
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in bifunctional synthesis gas conversion to chemicals and fuels with a comparison to monofunctional processes†
J. L.
Weber
,
C. Hernández
Mejía
 ,
K. P.
de Jong
and
P. E.
de Jongh
*
,
K. P.
de Jong
and
P. E.
de Jongh
*
Materials Chemistry and Catalysis, Universiteit Utrecht, Universiteitsweg 99, Utrecht, Netherlands. E-mail: p.e.dejongh@uu.nl
First published on 15th July 2024
Abstract
In order to meet the climate goals of the Paris Agreement and limit the potentially catastrophic consequences of climate change, we must move away from the use of fossil feedstocks for the production of chemicals and fuels. The conversion of synthesis gas (a mixture of hydrogen, carbon monoxide and/or carbon dioxide) can contribute to this. Several reactions allow to convert synthesis gas to oxygenates (such as methanol), olefins or waxes. In a consecutive step, these products can be further converted into chemicals, such as dimethyl ether, short olefins, or aromatics. Alternatively, fuels like gasoline, diesel, or kerosene can be produced. These two different steps can be combined using bifunctional catalysis for direct conversion of synthesis gas to chemicals and fuels. The synergistic effects of combining two different catalysts are discussed in terms of activity and selectivity and compared to processes based on consecutive reaction with single conversion steps. We found that bifunctional catalysis can be a strong tool for the highly selective production of dimethyl ether and gasoline with high octane numbers. In terms of selectivity bifunctional catalysis for short olefins or aromatics struggles to compete with processes consisting of single catalytic conversion steps.
1. Introduction
Increasing worldwide demand for chemicals and transportation fuels, combined with the urgent need to move to more sustainable production processes, has spurred research towards alternatives to the traditional crude oil-based processes. Implementation is driven by geopolitical, economic, and environmental considerations. Processes such as gas-to-liquids (GTL) and coal-to-liquids (CTL) have been developed as a result of these considerations in the course of the 20th century with more recent stimuli being i.e., the shale gas revolution in the USA, and the demand for transportation fuels and chemicals in China.1–3 GTL and CTL plants produce ultra-clean fuels and the possibility to shift to more sustainable feedstocks such as biomass or CO2 combined with renewable hydrogen.4–6Another advantage is the variety of products that can be selectively obtained from synthesis gas (Fig. 1), potentially playing a pivotal role in future chemical and energy industries. Synthesis gas can be directly or indirectly transformed to alcohols, long-chain hydrocarbons, olefins and aromatics, which constitute a sizable portion of industrial bulk chemicals and precursors for ultra-clean synthetic fuels.7–9 Currently, these transformations are performed in industry by thermally catalyzed processes (although other approaches such as electrochemical or plasma driven processes are being examined10,11) and largely rely on solid catalysts.7,12 A catalyst, typically a late-transition metal or metal carbide, that can hydrogenate molecules (a “hydrogenation function”) can help to selectively produce chemicals such as alcohols, olefins or paraffins. Addition of a second catalyst can be employed to couple reactions and expand the diversity of products to ethers, aromatics or branched hydrocarbons.9,13 Synergy between the functionalities in a catalyst mixture is particularly important for the desired performance, and is feasible by selecting the appropriate chemical properties and an optimal degree of intimacy.14–17 However, achieving the ideal composition while avoiding negative interference remains a challenge in these multifunctional catalytic systems.
 | ||
| Fig. 1 Schematic overview of pathways to convert synthesis gas (center) to dimethyl ether (DME), olefins, aromatics, or gasoline via oxygenate or Fischer–Tropsch intermediates. | ||
In this review, we highlight the developments over the past ten years in bifunctional catalysis systems for the transformation of synthesis gas. In particular, we compare in detail bifunctional catalysis approaches to processes comprising two or more reactors with individual conversion steps. We start with general considerations from an academic and fundamental point of view, followed by a discussion of the relevance of bifunctionally-catalyzed processes of the most important industrial fuels and chemicals. Dimethyl ether (DME), light olefins, aromatics, and liquid fuels (gasoline, kerosene, and diesel) were selected based on their high demand and maturity of their production process. For each of these product classes background information is given, followed by discussing the recent developments concerning the catalysts for their direct production from synthesis gas and the associated challenges. The advantages and drawbacks are highlighted, considering activity, selectivity, and stability, but also taking the resulting product quality into consideration. An overview and critical analysis of yield and conversion of the latest reported data is discussed in each section, contributing to a more quantitative comparison. Finally, we summarize the key points and give a perspective for the utilization of bifunctional systems.
1.1. Bifunctional catalysis
The process conditions and type of catalyst determine the products derived from synthesis gas. The initial products obtained after direct carbon monoxide hydrogenation (referred here as Primary conversion processes, section 1.4) vary according to the degree of hydrogen addition and carbon–carbon coupling. A strong hydrogenation catalyst such as nickel can yield methane, the smallest of hydrocarbons. This is interesting for the hydrogenation of captured carbon dioxide with hydrogen which is produced by electrolysis using renewable power to provide sustainable fuels.18–21 A hydrogenation catalyst like iron or cobalt that removes the oxygen to form water and enables carbon–carbon coupling, leads to the formation of long-chain hydrocarbons. These hydrocarbons can be further processed to fuel-range compounds such as gasoline, kerosene, or diesel (Fischer–Tropsch route Fig. 1), or to olefins. If the carbon–oxygen bond is maintained, for instance using a copper-based catalyst, this leads to methanol, or with polymerization to long-chain oxygenates (oxygenates route Fig. 1). Addition of a second functionality (typically an acid site) during reaction can further transform these initial products or intermediates. These subsequent reactions (referred here as “Secondary conversion processes”, section 1.5) can lead to ethers, olefines, carboxylic acids, aromatics, or branched hydrocarbons. A single catalyst combining these functionalities is referred to as a bifunctional catalyst.Applying a bifunctional catalyst or two different catalysts in a single reactor might reduce investment costs, energy requirements and complexity in comparison to two sequential reactors with individual monofunctional catalysts.22 Additionally, the combination of primary and secondary conversion catalysts can boost the overall synthesis gas conversion if the primary products are removed from this equilibrium effectively by the secondary conversion step. The combination of two catalytic functions in a single reactor can, however, also pose challenges. Undesired side reactions might emerge, for instance, the target products can further react on the primary catalytic function, or the feed might directly react on the secondary catalytic function. Examples of these side reactions are discussed in section 1.6, side reactions. The two catalytic functions can also negatively influence each other by electronic effects or migration of mobile species from one to the other. Another challenge is to find common reaction conditions in terms of reaction temperature, pressure, reaction atmosphere or space velocity for the two different catalysts. These challenges will be discussed more in detail throughout sections 2 to 5.
1.2. Relevant products
One of the potential products of bifunctional synthesis gas conversion is dimethyl ether (DME), which has a total annual production capacity of 10 million tons per year and a wide variety of applications (Fig. 2).23,24 More recently, DME is increasingly used to substitute liquefied petroleum gas, or as blend in a fuel mixture. The attractiveness of DME for use as a fuel lies in its excellent ignition and combustion properties (cetane number = 55–60), and ease of storage and handling as a liquid under a pressure of only 5–6 bar. Another advantage is that no soot is formed upon combustion. Major efforts are underway mostly in Asia and North America to further develop the infrastructure and broad introduction of DME as a clean transportation fuel.25 | ||
| Fig. 2 Overview of possible production pathways involving synthesis gas production, primary and secondary synthesis gas conversion processes. This scheme consists of synthesis gas conversion and the production of synthesis gas (light blue), the primary hydrocarbon products (methane, naphtha, wax, and diesel; given in green), oxygenates (red), short olefins as primary and secondary products (pink), other hydrocarbon secondary products (dark blue), examples of final product groups (grey), and final chemicals (purple). | ||
Light olefins, namely ethylene, propylene, and butylenes, are fundamental building blocks for the chemical industry.22 More than 50% of ethylene and 60% of propylene produced worldwide is used for fabrication of polyolefins (Fig. 2). Butadiene and 1-butene are used in the production of polymers and rubbers and as precursor of various chemicals.26 Light olefins are currently made from fossil resources, have a high energy demand and associated emission of pollutants.27,28 Several renewable alternatives to produce light olefines have been proposed.22,28 Also, the increase in C1 and C2 feedstocks derived from shale gas has promoted alternative pathways for olefins production.29
Aromatics like benzene, toluene, xylenes, and ethylbenzene are important precursors for intermediates and polymers (Fig. 2).26,30,31 The good anti-knocking properties of some aromatic compounds also makes them a good octane-enhancer for gasoline.32 The use as anti-knocking agent depends on availability and price, for instance toluene is blended in regularly, while this is less often the case for xylene, as the latter has a higher value for other chemical applications.26,33
Liquid transportation fuels (diesel, kerosine and gasoline) have a total annual consumption of ∼2.8 billion tons (in 2019).34 Diesel mainly consists of linear paraffins in the range of C10–C22 and a cetane number of 48–55 (the cetane number is an indicator for the willingness of diesel fuel to self-ignite).35–38 Kerosene consists of C8–C16 paraffins with a higher content of iso-paraffins than diesel, which decreases its freezing point and makes it suitable for application as aviation fuel.39,40 Gasoline usually comprises hydrocarbons in the range of C5–C11.41 The specifications for gasoline are that it should have a research octane number (RON, classification number for spark-ignition characteristics) between 91 and 102, a maximum olefin content of 10–18 vol% and maximum aromatics content of 35–40 vol%, depending on the category of the gasoline fuel.35 Liquid fuels are in general a blend to meet the needs of the transportation industry while adhering to the requirements of environmental regulations.42–44 The latter are especially stringent regarding sulfur content, requiring ultra-low sulfur concentrations of 10 ppm or less.45 Fuels derived from synthesis gas, via the Fischer–Tropsch synthesis (FTS) process do not contain significant amounts of sulfur.46 Large FTS plants with consecutive hydroprocessing have been operated for decades by Shell, SASOL, Chevron and others, each producing yearly between 500 kt and 7.5 Mt of synthetic hydrocarbons including high quality diesel and kerosene.47–51
1.3. Production of synthesis gas
Synthesis gas (or in short “syngas”) can be produced from virtually any carbon-containing source. The present production of synthesis gas is mainly based on coal52,53 and natural gas.54,55 In the past years biomass-derived synthesis gas (bio-syngas) has gained significance.56–59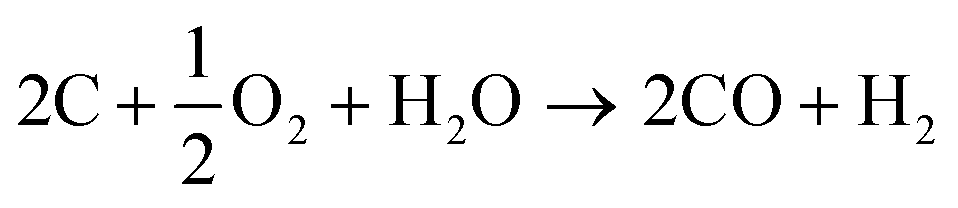 | (1) |
| CH4 + CO2 → 2CO + 2H2 | (2) |
 | (3) |
| CH4 + H2O → CO + 3H2 | (4) |
| (CxHyOz)n + H2O/O2 → CO + H2 + CO2 | (5) |
| CO + H2O ↔ CO2 + H2 | (6) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio.60 The production of synthesis gas from methane yields an H2:CO ratio between 1–3 mol mol−1, depending on the process: via dry reforming (eqn (2)) methane is converted together with carbon dioxide to synthesis gas with H2:CO = 1 mol mol−1,61,62 whereas via partial oxidation (eqn (3)) and steam reforming of methane (eqn (4)) show H2:CO ratios of 2 mol mol−1 and 3 mol mol−1, respectively.63–66 The gasification of biomass using either steam, oxygen, or a combination of both gives a high concentration of CO2 in the resulting bio-syngas (eqn (5)). This is related to the relatively high oxygen content in the biomass feedstock.67,68 The H2:CO ratio and presence of CO2 are relevant for the follow-up processes. If needed the composition of the synthesis gas can be adjusted using the (reverse) water-gas-shift reaction (eqn (6)).
:2 molar ratio.60 The production of synthesis gas from methane yields an H2:CO ratio between 1–3 mol mol−1, depending on the process: via dry reforming (eqn (2)) methane is converted together with carbon dioxide to synthesis gas with H2:CO = 1 mol mol−1,61,62 whereas via partial oxidation (eqn (3)) and steam reforming of methane (eqn (4)) show H2:CO ratios of 2 mol mol−1 and 3 mol mol−1, respectively.63–66 The gasification of biomass using either steam, oxygen, or a combination of both gives a high concentration of CO2 in the resulting bio-syngas (eqn (5)). This is related to the relatively high oxygen content in the biomass feedstock.67,68 The H2:CO ratio and presence of CO2 are relevant for the follow-up processes. If needed the composition of the synthesis gas can be adjusted using the (reverse) water-gas-shift reaction (eqn (6)).
Sulfur compounds (e.g., H2S or COS) in synthesis gas act as a poison to most synthesis gas conversion catalysts and might result from the feedstock.69,70 These can be removed using an absorber column with amine scrubbing.71 In contrast, removal of sulfur from heavier hydrocarbons in conventional refinery processes requires extensive effort. Here, the feedstock needs to be treated in the hydrodesulfurization (HDS) process.72
A major challenge with the production of synthesis gas from biomass is the competition with food and the impact on the environment by the use of monocropping and possible damage to the biodiversity.73 Furthermore, the availability of biomass for large scale synthesis gas production can be a hurdle considering the costs for transportation and the efficiency of land use.74 A technical challenge in bio-syngas production is catalyst deactivation by the formation of tar during biomass gasification. However, the use of a suitable catalyst in the steam reforming of biomass gives a tool to reduce the formation of tar drastically.68,75 Further impurities such as hydrochloric acid can be removed with amine scrubbing and an additional chloride guard bed.71,76
1.4. Primary conversion processes
| CO + 2H2 ↔ CH3OH | (7) |
| CO2 + 3H2 ↔ CH3OH + H2O | (8) |
Copper-based methanol synthesis catalysts are employed by the industry due to their high activity at milder reaction conditions.103 Recently, research has focused on finding a methanol synthesis catalyst for using CO2 as main carbon source.104 Compared to the traditional feed three challenges must be met: decreased catalyst stability due to the high-water concentrations, a less favorable equilibrium and hence driving force for the reaction, and the side reaction forming CO via the reverse water gas shift reaction. A wide variety of materials has been proposed as candidates: intermetallic compounds such as Ni–Ga (ref. 105) or In–Pd,106 supported metal oxide nanoparticles MnOx/Co3O4 (ref. 107) or In2O3/ZrO2,108 solid solutions of metal oxides ZnO–ZrO2 (ref. 109) and transition-metal phosphide catalysts such as MoP.110
Methanol can be used as the starting point to produce DME (see chapter 2) or hydrocarbons in processes generally known as methanol-to-hydrocarbons (MTH), methanol-to-olefins (MTO), methanol-to-gasoline (MTG) and methanol-to-aromatics (MTA).24,25
High temperature Fischer–Tropsch synthesis (HT-FTS) operates at 300–350 °C and about 20 bar utilizing an iron-based catalyst to produce hydrocarbons in the gasoline range (C5–C11) and light (C2–C4) olefins (eqn (9)).49 The active phase of catalyst is iron carbide.113–117 In industry, iron-based catalysts are often promoted with alkaline metals, such as potassium or sodium to increase activity, and selectivity to olefins.118 Additionally, copper is employed as promoter to increase the reducibility and SiO2 can be used as structural promoter.49 HT-FTS catalysts are also active in the water-gas-shift (WGS) reaction, converting CO and H2O into H2 and CO2 (eqn (6)).22 Promotion with sodium and sulfur allows decreasing the methane selectivity and increasing the C2–C4 olefin–paraffin ratio with respect to the ASF distribution.114,119–122 This enables 72% C2–C4 olefins formation, whereas according to the ASF 57% C2–C4 (olefins + paraffins) at maximum would be formed.121
| nCO + 2nH2 → CnH2n + nH2O | (9) |
| nCO + (2n + 2)H2 → CnH2n+2 + nH2O | (10) |
Ruthenium based FTS-catalysts can be operated at 140–220 °C and 15–100 bar.127–129 However, ruthenium is orders of magnitude more expensive than cobalt and less available.118,130,131 Nickel FTS-catalysts can display similar selectivity and activity as cobalt under similar reaction conditions.132,133 However, under high carbon monoxide partial pressure volatile nickel carbonyls are formed, leading to metal particle growth and/or nickel entrainment whereby the activity of the catalyst decreases over time.118 Bimetallic nickel–cobalt catalysts might be promising as they show increased activity and selectivity to C5+ hydrocarbons and better stability when supported on reducible oxide support materials.132
1.5. Secondary conversion processes
| 2CH3OH ↔ CH3OCH3 + H2O | (11) |
| nCH3OH → (CH2)n + nH2O | (12) |
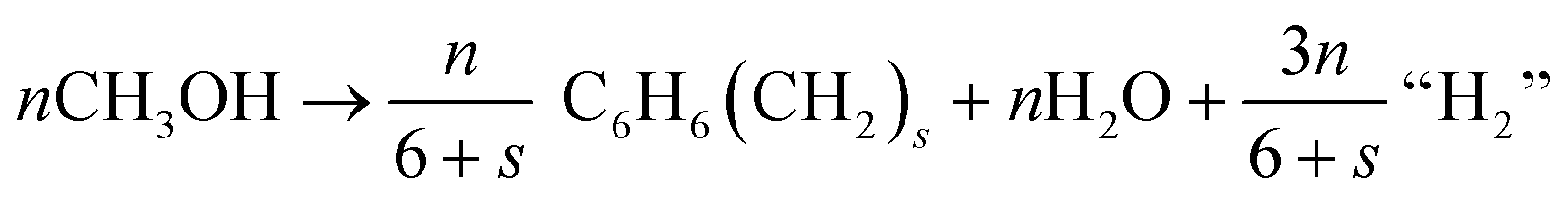 | (13) |
The conversion of methanol to olefins (MTO) and methanol to aromatics (MTA) is believed to follow the dual cycle mechanism (Fig. 3).134–137 In the alkene cycle, short olefins are alkylated by the addition of a CH2 group that is transferred from methanol, forming longer chain olefins and water. The products in the alkene cycle are higher olefins that either dealkylate forming short olefins or undergo aromatization and enter the aromatic cycle. In the aromatic cycle, light aromatics are alkylated by CH2 groups from methanol to form poly-alkylated aromatic species and water. These poly-alkylated aromatic species are protonated by the Brønsted acid sites of the catalyst, followed by dealkylation and consecutive deprotonation. During the dealkylation, short olefins are released which can either enter back into the alkene cycle or yield the final products.
 | ||
| Fig. 3 Illustration of the dual cycle mechanism consisting of the aromatic and alkene cycle.134 | ||
Zeolites with 8-membered ring pores such as SAPO-34 or H-SSZ-13 show high selectivities to short olefins (70% to 96%) at full methanol conversion,138,139 when operated at high temperature (300–450 °C) and atmospheric pressure.140,141 The zeolites used in the MTA reaction usually are 10-membered ring zeolites such as H-ZSM-5 and TNU-9 or 12-membered ring zeolite such as zeolite type beta or mordenite142–144 The MTA reaction yields a variety of products, ranging from 2–40% C2–C4 olefins, 8–51% aliphatic hydrocarbons with more five carbon atoms (C5+) to 17–50% aromatics.144,145 The strongly different product spectra of the MTO and the MTA process can be explained by the pore dimensions and topology of the zeolites.146–148 The aromatic species formed during the MTO reaction are retained in the small zeolite cavities and participate in alkylation and de-alkylation in the aromatic cycle. The pores of 10- and 12-membered ring zeolites used in the MTA process are wide enough to release the aromatic molecules.
During the MTO and MTA process the zeolite is rapidly deactivated mostly by formation of coke, blocking the active sites. Catalyst lifetimes vary from 20–200 h on stream, depending on material composition, crystallite size, acid site density, porosity and reaction conditions.138,141,145,149–153 Co-feeding water increases the lifetime of the catalyst, but reduces its activity due to co-adsorption on the acid sites.154 Also, here promoters can play a role. Partially replacing the protons of the Brønsted acid sites with zinc-ions in H-ZSM-5, led to increased selectivity towards aromatics and reduced paraffin selectivity in the MTA process.155–157
The mechanism of mono-functional acid catalyzed cracking and isomerization over zeolites involves carbocations at elevated temperatures (Fig. 4).160,161 A primary carbocation (positive charge on a carbon atom at the end of the chain) is relatively unstable and undergoes carbocation isomerization, resulting in a more stable secondary carbocation (positive charge being stabilized by two alkyl groups). Tertiary carbocations show even higher stability and are formed from skeletal rearrangement of secondary carbocations. These carbocations can be cracked at the β-position (one carbon atom further than positive charge, called β-scission) forming olefins and another carbocation. Acid cracking and isomerization yields few n-paraffins (alkanes) and gases, whereas high yields of aromatics, olefins and i-paraffins are produced.162 The majority of the hydrocarbon products is isomerized due to the high stability of the tertiary carbocation intermediate.163
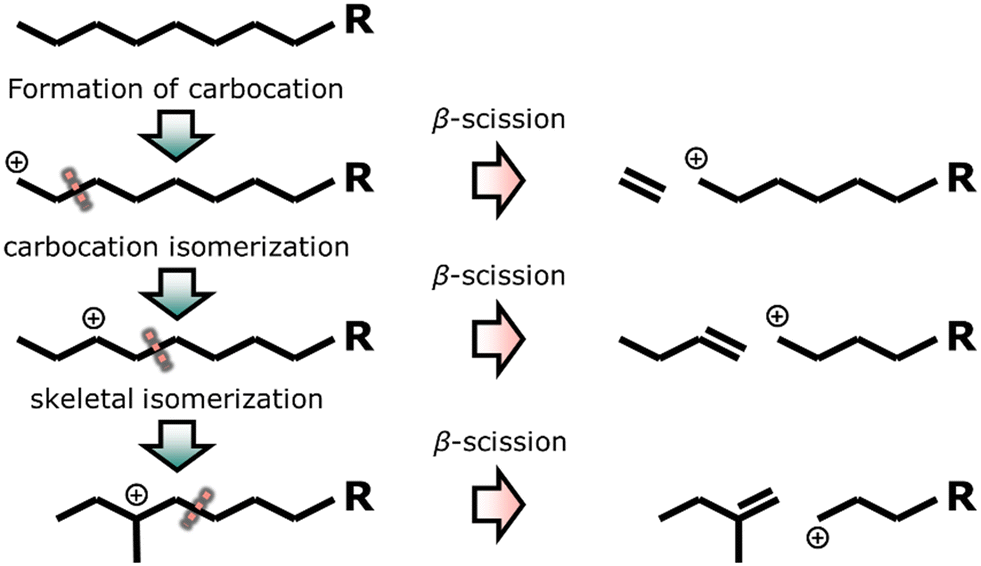 | ||
| Fig. 4 Catalytic cracking and isomerization with the formation of carbocations, isomerization and consecutive cracking with different positions of the positive charge.160,161 | ||
Alternatively, bifunctional catalysts can be used for cracking and isomerization at lower temperatures. The feed molecules are dehydrogenated on the metal sites forming olefins and hydrogen. The olefin molecules undergo carbocation formation, carbocation and skeletal isomerization, and β-scission to some extent similar to the mechanism of acid cracking and isomerization on the acid sites of the zeolite. The resulting olefins, however, are hydrogenated on the metal sites, forming paraffins.161 This means that transport of molecules between the two different sites is very important.
Bifunctional cracking and isomerization are performed in the presence of hydrogen and at pressures between 5–150 bar,14,164,165 in the so-called hydrocracking or hydroisomerization process. The product spectrum strongly depends on the reaction temperature. High reaction temperatures (between 300 °C and 400 °C) favor hydrocracking,165–168 whereas hydroisomerization is more likely at lower temperatures (between 200 °C and 260 °C).14,164,169–171 The distance between the metal and acid site is crucial to achieve high yields of branched isomers in the hydroisomerization reaction. Relatively large distances, in the range of micrometers, can cause strong concentration gradients of intermediates and reactants within the catalyst.172–174 Closest proximity, however, can lead to an increased degree of cracking forming more gaseous products.14
1.6. Side reactions
However, the WGS reaction is usually undesired when converting CO-based synthesis gas. Although the feed in this case does not contain water, product formation is often accompanied by water formation, especially in FT. The presence of both water and carbon monoxide can lead to the production of important concentrations of CO2, for instance in HT-FTS.180 This has a negative effect on the efficiency of the process, and hence a common challenge in these processes is to limit the WGS activity of the catalyst.
OX–ZEO catalysts consist of metal oxides combined with zeolites and can convert synthesis gas to olefins and aromatics.192 In the first step synthesis gas is converted into reactive intermediates such as methanol, dimethyl ether, or ketene over the metal oxide, followed by the formation of olefins on the zeolites. Olefins also act as intermediates in the OX–ZEO to aromatics process.193,194 Metal oxide functions with a high hydrogenation activity can cause secondary hydrogenation of olefins to paraffins.195 The hydrogenation of aromatics requires the presence noble metals such as platinum or palladium196,197 and does not take place in the OX–ZEO process with the commonly used catalysts.198 A detailed analysis of secondary hydrogenation can be found in chapter 3.3 and 4.3.
| 2CO(g) ⇄ CO2(g) + C(s) | (14) |
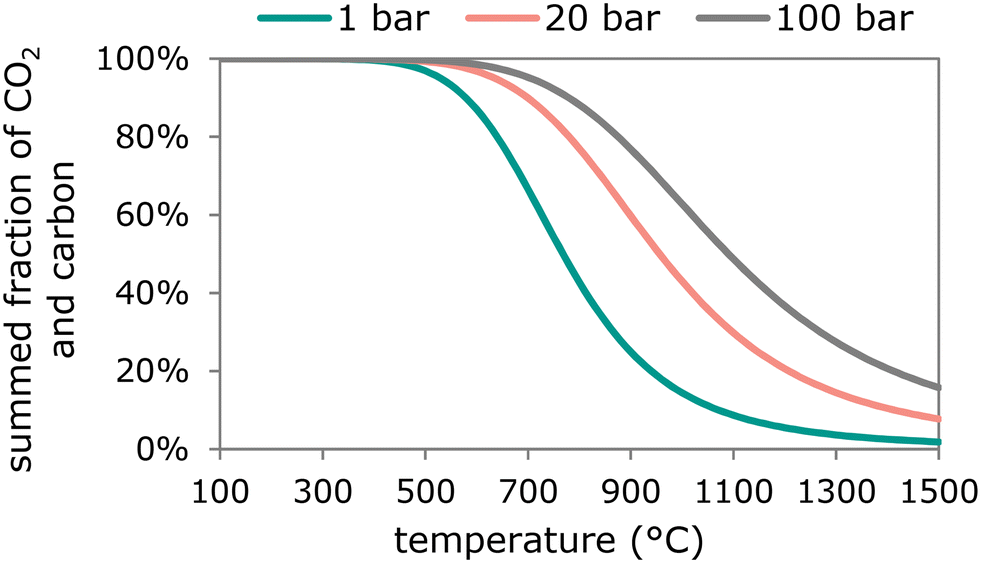 | ||
| Fig. 5 Fraction of CO2 + C at equilibrium as a function of temperature with CO, C and CO2 at either 1 bar, 20 bar or 100 bar pressure. | ||
Alternatively, heavy hydrocarbons formed by oligomerization in acid catalyzed cracking and isomerization can condense onto and hence deactivate active sites at temperatures below 200 °C.203 At high temperatures (350 °C and above) hydride transfer reactions take place causing the formation of polyaromatic hydrocarbons.203,204 The temperature for the formation of polyaromatic hydrocarbon species is reduced for hydrocracking and hydroisomerization, because (de)hydrogenation is catalyzed by metal sites.203
The mechanism of the MTO and MTA reactions is based on the alkylation and de-alkylation of light aromatic species inside zeolite crystals. The main cause for catalyst deactivation is the formation of large and heavy poly-aromatic hydrocarbons inside the zeolite pores or cavities, limiting accessibility to the acid sites of the zeolites.205,206 Using an H-SSZ-13 zeolite in the MTO process, it was shown that also at lower reaction temperatures pore filling of the zeolites with methylated bicyclic aromatics plays a role, whereas at higher temperatures the deactivation is caused by the formation of 3- and 4-cyclic aromatic species.207
In the following sections we analyze the recent literature of bifunctional catalysis for the conversion of synthesis gas to DME, short olefins, aromatics, and gasoline. Additionally, we compare the performance of these catalysts with established processes consisting of sequential individual catalytic steps in terms of overall selectivity and conversion.
2. DME
Methanol dehydration to DME is usually performed at atmospheric pressure, high space velocities and temperatures between 190 °C and 400 °C.208–210 The catalysts most widely used are solid oxide acids such as γ-Al2O3 or aluminosilicates, or zeolites.208,211 The active sites can be both Lewis and Brønsted acid sites.212 Processes using γ-Al2O3 catalysts proceed at the higher end of the mentioned temperature range.209,213 γ-Al2O3 has mainly Lewis acid sites, which might adsorb the formed water particularly at low temperatures, inhibiting the reaction with methanol.209 Increased reaction temperatures facilitate the desorption of water from the acid sites of the Al2O3 catalysts, but also decrease the maximum attainable one-pass DME yield due to equilibrium limitations (Fig. 6). | ||
| Fig. 6 Equilibrium composition of methanol dehydration to DME and water as function of reaction temperature calculated at 1 bar pressure (calculated with Outotec HSC 9.6.1). | ||
Although for methanol dehydration most commonly γ-Al2O3 is used, other acidic compounds can also be used as catalyst.214 Mixed metal oxides such as aluminosilicates and ZrO2/TiO2 have Brønsted acid sites next to Lewis acid sites, and display enhanced activity and stability compared to Al2O3 under the same reaction conditions.208,209 Zeolites have both Lewis and Brønsted acid sites and allow lower operation temperatures. Additionally, the strong Brønsted acid sites might allow sequential olefin formation at higher temperatures.214
2.1. Recent developments
Direct DME synthesis has attracted large interest, which is reflected by the extensive investments in direct DME synthesis pilot plants215–217 and several academic reviews on DME synthesis published in the recent years.211,218–223 Based on these reviews and several other publications, we give an overview of the optimal reaction conditions as well as catalysts.Colloidal synthesis of nanoparticles has emerged as a tool to prepare and understand catalytic model systems.224 Pre-forming the nanoparticles in solution and then depositing them onto a support material enables the preparation of monodisperse, single crystalline, and size-controlled nanoparticles, which is rather challenging for conventional synthesis techniques.225 For bifunctional catalysts, colloidal nanoparticles have been employed as a strategy to avoid structure sensitivity effects of the metal-based methanol synthesis catalysts and control its proximity to the acid sites.226 Monodisperse colloidal Cu–ZnO-based nanoparticles were either directly supported on the dehydration catalyst (γ-Al2O3) or mixed with the dehydration catalyst.227 Directly supporting the nanoparticles on the solid acid caused partial blockage of the acid sites and a slight decrease in DME selectivity (64% to 59% DME selectivity).
