
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceValence-to-core X-ray emission spectroscopy to resolve the size-dependent valence electronic structure of Pt nanoparticles†
David P.
Dean
 a,
Gaurav S.
Deshmukh
a,
Christopher K.
Russell
a,
Kuixin
Zhu
b,
Christina W.
Li
b,
Jeffrey P.
Greeley
a,
Denis
Leshchev
*c,
Eli
Stavitski
*c and
Jeffrey T.
Miller
*a
a,
Gaurav S.
Deshmukh
a,
Christopher K.
Russell
a,
Kuixin
Zhu
b,
Christina W.
Li
b,
Jeffrey P.
Greeley
a,
Denis
Leshchev
*c,
Eli
Stavitski
*c and
Jeffrey T.
Miller
*a
aDavidson School of Chemical Engineering, Purdue University, West Lafayette, IN 47907, USA. E-mail: jeffrey-t-miller@purdue.edu
bDepartment of Chemistry, Purdue University, West Lafayette, IN 47907, USA
cBrookhaven National Laboratory NSLS-II, Upton, NY 11973, USA. E-mail: dleschev@bnl.gov; istavitski@bnl.gov
First published on 26th March 2024
Abstract
X-ray characterization of catalyst materials using synchrotron radiation has become more widely available to the scientific community in recent decades. Techniques such as X-ray absorption spectroscopy (XAS) and resonant inelastic X-ray scattering (RIXS) have enabled in situ and in operando studies of dilute catalyst sites for heterogeneous catalyst materials in order to obtain information about local geometric and electronic structure and how it impacts chemical transformations. Non-resonant or valence-to-core X-ray emission spectroscopy (NR-XES or VtC-XES) is an emerging technique used to probe changes to the d-electron density of states (DOS) for various factors such as ligand environment or alloy formation. In this study, VtC-XES provides insights into the electronic structure of Pt nanoparticles of different sizes dispersed in a typical heterogeneous catalyst support of SiO2. The results experimentally verify that the d-band center of the Pt catalysts systematically increases with decreasing nanoparticle size. Transitioning from a fully coordinated Pt foil to Pt nanoparticles of 2 nm in diameter, the shift in the d-band center scales linearly with the proportion of surface Pt atoms to bulk Pt atoms. While VtC-XES shows that the filled Pt 5d states shift systematically to higher energy, XANES shows that the unfilled Pt 5d states also shift systematically to higher energy. These findings align with previous computational and experimental studies and confirm that in situ VtC-XES is a capable technique for assessing the d-DOS of catalyst materials with dilute metal loadings (<2 wt%). Moreover, a beamline with an in situ XAS capability can be coupled with VtC-XES to holistically assess both the geometric (EXAFS) and electronic structure (XANES, VtC-XES) of a catalyst under realistic reaction conditions. This capability is accessible to others to apply to their catalytic problems to help drive the design of future catalyst materials.
1. Introduction
The ability to characterize catalysts at the atomic level is critical to accelerated discovery and development of new materials. Significant improvements in characterization techniques such as synchrotron X-ray absorption spectroscopy (XAS), including improved synchrotron design and detector technology as well as accessibility, have enabled analyses such as X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) for characterization of active sites in metal catalysts.1,2 XANES provides useful information about the electronic structure and is often used to obtain oxidation states. EXAFS yields information about bond distances and local coordination, including the number and types of atoms.5The future of catalyst material discovery is expected to mirror future developments in characterization methods. Resonant inelastic X-ray scattering (RIXS) has been recently developed to assess the electronic structure of metallic catalyst materials.6,7 RIXS is a two-step process involving a resonant excitation of a core electron into an intermediate state and a subsequent decay into a final state, emitting an X-ray. Within the past two decades, RIXS has been used for in situ metallic catalyst studies to determine the structure of the valence electron band.5,22 More recently, the method has been extended to 5d metal catalysts such as Pt and Ir to obtain electronic information about the average energy of filled and unfilled 5d valence states.5,14,15,23–25 Despite the technique development, information about the entire filled density of states of 5d metal materials remains elusive. Using RIXS, the different intermediate states can interfere with one another because of the large number of resonant states.22 RIXS can circumvent core-hole lifetime broadening to achieve high energy resolution but is susceptible to considerable uncertainty incurred when subtracting the elastic scattering line from the spectra.14,22,48 This implies that only the average energy of the filled valence states can be ascertained. Valence-band X-ray or ultraviolet photoelectron spectroscopy (VB-XPS or UPS) experiments have been successfully used to determine the d-electron density of states (d-DOS) distribution for metals.18,20,26,33 Nevertheless, the photoemission of valence electrons is limited to a soft X-ray range, which often requires ultra-high vacuum. This is not compatible with in situ applications where the emitted X-rays must have sufficient energy to penetrate reacting gases, catalyst particles, cell windows, etc. In addition, this makes it difficult to discern whether an electronic change can be attributed to differences in surface adsorbates or a property inherent to the bulk material (e.g., surface oxidation of a nanoparticle or the formation of an alloy). Lastly, the subtraction of the background or the deconvolution of contributions of other elements in the sample may introduce significant uncertainty.20,34
Non-resonant X-ray emission spectroscopy (NR-XES), which focuses on the valence-to-core region (VtC-XES or V2C-XES), is an emerging technique that can probe the full distribution of the density of states while using a high flux of hard X-rays, which affords element-specific analysis.5 Given the highly penetrative power of hard X-rays, VtC-XES is better suited for in situ or in operando catalysis experiments which maintain the desired chemical environment (i.e., partial pressure of gases, temperature, pre-treatment) while being able to penetrate through a catalyst bed consisting of a bulk oxide such as SiO2 or Al2O3.28 Additionally, the setup can be used in tandem with XAS analysis to provide a holistic picture of the geometric and electronic properties of a material. XANES yields information about the unfilled valence states which do not participate in molecular activation or bond formation. VtC-XES yields information about the filled valence states, for certain elements, to procure information about the entire valence band.5,7,61 VtC-XES can decouple elastic and inelastic contributions to the X-ray emission by using an incident X-ray energy higher than that of the edge being studied, thereby exciting an electron into the continuum.5 Doing so turns the resulting decay of an electron to the core hole into a one-electron process whose specific energy can be selected using a crystal analyzer.28 VtC-XES has historically been applied to 3d or 4d compounds with a high weight loading of the desired metal, such as Mn, Fe, Cr, Cu, or Nb in order to probe the electronic structure with a different ligand environment.27,29,31,35,42 Due to recent improvements in synchrotron flux such as the development of 4th generation synchrotrons as well as the improvement of X-ray detector technology, VtC-XES has been extended to more dilute (∼1 to 2 wt%) catalysts with 3d metals, such as Cu/SSZ-13, Fe/ZSM-5, or Pt/CeOx (probing the Ce 3d states).30,32,36 The technique has proven to be useful for in situ and in operando studies to probe changes under reaction conditions.7,32
Nevertheless, there is significant opportunity for improvement of the VtC-XES technique. For example, there has been little application to 5d metals that are important catalyst materials such as Pt, Ir, or Au. This is in large part due to technological limitations for detecting the relatively weak Lβ5 emission line, which represents a 5d5/2 or 5d3/2 to 2p3/2 transition.39 While VtC-XES for the vast majority of 3d material studies typically probe Kβ emission lines,27,29–31,42 which represents a 3p to 1s transition or a 4p to 1s transition, VtC-XES for this study probes the structure of electrons in filled 5d valence orbitals which affords information about the 5d metal electrons involved in chemical transformations. This study focuses on Pt, which is widely used for important catalytic reactions in thermochemistry and electrochemistry such as alkane dehydrogenation or the electrochemical oxygen reduction reaction.37,38 VtC-XES furnishes the ability to obtain the structure of the Pt 5d-DOS for the filled states, while XANES furnishes information about the average energy of the Pt 5d unfilled states.39 Very few studies have attempted VtC-XES on Pt catalyst materials. One such study resolves VtC-XES to apply to Pt and Au compounds in order to study the electronic structure of the 5d metal with different ligands.12,43 Another recent study extends XES to Pt–Ni nanoparticles, but instead employs resonant X-ray emission spectroscopy (RXES), making it difficult to decouple resonant and non-resonant effects.34,41 Yet another study uses XES to explore Pt layers on a separate metal substrate, including alloys, but probes the oxygen K edge XES rather than Pt XES.44
Herein, VtC-XES is employed to investigate the 5d-DOS for Pt nanoparticles supported on SiO2 of various sizes like those commonly used as catalysts. Pt nanoparticles ranging from 2 to 5 nm are synthesized and characterized using STEM to confirm nanoparticle size. XAS and VtC-XES are employed to experimentally measure the average energy of the Pt filled and unfilled states, respectively. In this work, VtC-XES spectra were obtained for Pt nanoparticle samples of different sizes under in situ reducing conditions to assess changes in the d-DOS such as d-band center, d-band width, etc. The experimentally determined impact of Pt nanoparticle size on the 5d-DOS is shown to shift the DOS to higher energy with decreasing size. Density functional theory (DFT) calculations performed on clusters in this study serve as a direct analog to the VtC-XES results. DFT results show an analogous shift with respect to undercoordinated Pt sites, which aligns with previous literature results.20,21 This study serves to illustrate an example of applying a beamline capable of in situ VtC-XES to a 5d catalyst to procure insights into the electronic structure of the catalyst. The setup includes a plug flow reactor (PFR) cell that enables simultaneous gas and temperature treatment, enabling the study of VtC-XES under realistic reaction conditions. This study aims to be a template for others to leverage this new capability for their catalyst discovery.
2. Experimental methods
2.1. Materials and synthesis
Monometallic Pt nanoparticles of varying sizes on SiO2 supports were synthesized via incipient wetness impregnation (IWI). Three monometallic Pt samples were synthesized (shorthand names are given in parentheses): 2 wt% Pt/SiO2 (small), 2 wt% Pt/SiO2 (medium), and 10 wt% Pt/SiO2 (large) samples. In a typical synthesis, a tetraammineplatinum(II) nitrate [Pt(NH3)4(NO3)2, Sigma-Aldrich] is dissolved in ultrapure, deionized water to achieve a metal loading of 2 wt% or 10 wt%; 2 wt% was employed to achieve small and medium sized nanoparticles, while 10 wt% was used to make larger nanoparticles. The pH is adjusted to 11 using a minimal amount of 30% ammonium hydroxide solution [NH4OH, Thermo Fisher Scientific]. The solution is then added dropwise onto Davisil® Grade 646 [35–60 mesh, pore size 150 Å, Sigma Aldrich] silica gel support. The material is dried at 120 °C for 12 hours and then calcined in air for 3 hours. The small sample was calcined at 225 °C, the medium sample at 300 °C, and the large sample at 450 °C. Subsequently, each sample is reduced in dilute hydrogen [5% H2/He, Indiana Oxygen] for two hours. The small sample was reduced at 225 °C, the medium sample at 400 °C, and the large sample at 550 °C. The goal to synthesize common Pt catalyst materials with a dilute Pt content.A separate sample of Pt nanoparticles supported on silica was synthesized to serve as a standard reference to ensure energy calibration of the beamline setup. This sample was prepared similarly to the other samples with a weight loading of 3 wt% Pt, a calcination temperature of 250 °C, and a reduction temperature of 550 °C.
