
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Manganese catalysed reduction of nitriles with amine boranes†
Stefan
Weber
 *,
Ines
Blaha
and
Karl
Kirchner
*
*,
Ines
Blaha
and
Karl
Kirchner
*
Institute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9/163-AC, A-1060 Wien, Austria. E-mail: stefan.e163.weber@tuwien.ac.at; karl.kirchner@tuwien.ac.at
First published on 24th July 2024
Abstract
The room temperature reduction of various nitriles using amine boranes (ABs) catalysed by a manganese(I) alkyl complex is described. Based on experimental findings, a plausible mechanistic scenario is presented. This includes the presence of two catalytic cycles, one for productive reduction of nitriles and one for hydrogen evolution.
Introduction
The reduction of multiple bonds represents one of the most important transformations in organic chemistry. Apart from direct hydrogenation reactions employing hydrogen gas, transfer hydrogenation procedures can be utilised.1 Due to their high hydrogen content (up to 19.6 wt%), amine boranes (ABs) represent an interesting class of hydrogen donors for the reduction of unsaturated moieties.2 In order to avoid harsh reaction conditions or long reaction times, transition metal catalysts can be employed to facilitate increased reactivity.3 Noble metal complexes4 as well as base metal catalysts based on iron,5 cobalt,6 nickel,7 copper8 and very recently manganese9 may be utilized in such transformations. As far as the reduction of nitriles to primary amines is concerned, only little precedent is described using base-metal catalysts (Fig. 1). Zhou and Liu reported on the use of a PNN-based cobalt complex for the divergent reduction of nitriles to primary amines or secondary amines, depending on the choice of solvent.10 Later on, Wang employed a Mo(II) complex supported by an anionic PS-ligand.11 Notably, this complex showed high reactivity at room temperature. Zhou utilized Cu(OTf)2 as a simple precatalyst in combination with an in situ generated amine borane in the presence of a super-stoichiometric amount of KOtBu in aqueous media.12 Recently, Adhikari and Maji employed a Mn(I) complex, for the divergent reduction of nitriles to primary amines or secondary amines.13 A broad variety of nitriles could be successfully reduced under these conditions. However, three equivalents of AB had to be employed. Interestingly, all well-defined complexes shown in Fig. 1 contain a structural motif competent for metal–ligand cooperativity.14 | ||
| Fig. 1 Well-defined base metal catalysts for the reduction of nitriles to primary amines using ABs. | ||
Based on these examples, we questioned if such a motif is imperative for a productive reaction. Based on our previous findings on the reactivity of manganese alkyl carbonyl complexes for hydrogenation15 and hydrofunctionalization reactions16 we were curious if fac-[Mn(dippe)(CO)3(CH2CH2CH3)] (dippe = 1,2-bis(di-iso-propylphosphino)ethane) (Mn1) could be a competent catalyst for the afore described reaction. Herein we report on the application of Mn1 as a pre-catalyst for the reduction of nitriles under mild conditions and short reaction times. Furthermore, we present mechanistic studies and propose a plausible scenario.
Results and discussion