The same approach has been used to study Pd–Ga-based colloidal nanoparticles, as methanol synthesis catalyst from CO and H2, supported on γ-Al2O3.228 The interest in Pd–Ga systems arises from its activity in CO2 hydrogenation to methanol.229,230 The Pd–Ga/γ-Al2O3 system showed good stability. However, hydrocarbon selectivity remained an issue over the whole temperature range. Methane content increased from 12% at 250 °C to 43% at 300 °C, while Cu–ZnO-based catalyst produced only 1.2% at 250 °C and 8.8% at 300 °C. High methane yields has been reported to be a general problem of Pd-based DME catalysts.231
The use of core–shell systems is a popular strategy to circumvent Cu sintering. Typically, the metal-based core is encapsulated by a solid acid shell.232 This forces also the methanol formed on the core catalyst to pass through the acidic material before leaving the catalyst system, leading to a high DME selectivity.233 The catalytic performance (275 °C, 35 bar, H2:COx = 3 mol mol−1 and TOS = 24 h) of CuO–ZnO–ZrO2 and SAPO-11 was studied for a physical mixture of the two components, and for a core–shell catalyst with the Cu-based catalyst being covered by SAPO-11.234 The highest DME yields were obtained at a CuO–ZnO–ZrO2 to SAPO-11 weight ratio of 0.5. The core–shell catalyst showed a more stable catalytic performance than the physical mixture, having a relative decrease in DME yield at the end of the experiment (24 h time on stream) of 22% against 33% for the physical mixture. By comparing the acidity of the catalysts before and after reaction, it was observed that the core–shell catalysts lost around 10% of the initial acid sites after 24 h on stream, while the physical mixture lost around 26%. The loss of acid sites was identified as coke deposition, being lower for the core–shell catalyst. Physical separation of the metallic and acid functions by an intermediate silica layer contributes to reducing coke deposition on the SAPO-11, and therefore to preserve its acidity.
The strategy of a porous intermediate layer in a core–shell catalysts has been previously explored;235,236 a silica layer was deposited over the Cu-based catalyst to avoid damaging the integrity of the catalyst while depositing the solid acid overlayer. Alternatively, a mesoporous alumina interlayer has been also employed, on which silicotungstic acid is deposited to improve the shell's acidity and the catalysts DME selectivity.237
However, the use of a protective silica layer can cover part of the active sites of the methanol synthesis catalyst,233,238 resulting in lower CO conversion. To avoid this, a different coating method was reported in which various solvents (ethanol, water, methanol, and ethylene glycol) were used as binder to coat an H-ZSM-5 shell on a Cu–ZnO-based catalyst.239 Ethanol as a solvent showed the best performance, although this could not be explained by more exposed metal sites based on the characterization results.
The use of ultra-small (<5 nm) ZSM-5 zeolite crystals placed on a CuO–ZnO–Al2O3 methanol synthesis catalyst showed better activity, selectivity to DME, and stability than the methanol synthesis catalyst combined with amorphous aluminosilicate or ZSM-5 zeolites with 20–500 nm crystallite size.240 It was concluded that the ultra-small ZSM-5 nano particles had superior diffusion properties compared to larger crystals. Additionally, the medium strength of the Brønsted acid sites resulting from the small crystallite size did not facilitate further dehydration of DME to olefins.
Electrospinning has been employed for the synthesis of various fibrous high-performance materials,241,242 in this case for the design of a bifunctional catalysts. A fibrillar system has been reported which circumvents diffusion limitations while maintaining a close contact within functionalities.243 The Cu–ZnO/ZrO2–ZSM-5 fibrillar bifunctional catalyst was prepared by using an electrospinning technique. The polymeric filaments after calcination, resulted in this case in homogeneous zirconia-based fibers (with a diameter of 1.5 μm) and well-distributed Cu–ZnO and zeolite aggregates throughout the fibers of the bifunctional catalyst. The catalyst showed high DME yields (59–63%), with a low zeolite content of 10 wt%. This could be attributed to the high dispersion of the zeolite over the fibers, and the fact that the methanol synthesis function was not affected by the addition of the zeolite during synthesis. The pressure drop inside the fixed-bed reactor was theoretically calculated for the fibrillar structured catalyst with micrometric size and for the powder catalyst with the same effective dimension. The calculation results showed 5000 times less pressure-drop for the fibrillar packed bed than for a packed bed of spherical particles (0.3 vs. 1650 bar m−1). Longer tests than the reported 4 hours on stream might give more insight into the stability of this material.
In situ removal of water during DME synthesis, often referred as sorption enhanced dimethyl ether synthesis, has emerged as a relatively new approach to avoid the detrimental effects of water on the catalyst and boost the DME selectivity by inhibiting the water-gas-shift reaction. This idea is promising for process intensification and can be applied to different reactions in which water is a by-product as recently reviewed.244,245 Water can be removed from the catalyst bed using membrane technology or selective adsorption.246 The former requires large H2O partial pressures differences and high permselectivity of water over the reactants, the latter is preferred at low H2O partial pressures (<1 bar). Theoretical simulations have confirmed higher DME yields under H2O removal conditions, particularly upon addition of CO2 due to an increased methanol production and preventing the water-gas-shift reaction.247
Experimentally, enhanced DME production has been reported for a commercial copper-based catalyst mixed with a water absorbent material (commercial zeolite LTA-type with 3 Å pore size).245,248 Adsorption of water by the zeolite during DME synthesis led to a decrease in CO2 formation. The DME yield was 65% at around 70% CO conversion (275 °C, 25 bar and H2:CO = 2 v/v). Upon saturation of the zeolite after some minutes of the reaction, a regeneration step was carried out by switching to nitrogen, depressurizing to 1.7 bar and heating to 400 °C. More recently, the same concept has been studied using a Cu–ZnO-based catalyst in combination with γ-Al2O3 as solid acid and a zeolite 3A as water sorbent.249 The methanol catalyst alone showed a carbon conversion of 9.7% and 100% selectivity to methanol at 270 °C and 25 bar with a syngas composition of CO:CO2:H2 = 1:1.9:7.7 (v/v/v). When the methanol synthesis catalyst was combined with the methanol dehydration catalyst and the water sorbent zeolite at 275 °C, the carbon conversion increased to 83% (54% CO conversion and 97% CO2 conversion) with a DME selectivity of 99% in the early stage of the reaction (∼20 min). After the zeolite was saturated (after ∼100 min.) the carbon conversion dropped to 19% (41% CO conversion and 8% CO2 conversion) and DME was formed with 81% selectivity.
However, information on the stability of the catalysts in these studies is lacking, especially after regeneration cycles. Identifying sorbent materials that operate under DME synthesis conditions without suffering deactivation remains challenging.250 Research efforts have been focused on improving the regeneration procedure.251 It has been shown that a pressure swing (from 25 bar at reaction conditions to 1–3 bar) followed by purging with an inert gas can remove the water of the zeolite 3A and regenerate the activity without changing the temperature of the reactor. This swing process to remove the adsorbed water required 1 h, which is faster than the alternative thermal treatment at 400 °C which can require 6 h.
The conversion of CO2 or CO2-containing synthesis gas in the direct DME synthesis has attracted recently attention in research.252–258 Published data has shown that 48% CO2 conversion252,256 and high DME selectivities up to 100% (ref. 255 and 257) can be reached. Additionally, a comparison between CO2-rich and CO-rich synthesis gas revealed that a CO:CO2-ratio of 1:4 (v/v) in the synthesis gas led to higher conversion (65.6%, sum of CO + CO2) compared to a CO:CO2-ratio of 4:1 (v/v) (35.4% conversion) The DME selectivity resulting from the CO2-rich synthesis gas was slightly lower (73.2% compared to 88.7%), however, the yield of DME was higher (48% compared to 31.4%).237 Direct CO2 hydrogenation to DME also showed advantages in energy efficiency and net CO2 mitigation in a techno-economic study compared to different routes (indirect route via CO or direct CO2 hydrogenation).259 Compared to methanol synthesis from CO2, direct DME synthesis can result in higher CO2 conversions (+20%) and higher yields of valuable products (+70%).260
2.2. Benefits
Fig. 7 shows the CO conversion and yields of methanol, DME and CO2 as function of reaction temperature at 40 bar total pressure for methanol synthesis (Fig. 7-A), direct DME synthesis without WGS (Fig. 7-B) and direct DME synthesis with WGS (Fig. 7-C) in equilibrium (calculated with Outotec HSC 9.6.1). For the methanol synthesis the CO conversion is limited to 40% at 260 °C and 40 bar. If instead of pure CO (also) CO2 is added to the synthesis gas feed, the conversions are even lower. The direct DME synthesis shows an equilibrium conversion of 72%, whereas the direct DME synthesis with WGS shows a maximum CO conversion of 95% at these conditions. Removing methanol by subsequent dehydration hence increases the conversion of synthesis gas, reaching CO conversions as high as 96% and DME selectivities up to 87%.261,262 The additional removal of water via the WGS reaction drives the equilibrium to even higher conversions. Although the WGS reaction compromises the selectivity to DME, it increases the DME yield per single pass, especially at higher temperatures. | ||
| Fig. 7 Equilibrium composition (based on carbon atoms) and CO conversion as a function of temperature at 40 bar for carbon species involved in A: methanol synthesis, B: direct synthesis of DME without water-gas-shift reaction, and C: direct synthesis of DME with water-gas-shift reaction. The thermodynamic calculation considering all species in the gas phase was carried out with a synthesis gas composition of H2:CO = 2 v/v and as possible products methanol (A) and additionally DME (B) and DME and CO2 (C). HSC software from Outotec (v 7.14) was used to perform the calculations. | ||
Cu-based methanol synthesis catalysts display a high water-gas-shift activity.263–265 Water formed during dehydration can react with CO forming CO2 and H2 (see section 1.3 eqn (6)). This can be beneficial when using hydrogen-lean synthesis gas. Furthermore, the presence of a few percent of CO2 enhances the activity of the methanol synthesis catalyst.90 High water concentrations result in accelerated deactivation, which can be circumvented by water removal via the WGS reaction or via a membrane.266,267
2.3. Challenges
Methanol dehydration to form DME can be catalyzed by Brønsted as well as Lewis acid sites, and all acid strengths. However, particularly at high temperatures, strong acid sites can facilitate the further dehydration of DME to olefins and other hydrocarbons, compromising the DME yield.209,268,269 This does not represent a problem for the dual reactor process, since the methanol dehydration step can be operated at relatively low temperatures (≤200 °C, Fig. 6). At higher temperatures needed for methanol synthesis, less strong acid sites are preferred. Indeed, acid sites with a weak to medium strength have shown an excellent selectivity to DME under direct DME synthesis conditions.240,261,270Industrial methanol synthesis catalysts are copper-based.86 Copper nanoparticle growth and hence loss of active metal surface area, is the main deactivation mechanism.271 It is enhanced by higher water concentrations, to which it will be exposed when used in direct DME synthesis or in CO2 rich feeds.272,273 Faster deactivation was observed when co-feeding water using a Cu–ZnO methanol synthesis catalyst only.274,275 Recently, the stability of a Cu–ZnO catalyst physically mixed with a ZSM-5 zeolite was studied under DME synthesis conditions (260 °C, 20 bar, 90000 or 3600 cm3 gcat−1 h−1 and H2:CO = 2 v/v) by in situ synchrotron-based EXAFS and XRD experiments.276 Results show an increase of the copper crystallite size from 9 nm to 12 nm during the first hours under reaction conditions, while copper remained in the metallic state within the technique's detection limit. Decreasing the gas space velocity or co-feeding water led to larger crystallite sizes, 17 nm and 20 nm respectively. The authors concluded that the water generated during DME synthesis has a detrimental effect in the stability of the Cu–ZnO catalyst mainly by particle growth.
For the methanol dehydration catalysts, the challenges vary according to the nature of the material. Zeolites in the proton form typically have strong Brønsted acid sites which can lead to further DME dehydration to hydrocarbons, although some strategies have been developed to tune the zeolite acidity and to improve DME yields.277,278 Another main challenge is the microporous structure of zeolites which can limit the diffusion of reactants and products leading to hydrocarbon and coke formation, deactivating and blocking the active sites.279 H-ZSM-5 in a physical mixture with Cu–ZnO catalysts showed a decrease in activity of ∼20% due to accumulation of hydrocarbon species formed in the pores (250 °C, 10 bar and TOS = 100 h).280 The synthesis gas composition in this case was important for the zeolite stability, the presence of CO2 directly affected the partial pressure of water and hence aided to avoid accumulation of carbonaceous species in the pores.
γ-Al2O3 is a very selective solid acid catalyst to produce DME due to its mild Lewis acid sites, active for methanol dehydration. However, it can loss activity in the presence of water due to competitive water adsorption on the acid sites or by recrystallization.280 The γ-Al2O3 to boehmite phase transition has been investigated in the range of 250–400 °C and H2O partial pressures up to 15 bar.281,282 Results over γ-Al2O3 at 250 °C and water partial pressure of 13–14 bar led to the conversion of γ-Al2O3 into γ-AlO(OH). This was linked to a decrease in catalytic activity of methanol dehydration, from ∼60% methanol conversion to ∼15%. However, the phase transition was reversible under more standard reaction conditions or calcination at 350 °C, recovering its catalytic activity. Niobium oxide-based dehydration catalysts are less active but can form a stable NbO4–H2O phase and do not show water induced deactivation.270,283–288
The interaction between both catalytic materials can result in activity and/or selectivity loss, therefore the distance between functionalities is a key factor for the stability of the catalyst. Two distances in the micrometer range have been studied by co-tableting powders with different sieve fractions of a Cu–ZnO–Al2O3 methanol synthesis catalyst and silica-alumina dehydration catalyst.289 A fine sieve fraction of 50–100 μm of the individual catalysts and a coarse sieve fraction of 600–1000 μm were used to prepare bifunctional catalysts. During direct DME synthesis (285 °C, 60 bar, TOS = 700–800 h), the finer particle catalyst deactivated faster than the catalyst pelletized from larger particles. Characterization of the used catalysts revealed migration of zinc from the methanol synthesis catalyst into the dehydration component and silicon from the dehydration component diffused into the methanol synthesis catalyst particles. The authors concluded that the faster poisoning of the more finely sieved catalyst relates to the larger contact area between the two catalyst materials. Analog, it has been observed that species exchange between the solid acid and Cu–ZnO catalyst with H-ZSM-5 as solid acid.290–293 The extent of deactivation was linked to the amount of zeolite's extra-framework Al species and surface acid sites.292,294 Migration of copper from the methanol synthesis catalyst to niobium-based solid acids has also been observed after DME synthesis (260 °C, 40 bar, H2:CO = 2 v/v and TOS = 120 h).270
2.4. Process comparison
The direct production of DME from synthesis gas can be effectively carried out by use of bifunctional catalysts. Combining both functionalities in a single catalyst comes with clear advantages, in particular a higher conversion of CO in a single pass, over the dual reactor process. In Fig. 8 we have gathered experimental data for both types of processes from published literature, showing the DME yield as a function of CO conversion. The slope of the line corresponds to the overall selectivity with which CO is converted to DME. Complete data sets can be found in the ESI.† Values for the dual reactor process were obtained by combining the maximum reported conversion and selectivity values from the methanol synthesis and methanol dehydration reactions, respectively. The resulting slope of the trendline (in grey) shows an overall selectivity of 88% for the dual reactor process with the methanol synthesis being detached from the methanol dehydration reaction in separate reactors. | ||
| Fig. 8 DME yield as a function of CO conversion for the dual reactor process, bifunctional process, and bifunctional process with in situ removal of water. Data points were obtained from recent reports in which DME is the principal product, the complete data set with the corresponding references can be found in ESI.† | ||
For the bifunctional process, most catalysts follow a similar trend (in green) independently of the catalyst synthesis method, with a selectivity of 62%. This is lower than the dual reactor process due to the production of CO2 from the water-gas-shift reaction at the expense of DME yield. Still, the bifunctional process allows to reach much higher CO conversions hence also higher DME yields in a single one-pass conversion compared to the dual reactor process. Bifunctional systems have catalysts consisting of physical mixtures or core–shell catalysts. This uniform trend also indicates that the distance between functionalities does not have a strong effect on the selectivity to DME. This might be explained by fast diffusion of chemical species in this process (i.e., methanol and DME) in contrast to species in other bifunctional processes (e.g., long-chain paraffins or aromatic compounds). This in agreement with the recent work of Li et al.295 A physical mixture of methanol synthesis and dehydration catalysts displays an effective system for high DME yields. However, as discussed in the previous section, a very fine physical mixture might negatively affect the stability of the catalyst.289
Within the different catalysts' configurations for bifunctional process, the in situ water removal strategy248–250 follows a different trend (in red) and therefore has been plotted separately. Here the slope corresponds to a 98% DME selectivity, with also high CO conversion. By capturing the formed water, most of the water-gas-shift reaction is inhibited and thus CO is converted mainly to DME. This seemingly is the most attractive pathway for an efficient DME production. However, aspects such as cost, energy consumption and ease of operation might be significant disadvantages of this method.
Unfortunately, few studies report long time-on-stream results, which makes it difficult to assess the stability of the various catalysts configurations. At least 100 h-on-stream results would give a good indication of the stability of the catalysts. Sintering of the metal functionality in the methanol synthesis catalyst and ion migration within functionalities seem to be the main phenomena responsible for activity loss. Solid acids with mild acid strength are readily active and selective for DME synthesis. Their stability seems less problematic than that of the methanol synthesis catalyst, the copper and copper–zinc interphase in these catalysts are susceptible to crystallite growth in the presence of water.
3. Olefins
3.1. Recent developments
A process called OX–ZEO, developed by the groups of Prof. Bao and Prof. Wang, to convert synthesis gas to short olefins in the range of C2–C4 can be considered a breakthrough in bifunctional catalysis.192,296 The OX–ZEO catalysts consist of metal oxides (based on for instance zinc, zirconium and/or chromium oxides) and a zeolite. Synthesis gas is first converted over the CO activation catalyst (metal oxide) to reactive oxygenate intermediates such as methanol/dimethyl ether or ketene (Fig. 9-A).297–300 These intermediates are further converted to short olefins via C–C coupling over the acid sites of a zeolite with usually 8-membered ring pores,298,301–303 such as SAPO-34 or H-SSZ-13, which are well known in the methanol-to-olefins reaction for their high selectivity.138,304,305 | ||
| Fig. 9 A: General reaction scheme of the OX–ZEO process, whereas CO activation takes place on a metal oxide forming oxygenate intermediates followed by C–C coupling on a zeolite.298 B: Proposed mechanism of CO activation over a metal oxide catalyst forming ketene intermediates.306 C: Hydrocarbon product spectrum (CO2 free) when either feeding synthesis gas to an OX–ZEO catalyst (green bars), or ketene (red bars) or methanol (grey bars) to a MOR-zeolite with 8 membered ring pores and 12 membered ring pores being accessible. For the experiments with synthesis gas feed zinc–chromium oxide particles were attached to the MOR-zeolite for synthesis gas activation. Synthesis gas over ZnCrOx/MOR gave similar products as ketene fed over MOR-zeolite with high C2 selectivities.297 | ||
In general, the OX–ZEO process is operated at high temperatures (300–400 °C) and pressures (10–100 bar),192,195,307–311 achieving selectivities to C2–C4 olefins between 63% and 87% within the hydrocarbon products (excluding CO2)298,301 at 10–85% CO conversion.298,301,308,309,311 These very high selectivities are well beyond the maximum predicted for a single conversion process based on the Anderson–Schulz–Flory distribution of the C2–C4 fraction (sum of olefins and paraffins) of 58%,312 such as the Fischer–Tropsch synthesis. This is a clear example of how using a bifunctional catalyst can improve the selectivity towards a certain product by catalyst design, as is discussed in more detail in the following paragraphs. However, it was shown that with increasing CO conversion the selectivity towards short olefins decreases and it remains a challenge to combine high selectivity with high conversion/activity, albeit that progress in that direction has been made.308,309,311 The group of Prof. Bao demonstrated high CO conversion of 85%, while maintaining 83% selectivity to short olefins and reduced CO2 selectivity of 32%.311
The understanding of the underlaying mechanism in OX–ZEO catalysis is still incomplete. Oxygenate intermediates are key in this process, but there is still discussion about which species is the main intermediate diffusing from the metal oxide to the acid catalyst.313,314 The mechanism of the primary conversion of CO to these reactive oxygenate intermediate species on metal oxides was studied with in situ near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) and infrared (IR) spectroscopy.306 It was found that upon reduction and exposure to synthesis gas, 36% of the surface lattice oxygen was removed from a manganese oxide (MnO2) catalyst, resulting in a high density of oxygen vacancies near the surface. It was proposed that CO is dissociated on these vacancies, after which oxygen is transferred to another CO molecule, forming carbonate species from which CO2 is desorbed. The remaining carbon atom is hydrogenated to a CH2 species, followed by insertion of CO and desorption of ethenone (C2-ketene, Fig. 9-B). Ketenes are highly reactive molecules. The chain propagation (C–C coupling) in the zeolite is reported to follow a direct associative pathway; ketene adsorbs on the acid site of the zeolite and the CH2 group is transferred to an olefin in a consecutive step, leaving a CO molecule behind.315
However, ketene intermediates are thermodynamically unstable and therefore it might be argued that methanol and/or dimethyl ether are the actual reactive oxygenate intermediate.316,317 The group of Prof. He proposed methanol as the intermediate from quasi-CO2 hydrogenation using indium–zirconium oxide and SAPO-34 zeolite, based on DFT calculations.307 It was found that adsorbed CO formed an O–C–O species (quasi-CO2) on the catalyst surface with lattice oxygen, which was then hydrogenated forming the reactive intermediate. Furthermore, the pathway of side products formation such as methane and paraffins was investigated. The formation of methane was caused by hydrogenation of methanol (or surface O–C–O species) on the metal oxide catalysts. The C2+ paraffins were formed after methanol traveled from the metal oxide catalyst to the zeolite forming olefins, which were then hydrogenated to paraffins in a consecutive step on the metal oxide.
The nature of the reactive oxygenate intermediate in the OX–ZEO process is hence topic of ongoing debate. Using a zinc–chromium oxide catalyst mixed with a SAPO-34 zeolite, the group of Prof. Bao identified ketene as intermediate with synchrotron vacuum ultra-violet photoionization mass spectrometry (SVUV-PIMS).192 To provide more evidence, ketene was flowed over modified mordenite zeolite with only either 8-membered ring (MR) pores or 12-MR pores accessible.297 The product spectrum was similar to the spectrum obtained from experiments converting synthesis gas using a bifunctional catalyst consisting of zinc–chromium oxide and modified MOR zeolite (Fig. 9-C).
On the other hand, methanol and dimethyl ether were identified as intermediates in the OX–ZEO process using zinc-doped zirconia catalysts mixed with SSZ-13 zeolites with various degrees of sodium ion exchange to control the density of Brønsted acid sites.301 The mixture of zinc-doped zirconia with fully sodium exchanged SSZ-13 in the nano scale (∼250 nm), hence without Brønsted acid sites present, showed a low CO conversion of 5% and selectivities to methanol and dimethyl ether of 65%. The CO conversion and selectivity to C2–C4 olefins increased with increasing density of Brønsted acid sites, because the intermediates could be removed from the equilibrium and converted to short olefins. The influence of strength and concentration of acid sites of a ZnAlOx/CHA OX–ZEO catalyst on the conversion of synthesis gas to olefins was investigated.318 With increasing Si/Al ratio of the CHA zeolite from Si/Al = 20 to Si/Al = 308 the selectivity to paraffins decreased (from 32% to 6%, CO2 free), while olefin selectivity increased (from 59% to 85%, CO2 free). This was attributed to the low acid site density in combination with decreased acid site strength by the addition of boron during zeolite synthesis. Additionally, it was found that a high density and strength of acid sites in a ZnCrOx/SAPO-35 zeolite accelerated the catalyst deactivation by enhanced coke formation and can cause increased paraffin selectivity in large zeolite crystals.319–321
Alternatively to the OX–ZEO process, hydrocarbon intermediates can be used to convert synthesis gas to short olefins by combining an iron (carbide) based Fischer–Tropsch core catalyst with a SAPO-34 zeolite shell.322 Operating at temperatures of 325 °C, the iron FTS catalyst formed typical heavy hydrocarbon products, that were cracked on the acid sites of the SAPO-34 zeolite forming C2–C4 olefins with 53% selectivity within the hydrocarbons at 55% CO conversion. Remarkably, the CO2 selectivity was only 17%, which allows this approach to compete with the OX–ZEO process, although the olefin fraction in the hydrocarbon products is lower. Similar trends have been observed using a silicalite-1 encapsuled iron-based catalyst.323,324 Additionally, an iron-based FTO catalyst capsuled with an H-ZSM-5 zeolite or a hydrophobic SiO2 shell showed reduced CO2 selectivity (8.5–28%) and slightly increased C2–C4 olefins selectivity (41%–49%) compared to the FTO catalyst alone (30–39% CO2 and 25–38% olefins).325,326 A silica-coating of a manganese promoted cobalt carbide nano-prism FTO catalyst showed increased olefins selectivity (from 40% to 59%) and reduced CO2 selectivity (from 45% to 15%) compared to the uncoated catalyst.327 It was concluded that the silica-coating reduced the adsorption of water on the catalyst and promoted the diffusion of water away from the catalysts' active sites, thereby, lowering the actual concentration near the active FTO sites.327,328
In the Fischer–Tropsch to olefins process, CO activation and C–C–Coupling take place on the same catalyst component, while they are spatially separated in the OX–ZEO process (Fig. 9-C). The OX–ZEO process is operated at rather high temperatures, which shifts the equilibrium between synthesis gas and the reactive intermediates far to the side of synthesis gas (see section 3.2). This can partially be counteracted by operating at elevated pressures. However, it is essential to have the two functions for CO activation and C–C–Coupling in optimal proximity to effectively achieve removal of the intermediates, and hence high conversion.
The group of Prof. Wang investigated the influence of the distance between the two functions on the conversion and selectivity in the OX–ZEO process, by combining a zirconium–zinc binary oxide catalyst with a SAPO-34 zeolite in different mixing modes.298 Packing in stacked bed mode with the zeolite downstream of the metal oxide gave very low conversions. A physical mixture of catalyst granules with grain size of 250–600 μm (resulting distance between CO activation and C–C coupling catalyst in the range of ∼500 μm) led to 7% CO conversion with 75% selectivity to C2–C4 olefins. To achieve even closer proximity, the two individual catalysts were ground in a mortar, resulting in ∼500 nm distance between the two functions. The distance of the two functions could be decreased even further to ∼100 nm by a ball-milling procedure for 24 h. Bifunctional catalysts prepared with closer proximity using mortar-mixing and ball-milling techniques achieved CO conversions of 10–11%, with selectivities to C2–C4 olefins slightly decreasing to 63–70%. The decrease in selectivity was assigned to secondary hydrogenation of olefins to paraffins on the metal oxide catalyst. A ZnCrOx/SAPO-34 catalyst applied in the OX–ZEO to olefins reaction showed the best performance of 60% CO conversion and 76% C2–C4 olefin selectivity at medium proximity of 200–300 μm between the metal oxide and zeolite function.329 The authors concluded that with greater distance between the functions the removal of reactive intermediates suffered from mass transfer limitation. However, with increasing proximity zinc species migrated from the metal oxide to the zeolite, decreasing the activity of both functions. A MnOx/SAPO-34 catalyst did not show decreasing activity with increasing proximity.
The catalysts used in the OX–ZEO process are also active for the water-gas-shift (WGS) reaction and usually show CO2 selectivities between 32–45%, which is close to the equilibrium concentration of 45–49%, depending on the reaction conditions.192,301,306,330 This makes it possible to also convert hydrogen-lean synthesis gas obtained from coal or biomass, because one molecule of hydrogen is formed for every molecule CO that is converted. This comes at the expense of carbon atom economy because CO2 is being formed from CO. However, a high hydrogen partial pressure can also facilitate the secondary hydrogenation of olefins products over In2O3–ZrO2/SAPO-34 OX–ZEO catalysts.331 Hence, feeding hydrogen lean synthesis gas to the OX–ZEO process can circumvent unwanted side reactions and the WGS reaction can provide additional hydrogen further onwards in the catalyst bed.
The OX–ZEO catalysts can also be applied for the synthesis of short olefins from CO2 hydrogenation.83,321,331–337 Studies on a Mn2O3–ZnO/SAPO-34 catalyst showed that the alkaline character and CO2 activation over oxygen vacancies of the metal oxide function favor a high activity in CO2 conversion.338 By ball-milling the components together, hence decreasing the average distance between the two catalyst components, the performance of the OX–ZEO catalyst was further enhanced. The conversion of CO2 and selectivity to C2–C4 olefins increased from 20% to 30% and 49% to 80% (CO free), respectively, compared to a dual bed configuration. Additionally, the CO selectivity resulting from reverse WGS decreased from 90% to 55% upon ball-milling the catalyst.