2.2. Scanning transmission Electron microscopy (STEM) characterization
For scanning transmission electron microscopy (STEM), samples were prepared by drop-drying 10 μL of a 1 mg mL−1 supported catalyst suspension onto a Cu grid. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) were collected on an FEI Talos F200X S/TEM with a 200 kV X-FEG field-emission source.Nanoparticle size was assessed using ImageJ software.9 Assuming spherical nanoparticles, the percentage of surface atoms was calculated for each nanoparticle using a ratio of the volume of a shell with a thickness of δ to the total volume of the sphere, given in eqn (1).11 The thickness of the shell was estimated to be equal to the Pt–Pt bond distance obtained using XAS, which is analogous to the atomic diameter of Pt, and d is the nanoparticle diameter. Overall dispersion was estimated by dividing the total number of surface atoms by the total number of atoms across all the nanoparticles considered.
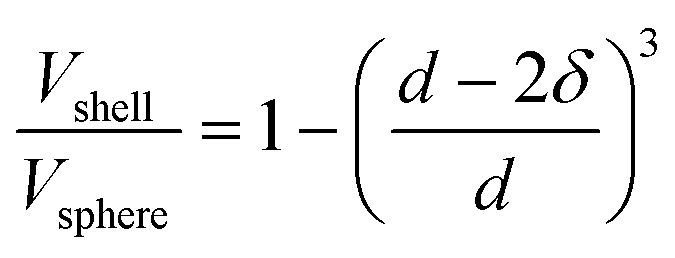 | (1) |
2.3. X-ray absorption spectroscopy (XAS) characterization
The XAS and XES measurements were conducted at the NSLS-II 8-ID Inner Shell Spectroscopy (ISS) beamline.3 XAS was conducted at the Pt LIII (11.564 keV) edge in transmission mode to obtain XANES and EXAFS. Three ion chambers were used for simultaneous measurement of a Pt foil energy calibration standard. The Pt foil is also used as a bulk metal (CN = 12) standard. Other Pt standards were diluted in boron nitride [BN, Thermo Fisher Scientific], ground into a fine powder, and pressed into wafers for ex situ scans in transmission mode. The Pt nanoparticle samples were inserted in capillaries, supported by a bed of quartz wool, and placed in an in situ cell for gas and temperature treatment. The in situ cell is a polyimide capillary tube that functions as a plug flow reactor (PFR). The Pt nanoparticle samples were scanned in fluorescence mode using a Passivated Implanted Planar Silicon (PIPS) detector placed at a 45° angle from the incident beam. The PIPS detector was equipped with a gallium oxide (Ga2O3) filter to filter out elastic scattering. The in situ cell is positioned to enable simultaneous fluorescence measurement of the sample as well as transmission measurement of the foil reference. In the in situ cell, the Pt nanoparticle samples were pre-treated at 250 °C in 10% H2 (balance He) for 30 minutes to remove surface oxygen from the nanoparticles, rendering them completely metallic. The sample was then cooled to 25 °C in He and scanned.All spectra were analyzed using WinXAS 4.0 software.4 The XANES of the Pt compounds was used for analysis of oxidation state. Standard EXAFS fitting procedures were applied to the Pt nanoparticle samples to estimate Pt coordination and bond distances. A Pt–Pt scattering path was generated using the Pt foil as a fully coordinated (CN = 12) metallic reference. Coordination number and % dispersion for Pt nanoparticles can be correlated by using the empirical relationship given in eqn (2) where D is percent dispersion.10
| log(D) = −0.13·CNPt–Pt + 2.58 | (2) |
2.4. Valence-to-core X-ray emission spectroscopy (VtC-XES) characterization
The in situ reactor is positioned such that simultaneous transmission, emission, and fluorescence are achievable. The incident X-ray energy was held at a constant value of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 eV, well above the edge energy of Pt LIII, thus constituting non-resonant X-ray emission spectroscopy. The spectrometer is a Johann spectrometer, equipped with a Si 〈933〉 spherically bent crystal analyzer (SBCA) with a radius of curvature of 0.5 m. The crystal analyzer diffracts the emitted X-rays to a Dectris Pilatus© detector positioned above the sample that moves in conjunction with the crystals, allowing the selection of a single emission energy that is focused to a single point on the detector. The crystal and detector move in a highly coordinated manner following a Rowland circle geometry.5,28 The spectrometer setup was tuned to accommodate the Lβ5 emission line. The energy resolution of the setup was estimated Based on the elastic scattering peak measured off the Pt foil measured at 11545 eV. The FWHM of the peak was found to be 2.45 eV which, after accounting for monochromator resolution, corresponds to ∼1.9 eV of the spectrometer. The scans were performed between 11542 and 11582 eV; the scan step was 0.5 eV around the VtC peak, i.e., between 11552 and 11572 eV, and 2 eV steps otherwise. XES intensity at each energy point was measured by exposing the detector for 1 s. A fast shutter was used to block the incident beam during the mechanical motions of the spectrometer. The intensity of the spectra is based on the X-ray count on the detector, and therefore has arbitrary units. Between 10 and 20 VtC-XES scans were averaged for each sample. The spectra were normalized using a linear fit of the baseline and the peak maximum.
900 eV, well above the edge energy of Pt LIII, thus constituting non-resonant X-ray emission spectroscopy. The spectrometer is a Johann spectrometer, equipped with a Si 〈933〉 spherically bent crystal analyzer (SBCA) with a radius of curvature of 0.5 m. The crystal analyzer diffracts the emitted X-rays to a Dectris Pilatus© detector positioned above the sample that moves in conjunction with the crystals, allowing the selection of a single emission energy that is focused to a single point on the detector. The crystal and detector move in a highly coordinated manner following a Rowland circle geometry.5,28 The spectrometer setup was tuned to accommodate the Lβ5 emission line. The energy resolution of the setup was estimated Based on the elastic scattering peak measured off the Pt foil measured at 11545 eV. The FWHM of the peak was found to be 2.45 eV which, after accounting for monochromator resolution, corresponds to ∼1.9 eV of the spectrometer. The scans were performed between 11542 and 11582 eV; the scan step was 0.5 eV around the VtC peak, i.e., between 11552 and 11572 eV, and 2 eV steps otherwise. XES intensity at each energy point was measured by exposing the detector for 1 s. A fast shutter was used to block the incident beam during the mechanical motions of the spectrometer. The intensity of the spectra is based on the X-ray count on the detector, and therefore has arbitrary units. Between 10 and 20 VtC-XES scans were averaged for each sample. The spectra were normalized using a linear fit of the baseline and the peak maximum.
2.5. Density functional theory (DFT) calculations
Pt nanoparticles were pre-relaxed using the effective medium theory (EMT) potential as implemented in the Python Atomic Simulation Environment (ASE).50 Geometry relaxations were performed on pre-relaxed nanoparticles using plane-wave density functional theory (DFT) calculations as implemented in the Vienna Ab initio Simulation Package (VASP) code51 with an energy cutoff of 400 eV and a single k-point. Relaxation was terminated when the Hellman–Feynman forces were below 0.02 eV Å−1. Vacuum space of 40 Å was added in the unit cell to prevent interaction between periodic images. Single-point energy calculations with an energy cutoff of 600 eV were performed on converged geometries. Electron states above the Fermi level were populated above 0 K using Gaussian smearing. Core-electron states were modeled using the frozen core approximation and expressed using the Projector Augmented Wave (PAW) method.52 Projected density of states (pDOS) were calculated for Pt atoms in a range of −10 eV to 5 eV referenced to the Fermi level with an energy resolution of 0.0065 eV. pDOS spectra were smoothed using the Hanning window as implemented in Scipy53 for visualization.The d-band center was evaluated computationally as the first moment of the pDOS (eqn (3)) where ELB = −10 eV, EUB = 5 eV, and ρ(E − Ef) is the pDOS. The integrals were evaluated numerically using the trapezoidal method.