We began our investigations by choosing 4-fluorobenzonitrile as a model substrate in combination with various boranes as hydrogen source and Mn1 as pre-catalyst. Selected optimization reactions are depicted in Table 1, for details see ESI.† High reactivity could be observed for various ABs at room temperature within 3 hours. This represents a rare example for the reduction of nitriles with ABs at room temperature in aprotic solvents.11 Dimethylamine borane (DMAB) performed best under these reaction conditions. We attribute this to high solubility of DMAB in organic solvents in comparison to methylamine borane and ammonia borane. Interestingly, no reactivity was observed for trimethylamine borane. This underscores the crucial role of the N–H motif in ABs for this reaction. Furthermore, no productivity was observed for pinacol borane (HBPin).|
|
||||
|---|---|---|---|---|
| Entry | Borane (equiv.) | Solvent | Conversionb (%) | Ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1ab :1ab |
| a Reaction conditions: 0.56 mmol 4-fluorobenzonitrile, Mn1 (2 mol%) solvent (0.56 mL, 1 M), 25 °C, 3 h, Ar, closed vial. b Determined by 19F{1H}-NMR spectroscopy. c 24 h. d 6 h. e In absence of Mn1. | ||||
| 1 | HNMe2BH3 (2) | Et2O | >99 | >99:1 |
| 2 | H2NMeBH3 (2) | Et2O | 43 | 98:2 |
| 3c | NMe3BH3 (2) | Et2O | — | — |
| 4c | H3NBH3 (2) | Et2O | 81 | 96:4 |
| 5c | HBPin (2) | Et2O | — | — |
| 6 | HNMe2BH3 (2) | THF | 82 | >99:1 |
| 7 | HNMe2BH3 (2) | C6H6 | >99 | >99:1 |
| 8 | HNMe2BH3 (2) | MeOH | — | — |
| 9 | HNMe2BH3 (1.3) | Et2O | >99 |
>99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
| 10d | HNMe2BH3 (1) | Et2O | 88 | >99:1 |
| 11e | HNMe2BH3 (2) | Et2O | — | — |

This is quite surprising, given our previous study wherein Mn1 showed high productivity in the hydroboration of alkenes and alkynes with HBPin.16a Silanes were also found to be unproductive for a reductive transformation. The implemented reduction procedure can be employed in a broad variety of solvents, whereas we found that alcohols lead to unproductive consumption of DMAB. Further optimization allowed us to decrease the equivalents of DMAB to 1.3 while still obtaining full conversion of nitrile. This is a significant lower amount than the vast majority of pervious reports in aprotic reaction media. Recently, Zhou and Liu reported on the Co catalysed reduction of nitriles, employing 1.6 equiv. of AB,10 whereas other reports utilize ≥2 equiv. of DMAB.11–13 In general, reducing the equivalents of reductant contributes to a more atom efficient reaction. However, lowering the amount of DMAB to one equivalent resulted in an incomplete reduction of the nitrile. Importantly, if the reaction is conducted in the absence of Mn1 no conversion of nitrile is observed.
We then investigated the generality of the introduced reduction protocol for a variety of aromatic and aliphatic nitriles (Table 2). Halides, the ester functionality as well as furan or thiophene heterocycles are well tolerated. Lower productivity was observed in the presence of a nitro group. No or low conversion was detected for an aniline derivative as well as a pyridine substrate. High yields were achieved for dinitrile giving diamine 16. Remarkably, no reduction of the alkyne group (17) and only trace reduction (<5%) of the conjugated double bond in 18 was observed. This is quite remarkable, since Mn1 was shown to efficiently reduce alkene15b and alkynes15c under hydrogenation conditions. This demonstrates that the choice of the hydrogen source can alter reactivity and selectivity of Mn1.
|
|
|---|