3.2. Benefits
A clear advantage of using bifunctional catalysis to convert synthesis gas to olefins is that CO conversions can be higher than for two separate reactors for which the maximum conversion is dictated by the equilibrium between the synthesis gas and oxygenates (Fig. 10). The thermodynamic limit for CO hydrogenation to methanol is only 0.07% (at 10 bar pressure) and 6.6% (at 100 bar pressure) at 390 °C, but recently 7.5-fold to 51-fold higher conversions were reported, reaching up to 59% conversion.308 These CO conversion levels are similar to those for methanol synthesis commonly operated at 260 °C.271,308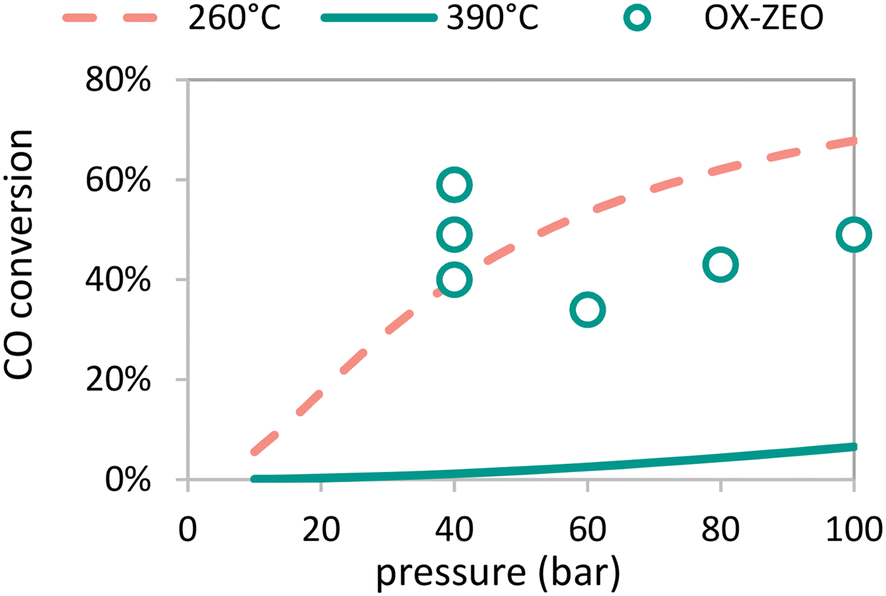 | ||
| Fig. 10 Equilibrium CO conversion for methanol synthesis from synthesis gas (H2:CO = 2 mol mol−1) as function of reaction pressure at 260 °C (dotted red line) and 390 °C (solid green line) and reported CO conversion as a function of reaction pressure of the OX–ZEO process operated at 390 °C (open green circles) exceeding the equilibrium CO conversion of the single pass methanol synthesis at 400 °C.308 | ||
Another clear advantage of operating the process with two different catalytic functions, whether in a single or double reactor, is the possibility to steer the selectivity. The OX–ZEO process showed a selectivity towards short olefins of up to 87% (ref. 301) caused by the highly selective MTO catalysts used for the C–C coupling step. In comparison, methanol or dimethyl ether feedstock in the DMTO (dimethyl ether or methanol to olefins) process also give 84–87% olefin selectivity.339,340 For Fischer Tropsch Synthesis only, the maximum selectivity according to the ASF distribution is limited to 58% C2–C4 olefins + paraffins312), while the FTO process, being able to break the ASF distribution, reaches 61% C2–C4 olefins.113
Furthermore, the product spectrum can be tuned by the choice or modifications of the C–C coupling catalyst.341,342 The zeolite pore size is critical. SAPO-34 or SSZ-13 zeolites as C–C coupling catalyst typically give a product spectrum with 13–20% C2, 40–59% C3 and 14–23% C4 products,192,309 which is similar to the product distribution in MTO.139,149 A modified MOR zeolite with selectively deactivated 12-MR pores and only 8-MR pores accessible as C–C coupling catalyst next to a ZnCrOx catalyst for CO activation showed a remarkably high selectivity to ethene of 73%.297
A last potential advantage is related to the stability of the catalyst. For the MTO process, the SAPO-34 or SSZ-13 catalyst lifetime is only a few hours due to severe coke formation.138 The OX–ZEO catalyst was reported to display much longer lifetimes, beyond 500 h.308 However, no clear explanation for the high stability has been offered so far. A possibility is that the productivity of OX–ZEO is lower (∼0.3 kgolefins kgcatalyst−1 h−1 (ref. 308)) compared to the MTO process (∼2 kgolefins kgcatalyst−1 h−1 for micro- and pilot-scale and ∼5 kgolefins kgcatalyst−1 h−1 for demo- and commercial scale304) which for OX–ZEO will give rise to much longer catalyst life times expressed in hours. Cheng et al. observed a high stability of their zinc-doped zirconia catalyst (CO activation) and H-ZSM-5 zeolite (C–C coupling and aromatization) over the course of 1000 h in the conversion of synthesis gas to aromatics.343 They postulated that the high silicon to aluminum ratio of the zeolite played a crucial role in its stability. Furthermore, the low partial pressure of methanol/dimethyl ether or ketene suppressed the excessive alkylation of aromatic species in the dual cycle mechanism that would eventually result in the formation of polycyclic aromatics hydrocarbons and hence catalyst deactivation.344 This low partial pressure of intermediates also allows to operate the OX–ZEO process with a zeolite that has a high silicon to aluminum ratio and hence low acid site density, which is beneficial for the zeolite stability.138 The reaction conditions of the OX–ZEO process with high temperatures and high hydrogen concentrations compared to the MTO process limit the formation of soft coke,344 which also mitigates catalyst deactivation.
3.3. Challenges
A first challenge is to realize an optimum hydrogenation activity of the metal oxide in the OX–ZEO catalyst, which is crucial for the selectivity.307,345 Metal oxide catalysts with high hydrogenation activity, such as zinc oxide, showed primary overhydrogenation of surface carbon species forming methane (Fig. 11-A) as well as secondary hydrogenation of re-adsorbed olefins (that were formed on the C–C coupling catalyst), which is detrimental to the olefin selectivity.195 A Mg-HZSM-5/Al2O3 catalyst applied in the DMTO reaction in the presence of synthesis gas did not show increased hydrogenation of olefins.346 Hence the metal oxides' hydrogenation activity of the OX–ZEO catalyst needs to be limited, but still sufficiently high for surface CO* species to undergo moderate hydrogenation to form the reactive oxygenate intermediates.309 The hydrogenation performance of the metal oxide catalysts can be influenced by a variety of parameters, such as the nature of the oxide,195 particle size,330,333 nature of dopants,347 promoters309 and proximity to the zeolite.348 Balancing the hydrogenation activity of the metal oxide catalyst without compromising the overall catalytic performance of the OX–ZEO process remains one of the main challenges for future work. Currently, zinc–chromium or zinc–zirconium binary oxide seem to be the most promising candidates as CO activation catalysts, whereas SAPO-34, SSZ-13, and ion exchanged AlPO-18 are promising solid acids.301,308,311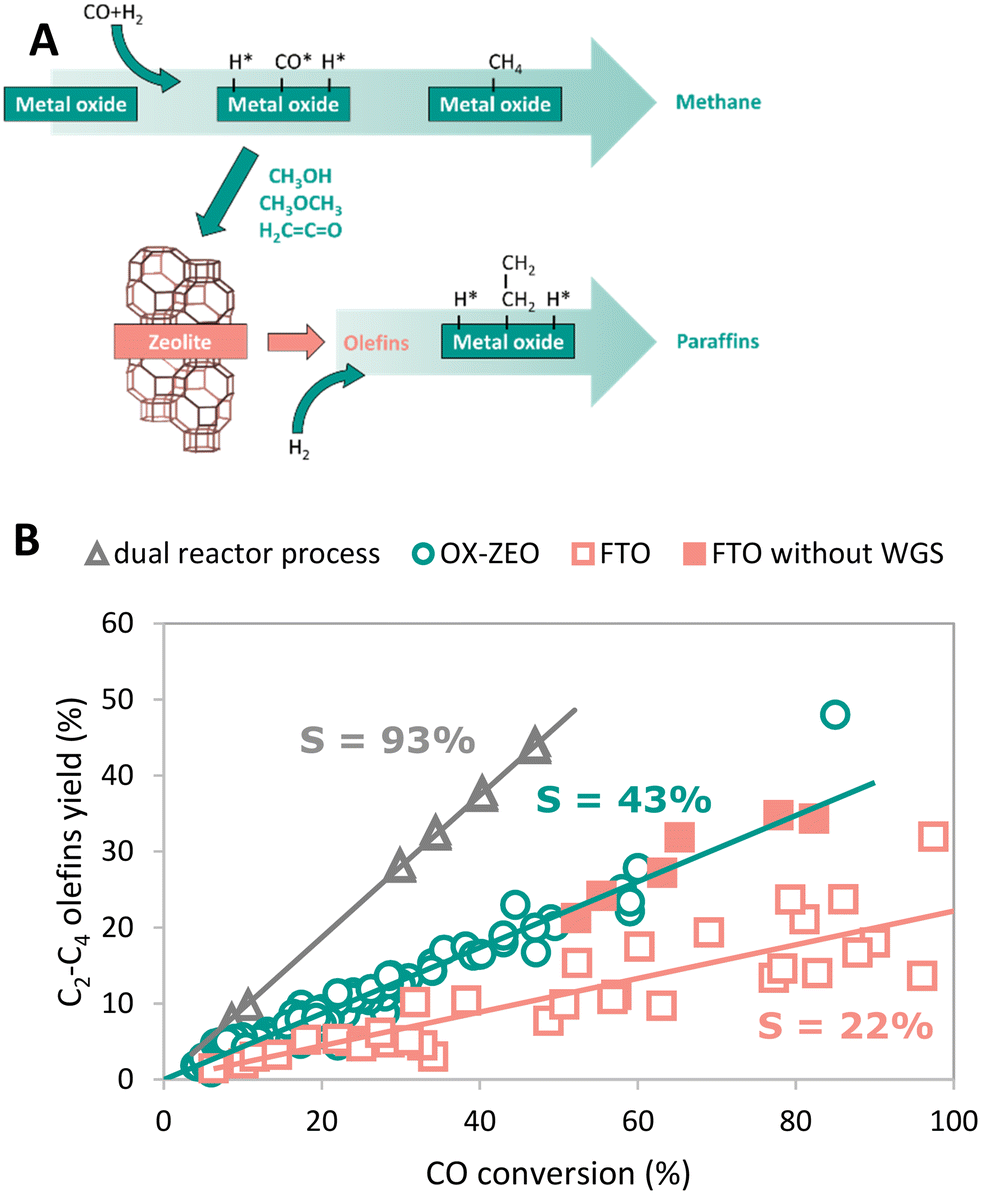 | ||
| Fig. 11 A: Proposed pathway for primary overhydrogenation forming methane on the metal oxide catalyst and secondary hydrogenation forming paraffins from olefins by re-adsorption of olefins on the metal oxide catalyst.195 B: Experimentally reported yields of C2–C4 olefins as function of CO conversion for a dual reactor process (single pass conversion over methanol catalyst and consecutive MTO process, gray triangles), OX–ZEO process (green circles) and FTO process (red squares, open symbols: fully WGS active, filled symbols: reduced WGS activity). The slopes of the fitted lines correspond to the overall selectivity to olefins. A detailed analysis of the catalytic data can be found in the ESI.† | ||
3.4. Process comparison
To generate a more global picture regarding attainable product selectivities, we compared the best reported catalytic performances for three different approaches to convert synthesis gas to short olefins. Fig. 11-B shows the yield to C2–C4 olefins for the FTO process, OX–ZEO and a dual reactor process as a function of the CO conversion. For the dual reactor process, we calculated the overall yields to C2–C4 olefins that we obtained from the combination of reported data for a methanol synthesis reactor and a reactor for the methanol-to-olefins (MTO) process. The dual reactor approach is based on two consecutive reactors, separating the CO activation catalyst (methanol synthesis) from the C–C coupling catalyst (MTO). The slopes of the yields plotted against the CO conversion per pass (once-through) correspond to the overall selectivity of the process and account for the formation of CO2.The FTO process shows a selectivity to C2–C4 olefins of ∼22%. The fraction of short olefins in the hydrocarbon products of the FTO process is reported up to 61%.349 However, the product stream is not only hydrocarbons, but also contains CO2 formed from CO via the WGS reaction, giving 30–50% CO2 in the product stream.119,350 This reduces the overall selectivity to short olefins to 30–42%. A successful strategy in the case of hydrogen-rich synthesis gas might be to mitigate the WGS reaction, as illustrated by the full red square in Fig. 11-B, which shows the yield of short olefins for a bifunctional catalyst consisting of an iron (carbide) based core and a SAPO-34 or silica shell, respectively.322,326 The increased selectivity is caused by the reduced WGS activity of this catalyst system and can compete with that in the OX–ZEO process. A selectivity to short olefins of ∼43% can be observed for the OX–ZEO process, caused by a high contribution of short olefins to the hydrocarbon products as high as 87% (ref. 298 and 301) in combination with high CO2 selectivities of 40–45% due to the WGS activity of the OX–ZEO catalysts.192,307
The approach with two separate reactors (methanol synthesis and MTO) gives an overall selectivity of ∼93%. This is caused by the high selectivity of the methanol synthesis (between 97% and 99.8%351,352) and the MTO process with 94–96% selectivity to short olefins.138 Furthermore, in this configuration the total selectivity towards CO2 from WGS (reaction of water and CO) is neglectable, because the water is mainly formed during the MTO process in the second reactor without CO being present.
Currently, a dual reactor approach to convert synthesis gas to olefins using methanol synthesis and an MTO process shows the most promising overall selectivity and carbon atom economy due to the absence of the WGS reactions and highly selective reactions. A challenge for the OX–ZEO process as well as for the FTO process is the suppression of the WGS reaction if hydrogen-rich synthesis gas is used to achieve higher yields of the desired products. This has been partially achieved with a zinc–cerium–zirconium oxide catalyst combined with a SAPO-34 zeolite.353 The selectivity towards carbon dioxide was reduced to 6% at a low CO conversion of 7%. However, the CO2 selectivity increased to 26% at 12% CO conversion by increasing reaction temperature. In terms of activity the hydrogenation strength of the metal oxide needs to be finely balanced to achieve higher total activity without intensifying secondary hydrogenation of products. For this, several strategies are already available, such as choice of material,354 dopants and promoters,355 or intimacy between the metal oxide and zeolite.356 Concerning catalysts stability important progress has been achieved with stable times on stream of 500 h and above.308,343 In brief, the two-step approach cannot compete with the two-reactor approach in terms of selectivity and carbon yield but does have clear advantages, such as exceeding conversion levels of the methanol synthesis catalysts dictated by the thermodynamic limits. Additionally, as it is a relatively new method, further development can be expected.
4. Aromatics
4.1. Recent developments
In the following paragraphs we introduce two different approaches to convert synthesis gas to aromatics (monoaromatics with a single aromatic ring) using bifunctional catalysis: (I) the combination of an FT catalyst with a zeolite and (II) the OX–ZEO process for aromatics (Fig. 12). One of the main differences between these two approaches is the location of the C–C coupling. In the combination of the FT catalyst and the zeolite (FT + zeolite) the C–C coupling takes place on the CO activation catalyst (the FT catalyst),357 while the zeolite is responsible for further oligomerization, cyclization and aromatization. In the OX–ZEO process the CO activation catalyst (metal oxide) forms carbon monomers,343,358 while the C–C coupling of these carbon monomers occurs on the zeolite.347 In both approaches, H-ZSM-5 zeolites with 10 membered ring pores are typically used, due to their excellent shape selectivity for aromatics.359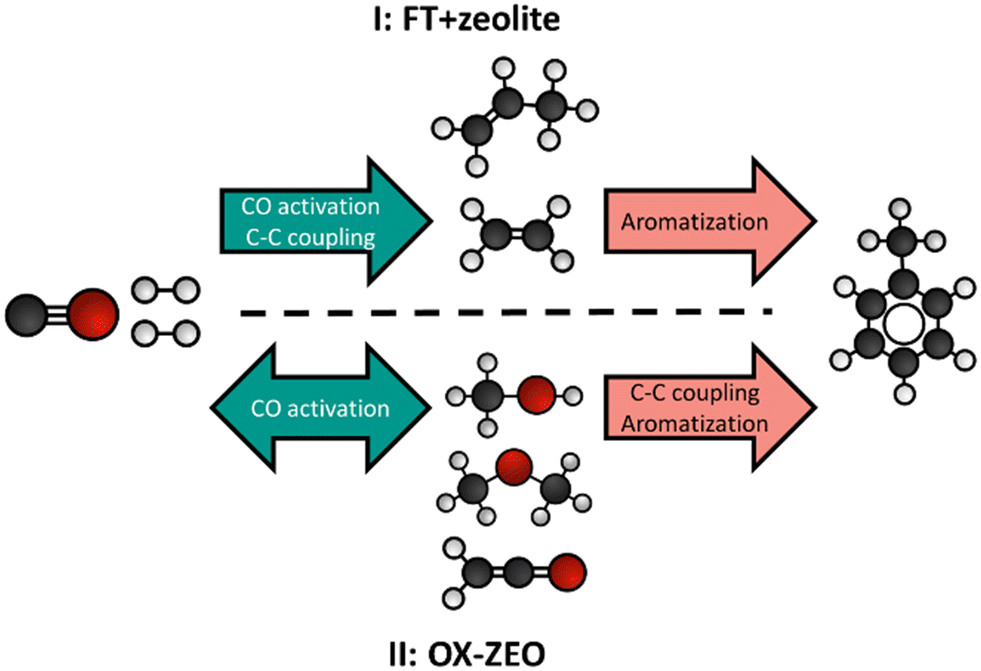 | ||
| Fig. 12 General reaction scheme for the conversion of synthesis gas to aromatics using a combination of an FT catalyst with a zeolite (I) or the OX–ZEO process (II). | ||
For the aromatization of olefins over H-ZSM-5 higher temperatures (350–480 °C) and lower pressures (1 bar–10 bar) are preferred.358,367–369 However, operating an FT catalyst under these conditions leads to rapid deactivation due to coke formation.113,121 Furthermore, more methane and less C2+ are formed.370 For example, a bifunctional catalyst consisting of a cobalt–manganese–aluminum oxide catalyst combined with an H-ZSM-5 zeolite operated at 270 °C and 10 bar led to 4% methane formation and 2–5% aromatics, while at 320 °C a 18–21% methane selectivity and 29–38% aromatics were formed. Alternatively, a tandem reactor design with the same cobalt–manganese–aluminum oxide catalyst upstream at 270 °C and the zeolite downstream at 320 °C allowed to maintain a low methane selectivity of 3% while increasing the selectivity to aromatics to 52%,357 albeit at the expense of CO conversion (32–40% at 270 °C compared to 65–72% at 320 °C).
The aromatization of olefins often follows a pathway that involves hydrogen transfer, forming three molecules of paraffins for every aromatic molecule formed (Fig. 13-A).358,371,372 However, a high operating temperature and/or low partial pressure of olefins shifts the aromatization towards dehydrogenation instead of hydrogen transfer.372 For example, operating a bifunctional catalyst consisting of a Fischer–Tropsch to olefins (FTO) catalyst and an H-ZSM-5 zeolite at 400 °C, 1 bar and low CO conversion (2%) facilitates aromatization of olefins via a dehydrogenation pathway with 17% aromatics selectivity and only 4% paraffins.113 Alternatively, a bifunctional catalyst consisting of a pyrolyzed iron containing metal–organic-framework (MOF) promoted with sodium and a modified H-ZSM-5 zeolite has been developed for the conversion of CO2 and hydrogen to aromatics.373 Catalytic tests performed at 320 °C, 30 bar and CO2 containing synthesis gas (H2:CO2 = 2.95 v/v) as feedstock and in granule stacking mode yielded 9.6% C2–C4 paraffins and 50.2% aromatics. The authors concluded that the olefin intermediates are converted into aromatics via dehydrogenative aromatization and that adsorbed CO2 on the pyrolyzed iron containing MOF acted as acceptor for hydrogen species formed during dehydrogenation.373
 | ||
| Fig. 13 A: Aromatization of higher olefins via hydrogen transfer134 (top), forming paraffins (C0) from olefins (C=) and via dehydrogenation372 (bottom) releasing hydrogen as by-product. B: Overview of different bifunctional catalyst configurations showing distances between the two catalytic functions varying from the meter scale in dual reactor processes to the nano mater scale for closest proximity. | ||
Both structure of the zeolite and acid site density have a major influence on the resulting product spectrum. For hierarchical ZSM-5 zeolites impregnated with iron, a correlation between the concentration of acid sites and the selectivity to aromatics was reported.374 Bifunctional catalysts with medium concentrations of acid sites (746 μmol g−1) gave a higher selectivity to aromatics (15%) then with a lower or higher acid site concentration. Additionally, increasing the fraction of mesopore volume within the total pore volume from 45% to 68% increased the selectivity to aromatics from 15% to 23%.374
A composite catalyst consisting of a copper-promoted bulk iron catalyst and an H-ZSM-5 zeolite was applied in the synthesis of aromatics from CO2 containing synthesis gas with H2:CO2 = 3 (v/v).375 At 320 °C, 30 bar, and the individual catalyst granules being mixed in the catalyst bed showed 57% conversion of CO2 and 57% selectivity to aromatics with only 3.5% CO selectivity resulting from the rWGS reaction. Increasing the content of copper promoter in the bulk iron catalyst from 6.25 wt% to 50 wt% caused the methane and short paraffin selectivity to increase (11% to 57% and 8% to 30%, respectively) and the aromatics selectivity to decrease (57% to 10%). This behavior indicates that the increasing copper content is responsible for secondary hydrogenation and methane formation of intermediate species. A similar behavior was also observed when promoting iron-based FT catalysts with copper in the conversion of CO and H2 to aromatics.376 With a copper content of 1.5%wt in the iron-based FT catalyst the methane selectivity was low (8%) and aromatics selectivity was high (42.5%). However, with lower (0.2% wt) or higher (5% wt) amounts of copper promoter the methane selectivity was higher (13% and 15.5%, respectively) and aromatics selectivity was lower (37% and 35% respectively).
The OX–ZEO process to convert synthesis gas to aromatics is usually operated at high temperatures (300–450 °C) and high pressures (20–60 bar),343,354,363,379 reaching CO conversions between 3% and 55% and selectivities to aromatics of 49–86% within the hydrocarbon products (29–67% selectivity to aromatics if the CO2 formation is taken into account),363,379,383 which is higher than FT + zeolite. The high temperature of the OX–ZEO process leads to a low equilibrium concentration of intermediates, which steers the aromatization towards a pathway that involves dehydrogenation rather than hydrogen transfer.372 Therefore, the formation of paraffins is limited and a high selectivity to aromatics can be achieved. However, the competition between aromatization via hydrogen transfer and via dehydrogenation strongly depends on the composition of the synthesis gas.363 An H2:CO ratio of 2 (v/v) showed a significantly higher C2–C4 paraffin fraction of 53.1% and low aromatic fraction of 35.2% in the hydrocarbons, compared to a hydrogen lean feed gas with H2:CO = 1 v/v, resulting in 20.1% C2–C4 paraffins and 56.3% aromatics in the hydrocarbon products.
The nature of the reactive intermediate of the OX–ZEO process to convert synthesis gas to aromatics is under debate. Using zinc chromium oxide, zinc manganese oxide, zinc–zirconium oxide, or zinc alumina catalysts for CO activation, a mixture of methanol and dimethyl ether was found as reactive intermediate.343,379,381,384,385 By feeding methanol and carbon monoxide over a zinc zirconium oxide catalyst mixed with an H-ZSM-5 zeolite it was found that the presence of CO showed a self-promoting effect in the conversion of methanol to aromatics, in which methanol was converted to short olefins on the zeolite. These olefins underwent aromatization via dehydrogenation, whereas the hydrogen was removed by carbon monoxide on the metal oxide catalyst resulting in the formation of methanol.343
Alternatively, ketene was proposed as reactive intermediate using zinc manganese oxide or zinc chromium oxide as CO activation catalyst.378,381 The zinc chromium oxide catalyst mixed with a mesoporous SAPO-34 zeolite already has been studied in the OX–ZEO to olefins process, where ketene was identified with synchrotron vacuum ultra-violet photoionization mass spectrometry.192 A cerium zirconium oxide catalyst in combination with an H-ZSM-5 zeolite showed improved oxygen vacancies on the surface of the CO activation catalyst and assisted the formation of C2+ oxygenates and C6+ olefins. This suggested a reactive intermediate other than methanol in the conversion of synthesis gas to aromatics.363
To remove the reactive intermediates effectively and hence increase the synthesis gas conversion, close proximity between the CO activation catalyst and the C–C coupling catalyst is crucial.384 With increasing proximity going from powder mixed bifunctional catalysts with ∼100 nm distance between the different catalytic functions to nanocomposites, the CO conversion and selectivity to aromatics increases, whereas the methane selectivity decreases.343,363 This indicates that a larger distance between the CO activation catalyst and the zeolite gives rise to secondary hydrogenation of reactive intermediates on the metal oxide catalyst, forming methane and C2+ paraffins. However, a reduced zeolite crystallite size from 1.5 μm to 200 nm in a physical mixture with a ZnCrOx catalyst showed reduced selectivity to aromatics and a 1.7-fold increase of side products386 which was assigned to enhanced secondary hydrogenation. Additionally, the zeolites owned different morphologies.
Generally, the formation of ortho- and meta-xylene and heavier aromatics takes place at the acid sites on the external surface of the zeolite by isomerization and alkylation.387 The kinetic diameter of para-xylene is smaller than those of ortho- and meta-xylene and only para-xylene can be formed inside the micropores of H-ZSM-5.379,380 Surface modification of the zeolites forming aromatics can have a significant influence on the product distribution within the aromatics fraction (Table 1). These modifications can be achieved by passivation of the external surface of the zeolites or by the growth of a shell that is free of acid sites (for example a silicalite-1 shell around H-ZSM-5 crystals), and generally lead to higher fractions of p-xylene in the aromatics.
| Reaction/catalyst | Zeolite modification | Aromatics selectivity | Reference | |
|---|---|---|---|---|
| Without modification | With modification | |||
| MTA | Chemical liquid deposition | 24% p-xylene in xylenes | 90% p-xylene in xylenes | 388 |
| H-ZSM-5 | ||||
| Disproportionation of toluene | Silicalite-1 shell | 80% p-xylene in xylenes | 389 | |
| H-ZSM-5 | ||||
| Fe + Z | Zn-promotion and silicalite-1 shell | 20–25% p-xylene in xylenes | 65–70% p-xylene in xylenes | 390 |
| Mn-promoted FT catalyst + H-ZSM-5 | ||||
| OX–ZEO | Zn-promotion and silicalite-1 shell | 77% p-xylene in xylenes | 379 | |
| CrZnOx + H-ZSM-5 | ||||
| OX–ZEO | USY zeolite downstream of OX–ZEO catalyst | 63% BTX in aromatics | 88% BTX in aromatics | 391 |
| SiO2-modified MnCrOx + ZSM-5 | ||||
OX–ZEO catalysts often show high CO2 selectivities due to their strong WGS activity. However, the group of Professor Tsubaki developed a catalyst that allows to convert CO2 containing synthesis gas (CO:CO2:H2 = 6.1:1:12.8 v/v/v) into aromatics.392 The rate of CO2 formation and consumption was kept in balance by adjusting the feed composition, hence this reaction was operated net-CO2 neutral. The OX–ZEO catalyst consisting of Cr2O3 as metal oxide and H-ZSM-5, or metal ion exchanged ZSM-5 zeolite with a silica coating showed Cox conversion between 17.4% and 24.6% (CO conversion: 18.3–28.4% and CO2 conversion: 1.5–13.1%, respectively) and aromatics selectivity between 65% and 76%. It is worth mentioning that Cr2O3 combined with a gallium exchanged and silica coated ZSM-5 zeolite performed with the highest Cox conversion (24.6% Cox, 28.4% CO, 1.5% CO2) and simultaneously with the highest selectivity to aromatics (76.4%) of the tested bifunctional catalysts. Additionally, the silica coating of the ZSM-5 zeolite reduced the alkylation of aromatics on the external acid sites of the zeolite, hence increasing the C6–C8 aromatics selectivity to 55%. p-Xylene was formed with 38.6% selectivity.
Analog to the OX–ZEO to olefins reaction, the production of aromatics can also be achieved by CO2 hydrogenation and reach CO2 conversion levels between 9% and 41% and selectivities to aromatics of up to 76%.393 Using a chromium-doped ZrO2 aerogel catalyst combined with an H-ZSM-5@SiO2 zeolite CO2 conversion of 14% was achieved with 77% aromatics selectivity, of which 2/3 were light aromatics (C6–C8).394 The methane selectivity was low with only 1.1%.