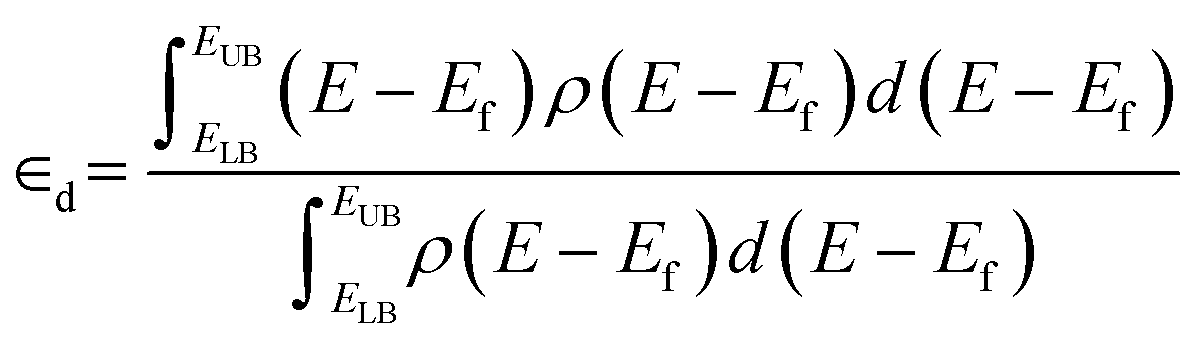 | (3) |
3. Results
Nanoparticle size distributions were determined using HAADF-STEM (Fig. 1). The small, medium, and large samples have an average diameter of 2.1 ± 0.5 nm, 3.0 ± 1.1 nm, and 5.1 ± 1.1 nm respectively. From these nanoparticle size distributions, dispersion was calculated based on an estimation of the total number of surface atoms relative to total nanoparticle volume,11 which increases with decreasing nanoparticle size, as shown in Table 1.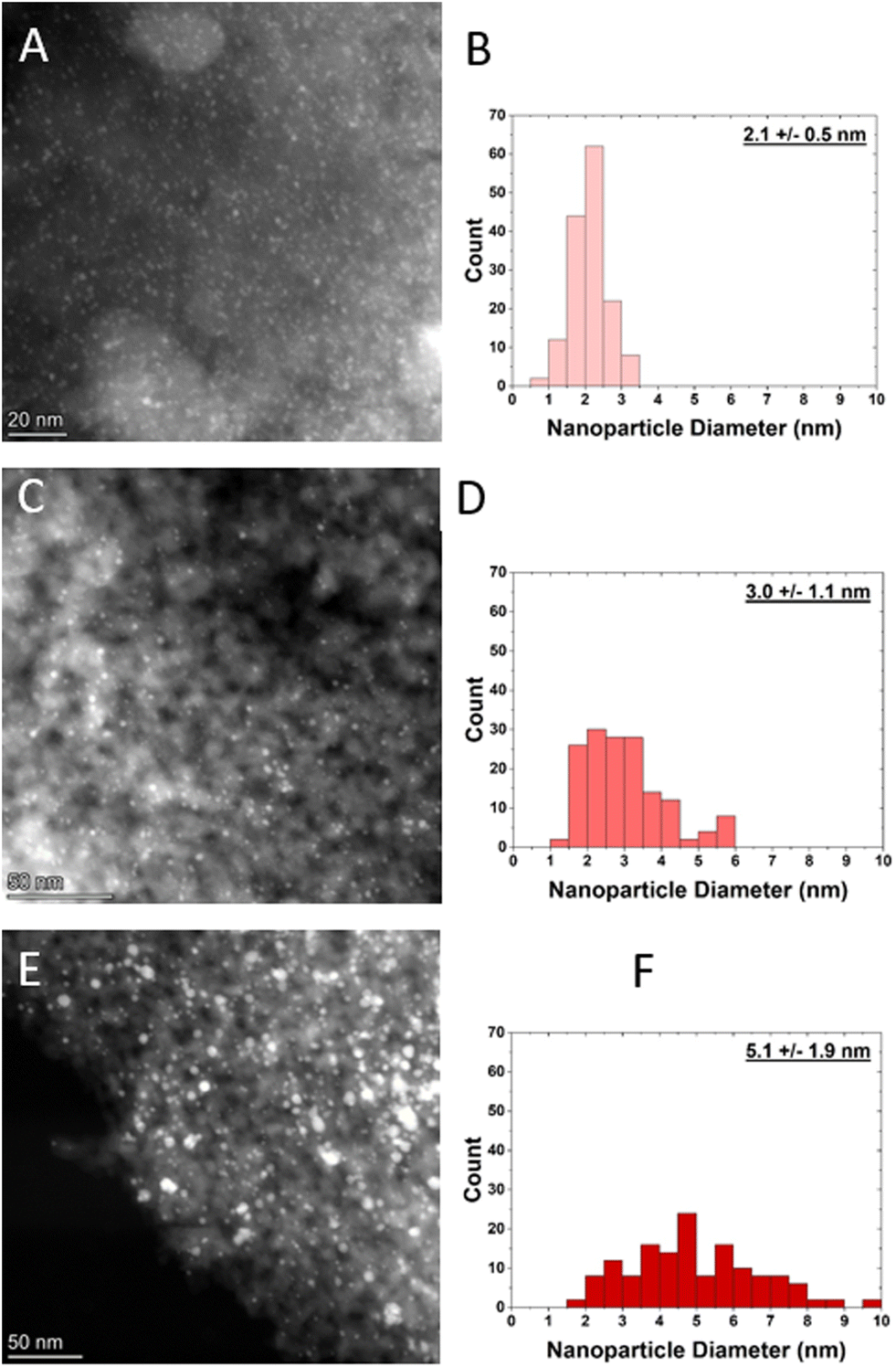 | ||
| Fig. 1 HAADF-STEM images and their corresponding size distributions of A and B) small Pt nanoparticle sample, C and D) medium Pt nanoparticle sample, and E and F) large Pt nanoparticle sample. | ||
| Sample | Nanoparticle diameter (nm) | Dispersion (%) |
|---|---|---|
| Pt foil | — | 0 |
| Large Pt NPs | 5.1 ± 1.9 | 23 |
| Medium Pt NPs | 3.0 ± 1.1 | 37 |
| Small Pt NPs | 2.1 ± 0.5 | 54 |
To corroborate the nanoparticle size distribution as determined by STEM, EXAFS is also employed to determine nanoparticle dispersion. Three different Pt nanoparticle samples were reduced in H2 at 250 °C to ensure no surface PtO is present. Subsequently, the samples were cooled to room temperature and scanned in flowing H2. The three-peak pattern for all the samples is indicative of metallic Pt (Fig. 2). There is no evidence of Pt–O scattering, which would occur at values of ∼1.7 Å (phase-uncorrected distance), meaning that all Pt in the samples has been reduced by the pre-treatment in dilute H2 at 250 °C. There is a systematic decrease in Fourier-transform magnitude with decreasing nanoparticle size, indicating a decrease in bulk average coordination. Additionally, the peaks shift to lower R with decreasing nanoparticle size, indicating bond contraction which is consistent with previous studies.8,10 The bond distance is assessed in the EXAFS fitting results, which are given in Table 2. The value of So,2 the amplitude reduction factor, was determined to be 0.81 based on the Pt foil reference. Using the phase and amplitude of an experimentally generated Pt–Pt scattering path from the Pt foil, the samples were found to have lower average Pt–Pt coordination relative to the foil. This is indicative of a higher proportion of undercoordinated, surface Pt atoms to fully coordinated Pt atoms, meaning the nanoparticles are smaller.
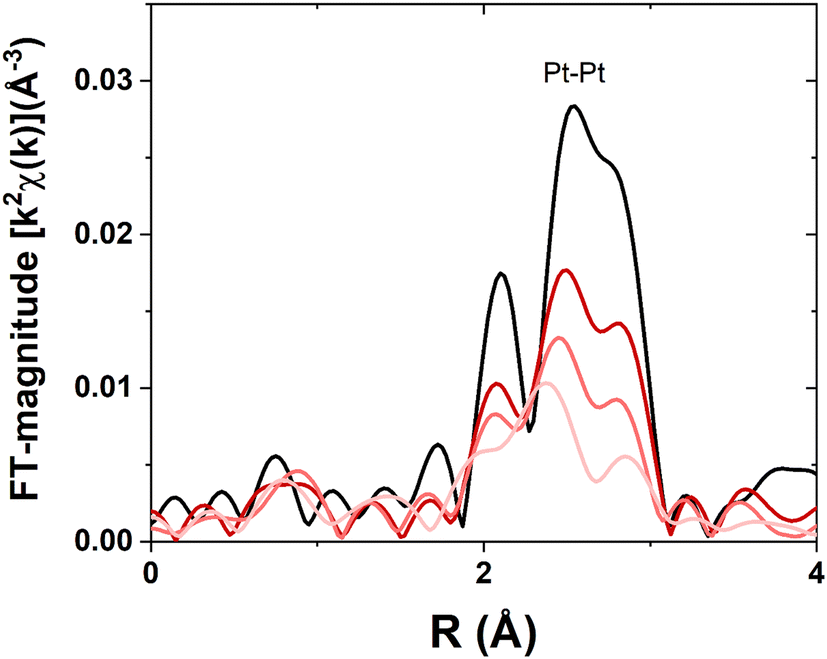 | ||
| Fig. 2 Pt LIII EXAFS for samples with Pt nanoparticles of different sizes. Pt foil (black), large sample (dark red), medium sample (moderate red), and small sample (light red). | ||
| Sample | Edge energy (eV) | Scattering pair | CN (±10%) | R (±0.02 Å) | Δσ2 (Å2) | Shift in Eo (eV) |
|---|---|---|---|---|---|---|
| Pt foil | 11564.4 |
Pt–Pt | 12.0 | 2.77 | 0.004 | 7.0 |
| Large Pt NPs | 11564.6 |
Pt–Pt | 10.1 | 2.76 | 0.006 | −0.6 |
| Medium Pt NPs | 11564.8 |
Pt–Pt | 8.3 | 2.74 | 0.007 | −1.5 |
| Small Pt NPs | 11565.0 |
Pt–Pt | 7.1 | 2.72 | 0.008 | −2.1 |
The coordination number for Pt has been shown to be correlated with nanoparticle dispersion; thus, the nanoparticle dispersion can be independently calculated to corroborate the STEM results (Table 1). Nanoparticle dispersion calculated from XAS is consistent with the dispersion calculated from STEM (Fig. 3).
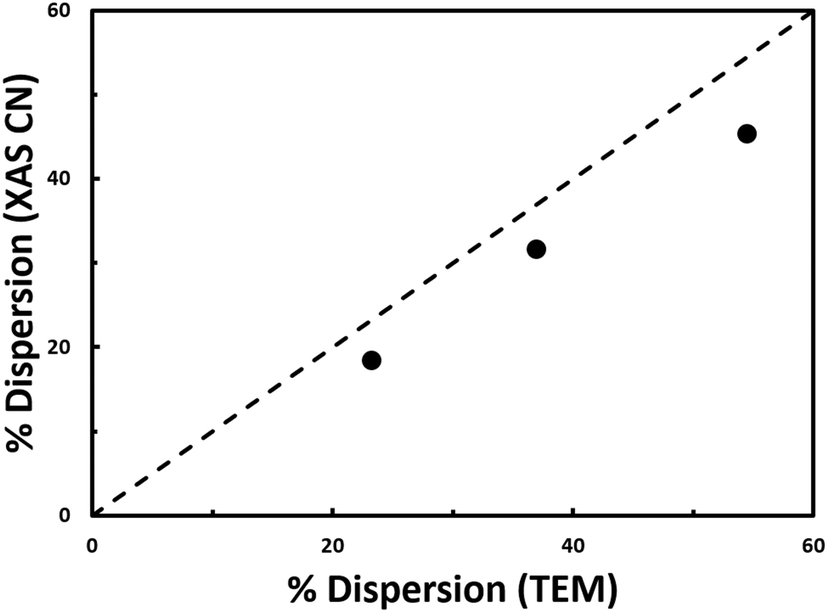 | ||
| Fig. 3 Dispersion determined from coordination number from EXAFS fitting versus dispersion determined from STEM. Results from the nanoparticle materials from this study (black circles) are plotted alongside the empirical relationship from literature (dashed black line).10 | ||
The XANES for the samples (Fig. 4) show a systematic increase in edge energy between 0.2 to 0.6 eV with decreasing nanoparticle size relative to the foil (Table 2), which is defined as the inflection point of the leading edge of the XANES spectrum.
