRef.
- P. S. Hammershøi, C. Negri, G. Berlier, S. Bordiga, P. Beato and T. V. W. Janssens, Temperature-programmed reduction with NO as a characterization of active Cu in Cu-CHA catalysts for NH3-SCR, Catal. Sci. Technol., 2019, 9, 2608–2619 RSC.
- C. Negri, E. Borfecchia, A. Martini, G. Deplano, K. A. Lomachenko, T. V. W. Janssens, G. Berlier and S. Bordiga, In situ X-ray absorption study of Cu species in Cu-CHA catalysts for NH3-SCR during temperature-programmed reduction in NO/NH3, Res. Chem. Intermed., 2021, 47, 357–375 CrossRef CAS.
- C. Negri, E. Borfecchia, M. Cutini, K. A. Lomachenko, T. V. W. Janssens, G. Berlier and S. Bordiga, Evidence of Mixed-Ligand Complexes in Cu−CHA by Reaction of Cu Nitrates with NO/NH3 at Low Temperature, ChemCatChem, 2019, 11, 3828–3838 CrossRef CAS.
- A. Vimont, F. Thibault-Starzyk and M. Daturi, Analysing and understanding the active site by IR spectroscopy, Chem. Soc. Rev., 2010, 39, 4928–4950 RSC.
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, Interaction of NH3 with Cu-SSZ-13 Catalyst: A Complementary FTIR, XANES, and XES Study, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS PubMed.
- L. M. A. Zecchina, S. Bordiga, C. Pazè and E. Gianotti, Vibrational Spectroscopy of NH4+ Ions in Zeolitic Materials: An IR Study, J. Phys. Chem. B, 1997, 101, 10128–10135 CrossRef.
- C. Li, M. Shen, T. Yu, J. Wang, J. Wang and Y. Zhai, The mechanism of ammonium bisulfate formation and decomposition over V/WTi catalysts for NH3-selective catalytic reduction at various temperatures, Phys. Chem. Chem. Phys., 2017, 19, 15194–15206 RSC.
- V. V. Mesilov, S. L. Bergman, S. Dahlin, X. Yang, S. B. Xi, Z. R. Ma, X. Lian, C. Wei, L. J. Pettersson and S. L. Bernasek, Differences in oxidation-reduction kinetics and mobility of Cu species in fresh and SO2-poisoned Cu-SSZ-13 catalysts, Appl. Catal., B, 2021, 284, 119756 CrossRef CAS.
- Y. Jangjou, D. Wang, A. Kumar, J. Li and W. S. Epling, SO2 Poisoning of the NH3-SCR Reaction over Cu-SAPO-34: Effect of Ammonium Sulfate versus Other S-Containing Species, ACS Catal., 2016, 6, 6612–6622 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00792a |
| ‡ Present address: Dipartimento di Ingegneria Elettronica, Chimica ed Ingegneria Industriale, University of Messina, V.le F. Stagno D'Alcontres 31, 98166 Messina, Italy. |
| This journal is © The Royal Society of Chemistry 2024 |