4.2. Benefits
Interestingly, combining a sodium and sulfur promoted FTO catalyst and an H-ZSM-5 zeolite in a physical mixture caused a 1.8-fold activity enhancement of the FTO catalyst compared to the FTO catalyst without zeolite or in stacked bed mode, where the two functions are spatially separated.113 Although not fully understood, Mößbauer spectroscopy measurements revealed an enhanced formation of iron carbide, which is the active phase in the FTO reaction for the physical mixture of FTO catalyst and zeolite. Here, 83% of the iron was transformed into an iron carbide phase after a 1 h carburization step at 290 °C and atmospheric pressure, whereas the FTO catalyst without zeolite only showed 57% carbide formation. Hence unexpected benefits can arise from the close coupling of the two functions.396
 | ||
| Fig. 14 Equilibrium CO conversion for methanol synthesis from synthesis gas (H2:CO = 2 mol mol−1) as function of reaction pressure for 260 °C (dotted red line) and 400 °C (solid green line) and reported CO conversion as function of pressure for the OX–ZEO process operated at 400 °C (open green circles) exceeding the equilibrium CO conversion of the single pass methanol synthesis at 400 °C.354,377,379 | ||
Furthermore, the OX–ZEO process has a high fraction of aromatics in the hydrocarbon products compared to FT + zeolite. The formation of aromatics from methanol follows a dual cycle mechanism in which higher olefins are formed in the alkene cycle that undergo aromatization to enter the aromatic cycle as shown in Fig. 3.134 The aromatization of higher olefins is based on the following steps: formation of dienes, cyclization to cyclic olefins, formation of cyclic dienes and formation of aromatics (Fig. 13-A). Commonly reported, the formation of dienes, formation of cyclic dienes and the aromatization is based on hydrogen transfer, in which also paraffins are formed at the expense of olefins. However, operating the OX–ZEO process at high temperatures and low reactive intermediate concentrations facilitates the aromatization via dehydrogenation, forming molecular hydrogen instead of paraffins.372 Therefore, the OX–ZEO shows higher selectivity than FT + zeolite, which operates at lower temperatures and higher concentrations of reactive intermediates. In FT + zeolite, the aromatization is more likely to follow hydrogen transfer and form undesirable paraffins.357,364 Furthermore, the product spectrum of the CO activation catalyst (FT catalyst) depends on the ASF distribution and the maximum selectivity of suitable intermediate products to be converted into aromatics is limited.397
The OX–ZEO process shows stabilities that exceed the stability of the methanol-to-aromatics process. The group of Prof. Wang presented an OX–ZEO catalyst system consisting of a zinc zirconium oxide catalyst and H-ZSM-5 zeolite, which showed stable performance with 80% selectivity to aromatics and CO conversion of 20% over the course of 1000 h at 400 °C and 30 bar.343 In the methanol-to-aromatics reaction the activity in methanol conversion drops significantly after 5–200 h, when operated at the same reaction temperature.155,398,399 Due to the low partial pressure of reactive intermediates, zeolites with high silicon-aluminum ratios and hence low density of strong acid sites can be used in the aromatization reaction, which is beneficial for the zeolite stability.278,343 Additionally, the low partial pressure of intermediates and the high reaction temperature contribute to the catalyst stability albeit at the expense of activity of the metal oxide catalyst.372 Another explanation for the stability expressed in hours could be the low productivity of the OX–ZEO catalysts (∼0.04 kgaromatics kgcatalyst−1 h−1) compared to the MTA process (∼0.4–1.4 kgaromatics kgcatalyst−1 h−1), which leads to a lower rate of coke formation.155,343,398
4.3. Challenges
The addition of sodium and sulfur promoters to a supported iron carbide based Fischer–Tropsch to olefins (FTO) catalyst decreased the methane production from synthesis gas and gave a high selectivity towards short olefins.403,404 Combining this promoted FTO catalyst with an H-ZSM-5 zeolite enabled the direct synthesis of aromatics from synthesis gas.113 However, in close proximity of the FTO catalyst to the zeolite, higher methane selectivities (15% in stacked bed mode and 30–35% in close proximity) and lower aromatics selectivities (12% in stacked bed mode and 5% in close proximity) were observed, probably due to migration of alkaline promoters from the FTO catalyst to the zeolite, which led to neutralization of the acid sites on the zeolite.15,405 The migration of promoters and the accompanying effects on the catalytic performance could be circumvented by placing the zeolite downstream of the FTO catalyst in a stacked bed mode. Alternatively, by using carbon nanofibers as support material, the migration of promoters was suppressed, despite close proximity of the two catalytic functions.15 This shows that controlling the mobility of mobile species, such as promoters or dopants, is crucial for the design of bifunctional catalysts for this process.
:Zr = 1:1000 mol mol−1 resulted in low CO conversion (14%) in combination with high selectivity to aromatics (76%), whereas the CO conversion increased (43%) and the selectivity to aromatics decreased (7%) with increasing zinc content (Zn:Zr = 1:5 mol mol−1). Furthermore, the selectivity to short paraffins increased from 18% to 52% with the same change in zinc fraction, indicating that a high zinc content enables secondary hydrogenation of reactive intermediates. Hence, a high hydrogenation activity is beneficial for the CO conversion but has a negative effect on the selectivity towards aromatics.
This hypothesis was supported by experiments performed with a cerium zirconium oxide catalyst mixed with an H-ZSM-5 zeolite, for which the selectivity towards aromatics and olefins decreased with increasing hydrogen content in the synthesis gas.363 Operating at 450 °C and 20 bar, the OX–ZEO catalyst showed 13.3% olefins in the range of C2 to C4 and 56.3% aromatics selectivity at a hydrogen to carbon monoxide ratio of 1 (v/v). Increasing the hydrogen content of the synthesis gas to H2:CO = 2 (v/v) led to decreased selectivity to short olefins of 3.4% and aromatics of 35.2%. This shows that for the OX–ZEO catalyst the hydrogenation activity needs to be carefully optimized.
4.4. Process comparison
In this section we focus on the overall selectivity to aromatics. Fig. 15-A shows the best reported aromatic yields as function of the CO conversion for the OX–ZEO process and for FT + zeolite. To compare, we added the aromatic yields of a dual reactor process in Fig. 15-B, calculated from a combination of reported data for a methanol synthesis reactor351,352 and a reactor for the methanol-to-aromatics (MTA) process.155,398,402,406 The slopes of the yields plotted against the CO conversion correspond to the overall selectivity of the processes (taking the formation of CO2 as one of the alternative products into account). | ||
| Fig. 15 A: Reported yields of aromatic hydrocarbons as function of CO conversion for the OX–ZEO process (green circles) and combination of FT catalyst and zeolite (red squares). The filled green circles show the yield to aromatics as function of CO conversion of the OX–ZEO process with reduced WGS activity B: Calculated overall aromatic selectivity resulting from the combination of a methanol synthesis reactor and a reactor for methanol aromatization via hydrogen transfer (open gray triangles) and dehydrogenation (solid gray triangles) in a dual reactor process. The slopes of the fitted lines correspond to the overall selectivity of the process. A detailed analysis of the catalytic data can be found in the ESI.† | ||
The combination of an FT catalyst with a zeolite showed an average selectivity to aromatics of ∼26%. The results are distributed over a range of 9–41%. Li et al. found that in the conversion of synthesis gas to aromatics the intimacy within the bifunctional catalysts played a crucial role for the selectivity, which can explain these wide-spread selectivities.407 The overall selectivity is rather low which can be explained by limited selectivity to suitable olefinic intermediates in the first step. Furthermore, the temperature needed for the CO activation catalyst favors the aromatization to follow the hydrogen transfer pathway, forming three molecules of paraffins from olefins for every aromatic molecule being formed.372
The OX–ZEO process for aromatics showed a higher overall selectivity of 41% to aromatics, resulting from a high fraction of aromatics in the hydrocarbon products between 49% and 86% but also high selectivities to CO2 in the range of 17% to 49%.363,378 The high aromatics fraction can be explained by the low partial pressure of reactive intermediates and the high reaction temperature, shifting the aromatization towards dehydrogenation.372 The OX–ZEO process shows a selectivity to paraffinic hydrocarbon side products as low as 6%.343,378 The high CO2 selectivity is caused by the WGS activity of the OX–ZEO catalysts and is in the same range as for the FT + zeolite systems, showing 16–49% CO2 selectivity.364 However, tailoring an OX–ZEO catalyst to reduced WGS activity and adapted feed compositions showed that selectivities to aromatics of ∼70% are possible (Fig. 15-A).392,408
The calculated overall aromatic selectivity resulting from the combination of a methanol synthesis reactor and a reactor for the MTA process shows a selectivity to aromatics of ∼41% (Fig. 15-B, open triangles), which is higher than FT + zeolite and in the same range as the OX–ZEO process. We based these calculations on reported catalytic data of single pass conversions of synthesis gas over methanol synthesis catalysts with CO conversion ranging from 9% to 47% and methanol selectivities of 97–99.8%.351,352 Consecutively, methanol is converted into aromatics via suitable zeolite catalysts in a separate MTA process. A moderate selectivity to aromatics from methanol was reported between 33% and 50%, which is in good agreement with the dual cycle mechanism in combination with hydrogen transfer, in which a substantial amount of paraffins of usually ∼40% is formed.155,398,402,406 This decreases the overall selectivity to aromatics, despite the absence of WGS activity and therefore no significant formation of CO2 in this approach. However, a zinc doped H-ZSM-5 zeolite operated at high temperature of 475 °C showed a selectivity to aromatics of 96% in the conversion of methanol, due to aromatization via dehydrogenation.156 The theoretically calculated maximum overall selectivity to aromatics from the combination of methanol synthesis and aromatization via only dehydrogenation in a dual reactor process was ∼95% (Fig. 15-B, solid triangles).
The high CO2 production in both the OX–ZEO process and FT + zeolite presents a great challenge for the conversion of hydrogen-rich synthesis gas to aromatics. According to the proposed reaction mechanism for the OX–ZEO process to form olefins with ketene intermediates, the formation of CO2 is inevitable, since the oxygen from carbon monoxide is removed from the surface of the metal oxide catalyst via CO oxidation.306 Iron carbide-based FT catalysts with high olefin selectivity commonly show high WGS activity and CO2 selectivities between 21% and 50%.350,409 The WGS of cobalt carbide-based FT catalysts is slower and leads to CO2 selectivities between 2% and 13%.360 However, for the cobalt carbide-based catalysts the selectivity to short olefins is rather low (17–30% in the hydrocarbons). Hence, an important challenge for bifunctional catalysis is the suppression of CO2 formation and the combination of cobalt carbide-based FT catalysts (with increased olefin selectivity) with a zeolite seems to have the potential to achieve this.410
However, selectivity is not the only important factor determining the feasibility of a process. To analyze the economic feasibility of a process for the direct conversion of synthesis gas to aromatics, technical-economical aspects were simulated by Song et al. using ASPEN software.411 Here, the direct conversion of synthesis gas to aromatics was compared to a dual reactor process, whereas synthesis gas was first converted to methanol and the methanol was further converted to aromatics. The simulation did not only include the reaction unit, but also units for quenching the reaction mixture, compression, distillation, cyclic absorption separation and pressure swing absorption. The catalytic data were based on 70% CO conversion per pass over the methanol synthesis catalyst and a fraction of 70–80% aromatics in the liquid products after passing an H-ZSM-5 zeolite. It was shown that a low CO conversion resulted in a low yield of aromatics and therefore low partial pressure, which gave additional challenges in product condensation and separation. To compete with a dual reactor approach with separate methanol synthesis and aromatization at individual reaction conditions, a novel bifunctional process needs to give a minimum of 66% CO conversion per pass with an aromatic fraction of 70–80% in the liquid products and very low CO2 selectivities. Both approaches, the OX–ZEO process and FT + zeolite, are currently not meeting these requirements.
5. Liquid fuels
The products formed in the Fischer–Tropsch synthesis are always a mixture of hydrocarbons with various chain lengths. The maximum selectivity to C5–C11 products in the Fischer–Tropsch synthesis is 48% for a chain growth probability of α = 0.76 according to the ASF distribution (Fig. 16-A).381 Increasing the production of liquid transportation fuels requires operating at higher α-values and cracking the resulting Fischer–Tropsch products with a too high chain length to the desired fraction. | ||
| Fig. 16 A: Anderson Schulz–Flory distribution of Fischer–Tropsch products showing a maximum selectivity to the C5–C11 fraction of 48% at chain growth probability of α = 0.76;381 B: equilibrium distribution of C5–C11 paraffin isomers at 250 °C and 20 bar. This represents the thermodynamic limitation for isomerization of n-paraffins (calculated with Outotec HSC 9.6.1). | ||
The octane number is relevant for gasoline and strongly increases with a smaller size and a higher degree of branching of hydrocarbons.412 In general, the octane numbers of a hydrocarbon molecule with the same number of carbon atoms follow the trend of paraffins < olefins < aromatics (e.g., n-hexane: 19, 1-hexene: 85 and benzene: 108).158,413–415Fig. 16-B shows the thermodynamic distribution of isomers according to the ASF distribution at 250 °C and 20 bar for C5–C11 paraffins, grouped by degree of branching. The maximum conversion of linear paraffins in the secondary isomerization is between 75% and 91%, leaving 9–15% n-paraffins un-isomerized. The main products are mono- or di-branched paraffins with 45–68% and 19–42% shares, respectively.412 The highly branched paraffins are key for a high-octane number, but only constitute 2–4% of the product mixture for tri-branched and 0.1–0.2% quad-branched paraffins. To convert paraffinic FT waxes into suitable gasoline fuels with octane numbers of ∼90 by the formation of aromatics, temperatures above 500 °C are applied.416
5.1. Recent developments
Different bifunctional catalysts, in particular for the direct production of gasoline from synthesis gas have been developed over the past years. These catalysts consist of iron- or cobalt-based Fischer–Tropsch catalysts, methanol or DME catalysts, or (mixed) metal oxides as the primary functional group and zeolites for the secondary conversion to gasoline. Some studies have also employed noble metals supported on zeolites, since these are industrially used as hydrocracking catalysts to upgrade paraffines by isomerization and cracking.417Fischer–Tropsch catalysts can be combined with different zeolites, such as H-ZSM-5, SAPO-11, SSZ-13, mordenite, Y or beta, in order to overcome the limitations for the selectivity to C5–C11 products according to the ASF model.418–421 Both pore structure and acidity of these zeolites play a crucial role in the final product distribution.418,422–424 Acidity plays a major role for primary cracking and isomerization, whereas porosity affects the secondary olefin isomerization by micropore diffusion limitations.425 Larger pore sizes of zeolites facilitate the formation of multi-branched isomer products and strong acidity can cause over-cracking to lighter products.426 Hydrocracking catalysts such as Pt/ZSM-5 were also effective for the secondary conversion of the Fischer–Tropsch products.417,427
Co/SiO2 catalysts with different average pore diameters (10 nm and 50 nm) were used as silicon source for a Co containing zeolite catalyst with a hierarchical pore structure for the direct conversion of syngas to gasoline fuel.41 The resulting catalysts with the zeolite in Na-form showed high selectivities towards C5–C11 of 65–68% and 14–25% iso-paraffins in the hydrocarbon products, whereas the Co/SiO2 catalyst alone displayed 48–49% selectivity to C5–C11 products with 11–19% iso-paraffins. After ion-exchange to convert the zeolite into the proton form, the C5–C11 selectivity remained at the same level, but the fraction of iso-paraffins increased to 35–37%. The selectivity to C5–C11 products was further increased by the introduction of mesopores to the catalyst.428 Additionally, the presence of mesopores in an H-ZSM-5 support can increase the dispersion of the Co nanoparticles and hence the overall activity.429 Co/Al2O3 catalyst with multimodal porosity in a dual bed configuration with a Pt/nano-ZSM-5 hydrocracking catalyst showed a 2-fold increase of hydrocarbon products in the middle distillate fraction (C10–C24) compared to the configuration with a mono-modal Co/Al2O3 FT catalyst.427
Experiments with different average distances between Co and acid sites of an H-ZSM-5 zeolite showed a maximum selectivity to C5–C11 products for proximity in the μm-range.430 C5–C11 products were formed with 89.5% selectivity and 35.5% isomers in the C5+ products at 270 °C, 20 bar and conversion between 71% and 92%. Additionally, mesoporous H-ZSM-5 zeolite coated with a pyrolytic carbon layer prior to impregnation with Co precursor showed enhanced reducibility of the cobalt oxide, low CH4 selectivity and higher selectivity to C5–C11 than the catalyst system without carbon layer, due to reduced metal–support-interaction.431
The influence of the amount of acid sites has also been studied. Co/MCF and nano-sized H-ZSM-5, with different mass ratios in the physical mixture, showed that with increasing zeolite mass content, the selectivity to C12+ products decreased from 50% (Co/MCF alone) to 6% for Co/MCF:Z = 1:4 m/m.432 Also, the C2–C4 selectivity increased from 6% to 23% and the sum of iso-paraffins and olefins increased from 17% to 52% in the hydrocarbon products. The C5–C11 selectivity showed a plateau at medium zeolite content (34% for Co/MCF, 54% for Co/MCF:Z = 1:1 m/m, 45% for Co/MCF:Z = 1:4 m/m).
Iron-based Fischer–Tropsch catalysts allow to form a larger fraction of olefins in the products and are usually operated at higher temperatures compared to cobalt-based Fischer–Tropsch catalysts.433 The addition of a zeolite to an iron-based Fischer–Tropsch catalyst can promote the formation of aromatics, which significantly raises the octane number of the C5–C11 product fraction. A co-precipitated iron-based Fischer–Tropsch catalyst containing Cu, Mg and K as promoters showed 53% selectivity to C5–C11 products of which 4% were aromatics (300 °C, 10 bar, CO conversion 70–90%).434 The addition of an H-ZSM-5 with medium concentration of acid sites (Si/Al = 240) by physical mixing increased the selectivity to C5–C11 products to 67% with 73% aromatics in this fraction. A physical mixture of the iron-based Fischer–Tropsch catalyst with an H-ZSM-5 with high acid site concentration (Si/Al = 40) increased content of aromatics in the C5–C11 hydrocarbon fraction to 90%, however, the total C5–C11 fraction decreased to 58% due to over-cracking and increased formation of C1–C4 products. Additionally, the olefins/paraffin ratio increased 3–7-fold upon zeolite addition compared to the Fischer–Tropsch catalyst alone. All experiments showed high water-gas-shift activities with 40–44% CO2 formed.
Coating the iron-based catalyst with a hydrophobic methylated silica layer decreased the formation of CO2.435 Further addition of an HZSM-5 zeolite packed below the FTS catalyst in the reactor led to a high C5–C11 selectivity (62.5% at 260 °C, 20 bar and 50% CO conversion) and low CO2 selectivity (14.3%). The authors showed that the diffusion of water through the hydrophobic layer was unidirectional, which led to a reduced CO2 formation by hampering the water-gas shift reaction on the iron-based catalyst.
An Fe/SiO2 core/shell catalyst was tested in the direct conversion of synthesis gas to gasoline.436 The silicalite-1 membrane applied onto the core catalyst served as protection as well as anchor point for the functional H-ZSM-5 membrane. This catalyst showed similar CO conversions (55–60%) and C5–C11 selectivities (49–53%, CO2-free) as the base core catalyst or the core catalyst in physical mixture with H-ZSM-5 at 280 °C and 10 bar. The selectivity to iso-paraffins was 30% higher for the core/shell catalyst than for the core catalyst alone and the physical mixture with hereof, which was ascribed to hydrogenation and isomerization of olefins, next to hydrocracking and isomerization of C12+ hydrocarbons.
Combining a zinc–manganese-oxide catalyst with different 10-membered ring zeolites revealed that the OX–ZEO process allows to form C5–C11 products with a high selectivity of up to 77%.381 The product spectrum of the ZnMnOx catalyst mixed with SAPO-11 showed only 6% n-paraffins in the C5–C11 aliphatics as well as 16% aromatics in C5–C11. The CH4 selectivity was remarkably low (2.3%). Introducing mesopores into an H-ZSM-5 zeolite of a zinc–chromium-oxide containing OX–ZEO catalyst enhanced the selectivity to C5+ from 20% to 61%, while maintaining the low CH4 selectivity.194 Conversion of ketene, which is thought to be an intermediate in OX–ZEO catalysts, using H-SAPO-11 has been studied to elucidate the reaction mechanism to form C5–C11 hydrocarbons.437 The authors showed by in situ IR and quasi-in situ ssNMR spectroscopy that ketene transforms via either an acetic acid ketonization pathway or an acetoacetic acid decarboxylation pathway to acetone, butene, and C5–C11 hydrocarbons.
A DME catalyst (Cu/ZnO/Al2O3+γ-Al2O3) allows to convert synthesis gas to DME with 90% selectivity (CO2 free) at 300 °C, 30 bar and 65% CO conversion.438 In a physical mixture with a nano-sized H-ZSM-5, a DME-to-gasoline (DTG) catalyst, the CO conversion increased to 75%, C5–C11 products were formed with 26% selectivity and C1–C2 products with a selectivity of 20%. Fig. 17 illustrates the difference between the dual bed and dual reactor configurations. The dual bed configuration can have dedicated temperatures for the individual catalyst beds applied in a single reactor. The dual reactor configuration consists of two consecutive reactors with removal of intermediates between the reactors.357,438 Placing the two different catalysts in a dual bed configuration (Fig. 17-A) with the DME catalyst upstream led to an increase of C5–C11 selectivity to 76% and only 5% C1–C2. When the dual bed configuration was operated with dedicated temperatures for each catalyst (DME catalyst at 260 °C and DTG catalyst at 320 °C) the CO conversion increased to 87% and the C1–C2 selectivity decreased to 1.7%. C5–C11 products were formed with 79% selectivity, of which 34% were aromatics. It was also found that C5–C11 aliphatics consisted of 95% isomerized products. Increasing the temperature of the DTG catalyst bed caused the C5–C11 and aromatics selectivity to decrease. Additionally, nano-sized H-ZSM-5 DTG catalysts with medium Si/Al ratio and medium acid sites concentration show superior stability in the DME conversion to C5–C11 products, compared to zeolites with a higher acid site concentration due to reduced coke formation on the zeolite.
 | ||
| Fig. 17 Illustration of bifunctional catalysis performed in (A) dual bed and (B) dual reactor configuration. | ||
5.2. Benefits
In the following paragraphs we illustrate the potential benefits of bifunctional catalyst systems for the direct production of fuels compared to the operation in multiple individual reactors.An OX–ZEO catalyst capable of converting synthesis gas into gasoline showed a high content of 95% of branched isomers in the C5–C11 aliphatics (non-aromatic molecules) fraction.381 In the OX–ZEO process branched molecules are predominantly by the alkylation of hydrocarbons with oxygenates such as MeOH, DME, or ketene.439–441 The dual bed process with a methanol synthesis or FTO catalyst in the first bed and zeolite at high temperature (320 °C) in the bed downstream showed a low selectivity to linear C5–C11 paraffins of 3% and 4%, respectively.357,438 At high temperatures, in the zeolite bed oligomerization of short iso-olefins acts as an additional source of C5–C11 branched molecules next to isomerization of linear aliphatics, which also holds for medium- and high-temperature operation of iron-based Fischer–Tropsch catalysts combined with zeolites.397
Fischer–Tropsch catalysts can form a liquid product layer around the metal particles, inside the support's pores or in the void space between the catalyst particles. This may result in H2 and in particular CO profiles as a function of distance to the catalyst surface as schematically illustrated in Fig. 18.442–449 Hydrogen diffuses 2–3 times faster through FT wax than carbon monoxide,450–452 although the latter has a ∼20% higher solubility.453–456 As a result, the H2/CO ratio at the catalyst surface can be significantly higher than in the bulk of the reactor, leading to lower C5+ and higher CH4 selectivities with increasing catalyst particle size and liquid layer thickness.457–459 Experiments with a core/shell catalyst consisting of a Co/SiO2 core and H-ZSM-5 shell showed a reduced CH4 selectivity compared to the Co/SiO2 alone (10.3% vs. 25.7%) together with a reduced selectivity to C11+ (0.3% vs. 15.3%) at 280 °C, 10 bar and full conversion.236
 | ||
| Fig. 18 Illustration of the concentration profile of hydrogen and carbon monoxide through the gas phase and the liquid product layer to the catalyst surface with different liquid product layer thicknesses. A: A thick layer of liquid products increases the effective H2:CO ratio on the catalyst surface. B: Reduction of the liquid layer thickness leads to a lower H2:CO ratio on the catalyst surface. | ||
An often claimed benefit for the conversion of synthesis gas to gasoline using bifunctional catalysis is the lower investment costs for a single reactor.430 However, Fischer–Tropsch reactors may have higher costs per installed unit compared to a dedicated reactor filled with a hydrocracking/isomerisation catalyst.460 Hence, the lower investment costs for a larger Fischer–Tropsch reactor (to accommodate the Fischer–Tropsch catalyst and the zeolite) compared to two separate reactors seems to be limited.
5.3. Challenges
Catalysts containing noble metals for hydrocracking are widely employed and their performances maximized. However, when employed for in situ hydrocracking of Fischer–Tropsch products in bifunctional catalysts, poisoning of the noble metal with carbon monoxide reduces the hydrotreating performance of these catalysts.417,461–463 The conversion of long chain paraffins is reduced 4.3-fold over a Pt/ZSM-5 catalyst in the presence of synthesis gas compared to H2 atmosphere.417 Olefins still undergo isomerization and cracking on the acid sites of the zeolite.The liquid wax filling the FT catalyst pores can reduce catalyst activity by causing mass transfer limitation for synthesis gas.442–449 Removal or reduction of this product layer by operating alternatingly at Fischer–Tropsch and hydrogenolysis conditions can lead to enhanced activity and stability of the Fischer–Tropsch catalysts.464 However, a combination of Fischer–Tropsch catalyst with a zeolite to reduce the product layer by cracking did not show significant activity enhancement and displayed a similar turn-over-frequency as the FT catalyst alone,422,429,465,466 despite an altered ASF distribution.431
5.4. Process comparison
Fig. 19-A and B show the yield to C5–C11 products for the bifunctional catalysts consisting of Co-based Fischer–Tropsch catalysts and solid acids (Co + Z), iron-based Fischer–Tropsch catalysts with zeolite (Fe + Z), the OX–ZEO process for the synthesis of gasoline (OX–ZEO) as function of CO conversion. Additionally, the C5–C11 yield of dual bed approaches is shown. Furthermore, Fig. 19-B contains the yield of C5–C11 hydrocarbons as function of CO conversion of two separate processes (dual reactor process, Fig. 17-B) that involves methanol synthesis and separation in the first process and the methanol to gasoline (MTG) as the second process.357,438 The slopes for the individual approaches correspond to the overall C5–C11 selectivity and take the formation of CO2 into account. As a reference the maximum yield as function of CO conversion resulting from the ASF distribution (48% selectivity for C5–C11) is shown as well. | ||
| Fig. 19 A: Yield of C5–C11 hydrocarbons as function of CO conversion for Co + Z and Fe + Z; B: yield of C5–C11 hydrocarbons as function of CO conversion for OX–ZEO, dual bed configuration and separated dual reactor processes C: the calculated octane number and D: methane selectivity for the combination of cobalt-based FT catalysts with zeolites (Co + Z), whereas zeolites were 12-membered ring or 10-membered ring zeolites or non-micro-porous solid acids (NMPA), iron-based FT catalysts with zeolite (Fe + Z), the OX–ZEO process and the combination of DME or FTO catalysts with zeolites in a dual bed configuration. The solid diamond in A indicates a combination of iron-based FT catalyst with zeolite showing reduced CO2 selectivity of 14.3%.435 A cobalt-carbide based FTO catalyst combined with a zeolite is shown in C and D as red triangle (first point of the Fe + Z series).410 The horizontal grey bars in C and D highlight the octane numbers needed to use the C5–C11 fraction directly as gasoline fuel and the typical methane selectivities of the HT-FTS for comparison, respectively. | ||
Co + Z, Fe + Z and OX–ZEO show overall selectivities to C5–C11 of 54%, 22% and 39%, respectively. The dual bed process allows to produce C5–C11 products with 51% selectivity. The scattering is due to the fact that the C5–C11 selectivity is influenced by many parameters, such as reaction conditions, type of bifunctional catalyst, and nature of the zeolite. Fe + Z shows a low overall selectivity to C5–C11 products (22%) resulting from a moderate fraction of C5–C11 (10–55%) in the hydrocarbon products and much CO2 production due to the high WGS activity.374,397 Inhibiting the WGS can increase the C5–C11 selectivity (62%).435 Cobalt-based FT catalysts show low WGS activity, (only 1–4% CO2) thus Co + Z can have a higher overall selectivity to C5–C11 products (56%).420 The OX–ZEO process displays a large fraction of C5–C11 in the hydrocarbons (67–77%), but with a high WGS activity (∼50% CO2) the overall selectivity is reduced to 39%.305,381
The dual bed processes show a C5–C11 selectivity of 51%, which is comparable to the highest possible selectivity predicted by the ASF distribution. This selectivity results from a high fraction of C5–C11 in the hydrocarbon products (70–78%) and a significant WGS activity (32–38% CO2 formed) which reduces the overall C5–C11 selectivity.357,438 A combination of separate processes (MeOH synthesis, MeOH recovery, MTG process) could potentially show much higher overall selectivity of 96–99%, due to high selectivities of the MeOH synthesis (97–99%)351,352 and the MTG process (up to 99%)441 (data can be found in the ESI†).
Based on reported catalytic performance of bifunctional catalysts for the direct conversion of synthesis gas to gasoline we calculated the octane number of the corresponding C5–C11 fraction (Fig. 19-C) for different approaches: Co + Z, Fe + Z and OX–ZEO. Additionally, we added dual bed processes with direct DME synthesis or FTO catalyst in the first bed and a zeolite in the bed downstream. These processes have different temperatures for the individual catalyst beds but do not separate the intermediate products after the first catalytic conversion from unreacted reactants or formed side products. The detailed analysis and calculation of the octane numbers can be found in the ESI.†
The linear C5–C11 paraffin products of a Co-based FTS catalyst with ASF product distribution at α = 0.76 without zeolite have a low octane number (∼1.5). The combination of Co-based Fischer–Tropsch catalysts and a zeolite increases octane numbers to between 13 and 72. The use of zeolites with different pore dimensions, namely 10- or 12-membered-ring pores, does not affect the resulting octane number significantly. Iron-based Fischer–Tropsch catalyst mixed with zeolites show higher octane numbers of the C5–C11 products of 65–91. Also, the OX–ZEO process allows high octane products with an octane number of 73–89. The dual bed processes with different temperatures for the catalyst beds exhibit high octane numbers between 88 and 96. The horizontal grey bar in Fig. 19-C highlights the octane numbers needed to use the C5–C11 fraction directly as gasoline fuel. However, the octane number is often boosted by fuel additives, such as MTBE or ethanol.
The relatively low octane number of Co + Z can be explained by the mainly paraffinic products of Co-FTS, which hardly undergo isomerization, oligomerization or aromatization under typical reaction conditions.467–469 Fe + Z on the other hand is usually applied at higher temperatures, hence iron-based Fischer–Tropsch catalysts produce a higher fraction of olefins, allowing to form aromatics, boosting the octane number of the C5–C11 fraction drastically. OX–ZEO is not thermodynamically limited in the iso-paraffin fraction in the product and can additionally form aromatics and olefins with moderate selectivity. The analyzed OX–ZEO catalysts showed selectivity to iso-paraffin of 52–78%, olefin of 17–28% and aromatics of up to 16% in the C5–C11 hydrocarbon fraction.305,381 The dual bed process allows to operate the first bed (CO activation) at lower temperatures (260–270 °C), reducing CH4 selectivity and boosting CO conversion to 88% for the DME or FTO catalysts. Operating the second catalyst bed accommodating the zeolite (for gasoline synthesis) at higher temperatures of 320 °C enables the formation of aromatics, oligomerization of short olefins and eventually produce high-octane gasoline with high yields.438
Next to the octane number, the methane content is also important. In the past decades, Fischer–Tropsch catalysts and their process conditions have been optimized to reduce the CH4 selectivity. However, the addition of a zeolite leads to more methane.374,419,421,432,470,471 In Fig. 19-D the reported methane selectivities for Co + Z, Fe + Z, OX–ZEO and the dual bed processes are shown. The horizontal grey bar shows the typical methane selectivities of the HT-FTS for comparison. Co + Z and Fe + Z catalysts show high methane selectivities (7–30%), 2- to 3-fold higher than without the zeolite.432,436 OX–ZEO and the dual reactor processes only produce 2–3% methane similar to the values obtained for high-alpha FT catalysts.