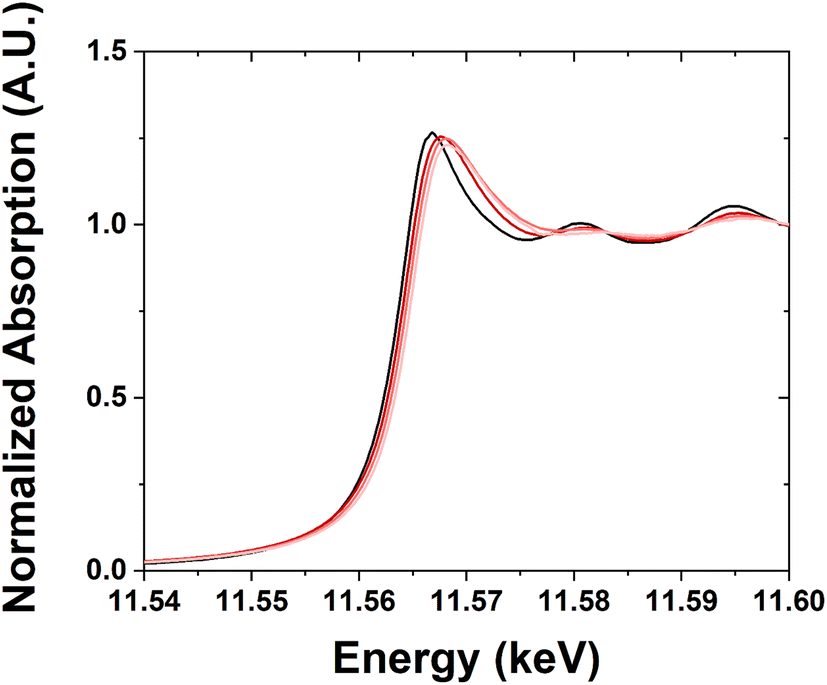 | ||
| Fig. 4 Pt LIII XANES for samples with Pt nanoparticles of different sizes. Pt foil (black), large sample (dark red), medium sample (moderate red), and small sample (light red). | ||
The standard Pt foil reference was scanned periodically throughout the experiment, without gas or temperature treatment, to ensure the energy calibration of the beamline setup (Fig. 5a). The spectra for this sample are identical within ±0.1 eV. The spectra were normalized using the peak maximum and the baseline. The peaks are slightly skewed toward having a higher density at a higher energy level.
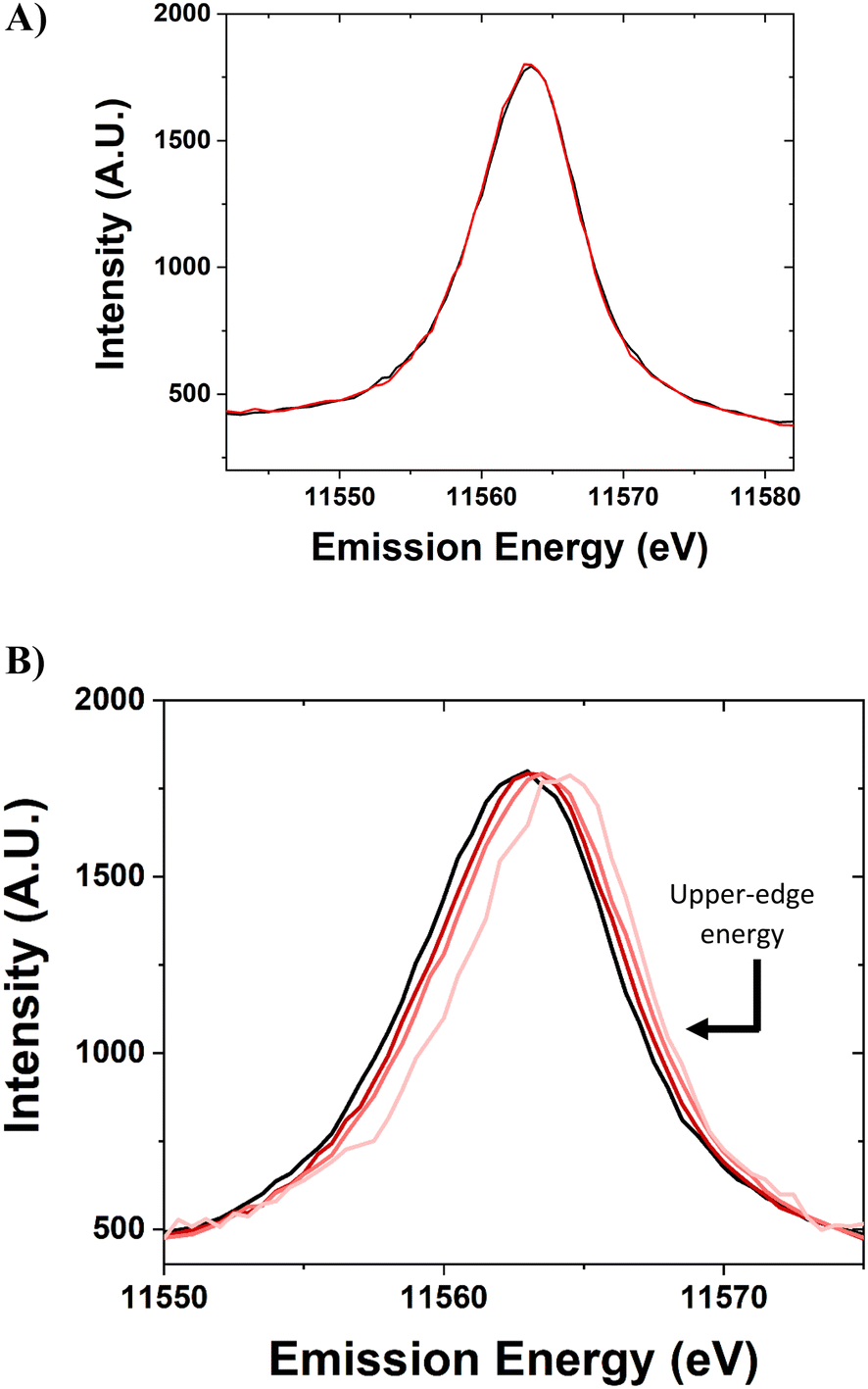 | ||
| Fig. 5 A) Day #1 (black) and day #3 (red) VtC-XES calibration spectra. B) VtC-XES spectra for samples with Pt nanoparticles of different sizes. Pt foil (black), large sample (dark red), medium sample (moderate red), and small sample (light red). | ||
The X-ray emission spectroscopy setup at the 8-ID beamline at the NSLS-II has the capability to detect the Lβ5 VtC-XES spectra for 5d nanomaterials and Pt foil (Fig. 5b). The Pt nanoparticle samples were scanned for VtC-XES after being reduced in H2 at 250 °C. The VtC-XES spectra show a systematic increase in emission energy with decreasing nanoparticle size. The spectrum is analogous to a density of states (5d-DOS) distribution and occurs at a similar energy (∼11565 eV) compared to previous study of Pt compounds.12 In this case, the VtC-XES features at higher energy are closer to the Fermi level. Two metrics have been previously employed to assess the spectra: the d-band center and the full width at half maximum. The d-band center is defined as the first moment of the area underneath the distribution (relative to the baseline).16 The full width at half maximum (FWHM, relative to baseline) intensity is a measure of the broadness of the spectra. However, as evidenced in Fig. 5b, the spectra become increasingly shifted towards the Fermi level for smaller nanoparticles (i.e., shifted to higher energies). Thus, a third term to describe VtC-XES spectra is the upper-edge energy. The proposed definition of the upper-edge energy is the energy of the right edge (high energy) of the VtC-XES spectrum at half maximum intensity (where the FWHM was determined). For an approximately Gaussian distribution, this would represent a point where roughly 10% of the density has higher energy, which provides a reasonable estimate for the highest occupied molecular orbitals (HOMO) involved in metal-adsorbate binding and catalytic transformations.
The metrics for VtC-XES are listed in Table 3 along with the XANES edge energy. The FWHM of the small sample has a slightly narrower XES spectrum compared to the other samples, which has been observed computationally for the d-DOS of small Pt nanoparticles.21 The FWHM metric value of ∼7 to 8 eV is similar compared to computational predictions for the 5d-DOS of Pt, computational predictions of the VtC-XES spectra (Fig. S2†), and experimental predictions using VB-XPS (∼6 eV for similarly sized Pt nanoparticles).13,15,17,18,20
| Sample | Upper-edge energy (eV) | d-Band center energy (eV) | FWHM (eV) | XANES edge energy (eV) |
|---|---|---|---|---|
| Pt foil | 11566.6 |
11562.5 |
8.0 | 11564.4 |
| Large Pt NPs | 11567.0 |
11562.8 |
8.2 | 11564.6 |
| Medium Pt NPs | 11567.3 |
11563.1 |
8.0 | 11564.8 |
| Small Pt NPs | 11567.6 |
11563.6 |
7.4 | 11565.0 |
To probe the effect of changing nanoparticle size on the electronic structure and, specifically, the d-band center of Pt, density functional theory (DFT) calculations are performed on four octahedral nanoparticle clusters—Pt-38, Pt-79, Pt-201, and Pt-314 (Fig. 6a–d, where the number indicates the number of atoms in the cluster). These correspond to nanoparticles with sizes ranging from 0.8–2.0 nm. While these sizes are smaller compared to the experimental samples, the trends from our analyses are expected to be generalizable to larger nanoparticles. The dispersion for each nanoparticle is calculated as the ratio of the number of atoms with a coordination number less than 12 (bulk FCC coordination) to the total number of atoms. Further, the d-band center is calculated by evaluating the first moment of the projected density of 5d states for all atoms in the nanoparticle. There is a linear relationship between the d-band center and dispersion (Fig. 6e) and between the upper-edge and the dispersion (Fig. S3†). Additionally, the intercept of linear relation can be interpreted as the d-band center of a bulk Pt atom in the limiting case of zero dispersion.