| a Reaction conditions: 0.56 mmol nitrile (1 equiv.), 0.73 mmol DMAB (1.3 equiv.), Mn1 (2 mol%), Et2O (1 M), 25 °C, 3 h Ar, closed vial. Conversion determined by GC-MS, isolated yields as ammonium chloride given in paratheses. b THF, 50 °C. c 1.4 mmol DMAB (2.5 equiv.), Mn1 (2.5 mol%), 18 h. n.d. = not determined. |
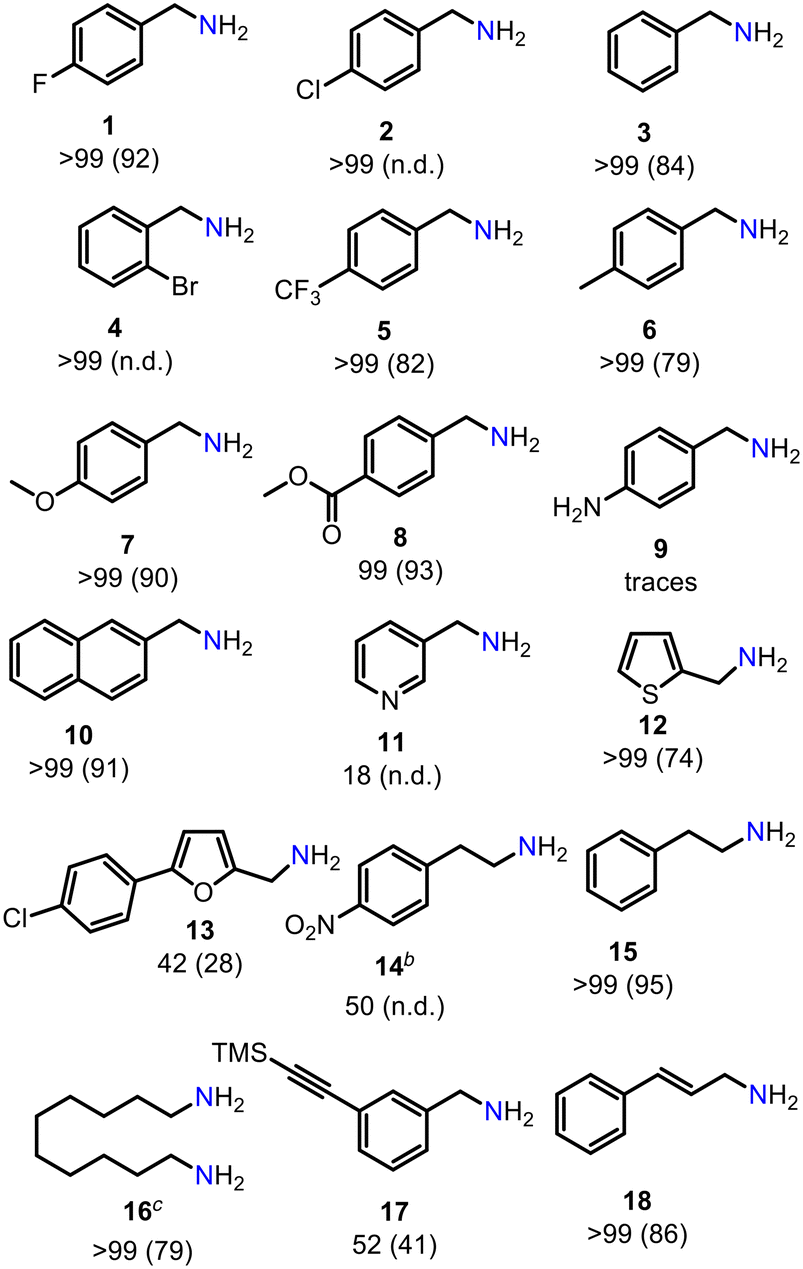
|

In order to gain insights in the reaction mechanism of the title reaction a series of experiments were conducted (Scheme 1). At first, analysis of the reaction mixture prior quenching was done. By employing 11B-NMR analysis, we were able to detect the cyclic dimer [Me2NBH2]2,17 a typical dehydrogenation product (triplet at 5.3 ppm) and a novel species, giving rise to a doublet centred at 29.0 ppm. Based on multinuclear NMR analysis and comparison with similar compounds presented in the literature,18 we assign the structure of the primary product (P) as depicted in Scheme 1. Unfortunately, we were unable to isolate P from the reaction mixture. This is attributed to complicated separation and its high sensitivity to moisture and air. It should be noted that the formation P (or related structures) is thus far unprecedented by the reduction of nitriles. Given the high sensitivity of P, it can be swiftly converted to free amines and eventually to ammonium salts, which were isolated in the substrate scope.
 | ||
| Scheme 1 Mechanistic experiments for the reduction of nitriles by Mn1. | ||
Next, we turned our attention to the interaction of Mn1 with the reactants. While no reaction was observed between nitriles and Mn1, it reacts with an excess of DMAB to give the borohydride complex cis-[Mn(dippe)(CO)3(κ2-BH4)] (MnBH4)19 under release of hydrogen gas accompanied by the formation of typical amine–borane products upon dehydrogenation.17,20 Interestingly, this reaction is significantly slower (ca. 36 h for >95% conversion) than the reduction of nitriles (3 h for >99% conversion). If MnBH4 was directly used as pre-catalyst a similar reactivity was observed for the model reaction.
When the reaction was carried out in an open system, 91% conversion of nitrile was observed. This strongly favours a hydrogen transfer pathway over a dehydrogenation of DMAB followed by hydrogenation with released H2 gas. This is in line with an earlier report from our group, since direct hydrogenation requires harsher reaction conditions (100 °C, 50 bar H2) employing a similar manganese alkyl carbonyl complex.15a Furthermore, formation of P is only consistent with a hydrogen- (and boron-) transfer mechanism and not with a classic hydrogenation reaction. Surprisingly, if Imine I was employed as substrate, no reaction was detected. This is consistent with reduction of nitriles to P within one catalytic cycle. Noteworthy, the presence of Imine I does not interfere with the dehydrogenation of DMAB. Given these observations, a stepwise reduction with substrate dissociation followed by recoordination seems unlikely. It should be noted that subjecting the cyclic dimer [Me2NBH2]2 as potential reductant, no conversion of neither nitrile nor Imine I could be observed. All of the above indicates that the dehydrogenation of DMAB is likely not coupled to productive nitrile reduction. We conclude that an interaction of nitrile, DMAB and catalyst is required for a productive process. Furthermore, deuterium labelling experiments were carried out. If DMAB-ND was employed as reductant, only traces of deuterium incorporation in the benzylic position was observed. The utilization of DMAB-BD3 gave a deuterium content of 66% in the benzylic position.