The higher operating temperature and the resulting shift to lower alpha-values in the ASF distribution contributes to the high methane selectivity of FT + Z. For Co-based catalysts, the (acidity of the) support plays an important role.472 Methane production increases 3–4-fold than that predicted by the ASF model for catalysts supported on oxides with higher acidic character.473 XPS studies revealed that cobalt supported on a zeolite showed higher binding energy for Co 2p3/2 electrons compared to cobalt on a zeolite that has been covered with a layer of carbon prior to Co impregnation.431 Hence, the increased methane selectivity for cobalt catalysts supported directly on a zeolite might be explained by electronic effects.474 A reduced electron density of the cobalt particles causes weaker binding of hydrogen and stronger bonds between carbon and hydrogen of adsorbed CHx species475 making the hydrogenation of CHx species to CH4 energetically favored. Alternatively, the increased methane formation can be explained by a decreased reducibility of cobalt particles supported on zeolites476,477 or by small cobalt particles inside the zeolite pores, which also give rise to high methane selectivity.478,479
Co + Z shows the highest selectivity to the C5–C11 fraction (55%). However, the resulting octane number of this fraction is very low, hence it cannot be used as gasoline fuel directly. It has to be blended with a high fraction of additives. In terms of octane number for bifunctional processes, OX–ZEO and Fe + Z are the most promising, as they allow C5–C11 products with high octane numbers of up to ∼91 to form. Regarding the methane selectivity, only OX–ZEO and the dual bed processes can compete with the low methane selectivities achieved in FTS which are necessary for industrial application. In summary, the OX–ZEO process can form C5–C11 fuels with high octane number and little methane. If the WGS activity can be further reduced, this bifunctional catalyst has potential for the direct conversion of synthesis gas to gasoline fuel.
6. Summary and perspective
The transition to a more sustainable society is forcing a change in the current production processes of chemicals and fuels. A key step is the use of alternative feedstocks to the traditional fossil-based ones. Synthesis gas plays a crucial role due to the versatility of its sources and the various products it can create. In this regard, carbon sources from feedstocks such as CO2, organic waste, or biomass, together with the implementation of hydrogen production from renewable energy sources, can become central in this transition.480 Additionally, synthesis gas operation (production and conversion) is also feasible on a smaller scale, allowing a targeted production in remote locations.76 The use of bifunctional catalysts can expand the variety of products directly obtained from these sources.In the past years, great progress has been realized in the field of synthesis gas conversion using bifunctional catalysts. A major advantage is that when combining different catalytic functions in a single reactor, synthesis gas conversion levels can lay far beyond the thermodynamic limitations of a first conversion step in a two-reactor system (Fig. 10 and 14).
In many cases in the first conversion step (e.g., in methanol synthesis) no water is formed. Combining with a second conversion step can enhance the water content in the proximity of the CO activation catalyst, which can lead to the formation of CO2via the WGS reaction. This has been observed for the OX–ZEO process and DME synthesis, leading to CO2 selectivities up to 50% and 33%, respectively. The degree to which this happens is an important parameter. On the one hand CO2 formation lowers the carbon atom economy. On the other hand, the removal of water by the WGS reaction can also increase the catalyst lifetime and facilitate in situ production of additional hydrogen, favoring the utilization of carbon-rich synthesis gas over bifunctional catalysts. For the use of hydrogen-rich synthesis gas, the water-gas-shift activity of the OX–ZEO catalysts should be reduced, for example by recycling CO2. Our detailed analysis of recently published data for the direct synthesis of DME using bifunctional catalysts revealed an average DME selectivity of 62%. In contrast, a process consisting of two consecutive reactors (methanol synthesis and methanol dehydration) can achieve an overall DME selectivity of 88%. The lower selectivity for the bifunctional catalysts relates to the formation of CO2. In situ water removal by adsorption has proven an effective strategy to circumvent this limitation. The DME selectivity of bifunctional catalysts can be enhanced to 98% by in situ water removal by adsorption. Although the application in an industrial production scale still needs to be demonstrated, the Netherlands Organization for Applied Scientific Research (TNO) has taken the first steps in 2022 by building a containerized pilot reactor for sorption enhanced DME synthesis (SEDMES).245,251,481–483
In terms of C2–C4 olefins selectivity, neither OX–ZEO nor FTO can compete with a dual reactor process with methanol or DME synthesis in the first reactor and (D)MTO in a consecutive reactor (93% to C2–C4 olefins). The OX–ZEO process allows to form a high olefin fraction in the hydrocarbon products, but the high WGS activity and hence CO2 production reduces the overall selectivity to short olefins to 43% on average. FTO catalysts enable the production of short olefins with an average selectivity of 22% due to the high WGS activity and a limited high fraction of short olefins in the hydrocarbon products. Specific reduction of the WGS activity of FTO catalysts can increase the overall selectivity to 44%.
The situation is different for direct synthesis of aromatics from synthesis gas. OX–ZEO and the combination of iron-based Fischer–Tropsch catalysts with a zeolite (FT + Z) show selectivities to aromatics of 41% and 26%, respectively. Especially for the OX–ZEO process, this is only slightly lower than for the dual reactor process that comprises the synthesis of methanol or DME accompanied by aromatization based on hydrogen transfer (41%). This can make OX–ZEO competitive to this type of dual reactor process based on hydrogen transfer. A dual reactor process with aromatization based on dehydrogenation could theoretically achieve selectivities as high as 95%.
Three factors were considered for the analysis of the direct production of gasoline fuels using bifunctional catalysts: the overall selectivity to hydrocarbons in the gasoline range (C5–C11), the methane selectivity and the octane number of the resulting C5–C11 fraction. The combination of cobalt-based Fischer–Tropsch catalysts and zeolites (Co + Z) showed C5–C11 selectivities of 54%, which is beyond the maximum predicted by the ASF distribution. However, the methane selectivity was between 7% and 30% and octane numbers were rather low (30–50). Iron-based Fischer–Tropsch catalysts combined with zeolites (Fe + Z) yielded higher octane numbers (65–91). However, the methane selectivity was high (7–28%) and the overall selectivity to C5–C11 hydrocarbons was reduced to 22%, due to the high WGS activity. OX–ZEO produced a high-octane number of 73–89, low methane selectivity (only 1–2%) and medium overall selectivity to C5–C11 hydrocarbons (39%). This is slightly less than for a dual bed process with a zeolite downstream of a DME or FTO catalyst and dedicated reaction conditions for every catalyst bed: octane numbers between 88 and 96, methane selectivity 2–3% and medium C5–C11 selectivity (51%). Further efforts are then necessary to make gasoline production with bifunctional catalysis more attractive, particularly by reducing the water gas shift activity (and hence CO2 production).
Interest for bifunctional catalysts at industrial scale has been leaning towards DME and C2–C4 olefins production. The synthesis of DME from synthesis gas is mainly performed via the indirect route with a total annual production capacity of 107 t per year.23 In 2003 the JFE Group in Japan finished the construction of a pilot plant designed for the direct DME synthesis with a capacity of 3.6 × 104 t per year in Shiranuka-cho, Hokkaido, Japan using a slurry phase reactor.484,485 The Korea Gas Corporation launched a demonstration plant for the direct synthesis of DME with a capacity of 10 t per day already in 2004 at the Incheon KOGAS LNG terminal based on the KOGAS DME process.23,222,486 After successful operation, the design of a commercial production plant with 3 × 105 t per year is in progress.486
From 2002 to 2007, a 100 ton per day demonstration plant project was successfully conducted by DME Development Corp. funded by 10 companies. Process performance analysis, catalyst life and long-term stable operation were assessed. Building on the technical data, the feasibility studies of commercial scale DME production from natural gas or coal were explored. Total Energies, JAPEX, INPEX and Toyota Tsusho, former members of the DME Development Corp., developed the technology in 2010. In 2016, those four companies transferred the technology patents to RenFud Corporation, which licenses the DME synthesis process technology and supplies proprietary catalysts. The first demonstrating operations with coke oven gas feed showed promising results, with 96% synthesis gas conversion, 93% selectivity to DME and 99.6% purity of DME.
In September 2019, the Dalian Institute of Chemical Physics and the Bureau of Major R&D Programs (Chinese Academy of Science) announced a cooperation between the Dalian Institute of Chemical Physics, the Chinese Academy of Sciences and Shaanxi Yanchang Petroleum (Group) Co., Ltd to perform industrial pilot trials with the aim to produce short olefins via the OX–ZEO process (Fig. 20). The capacity of this demonstration plant was 1000 t of short olefins per year and it is located in Shaanxi, China.487,488 In a first step synthesis gas is produced from coal, followed by the OX–ZEO process, converting 50% of the synthesis gas to C2–C4 olefins with 75% selectivity in a single pass.
 | ||
| Fig. 20 Photo of the industrial pilot plant to produce short olefins via the OX–ZEO to olefins (OXZEO©-TO) process, located in Shaanxi, China.489 | ||
At the moment research also focuses increasingly on the direct conversion of CO2 to chemicals and fuels.490–495 Bifunctional heterogeneous catalysts can be key to integrating CO2 activation and subsequent conversion to chemicals and fuels in an efficient way, contributing to carbon capture and utilization efforts. Despite the challenges for applications such as high costs, development of bifunctional catalysts for effective CO2 conversion is a promising topic with potentially beneficial environmental effects.496
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
JLW and CHM contributed to this work by performing formal analysis, investigation, visualization and writing of the original draft. KPdJ and PEdJ contributed by supervision, validation, project administration and writing (review and editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC), an NWO Gravitation program funded by the Ministry of Education, Culture and Science of the government of the Netherlands. Companhia Brasileira de Metalurgia is thanked for financial support. KPdJ acknowledges the European Research Council, EU FP7 ERC Advanced Grant no. 338846.References
- D. A. Wood, C. Nwaoha and B. F. Towler, Gas-to-liquids (GTL): A review of an industry offering several routes for monetizing natural gas, J. Nat. Gas Sci. Eng., 2012, 9, 196–208 CrossRef CAS.
- Z. Kong, X. Dong and Q. Jiang, Forecasting the development of China's coal-to-liquid industry under security, economic and environmental constraints, Energy Econ., 2019, 80, 253–266 CrossRef.
- Z. Kong, X. Dong and Q. Jiang, The net energy impact of substituting imported oil with coal-to-liquid in China, J. Cleaner Prod., 2018, 198, 80–90 CrossRef.
- M. M. B. Noureldin, N. O. Elbashir and M. M. El-Halwagi, Optimization and selection of reforming approaches for syngas generation from natural/shale gas, Ind. Eng. Chem. Res., 2014, 53, 1841–1855 CrossRef CAS.
- D. Hildebrandt, D. Glasser, B. Hausberger, B. Patel and B. J. Glasser, Producing Transportation Fuels with Less Work, Science, 2009, 323, 1680–1681 CrossRef PubMed.
- G. Iaquaniello, G. Centi, A. Salladini, E. Palo and S. Perathoner, Waste to Chemicals for a Circular Economy, Chem. – Eur. J., 2018, 24, 11831–11839 CrossRef CAS PubMed.
- K. Cheng, et al., Advances in Catalysis for Syngas Conversion to Hydrocarbons. Advances in Catalysis, Elsevier Inc., 2017, vol. 60 Search PubMed.
- J. Sun, et al., Beyond Cars: Fischer-Tropsch Synthesis for Non-Automotive Applications, ChemCatChem, 2019, 11, 1412–1424 CrossRef CAS.
- J. Bao, G. Yang, Y. Yoneyama and N. Tsubaki, Significant advances in C1 catalysis: highly efficient catalysts and catalytic reactions, ACS Catal., 2019, 9, 3026–3053 CrossRef CAS.
- P. De Luna, et al., What would it take for renewably powered electrosynthesis to displace petrochemical processes?, Science, 2019, 364, 351–360 CrossRef PubMed.
- D. Corral, et al., Advanced manufacturing for electrosynthesis of fuels and chemicals from CO2, Energy Environ. Sci., 2021, 14, 3064–3074 RSC.
- A. Y. Khodakov, W. Chu and P. Fongarland, Advances in the Development of Novel Cobalt Fischer−Tropsch Catalysts for Synthesis of Long-Chain Hydrocarbons and Clean Fuels, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed.
- W. Zhou, et al., New horizon in C1 chemistry: Breaking the selectivity limitation in transformation of syngas and hydrogenation of CO2 into hydrocarbon chemicals and fuels, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- J. Zecevic, G. Vanbutsele, K. P. De Jong and J. A. Martens, Nanoscale intimacy in bifunctional catalysts for selective conversion of hydrocarbons, Nature, 2015, 528, 245–254 CrossRef CAS PubMed.
- J. L. Weber, et al., Effect of proximity and support material on deactivation of bifunctional catalysts for the conversion of synthesis gas to olefins and aromatics, Catal. Today, 2020, 342, 161–166 CrossRef CAS.
- H. Ham, et al., Enhanced Stability of Spatially Confined Copper Nanoparticles in an Ordered Mesoporous Alumina for Dimethyl Ether Synthesis from Syngas, ACS Catal., 2016, 6, 5629–5640 CrossRef CAS.
- C. Jeong, et al., Facile Structure Tuning of a Methanol-Synthesis Catalyst towards the Direct Synthesis of Dimethyl Ether from Syngas, ChemCatChem, 2017, 9, 4484–4489 CrossRef CAS.
- A. Nemmour, A. Inayat, I. Janajreh and C. Ghenai, Green hydrogen-based E-fuels (E-methane, E-methanol, E-ammonia) to support clean energy transition: A literature review, Int. J. Hydrogen Energy, 2023, 48, 29011–29033 CrossRef CAS.
- M. Thema, F. Bauer and M. Sterner, Power-to-Gas: Electrolysis and methanation status review, Renewable Sustainable Energy Rev., 2019, 112, 775–787 CrossRef CAS.
- C. Wulf, J. Linßen and P. Zapp, Review of Power-to-Gas Projects in Europe, Energy Procedia, 2018, 155, 367–378 CrossRef.
- C. Tregambi, et al., Techno-economic assessment of a synthetic methane production process by hydrogenation of carbon dioxide from direct air capture, Int. J. Hydrogen Energy, 2023, 48, 37594–37606 CrossRef CAS.
- A. Review, H. M. T. Galvis and K. P. Jong, Catalysts for Production of Lower Ole fi ns from Synthesis Gas, ACS Catal., 2013, 3, 2130–2149 CrossRef.
- T. H. Fleisch, A. Basu and R. A. Sills, Introduction and advancement of a new clean global fuel: The status of DME developments in China and beyond, J. Nat. Gas Sci. Eng., 2012, 9, 94–107 CrossRef CAS.
- M. Müller and U. Hübsch, Dimethyl Ether, Ullmann's Encyclopedia of Industrial Chemistry, 2012, DOI:10.1002/14356007.a08.
- B. Elvers and A. Schutze, Handbook of Fuels, Energy Sources for Transportation, 2021 Search PubMed.
- S. Chemicals, Product Stewardship Summaries, 2017 Search PubMed.
- A. Corma, et al., Crude oil to chemicals: Light olefins from crude oil, Catal. Sci. Technol., 2017, 7, 12–46 RSC.
- Y. Gao, et al., Recent Advances in Intensified Ethylene Production - A Review, ACS Catal., 2019, 9, 8592–8621 CrossRef CAS.
- U. S. E. I. Administration, Monthly Energy Review: January 2022, 2022 Search PubMed.
- W. Fruscella, Kirk-Othmer Encycl. Chem. Technol., 2000 Search PubMed.
- E. D. Ozokwelu, Kirk-Othmer Encycl. Chem. Technol., 2000 Search PubMed.
- J. S. Ninomiya and B. Biggers, Effects of toluene content in fuel on aromatic emissions in the exhaust, J. Air Pollut. Control Assoc., 1970, 20, 609–611 CrossRef CAS PubMed.
- W. J. Cannella, in Van Nostrand's Encyclopedia of Chemistry, Wiley Online Library, 2007 Search PubMed.
- B. Looney, Statistical Review of World Energy. BP, 2021, vol. 70 Search PubMed.
- E. M. Huitema, D. Schwietert, J. R. Mandel and S. Nagatsuka, Worldwide Fuel Charter, Gasoline and diesel fuel, 6th edn, 2019 Search PubMed.
- A. Bemani, et al., Modeling of cetane number of biodiesel from fatty acid methyl ester (FAME) information using GA-, PSO-, and HGAPSO-LSSVM models, Renewable Energy, 2020, 150, 924–934 CrossRef CAS.
- D. Leckel, Diesel production from fischer - Tropsch: The past, the present, and new concepts, Energy Fuels, 2009, 23, 2342–2358 CrossRef CAS.
- Y. Ohtsuka, T. Arai, S. Takasaki and N. Tsubouchi, Fischer-Tropsch synthesis with cobalt catalysts supported on mesoporous silica for efficient production of diesel fuel fraction, Energy Fuels, 2003, 17, 804–809 CrossRef CAS.
- A. Meurer and J. Kern, Fischer–Tropsch Synthesis as the Key for Decentralized Sustainable Kerosene Production, Energies, 2021, 14, 1836 CrossRef CAS.
- Z. Wang, et al., Hierarchical ZSM-5 Supported CoMn Catalyst for the Production of Middle Distillate from Syngas, Ind. Eng. Chem. Res., 2021, 60, 5783–5791 CrossRef CAS.
- B. Mazonde, et al., A solvent-free: In situ synthesis of a hierarchical Co-based zeolite catalyst and its application to tuning Fischer-Tropsch product selectivity, Catal. Sci. Technol., 2018, 8, 2802–2808 RSC.
- B. P. Vempatapu and P. K. Kanaujia, Monitoring petroleum fuel adulteration: A review of analytical methods, TrAC, Trends Anal. Chem., 2017, 92, 1–11 CrossRef CAS.
- C. Lamonier, Transportation fuels: Desulfurizing diesel, Nat. Energy, 2017, 2, 1–2 Search PubMed.
- P. Sikarwar, V. Gosu and V. Subbaramaiah, An overview of conventional and alternative technologies for the production of ultra-low-sulfur fuels, Rev. Chem. Eng., 2019, 35, 669–705 CAS.
- C. of the E. U. European Parliament, Directive 2009/30/EC of the European Parliament and of The Council, 2009 Search PubMed.
- A. J. Beyersdorf, et al., Reductions in aircraft particulate emissions due to the use of Fischer-Tropsch fuels, Atmos. Chem. Phys., 2014, 14, 11–23 CrossRef.
- M. E. Dry, High quality diesel via the Fischer-Tropsch process - A review, J. Chem. Technol. Biotechnol., 2002, 77, 43–50 CrossRef CAS.
- J. Eilers, S. A. Posthuma and S. T. Sie, The shell middle distillate synthesis process (SMDS), Catal. Lett., 1990, 7, 253–269 CrossRef CAS.
- M. E. Dry, The Fischer–Tropsch process: 1950-2000, Catal. Today, 2002, 71, 227–241 CrossRef CAS.
- D. Martínez del Monte, A. J. Vizcaíno, J. Dufour and C. Martos, Potential pathways for syngas transformation towards kerosene range hydrocarbons in a dual Fischer-Tropsch-zeolite bed, Int. J. Energy Res., 2022, 46, 5280–5287 CrossRef.
- H. W. Schablitzky, et al., Hydroprocessing of Fischer-Tropsch biowaxes to second-generation biofuels, Biomass Convers. Biorefin., 2011, 1, 29–37 CrossRef CAS.
- J. C. van Dyk, M. J. Keyser and M. Coertzen, Syngas production from South African coal sources using Sasol-Lurgi gasifiers, Int. J. Coal Geol., 2006, 65, 243–253 CrossRef CAS.
- F. Li and L. S. Fan, Clean coal conversion processes - Progress and challenges, Energy Environ. Sci., 2008, 1, 248–267 RSC.
- J. R. Rostrup-Nielsen, Syngas in perspective, Catal. Today, 2002, 71, 243–247 CrossRef CAS.
- S. S. Bharadwaj and L. D. Schmidt, Catalytic partial oxidation of natural gas to syngas, Fuel Process. Technol., 1995, 42, 109–127 CrossRef CAS.
- J. J. Hernández, G. Aranda-Almansa and C. Serrano, Co-gasification of biomass wastes and coal-coke blends in an entrained flow gasifier: An experimental study, Energy Fuels, 2010, 24, 2479–2488 CrossRef.
- J. J. Hernández, G. Aranda-Almansa and A. Bula, Gasification of biomass wastes in an entrained flow gasifier: Effect of the particle size and the residence time, Fuel Process. Technol., 2010, 91, 681–692 CrossRef.
- S. Maisano, F. Urbani, F. Cipitì, F. Freni and V. Chiodo, Syngas production by BFB gasification: Experimental comparison of different biomasses, Int. J. Hydrogen Energy, 2019, 44, 4414–4422 CrossRef CAS.
- R. Anghilante, et al., Innovative power-to-gas plant concepts for upgrading of gasification bio-syngas through steam electrolysis and catalytic methanation, Energy Convers. Manage., 2019, 183, 462–473 CrossRef CAS.
- P. J. Hancox and A. E. Götz, South Africa's coalfields - A 2014 perspective, Int. J. Coal Geol., 2014, 132, 170–254 CrossRef CAS.
- N. Laosiripojana and S. Assabumrungrat, Catalytic dry reforming of methane over high surface area ceria, Appl. Catal., B, 2005, 60, 107–116 CrossRef CAS.
- J. Guo, H. Lou, H. Zhao, D. Chai and X. Zheng, Dry reforming of methane over nickel catalysts supported on magnesium aluminate spinels, Appl. Catal., A, 2004, 273, 75–82 CrossRef CAS.
- A. T. Ashcroft, et al., Selective oxidation of methane to synthesis gas using transition metal catalysts, Nature, 1990, 344, 319–321 CrossRef CAS.
- D. A. Hickman, E. A. Haupfear and L. D. Schmidt, Synthesis gas formation by direct oxidation of methane over Rh monoliths, Catal. Lett., 1992, 138, 267–282 CrossRef CAS.
- Y. Matsumura and T. Nakamori, Steam reforming of methane over nickel catalysts at low reaction temperature, Appl. Catal., A, 2004, 258, 107–114 CrossRef CAS.
- K. Hou and R. Hughes, The kinetics of methane steam reforming over a Ni/alpha -Al2O3 catalyst, Chem. Eng. J., 2001, 82, 311–328 CrossRef CAS.
- P. Lv, et al., Bio-syngas production from biomass catalytic gasification, Energy Convers. Manage., 2007, 48, 1132–1139 CrossRef CAS.
- M. Shahbaz, S. Yusup, A. Inayat, D. O. Patrick and M. Ammar, The influence of catalysts in biomass steam gasification and catalytic potential of coal bottom ash in biomass steam gasification: A review, Renewable Sustainable Energy Rev., 2017, 73, 468–476 CrossRef CAS.
- W. Torres, S. S. Pansare and J. G. Goodwin, Hot gas removal of tars, ammonia, and hydrogen sulfide from biomass gasification gas, Catal. Rev.: Sci. Eng., 2007, 49, 407–456 CrossRef CAS.
- P. J. Woolcock and R. C. Brown, A review of cleaning technologies for biomass-derived syngas, Biomass Bioenergy, 2013, 52, 54–84 CrossRef CAS.
- D. Chiche, C. Diverchy, A. C. Lucquin, F. Porcheron and F. Defoort, Synthesis Gas Purification D, Oil Gas Sci. Technol., 2013, 68, 707–723 CrossRef CAS.
- S. Brunet, D. Mey, G. Pérot, C. Bouchy and F. Diehl, On the hydrodesulfurization of FCC gasoline: A review, Appl. Catal., A, 2005, 278, 143–172 CrossRef CAS.
- G. Van Oost, et al., Pyrolysis/gasification of biomass for synthetic fuel production using a hybrid gas–water stabilized plasma torch, Vacuum, 2008, 83, 209–212 CrossRef CAS.
- J. R. Hess, K. L. Kenney, C. T. Wright, R. Perlack and A. Turhollow, Corn stover availability for biomass conversion: situation analysis, Cellulose, 2009, 16, 599–619 CrossRef CAS.
- G. Guan, M. Kaewpanha, X. Hao and A. Abudula, Catalytic steam reforming of biomass tar: Prospects and challenges, Renewable Sustainable Energy Rev., 2016, 58, 450–461 CrossRef CAS.
- A. Kurkjian, S. C. Leviness, K. J. Massie and J. Nighswander Remote micro-scale gtl products for uses in oil- and gas-field and pipeline applications. 1, US 2010/0000153 A1, 2008.
- K. C. Waugh, Methanol Synthesis, Catal. Today, 1992, 15, 51–75 CrossRef CAS.
- D. Sheldon, Methanol production - A technical history, Johnson Matthey Technol. Rev., 2017, 61, 172–182 CrossRef CAS.
- G. Ertl, H. Knözinger and J. Weitkamp, Handbook of Heterogeneous Catalysis, 2008, DOI:10.1524/zpch.1999.208.part_1_2.274.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Chemical recycling of carbon dioxide to methanol and dimethyl ether: From greenhouse gas to renewable, environmentally carbon neutral fuels and synthetic hydrocarbons, J. Org. Chem., 2009, 74, 487–498 CrossRef CAS PubMed.
- A. Álvarez, et al., Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed.
- B. de Oliveira Campos, et al., Surface reaction kinetics of the methanol synthesis and the water gas shift reaction on Cu/ZnO/Al2O3, React. Chem. Eng., 2021, 6, 868–887 RSC.
- K. B. Tan, G. Zhan, D. Sun, J. Huang and Q. Li, The development of bifunctional catalysts for carbon dioxide hydrogenation to hydrocarbons via the methanol route: from single component to integrated components, J. Mater. Chem. A, 2021, 9, 5197–5231 RSC.
- M. Sahibzada, I. S. Metcalfe and D. Chadwick, Methanol synthesis from CO/CO2/H2 over Cu/ZnO/Al2O3 at differential and finite conversions, J. Catal., 1998, 174, 111–118 CrossRef CAS.
- N. J. Azhari, et al., Methanol synthesis from CO2: A mechanistic overview, Results Eng., 2022, 16, 100711 CrossRef CAS.
- J. Sehested, Industrial and scientific directions of methanol catalyst development, J. Catal., 2019, 371, 368–375 CrossRef CAS.
- R. Van Den Berg, et al., Structure sensitivity of Cu and CuZn catalysts relevant to industrial methanol synthesis, Nat. Commun., 2016, 7, 13057 CrossRef CAS PubMed.
- F. Chen, et al., Structure–Performance Correlations over Cu/ZnO Interface for Low-Temperature Methanol Synthesis from Syngas Containing CO2, ACS Appl. Mater. Interfaces, 2021, 13, 8191–8205 CrossRef CAS PubMed.
- X. Cui, et al., Preserving the Active Cu–ZnO Interface for Selective Hydrogenation of CO2 to Dimethyl Ether and Methanol, ACS Sustainable Chem. Eng., 2021, 9, 2661–2672 CrossRef CAS.
- M. Behrens, Promoting the Synthesis of Methanol: Understanding the Requirements for an Industrial Catalyst for the Conversion of CO 2, Angew. Chem., Int. Ed., 2016, 55, 14906–14908 CrossRef CAS PubMed.
- T. Lunkenbein, J. Schumann, M. Behrens, R. Schlögl and M. G. Willinger, Formation of a ZnO Overlayer in Industrial Cu/ZnO/Al2O3 Catalysts Induced by Strong Metal-Support Interactions, Angew. Chem., Int. Ed., 2015, 54, 4544–4548 CrossRef CAS PubMed.
- S. A. Kondrat, et al., Stable amorphous georgeite as a precursor to a high-activity catalyst, Nature, 2016, 531, 83–87 CrossRef CAS PubMed.
- M. Behrens, et al., The Potential of Microstructural Optimization in Metal/Oxide Catalysts: Higher Intrinsic Activity of Copper by Partial Embedding of Copper Nanoparticles, ChemCatChem, 2010, 2, 816–818 CrossRef CAS.
- O. Martin, et al., Operando Synchrotron X-ray Powder Diffraction and Modulated-Excitation Infrared Spectroscopy Elucidate the CO2Promotion on a Commercial Methanol Synthesis Catalyst, Angew. Chem., Int. Ed., 2016, 55, 11031–11036 CrossRef CAS PubMed.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- M. Behrens, et al., The active site of methanol synthesis over Cu/ZnO/Al2O 3 industrial catalysts, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- S. Kuld, et al., Quantifying the promotion of Cu catalysts by ZnO for methanol synthesis, Science, 2016, 352, 969–974 CrossRef CAS PubMed.
- S. Kuld, C. Conradsen, P. G. Moses, I. Chorkendorff and J. Sehested, Quantification of zinc atoms in a surface alloy on copper in an industrial-type methanol synthesis catalyst, Angew. Chem., Int. Ed., 2014, 53, 5941–5945 CrossRef CAS PubMed.
- J. Nakamura, Y. Choi and T. Fujitani, On the issue of the active site and the role of ZnO in Cu/ZnO methanol synthesis catalysts, Top. Catal., 2003, 22, 277–285 CrossRef CAS.
- H. Topsøe, J.-D. Grunwaldt, B. Clausen, A. Molenbroek and N.-Y. Topsøe, In Situ Investigations of Structural Changes in Cu/ZnO Catalysts, J. Catal., 2002, 194, 452–460 Search PubMed.
- R. Dalebout, N. L. Visser, C. E. L. Pompe, K. P. de Jong and P. E. de Jongh, Interplay between carbon dioxide enrichment and zinc oxide promotion of copper catalysts in methanol synthesis, J. Catal., 2020, 392, 150–158 CrossRef CAS.
- R. Dalebout, et al., Insight into the Nature of the ZnOx Promoter during Methanol Synthesis, ACS Catal., 2022, 12, 6628–6639 CrossRef CAS PubMed.
- G. Bozzano and F. Manenti, Efficient methanol synthesis: Perspectives, technologies and optimization strategies, Prog. Energy Combust. Sci., 2016, 56, 71–105 CrossRef.
- M. D. Porosoff, B. Yan and J. G. Chen, Catalytic reduction of CO2by H2for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- F. Studt, et al., Discovery of a Ni-Ga catalyst for carbon dioxide reduction to methanol, Nat. Chem., 2014, 6, 320–324 CrossRef CAS PubMed.
- J. L. Snider, et al., Revealing the Synergy between Oxide and Alloy Phases on the Performance of Bimetallic In–Pd Catalysts for CO 2 Hydrogenation to Methanol, ACS Catal., 2019, 3399–3412, DOI:10.1021/acscatal.8b04848.