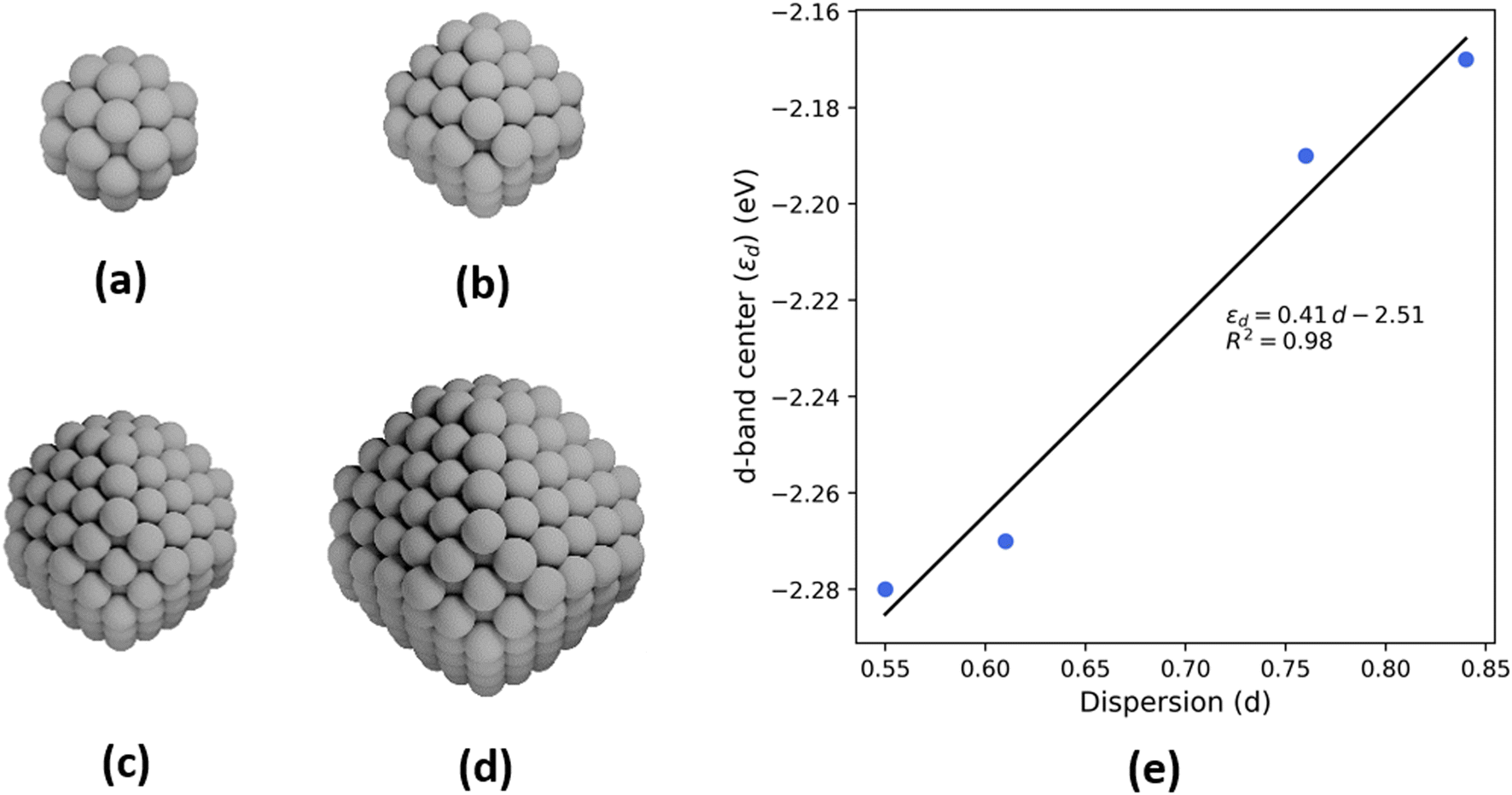 | ||
| Fig. 6 Schematics of octahedral nanoparticle clusters in the order of decreasing dispersion (a) Pt-38, (b) Pt-79, (c) Pt-201, (d) Pt-314. (e) Plot of d-band centers of the nanoparticle clusters against dispersion. A linear fit is also shown along with the slope (0.41), intercept (−2.51 eV), and coefficient of determination (R2 = 0.98). | ||
4. Discussion
Both the XANES energy shift (Fig. 4) and the systematic decrease in FT magnitude of the first three EXAFS oscillations (Fig. 3) are characteristic of metallic Pt nanoparticles with decreasing size.8,10,19 The white line intensity of the XANES, defined as the intense absorption above the normalized value of 1 in the near edge, is the same or slightly lower for all the samples compared to the Pt foil, which is typical of Pt nanoparticles.8 As the energy of the 5d orbitals changes with size, these states partially mix with the 6s and 6p orbitals leading to changes in the white line intensity and the shape of the XANES.45 Additionally, there is no evidence in the XANES of Pt2+ or Pt4+ from PtO or PtO2 phases, respectively, which would manifest as a higher white line intensity. Fig. 3 shows a reasonable agreement based on previous literature for metallic Pt nanoparticles and the dispersion determined from EXAFS fitting or STEM.10 Therefore, the method used to calculate dispersion is a reasonable estimate for the proportion of fully coordinated (bulk) Pt atoms to undercoordinated (surface) Pt atoms in the nanoparticle samples. Because XANES corresponds with the Pt LIII edge, it represents the transition of an electron from a 2p3/2 to an unfilled 5d3/2 or 5d5/2 state, meaning that it can be used to assess properties about the valence electronic structure of Pt. Specifically, it can be used to quantify the average energy of the Pt unfilled 5d states.14,15 Therefore, with decreasing nanoparticle size, the average energy of the unfilled 5d states increases.While XANES yields information about the unfilled 5d states, VtC-XES yields information about the filled 5d states.61 The narrowing of the d-DOS (Fig. 5b) has also been observed for nanoparticles with decreasing size using high resolution VB-XPS, and the study goes on to attribute the narrowing to the hybridization of fewer wave functions in smaller Pt nanoparticles.20,62 This could also be due to the narrower nanoparticle size distribution (determined from the STEM imagery). The d-band center shift assessed is also similar to the shift from VB-XPS. Going from nanoparticles of 5.3 nm to 2.4 nm in diameter, there is an increase in d-band center of 0.4 eV for VB-XPS.20 In this study, going from nanoparticles 5.1 nm to 2.1 nm in diameter, there is a comparable increase in d-band center of 0.8 eV. Slight differences may be attributed to differences in the oxidation state of each material. While all Pt atoms in this in situ study are metallic (Pt0), the Pt atoms in ex situ VB-XPS located near the surface of the nanoparticles may be oxidized, leading to a different electronic environment with Pt–O bonds as opposed to only Pt–Pt bonds.20,47,62 The FWHM width of the VtC-XES peaks is approximately 8 eV, which is similar to the FWHM (6 to 7 eV) for Pt nanoparticles of similar size.20 The same study witnesses a similar effect of spectral narrowing for the small sample (∼0.7 eV narrower than the other samples when considering FWHM).20 The experimental and calculated VtC-XES spectra (Fig. 5b and S2†) have a very similar FWHM. There are a few reasons that experimental spectra may appear broader than computational DOS calculations. VtC-XES inherently experiences core-hole broadening, due to the short, finite lifetime of the core hole created by the initial excitation, as well as broadening due to the energy resolution of the spectrometer which was estimated to be 1.9 eV (i.e., ±0.85 eV) using elastic scattering. Other broadening may result from the distribution of nanoparticle sizes. Still other uncertainty may arise from the fact that supported Pt nanoparticles are not all perfectly spherical49 and consequently the number of surface Pt atoms with different coordination numbers can vary.
Fig. 7a and b show the VtC-XES d-band center as well as the energy of the upper-edge of the 5d DOS linearly increased with increasing dispersion. There is a strong correlation in both cases, reporting an R-value of 0.981 and 0.999 respectively. In other words, the proportion of surface atoms to bulk atoms is directly proportional to the shift in the upper-edge energy of the d-DOS. Computationally, similar shifts in the d-band center to higher energy for smaller nanoparticles have been predicted.21 A previous study using computational FEFF modeling for Au nanoparticles of different sizes reports a narrower d band in small Au nanoparticles, with the d-DOS being shifted toward the Fermi level for the smallest nanoparticles.45 This study shows a similar trend for Pt, where the spectrum narrows and shifts closer to the Fermi level. Using VB-XPS, similar shifts of the d-band center to higher energy have been observed for nanoparticles of decreasing size. For example, it is reported that for Pt nanoparticles of 5.3 nm and 2.4 nm average diameter, there is a d-band center increase in energy of ∼0.4 eV.20 This shift corresponds to the d-band energy increase observed from the large sample to the medium/small samples of between 0.3 and 0.8 eV.
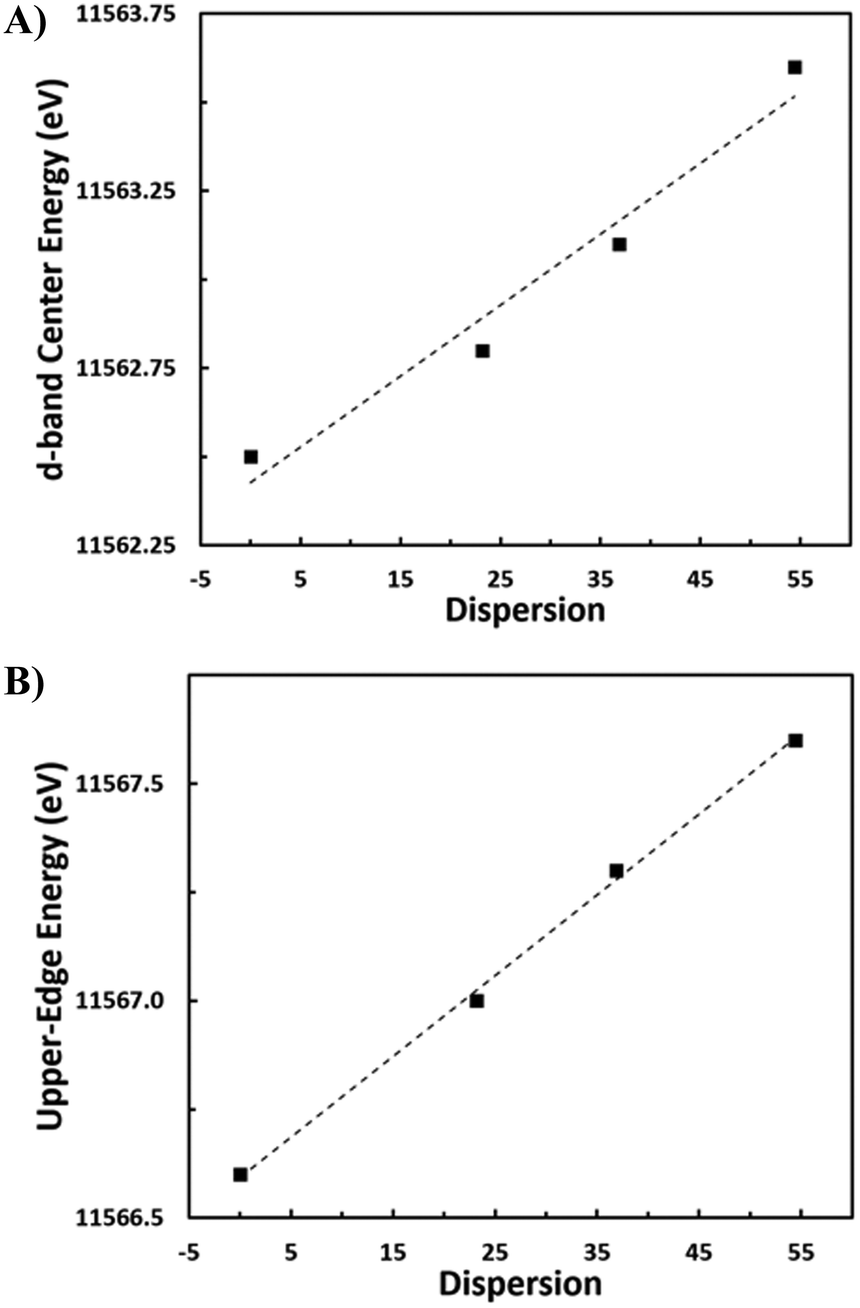 | ||
| Fig. 7 A) d-Band center energy versus nanoparticle dispersion. B) Upper-edge energy versus nanoparticle dispersion. | ||
While the XANES (unfilled 5d states) and VtC-XES (filled 5d states) energies both shift to higher energies with decreasing nanoparticle size, this result is not generally observed. For example, for Pt alloys, the XANES and the XES (determined by RIXS) shift in opposite directions.14,15,25,61 Whereas XANES and RIXS determine the average energy of the filled states, VtC-XES determines the energy of the entire 5d filled states, which can now be determined directly under realistic in situ or in operando conditions.14,22,48 Pt VtC-XES can also be applied to Pt compounds where the filled and unfilled 5d states are more complex.