We then turned our attention to in situ NMR spectroscopy. Monitoring the reaction progress gave rise to a pronounced induction period followed by a pseudo-first order regime. This is consistent with a preequilibrium for the activation of Mn1. During the course of the reaction, two new sets of resonances for manganese-species were detected by 31P{1H} NMR spectroscopy. A singlet at 116.1 ppm corresponds to known tricarbonyl-hydride fac-[Mn(dippe)(CO)3(H)],21 which does not show any reactivity in the reduction of nitriles. The other manganese compound was tentatively assigned to cis-[Mn(dippe)(CO)2(nitrile)(H)] (MnH) which gives rise to two doublets centred at 129.3 ppm and 99.1 ppm in the 31P{1H}-NMR spectrum and a pseudo-triplet at −4.68 ppm in the 1H-NMR spectrum. This is in line with the related previously reported manganese hydride complex cis-[Mn(dippe)(CO)2(dmso)(H)].22 Deuterium labelling studies established that the B–H moiety in DMAB is the origin of the hydride rather than the proton of the N–H bond. Interestingly, the onset of catalysis was found to be dependent on the employed nitriles. More electron deficient nitriles initiated the catalytic reaction faster than electron-rich substrates. Furthermore, the reaction proceeds significantly faster with electron-poor nitriles.
These observations are in line with hydride transfer to the coordinated nitrile being the rate determining step during catalysis. This is also supported by a built up in the concentration of MnH during catalysis. Surprisingly, we could not detect MnBH4 during the course of the reaction, which seems to be an off-cycle species. However, we were able to demonstrate that MnBH4 reacts with nitriles to give MnH as an on-cycle intermediate. This interconversion may be fast under catalytic conditions, given the high concentration of substrate relative to MnBH4. Upon high conversion the fate of the active species is a variety of borohydride species.16a
Based on these studies we suggest that two catalytic cycles are operative throughout catalysis (Scheme 2). One cycle presents the reduction of nitriles, whereas the other depicts the pathway for hydrogen evolution reaction (HER). Based on our experience with manganese alkyl carbonyl complexes, we propose activation of the catalyst by migratory insertion of the alkyl ligand into the adjacent CO.15,16 This can be initiated by DMAB alone, activating the catalyst for the hydrogen evolution pathway (Scheme 2, right cycle). Notably, the activation of Mn1 solely by DMAB and the consecutive HER were shown to be rather sluggish. Alternatively, Mn1 may also be activated by a combination of DMAB and nitrile substrate (Scheme 2, left side). This feeds into a productive cycle with superior turnover frequencies in comparison to the HER cycle. Within this pathway, MnH was found to be the resting state, presumably due to turnover-limiting hydride transfer to nitrile. Crossing over of the active species from the nitrile reduction cycle to the HER cycle (and vice versa) seems feasible. This is consistent with an earlier onset of HER and faster HER in presence of nitriles.