- C.-S. Li, et al., High-performance hybrid oxide catalyst of manganese and cobalt for low-pressure methanol synthesis, Nat. Commun., 2015, 6, 6538 CrossRef PubMed.
- O. Martin, et al., Indium Oxide as a Superior Catalyst for Methanol Synthesis by CO 2 Hydrogenation, Angew. Chemie Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed.
- J. Wang, et al., A highly selective and stable ZnO-ZrO 2 solid solution catalyst for CO 2 hydrogenation to methanol, Sci. Adv., 2017, 3, e1701290 CrossRef PubMed.
- M. S. Duyar, et al., A Highly Active Molybdenum Phosphide Catalyst for Methanol Synthesis from CO and CO2, Angew. Chem., Int. Ed., 2018, 57, 15045–15050 CrossRef CAS PubMed.
- X. Liu, A. Hamasaki, T. Honma and M. Tokunaga, Anti-ASF distribution in Fischer-Tropsch synthesis over unsupported cobalt catalysts in a batch slurry phase reactor, Catal. Today, 2011, 175, 494–503 CrossRef CAS.
- N. Tsubaki, K. Yoshii and K. Fujimoto, Anti-ASF Distribution of Fischer–Tropsch Hydrocarbons in Supercritical-Phase Reactions, J. Catal., 2002, 207, 371–375 CrossRef CAS.
- J. L. L. Weber, I. Dugulan, P. E. P. E. de Jongh and K. P. K. P. de Jong, Bifunctional Catalysis for the Conversion of Synthesis Gas to Olefins and Aromatics, ChemCatChem, 2018, 10, 1107–1112 CrossRef CAS.
- H. M. Torres Galvis, et al., Iron particle size effects for direct production of lower olefins from synthesis gas, J. Am. Chem. Soc., 2012, 134, 16207–16215 CrossRef CAS PubMed.
- F. Lu, X. Chen, Z. Lei, L. Wen and Y. Zhang, Revealing the activity of different iron carbides for Fischer-Tropsch synthesis, Appl. Catal., B, 2021, 281, 119521 CrossRef CAS.
- F. Lu, X. Chen, L. Wen, Q. Wu and Y. Zhang, The Synergic Effects of Iron Carbides on Conversion of Syngas to Alkenes, Catal. Lett., 2021, 151, 2132–2143 CrossRef CAS.
- M. Claeys, et al., Oxidation of Hägg Carbide during High-Temperature Fischer–Tropsch Synthesis: Size-Dependent Thermodynamics and In Situ Observations, ACS Catal., 2021, 11, 13866–13879 CrossRef CAS.
- M. E. Dry, Fischer-Tropsch synthesis over iron catalysts, Catal. Lett., 1990, 7, 241–251 CrossRef CAS.
- H. M. Torres Galvis, et al., Supported iron nanoparticles as catalysts for sustainable production of lower olefins, Science, 2012, 335, 835–838 CrossRef CAS PubMed.
- H. M. Torres Galvis, et al., Effects of sodium and sulfur on catalytic performance of supported iron catalysts for the Fischer-Tropsch synthesis of lower olefins, J. Catal., 2013, 303, 22–30 CrossRef CAS.
- J. Xie, et al., Size and promoter effects on stability of carbon-nanofiber-supported iron-based Fischer-Tropsch catalysts, ACS Catal., 2016, 6, 4017–4024 CrossRef CAS PubMed.
- X. Yang, et al., Promotional effects of sodium and sulfur on light olefins synthesis from syngas over iron-manganese catalyst, Appl. Catal., B, 2022, 300, 120716 CrossRef CAS.
- R. Oukaci, A. H. Singleton and J. G. Goodwin, Comparison of patented Co F–T catalysts using fixed-bed and slurry bubble column reactors, Appl. Catal., A, 1999, 186, 129–144 CrossRef CAS.
- F. G. Botes, Influences of Water and Syngas Partial Pressure on the Kinetics of a Commercial Alumina-Supported Cobalt Fischer−Tropsch Catalyst, Ind. Eng. Chem. Res., 2009, 48, 1859–1865 CrossRef CAS.
- B. H. Davis, Fischer−Tropsch Synthesis: Comparison of Performances of Iron and Cobalt Catalysts, Ind. Eng. Chem. Res., 2007, 46, 8938–8945 CrossRef CAS.
- J. P. Den Breejen, et al., On the origin of the cobalt particle size effects in Fischer-Tropsch catalysis, J. Am. Chem. Soc., 2009, 131, 7197–7203 CrossRef CAS PubMed.
- M. Claeys and E. Van Steen, On the effect of water during Fischer-Tropsch synthesis with a ruthenium catalyst, Catal. Today, 2002, 71, 419–427 CrossRef CAS.
- W. Z. Li, et al., Chemical Insights into the Design and Development of Face-Centered Cubic Ruthenium Catalysts for Fischer-Tropsch Synthesis, J. Am. Chem. Soc., 2017, 139, 2267–2276 CrossRef CAS PubMed.
- Y. Zhang, et al., Ru/TiO2 Catalysts with Size-Dependent Metal/Support Interaction for Tunable Reactivity in Fischer–Tropsch Synthesis, ACS Catal., 2020, 10, 12967–12975 CrossRef CAS.
- Q. Zhang, K. Cheng, J. Kang, W. Deng and Y. Wang, Fischer-tropsch catalysts for the production of hydrocarbon fuels with high selectivity, ChemSusChem, 2014, 7, 1251–1264 CrossRef CAS PubMed.
- R. Y. Abrokwah, M. M. Rahman, V. G. Deshmane and D. Kuila, Effect of titania support on Fischer-Tropsch synthesis using cobalt, iron, and ruthenium catalysts in silicon-microchannel microreactor, Mol. Catal., 2019, 478, 110566 CrossRef CAS.
- C. Hernández Mejía, J. E. S. van der Hoeven, P. E. de Jongh and K. P. de Jong, Cobalt–Nickel Nanoparticles Supported on Reducible Oxides as Fischer–Tropsch Catalysts, ACS Catal., 2020, 10, 7343–7354 CrossRef PubMed.
- C. Hernández Mejía, C. Vogt, B. M. Weckhuysen and K. P. de Jong, Stable niobia-supported nickel catalysts for the hydrogenation of carbon monoxide to hydrocarbons, Catal. Today, 2020, 343, 56–62 CrossRef.
- M. Zhang, et al., Changing the balance of the MTO reaction dual-cycle mechanism: Reactions over ZSM-5 with varying contact times, Cuihua Xuebao, 2016, 37, 1413–1422 CAS.
- S. Svelle, et al., Conversion of methanol into hydrocarbons over zeolite H-ZSM-5: Ethene formation is mechanistically separated from the formation of higher alkenes, J. Am. Chem. Soc., 2006, 128, 14770–14771 CrossRef CAS PubMed.
- S. Svelle, U. Olsbye, F. Joensen and M. Bjørgen, Conversion of methanol to alkenes over medium- and large-pore acidic zeolites: Steric manipulation of the reaction intermediates governs the ethene/propene product selectivity, J. Phys. Chem. C, 2007, 111, 17981–17984 CrossRef CAS.
- M. Bjørgen, et al., Conversion of methanol to hydrocarbons over zeolite H-ZSM-5: On the origin of the olefinic species, J. Catal., 2007, 249, 195–207 CrossRef.
- L. Wu and E. J. M. Hensen, Comparison of mesoporous SSZ-13 and SAPO-34 zeolite catalysts for the methanol-to-olefins reaction, Catal. Today, 2014, 235, 160–168 CrossRef CAS.
- B. P. C. Hereijgers, et al., Product shape selectivity dominates the Methanol-to-Olefins (MTO) reaction over H-SAPO-34 catalysts, J. Catal., 2009, 264, 77–87 CrossRef CAS.
- F. Bleken, et al., The effect of acid strength on the conversion of methanol to olefins over acidic microporous catalysts with the CHA topology, Top. Catal., 2009, 52, 218–228 CrossRef CAS.
- G. Chen, Q. Sun and J. Yu, Nanoseed-assisted synthesis of nano-sized SAPO-34 zeolites using morpholine as the sole template with superior MTO performance, Chem. Commun., 2017, 53, 13328–13331 RSC.
- F. Goodarzi, et al., Synthesis of mesoporous ZSM-5 zeolite encapsulated in an ultrathin protective shell of silicalite-1 for MTH conversion, Microporous Mesoporous Mater., 2020, 292, 109730 CrossRef CAS.
- D. Rojo-Gama, et al., Time- and space-resolved study of the methanol to hydrocarbons (MTH) reaction-influence of zeolite topology on axial deactivation patterns, Faraday Discuss., 2017, 197, 421–446 RSC.
- M. García-Ruiz, Synthesis of 10 and 12 Ring Zeolites (MCM-22, TNU-9 and MCM-68) Modified with Zn and Its Potential Application in the Reaction of Methanol to Light Aromatics and Olefins, Top. Catal., 2020, 63, 451–467 CrossRef.
- Z. Wei, et al., Alkaline modification of ZSM-5 catalysts for methanol aromatization: The effect of the alkaline concentration, Front. Chem. Sci. Eng., 2015, 9, 450–460 CrossRef CAS.
- J. Zhong, et al., Tuning the product selectivity of SAPO-18 catalysts in MTO reaction via cavity modification, Chin. J. Catal., 2019, 40, 477–485 CrossRef CAS.
- P. Ferri, et al., Chemical and Structural Parameter Connecting Cavity Architecture, Confined Hydrocarbon Pool Species, and MTO Product Selectivity in Small-Pore Cage-Based Zeolites, ACS Catal., 2019, 9, 11542–11551 CrossRef CAS.
- U. Olsbye, et al., Conversion of methanol to hydrocarbons: How zeolite cavity and pore size controls product selectivity, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed.
- N. Nishiyama, et al., Size control of SAPO-34 crystals and their catalyst lifetime in the methanol-to-olefin reaction, Appl. Catal., A, 2009, 362, 193–199 CrossRef CAS.
- Z. Li, M. T. Navarro, J. Martínez-Triguero, J. Yu and A. Corma, Synthesis of nano-SSZ-13 and its application in the reaction of methanol to olefins, Catal. Sci. Technol., 2016, 6, 5856–5863 RSC.
- X. Zhu, et al., Fluoride-assisted synthesis of bimodal microporous SSZ-13 zeolite, Chem. Commun., 2016, 52, 3227–3230 RSC.
- Z. Xu, et al., Size control of SSZ-13 crystals with APAM and its influence on the coking behaviour during MTO reaction, Catal. Sci. Technol., 2019, 9, 2888–2897 RSC.
- W. Skistad, et al., Methanol conversion to hydrocarbons (MTH) over H-ITQ-13 (ITH) zeolite, Top. Catal., 2014, 57, 143–158 CrossRef CAS.
- J. Valecillos, G. Elordi, A. T. Aguayo and P. Castaño, The intrinsic effect of co-feeding water on the formation of active/deactivating species in the methanol-to-hydrocarbons reaction on ZSM-5 zeolite, Catal. Sci. Technol., 2021, 11, 1269–1281 RSC.
- X. Niu, et al., Influence of crystal size on the catalytic performance of H-ZSM-5 and Zn/H-ZSM-5 in the conversion of methanol to aromatics, Fuel Process. Technol., 2017, 157, 99–107 CrossRef CAS.
- J. Zhang, et al., Solvent-Free Synthesis of Core-Shell Zn/ZSM-5@Silicalite-1 Catalyst for Selective Conversion of Methanol to BTX Aromatics, Ind. Eng. Chem. Res., 2019, 58, 15453–15458 CrossRef CAS.
- Y. Wang, et al., Direct synthesis of Zn-incorporated nano-ZSM-5 zeolite by a dry gel conversion method for improving catalytic performance of methanol to aromatics reaction, J. Porous Mater., 2021, 28, 1609–1618 CrossRef CAS.
- A. Perdih and F. Perdih, Chemical interpretation of octane number, Acta Chim. Slov., 2006, 53, 306–315 CAS.
- B. Creton, C. Dartiguelongue, T. De Bruin and H. Toulhoat, Prediction of the cetane number of diesel compounds using the quantitative structure property relationship, Energy Fuels, 2010, 24, 5396–5403 CrossRef CAS.
- R. Sadeghbeigi, Chemistry of FCC Reactions, Fluid Catal. Crack. Handb., 2012, pp. 125–135, DOI:10.1016/b978-0-12-386965-4.00006-9.
- C. S. Hsu and P. R. Robinson, Cracking, Pet. Sci. Technol., 2019, 211–244, DOI:10.1007/978-3-030-16275-7_11.
- J. S. Buchanan, J. G. Santiesteban and W. O. Haag, Mechanistic considerations in acid-catalyzed cracking of olefins, J. Catal., 1996, 158, 279–287 CrossRef CAS.
- J. G. Speight, Catalytic Cracking, Heavy Extra-heavy Oil Upgrad. Technol., 2013, pp. 39–67, DOI:10.1016/b978-0-12-404570-5.00003-x.
- N. Batalha, L. Pinard, C. Bouchy, E. Guillon and M. Guisnet, N-Hexadecane hydroisomerization over Pt-HBEA catalysts. Quantification and effect of the intimacy between metal and protonic sites, J. Catal., 2013, 307, 122–131 CrossRef CAS.
- A. Galadima and O. Muraza, Hydrocracking catalysts based on hierarchical zeolites: A recent progress, J. Ind. Eng. Chem., 2018, 61, 265–280 CrossRef CAS.
- S. F. Anis, G. Singaravel and R. Hashaikeh, Hierarchical nano zeolite-Y hydrocracking composite fibers with highly efficient hydrocracking capability, RSC Adv., 2018, 8, 16703–16715 RSC.
- I. Nam, et al., Production of middle distillate from synthesis gas in a dual-bed reactor through hydrocracking of wax over mesoporous Pd-Al2O3 composite catalyst, Catal. Lett., 2009, 130, 192–197 CrossRef CAS.
- K. Kohli, R. Prajapati, M. Sau and S. K. Maity, Mesoporous Alumina Supported NiMo Catalysts for Residue Conversion, Procedia Earth Planet. Sci., 2015, 11, 325–331 CrossRef CAS.
- Y. Chen, et al., Synthesis and Characterization of Iron-Substituted ZSM-23 Zeolite Catalysts with Highly Selective Hydroisomerization of n-Hexadecane, Ind. Eng. Chem. Res., 2018, 57, 13721–13730 CrossRef CAS.
- A. Dhar, A. Dutta, P. Sharma, B. Panda and S. Roy, Synthesis and characterization of solid-phase super acid catalysts and their application for isomerization of n-alkanes, Chem. Eng. Commun., 2017, 204, 1341–1356 CrossRef CAS.
- W. Wang, C. J. Liu and W. Wu, Bifunctional catalysts for the hydroisomerization of n -alkanes: The effects of metal-acid balance and textural structure, Catal. Sci. Technol., 2019, 9, 4162–4187 RSC.
- P. B. Weisz, Polyfunctional Heterogeneous Catalysis, Adv. Catal., 1962, 13, 137–190 CAS.
- P. Wang, et al., The controlled synthesis of Pt/Hβ catalysts with intimate metal-acid sites for n-butane isomerization, Microporous Mesoporous Mater., 2020, 309, 110547 CrossRef CAS.
- C. Guo, et al., Influences of the metal-acid proximity of Pd-SAPO-31 bifunctional catalysts for n-hexadecane hydroisomerization, Fuel Process. Technol., 2021, 214, 106717 CrossRef CAS.
- US Department of Energy, Report of the Hydrogen Production Expert Panel : A Subcommittee of the Hydrogen & Fuel Cell Technical Advisory Committee, DoE Fuel Cell Technologies Office, 2013 Search PubMed.
- T. L. Levalley, A. R. Richard and M. Fan, The progress in water gas shift and steam reforming hydrogen production technologies - A review, Int. J. Hydrogen Energy, 2014, 39, 16983–17000 CrossRef CAS.
- D. C. Grenoble, M. M. Estadt and D. F. Ollis, The chemistry and catalysis of the water gas shift reaction. 1. The kinetics over supported metal catalysts, J. Catal., 1981, 67, 90–102 CrossRef CAS.
- D. B. Pal, R. Chand, S. N. Upadhyay and P. K. Mishra, Performance of water gas shift reaction catalysts: A review, Renewable Sustainable Energy Rev., 2018, 93, 549–565 CrossRef CAS.
- D. Damma and P. G. Smirniotis, Recent advances in iron-based high-temperature water-gas shift catalysis for hydrogen production, Curr. Opin. Chem. Eng., 2018, 21, 103–110 CrossRef.
- M. Javed, et al., From hydrophilic to hydrophobic: A promising approach to tackle high CO2 selectivity of Fe-based Fischer-Tropsch microcapsule catalysts, Catal. Today, 2019, 330, 39–45 CrossRef CAS.
- B. Todic, et al., Kinetic Modeling of Secondary Methane Formation and 1-Olefin Hydrogenation in Fischer–Tropsch Synthesis over a Cobalt Catalyst, Int. J. Chem. Kinet., 2017, 49, 859–874 CrossRef CAS.
- M. K. Gnanamani, R. A. Keogh, W. D. Shafer and B. H. Davis, Deutero-1-pentene tracer studies for iron and cobalt Fischer-Tropsch synthesis, Appl. Catal., A, 2011, 393, 130–137 CrossRef CAS.
- J. Patzlaff, Y. Liu, C. Graffmann and J. Gaube, Interpretation and kinetic modeling of product distributions of cobalt catalyzed Fischer-Tropsch synthesis, Catal. Today, 2002, 71, 381–394 CrossRef CAS.
- B. Shi, G. Jacobs, D. Sparks and B. H. Davis, Fischer-Tropsch synthesis: 14C labeled 1-alkene conversion using supercritical conditions with Co/A12O3, Fuel, 2005, 84, 1093–1098 CrossRef CAS.
- F. G. Botes, Proposal of a new product characterization model for the iron-based low-temperature Fischer-Tropsch synthesis, Energy Fuels, 2007, 21, 1379–1389 CrossRef CAS.
- H. Schulz and M. Claeys, Reactions of α-olefins of different chain length added during Fischer-Tropsch synthesis on a cobalt catalyst in a slurry reactor, Appl. Catal., A, 1999, 186, 71–90 CrossRef CAS.
- R. Kosol, et al., Iron catalysts supported on nitrogen functionalized carbon for improved CO2 hydrogenation performance, Catal. Commun., 2021, 149, 106216 CrossRef CAS.
- C. G. Visconti, et al., CO2 hydrogenation to lower olefins on a high surface area K-promoted bulk Fe-catalyst, Appl. Catal., B, 2017, 200, 530–542 CrossRef CAS.
- L. Guo, et al., Selective formation of linear-alpha olefins (LAOs) by CO2 hydrogenation over bimetallic Fe/Co-Y catalyst, Catal. Commun., 2019, 130, 105759 CrossRef.
- H. Schulz, Selforganization in Fischer-Tropsch synthesis with iron- and cobalt catalysts, Catal. Today, 2014, 228, 113–122 CrossRef CAS.
- H. Schulz, G. Schaub, M. Claeys and T. Riedel, Transient initial kinetic regimes of Fischer-Tropsch synthesis, Appl. Catal., A, 1999, 186, 215–227 CrossRef CAS.
- F. Jiao, et al., Selective conversion of syngas to light olefins, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed.
- X. Yang, X. Su, D. Chen, T. Zhang and Y. Huang, Direct conversion of syngas to aromatics: A review of recent studies, Chin. J. Catal., 2020, 41, 561–573 CrossRef CAS.
- X. Yang, et al., The influence of alkali-treated zeolite on the oxide-zeolite syngas conversion process, Catal. Sci. Technol., 2018, 8, 4338–4348 RSC.
- A. V. Kirilin, et al., Conversion of Synthesis Gas to Light Olefins: Impact of Hydrogenation Activity of Methanol Synthesis Catalyst on the Hybrid Process Selectivity over Cr-Zn and Cu-Zn with SAPO-34, Ind. Eng. Chem. Res., 2017, 56, 13392–13401 CrossRef CAS.
- A. Stanislaus and H. C. Barry, Aromatic hydrogenation catalysis: A review, Catal. Rev.: Sci. Eng., 1994, 36, 75–123 CrossRef CAS.
- N. Batalha, J.-D. Comparot, A. Le Valant and L. Pinard, In-situ FTIR to unravel the bifunctional nature of aromatics hydrogenation synergy on zeolite / metal catalysts, Catal. Sci. Technol., 2022, 12, 1117–1129 RSC.
- C. Du, P. Lu and N. Tsubaki, Efficient and New Production Methods of Chemicals and Liquid Fuels by Carbon Monoxide Hydrogenation, ACS Omega, 2020, 5, 49–56 CrossRef CAS PubMed.
- J. Zhou, et al., Regeneration of catalysts deactivated by coke deposition: A review, Chin. J. Catal., 2020, 41, 1048–1061 CrossRef CAS.
- P. P. Paalanen, S. H. Van Vreeswijk and B. M. Weckhuysen, Combined in Situ X-ray Powder Diffractometry/Raman Spectroscopy of Iron Carbide and Carbon Species Evolution in Fe(-Na-S)/α-Al2O3Catalysts during Fischer-Tropsch Synthesis, ACS Catal., 2020, 10, 9837–9855 CrossRef CAS.
- D. S. Kalakkad, M. D. Shroff, S. Köhler, N. Jackson and A. K. Datye, Appl. Catal., A, 1995, 133, 335–350 CrossRef CAS.
- Y. Chen, et al., Sintered precipitated iron catalysts with enhanced fragmentation-resistance ability for Fischer-Tropsch synthesis to lower olefins, Catal. Sci. Technol., 2018, 8, 5943–5954 RSC.
- M. Guisnet and P. Magnoux, Organic chemistry of coke formation, Appl. Catal., A, 2001, 212, 83–96 CrossRef CAS.
- S. Shao, H. Zhang, R. Xiao, X. Li and Y. Cai, Evolution of coke in the catalytic conversion of biomass-derivates by combined in-situ DRIFTS and ex-situ approach: Effect of functional structure, Fuel Process. Technol., 2018, 178, 88–97 CrossRef CAS.
- S. Gao, et al., Insight into the deactivation mode of methanol-to-olefins conversion over SAPO-34 : Coke , diffusion , and acidic site accessibility, J. Catal., 2018, 367, 306–314 CrossRef CAS.
- J. F. Haw, W. Song, D. M. Marcus and J. B. Nicholas, The mechanism of methanol to hydrocarbon catalysis, Acc. Chem. Res., 2003, 36, 317–326 CrossRef CAS PubMed.
- E. Borodina, et al., Influence of the reaction temperature on the nature of the active and deactivating species during methanol to olefins conversion over H-SSZ-13, ACS Catal., 2015, 5, 992–1003 CrossRef CAS.
- F. Yaripour, F. Baghaei, I. Schmidt and J. Perregaard, Catalytic dehydration of methanol to dimethyl ether (DME) over solid-acid catalysts, Catal. Commun., 2005, 6, 147–152 CrossRef CAS.
- M. Xu, J. H. Lunsford, D. W. Goodman and A. Bhattacharyya, Synthesis of dimethyl ether (DME) from methanol over solid-acid catalysts, Appl. Catal., A, 1997, 149, 289–301 CrossRef CAS.
- B. Sabour, M. H. Peyrovi, T. Hamoule and M. Rashidzadeh, Catalytic dehydration of methanol to dimethyl ether (DME) over Al-HMS catalysts, J. Ind. Eng. Chem., 2014, 20, 222–227 CrossRef CAS.
- Z. Azizi, M. Rezaeimanesh, T. Tohidian and M. R. Rahimpour, Dimethyl ether: A review of technologies and production challenges, Chem. Eng. Process., 2014, 82, 150–172 CrossRef CAS.
- M. Stiefel, R. Ahmad, U. Arnold and M. Döring, Direct synthesis of dimethyl ether from carbon-monoxide-rich synthesis gas: Influence of dehydration catalysts and operating conditions, Fuel Process. Technol., 2011, 92, 1466–1474 CrossRef CAS.
- L. Liu, et al., Conversion of syngas to methanol and DME on highly selective Pd/ZnAl2O4 catalyst, J. Energy Chem., 2021, 58, 564–572 CrossRef CAS.
- J. S. Martinez-Espin, et al., New insights into catalyst deactivation and product distribution of zeolites in the methanol-to-hydrocarbons (MTH) reaction with methanol and dimethyl ether feeds, Catal. Sci. Technol., 2017, 7, 2700–2716 RSC.
- Air Products and Chemicals, I. Liquid phase Dimethyl Ether demonstration in the Laporte alternative fuels development unit, 2001 Search PubMed.
- S. H. Lee, W. Cho, T. Song and Y. J. Ra, Scale up study of direct DME synthesistechnology, in Proceedings of 24th World Gas Conference, 2009 Search PubMed.
- BASF & Sichuan Lutianhua Co., L., BASF and Lutianhua plan to pilot a new production process that significantly reduces CO2 emissions, Joint News Release, 2019 Search PubMed.
- J. Sun, G. Yang, Y. Yoneyama and N. Tsubaki, Catalysis chemistry of dimethyl ether synthesis, ACS Catal., 2014, 4, 3346–3356 CrossRef CAS.
- U. Mondal and G. D. Yadav, Perspective of dimethyl ether as fuel: Part I, Catalysis, J. CO2 Util., 2019, 32, 299–320 CrossRef CAS.
- S. N. Khadzhiev, N. N. Ezhova and O. V. Yashina, Catalysis in the dispersed phase: Slurry technology in the synthesis of dimethyl ether (Review), Pet. Chem., 2017, 57, 553–570 CrossRef CAS.
- U. Mondal and G. D. Yadav, Perspective of dimethyl ether as fuel: Part II- analysis of reactor systems and industrial processes, J. CO2 Util., 2019, 32, 321–338 CrossRef CAS.
- V. Dieterich, A. Buttler, A. Hanel, H. Spliethoff and S. Fendt, Power-to-liquid via synthesis of methanol, DME or Fischer–Tropsch-fuels: a review, Energy Environ. Sci., 2020, 13, 3207–3252 RSC.
- K. Saravanan, H. Ham, N. Tsubaki and J. W. Bae, Recent progress for direct synthesis of dimethyl ether from syngas on the heterogeneous bifunctional hybrid catalysts, Appl. Catal., B, 2017, 217, 494–522 CrossRef CAS.
- P. Munnik, P. E. De Jongh and K. P. De Jong, Recent Developments in the Synthesis of Supported Catalysts, Chem. Rev., 2015, 115, 6687–6718 CrossRef CAS PubMed.
- K. An and G. A. Somorjai, Size and Shape Control of Metal Nanoparticles for Reaction Selectivity in Catalysis, ChemCatChem, 2012, 4, 1512–1524 CrossRef CAS.
- M. Gentzen, et al., Bifunctional catalysts based on colloidal Cu/Zn nanoparticles for the direct conversion of synthesis gas to dimethyl ether and hydrocarbons, Appl. Catal., A, 2018, 557, 99–107 CrossRef CAS.
- M. Gentzen, et al., Bifunctional hybrid catalysts derived from Cu/Zn-based nanoparticles for single-step dimethyl ether synthesis, Catal. Sci. Technol., 2016, 6, 1054–1063 RSC.
- M. Gentzen, et al., An intermetallic Pd2Ga nanoparticle catalyst for the single-step conversion of CO-rich synthesis gas to dimethyl ether, Appl. Catal., A, 2018, 562, 206–214 CrossRef CAS.
- A. Ota, et al., Comparative study of hydrotalcite-derived supported Pd 2Ga and PdZn intermetallic nanoparticles as methanol synthesis and methanol steam reforming catalysts, J. Catal., 2012, 293, 27–38 CrossRef CAS.
- E. M. Fiordaliso, et al., Intermetallic GaPd2 Nanoparticles on SiO2 for Low-Pressure CO2 Hydrogenation to Methanol: Catalytic Performance and in Situ Characterization, ACS Catal., 2015, 5, 5827–5836 CrossRef CAS.
- G. Yang, D. Wang, Y. Yoneyama, Y. Tan and N. Tsubaki, Facile synthesis of H-type zeolite shell on a silica substrate for tandem catalysis, Chem. Commun., 2012, 48, 1263–1265 RSC.
- X. H. Gao, Q. X. Ma, T. S. Zhao, J. Bao and N. Tsubaki, Recent advances in multifunctional capsule catalysts in heterogeneous catalysis, Chin. J. Chem. Phys., 2018, 31, 393–403 CrossRef CAS.
- G. Yang, N. Tsubaki, J. Shamoto, Y. Yoneyama and Y. Zhang, Confinement effect and synergistic function of H-ZSM-5/Cu-ZnO-Al 2O3 capsule catalyst for one-step controlled synthesis, J. Am. Chem. Soc., 2010, 132, 8129–8136 CrossRef CAS PubMed.
- M. Sánchez-Contador, A. Ateka, A. T. Aguayo and J. Bilbao, Direct synthesis of dimethyl ether from CO and CO2 over a core-shell structured CuO-ZnO-ZrO2@SAPO-11 catalyst, Fuel Process. Technol., 2018, 179, 258–268 CrossRef.
- K. Pinkaew, et al., A new core-shell-like capsule catalyst with SAPO-46 zeolite shell encapsulated Cr/ZnO for the controlled tandem synthesis of dimethyl ether from syngas, Fuel, 2013, 111, 727–732 CrossRef CAS.
- G. Yang, et al., Tandem catalytic synthesis of light isoparaffin from syngas via Fischer-Tropsch synthesis by newly developed core-shell-like zeolite capsule catalysts, Catal. Today, 2013, 215, 29–35 CrossRef CAS.
- B. P. Karaman, N. Oktar, G. Doğu and T. Dogu, Heteropolyacid Incorporated Bifunctional Core-Shell Catalysts for Dimethyl Ether Synthesis from Carbon Dioxide/Syngas, Catalysts, 2022, 12, 1102 CrossRef CAS.
- G. Baracchini, et al., Structured catalysts for the direct synthesis of dimethyl ether from synthesis gas: A comparison of core@shell versus hybrid catalyst configuration, Catal. Today, 2020, 342, 46–58 CrossRef CAS.
- Y. Guo and Z. Zhao, Ethanol as a Binder to Fabricate a Highly-Efficient Capsule-Structured CuO−ZnO−Al2O3@HZSM-5 Catalyst for Direct Production of Dimethyl Ether from Syngas, ChemCatChem, 2020, 12, 999–1006 CrossRef CAS.