DFT calculations are employed in this study to examine the d-band centers of individual step, terrace, and bulk atoms in Pt-314 to elucidate the cause of the upshift in d-band center with increase in dispersion. In Fig. 8a, we plot the d-band centers of a row of bulk atoms along the [100] axis of the nanoparticle capped by terrace atoms on the short (100) facet. The d-band center first shifts downward and then slightly upward on moving from the (100) terrace to the center of the nanoparticle. The d-band center of the center bulk atoms is −2.51 eV, which is equal to the intercept of the linear relationship between d-band center and dispersion. This is consistent with the previous interpretation of the intercept. The overall difference between the d-band center of the terrace atom and the center bulk atom is 0.59 eV. Furthermore, in Fig. 8b, the d-band centers of a row of atoms on the (111) terrace capped by step atoms on either side is plotted. The d-band center shifts downward from the step edge to the center of the terrace, with the overall difference equaling 0.15 eV.
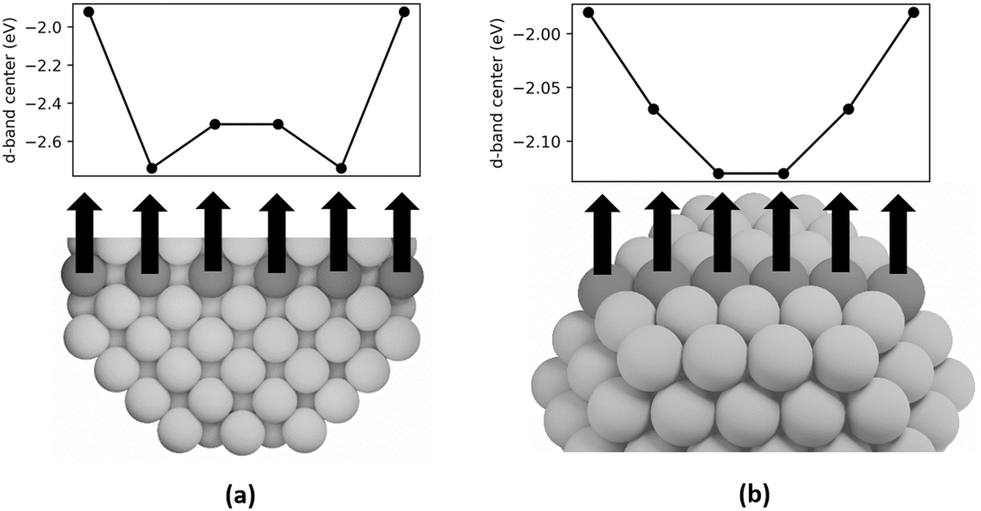 | ||
| Fig. 8 a) Plot of d-band center for each shaded atom along the cross-section of the Pt-314 nanoparticle. The first and last atoms are present on the terraces (CN = 9), and the remaining atoms are present in the bulk (CN = 12). b) Plot of d-band center for each shaded atom on the terrace of the Pt-314 nanoparticle. The first and last atoms correspond to undercoordinated steps (CN = 7) and the remaining atoms correspond to terrace atoms (CN = 9). | ||
The downshift in d-band center from the step to the terrace and from the terrace to the bulk can be rationalized based on coordination. Nørskov and coworkers54,55 have shown that the d-band center shifts to a lower energy with an increase in the coordination number for single crystal facets of Pt. Moreover, the downshift in d-band center of the subsurface atoms closer to the terrace compared to the center bulk atoms can be explained by the presence of compressive strain at the surface. Many prior studies have shown that strain can lead to shifts in the d-band center, with compressive strain leading to a downshift and tensile strain leading to an upshift.54,56–59 Since subsurface atoms have the same coordination as the center bulk atoms, the coordination effect should be negligible. However, since the subsurface atoms have contracted bonds near the surface (see Fig. S1 and Tables S1–S3), their d-band center is lower than the bulk atoms. To summarize, since an increase in dispersion corresponds to an increase in the fraction of step and terrace atoms that show higher d-band centers compared to bulk atoms, there is an overall increase in the averaged d-band center of the nanoparticle. The projected density of states is plotted in Fig. 9, which is smoothed using Hanning smoothing as implemented in SciPy.53 The nature of the states transforms from discrete-like to continuous as the nanoparticle size (i.e., the number of atoms) increases.
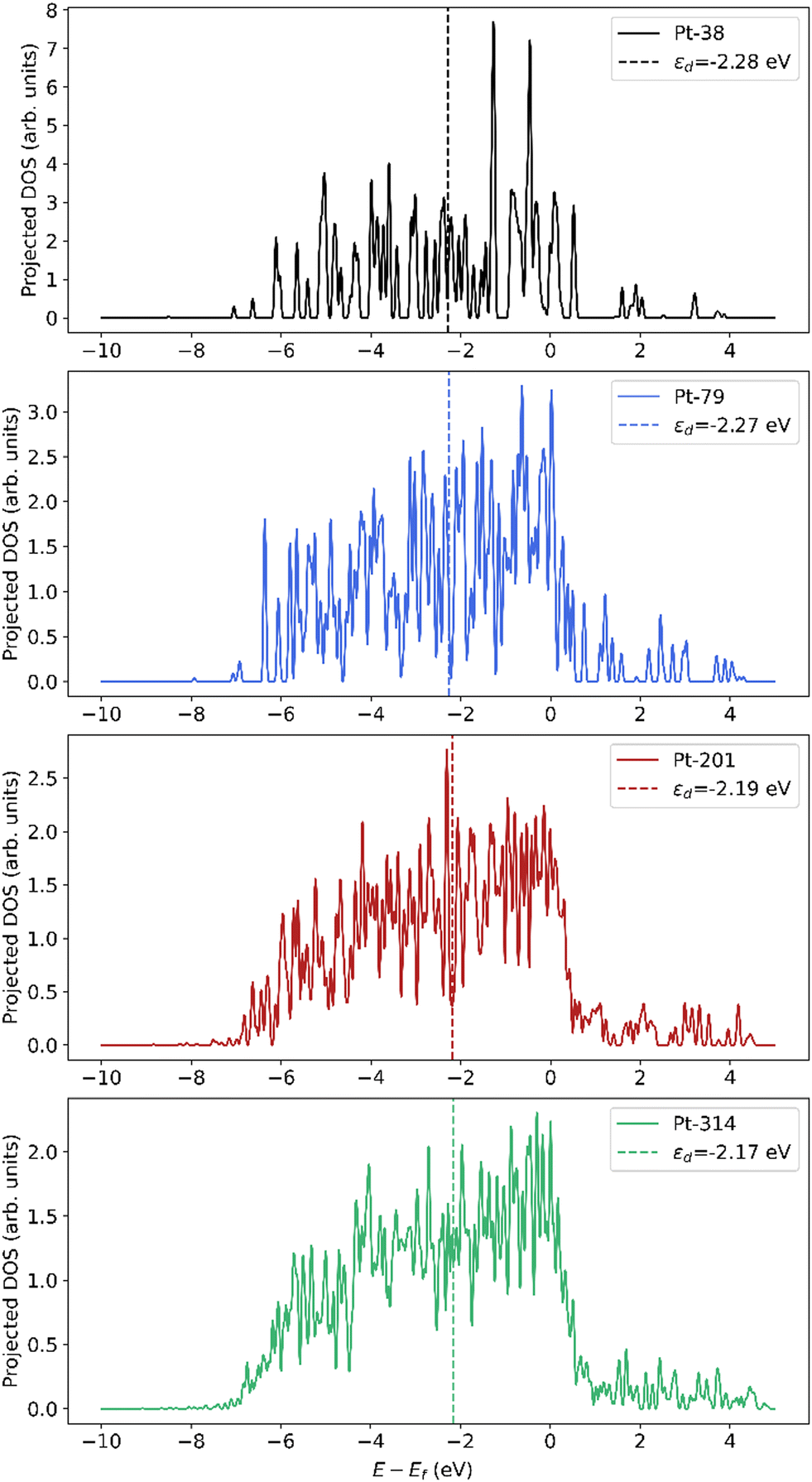 | ||
| Fig. 9 Smoothed projected density of states (pDOS) of Pt atoms in nanoparticles of different sizes. The pDOS is normalized by the number of atoms. | ||
An estimation of calculated VtC-XES spectra was conducted based on the methodology recently used in a study by Bukowski et al.61 The methodology modifies a calculation of the intensity of RIXS planes to apply to VtC-XES (ESI†) on simulated octahedral Pt nanoparticles. The results determine that the peaks skew toward the Fermi level (higher energy) with decreasing nanoparticle size and increased dispersion (Fig. S2†). This manifests in the increased peak position (Table S4†) as well as an increased upper-edge energy (Table S6†) with decreasing nanoparticle size. This trend is the same as for the pDOS in Fig. 9. Simultaneously, the peaks appear to narrow as given by the FWHM (Table S5†). The result is that the nanoparticle dispersion and the calculated upper edge energy is highly correlated (R = 0.99), which matches the result from Fig. 7b. It is important to note that the calculated VtC-XES intensities have smaller changes compared to experimental intensities because the range of nanoparticle sizes for which DFT calculations are performed is smaller than the range of sizes used for the experimental study.
The increase in d-band center energy, which represents the filled Pt 5d states as determined using VtC-XES and DFT, is accompanied by an increase in the average energy of the XANES, which represents the unfilled Pt 5d states. Both the bonding (VtC-XES) and antibonding (XANES) orbitals shift to a higher energy with decreasing nanoparticle size. Changes in the d-DOS are directly related to the catalytic properties of a catalyst material.13,46 By measuring the energy of the 5d valence states, which are important for bond activation and metal-adsorbate heats of formation, in situ or in operando VtC-XES can directly correlate electronic structure with catalyst performance. While this study will not draw conclusions for a particular catalyst system, the VtC-XES technique can hopefully contribute to catalyst design in other systems. As an example, an increase in the average energy of the filled orbitals may help explain an order of magnitude increase in coking rate for high temperature alkane dehydrogenation with decreasing nanoparticle size.40 As the energy of the orbitals increases, Pt forms stronger bonds with adsorbates, making it more difficult for species to desorb, which leads to the formation of deeply dehydrogenated species.14,15,60 Just as nanoparticle size can impact selectivity, it can also affect the rate of reaction by more than an order of magnitude.20 VtC-XES can also potentially be used in tandem with DFT calculations as experimental validation for computationally-determined heats of formation; consequently, VtC-XES can help refine calculations to be more accurate and reliable. VtC-XES is an accessible technique that can provide useful information about the electronic structure of a specific element in a catalyst to drive the discovery of future catalyst materials.