 | ||
| Scheme 2 Proposed mechanistic scenario for the reduction of nitriles by DMAB catalysed by Mn1. | ||
Conclusions
In conclusion, we have introduced a manganese catalysed reduction of nitriles with amine boranes. This reaction takes place under mild reaction conditions and requires a reduced amount of ABs in comparison to previous reports. Based on mechanistic studies, we presented plausible reaction pathways. We propose two distinct catalytic cycles, one being productive for the reduction of nitriles by hydrogen- (and boron-) transfer, whereas the other represents a hydrogen evolution reaction. Future studies will be dedicated to the investigation of hydrogen release from amine boranes and (transfer) hydrogenation of unsaturated substrates by in situ generated hydrogen gas.Data availability
The authors of the manuscript entitled manganese catalysed reduction of nitriles with amine boranes by Stefan Weber, Ines Blaha and Karl Kirchner that all the data is available in ESI.† Original and processed data available in specific data formats can be provided on request from the corresponding authors. Raw NMR data associated with publication: Manganese Catalysed Reduction of Nitriles with Amine Boranes Stefan Weber, Ines Blaha and Kirchner, K. data can be directly processed Created with Bruker NMR software and will be deposited at the TU Wien repository (https://researchdata.tuwien.ac.at/).Author contributions
Stefan Weber contributed to conceptualization, investigation, methodology, visualization, and writing the original draft. Ines Blaha carried out experiments and the physical–chemical characterization. Karl Kirchner contributed to supervision, writing, review & editing, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Austrian Science Fund (FWF) is gratefully acknowledged (Project No. P 33016-N).References
- (a) H.-U. Blaser, F. Spindler and M. Thommen, in The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2008, ch. 37, pp. 1279–1324 Search PubMed; (b) P. N. Rylander, Best Synthetic Methods: Hydrogenation Methods, Academic Press, Orlando. Florida, 1985, ch. 3, pp. 53–65 Search PubMed; (c) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- (a) D. J. Heldebrant, A. Karkamkar, J. C. Linehan and T. Autrey, Energy Environ. Sci., 2008, 1, 156–160 RSC; (b) C. Y. Alpaydın, S. K. Gülbay and C. O. Colpan, Int. J. Hydrogen Energy, 2020, 45, 3414–3434 CrossRef.
- (a) M. Ding, J. Chang, J.-X. Mao, J. Zhang and X. Chen, J. Org. Chem., 2022, 87, 16230–16235 CrossRef CAS PubMed; (b) X. Yang, L. Zhao, T. Fox, Z.-X. Wang and H. Berke, Angew. Chem., Int. Ed., 2010, 49, 2058–2062 CrossRef CAS PubMed.
- (a) T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Tetrahedron Lett., 2011, 52, 6652–6654 CrossRef CAS; (b) C. E. Hartmann, V. Jurcik, O. Songis and C. S. J. Cazin, Chem. Commun., 2013, 49, 1005–1007 RSC; (c) T. Bhatt and K. Natte, Org. Lett., 2024, 26, 866–871 CrossRef CAS PubMed; (d) A. Kumar, J. Eyyathiyil and J. Choudhury, Inorg. Chem., 2021, 60, 11684–11692 CrossRef CAS PubMed; (e) J. Chang, J.-X. Mao, M. Ding, J. Zhang and X. Chen, Inorg. Chem., 2023, 62, 4971–4979 CrossRef CAS PubMed.
- M. Espinal-Viguri, S. E. Neale, N. T. Coles, A. Stuart, S. A. Macgregor and L. R. Webster, J. Am. Chem. Soc., 2019, 141, 572–582 CrossRef CAS PubMed.
- (a) T. M. Maier, S. Sandl, I. G. Shenderovich, A. Jacobi von Wangelin, J. J. Weigand and R. Wolf, Chem. – Eur. J., 2019, 25, 238–245 CrossRef CAS PubMed; (b) D. M. Sharmaa and B. Punji, Adv. Synth. Catal., 2019, 361, 3930–3936 CrossRef; (c) M. Pang, J.-Y. Chen, S. Zhang, R.-Z. Liao, C.-H. Tung and W. Wang, Nat. Commun., 2020, 11, 1249–1258 CrossRef CAS PubMed; (d) T. P. Lin and J. C. Peter, J. Am. Chem. Soc., 2013, 135, 15310–15313 CrossRef CAS PubMed; (e) S. Fu, N.-Y. Chen, X. Liu, Z. Shao, S.-P. Luo and Q. Liu, J. Am. Chem. Soc., 2016, 138, 8588–8594 CrossRef CAS PubMed; (f) X. Zhuang, J.-Y. Chen, Z. Yang, M. Jia, C. Wu, R.-Z. Liao, C.-H. Tung and W. Wang, Organometallics, 2019, 38, 3752–3759 CrossRef CAS.
- (a) V. Vermaak, H. C. M. Vosloo and A. J. Swarts, Adv. Synth. Catal., 2020, 362, 5788–5793 CrossRef CAS; (b) V. Vermaak, H. C. M. Vosloo and A. J. Swarts, Mol. Catal., 2021, 511, 111738–111745 CrossRef CAS.
- (a) M. Das, T. Kaicharla and J. F. Teichert, Org. Lett., 2018, 20, 4926–4929 CrossRef CAS PubMed; (b) E. Korytiaková, N. O. Thiel, F. Pape and J. F. Teichert, Chem. Commun., 2017, 53, 732–735 RSC.