- A. Palčić, et al., Embryonic zeolites for highly efficient synthesis of dimethyl ether from syngas, Microporous Mesoporous Mater., 2021, 322, 111138 CrossRef.
- V. Thavasi, G. Singh and S. Ramakrishna, Electrospun nanofibers in energy and environmental applications, Energy Environ. Sci., 2008, 1, 205–221 RSC.
- S. F. Anis, A. Khalil, S. Saepurahman, G. Singaravel and R. Hashaikeh, A review on the fabrication of zeolite and mesoporous inorganic nanofibers formation for catalytic applications, Microporous Mesoporous Mater., 2016, 236, 176–192 CrossRef CAS.
- J. Palomo, M. Á. Rodríguez-Cano, J. Rodríguez-Mirasol and T. Cordero, ZSM-5-decorated CuO/ZnO/ZrO2 fibers as efficient bifunctional catalysts for the direct synthesis of DME from syngas, Appl. Catal., B, 2020, 270, 118893 CrossRef CAS.
- J. van Kampen, J. Boon, F. van Berkel, J. Vente and M. van Sint Annaland, Steam separation enhanced reactions: Review and outlook, Chem. Eng. J., 2019, 374, 1286–1303 CrossRef CAS.
- J. van Kampen, J. Boon, J. Vente and M. van Sint Annaland, Sorption enhanced dimethyl ether synthesis for high efficiency carbon conversion: Modelling and cycle design, J. CO2 Util., 2020, 37, 295–308 CrossRef CAS.
- P. Rodriguez-Vega, et al., Experimental implementation of a catalytic membrane reactor for the direct synthesis of DME from H2+CO/CO2, Chem. Eng. Sci., 2021, 234, 116396 CrossRef CAS.
- I. Iliuta, M. C. Iliuta and F. Larachi, Sorption-enhanced dimethyl ether synthesis-Multiscale reactor modeling, Chem. Eng. Sci., 2011, 66, 2241–2251 CrossRef CAS.
- J. Boon and F. P. F. Berkel, Separation Enhanced Dimethyl Ether Synthesis, Fifth Int. Conf. 2017 Tailor Made Fuels from Biomass, 2017 Search PubMed.
- D. Liuzzi, et al., Increasing dimethyl ether production from biomass-derived syngas by in situ steam adsorption, Sustainable Energy Fuels, 2020, 4, 5674–5681 RSC.
- A. Ateka, J. Ereña, J. Bilbao and A. T. Aguayo, Strategies for the Intensification of CO2 Valorization in the One-Step Dimethyl Ether Synthesis Process, Ind. Eng. Chem. Res., 2020, 59, 713–722 CrossRef CAS.
- J. van Kampen, S. Booneveld, J. Boon, J. Vente and M. van Sint Annaland, Experimental validation of pressure swing regeneration for faster cycling in sorption enhanced dimethyl ether synthesis, Chem. Commun., 2020, 56, 13540–13542 RSC.
- A. A. Al-Qadri, G. A. Nasser, H. Adamu, O. Muraza and T. A. Saleh, CO2 utilization in syngas conversion to dimethyl ether and aromatics: Roles and challenges of zeolites-based catalysts, J. Energy Chem., 2023, 79, 418–449 CrossRef CAS.
- S. Poto, M. A. Llosa Tanco, D. A. Pacheco Tanaka, M. F. N. d'Angelo and F. Gallucci, Experimental investigation of a packed bed membrane reactor for the direct conversion of CO2 to dimethyl ether, J. CO2 Util., 2023, 72, 102513 CrossRef CAS.
- X. Wang, et al., Catalytic activity for direct CO2 hydrogenation to dimethyl ether with different proximity of bifunctional Cu-ZnO-Al2O3 and ferrierite, Appl. Catal., B, 2023, 327, 122456 CrossRef CAS.
- K. B. Tan, K. Xu, D. Cai, J. Huang and G. Zhan, Rational design of bifunctional catalysts with proper integration manners for CO and CO2 hydrogenation into value-added products: A review, Chem. Eng. J., 2023, 463, 142262 CrossRef CAS.
- S. Xia, et al., Effects of precursor phase distribution on the performance of Cu-based catalysts for direct CO2 conversion to dimethyl ether, J. Energy Inst., 2023, 109, 101302 CrossRef CAS.
- M. Y. Mohamud, et al., Direct Synthesis of Dimethyl Ether from CO2 Hydrogenation over Core-Shell Nanotube Bi-Functional Catalyst, Catalysts, 2023, 13 Search PubMed.
- D. Kubas, et al., From CO2 to DME: Enhancement through Heteropoly Acids from a Catalyst Screening and Stability Study, ACS Omega, 2023, 8, 15203–15216 CrossRef CAS PubMed.
- R. Gao, et al., Conceptual design of full carbon upcycling of CO2 into clean DME fuel: Techno-economic assessment and process optimization, Fuel, 2023, 344, 128120 CrossRef CAS.
- D. Kubas, M. Semmel, O. Salem and I. Krossing, Is Direct DME Synthesis Superior to Methanol Production in Carbon Dioxide Valorization? From Thermodynamic Predictions to Experimental Confirmation, ACS Catal., 2023, 13, 3960–3970 CrossRef CAS.
- Y. Sun and Z. Zhao, Implanting Copper−Zinc Nanoparticles into the Matrix of Mesoporous Alumina as a Highly Selective Bifunctional Catalyst for Direct Synthesis of Dimethyl Ether from Syngas, ChemCatChem, 2020, 12, 1276–1281 CrossRef CAS.
- D. Mao, et al., Highly effective hybrid catalyst for the direct synthesis of dimethyl ether from syngas with magnesium oxide-modified HZSM-5 as a dehydration component, J. Catal., 2005, 230, 140–149 CrossRef CAS.
- F. Samimi, D. Karimipourfard and M. R. Rahimpour, Green methanol synthesis process from carbon dioxide via reverse water gas shift reaction in a membrane reactor, Chem. Eng. Res. Des., 2018, 140, 44–67 CrossRef.
- J. Yoshihara and C. T. Campbell, Methanol synthesis and reverse water-gas shift kinetics over Cu(110) model catalysts: Structural sensitivity, J. Catal., 1996, 161, 776–782 CrossRef CAS.
- G. C. Chinchen, M. S. Spencer, K. C. Waugh and D. A. Whan, Promotion of methanol synthesis and the water-gas shift reactions by adsorbed oxygen on supported copper catalysts, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 2193–2212 RSC.
- C. H. Bartholomew, Mechanisms of catalyst deactivation, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
- F. Dadgar, R. Myrstad, P. Pfeifer, A. Holmen and H. J. Venvik, Direct dimethyl ether synthesis from synthesis gas: The influence of methanol dehydration on methanol synthesis reaction, Catal. Today, 2016, 270, 76–84 CrossRef CAS.
- J. J. Spivey, Review: Dehydration catalysts for the methanol/dimethyl ether reaction, Chem. Eng. Commun., 1991, 110, 123–142 CrossRef CAS.
- A. Tariq, et al., Combination of Cu / ZnO Methanol Synthesis Catalysts and ZSM - 5 Zeolites to Produce Oxygenates from CO 2 and H 2, Top. Catal., 2021, 64, 965–973 CrossRef CAS.
- C. H. Mejía, D. M. A. Verbart and K. P. de Jong, Niobium-based solid acids in combination with a methanol synthesis catalyst for the direct production of dimethyl ether from synthesis gas, Catal. Today, 2021, 369, 77–87 CrossRef.
- G. Prieto, J. Zečević, H. Friedrich, K. P. De Jong and P. E. De Jongh, Towards stable catalysts by controlling collective properties of supported metal nanoparticles, Nat. Mater., 2013, 12, 34–39 CrossRef CAS PubMed.
- M. S. Spencer and M. V. Twigg, Metal catalyst design and preparation in control of deactivation, Annu. Rev. Mater. Res., 2005, 35, 427–464 CrossRef CAS.
- X. Fan, et al., Roles of interaction between components in CZZA/HZSM-5 catalyst for dimethyl ether synthesis via CO2 hydrogenation, AIChE J., 2021, 67, e17353 CrossRef CAS.
- M. B. Fichtl, et al., Kinetics of deactivation on Cu/ZnO/Al2O3 methanol synthesis catalysts, Appl. Catal., A, 2015, 502, 262–270 CrossRef CAS.
- T. Lunkenbein, et al., Bridging the Time Gap: A Copper/Zinc Oxide/Aluminum Oxide Catalyst for Methanol Synthesis Studied under Industrially Relevant Conditions and Time Scales, Angew. Chem., Int. Ed., 2016, 55, 12708–12712 CrossRef CAS PubMed.
- A. Y. Khodakov, et al., Assessment of metal sintering in the copper-zeolite hybrid catalyst for direct dimethyl ether synthesis using synchrotron-based X-ray absorption and diffraction, Catal. Today, 2020, 343, 199–205 CrossRef CAS.
- V. Vishwanathan, K. W. Jun, J. W. Kim and H. S. Roh, Vapour phase dehydration of crude methanol to dimethyl ether over Na-modified H-ZSM-5 catalysts, Appl. Catal., A, 2004, 276, 251–255 CrossRef CAS.
- Y. Wei, et al., Enhanced catalytic performance of zeolite ZSM-5 for conversion of methanol to dimethyl ether by combining alkaline treatment and partial activation, Appl. Catal., A, 2015, 504, 211–219 CrossRef CAS.
- M. Choi, et al., Stable single-unit-cell nanosheets of zeolite MFI as active and long-lived catalysts, Nature, 2009, 461, 246–249 CrossRef CAS PubMed.
- F. Dadgar, R. Myrstad, P. Pfeifer, A. Holmen and H. J. Venvik, Catalyst Deactivation During One-Step Dimethyl Ether Synthesis from Synthesis Gas, Catal. Lett., 2017, 147, 865–879 CrossRef CAS.
- J. Boon, et al., Reversible deactivation of γ-alumina by steam in the gas-phase dehydration of methanol to dimethyl ether, Catal. Commun., 2019, 119, 22–27 CrossRef CAS.
- S. Sahebdelfar, P. M. Bijani and F. Yaripour, Deactivation kinetics of γ-Al2O3 catalyst in methanol dehydration to dimethyl ether, Fuel, 2022, 310, 122443 CrossRef CAS.
- S. Hayashi, et al., Nb2O5.nH2O as a Heterogeneous Catalyst with Water-Tolerant Lewis Acid Sites, J. Am. Chem. Soc., 2011, 133, 4224–4227 CrossRef PubMed.
- Q. Sun, Y. Fu, H. Yang, A. Auroux and J. Shen, Dehydration of methanol to dimethyl ether over Nb2O5and NbOPO4catalysts: Microcalorimetric and FT-IR studies, J. Mol. Catal. A: Chem., 2007, 275, 183–193 CrossRef CAS.
- D. Liu, C. Yao, J. Zhang, D. Fang and D. Chen, Catalytic dehydration of methanol to dimethyl ether over modified γ-Al2O3catalyst, Fuel, 2011, 90, 1738–1742 CrossRef CAS.
- R. Ladera, E. Finocchio, S. Rojas, J. L. G. Fierro and M. Ojeda, Supported niobium catalysts for methanol dehydration to dimethyl ether: FTIR studies of acid properties, Catal. Today, 2012, 192, 136–143 CrossRef CAS.
- A. S. Rocha, A. M. Aline, E. R. Lachter, E. F. Sousa-Aguiar and A. C. Faro, Niobia-modified aluminas prepared by impregnation with niobium peroxo complexes for dimethyl ether production, Catal. Today, 2012, 192, 104–111 CrossRef CAS.
- S. H. Lima, A. M. S. Forrester, L. A. Palacio and A. C. Faro, Niobia-alumina as methanol dehydration component in mixed catalyst systems for dimethyl ether production from syngas, Appl. Catal., A, 2014, 488, 19–27 CrossRef CAS.
- B. Voss, A. Katerinopoulou, R. Montesano and J. Sehested, Zn and Si transport in dual-functioning catalyst for conversion of synthesis gas to dimethyl ether, Chem. Eng. J., 2019, 121940, DOI:10.1016/j.cej.2019.121940.
- A. García-Trenco, A. Vidal-Moya and A. Martínez, Study of the interaction between components in hybrid CuZnAl/HZSM-5 catalysts and its impact in the syngas-to-DME reaction, Catal. Today, 2012, 179, 43–51 CrossRef.
- A. García-Trenco, S. Valencia and A. Martínez, The impact of zeolite pore structure on the catalytic behavior of CuZnAl/zeolite hybrid catalysts for the direct DME synthesis, Appl. Catal., A, 2013, 468, 102–111 CrossRef.
- A. García-Trenco and A. Martínez, The influence of zeolite surface-aluminum species on the deactivation of CuZnAl/zeolite hybrid catalysts for the direct DME synthesis, Catal. Today, 2014, 227, 144–153 CrossRef.
- V. V. Ordomsky, et al., The role of external acid sites of ZSM-5 in deactivation of hybrid CuZnAl/ZSM-5 catalyst for direct dimethyl ether synthesis from syngas, Appl. Catal., A, 2014, 486, 266–275 CrossRef CAS.
- M. Cai, et al., Direct dimethyl ether synthesis from syngas on copper-zeolite hybrid catalysts with a wide range of zeolite particle sizes Dedicated to Professor Jean-Pierre Gilson on the occasion of his 60th birthday, J. Catal., 2016, 338, 227–238 CrossRef CAS.
- Y. Li, et al., Distance for Communication between Metal and Acid Sites for Syngas Conversion, ACS Catal., 2022, 12, 8793–8801 CrossRef CAS.
- Y. Wang, A new horizontal in C1 chemistry: Highly selective conversion of syngas to light olefins by a novel OX-ZEO process, J. Energy Chem., 2016, 25, 169–170 CrossRef.
- F. Jiao, et al., Shape-Selective Zeolites Promote Ethylene Formation from Syngas via a Ketene Intermediate, Angew. Chem., Int. Ed., 2018, 57, 4692–4696 CrossRef CAS PubMed.
- K. Cheng, et al., Direct and Highly Selective Conversion of Synthesis Gas into Lower Olefins: Design of a Bifunctional Catalyst Combining Methanol Synthesis and Carbon-Carbon Coupling, Angew. Chem., 2016, 128, 4803–4806 CrossRef.
- F. Meng, B. Li, J. Zhang, L. Wang and Z. Li, Role of Zn-Al oxide structure and oxygen vacancy in bifunctional catalyst for syngas conversion to light olefins, Fuel, 2023, 346, 128351 CrossRef CAS.
- F. Meng, et al., Unraveling the role of GaZrOx structure and oxygen vacancy in bifunctional catalyst for highly active and selective conversion of syngas into light olefins, Chem. Eng. J., 2023, 467, 143500 CrossRef CAS.
- X. Liu, et al., Design of efficient bifunctional catalysts for direct conversion of syngas into lower olefins: Via methanol/dimethyl ether intermediates, Chem. Sci., 2018, 9, 4708–4718 RSC.
- F. Meng, et al., Effect of zeolite topological structure in bifunctional catalyst on direct conversion of syngas to light olefins, Microporous Mesoporous Mater., 2023, 362, 112792 CrossRef CAS.
- J. Xie and U. Olsbye, The Oxygenate-Mediated Conversion of COx to Hydrocarbons—On the Role of Zeolites in Tandem Catalysis, Chem. Rev., 2023, 123, 11775–11816 CrossRef CAS PubMed.
- P. Tian, Y. Wei, M. Ye and Z. Liu, Methanol to olefins (MTO): From fundamentals to commercialization, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- M. Wang, et al., Effect of zeolite topology on the hydrocarbon distribution over bifunctional ZnAlO/SAPO catalysts in syngas conversion, Catal. Today, 2021, 371, 85–92 CrossRef CAS.
- Y. Zhu, et al., Role of Manganese Oxide in Syngas Conversion to Light Olefins, ACS Catal., 2017, 7, 2800–2804 CrossRef CAS.
- J. Su, et al., Direct Conversion of Syngas into Light Olefins over Zirconium-Doped Indium(III) Oxide and SAPO-34 Bifunctional Catalysts: Design of Oxide Component and Construction of Reaction Network, ChemCatChem, 2018, 10, 1536–1541 CrossRef CAS.
- J. Su, et al., Syngas to light olefins conversion with high olefin/paraffin ratio using ZnCrO x /AlPO-18 bifunctional catalysts, Nat. Commun., 2019, 10, 1297 CrossRef PubMed.
- H. Zhou, et al., Light Olefin Synthesis from Syngas over Sulfide-Zeolite Composite Catalyst, Ind. Eng. Chem. Res., 2018, 57, 6815–6820 CrossRef CAS.
- Q. Zhang, J. Yu and A. Corma, Applications of Zeolites to C1 Chemistry: Recent Advances, Challenges, and Opportunities, Adv. Mater., 2020, 32, 2002927 CrossRef PubMed.
- F. Jiao, et al., Disentangling the activity-selectivity trade-off in catalytic conversion of syngas to light olefins, Science, 2023, 380, 727–730 CrossRef CAS PubMed.
- H. M. Torres Galvis and K. P. De Jong, ACS Catal., 2013, 3, 2130–2149 CrossRef CAS.
- Z. Lai, et al., Resolving the Intricate Mechanism and Selectivity of Syngas Conversion on Reduced ZnCr2Ox: A Quantitative Study from DFT and Microkinetic Simulations, ACS Catal., 2021, 11, 12977–12988 CrossRef CAS.
- H. Chen, et al., A Mechanistic Study of Syngas to Light Olefins over OXZEO Bifunctional Catalysts: Insights into the Initial Carbon-Carbon Bond Formation on the Oxide, Catal. Sci. Technol., 2022, 12, 1289–1295 RSC.
- C. M. Wang, Y. D. Wang and Z. K. Xie, Methylation of olefins with ketene in zeotypes and its implications for the direct conversion of syngas to light olefins: A periodic DFT study, Catal. Sci. Technol., 2016, 6, 6644–6649 RSC.
- A. D. Chowdhury and J. Gascon, The Curious Case of Ketene in Zeolite Chemistry and Catalysis, Angew. Chem., Int. Ed., 2018, 57, 14982–14985 CrossRef CAS PubMed.
- K. P. de Jong, Surprised by selectivity, Science, 2016, 351, 1030–1031 CrossRef CAS PubMed.
- L. Ren, et al., Syngas to light olefins over ZnAlOx and high-silica CHA prepared by boron-assisted hydrothermal synthesis, Fuel, 2022, 307, 121916 CrossRef CAS.
- Y. Huang, et al., Utilization of SAPO-18 or SAPO-35 in the bifunctional catalyst for the direct conversion of syngas to light olefins, RSC Adv., 2021, 11, 13876–13884 RSC.
- Y. Huang, et al., Direct Conversion of Syngas to Light Olefins over a ZnCrOx + H-SSZ-13 Bifunctional Catalyst, ACS Omega, 2021, 6, 10953–10962 CrossRef CAS PubMed.
- M. Tong, et al., Tandem catalysis over tailored ZnO-ZrO2/MnSAPO-34 composite catalyst for enhanced light olefins selectivity in CO2 hydrogenation, Microporous Mesoporous Mater., 2021, 320, 111105 CrossRef CAS.
- Z. Di, T. Zhao, X. Feng and M. Luo, A Newly Designed Core-Shell-Like Zeolite Capsule Catalyst for Synthesis of Light Olefins from Syngas via Fischer–Tropsch Synthesis Reaction, Catal. Lett., 2019, 149, 441–448 CrossRef CAS.
- C. Zhu, M. Zhang, C. Huang, Y. Han and K. Fang, Controlled Nanostructure of Zeolite Crystal Encapsulating FeMnK Catalysts Targeting Light Olefins from Syngas, ACS Appl. Mater. Interfaces, 2020, 12, 57950–57962 CrossRef CAS PubMed.
- R. Hu, et al., Nano-Hollow Zeolite-Encapsulated Highly Dispersed Ultra-Fine Fe Nanoparticles as Fischer–Tropsch Catalyst for Syngas-to-Olefins, Catalysts, 2023, 13 Search PubMed.
- F. Song, et al., FeMn@HZSM-5 capsule catalyst for light olefins direct synthesis via Fischer-Tropsch synthesis: Studies on depressing the CO2 formation, Appl. Catal., B, 2022, 300, 120713 CrossRef CAS.
- X. Liu, T. Lin, P. Liu and L. Zhong, Hydrophobic interfaces regulate iron carbide phases and catalytic performance of FeZnOx nanoparticles for Fischer-Tropsch to olefins, Appl. Catal., B, 2023, 331, 122697 CrossRef CAS.
- T. Lin, et al., Designing silica-coated CoMn-based catalyst for Fischer-Tropsch synthesis to olefins with low CO2 emission, Appl. Catal., B, 2021, 299, 120683 CrossRef CAS.
- P. Liu, et al., Tuning cobalt carbide wettability environment for Fischer-Tropsch to olefins with high carbon efficiency, Chin. J. Catal., 2023, 48, 150–163 CrossRef CAS.
- Y. Ding, et al., Effects of Proximity-Dependent Metal Migration on Bifunctional Composites Catalyzed Syngas to Olefins, ACS Catal., 2021, 11, 9729–9737 CrossRef CAS.
- N. Li, et al., Size Effects of ZnO Nanoparticles in Bifunctional Catalysts for Selective Syngas Conversion, ACS Catal., 2019, 9, 960–966 CrossRef CAS.
- A. Portillo, A. Ateka, J. Ereña, A. T. Aguayo and J. Bilbao, Conditions for the Joint Conversion of CO2 and Syngas in the Direct Synthesis of Light Olefins Using In2O3–ZrO2/SAPO-34 Catalyst, Ind. Eng. Chem. Res., 2022, 61, 10365–10376 CrossRef CAS PubMed.
- T. Numpilai, et al., CO2 Hydrogenation to Light Olefins Over In2O3/SAPO-34 and Fe-Co/K-Al2O3 Composite Catalyst, Top. Catal., 2021, 64, 316–327 CrossRef CAS.
- S. Lu, et al., Effect of In2O3 particle size on CO2 hydrogenation to lower olefins over bifunctional catalysts, Chin. J. Catal., 2021, 42, 2038–2048 CrossRef CAS.
- S. Wang, et al., Selective Conversion of CO2 into Propene and Butene, Chem, 2020, 6, 3344–3363 CAS.
- W. Li, K. Wang, G. Zhan, J. Huang and Q. Li, Design and Synthesis of Bioinspired ZnZrOx&Bio-ZSM-5 Integrated Nanocatalysts to Boost CO2 Hydrogenation to Light Olefins, ACS Sustainable Chem. Eng., 2021, 9, 6446–6458 CrossRef CAS.
- L. Zhang, et al., Boosting Conversion of CO2 to Light Olefins over MgO-Promoted ZnZrO/SAPO-34 Bifunctional Catalyst, Ind. Eng. Chem. Res., 2023, 62, 9123–9133 CrossRef CAS.
- Y. Wang, et al., Direct conversion of carbon dioxide into light olefins over ZnZrOx/ZSM-5@n-ZrO2 tandem catalyst, Fuel, 2024, 357, 129727 CrossRef CAS.
- J. Mou, et al., CO2 hydrogenation to lower olefins over Mn2O3-ZnO/SAPO-34 tandem catalysts, Chem. Eng. J., 2021, 421, 129978 CrossRef CAS.
- M. Ghavipour, R. M. Behbahani, R. B. Rostami and A. S. Lemraski, Methanol/dimethyl ether to light olefins over SAPO-34: Comprehensive comparison of the products distribution and catalyst performance, J. Nat. Gas Sci. Eng., 2014, 21, 532–539 CrossRef CAS.
- Y. Li, M. Zhang, D. Wang, F. Wei and Y. Wang, Differences in the methanol-to-olefins reaction catalyzed by SAPO-34 with dimethyl ether as reactant, J. Catal., 2014, 311, 281–287 CrossRef CAS.
- J. Su, et al., High Conversion of Syngas to Ethene and Propene on Bifunctional Catalysts via the Tailoring of SAPO Zeolite Structure, Cell Rep. Phys. Sci., 2021, 2, 100290 CrossRef CAS.
- S. Wang, et al., Catalytic roles of the acid sites in different pore channels of H-ZSM-5 zeolite for methanol-to-olefins conversion, Chin. J. Catal., 2021, 42, 1126–1136 CrossRef CAS.
- K. Cheng, et al., Bifunctional Catalysts for One-Step Conversion of Syngas into Aromatics with Excellent Selectivity and Stability, Chem, 2017, 3, 334–347 CAS.
- D. L. S. Nieskens, J. D. Lunn and A. Malek, Understanding the Enhanced Lifetime of SAPO-34 in a Direct Syngas-to-Hydrocarbons Process, ACS Catal., 2019, 9, 691–700 CrossRef CAS.
- M. Wang, et al., Synthesis of hierarchical SAPO-34 to improve the catalytic performance of bifunctional catalysts for syngas-to-olefins reactions, J. Catal., 2021, 394, 181–192 CrossRef CAS.
- E. G. Galanova, M. V. Magomedova, M. I. Afokin, A. V. Starozhitskaya and A. L. Maximov, Synthesis of olefins from dimethyl ether in a synthesis gas atmosphere, Catal. Commun., 2021, 153, 106297 CrossRef CAS.
- W. Zhou, et al., Selective Conversion of Syngas to Aromatics over a Mo−ZrO 2 /H-ZSM-5 Bifunctional Catalyst, ChemCatChem, 2019, 11, 1681–1688 CrossRef CAS.
- X. Liu, et al., Selective transformation of carbon dioxide into lower olefins with a bifunctional catalyst composed of ZnGa2O4 and SAPO-34, Chem. Commun., 2017, 54, 140–143 RSC.
- L. Zhong, et al., Cobalt carbide nanoprisms for direct production of lower olefins from syngas, Nature, 2016, 538, 84–87 CrossRef CAS PubMed.
- B. Gu, et al., Effects of the promotion with bismuth and lead on direct synthesis of light ole fi ns from syngas over carbon nanotube supported iron catalysts, Appl. Catal., B, 2018, 234, 153–166 CrossRef CAS.
- C. E. Pompe, et al., Stability of mesocellular foam supported copper catalysts for methanol synthesis, Catal. Today, 2019, 334, 79–89 CrossRef CAS.
- P. Reubroycharoen, et al., Continuous low-temperature methanol synthesis from syngas using alcohol promoters, Energy Fuels, 2003, 17, 817–821 CrossRef CAS.
- S. Wang, et al., Direct Conversion of Syngas into Light Olefins with Low CO2 Emission, ACS Catal., 2020, 10, 2046–2059 CrossRef CAS.
- J. Liu, et al., Nano-ZrO2 as hydrogenation phase in bi-functional catalyst for syngas aromatization, Fuel, 2020, 263, 116803 CrossRef CAS.
- J. Li, T. Yu, D. Miao, X. Pan and X. Bao, Carbon dioxide hydrogenation to light olefins over ZnO-Y2O3 and SAPO-34 bifunctional catalysts, Catal. Commun., 2019, 129, 105711 CrossRef.
- G. Li, et al., Selective conversion of syngas to propane over ZnCrOx-SSZ-39 OX-ZEO catalysts, J. Energy Chem., 2019, 36, 141–147 CrossRef.
- Y. S. T. Sun, T. Lin, Y. An, K. Gong and L. Zhong, Syngas Conversion to Aromatics over Co 2 C- based Catalyst and HZSM-5 via Tandem System, Ind. Eng. Chem. Res., 2020, 59, 4419–4427 CrossRef.
- L. Zhang, et al., Insight into the impact of Al distribution on the catalytic performance of 1-octene aromatization over ZSM-5 zeolite, Catal. Sci. Technol., 2019, 9, 7034–7044 RSC.
- C. Wang, et al., Impact of temporal and spatial distribution of hydrocarbon pool on methanol conversion over H-ZSM-5, J. Catal., 2017, 354, 138–151 CrossRef CAS.
- J. Xie, et al., Promoted cobalt metal catalysts suitable for the production of lower olefins from natural gas, Nat. Commun., 2019, 10, 1–10 CrossRef PubMed.
- Y. Xu, et al., Effect of iron loading on acidity and performance of Fe/HZSM-5 catalyst for direct synthesis of aromatics from syngas, Fuel, 2018, 228, 1–9 CrossRef.
- A. Ramirez, et al., Selectivity descriptors for the direct hydrogenation of CO2 to hydrocarbons during zeolite-mediated bifunctional catalysis, Nat. Commun., 2021, 12, 5914 CrossRef CAS PubMed.
- Z. Huang, et al., Ceria-Zirconia/Zeolite Bifunctional Catalyst for Highly Selective Conversion of Syngas into Aromatics, ChemCatChem, 2018, 10, 4519–4524 CrossRef CAS.
- Y. Xu, D. Liu and X. Liu, Conversion of syngas toward aromatics over hybrid Fe-based Fischer-Tropsch catalysts and HZSM-5 zeolites, Appl. Catal., A, 2018, 552, 168–183 CrossRef CAS.
- S. C. Kang, et al., Enhancing selectivity of aromatics in direct conversion of syngas over K/FeMn and HZSM-5 bifunctional catalysts, Mol. Catal., 2022, 533, 112790 CrossRef CAS.
- K. Fujimoto, I. Nakamura, K. Yokota and K. Aimoto, On the Role of Gallium for the Aromatization of Lower Paraffins with Ga-Promoted ZSM-5 Catalysts, Bull. Chem. Soc. Jpn., 1991, 64, 2275–2280 CrossRef CAS.
- Y. Fang, et al., Aromatization over nanosized Ga-containing ZSM-5 zeolites prepared by different methods: Effect of acidity of active Ga species on the catalytic performance, J. Energy Chem., 2017, 26, 768–775 CrossRef.
- X. Su, W. Zan, X. Bai, G. Wang and W. Wu, Synthesis of microscale and nanoscale ZSM-5 zeolites: Effect of particle size and acidity of Zn modified ZSM-5 zeolites on aromatization performance, Catal. Sci. Technol., 2017, 7, 1943–1952 RSC.
- A. Kostyniuk, D. Key and M. Mdleleni, Effect of Fe-Mo promoters on HZSM-5 zeolite catalyst for 1-hexene aromatization, J. Saudi Chem. Soc., 2019, 23, 612–626 CrossRef CAS.