5. Conclusions
This study has synthesized and characterized a series of Pt nanoparticle samples supported on SiO2 typical of a Pt catalyst material. Using TEM and EXAFS, nanoparticles ranging from 2 nm to 5 nm were quantified with a dispersion ranging from 0% (Pt foil) 50% (small nanoparticle sample). It was found, using VtC-XES at the BNL NSLS-II 8-ID beamline, that the d-band center and upper-edge (near – HOMO) energy both shift to higher energy with decreasing nanoparticle size. The assessed changes correlate well with the proportion of surface atoms to bulk atoms, pointing to the conclusion that undercoordinated Pt atoms have a different electronic structure than bulk atoms. Differences in the catalyst metal's electronic structure may affect rates and selectivity in reaction systems by dictating the adsorption/desorption of reactants and products, thereby emphasizing certain pathways or mitigating others. Developments in synchrotron and X-ray detector technology have enabled the resolution of small changes to the d-DOS of Pt atoms in a dilute catalyst material Pt. VtC-XES is a capable technique that is element-specific, can be used for in situ or in operando experiments, and can be used to explain and predict performance of catalyst sites for a variety of reactions. The technique is becoming increasingly available to users at BNL and across the world and hopes to enhance catalyst characterization capabilities to obtain unparalleled insights into catalyst materials.Author contributions
David P. Dean: Writing – Original Draft; Writing – Review & Editing; Formal Analysis. Gaurav S. Deshmukh: Writing – Original Draft; Writing – Review & Editing; Software. Christopher K. Russell: Writing – Review & Editing. Kuixin Zhu: Writing – Review & Editing. Christina W. Li: Investigation. Jeffrey P. Greeley: Investigation. Denis Leshchev: Investigation; Conceptualization; Data Curation; Writing – Review & Editing. Eli Stavitski: Investigation; Conceptualization; Data Curation. Jeffrey T. Miller: Investigation; Writing – Review & Editing; Conceptualization.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research used resources at the 8-ID Beamline of the National Synchrotron Light Source II, a US Department of Energy Office of Science User Facility operated by Brookhaven National Laboratory under contract no. DE-SC0012704. D. P. D., C. K. R., K. Z., C. W. L., and J. T. M were funded in part by the National Science Foundation (NSF) under Cooperative Agreement No. EEC-1647722. G. S. D. and J. P. G. acknowledge the United States Department of Energy through the Office of Science, Office of Basic Energy Sciences (BES), Chemical, Biological, and Geosciences Division, Data Science Initiative, grant DE-SC0010379. Use of the Center for Nanoscale Materials, U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences User Facility under Contract No. DE-AC02-06CH11357, and of the computational resources from the National Energy Research Scientific Computing Center, is also acknowledged. K. Z. and C. W. L. would like to acknowledge funding from the National Science Foundation grant CHE-2106450.References
- M. Filez, E. A. Redekop, J. Dendooven, R. K. Ramachandran, E. Solano, U. Olsbye, B. M. Weckhuysen, V. V. Galvita, H. Poelman, C. Detavernier and G. B. Marin, Formation and functioning of bimetallic nanocatalysts: the power of X-ray probes, Angew. Chem., Int. Ed., 2019, 58, 13220–13230 CrossRef CAS PubMed.
- P. Zimmermann, S. Peredkov, P. M. Abdala, S. DeBeer, M. Tromp, C. Müller and J. A. van Bokhoven, Modern X-ray spectroscopy: XAS and XES in the laboratory, Coord. Chem. Rev., 2020, 423, 213466 CrossRef CAS.
- D. Leshchev, M. Rakitin, B. Luvizotto, R. Kadyrov, B. Ravel, K. Attenkofer and E. Stavitski, The Inner Shell Spectroscopy beamline at NSLS-II: a facility for in situ and operando X-ray absorption spectroscopy for materials research, J. Synchrotron Radiat., 2022, 29, 1095–1106 CrossRef CAS PubMed.
- T. Ressler, WinXAS: a program for X-ray absorption spectroscopy data analysis under MS-Windows, J. Synchrotron Radiat., 1998, 5, 118–122 CrossRef CAS PubMed.
- P. Glatzel, M. Sikora, G. Smolentsev and M. Fernández-García, Hard X-ray photon-in photon-out spectroscopy, Catal. Today, 2009, 145, 294–299 CrossRef CAS.
- L. J. P. Ament, M. van Veenendaal, T. P. Devereaux, J. P. Hill and J. van den Brink, Resonant inelastic X-ray scattering studies of elementary reactions, Rev. Mod. Phys., 2011, 83(2), 705–767 CrossRef CAS.
- J. Singh, C. Lamberti and J. A. van Bokhoven, Advanced X-ray absorption and emission spectroscopy: in situ catalytic studies, Chem. Soc. Rev., 2010, 39, 4754–4766 RSC.
- Y. Lei, J. Jelic, L. C. Nitsche, R. Meyer and J. T. Miller, Effect of particle size and adsorbates on the L3, L2, and L1 X-ray absorption near edge structure of supported Pt nanoparticles, Top. Catal., 2011, 54, 334–348 CrossRef CAS.
- C. A. Schneider, W. S. Resband and K. W. Eliceiri, NIH Image to ImageJ: 25 years of image analysis, Nat. Methods, 2012, 9(7), 671–675 CrossRef CAS PubMed.
- J. T. Miller, A. J. Kropf, Y. Zha, J. R. Regalbuto, L. Delannoy, C. Louis, E. Bus and J. A. van Bokhoven, The effect of gold particle size on Au-Au bond length and reactivity toward oxygen in supported catalysts, J. Catal., 2006, 240, 222–234 CrossRef CAS.
- D. Vollath, Nanoparticles-nanocomposites-nanomaterials: an introduction for beginners, Wiley-VCH Verlag GmbH & Co., Weinheim, Germany, 2013 Search PubMed.
- V. V. Pryadchenko, V. V. Srabionyan, L. A. Avakyan, J. A. van Bokhoven and L. A. Bugaev, Electronic structure of Pt and Au compounds measured by X-ray emission and X-ray absorption spectroscopy, J. Phys. Chem. C, 2012, 116, 25790–25796 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Electronic factors determining the reactivity of metal surfaces, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- S. C. Purdy, P. Ghanekar, G. Mitchell, A. J. Kropf, D. Y. Zemlyanov, Y. Ren, F. Riveiro, W. N. Delgass, J. Greeley and J. T. Miller, Origin of electronic modification of platinum in a Pt3V alloy and its consequences for propane dehydrogenation catalysis, ACS Appl. Energy Mater., 2020, 3, 1410–1422 CrossRef CAS.
- V. J. Cybulskis, B. C. Bukowski, H. Tseng, J. R. Gallagher, Z. Wu, E. Wegener, A. J. Kropf, B. Ravel, F. H. Ribeiro, J. Greeley and J. T. Miller, Zinc promotion of platinum for catalytic light alkane dehydrogenation: insights into geometric and electronic effects, ACS Catal., 2017, 7, 4173–4181 CrossRef CAS.
- C. Kittel, Introduction to solid state physics, John Wiley & Sons, 8th edn, 2004 Search PubMed.
- M. Yang, Y. Zhu, X. Zhou, Z. Sui and D. Chen, First-principles calculations of propane dehydrogenation over PtSn catalysts, ACS Catal., 2012, 2, 1247–1258 CrossRef CAS.
- N. V. Smith, G. K. Wertheim, S. Hüfner and M. M. Traum, Photoemission spectra and band structures of d-band metals. IV. X-ray photoemission spectra and densities of states in Rh, Pd, Ag, Ir, Pt, and Au.
- F. Behafarid, L. K. Ono, S. Mostafa, J. R. Croy, G. Shafai, S. Hong, T. S. Rahman, S. R. Bare and B. R. Cuenya, Electronic properties and charge transfer phenomena in Pt nanoparticles on γ-Al2O3: size, shape, support, and adsorbate effects.
- L. Bai, X. Wang, Q. Chen, Y. Ye, H. Zheng, J. Guo, Y. Yin and C. Gao, Explaining the size dependence in Platinum-nanoparticle-catalyzed hydrogenation reactions, Angew. Chem., Int. Ed., 2016, 55, 15656–15661 CrossRef CAS PubMed.
- Y. Sun, L. Zhuang, J. Lu, X. Hong and P. Liu, Collapse in crystalline structure and decline in catalytic activity of Pt nanoparticles on reducing particle size to 1 nm, J. Am. Chem. Soc., 2007, 129, 15465–15467 CrossRef CAS PubMed.
- P. Glatzel and U. Bergmann, High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes – electronic and structural information, Coord. Chem. Rev., 2005, 249, 65–95 CrossRef CAS.
- J. Kim, D. Casa, M. H. Upton, T. Gog, Y. Kim, J. F. Mitchell, M. van Veenendaal, M. Daghofer, J. van den Brink, G. Khaliullin and B. J. Kim, Magnetic excitation spectra of Sr2IrO4 probed by resonant inelastic X-ray scattering: establishing links to cuprate superconductors, Phys. Rev. Lett., 2012, 108, 177003 CrossRef PubMed.
- P. Glatzel, J. Singh, K. O. Kvashnina and J. A. van Bokhoven, In situ characterization of the 5d density of states of Pt nanoparticles upon adsorption of CO, J. Am. Chem. Soc., 2010, 132, 2555–2557 CrossRef CAS PubMed.
- E. C. Wegener, B. C. Bukowski, D. Yang, Z. Wu, A. J. Kropf, W. N. Delgass, J. Greeley, G. Zhang and J. T. Miller, Intermetallic compounds as an alternative to single-atom alloy catalysts: geometric and electronic structures from advanced X-ray spectroscopies and computational studies, ChemCatChem, 2020, 12, 1325–1333 CrossRef CAS.
- K. Kobayashi, Hard X-ray photoemission spectroscopy, Nucl. Instrum. Methods Phys. Res., 2009, 601, 32–47 CrossRef CAS.
- G. Smolentsev, A. V. Soldatov, J. Messinger, K. Merz, T. Weyhermüller, U. Bergmann, Y. Pushkar, J. Yano, V. K. Yachandra and P. Glatzel, X-ray emission spectroscopy to study ligand valence orbitals in Mn coordination complexes, J. Am. Chem. Soc., 2009, 131, 13161–13167 CrossRef CAS PubMed.