- (a) L. Wang, J. Lin, C. Xia and W. Sun, J. Catal., 2022, 413, 487–497 CrossRef CAS; (b) A. Maji, S. Gupta, D. Panja, S. Sutradhar and S. Kundu, Organometallics, 2023, 42, 3385–3396 CrossRef CAS; (c) X. Chu, G. Zhou, M. Pang and H. Zhang, Eur. J. Org. Chem., 2023, 26, e202300597 CrossRef CAS; (d) A. Brzozowska, L. M. Azofra, V. Zubar, I. Atodiresei, L. Cavallo, M. Rueping and O. El-Sepelgy, ACS Catal., 2018, 8, 4103–4109 CrossRef CAS; (e) Y.-P. Zhou, Z. Mo, M.-P. Luecke and M. Driess, Chem. – Eur. J., 2018, 24, 4780–4784 CrossRef CAS PubMed.
- Z. Shao, S. Fu, M. Wie, S. Zhou and Q. Liu, Angew. Chem., Int. Ed., 2016, 55, 14653–14657 CrossRef CAS PubMed.
- S.-F. Hou, J.-Y. Chen, M. Xue, M. Jia, X. Zhai, R.-Z. Liao, C.-H. Tung and W. Wang, ACS Catal., 2020, 10, 380–390 CrossRef CAS.
- H. Song, Y. Xiao, Z. Zhang, W. Xiong, R. Wang, L. Guo and T. Zhou, J. Org. Chem., 2022, 87, 790–800 CrossRef CAS PubMed.
- K. Sarkar, K. Das, A. Kundu, D. Adhikari and B. Maji, ACS Catal., 2021, 11, 2786–2794 CrossRef CAS.
- J. R. Khusnutdinov and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef PubMed.
- (a) S. Weber, L. F. Veiros and K. Kirchner, Adv. Synth. Catal., 2019, 361, 5412–5420 CrossRef CAS PubMed; (b) S. Weber, B. Stöger, L. F. Veiros and K. Kirchner, ACS Catal., 2019, 9, 9715–9720 CrossRef CAS; (c) R. A. Farrar-Tobar, S. Weber, Z. Csendes, A. Ammaturo, S. Fleissner, H. Hoffmann and K. Kirchner, ACS Catal., 2022, 12, 2253–2260 CrossRef CAS PubMed; (d) S. Weber, J. Brünig, L. F. Veiros and K. Kirchner, Organometallics, 2021, 40, 1388–1394 CrossRef CAS PubMed.
- (a) S. Weber, D. Zobernig, B. Stöger, L. F. Veiros and Kirchner, Angew. Chem., Int. Ed., 2021, 60, 24488–24492 CrossRef CAS PubMed; (b) S. Weber, M. Glavic, B. Stöger, E. Pittenauer, M. Podewitz, L. F. Veiros and K. Kirchner, J. Am. Chem. Soc., 2021, 143, 17825–17832 CrossRef CAS PubMed; (c) S. Weber, L. F. Veiros and K. Kirchner, ACS Catal., 2021, 11, 6474–6483 CrossRef CAS PubMed; (d) S. Weber and K. Kirchner, Acc. Chem. Res., 2022, 55, 2740–2751 CrossRef CAS PubMed.
- C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424–9434 CrossRef CAS PubMed.
- D. A. Resendiz-Lara, G. R. Whittell, E. M. Leitao and I. Manners, Macromolecules, 2019, 52, 7052–7064 CrossRef CAS.
- I. Blaha, S. Weber, R. Dülger, L. F. Veiros and K. Kirchner, ChemRxiv, preprint, 2024, DOI:10.26434/chemrxiv-2024-vvxlx.
- D. J. Liptrot, M. D. Hill, M. F. Mahon and D. MacDougall, Chem. – Eur. J., 2010, 16, 8508–8515 CrossRef CAS PubMed.
- J. A. Garduño and J. J. Garcia, ACS Catal., 2018, 9, 392–401 CrossRef.
- S. Kostera, S. Weber, I. Blaha, M. Peruzzini, K. Kirchner and L. Gonsalvi, ACS Catal., 2023, 13, 5236–5244 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00813h |
| This journal is © The Royal Society of Chemistry 2024 |