- Y. Xu, et al., Synthesis of aromatics from syngas over FeMnK/SiO2 and HZSM-5 tandem catalysts, Mol. Catal., 2018, 454, 104–113 CrossRef CAS.
- Z. Chen, et al., Temperature-dependent secondary conversion of primary products from methanol aromatization in a two-stage fluidized bed, Fuel, 2020, 267, 117204 CrossRef CAS.
- E. A. Uslamin, et al., Aromatization of ethylene over zeolite-based catalysts, Catal. Sci. Technol., 2020, 10, 2774–2785 RSC.
- Y. Wang, et al., Direct conversion of CO2 to aromatics with high yield via a modified Fischer-Tropsch synthesis pathway, Appl. Catal., B, 2020, 269, 118792 CrossRef CAS.
- C. Wen, et al., Effect of hierarchical ZSM-5 zeolite support on direct transformation from syngas to aromatics over the iron-based catalyst, Fuel, 2019, 244, 492–498 CrossRef CAS.
- G. Song, M. Li, P. Yan, M. A. Nawaz and D. Liu, High Conversion to Aromatics via CO2-FT over a CO-Reduced Cu-Fe2O3 Catalyst Integrated with HZSM-5, ACS Catal., 2020, 10, 11268–11279 CrossRef CAS.
- C. Wen, et al., Insight into the direct conversion of syngas toward aromatics over the Cu promoter Fe-zeolite tandem catalyst, Fuel, 2023, 331, 125855 CrossRef CAS.
- S. Wang, Y. Fang, Z. Huang, H. Xu and W. Shen, The Effects of the Crystalline Phase of Zirconia on C – O Activation and C – C Coupling in Converting Syngas into Aromatics, Catalysts, 2020, 10, 262 CrossRef CAS.
- J. Yang, X. Pan, F. Jiao, J. Li and X. Bao, Direct conversion of syngas to aromatics, Chem. Commun., 2017, 53, 11146–11149 RSC.
- P. Zhang, L. Tan, G. Yang and N. Tsubaki, One-pass selective conversion of syngas to para-xylene, Chem. Sci., 2017, 8, 7941–7946 RSC.
- T. Yang, L. Cheng, N. Li and D. Liu, Effect of Metal Active Sites on the Product Distribution over Composite Catalysts in the Direct Synthesis of Aromatics from Syngas, Ind. Eng. Chem. Res., 2017, 56, 11763–11772 CrossRef CAS.
- N. Li, et al., High-Quality Gasoline Directly from Syngas by Dual Metal Oxide–Zeolite (OX-ZEO) Catalysis, Angew. Chem., 2019, 131, 7478–7482 CrossRef.
- G. Tian, et al., Accelerating syngas-to-aromatic conversion via spontaneously monodispersed Fe in ZnCr2O4 spinel, Nat. Commun., 2022, 13, 5567 CrossRef CAS PubMed.
- D. Ma, et al., The Direct Synthesis of Aromatic Hydrocarbons from Syngas over Bifunctional MgZrOx/HZSM-5 Catalysts, Catalysts, 2023, 13 Search PubMed.
- X. Yang, et al., The influence of intimacy on the ‘iterative reactions’ during OX-ZEO process for aromatic production, J. Energy Chem., 2019, 35, 60–65 CrossRef.
- Y. Ji, et al., Oxygenate-based routes regulate syngas conversion over oxide–zeolite bifunctional catalysts, Nat. Catal., 2022, 5, 594–604 CrossRef CAS.
- Z. Ma, et al., Catalytic roles of acid property in different morphologies of H-ZSM-5 zeolites for syngas-to-aromatics conversion over ZnCrOx/H-ZSM-5 catalysts, Microporous Mesoporous Mater., 2023, 349, 112420 CrossRef CAS.
- Y. Fang, et al., Enhancing BTX selectivity of the syngas to aromatics reaction through silylation of CTAB pretreated ZSM-5, Catal. Sci. Technol., 2021, 11, 4944–4952 RSC.
- J. Zhang, W. Qian, C. Kong and F. Wei, Increasing para-Xylene Selectivity in Making Aromatics from Methanol with a Surface-Modified Zn/P/ZSM-5 Catalyst, ACS Catal., 2015, 5, 2982–2988 CrossRef CAS.
- D. Chen, J. Wang, X. Ren, H. Teng and H. Gu, Silicalite-1 shell synthesized onto cylinder-shaped ZSM-5 extrudate for disproportionation of toluene into para-xylene, Catal. Lett., 2010, 136, 65–70 CrossRef CAS.
- T. Wang, et al., Direct production of aromatics from syngas over a hybrid FeMn Fischer-Tropsch catalyst and HZSM-5 zeolite: Local environment effect and mechanism-directed tuning of the aromatic selectivity, Catal. Sci. Technol., 2019, 9, 3933–3946 RSC.
- D. Miao, et al., Selective Synthesis of Benzene, Toluene, and Xylenes from Syngas, ACS Catal., 2020, 10, 7389–7397 CrossRef CAS.
- Y. Wang, et al., Boosting the synthesis of value-added aromatics directly from syngas via a Cr2O3 and Ga doped zeolite capsule catalyst, Chem. Sci., 2021, 12, 7786–7792 RSC.
- Z. Hua, Y. Yang and J. Liu, Direct hydrogenation of carbon dioxide to value-added aromatics, Coord. Chem. Rev., 2023, 478, 214982 CrossRef CAS.
- L. Zhang, et al., Highly selective synthesis of light aromatics from CO2 by chromium-doped ZrO2 aerogels in tandem with HZSM-5@SiO2 catalyst, Appl. Catal., B, 2023, 328, 122535 CrossRef CAS.
- N. A. Krans, et al., Influence of Promotion on the Growth of Anchored Colloidal Iron Oxide Nanoparticles during Synthesis Gas Conversion, ACS Catal., 2020, 10, 1913–1922 CrossRef CAS PubMed.
- H. Liu, et al., Activated carbon templated synthesis of hierarchical zeolite Y-encapsulated iron catalysts for enhanced gasoline selectivity in CO hydrogenation, J. Mater. Chem. A, 2021, 9, 8663–8673 RSC.
- J. L. Weber, et al., Conversion of synthesis gas to aromatics at medium temperature with a fischer tropsch and ZSM-5 dual catalyst bed, Catal. Today, 2021, 369, 175–183 CrossRef CAS.
- Y. Jia, et al., Hierarchical ZSM-5 zeolite synthesized via dry gel conversion-steam assisted crystallization process and its application in aromatization of methanol, Powder Technol., 2018, 328, 415–429 CrossRef CAS.
- S. Teketel, U. Olsbye, K. P. Lillerud, P. Beato and S. Svelle, Co-conversion of methanol and light alkenes over acidic zeolite catalyst H-ZSM-22: Simulated recycle of non-gasoline range products, Appl. Catal., A, 2015, 494, 68–76 CrossRef CAS.
- A. Aloise, E. Catizzone, M. Migliori, J. B. Nagy and G. Giordano, Catalytic behavior in propane aromatization using GA-MFI catalyst, Chin. J. Chem. Eng., 2017, 25, 1863–1870 CrossRef.
- M. W. Schreiber, et al., Lewis-Brønsted Acid Pairs in Ga/H-ZSM-5 to Catalyze Dehydrogenation of Light Alkanes, J. Am. Chem. Soc., 2018, 140, 4849–4859 CrossRef CAS PubMed.
- P. Gao, et al., A Mechanistic Study of Methanol-to-Aromatics Reaction over Ga-Modified ZSM-5 Zeolites: Understanding the Dehydrogenation Process, ACS Catal., 2018, 8, 9809–9820 CrossRef CAS.
- M. Oschatz, et al., Effects of calcination and activation conditions on ordered mesoporous carbon supported iron catalysts for production of lower olefins from synthesis gas, Catal. Sci. Technol., 2016, 6, 8464–8473 RSC.
- M. Casavola, et al., Promoted Iron Nanocrystals Obtained via Ligand Exchange as Active and Selective Catalysts for Synthesis Gas Conversion, ACS Catal., 2017, 7, 5121–5128 CrossRef CAS PubMed.
- M. Li, M. A. Nawaz, G. Song, W. Q. Zaman and D. Liu, Influential role of elemental migration in composite iron- zeolite catalyst for aromatics synthesis from syngas Influential role of elemental migration in composite iron-zeolite catalyst for aromatics synthesis from syngas, Ind. Eng. Chem. Res., 2020, 59, 9043–9054 CrossRef CAS.
- S. Kim, Y. T. Kim, A. Hwang, K. W. Jun and G. Kwak, Coke-Tolerant Gadolinium-Promoted HZSM-5 Catalyst for Methanol Conversion into Hydrocarbons, ChemCatChem, 2017, 9, 1569–1573 CrossRef CAS.
- Y. Li, Y. Zhang, K. Qian and W. Huang, Metal–Support Interactions in Metal/Oxide Catalysts and Oxide–Metal Interactions in Oxide/Metal Inverse Catalysts, ACS Catal., 2022, 12, 1268–1287 CrossRef CAS.
- W. Zhou, et al., Direct conversion of syngas into aromatics over a bifunctional catalyst: Inhibiting net CO2release, Chem. Commun., 2020, 56, 5239–5242 RSC.
- P. Zhai, et al., Highly Tunable Selectivity for Syngas-Derived Alkenes over Zinc and Sodium-Modulated Fe5C2Catalyst, Angew. Chem., Int. Ed., 2016, 55, 9902–9907 CrossRef CAS PubMed.
- H. Wang, et al., Bifunctional catalysts with versatile zeolites enable unprecedented para-xylene productivity for syngas conversion under mild conditions, Chem Catal., 2022, 2, 779–796 CrossRef CAS.
- W. Song, Y. Hou, Z. Chen, D. Cai and W. Qian, Process simulation of the syngas-to-aromatics processes: Technical economics aspects, Chem. Eng. Sci., 2020, 212, 115328 CrossRef CAS.
- D. X. Martínez-Vargas, et al., Recent Advances in Bifunctional Catalysts for the Fischer-Tropsch Process: One-Stage Production of Liquid Hydrocarbons from Syngas, Ind. Eng. Chem. Res., 2019, 58, 15872–15901 CrossRef.
- G. Eglof and P. M. Van Arsdell, Octane rating relationships of aliphatic, alicyclic, mononuclear aromatic, Inst. Pet., 1941, 27, 121–138 Search PubMed.
- L. Cai, R. Tripathi, R. Broda and H. Pitsch, A property database of fuel compounds with emphasis on spark-ignition engine applications, Appl. Energy Combust. Sci., 2021, 5, 100018 Search PubMed.
- V. L. Dagle, et al., Production, fuel properties and combustion testing of an iso-olefins blendstock for modern vehicles, Fuel, 2022, 310, 122314 CrossRef CAS.
- M. Yang, et al., Fischer-Tropsch wax catalytic cracking for the production of low olefin and high octane number gasoline: Experiment and molecular level kinetic modeling study, Fuel, 2021, 303, 121226 CrossRef CAS.
- N. Duyckaerts, I. T. Trotuş, A. C. Swertz, F. Schüth and G. Prieto, In Situ Hydrocracking of Fischer-Tropsch Hydrocarbons: CO-Prompted Diverging Reaction Pathways for Paraffin and α-Olefin Primary Products, ACS Catal., 2016, 6, 4229–4238 CrossRef CAS.
- R. E. Yakovenko, et al., Bifunctional Cobalt-Containing Catalytic Systems Based on SAPO-11 Molecular Sieves in Fischer–Tropsch Synthesis of Fuels, Pet. Chem., 2021, 61, 378–387 CrossRef CAS.
- J. Li, et al., Integrated tuneable synthesis of liquid fuels via Fischer–Tropsch technology, Nat. Catal., 2018, 1, 787–793 CrossRef CAS.
- C. Xing, et al., Hierarchical zeolite y supported cobalt bifunctional catalyst for facilely tuning the product distribution of Fischer-Tropsch synthesis, Fuel, 2015, 148, 48–57 CrossRef CAS.
- C. Xing, et al., Completed encapsulation of cobalt particles in mesoporous H-ZSM-5 zeolite catalyst for direct synthesis of middle isoparaffin from syngas, Catal. Commun., 2014, 55, 53–56 CrossRef CAS.
- G. Park, et al., Diffusion-dependent upgrading of hydrocarbons synthesized by Co/zeolite bifunctional Fischer–Tropsch catalysts, Appl. Catal., A, 2020, 607, 117840 CrossRef CAS.
- Y. Zhuo, L. Zhu, J. Liang and S. Wang, Selective Fischer-Tropsch synthesis for gasoline production over Y, Ce, or La-modified Co/H-β, Fuel, 2020, 262, 116490 CrossRef CAS.
- J. Feng, et al., Direct Synthesis of Isoparaffin-rich Gasoline from Syngas, ACS Energy Lett., 2022, 7, 1462–1468 CrossRef CAS.
- A. Straß-Eifert, et al., Bifunctional Co-based Catalysts for Fischer-Tropsch Synthesis: Descriptors affecting the product distribution, ChemCatChem, 2021, 13, 2726–2742 CrossRef.
- C. Xing, et al., Syngas to isoparaffins: Rationalizing selectivity over zeolites assisted by a predictive isomerization model, Fuel, 2021, 285, 119233 CrossRef CAS.
- N. Duyckaerts, et al., Intermediate Product Regulation in Tandem Solid Catalysts with Multimodal Porosity for High-Yield Synthetic Fuel Production, Angew. Chem., 2017, 129, 11638–11642 CrossRef.
- C. Zhu, D. P. Gamliel, J. A. Valla and G. M. Bollas, Fischer-Tropsch synthesis in monolith catalysts coated with hierarchical ZSM-5, Appl. Catal., B, 2021, 284, 119719 CrossRef CAS.
- J. E. Min, et al., Role of mesopores in Co/ZSM-5 for the direct synthesis of liquid fuel by Fischer-Tropsch synthesis, Catal. Sci. Technol., 2018, 8, 6346–6359 RSC.
- X. Li, et al., Enhanced gasoline selectivity through Fischer-Tropsch synthesis on a bifunctional catalyst: Effects of active sites proximity and reaction temperature, Chem. Eng. J., 2021, 416, 129180 CrossRef CAS.
- M. J. Valero-Romero, et al., Carbon/H-ZSM-5 composites as supports for bi-functional Fischer-Tropsch synthesis catalysts, Catal. Sci. Technol., 2016, 6, 2633–2646 RSC.
- Y. Chen, et al., Nano-ZSM-5 decorated cobalt based catalysts for Fischer-Tropsch synthesis to enhance the gasoline range products selectivity, J. Taiwan Inst. Chem. Eng., 2020, 116, 153–159 CrossRef CAS.
- W. Ma, W. D. Shafer, M. Martinelli, D. E. Sparks and B. H. Davis, Fischer-Tropsch synthesis: Using deuterium tracer coupled with kinetic approach to study the kinetic isotopic effects of iron, cobalt and ruthenium catalysts, Catal. Today, 2020, 343, 137–145 CrossRef CAS.
- J. Plana-Pallejà, S. Abelló, C. Berrueco and D. Montané, Effect of zeolite acidity and mesoporosity on the activity of Fischer-Tropsch Fe/ZSM-5 bifunctional catalysts, Appl. Catal., A, 2016, 515, 126–135 CrossRef.
- Y. Xu, et al., Insights into the Diffusion Behaviors of Water over Hydrophilic/Hydrophobic Catalysts During the Conversion of Syngas to High-Quality Gasoline, Angew. Chem., 2023, 62, e202306786 CrossRef CAS PubMed.
- Y. Jin, et al., Development of dual-membrane coated Fe/SiO2 catalyst for efficient synthesis of isoparaffins directly from syngas, J. Membr. Sci., 2015, 475, 22–29 CrossRef CAS.
- Y. Zhang, et al., Chemistry of Ketene Transformation to Gasoline Catalyzed by H-SAPO-11, J. Am. Chem. Soc., 2022, 144, 18251–18258 CrossRef CAS PubMed.
- Y. Ni, et al., Realizing high conversion of syngas to gasoline-range liquid hydrocarbons on a dual-bed-mode catalyst, Chem Catal., 2021, 1, 383–392 CrossRef CAS.
- E. Kianfar, M. Salimi, V. Pirouzfar and B. Koohestani, Synthesis of modified catalyst and stabilization of CuO/NH4-ZSM-5 for conversion of methanol to gasoline, Int. J. Appl. Ceram. Technol., 2018, 15, 734–741 CrossRef CAS.
- A. K. Jamil, et al., Stable Production of Gasoline-Ranged Hydrocarbons from Dimethyl Ether over Iron-Modified ZSM-22 Zeolite, Energy Fuels, 2018, 32, 11796–11801 CrossRef CAS.
- E. Kianfar, S. Hajimirzaee, S. Mousavian and A. S. Mehr, Zeolite-based catalysts for methanol to gasoline process: A review, Microchem. J., 2020, 156, 104822 CrossRef CAS.
- A. Duerksen, J. Thiessen, C. Kern and A. Jess, Fischer-Tropsch synthesis with periodical draining of a liquid-filled catalyst by hydrogenolysis, Sustainable Energy Fuels, 2020, 4, 2055–2064 RSC.
- C. Sun, Z. Luo, A. Choudhary, P. Pfeifer and R. Dittmeyer, Influence of the Condensable Hydrocarbons on an Integrated Fischer-Tropsch Synthesis and Hydrocracking Process: Simulation and Experimental Validation, Ind. Eng. Chem. Res., 2017, 56, 13075–13085 CrossRef CAS.
- A. Nanduri and P. L. Mills, Effect of catalyst shape and multicomponent diffusion flux models on intraparticle transport-kinetic interactions in the gas-phase Fischer–Tropsch synthesis, Fuel, 2020, 278, 118117 CrossRef.
- J. Yang, X. Fang, Y. Xu and X. Liu, Investigation of the deactivation behavior of Co catalysts in Fischer-Tropsch synthesis using encapsulated Co nanoparticles with controlled SiO2 shell layer thickness, Catal. Sci. Technol., 2020, 10, 1182–1192 RSC.
- C. Niu, et al., Mass transfer advantage of hierarchical structured cobalt-based catalyst pellet for Fischer–Tropsch synthesis, AIChE J., 2021, n/a, e17226 CrossRef.
- A. P. Savost'yanov, G. B. Narochnyi, R. E. Yakovenko, V. N. Soromotin and I. N. Zubkov, Effect of Diffusion Limitations on the Fischer–Tropsch Synthesis of Long-Chain Hydrocarbons on a Cobalt–Alumina Silica Gel Catalyst, Catal. Ind., 2018, 10, 181–184 CrossRef.
- D. B. Bukur, M. Mandić, B. Todić and N. Nikačević, Pore diffusion effects on catalyst effectiveness and selectivity of cobalt based Fischer-Tropsch catalyst, Catal. Today, 2020, 343, 146–155 CrossRef CAS.
- A. P. Savost'yanov, et al., Deactivation of Co-Al2O3/SiO2 Fischer–Trospch Synthesis Catalyst in Industrially Relevant Conditions, Catal. Lett., 2020, 150, 1932–1941 CrossRef.
- H. Li, et al., Effects of macropores on reducing internal diffusion limitations in Fischer-Tropsch synthesis using a hierarchical cobalt catalyst, RSC Adv., 2017, 7, 9436–9445 RSC.
- Q. Zheng, et al., Experimental Determination of H2 and CO Diffusion Coefficients in a Wax Mixture Confined in a Porous Titania Catalyst Support, J. Phys. Chem. B, 2020, 124, 10971–10982 CrossRef CAS PubMed.
- C. Erkey, J. B. Rodden and A. Akgerman, Diffusivities of Synthesis Gas and n-Alkanes in Fischer–Tropsch Wax, Energy Fuels, 1990, 4, 275–276 CrossRef CAS.
- J. S. Chou and K. C. Chao, Solubility of Synthesis and Product Gases in a Fischer-Tropsch SASOL Wax, Ind. Eng. Chem. Res., 1992, 31, 621–623 CrossRef CAS.
- S. H. Huang, H. M. Lin, K. C. Chao and F. N. Tsai, Solubility of Synthesis Gases in Heavy N-Paraffins and Fischer-Tropsch Wax, Ind. Eng. Chem. Res., 1988, 27, 162–169 CrossRef CAS.
- S. H. Huang, H. M. Lin and K. C. Chao, Experimental Investigation of Synthesis Gas Solubility in Fischer–Tropsch Reactor Slurry, Fluid Phase Equilib., 1987, 36, 141–148 CrossRef CAS.
- D. S. van Vuuren, J. R. Hunter and M. D. Heydenrych, The solubility of various gases in Fischer-Tropsch reactor wax, Chem. Eng. Sci., 1988, 43, 1291–1296 CrossRef CAS.
- E. Rytter, et al., Catalyst particle size of cobalt/rhenium on porous alumina and the effect on Fischer - Tropsch catalytic performance, Ind. Eng. Chem. Res., 2007, 46, 9032–9036 CrossRef CAS.
- L. Schurm, C. Kern and A. Jess, Accumulation and distribution of higher hydrocarbons in the pores of a cobalt catalyst during low-temperature Fischer–Tropsch fixed-bed synthesis, Catal. Sci. Technol., 2021, 11, 6143–6154 RSC.
- P. Hazemann, et al., Selectivity loss in Fischer-Tropsch synthesis: The effect of carbon deposition, J. Catal., 2021, 401, 7–16 CrossRef CAS.
- R. C. Baliban, J. A. Elia, V. Weekman and C. A. Floudas, Process synthesis of hybrid coal, biomass, and natural gas to liquids via Fischer-Tropsch synthesis, ZSM-5 catalytic conversion, methanol synthesis, methanol-to-gasoline, and methanol-to-olefins/distillate technologies, Comput. Chem. Eng., 2012, 47, 29–56 CrossRef CAS.
- A. Medina Ramirez, B. Ruiz Camacho, M. Villicaña Aguilera, I. R. Galindo Esquivel and J. J. Ramírez-Minguela, Effect of different zeolite as Pt supports for methanol oxidation reaction, Appl. Surf. Sci., 2018, 456, 204–214 CrossRef CAS.
- H. Igarashi, H. Uchida, M. Suzuki, Y. Sasaki and M. Watanabe, Removal of carbon monoxide from hydrogen-rich fuels by selective oxidation over platinum catalyst supported on zeolite, Appl. Catal., A, 1997, 159, 159–169 CrossRef CAS.
- M. K. Jeon and P. J. McGinn, Effect of Ti addition to Pt/C catalyst on methanol electro-oxidation and oxygen electro-reduction reactions, J. Power Sources, 2010, 195, 2664–2668 CrossRef CAS.
- T. S. Zhao, J. Chang, Y. Yoneyama and N. Tsubaki, Selective synthesis of middle isoparaffins via a two-stage Fischer-Tropsch reaction: Activity investigation for a hybrid catalyst, Ind. Eng. Chem. Res., 2005, 44, 769–775 CrossRef CAS.
- L. V. Sineva, E. O. Gorokhova, K. O. Gryaznov, I. S. Ermolaev and V. Z. Mordkovich, Zeolites as a tool for intensification of mass transfer on the surface of a cobalt Fischer–Tropsch synthesis catalyst, Catal. Today, 2021, 378, 140–148 CrossRef CAS.
- S. Mohammadnasabomran, C. Márquez-Álvarez, J. Pérez-Pariente and A. Martínez, Short-channel mesoporous SBA-15 silica modified by aluminum grafting as a support for CoRu Fischer–Tropsch synthesis catalysts, Catal. Sci. Technol., 2021, 11, 4245–4258 RSC.
- I. Nakamura and K. Fujimoto, On the role of gallium for the aromatization of lower paraffins with Ga-promoted ZSM-5 catalysts, Catal. Today, 1996, 31, 335–344 CrossRef CAS.
- M. Janardanarao, Direct catalytic conversion of synthesis gas to lower olefins, Ind. Eng. Chem. Res., 1990, 29, 1735–1753 CrossRef CAS.
- Y. Song, X. Zhu, S. Xie, Q. Wang and L. Xu, The effect of acidity on olefin aromatization over potassium modified ZSM-5 catalysts, Catal. Lett., 2004, 97, 31–36 CrossRef CAS.
- X. Peng, et al., Impact of Hydrogenolysis on the Selectivity of the Fischer–Tropsch Synthesis: Diesel Fuel Production over Mesoporous Zeolite-Y-Supported Cobalt Nanoparticles, Angew. Chem., Int. Ed., 2015, 54, 4553–4556 CrossRef CAS PubMed.
- S. Sartipi, J. E. Van Dijk, J. Gascon and F. Kapteijn, Toward bifunctional catalysts for the direct conversion of syngas to gasoline range hydrocarbons: H-ZSM-5 coated Co versus H-ZSM-5 supported Co, Appl. Catal., A, 2013, 456, 11–22 CrossRef CAS.
- J. H. Sinfelt, Catalytic Hydrogenolysis over Supported Metals, Catal. Rev., 1970, 3, 175–205 CrossRef.
- G. Prieto, et al., Cobalt-catalyzed Fischer-Tropsch synthesis: Chemical nature of the oxide support as a performance descriptor, ACS Catal., 2015, 5, 3323–3335 CrossRef CAS.
- S. S. Gupta, P. M. Shenai, J. Meeuwissen, G. L. Bezemer and S. Shetty, Electronic Promotion Effects of Metal Oxides: A Case Study of MnO Impact on Fischer–Tropsch Catalysis, J. Phys. Chem. C, 2021, 125, 21390–21401 CrossRef CAS.
- J. Xie, et al., Size and Promoter Effects in Supported Iron Fischer-Tropsch Catalysts: Insights from Experiment and Theory, ACS Catal., 2016, 6, 3147–3157 CrossRef CAS.
- R. Sadek, et al., Fischer-Tropsch reaction on Co-containing microporous and mesoporous Beta zeolite catalysts: the effect of porous size and acidity, Catal. Today, 2020, 354, 109–122 CrossRef CAS.
- K. A. Chalupka, et al., The Impact of Reduction Temperature and Nanoparticles Size on the Catalytic Activity of Cobalt-Containing BEA Zeolite in Fischer–Tropsch Synthesis, Catalysts, 2020, 10 Search PubMed.
- A. Y. Khodakov, A. Griboval-Constant, R. Bechara and V. L. Zholobenko, Pore Size Effects in Fischer Tropsch Synthesis over Cobalt-Supported Mesoporous Silicas, J. Catal., 2002, 206, 230–241 CrossRef CAS.
- X. Fang, et al., Particle-Size-Dependent Methane Selectivity Evolution in Cobalt-Based Fischer–Tropsch Synthesis, ACS Catal., 2020, 10, 2799–2816 CrossRef CAS.
- G. Zang, et al., Synthetic Methanol/Fischer–Tropsch Fuel Production Capacity, Cost, and Carbon Intensity Utilizing CO2 from Industrial and Power Plants in the United States, Environ. Sci. Technol., 2021, 55, 7595–7604 CrossRef CAS PubMed.
- S. Guffanti, C. G. Visconti, J. van Kampen, J. Boon and G. Groppi, Reactor modelling and design for sorption enhanced dimethyl ether synthesis, Chem. Eng. J., 2021, 404, 126573 CrossRef CAS.
- J. van Kampen, J. Boon, J. Vente and M. van Sint Annaland, Sorption enhanced dimethyl ether synthesis under industrially relevant conditions, experimental validation of pressure swing regeneration, React. Chem. Eng., 2021, 6, 244–257 RSC.
- G. Skorikova, et al., The Techno-Economic Benefit of Sorption Enhancement: Evaluation of Sorption-Enhanced Dimethyl Ether Synthesis for CO2 Utilization, Front. Chem. Eng., 2020, 2, 594884 CrossRef.
- H. Yagi, Y. Ohno, N. Inoue, K. Okuyama and S. Aoki, Slurry phase reactor technology for DME direct synthesis, Int. J. Chem. React. Eng., 2010, 8, A109 Search PubMed.
- Y. Ohno, et al., New direct synthesis technology for DME (dimethyl ether) and its application technology, JFE Tech. Rep., 2006, 8, 34–40 Search PubMed.
- J. Chung, W. Cho, Y. Baek and C. Lee, Optimization of KOGAS DME Process From Demonstration Long-Term Test, Trans. Korean Hydrogen New Energy Soc., 2012, 23, 559–571 CrossRef.
- X. Pan, F. Jiao, D. Miao and X. Bao, Oxide − Zeolite-Based Composite Catalyst Concept That Enables Syngas Chemistry beyond Fischer − Tropsch Synthesis, Chem. Rev., 2021, 121, 6588–6609 CrossRef CAS PubMed.
- Y. Liu, D. Deng and X. Bao, Catalysis for Selected C1 Chemistry, Chem, 2020, 6, 2497–2514 CAS.
- What is the magic of turning coal into plastic? | Bi-Carbon Pathways to Infinite Possibilities. accessed 15.02.2024 https://www.nengyuanjie.net/article/68797.html.
- J. Wei, R. Yao, Y. Han, Q. Ge and J. Sun, Towards the development of the emerging process of CO 2 heterogenous hydrogenation into high-value unsaturated heavy hydrocarbons, Chem. Soc. Rev., 2021, 50, 10764–10805 RSC.
- Y. Wang, et al., Direct Conversion of CO2 to Ethanol Boosted by Intimacy-Sensitive Multifunctional Catalysts, ACS Catal., 2021, 11, 11742–11753 CrossRef CAS.
- I. Nezam, et al., Direct aromatization of CO2 via combined CO2 hydrogenation and zeolite-based acid catalysis, J. CO2 Util., 2021, 45, 101405 CrossRef CAS.
- R. P. Ye, et al., CO2 hydrogenation to high-value products via heterogeneous catalysis, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- K. Li and J. G. Chen, CO2 Hydrogenation to Methanol over ZrO2-Containing Catalysts: Insights into ZrO2 Induced Synergy, ACS Catal., 2019, 9, 7840–7861 CrossRef CAS.
- C. Panzone, R. Philippe, A. Chappaz, P. Fongarland and A. Bengaouer, Power-to-Liquid catalytic CO2 valorization into fuels and chemicals: focus on the Fischer-Tropsch route, J. CO2 Util., 2020, 38, 314–347 CrossRef CAS.
- O. Kraan, G. J. Kramer, M. Haigh and C. Laurens, An Energy Transition That Relies Only on Technology Leads to a Bet on Solar Fuels, Joule, 2019, 3, 2286–2290 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00437j |
| This journal is © The Royal Society of Chemistry 2024 |