- M. Bauer, HERFD-XAS and valence-to-core-XES: new tools to push the limits in research with hard X-rays?, Phys. Chem. Chem. Phys., 2014, 16, 13827–13837 RSC.
- C. J. Pollock and S. DeBeer, Valence-to-core X-ray emission spectroscopy: a sensitive probe of the nature of a bound ligand, J. Am. Chem. Soc., 2011, 133, 5594–5601 CrossRef CAS PubMed.
- A. Boubnov, H. W. P. Carvalho, D. E. Doronkin, T. Günter, E. Gallo, A. J. Atkins, C. R. Jacob and J. Grunwaldt, Selective catalytic reduction of NO over Fe-ZSM-5: mechanistic insights by operando HERFD-XANES and valence-to-core X-ray emission spectroscopy, J. Am. Chem. Soc., 2014, 136(37), 13006–13015 CrossRef CAS PubMed.
- V. A. Safonov, L. N. Vykhodtseva, Y. M. Polukarov, O. V. Safonova, G. Smolentsev, M. Sikora, S. G. Eeckhout and P. Glatzel, Valence-to-core X-ray emission spectroscopy identificaiton of carbide compounds in nanocrystalline Cr coatings deposited from Cr(III) electrolytes containing organic substances, J. Phys. Chem. B, 2006, 110, 23192–23196 CrossRef CAS PubMed.
- R. Kopelent, J. A. van Bokhoven, M. Nachtegaal, J. Szlachetko and O. V. Safonova, X-ray emission spectroscopy: highly sensitive techniques for time-resolved probing of cerium reactivity under catalytic conditions, Phys. Chem. Chem. Phys., 2016, 18, 32486 RSC.
- Z. Bayindir, P. N. Duchesne, S. C. Cook, M. A. MacDonald and P. Zhang, X-ray spectroscopy studies on the surface structural characteristics and electronic properties of platinum nanoparticles, J. Chem. Phys., 2009, 131, 244716 CrossRef CAS PubMed.
- J. Chen, Y. Z. Finfrock, Z. Wang and T. Sham, Strain and ligand effects in Pt-Ni alloys studies by valence-to-core X-ray emission spectroscopy, Sci. Rep., 2021, 11, 13698 CrossRef CAS PubMed.
- B. L. Geoghegan, Y. Liu, S. Peredkov, S. Dechert, F. Meyer, S. DeBeer and G. E. Cutsail III, Combining valence-to-core X-ray emission and Cu K-edge X-ray absorption spectroscopies to experimentally assess oxidation state of organometallic Cu(I)/(II)/(III) complexes, J. Am. Chem. Soc., 2022, 144, 2520–2534 CrossRef CAS PubMed.
- F. Giordanino, E. Borfecchia, K. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. Soldatov, P. Beato, S. Bordiga and C. Lamberti, Interaction of NH3 with Cu-SSZ-13 catalyst: a complementary FTIR, XANES, and XES study, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS PubMed.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Catalytic dehydrogenation of light alkanes on metals and metal oxides, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- N. M. Marković, T. J. Schmidt, V. Stamenković and P. N. Ross, Oxygen reduction reaction on Pt and Pt bimetallic surfaces: a selective review, Fuel Cells, 2001, 1(2), 105–116 CrossRef.
- C. Garino, E. Borfecchia, R. Gobetto, J. A. van Bokhoven and C. Lamberti, Determination of the electronic and structural configuration of coordination compounds by synhcrotron-radiation techniques, Coord. Chem. Rev., 2014, 277–278, 130–186 CrossRef CAS.
- J. Zhu, M. Yang, Y. Yu, Y. Zhu, Z. Sui, X. Zhou, A. Holmen and D. Chen, Size-dependent reaction mechanism and kinetics for propane dehydrogenation over Pt catalysts, ACS Catal., 2015, 5, 6310–6319 CrossRef CAS.
- Y. Zhou, D. E. Doronkin, M. Chen, S. Wei and J. Grunwaldt, Interplay of Pt and crystal facets of TiO2: CO oxidation activity and operando XAS/DRIFTS studies, ACS Catal., 2016, 6, 7799–7809 CrossRef CAS.
- B. Ravel, A. J. Kropf, D. Yang, M. Wang, M. Topsakal, D. Lu, M. C. Stennett and N. C. Hyatt, Nonresonant valence-to-core X-ray emission spectroscopy of niobium, Phys. Rev. B, 2018, 97, 125139 CrossRef CAS PubMed.
- C. Garino, E. Gallo, N. Smolentsev, P. Glatzel, R. Gobetto, C. Lamberti, P. J. Sadler and L. Salassa, Resonant X-ray emission spectroscopy reveals d-d ligand-field states involved in the self-assembly of a square-planar platinum complex, Phys. Chem. Chem. Phys., 2012, 14, 15278–15281 RSC.
- S. Kaya, D. Friebel, H. Ogasawara, T. Anniyev and A. Nilsson, Electronic structure effects in catalysis probed by X-ray and electron spectroscopy, J. Electron Spectrosc. Relat. Phenom., 2013, 190, 113–124 CrossRef CAS.
- J. A. Van Bokhoven and J. T. Miller, d electron density and reactivity of the d band as a function of particle size in supported gold catalysts, J. Phys. Chem. C, 2007, 9245–9249 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Theoretical surface science and catalysis – calculations and concepts, Adv. Catal., 2000, 45, 71–129 CAS.
- N. J. LiBretto, C. Yang, Y. Ren, G. Zhang and J. T. Miller, Identification of surface structures in Pt3Cr intermetallic nanocatalysts, Chem. Mater., 2019, 1597–1609 CrossRef CAS.
- J. Singh, R. C. Nelson, B. C. Vicente, S. L. Scott and J. A. van Bokhoven, Electronic structure of alumina-supported monometallic Pt and bimetallic PtSn catalysts under hydrogen and carbon monoxide environment, Phys. Chem. Chem. Phys., 2010, 12, 5668–5677 RSC.
- S. B. Simonsen, I. Chorkendorff, S. Dahl, M. Skoglundh, K. Meinander, T. N. Jensen, J. V. Lauritsen and S. Helveg, Effect of particle morphology on the ripening of supported Pt nanoparticles, J. Phys. Chem. C, 2012, 116(9), 5646–5653 CrossRef CAS.
- H. A. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, The atomic simulation environment – a Python library for working with atoms, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- G. Kresse and J. Furthmü, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser, J. Bright, S. J. van der Walt, M. Brett, J. Wilson, K. J. Millman, N. Mayorov, A. R. J. Nelson, E. Jones, R. Kern, E. Larson, C. J. Carey, İ. Polat, Y. Feng, E. W. Moore, J. VanderPlas, D. Laxalde, J. Perktold, R. Cimrman, I. Henriksen, E. A. Quintero, C. R. Harris, A. M. Archibald, A. H. Ribeiro, F. Pedregosa, P. van Mulbregt, A. Vijaykumar, A. P. Bardelli, A. Rothberg, A. Hilboll, A. Kloeckner, A. Scopatz, A. Lee, A. Rokem, C. N. Woods, C. Fulton, C. Masson, C. Häggström, C. Fitzgerald, D. A. Nicholson, D. R. Hagen, D. V. Pasechnik, E. Olivetti, E. Martin, E. Wieser, F. Silva, F. Lenders, F. Wilhelm, G. Young, G. A. Price, G. L. Ingold, G. E. Allen, G. R. Lee, H. Audren, I. Probst, J. P. Dietrich, J. Silterra, J. T. Webber, J. Slavič, J. Nothman, J. Buchner, J. Kulick, J. L. Schönberger, J. V. de Miranda Cardoso, J. Reimer, J. Harrington, J. L. C. Rodríguez, J. Nunez-Iglesias, J. Kuczynski, K. Tritz, M. Thoma, M. Newville, M. Kümmerer, M. Bolingbroke, M. Tartre, M. Pak, N. J. Smith, N. Nowaczyk, N. Shebanov, O. Pavlyk, P. A. Brodtkorb, P. Lee, R. T. McGibbon, R. Feldbauer, S. Lewis, S. Tygier, S. Sievert, S. Vigna, S. Peterson, S. More, T. Pudlik, T. Oshima, T. J. Pingel, T. P. Robitaille, T. Spura, T. R. Jones, T. Cera, T. Leslie, T. Zito, T. Krauss, U. Upadhyay, Y. O. Halchenko and Y. Vázquez-Baeza, SciPy 1.0: fundamental algorithms for scientific computing in Python, Nat. Methods, 2020, 17, 261–272 CrossRef CAS PubMed.
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Effect of strain on the reactivity of metal surfaces, Phys. Rev. Lett., 1998, 81, 2819 CrossRef.
- T. Jiang, D. J. Mowbray, S. Dobrin, H. Falsig, B. Hvolbœk, T. Bligaard and J. K. Nørskov, Trends in CO oxidation rates for metal nanoparticles and close-packed, stepped, and kinked surfaces, J. Phys. Chem. C, 2009, 113(24), 10548–10553 CrossRef CAS.
- L. Wang, Z. Zeng, W. Gao, T. Maxson, D. Raciti, M. Giroux, X. Pan, C. Wang and J. P. Greeley, Tunable intrinsic strain in two-dimensional transition metal electrocatalysts, Science, 2019, 363(6429), 870–874 CrossRef CAS PubMed.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, Role of strain and ligand effects in the modification of the electronic and chemical properties of bimetallic surfaces, Phys. Rev. Lett., 2004, 93, 156801 CrossRef CAS PubMed.
- F. Abild-Pedersen, J. P. Greeley and J. K. Nørskov, Understanding the effect of steps, strain, poisons, and alloying: methane activation on Ni surfaces, Catal. Lett., 2005, 105, 9–13 CrossRef CAS.
- P. Strasser, S. Koh, T. Anniyev, J. P. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Lattice-strain control of the activity in dealloyed core-shell fuel cell catalysts, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- R. Alcala, D. P. Dean, I. Chavan, C. Chang, B. Burnside, H. N. Pham, E. Peterson, J. T. Miller and A. K. Datye, Strategies for regeneration of Pt-alloy catalysts supported on silica for propane dehydrogenation, Appl. Catal., A, 2023, 658, 119157 CrossRef CAS.
- B. C. Bukowski, S. C. Purdy, E. C. Wegener, Z. Wu, A. J. Kropf, G. Zhang, J. T. Miller and J. Greeley, Intermetallic Alloy Structure-Activity Descriptors Derived from Inelastic X-ray Scattering, Phys. Chem. Chem. Phys., 2023, 25, 11216–11226 RSC.
- L. Bai, S. Zhang, Q. Chen and C. Gao, Synthesis of Ultrasmall Platinum Nanoparticles on Polymer Nanoshells for Size-Dependent Catalytic Oxidation Reactions, ACS Appl. Mater. Interfaces, 2017, 9, 9710–9717 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01554h |
| This journal is © The Royal Society of Chemistry 2024 |